

Abstract
Recent studies show a connection between glycolysis and inflammatory response in rheumatoid arthritis (RA) macrophages (MΦs) and fibroblasts (FLS). Yet, it is unclear which pathways could be targeted to rebalance RA MΦs and FLS metabolic reprogramming. To identify novel targets that could normalize RA metabolic reprogramming, TLR7-mediated immunometabolism was characterized in RA MΦs, FLS and experimental arthritis. We uncovered that GLUT1, HIF1α, cMYC, LDHA and lactate were responsible for the TLR7-potentiated metabolic rewiring in RA MΦs and FLS, which was negated by IRAKi. While in RA FLS, HK2 was uniquely expanded by TLR7 and negated by IRAK4i. Conversely, TLR7-driven hypermetabolism, non-oxidative PPP (CARKL) and oxidative phosphorylation (PPARγ) were narrowly dysregulated in TLR7-activated RA MΦs and FLS and was reversed by IRAK4i. Consistently, IRAK4i therapy disrupted arthritis mediated by miR-Let7b/TLR7 along with impairing a broad-range of glycolytic intermediates, GLUT1, HIF1α, cMYC, HK2, PFKFB3, PKM2, PDK1 and RAPTOR. Notably, inhibition of the mutually upregulated glycolytic metabolites, HIF1α and cMYC, was capable of mitigating TLR7-induced inflammatory imprint in RA MΦs and FLS. In keeping with IRAK4i, treatment with HIF1i and cMYCi intercepted TLR7-enhanced IRF5 and IRF7 in RA MΦs, distinct from RA FLS. Interestingly, in RA MΦs and FLS, IRAKi counteracted TLR7-induced CARKL reduction in line with HIF1i. Whereas, cMYCi in concordance with IRAK4i, overturned oxidative phosphorylation via PPARγ in TLR7-activated RA MΦs and FLS. The blockade of IRAK4 and its interconnected intermediates can rebalance the metabolic malfunction by obstructing glycolytic and inflammatory phenotypes in RA MΦs and FLS.
Keywords: RA macrophages, RA FLS, TLR7-induced arthritis, IRAK4, cMYC, HIF1α
Graphical Abstract
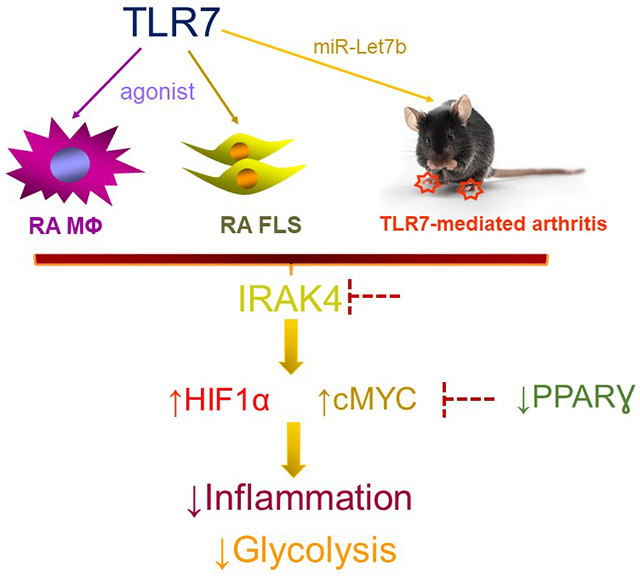
INTRODUCTION
Rheumatoid arthritis (RA) is an autoimmune inflammatory disease in which the crosstalk between joint myeloid cells and fibroblast-like synoviocytes (FLS) orchestrates pannus formation and bone erosion (McInnes and Schett, 2017). Increasing evidence links metabolic misregulation in the RA joint to chronically activated macrophages and FLS (Garcia-Carbonell et al., 2016, McGarry et al., 2017, McGarry et al., 2021). In RA synovium, CD68+ macrophages (MΦs) are characterized by mitochondrial misfunction and oxidative stress(Harty et al., 2012). Consistently, hypoxia-induced metabolic stress, LPS/IFNγ and IL-34 remodel joint myeloid cells into glycolytic MΦs along with a dysregulated TCA cycle(Krawczyk et al., 2010, Van Raemdonck et al., 2021, Xue et al., 2014). Pyruvate kinase M2 (PKM2) in MΦs, fuels glucose uptake and breakdown, resulting in lactate secretion (Palsson-McDermott et al., 2015). Synovial tissue MΦs reshape their milieu by oxygen and glucose reduction in coordination with lactate elevation (Zeisbrich et al., 2018). Hence glycolytic intermediates, function as bioenergy sensors that potentiate FLS migration, proliferation, and immunometabolism (Ahn et al., 2016, Falconer et al., 2018, Volchenkov et al., 2017). Additionally, hypoxia, oxidative stress, PDGF and TNF also participate in rewiring the metabolic profile of RA FLS towards glycolysis and the pentose phosphate pathway (PPP) and away from oxidative phosphorylation (Garcia-Carbonell et al., 2016). Therefore recent studies have explored the feasibility of rebalancing oxidative phosphorylation over glycolysis to mitigate RA MΦ and FLS immunopathology.
Interestingly, some of the anti-rheumatic compounds such as Methotrexate have been implicated in expanding Aminoimidazole-4-carboxamide ribonucleotide (AICAR)-induced AMPK, cultivating M2 MΦ differentiation while constraining FLS proliferation, intracellular oxidative stress and inflammatory cytokines (Biniecka et al., 2016, Huang et al., 2005, Palsson-McDermott and O’Neill, 2020, Phillips et al., 2003). TNFi treatment curtailed glycolytic intermediates (GLUT1, PKM2 & GAPDH) in RA synovium (Biniecka et al., 2016); while Tofacitinib had no impact on RA MΦs and FLS glycolytic activity but reversed oxidative phosphorylation in these cells (McGarry et al., 2018, Palasiewicz et al., 2021). Moreover Metformin or IACS, a complex1i was capable of restricting RA FLS glycolysis and amplifying RA MΦ and FLS oxidative phosphorylation (Chen et al., 2019, Chen et al., 2020). Another attractive anti-inflammatory compound that intercepts metabolism is dimethyl fumarate (DMF) that activates NRF2 and targets TLR-related IRAK4-MyD88 complex in MΦs (Zaro et al., 2019).
Toll-like receptors have been linked to changes in metabolism. Ligation of TLR2/4 galvanizes metabolic switch to glycolysis that is coupled with upregulation of GLUT1, PFK2/PFKBF3, lactate, iNOS, AKT/mTOR, HIF1α, and IKKε to support inflammation in myeloid cells (Geeraerts et al., 2017, Krawczyk et al., 2010, Murray et al., 2014, Qualls et al., 2012). While the function of TLR7/9 is less clear and has been described as a “double-edged sword”. Administration of TLR7 agonist, imiquimod, represses tumor antigen immune response and has shown to be more effective in combination with indoleamine 2,3-dioxygenase (IDO) inhibitor (Ito et al., 2015). Others have shown that TLR9-mediated type 1 IFN expression is responsible for escalating oxidative phosphorylation and fatty acid oxidation (FAO) in dendritic cells and AMPK inhibitor can block this function (Wu et al., 2016). In plasmacytoid dendritic cells, TLR7 and TLR9 escalate both glycolysis and lipid metabolism (Saas et al., 2017).
Recent studies have revealed that the miR-Let7b/TLR7-mediated inflammatory imprint in RA explants, MΦs, FLS, and collagen-induced arthritis (CIA) was obstructed by IRAK4i therapy. In this study, we sought to elucidate the metabolic pathways that are responsible for TLR7-modulated RA MΦ and FLS activation and determine whether treatment with IRAK4i can subside the metabolic malfunction. We found that TLR7 signaling shifts RA MΦ and FLS metabolic profile towards glycolysis and away from oxidative phosphorylation which can be reversed by IRAK4i therapy. We uncovered that GLUT1, HIF1α, cMYC, LDHA and lactate were accountable for the hypermetabolic activity instigated by TLR7 activation in RA MΦs and FLS and was counteracted by IRAK4i therapy. Notably, the TLR7 agonist was incapable of downregulating the secretion of TCA metabolites, citrate or succinate, in RA MΦs and FLS, while transcription of PPARγ was compromised in these cells. Moreover, miR-Let7b-induced joint inflammation was accompanied by a broader metabolic phenotype compared to RA joint cells. Nevertheless, HIF1α & cMYC are the common metabolites whose expression was upregulated by TLR7 and repressed by IRAK4 therapy in both RA cells and a preclinical model. These results indicate that joint TLR7 endogenous miRNA can foster metabolic malfunction that can be rebalanced by IRAK4i therapy in RA patients.
MATERIALS AND METHODS
RA cells:
Studies were approved by the University of Illinois at Chicago (UIC) Institutional Ethics Review Board and all donors gave informed written consent. RA patients were diagnosed according to the 1987 revised criteria from the ACR(Arnett et al., 1988). Mononuclear cells were isolated by Histopaque gradient centrifugation and monocytes were isolated from normal or RA peripheral blood (PB) using a negative selection kit according to the manufacturer’s instruction (StemCell Technology) (Kim et al., 2020, Kim et al., 2017). FLS from fresh RA ST were isolated by mincing and digestion in a solution of dispase, collagenase, and DNase. Cells were used between passages 3 and 9 (Chamberlain et al., 2012, Elshabrawy et al., 2018, Pickens et al., 2011a, Pickens et al., 2011b). For the in vitro studies, IRAK4i (cat# PZ0327), cMYCi (cat# 475956), and HIF1i (cat#500498) were purchased from Sigma.
Preclinical studies:
All animal studies were approved by UIC Animal Care and Use Committee. Eight-week-old DBA1 mice were immunized with collagen type II (Chondrex, Redmond, WA) on days 0 and 21. In this model, CIA mice received i.a. injection of adenovirus (Ad)-Let7b (107 PFU; Welgen Inc) on days 28 and 42. miR-Let7b arthritic mice were fed with IRAK4i (10g IRAK4i/1kg chow, PF-06426779 (Cushing et al., 2014, Lee et al., 2017) Research Diets Inc.) or control formulated chow from day 25-52 and non-arthritic DBA1 mice (Ctl) were also on control formulated chow.
Real-time RT-PCR:
RNA isolated using TRIzol (Life Technologies) was reverse transcribed to cDNA using the high-capacity cDNA reverse transcription kit (Applied Biosystems) for subsequent qRT-PCR analysis. Taqman primer/probe mixes predesigned by Integrated DNA Technologies and Taqman gene expression master mix (Applied Biosystems) was used to perform qRT-PCR. Data are presented as fold changes in RNA levels compared to control treatment, calculated following the 2−ΔΔCt method.
Metabolite quantification:
RA MΦs or RA FLS were stimulated for 24h under starvation conditions (0% FBS). Metabolites including L-lactate, citrate and succinate concentrations were measured in the collected conditioned media using colorimetric assays (Sigma-Aldrich) according to the manufacturer’s instructions.
Seahorse cell energy phenotype test:
We tested the glycolytic capacity and oxygen consumption of control and (10μg/ml) R837-stimulated RAW 264.7 cells (5x103 cells/well) in presence of IRAK4i (10μM) using the XF Cell Mito Stress Test kit (103015-100; Agilent Technologies) as per manufacturer’s instructions. Cells were pre-conditioned with the inhibitors and stimuli in 0% FBS/DMEM for 24h before ECAR and OCR evaluation.
Immunohistochemistry:
Ankles were decalcified, formalin-fixed, paraffin-embedded, and sectioned. Briefly, slides were deparaffinized in xylene, and antigens were unmasked by either incubating slides in Proteinase K digestion buffer (F480), heat-induced epitope retrieval in 10mM citrate (vimentin). Ankles were stained with F480 (1:100; GeneTex), vimentin (Lab vision, 1:2500), HIF1α (1:50, Santa Cruz Biotechnology), cMYC (1:50, Thermofisher) antibodies, and control IgG. Sections were scored on a 0-5 scale. Where 0= normal appearance, 1= minimal changes, 2= mixed appearance, 3= moderate changes, 4= marked changes, and 5= severe changes (Pickens et al., 2011a).
Statistical Analysis:
For comparison among multiple groups, one-way ANOVA followed by Tukey’s multiple comparison test was employed, using Graph Pad Prism9 software. The data were also analyzed using a two-tailed Student’s t-test for paired or unpaired comparisons between two groups. Values of p < 0.05 were considered significant.
RESULTS
TLR7 ligation activates glycolysis and has a restrained effect on oxidative phosphorylation in RA macrophages.
To elucidate the significance of TLR7 on MΦ immunometabolism and the efficiency of IRAK4i therapy on counteracting the identified function, biogenesis was evaluated in RA myeloid cells. We found that in RA MΦs, IRAK4i treatment reverses inflammatory transcription factors, IRF5 and IRF7 that were enriched 3-6 fold by the TLR7 agonist, R837 stimulation (Fig. 1A). Myeloid cells exposed to R837 were metabolically active and had a higher ECAR compared to the control group and this phenomenon was overturned by IRAK4i therapy (Fig. 1B). Moreover, we showed that while transcriptional upregulation of HIF1α, cMYC, LDHA, and secretion of lactate was responsible for R837-activated glycolytic reprogramming in RA MΦs (Figs. 1C–E), PFKBF3, PKM2, ENO1, PDK1 and RAPTOR were uninvolved in this manifestation (Fig. 1D). Surprisingly, transcription of non-oxidative PPP intermediate, CARKL, was diminished by R837 and rescued above the baseline levels by IRAK4i therapy (Fig. 1C).
Figure 1. In RA MΦs, TLR7-fostered hypermetabolic activity is reversed by IRAK4i.
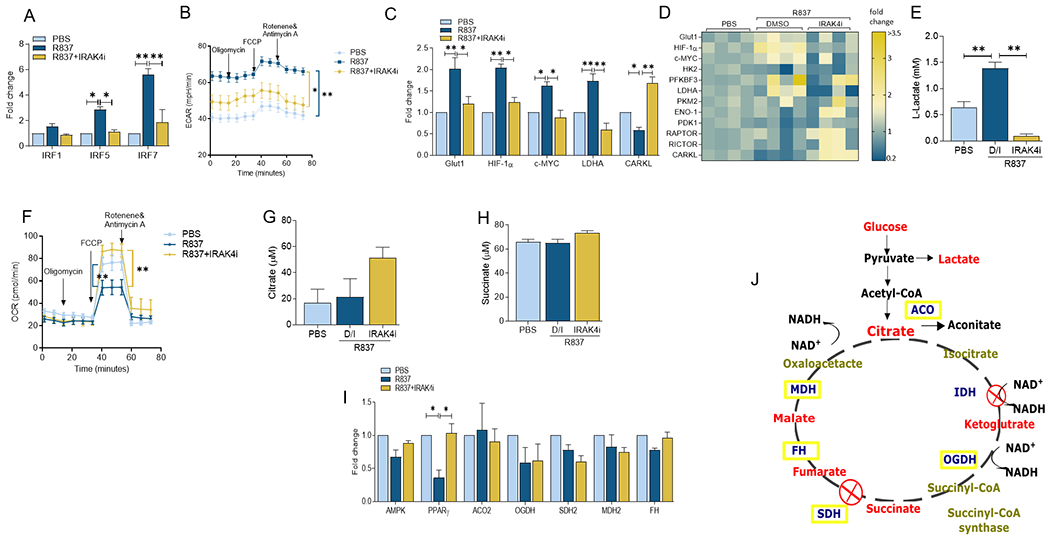
RA MΦs were untreated (PBS) or pretreated with IRAK4i (1μM) o/n before R837 (1μg/ml) stimulation for 6h and quantification of IRF1, IRF5 or IRF7 (A), metabolic intermediates including GLUT1, HIF1α, cMYC, LDHA, CARKL (C-D) or oxidative enzymes such as ACO2, OGDH, SDH2, MDH2 and FH (I) by realtime RT-PCR, n=4. Effect of IRAK4i (10 μM) was examined on the regulation of TLR7-mediated glycolytic capacity (ECAR, B) and oxygen consumption (OCR, F) in R837 (10μg/ml) stimulated RAW cells (5×103 cells/well) using the XF Cell Mito Stress Test kit (103015-100; Agilent Technologies) per manufacturer’s instructions. Cells were pre-conditioned with the inhibitors and stimuli in serum-free DMEM for 24h before taking ECAR and OCR measurements for 0-75min by Seahorse Bioscience XF96 analyzer n=5-6. Conditioned media were obtained from RA MΦs untreated (PBS) or pretreated with IRAK4i (1μM) before R837 (1μg/ml) stimulation for 24h and quantification of Lactate (E) Citrate (G) and Succinate (H) by colorimetric assay, n=4. J. Demonstrates a schematic illustration of TCA cycle and the involved metabolites and enzymes. The data are shown as mean ± SEM, * represents p<0.05, ** denotes p<0.01.
Corroborating the ECAR finding, TLR7 expansion of HIF1α, cMYC and LDHA transcription and lactate secretion was negated by IRAK4i therapy in RA MΦs (50-60%, Figs. 1C–E). In contrast, the inhibitory effect of R837 on OCR and the ability of IRAK4i to reverse its function were inconsistent with the unaltered production of citrate or succinate or transcription of oxidative enzymes, aconitase (ACO2), oxoglutarate dehydrogenase (OGDH), succinate dehydrogenase (SDH2), and fumarate hydratase (FH), malate dehydrogenase (MDH2) and in response to TLR7-activated RA MΦs (Figs. 1F–I). However, the expression of an other oxidative mediator, PPARγ, was compromised in R837-stimulated RA MΦs and impaired by IRAK4i (Fig. 1I). Taken together, IRAK4i can strongly interfere with TLR7-amplified glycolytic rewiring in RA MΦs, whereas its capacity to potentiate oxidative phosphorylation is rather limited in these cells.
R837 triggers hypermetabolic activity in RA FLS which is dysregulated by IRAK4i treatment.
To determine the impact of TLR7 on RA FLS metabolic reprogramming, cells activated by R837 in the presence or absence of IRAK4i were analyzed for M1 TFs, glycolytic and oxidative regulators. We observed that IRF1 and IRF7 transcription levels were unchanged in response to R837 and IRAK4i treatment was inconsequential on its regulation in RA FLS (Fig. 2A). We revealed that compared to RA MΦs, TLR7-activated FLS exhibited a broader glycolytic profile that comprised of potentiated GLUT1, HIF1α, cMYC, HK2 and LDHA transcription followed by higher lactate production that was nullified by IRAK4i (2 fold, Figs. 2B–D). Consistent with RA MΦs, PFKBF3, PKM2, ENO1, PDK1 and RAPTOR were uninvolved in the metabolic rewiring of RA FLS expanded by TLR7 ligation (Fig. 2C). Remarkedly, while TLR7 activation was ineffective on citrate and succinate secretion as well as ACO2, OGDH, SDH2, FH, and MDH2 transcriptome, the expression level of PPARγ was restricted in R837-treated RA FLS and was reversed by IRAK4i (Figs. 2E–G). Collectively, these results suggest that the TLR7 has a similar metabolic profile in RA MΦs and FLS.
Figure 2. IRAK4i can counteract TLR7-potentiated immunometabolism in RA FLS.
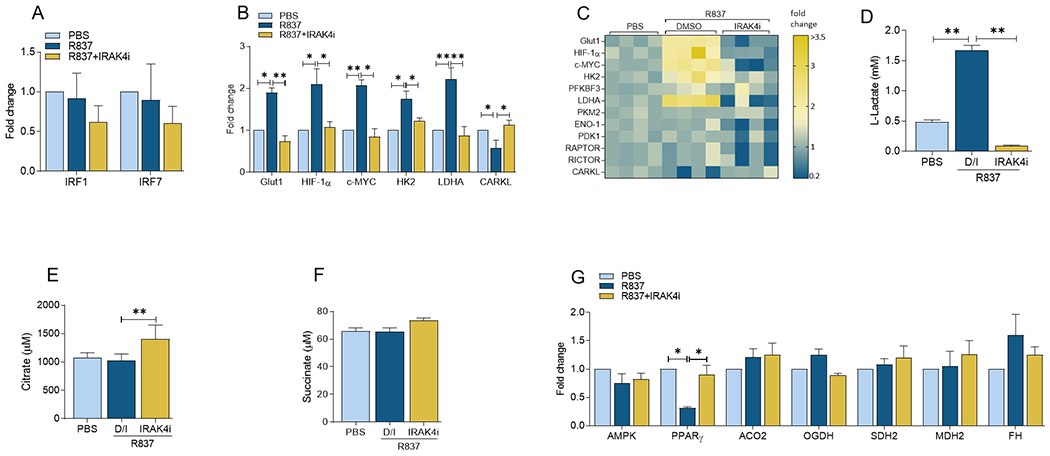
RA FLS were pretreated with IRAK4i (1μM) o/n before R837 (1μg/ml) stimulation for 6h and quantification of IRF1, IRF5 or IRF7 (A), metabolic intermediates including GLUT1, HIF1α, cMYC, LDHA, CARKL (B-C) or oxidative enzymes such as ACO2, OGDH, SDH2, MDH2 and FH (G) by realtime RT-PCR, n=4. Conditioned media were obtained from RA FLS untreated (PBS) or pretreated with IRAK4i (1μM) before R837 (1μg/ml) stimulation for 24h and quantification of Lactate (D) Citrate (E) and Succinate (F) by colorimetric assay, n=4. The data are shown as mean ± SEM, * represents p<0.05, ** denotes p<0.01.
miR-Let7b-induced arthritis promotes joint metabolic hyperactivity that is mitigated by IRAK4i therapy.
To solidify the contribution of immunometabolism in TLR7-mediated arthritis and the therapeutic significance of IRAK4 inhibition in this process, the glycolytic and oxidative regulators were phenotyped in experimental arthritis. miR-Let7b-induced joint inflammation coincided with the expansion of an extensive number of glycolic metabolites including GLUT1, HIF1α, cMYC, PFKBF3, PKM2, ENO1, PDK1, and RAPTOR which were ablated along with arthritis resolution by IRAK4i therapy (Figs. 3A–C). Interestingly, HIF1α and cMYC immunostaining were markedly advanced in miR-Let7b arthritic joints and its immunophenotype was dysregulated by IRAK4i therapy in joint myeloid cells (Figs. 3D–E). In short, these findings support the central role of HIF1α and cMYC in arthritis as well as in the metabolic remodeling of RA synoviocytes.
Figure 3. IRAK4i attenuates TLR7-induced arthritis through restrained joint HIF1α and cMYC expression.
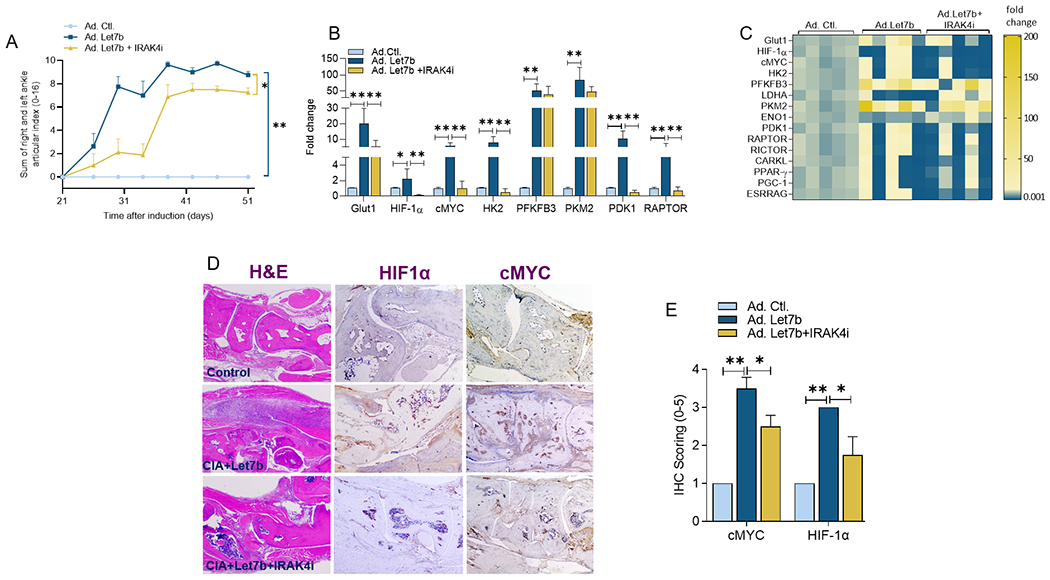
A. Articular index score was assessed in non-arthritic (Ad-Control) and the CIA mice that received Ad-Let7b (107 PFU; Welgen Inc) on days 28 and 42 and were treated with placebo or IRAK4i chow (10mg/kg BW) from day 25-51, n=7 mice (14 ankles). Harvested ankles were homogenized and mRNA expression of glycolytic genes was quantified by real-time RT-PCR (B-C), n=5. Ankles harvested on day 52 were stained with H&E, HIF1α (1:50, Santa Cruz), cMYC (1:50, Novus Bio) and the immunophenotype was scored on a 0-5 scale (D-E), n=4. The data are shown as mean ± SEM, * represents p<0.05 and ** denotes p<0.01.
HIF1α and cMYC are the master regulators that are mutually amplified by TLR7 and disrupted by IRAK4i therapy in RA MΦs and FLS.
To elucidate the influence of HIF1α and cMYC signaling in TLR7-instigated inflammation and immunometabolism, inhibitors for these factors were used to characterize their mechanism of action in RA MΦs and FLS.
We uncovered that dysregulation of cMYC or HIF1α function abrogates IL-6, IL-1β, TNFα and CCL2 transcription levels escalated by TLR7 (50-80%, Fig. 4A). Consistently, enriched IRF5 and IRF7 expression together with baseline IRF1 levels were repressed by cMYC and HIF1α blockade in TLR7-activated RA MΦs (Fig. 4B). However, cMYCi and HIF1i were capable of impeding TLR7 metabolic imprints that were also disrupted by IRAK4i including GLUT1, LDHA and lactate in RA MΦs (Figs. 4C–D). Interestingly, like IRAK4i therapy, deregulation of HIF1α but not cMYC in RA MΦs could reverse the TLR7-driven CARKL transcriptional downregulation (Fig. 4C). In contrast to HIF1i, cMYCi therapy rescues R837-reduced PPARγ expression in RA MΦs without impacting levels of citrate, succinate, or the oxidative enzymes (Figs. 4E–G).
Figure 4. cMYC and HIF1α inhibitors mitigate TLR7-driven inflammatory and metabolic imprints in RA MΦs.
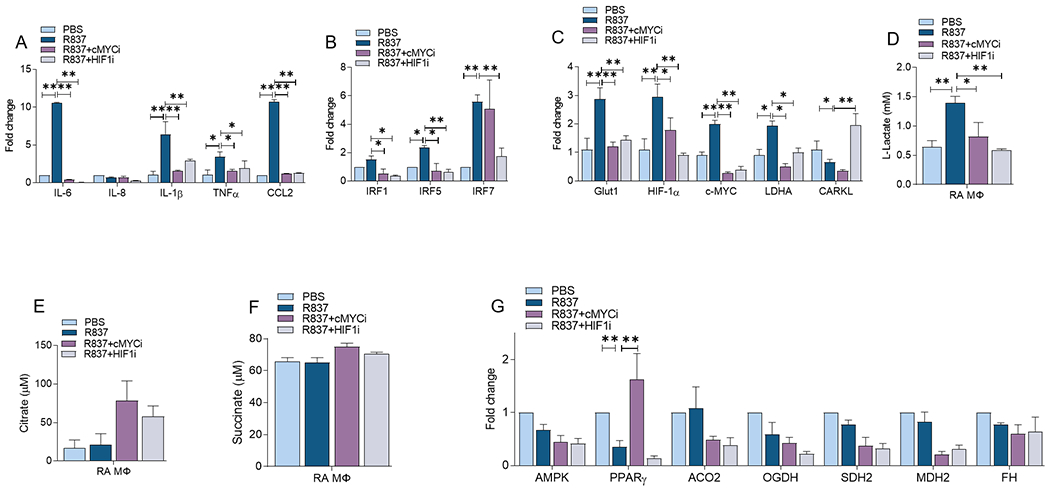
RA MΦs were untreated (PBS) or pretreated with cMYCi (50μM) or HIFi (2μM) o/n before R837 (1μg/ml) stimulation for 6h and transcriptional regulation of monokines (A), transcriptional factors (B), metabolic intermediates (C) or oxidative enzymes (G) were determined by realtime RT-PCR, n=4. Conditioned media were obtained from RA MΦs untreated (PBS) or pretreated with cMYCi (50μM) or HIFi (2μM) before R837 (1μg/ml) activation for 24h and measuring of Lactate (D) Citrate (E) and Succinate (F) levels by colorimetric assay, n=4. The data are shown as mean ± SEM, * represents p<0.05 and ** denotes p<0.01.
In coordination with our findings in RA MΦs, cMYCi and HIF1i could effectively impair TLR7-triggered IL-6 and CCL2 transcription in RA FLS (Fig. 5A). However, unlike RA MΦs, IRF1 and IRF7 were unaltered in RA FLS by R837 stimulation or cMYCi & HIF1i treatment (Fig. 5B). Remarkably, TLR7-activated glycolytic regulators were uniformly negated by cMYCi and HIF1i in RA FLS (Figs. 5C–D). Yet, while cMYCi rebalances reduction of PPARγ through R837, HIF1i expands citrate and succinate in RA FLS (Figs. 5E–G). In conclusion, inhibition of HIF1 or cMYC restrains TLR7-driven glycolysis in RA MΦs & FLS through a similar mechanism of action. Nevertheless in RA MΦs & FLS, HIF1i and cMYCi, have unique features on non-oxidative PPP and oxidative pathways.
Figure 5. Inhibition of cMYC and HIF1α pathways can diminish TLR7-instigated inflammatory and glycolytic regulators in RA FLS.
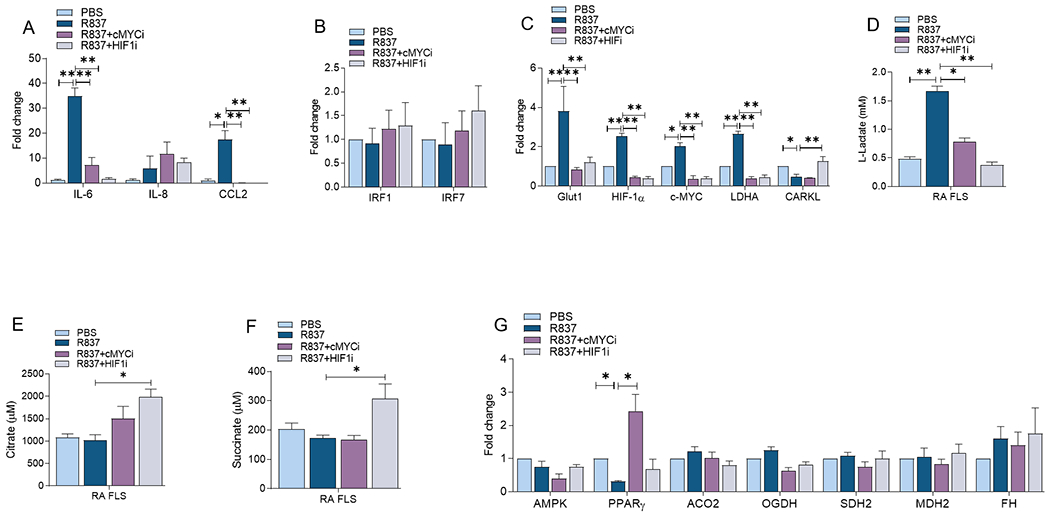
RA FLS were untreated (PBS) or pretreated with cMYCi (50μM) or HIFi (2μM) o/n before R837 (1μg/ml) stimulation for 6h. Thereafter transcriptional regulation of monokines (A), transcriptional factors (B), metabolic intermediates (C) or oxidative enzymes (G) were determined by realtime RT-PCR, n=4. Conditioned media were obtained from RA FLS untreated (PBS) or pretreated with cMYCi (50μM) or HIFi (2μM) before R837 (1μg/ml) activation for 24h and measuring of Lactate (D) Citrate (E) and Succinate (F) levels by colorimetric assay, n=4. The data are shown as mean ± SEM, * represents p<0.05 and ** denotes p<0.01.
DISCUSSION
This study characterizes a distinct imprint of TLR7 activation in RA MΦs & FLS and guides the mechanism by which hyperglycolysis can be rebalanced in joint cells and arthritic models. We exhibited that TLR7 ligation cultivates a strong RA MΦ & FLS glycolytic profile along with a narrow suppression on non-oxidative PPP and oxidative regulators. We uncovered that IRAK4i therapy mitigates miR-Let7b-induced arthritis by dysregulating joint glycolysis in part through impairing HIF1α and cMYC expression. We noted that in line with IRAK4i treatment, HIF1 and cMYC inhibitors reverse the metabolic reprogramming in RA MΦs & FLS, however, display distinctive characteristics on non-oxidative PPP and oxidative modulators.
Earlier studies have shown that ligation of GU-rich miR-Let7b to TLR7 transforms RA and murine M0 and M2 cells into M1 MΦs that express arthritogenic monokines, including TNFα, IL-6, IL-1β, IL-12, CCL2 and CCL5 (Kim et al., 2016, Umar et al., 2020, Van Raemdonck et al., 2020). Potentiation of CCL2 and CCL5 by TLR7 ligation exacerbates joint monocyte migration (Umar et al., 2020). Supporting these observations, the expression of TLR7 on RA monocytes is closely linked to disease activity score (DAS28) and TNFα levels (Chamberlain et al., 2013). Moreover, TLR7 polarization of RA MΦs was accompanied by IRAK4 and IRF5 phosphorylation, unlike IRF7. Further, it was shown that IRF5 and IRF7 transcription were amplified in TLR7-activated RA MΦs and inhibited by IRAK4i and cMYCi but not HIF1i.
Intriguingly, TLR7-instigated inflammatory response in RA FLS was accompanied by phosphorylation of IRAK and IRF5 and was disconnected from pIRF7 profile (Umar et al., 2020). Nonetheless, R837 was incapable of modulating transcription IRF1 and IRF7 in RA FLS; and consequentially their levels were unaffected by IRAK4i, cMYC or HIF1i therapy. Unlike RA monocyte homing that was indirectly fostered by TLR7-induced CCL2 and CCL5 secretion, R837 directly attracted RA FLS migration that was deregulated by IRAK4i therapy (Umar et al., 2020). Moreover, enrichment of TLR7 on RA compared to normal synovial tissues (STs) lining MΦs and FLS corroborates with the implication of IRAK4i therapy on downregulation of monokines and fibrokines secreted from RA explant (Umar et al., 2020).
We next examined the RA MΦs and FLS metabolic reprogramming orchestrated by TLR7 ligation. Interestingly, we illustrated that RA MΦs-polarized by LPS/IFNγ has overlapping and unique characteristics relative to TLR7 differentiated cells (Cellular & Molecular Life Science, CMLS-D-21-00542R1 MS in press). In classical RA M1 MΦs, amplification of IRF1 and IRF5 was disrupted by 2-DG (CMLS in press), suggesting that IRF5 is the common denominator that was modulated by LPS/IFNγ and R837 and restrained by 2-DG, IRAK4i, cMYci or HIF1i. Substantiating this rationale, GLUT1, HIF1α, LDHA and lactate levels as well as the ECAR activity were escalated by LPS/IFNγ or R837 exposure in RA MΦs and this glycolytic phenotype was hampered by 2-DG (CMLS in press), IRAK4i, cMYci or HIF1i. On the other hand, PFKFB3 or cMYC elevation was exclusive to RA MΦs exposed to LPS/IFNγ (CMLS in press) or R837, respectively. Others have shown that expression of HIF1α and PFKFB3 is highly elevated in monocytes of RA patients compared to non-symptomatic or healthy donors (McGarry et al., 2021). More recently, it was uncovered that IL-34 reprograms RA naïve cells into CD14+CD86+GLUT1 M34 MΦs that display amplified glycolytic imprint accompanied by GLUT1, cMYC, HIF1α and lactate (Van Raemdonck et al., 2021). Intriguingly, 2-DG treatment nullifies IL-34-induced arthritis via cMYC and HIF1α pathways (Van Raemdonck et al., 2021). Extending this notion, treatment with 2-DG or HIF1i could effectively abrogate the frequency of F480+CD80+ MΦs and inflammatory response to R837 (Van Raemdonck et al., 2020). Others have shown that the primed CD8+ T cells are metabolically remodeled by TLR7 through AKT/mTOR signaling (Li et al., 2019).
OCR is severely impaired by LPS/IFNγ or R837 stimulation in RA MΦs due to TCA truncation (Kelly and O’Neill, 2015) (Diskin and Palsson-McDermott, 2018, Williams and O’Neill, 2018). However, while 2-DG can not overturn OCR suppression facilitated by LPS/IFNγ exposure; IRAK4i and cMYCi therapies rescue the TLR7-mediated malfunction through PPARγ (Ryan and O’Neill, 2017, Williams and O’Neill, 2018). Diverse from 2-DG, Tofacitinib rebalances LPS/IFNγ-mediated OCR suppression in RA MΦs via PPARγ induction (Palasiewicz et al., 2021).
In RA FLS, transcription of IRF1, IRF5 and IRF7 was accentuated by LPS/IFNγ but not R837 stimulation and this process was blocked by 2-DG treatment. Several metabolic markers including GLUT1, HK2, HIF1α, LDHA and lactate were mutually amplified in RA FLS exposed to LPS/IFNγ and R837 and markedly reduced by 2-DG, IRAK4i, cMYci or HIF1i (Bustamante et al., 2018, Garcia-Carbonell et al., 2016, Palasiewicz et al., 2021). In line with RA MΦs, selected glycolytic regulators such as PFKFB3 or cMYC were augmented in RA FLS in response to LPS/IFNγ or R837 respectively (CMLS in press). Interestingly in RA FLS, blockade of IRAK4i has a broader impact on restructuring oxidative mediators, PPARγ and citrate, compared to cMYCi or HIF1i that reverse either PPARγ or succinate suppression. Previous studies have also demonstrated that hypoxia, oxidative stress, TLR2, and TNFα advance hypermetabolic activity in RA FLS through PFKFB3, PKM2 and GLUT1 (Biniecka et al., 2016, Garcia-Carbonell et al., 2016, McGarry et al., 2017). As such others have shown that inhibitors to PFKFB3 and HK2 downregulate RA FLS infiltration and inflammatory response (Biniecka et al., 2016, Bustamante et al., 2018).
It was illustrated that treatment with IRAK4i attenuates miR-Let7b-induced arthritis by restricting joint infiltration of F480+iNOS+ and Vimentin+FLS MΦs (Umar et al., 2020). Supporting these findings, amelioration of miR-Let7b-induced arthritis by IRAK4i was accompanied by a reduction of several monokines, fibrokines and erosive factors (Umar et al., 2020). Consist with the robust TLR7-driven inflammatory phenotype in RA MΦs and FLS, expression of numerous metabolic factors including GLUT1, HIF1α, cMYC, HK2, PFKFB3, PKM2, PDK1, and RAPTOR were expanded in miR-Let7b-induced arthritis and markedly impaired by IRAK4i. Notably, HIF1α and cMYC were the 2 common factors that were upregulated via TLR7 activation in RA MΦs, FLS, and experimental model and were repressed by IRAK4i therapy. Further, our findings indicate that HIF1α and cMYC are both involved in the TLR7 and its ability to expand the IL-1β mechanism of action.
In contrast with the effectiveness of IRAK4i in miR-Let7b arthritic mice, treatment with TNFi and IL-6R Ab was inconsequential on this model and the frequency of joint F480+iNOS+ MΦs and Vimentin+FLS remained unchanged (Umar et al., 2020). Since metabolic dysregulation can more efficiently alleviate arthritic manifestations compared to oxidative expansion (Abboud et al., 2018), and TNFi and IL-6R Ab therapies are ineffective in TLR7-induced arthritis (Umar et al., 2020), IRAK4i or its downstream pathways may provide a promising target for RA patients.
Our findings are specifically important as phase 1 studies reveal that IRAK4i therapy is well-tolerated and reduces type I IFN gene signature in healthy participates (Winkler et al., 2021).
CONCLUSION
In conclusion, our results indicate that inhibition of IRAK4 and its interconnected metabolic factors, HIF1α and cMYC, counteract TLR7-enhanced metabolic activity in RA MΦs and FLS to resolve inflammatory imprints.
HIGHLIGHTS.
TLR7 shifts RA macrophage (MΦs) and fibroblast-like synoviocytes (FLS) metabolism towards glycolysis and away from oxidative phosphorylation via potentiation of GLUT1, HIF1α, cMYC, LDHA, and lactate
Dysregulation of IRAK4, HIF1α and cMYC is capable of reversing TLR7-induced hypermetabolism in RA MΦs and FLS
Inhibition of IRAK4, HIF1α, or cMYC has a modest impact on baseline oxidative phosphorylation
IRAK4 inhibitor (i) alleviates TLR7-induced arthritis by metabolic dysregulation in part through constraining joint HIF1α and cMYC expression
ACKNOWLEDGEMENT:
This work was supported in part by awards from the Department of Veteran’s Affairs MERIT Award BX002286, the National Institutes of Health NIH AI147697, the National Psoriasis Foundation (NPF), Pfizer Investigator-Initiated Research (IIR) Program and Chicago Biomedical Consortium (CBC) Accelerator Award.
Footnotes
CONFLICT OF INTEREST: The authors have declared that no commercial or financial conflict of interest exists.
Ethics approval and consent to participate: All peripheral blood (PB) was collected following our protocol approved by the University of Illinois at Chicago Institutional Ethics Review Board. All the RA patients have consented to participate in this study and have provided written consent. Furthermore, all animal studies were approved by the University of Illinois at Chicago Animal Care and Use Committee following the legal requirements and guidelines of the state of Illinois in the USA and NIH.
Author’s Consent to publication: All authors have been involved in writing the manuscript and consented to publication.
AVAILABILITY OF DATA AND MATERIAL:
All findings are exhibited in the paper and the material and data are available for transparency.
REFERENCES
- Abboud G, Choi SC, Kanda N, Zeumer-Spataro L, Roopenian DC, Morel L. Inhibition of Glycolysis Reduces Disease Severity in an Autoimmune Model of Rheumatoid Arthritis. Front Immunol. 2018;9:1973. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Ahn JK, Kim S, Hwang J, Kim J, Kim KH, Cha HS. GC/TOF-MS-based metabolomic profiling in cultured fibroblast-like synoviocytes from rheumatoid arthritis. Joint Bone Spine. 2016;83:707–13. [DOI] [PubMed] [Google Scholar]
- Arnett FC, Edworthy SM, Bloch DA, McShane DJ, Fries JF, Cooper NS, et al. The American Rheumatism Association 1987 revised criteria for the classification of rheumatoid arthritis. Arthritis Rheum. 1988;31:315–24. [DOI] [PubMed] [Google Scholar]
- Biniecka M, Canavan M, McGarry T, Gao W, McCormick J, Cregan S, et al. Dysregulated bioenergetics: a key regulator of joint inflammation. Ann Rheum Dis. 2016;75:2192–200. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Bustamante MF, Oliveira PG, Garcia-Carbonell R, Croft AP, Smith JM, Serrano RL, et al. Hexokinase 2 as a novel selective metabolic target for rheumatoid arthritis. Ann Rheum Dis. 2018;77:1636–43. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Chamberlain ND, Kim SJ, Vila OM, Volin MV, Volkov S, Pope RM, et al. Ligation of TLR7 by rheumatoid arthritis synovial fluid single strand RNA induces transcription of TNFalpha in monocytes. Annals of the Rheumatic Diseases. 2013;72:418–26. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Chamberlain ND, Vila OM, Volin MV, Volkov S, Pope RM, Swedler W, et al. TLR5, a novel and unidentified inflammatory mediator in rheumatoid arthritis that correlates with disease activity score and joint TNF-alpha levels. J Immunol. 2012;189:475–83. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Chen K, Lin ZW, He SM, Wang CQ, Yang JC, Lu Y, et al. Metformin inhibits the proliferation of rheumatoid arthritis fibroblast-like synoviocytes through IGF-IR/PI3K/AKT/m-TOR pathway. Biomed Pharmacother. 2019;115:108875. [DOI] [PubMed] [Google Scholar]
- Chen Y, Qiu F, Yu B, Chen Y, Zuo F, Zhu X, et al. Metformin, an AMPK Activator, Inhibits Activation of FLSs but Promotes HAPLN1 Secretion. Mol Ther Methods Clin Dev. 2020;17:1202–14. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Cushing L, Stochaj W, Siegel M, Czerwinski R, Dower K, Wright Q, et al. Interleukin 1/Toll-like receptor-induced autophosphorylation activates interleukin 1 receptor-associated kinase 4 and controls cytokine induction in a cell type-specific manner. J Biol Chem. 2014;289:10865–75. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Diskin C, Palsson-McDermott EM. Metabolic Modulation in Macrophage Effector Function. Front Immunol. 2018;9:270. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Elshabrawy HA, Volin MV, Essani AB, Chen Z, McInnes IB, Van Raemdonck K, et al. IL-11 facilitates a novel connection between RA joint fibroblasts and endothelial cells. Angiogenesis. 2018;21:215–28. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Falconer J, Murphy AN, Young SP, Clark AR, Tiziani S, Guma M, et al. Review: Synovial Cell Metabolism and Chronic Inflammation in Rheumatoid Arthritis. Arthritis Rheumatol. 2018;70:984–99. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Garcia-Carbonell R, Divakaruni AS, Lodi A, Vicente-Suarez I, Saha A, Cheroutre H, et al. Critical Role of Glucose Metabolism in Rheumatoid Arthritis Fibroblast-like Synoviocytes. Arthritis Rheumatol. 2016;68:1614–26. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Geeraerts X, Bolli E, Fendt SM, Van Ginderachter JA. Macrophage Metabolism As Therapeutic Target for Cancer, Atherosclerosis, and Obesity. Front Immunol. 2017;8:289. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Harty LC, Biniecka M, O’Sullivan J, Fox E, Mulhall K, Veale DJ, et al. Mitochondrial mutagenesis correlates with the local inflammatory environment in arthritis. Ann Rheum Dis. 2012;71:582–8. [DOI] [PubMed] [Google Scholar]
- Huang C, Hsu P, Hung Y, Liao Y, Liu C, Hour C, et al. Ornithine decarboxylase prevents methotrexate-induced apoptosis by reducing intracellular reactive oxygen species production. Apoptosis. 2005;10:895–907. [DOI] [PubMed] [Google Scholar]
- Ito H, Ando T, Arioka Y, Saito K, Seishima M. Inhibition of indoleamine 2,3-dioxygenase activity enhances the anti-tumour effects of a Toll-like receptor 7 agonist in an established cancer model. Immunology. 2015;144:621–30. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Kelly B, O’Neill LA. Metabolic reprogramming in macrophages and dendritic cells in innate immunity. Cell Res. 2015;25:771–84. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Kim SJ, Chang HJ, Volin MV, Umar S, Van Raemdonck K, Chevalier A, et al. Macrophages are the primary effector cells in IL-7-induced arthritis. Cell Mol Immunol. 2020;17:728–40. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Kim SJ, Chen Z, Essani AB, Elshabrawy HA, Volin MV, Fantuzzi G, et al. Differential impact of obesity on the pathogenesis of RA or preclinical models is contingent on the disease status. Ann Rheum Dis. 2017;76:731–9. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Kim SJ, Chen Z, Essani AB, Elshabrawy HA, Volin MV, Volkov S, et al. Identification of a Novel Toll-like Receptor 7 Endogenous Ligand in Rheumatoid Arthritis Synovial Fluid That Can Provoke Arthritic Joint Inflammation. Arthritis Rheumatol. 2016;68:1099–110. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Krawczyk CM, Holowka T, Sun J, Blagih J, Amiel E, DeBerardinis RJ, et al. Toll-like receptor-induced changes in glycolytic metabolism regulate dendritic cell activation. Blood. 2010;115:4742–9. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Lee KL, Ambler CM, Anderson DR, Boscoe BP, Bree AG, Brodfuehrer JI, et al. Discovery of Clinical Candidate 1-{[(2S,3S,4S)-3-Ethyl-4-fluoro-5-oxopyrrolidin-2-yl]methoxy}-7-methoxyisoquinoli ne-6-carboxamide (PF-06650833), a Potent, Selective Inhibitor of Interleukin-1 Receptor Associated Kinase 4 (IRAK4), by Fragment-Based Drug Design. J Med Chem. 2017;60:5521–42. [DOI] [PubMed] [Google Scholar]
- Li Q, Yan Y, Liu J, Huang X, Zhang X, Kirschning C, et al. Toll-Like Receptor 7 Activation Enhances CD8+ T Cell Effector Functions by Promoting Cellular Glycolysis. Front Immunol. 2019;10:2191. [DOI] [PMC free article] [PubMed] [Google Scholar]
- McGarry T, Biniecka M, Gao W, Cluxton D, Canavan M, Wade S, et al. Resolution of TLR2-induced inflammation through manipulation of metabolic pathways in Rheumatoid Arthritis. Sci Rep. 2017;7:43165. [DOI] [PMC free article] [PubMed] [Google Scholar]
- McGarry T, Hanlon MM, Marzaioli V, Cunningham CC, Krishna V, Murray K, et al. Rheumatoid arthritis CD14(+) monocytes display metabolic and inflammatory dysfunction, a phenotype that precedes clinical manifestation of disease. Clin Transl Immunology. 2021;10:e1237. [DOI] [PMC free article] [PubMed] [Google Scholar]
- McGarry T, Orr C, Wade S, Biniecka M, Wade S, Gallagher L, et al. JAK/STAT Blockade Alters Synovial Bioenergetics, Mitochondrial Function, and Proinflammatory Mediators in Rheumatoid Arthritis. Arthritis Rheumatol. 2018;70:1959–70. [DOI] [PubMed] [Google Scholar]
- McInnes IB, Schett G. Pathogenetic insights from the treatment of rheumatoid arthritis. Lancet. 2017;389:2328–37. [DOI] [PubMed] [Google Scholar]
- Murray PJ, Allen JE, Biswas SK, Fisher EA, Gilroy DW, Goerdt S, et al. Macrophage activation and polarization: nomenclature and experimental guidelines. Immunity. 2014;41:14–20. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Palasiewicz K, Umar S, Romay B, Zomorrodi RK, Shahrara S. Tofacitinib therapy intercepts macrophage metabolic reprogramming instigated by SARS-CoV-2 Spike protein. Eur J Immunol. 2021. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Palsson-McDermott EM, Curtis AM, Goel G, Lauterbach MAR, Sheedy FJ, Gleeson LE, et al. Pyruvate Kinase M2 Regulates Hif-1alpha Activity and IL-1beta Induction and Is a Critical Determinant of the Warburg Effect in LPS-Activated Macrophages. Cell Metab. 2015;21:347. [DOI] [PubMed] [Google Scholar]
- Palsson-McDermott EM, O’Neill LAJ. Targeting immunometabolism as an anti-inflammatory strategy. Cell Res. 2020;30:300–14. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Phillips DC, Woollard KJ, Griffiths HR. The anti-inflammatory actions of methotrexate are critically dependent upon the production of reactive oxygen species. Br J Pharmacol. 2003;138:501–11. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Pickens SR, Chamberlain ND, Volin MV, Pope RM, Mandelin AM, 2nd, Shahrara S. Characterization of CCL19 and CCL21 in rheumatoid arthritis. Arthritis Rheum. 2011a;63:914–22. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Pickens SR, Chamberlain ND, Volin MV, Pope RM, Talarico NE, Mandelin AM 2nd, et al. Characterization of interleukin-7 and interleukin-7 receptor in the pathogenesis of rheumatoid arthritis. Arthritis Rheum. 2011b;63:2884–93. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Qualls JE, Subramanian C, Rafi W, Smith AM, Balouzian L, DeFreitas AA, et al. Sustained generation of nitric oxide and control of mycobacterial infection requires argininosuccinate synthase 1. Cell Host Microbe. 2012;12:313–23. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Ryan DG, O’Neill LAJ. Krebs cycle rewired for macrophage and dendritic cell effector functions. FEBS Lett. 2017;591:2992–3006. [DOI] [PubMed] [Google Scholar]
- Saas P, Varin A, Perruche S, Ceroi A. Recent insights into the implications of metabolism in plasmacytoid dendritic cell innate functions: Potential ways to control these functions. F1000Res. 2017;6:456. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Umar S, Palasiewicz K, Van Raemdonck K, Volin MV, Romay B, Amin MA, et al. IRAK4 inhibition: a promising strategy for treating RA joint inflammation and bone erosion. Cell Mol Immunol. 2020. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Van Raemdonck K, Umar S, Palasiewicz K, Romay B, Volkov S, Arami S, et al. TLR7 endogenous ligands remodel glycolytic macrophages and trigger skin-to-joint crosstalk in psoriatic arthritis. Eur J Immunol. 2020. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Van Raemdonck K, Umar S, Palasiewicz K, Volin MV, Elshabrawy HA, Romay B, et al. IL-34 reprograms glycolytic and osteoclastic RA macrophages via Syndecan-1 and M-CSFR. Arthritis Rheumatol. 2021. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Volchenkov R, Dung Cao M, Elgstoen KB, Goll GL, Eikvar K, Bjorneboe O, et al. Metabolic profiling of synovial tissue shows altered glucose and choline metabolism in rheumatoid arthritis samples. Scand J Rheumatol. 2017;46:160–1. [DOI] [PubMed] [Google Scholar]
- Williams NC, O’Neill LAJ. A Role for the Krebs Cycle Intermediate Citrate in Metabolic Reprogramming in Innate Immunity and Inflammation. Front Immunol. 2018;9:141. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Winkler A, Sun W, De S, Jiao A, Nusrat Sharif M, Symanowicz PT, et al. The IRAK4 kinase inhibitor PF-06650833 blocks inflammation in preclinical models of rheumatologic disease and in humans enrolled in a randomized clinical trial. Arthritis Rheumatol. 2021. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Wu D, Sanin DE, Everts B, Chen Q, Qiu J, Buck MD, et al. Type 1 Interferons Induce Changes in Core Metabolism that Are Critical for Immune Function. Immunity. 2016;44:1325–36. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Xue J, Schmidt SV, Sander J, Draffehn A, Krebs W, Quester I, et al. Transcriptome-based network analysis reveals a spectrum model of human macrophage activation. Immunity. 2014;40:274–88. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Zaro BW, Vinogradova EV, Lazar DC, Blewett MM, Suciu RM, Takaya J, et al. Dimethyl Fumarate Disrupts Human Innate Immune Signaling by Targeting the IRAK4-MyD88 Complex. J Immunol. 2019;202:2737–46. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Zeisbrich M, Yanes RE, Zhang H, Watanabe R, Li Y, Brosig L, et al. Hypermetabolic macrophages in rheumatoid arthritis and coronary artery disease due to glycogen synthase kinase 3b inactivation. Ann Rheum Dis. 2018;77:1053–62. [DOI] [PMC free article] [PubMed] [Google Scholar]
Associated Data
This section collects any data citations, data availability statements, or supplementary materials included in this article.
Data Availability Statement
All findings are exhibited in the paper and the material and data are available for transparency.