

Abstract
TNF mediates a variety of biological processes including cellular proliferation, inflammatory responses and cell death, and is therefore associated with numerous pathologies including auto-inflammatory diseases and septic shock. The inflammatory and cell death responses to TNF have been studied extensively downstream of TNF-R1 and are believed to rely on the formation of pro-inflammatory complex I and pro-death complex II respectively. We recently identified a similar multimeric complex downstream of TLR4, termed the TRIFosome that regulates inflammation and cell death in response to LPS or Yersinia pseudotuberculosis. Herein, we present evidence of a role for the TRIFosome downstream of TNF-R1, independent of TLR3 or TLR4 engagement. Specifically, TNF-induced cell death and inflammation in murine macrophages were driven by the TLR4 adaptor TRIF and the LPS co-receptor CD14, highlighting an important role for these proteins beyond TLR-mediated immune responses. Via immunoprecipitation and visualization of TRIF specific puncta, we demonstrated TRIF− and CD14-dependent formation of pro-death and pro-inflammatory complexes in response to TNF. Extending these findings, in a murine TNF-induced sepsis model, TRIF and CD14-deficiency decreased systemic inflammation, reduced organ pathology and improved survival. The outcome of TRIF activation was cell specific, as TNF-induced lethality was mediated by neutrophils and macrophages responding to TNF in a TRIF-dependent manner. Our findings suggest that in addition to their crucial role in TNF production, myeloid cells are central to TNF-toxicity, and position TRIF and CD14 as universal components of receptor-mediated immune responses.
One Sentence Summary:
TRIFosome formation in neutrophils and macrophages mediates TNF-induced lethality.
INTRODUCTION
Tumor necrosis factor α (TNF) elicits a wide array of responses ranging from the production of inflammatory cytokines to the initiation of cell death pathways (1). Given these wide-reaching effects, elevated and sustained TNF mediates numerous auto-inflammatory diseases such as rheumatoid arthritis, Crohn’s disease, psoriasis, and many others (2–4). Furthermore, TNF associates with disease severity in COVID-patients (5), and drives neurodegeneration and acute brain injury mediated by CD14+ microglia (6). Given the debilitating and diverse nature of these diseases, understanding the ever-expanding pathways triggered in response to TNF is critical. During the acute phase of the immune response, TNF is central to septic shock, which has been modeled for the last 60 years using LPS-induced toxicity in mice (7). In this well-established model, macrophages are the main producers of the TNF required for the upregulation of receptors and mediators of the response to intracellular LPS (8). In contrast to LPS-toxicity, TNF-induced toxicity in vivo has been shown to target intestinal epithelial cells (9), but the role of myeloid cells and specifically macrophages has not been elucidated. In fact, the mechanisms that confer pathology and lethality in response to TNF remain poorly understood.
This is in contrast to the well characterized cellular mechanism of activation and regulation of pro-inflammatory and pro-death responses downstream of TNF-R1 in vitro (10, 11). During this process, formation of pro-inflammatory complex I relies on ubiquitination of receptor interacting protein kinase-1 (RIPK1) to recruit TGF-β activated kinase 1 (TAK1) and the IKK complex via NF-κB essential modulator (NEMO), leading to the activation of downstream MAPK and NF-κB signaling cascades respectively (12–15). However, if RIPK1 modification is perturbed, RIPK1 associates with FADD and caspase-8 to form pro-death complex II, leading to apoptosis (16–18). Recently, an alternative lytic form of caspase-8-mediated pyroptotic cell death was reported in macrophages in response to infection with Yersinia species bacteria or treatment with TNF or LPS in the context of TAK1 inhibition (19–21). In these reports, cell death and the formation of a pro-death TRIFosome complex similar to complex II relied on the TLR4-adaptor TRIF (22, 23). Given the similarities between the TRIFosome and complex II, as well as the role of TNF/TNF-R1 signaling in Yersinia-induced cell death (24), the possibility of TRIF mediating responses to TNF warranted further investigation.
Using a mouse model of TNF-lethality, which mimics the hyperinflammatory cytokine storm associated with severe sepsis, we showed that TRIF and CD14 conferred lethality to TNF. Additionally, we showed that macrophages and neutrophils also play a crucial role in TNF-lethality. In this role, these CD14+ cells uniquely utilized CD14 to recruit the endosomal adaptor TRIF to RIPK1 upon TNF receptor ligation. Thus, in addition to a well-established central role of macrophages in TNF-production in response to LPS, these data positioned macrophages as main effectors of TNF-induced toxicity as well. Furthermore, the activation pathway that uniquely engages TNF-induced inflammatory and cell death responses relied on formation of the TRIFosome. Together with our earlier characterization of TRIFosome activation downstream of TLR4 (22, 23), these findings positioned TRIF and CD14 as core components of the TNF-R1-pathway, raising the possibility that this cross-talk may apply to other members of the TNF receptor superfamily.
RESULTS
The TRIFosome potentiates TNF-induced inflammation and cell death in macrophages
In response to Yersinia pseudotuberculosis (Y.p.) infection, macrophages activate caspase-8-mediated cell death downstream of the TLR4 adaptor TRIF, which is further sustained via autocrine TNF (20, 22, 23, 25). Accordingly, comparison of Y.p.-induced cell death in Tlr4−/−, Tnfr1−/−, and Trif−/− macrophages revealed that while TLR4- and TNFR1-deficiency offered comparable protection from cell death, Trif−/− cells showed more resistance than either Tlr4−/− or Tnfr1−/− cells (Fig. 1A), suggesting additional contribution of TRIF in mediating cell death responses to Y.p.. Indeed, analysis of pro-death complex formation in response to Y.p. infection via immunoprecipitation of FADD (Fig. 1B) revealed delayed association of RIPK1 and FADD in Trif−/− bone marrow derived macrophages (BMDMs) compared to B6 and even Tnfr1−/− BMDMs. Conversely, complex formation appeared to be attenuated at later timepoints in the absence of TNF-R1, supporting the model that autocrine TNF sustains and amplifies Y.p.-induced cell death initiated by TRIF (Fig. 1B). As a result, self-processing of caspase-8 to the p18 subunit was also delayed in Trif−/− cells compared to B6 and Tnfr1−/− cells.
Fig. 1. The TRIFosome potentiates TNF-induced inflammation and cell death in macrophages.
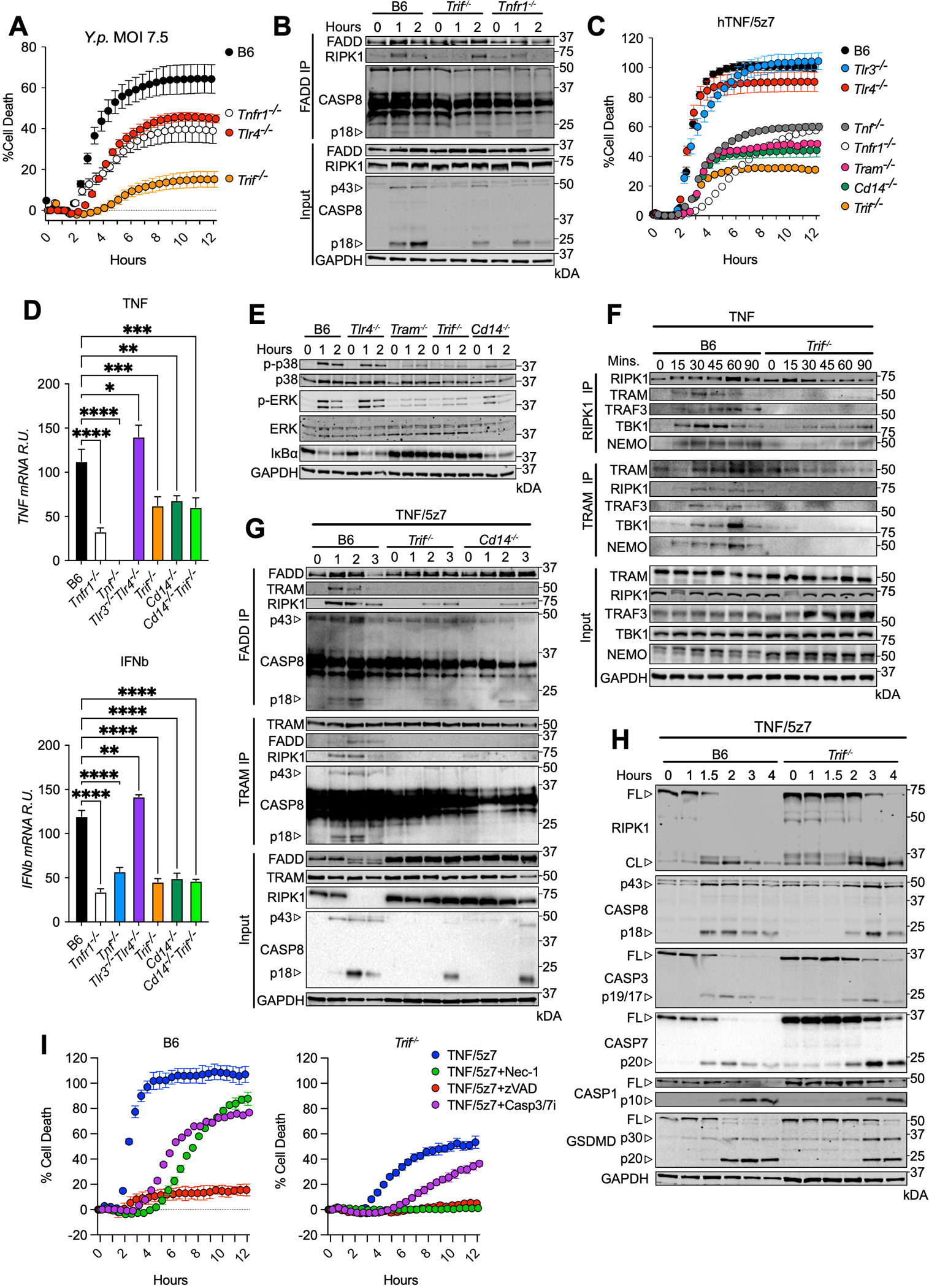
(A) Cell death overtime as measured by propidium iodide incorporation in indicated bone marrow derived macrophages (BMDMs) infected with Yersinia pseudotuberculosis (Y.p.). (B) FADD immunoprecipitation in B6, Trif−/−, and Tnfr1−/− BMDMs infected with Y.p. (7.5 MOI) for indicated timepoints. (C) Cell death overtime as measured by propidium iodide incorporation in indicated BMDMs activated with hTNF/5z7. (D) Relative TNF and IFNb mRNA levels, normalized to β-Actin in response to mTNF for 4 hours in indicated BMDMs. (E) Levels of total and phospho-p38, -ERK and IκBα over time in indicated BMDMs stimulated with mTNF for indicated timepoints. (F) RIPK1 and TRAM immunoprecipitations in B6 and Trif−/− BMDMs stimulated with mTNF for indicated timepoints. (G) FADD and TRAM immunoprecipitation in B6, Trif−/−, and Cd14−/− BMDMs stimulated with mTNF/5z7 for indicated timepoints. (H) B6 and Trif−/− BMDMs stimulated with mTNF/5z7 for indicated timepoints and probed for RIPK1, indicated caspases and GSDMD. (I) Cell death overtime as measured by propidium iodide incorporation in B6 and Trif−/− BMDMs stimulated as indicated. Data from cell death assays and immunoblots are representative of three or more independent experiments, cell death data are presented as the mean ± SD of triplicate wells (~10,000 cells/field of view). qPCR data are presented as the mean ± SD for triplicate wells from three or more independent experiments. Analysis of variance (ANOVA) was used for comparison between groups: ns, nonsignificant (p>0.05); *p<0.05; **p<0.01; ***p<0.001; ****p<0.0001.
To see whether TRIF was directly involved in TNF-R1-mediated cell death, we activated BMDMs with TNF in the presence of 5z7 (Fig. 1C), which like treatment with LPS/5z7, recapitulates the cell death driven by Y.p. infection dependent on caspase-8, RIPK1 kinase activity and the effector of pyroptosis Gasdermin-D (GSDMD) (Fig. S1A). To simplify interpretation of the data, we used recombinant human TNF (hTNF) that specifically activates TNF-R1, eliminating possible contributions from TNF-R2, which may induce cell death via divergent mechanisms. Interestingly, in addition to its dependence on TNF-R1 and TNF, hTNF/5z7-induced cell death was also strikingly dependent on TRIF, TRIF-related adaptor molecule TRAM and the LPS co-receptor CD14 (Fig. 1C, S1B). In contrast, TLR3 and TLR4 were not required for hTNF/5z7-induced cell death (Fig. 1C, S1B), thus suggesting a novel role for TRIF in mediating cellular responses to TNF. Before extending our investigation of the role of TRIF in TNF responses further, we ensured that our TRIF-dependent phenotype was not due to endotoxin contamination of our recombinant TNF, or differences in the levels of TNF-R1 in TRIF− and CD14-deficient BMDMs (Fig. S1C, D).
To determine if TRIF and CD14 were important for other modes of TNF-induced cell death, we treated BMDMs with hTNF in the presence of cycloheximide (CHX) or SMAC mimetic (SMAC) and the pan-caspase inhibitor zVAD to drive apoptosis or necroptosis respectively (Fig. S1E). Strikingly, deficiency in TRIF and/or CD14 but not TLR3 and/or TLR4 offered protection from TNF-induced apoptosis and necroptosis (Fig. S1E), demonstrating a role for these proteins beyond the caspase-8-mediated pyroptosis induced by hTNF/5z7.
In further characterization of this pathway, transcriptional activation of Tnf and Ifnb genes was abrogated in Trif−/−, and Cd14−/−, but not in Tlr3−/−Tlr4−/− BMDMs (Fig. 1D). This defect in the inflammatory signature was explained mechanistically by defective activation of MAPKs such as p38 and ERK in Tram−/−, Trif−/− and Cd14−/− but not Tlr4−/− macrophages activated with TNF or TNF/5z7 (Fig. 1E, S1F). Since 5z7 inhibits the upstream MAPK TAK1, the inflammatory phenotype was severely dampened in response to TNF/5z7 (Fig. S1F). However, deficiency in TRAM, TRIF and CD14 further attenuated p38 and ERK activation. In further support, IκBα degradation indicative of NF-κB activation was absent in Trif−/− and Tram−/− macrophages, and to a lesser extent in Cd14−/− BMDMs (Fig. 1E). These results stressed the critical role of TRIF and TRAM in activating MAPK and NF-κB pathways downstream of TNF, and identified an additional non-redundant role for CD14 in this process.
To further characterize the signaling pathways activated by TNF in a TRIF-dependent manner, we immunoprecipitated (IP) RIPK1 in TNF-activated macrophages and probed for pro-inflammatory complex components that are involved in TRIF-mediated signaling in response to LPS (Fig. 1F). Strikingly, we detected TRAF3, TBK1 and NEMO bound to RIPK1 in response to TNF, which was attenuated in the absence of TRIF, suggesting a critical role for TRIF in regulating pro-inflammatory complex formation in response to TNF (Fig. 1F). This was further supported by the TNF-mediated binding of the TRIF adaptor protein TRAM to RIPK1. Due to the fact that TRAF3 does not localize to the plasma membrane and is only accessible to receptors localized within the endosomal compartment (26), these results indicated that this TRIF-dependent pro-inflammatory complex induced by TNF occurred at the endosome. This finding was particularly interesting, since engagement of TRAF3 may explain how type I interferons can be induced downstream of TNF-R1 in a TRIF dependent manner.
While binding of pro-inflammatory components to RIPK1 was largely abrogated in the absence of TRIF, weak binding of TBK1 and NEMO persisted in TNF-activated cells (Fig. 1F). Since TBK1 and NEMO are known to function within TNF-R1-mediated complex I, this suggested that RIPK1 participates in TRIF-dependent and TRIF-independent pro-inflammatory complexes in response to TNF. In order to isolate the TRIF-dependent complex, we performed a TRAM-specific IP in TNF stimulated BMDMs using TRAM as a proxy of TRIF. Similar to the RIPK1-specific IP, we observed binding of RIPK1, TRAF3, TBK1 and NEMO in response to TNF, which was completely abrogated in the absence of TRIF (Fig. 1F). These results indicated a critical role for TRIF/TRAM in the formation of a RIPK1-containing pro-inflammatory complex in response to TNF that was distinct from TNF-R1-mediated complex I. For this reason, we have termed this complex the ‘pro-inflammatory TRIFosome’.
To confirm that similar requirements for TRIF and CD14 hold true for pro-death responses downstream of TNF-R1, we used TNF/5z7 to induce cell death, immunoprecipitated FADD, and probed for RIPK1 and caspase-8 indicative of pro-death complex formation. Formation of this complex was delayed and attenuated in the absence of TRIF or CD14 (Fig. 1G). As in the “pro-inflammatory TRIFosome” (Fig. 1F), we observed TRAM recruitment to the pro-death complex upon treatment with TNF/5z7, supporting interaction between the complex components and TRIF (Fig. 1G), and providing an explanation for the resistance to TNF/5z7-induced cell death observed in the absence of TRAM (Fig. 1C). In order to isolate the TRIF-dependent pro-death complex, we performed a TRAM-specific IP in TNF/5z7-stimulated macrophages (Fig. 1G). We observed binding of FADD, RIPK1 and caspase-8 to TRAM, which was absent in Trif−/− and Cd14−/− macrophages. Supporting the relevance of these TRIF-mediated interactions, deficiency in TRIF delayed and attenuated the cleavage of the apoptotic caspases-3 and-7, as well as the pyroptotic effectors caspase-1 and GSDMD downstream of caspase-8 cleavage (Fig. 1H). This suggested that TRIF-deficiency did not alter the mode of cell death induced by TNF/5z7, but delayed and dampened the response. In support of this, TNF/5z7-induced cell death in Trif−/− BMDMs was dependent on caspase-8 and RIPK1 and partially dependent on casapase-3 and -7 like B6 (Fig. 1I). As we have seen previously in the context of LPS/5z7-induced death (25), TNF/5z7-induced cell death in Trif−/− was entirely dependent on the kinase activity of RIPK1, given the protection offered by treatment with Nec-1 (Fig. 1I). These findings support a model in which, in the absence of TAK1 activity, TRIF-mediated activation downstream of TNF-R1 leads to the formation of a TRIF-dependent pro-death complex (pro-death TRIFosome) containing FADD, RIPK1 and caspase-8, which amplifies cell death via enhanced activation of GSDMD and executioner caspases (Fig. 1G, H). To date, TRIF has been described to function solely downstream of TLR3/4, however, these data point to a role for TLR pathway components downstream of activated TNF-R1.
In macrophages, CD14-mediated internalization of TNF-R1 promotes TRIF-RIPK1 colocalization
Upon LPS induction, CD14 mediates the endocytosis of TLR4 to promote TRIF activation. We hypothesized that CD14 may similarly regulate TRIF activation in response to TNF. Indeed, TNF simulation resulted in the internalization of CD14 within one hour (Fig. S2A), and led to the colocalization of TNF-R1 with RAB5A+ endosomes as early as 10 minutes after treatment (Fig. S2B, C). Strikingly, TNF-R1 endosomal localization was at least partially dependent on CD14, but occurred independently of TRAM, indicating a specific role for CD14 in the internalization of TNF-R1, and suggesting potential undiscovered roles for CD14 in the internalization and activation of additional immune receptors within the TLR and TNF superfamily (Fig. S2B, C). Although TRIF is notoriously difficult to detect by western blot, TRIF-specific puncta were detectable in TNF-activated macrophages (Fig. S2D, E). These puncta colocalized with RAB5A+ endosomes but were only detectable 10–30 minutes after exposure to TNF (Fig. S2D–F). TRIF-specific puncta failed to form in the absence of CD14 or TRAM (Fig. S2D–F). These results suggested that CD14 was required for TRIF endosomal localization and oligomerization. Given the ability of TRAM to localize to regions of the plasma membrane enriched for CD14 in response to LPS, yet independently of TLR4 (27), it is possible that in response to TNF, CD14 enrichment within the endosome may be sufficient for TRAM/TRIF endosomal localization and activation.
Given the integral role that RIPK1 plays in TRIF-dependent death and inflammatory signaling in response to TNF, and the fact that we detected RIPK1 bound to the TRIF trafficking adaptor TRAM in response to both mTNF or mTNF/5z7 (Fig. 1F, G), we tested whether TRIF-RIPK1 interactions were possible to visualize via immunofluorescence. Accordingly, we used our validated anti-TRIF antibody to look for colocalization of RIPK1 with TRIF in TNF- or TNF/5z7-stimulated macrophages (Fig. 2A–E). In response to both mTNF and mTNF/5z7, TRIF puncta were observed 10–30 minutes after stimulation (Fig. 2A–C). However, puncta were larger but less numerous in response to TNF alone, as compared to TNF/5z7, thus suggesting that polarization of cells towards pro-inflammatory signaling sustained TRIF:RIPK1 interactions and complex formation (Fig. 2A–C). After 20 minutes of stimulation with mTNF or mTNF/5z7, TRIF and RIPK1 were highly colocalized (Fig. 2A, D, E). However, the formation and colocalization of TRIF and RIPK1 specific puncta were abrogated in the absence of CD14, suggesting that CD14 functions upstream of TRIF oligomerization and recruitment to RIPK1 (Fig. 2A–E). Further, TNFR1-deficient cells facilitated, albeit much weaker, aggregation of TRIF and its co-localization with RIPK1 (Fig. 2A–E).
Fig. 2. CD14-mediated internalization of TNF-R1 promotes TRIF-RIPK1 colocalization in macrophages.
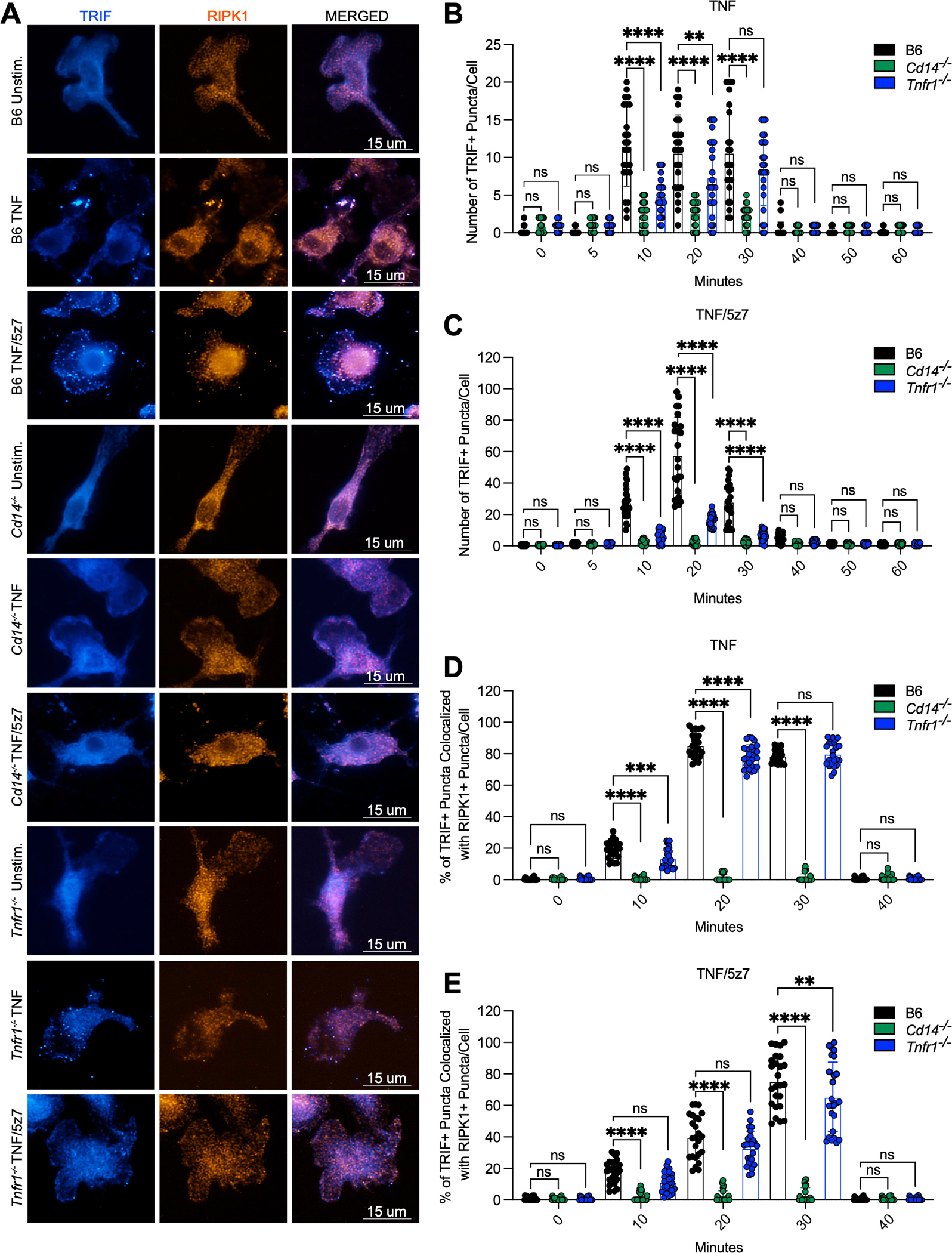
(A) Representative 60X images of TRIF or RIPK1 staining and colocalization in B6, Cd14−/− and Tnfr1−/−macrophages stimulated with mTNF or mTNF/5z7 for 20 minutes. (B, C) Quantification of the number of TRIF+ puncta/cell over time in response to (B) mTNF or (C) mTNF/5z7 in B6, Cd14−/− and Tnfr1−/−macrophages. (D, E) Quantification of the percentage of TRIF+ puncta colocalized with RIPK1+ puncta/cell over time in response to (D) mTNF or (E) mTNF/5z7 in B6, Cd14−/− and Tnfr1−/−macrophages. Data from imaging experiments are representative of three or more independent experiments, data points indicate individual cells, n=25. Analysis of variance (ANOVA) was used for comparison between groups: ns, nonsignificant (p>0.05); *p<0.05; **p<0.01; ***p<0.001; ****p<0.0001.
TRIF and CD14 mediate TNF-induced lethality
To look for a role for TRIF-mediated responses to TNF in a physiological context, we considered that both caspase-8 and GSDMD play crucial roles in mouse models of LPS- and TNF-lethality (28–32), suggesting that responses to TNF in vivo might also be regulated by TRIF. To this end, we challenged wildtype B6, Trif−/−, Cd14−/−, and several other mice with deficiencies for genes of interest, with murine TNF (mTNF, 9ug, i.v.) and monitored body temperature hourly (Fig. 3A). While B6 mice succumbed to TNF-induced lethality within 4 to 8 hours, Trif−/− and Cd14−/− mice were resistant to TNF-induced hypothermia, with ~70% of animals surviving for at least 24 hours after TNF injection (Fig. 3B). Strikingly, similar to Tnfr1−/− mice, 100% of Cd14−/−Trif−/− double-knockout mice were protected from TNF-lethality (Fig. 3A, B). However, TNF-deficiency provided only a slight delay in lethality (Fig. 3A, B). TNF injection induces rampant intestinal epithelial cell (IEC) death, disrupting the epithelial barrier and allowing for leakage of the gut microbiome (33). Therefore, it was possible that microbial leakage from the gut, as well as the release of endogenous damage signals could be responsible for driving our TRIF-dependent phenotype in response to TNF. However, Tlr3−/−, Tlr4−/− and Tlr3−/−Tlr4−/− double-knockout mice were similarly susceptible to TNF-induced hypothermia compared to B6 (Fig. 3A, B), thus dismissing the possibility that TRIF drove TNF-induced responses downstream of TLR3/4, and alluding to a novel mode of activation of the endosomal adaptor TRIF.
Fig. 3. TRIF and CD14 mediate TNF-induced lethality.
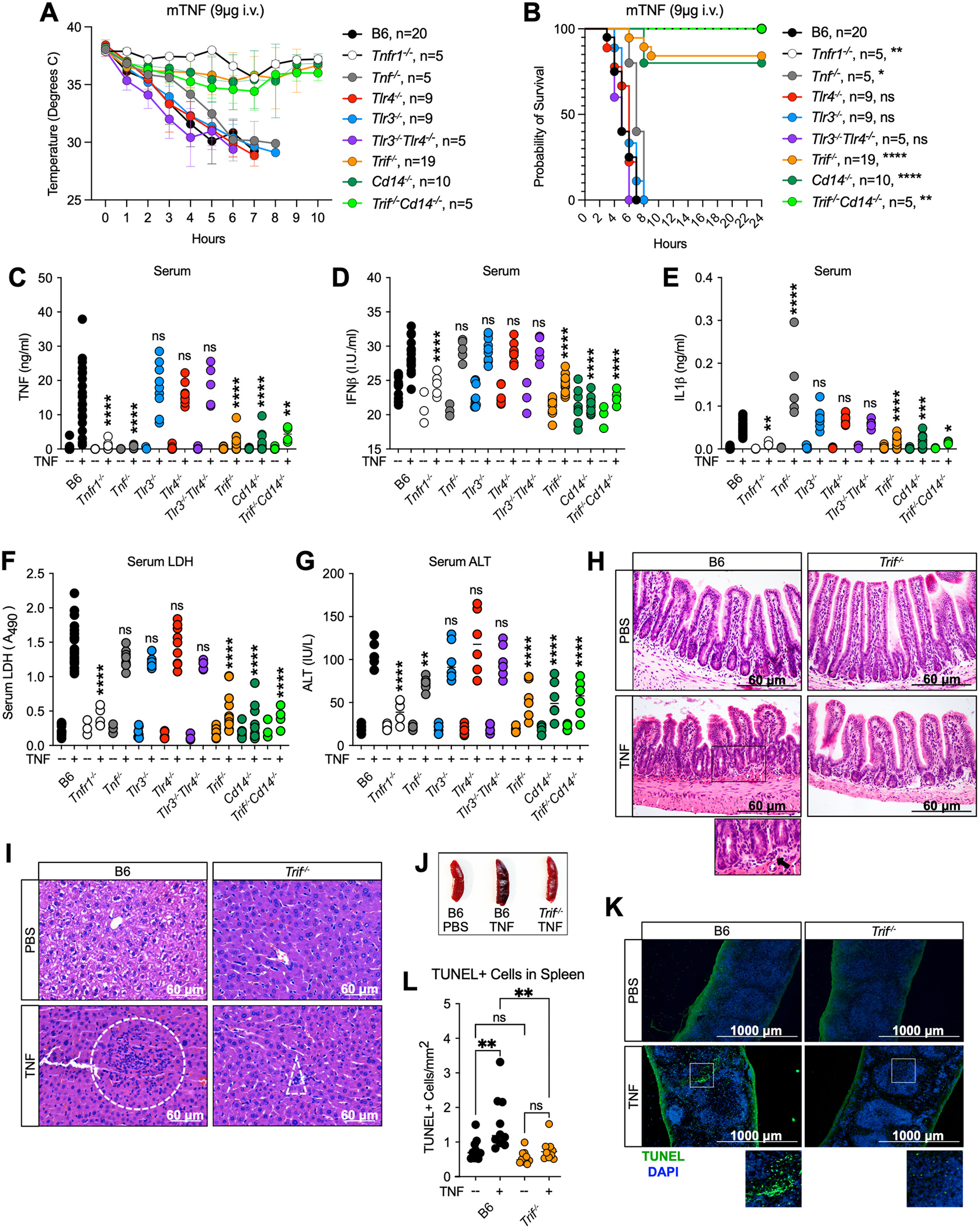
(A, B) Mouse body temperature (A) and survival (B) over time in B6, Tnfr1−/−, Tnf−/−, Trif−/−, Cd14−/−, Trif−/−Cd14−/− Tlr3−/−, Tlr4−/− and Tlr3−/−Tlr4−/− mice after intravenous injection with 9ug mTNF. (C-G) Serum levels of (C) TNF, (D) IFNβ, (E) IL-1β, (F) relative lactate dehydrogenase (LDH) and (G) Alanine Aminotransferase (ALT) in indicated mice 4 hours after i.v. injection with 9ug mTNF. Data points indicate individual mice tested (B6 PBS, n=13; Tnfr1−/− PBS, n=3; Tnf−/− PBS, n=3; Trif−/− PBS, n=10; Cd14−/− PBS, n=7; Trif−/−Cd14−/− PBS, n=3; Tlr4−/− PBS, n=6; Tlr3−/− PBS; n=9; Tlr3−/−Tlr4−/− PBS, n=3 B6 mTNF, n=22; Tnfr1−/− TNF, n=5; Tnf−/− TNF, n=5, Trif−/− mTNF, n=14; Cd14−/− mTNF, n=10; Trif−/−Cd14−/− TNF, n=5; Tlr4−/− mTNF, n=9; Tlr3−/− mTNF, n=9; Tlr3−/−Tlr4−/− TNF, n=5 ). (H, I) H&E staining of (H) ileum and (I) liver sections from B6 and Trif−/− mice 4 hours after injection with 9ug mTNF or PBS. Dotted boxes indicate areas of leukocyte recruitment. (J) Representative image of spleens from B6 and Trif−/− mice injected with 9ug TNF or PBS 6 hours after injection. (K) TUNEL staining and (L) quantification of TUNEL+cells/mm2 in the spleen of B6 and Trif−/− mice 4 hours after injection with 9ug mTNF. Each point represents 1 field of view across 3 biological replicates. Analysis of variance (ANOVA) was used for comparison between groups: ns, nonsignificant (p>0.05); *p<0.05; **p<0.01; ***p<0.001; ****p<0.0001.
In further mechanistic inquiry of the phenotype, Trif−/−, Cd14−/− and Cd14−/−Trif−/−, but not Tlr3−/−, Tlr4−/− or Tlr3−/−Tlr4−/− mice exhibited attenuated accumulation of TNF, IFNβ and IL1β in the serum 4 hours after TNF injection (Fig. 3C–E), indicating that TRIF and CD14, like TNFR1 are critical for mounting a pro-inflammatory response to TNF in vivo. Lower levels of IL-1β released in the serum of Trif−/− and Cd14−/− mice as a consequence of inflammatory cell death implied that TRIF and CD14 might also regulate cell death in response to TNF. Indeed, serum lactate dehydrogenase (LDH) and alanine aminotransferase (ALT) levels indicative of lytic cell death and liver damage respectively were decreased in the absence of TRIF and CD14 (Fig. 3F, G). Villus blunting and IEC death is a hallmark of the TNF-toxicity in mice (9). In support of this, we observed epithelial sloughing, villus blunting and edema of the lamina propria in the ileum of B6 that was lessened in Trif−/− mice (Fig. 3H, S3A). Notably, mucosal edema was identified in both PBS and TNF injected Trif−/− mice and was interpreted as a potential background change in mice with TRIF-deficiency. However, unlike B6, TNF-injection did not increase the severity or incidence of edema in the absence of TRIF (Fig. S3A). Further magnification of the B6 intestine 4 hours after TNF injection showed increased leukocyte (mainly neutrophil) infiltration that was absent in the TNF-treated TRIF-deficient animals (Fig. 3H, S3A). Similarly, increased leukocyte infiltration was observed in the liver of B6, and to a lesser extent Trif−/− mice in response to TNF (Fig. 3I, S3B). Additionally, while the spleens from Trif−/− mice appeared grossly normal 6 hours after TNF injection, B6 spleens exhibited considerable darkening indicative of tissue damage (Fig. 3J). To this end, B6 spleens exhibited evidence of red pulp congestion that was lessened in the absence of TRIF (Fig. S3C, D). Additionally, TNF-induced splenic cell death was markedly decreased in Trif−/− mice as evidenced by TUNEL staining (Fig. 3K, L). In support of a role for the TRIF pathway in the regulation of not only cell death but also inflammatory responses in the spleen, spleen TNF and IFNβ levels were significantly attenuated in Trif−/−, Cd14−/− and Trif−/−Cd14−/− mice (Fig. S3E). Interestingly, Tlr3−/−, Tlr4−/− and Tlr3−/−Tlr4−/− mice exhibited slightly elevated cytokine levels in the spleen compared to B6 (Figure S3E), suggesting that TLR3 and TLR4 may sequester stores of TRIF, dampening the response to TNF.
A cell-specific requirement for TRIF in the regulation of immune responses to TNF
To identify cells that rely on TRIF to mediate TNF-induced splenic morphology and immune cell infiltration (Fig. 3J, S3C, D), we performed single cell RNA sequencing (SCS) on the spleens of mice 4-hours after injection with TNF. We identified 8 cellular clusters corresponding to discrete immune cell populations, including a cluster of cells with a combination of markers from splenic red pulp macrophages (RPMs) and erythrocytes (Fig. 4A, S4A). RPMs play a critical role in phagocytosis of damaged and senescent erythrocytes and produce type I interferons in response to various blood-borne pathogens (34). Together with the fact that erythrocytes present in our splenic samples were lysed prior to sequencing, this cluster likely represents RPMs containing phagocytosed erythrocytes (Fig. 4A). Proportions of B cell, T cell and dendritic cell populations remained largely unchanged in response to TNF administration (Fig. 4B, C). However, proportions of RPM, neutrophil, and macrophage populations were expanded in response to TNF in Trif−/− but not B6 spleens, supporting a role for TRIF in TNF-responses in vivo in these cellular subsets, and suggesting immune cell recruitment to and/or proliferation in the spleen in the absence of overwhelming cell death (Fig. 4B, C). Some cellular subsets such as monocytes and NK cells responded to TNF in a TRIF-independent manner, further suggesting that the TRIF-dependent response to TNF is likely cell-type specific (Fig. 4B, C).
Fig. 4. A cell-specific requirement for TRIF in the regulation of immune responses to TNF.
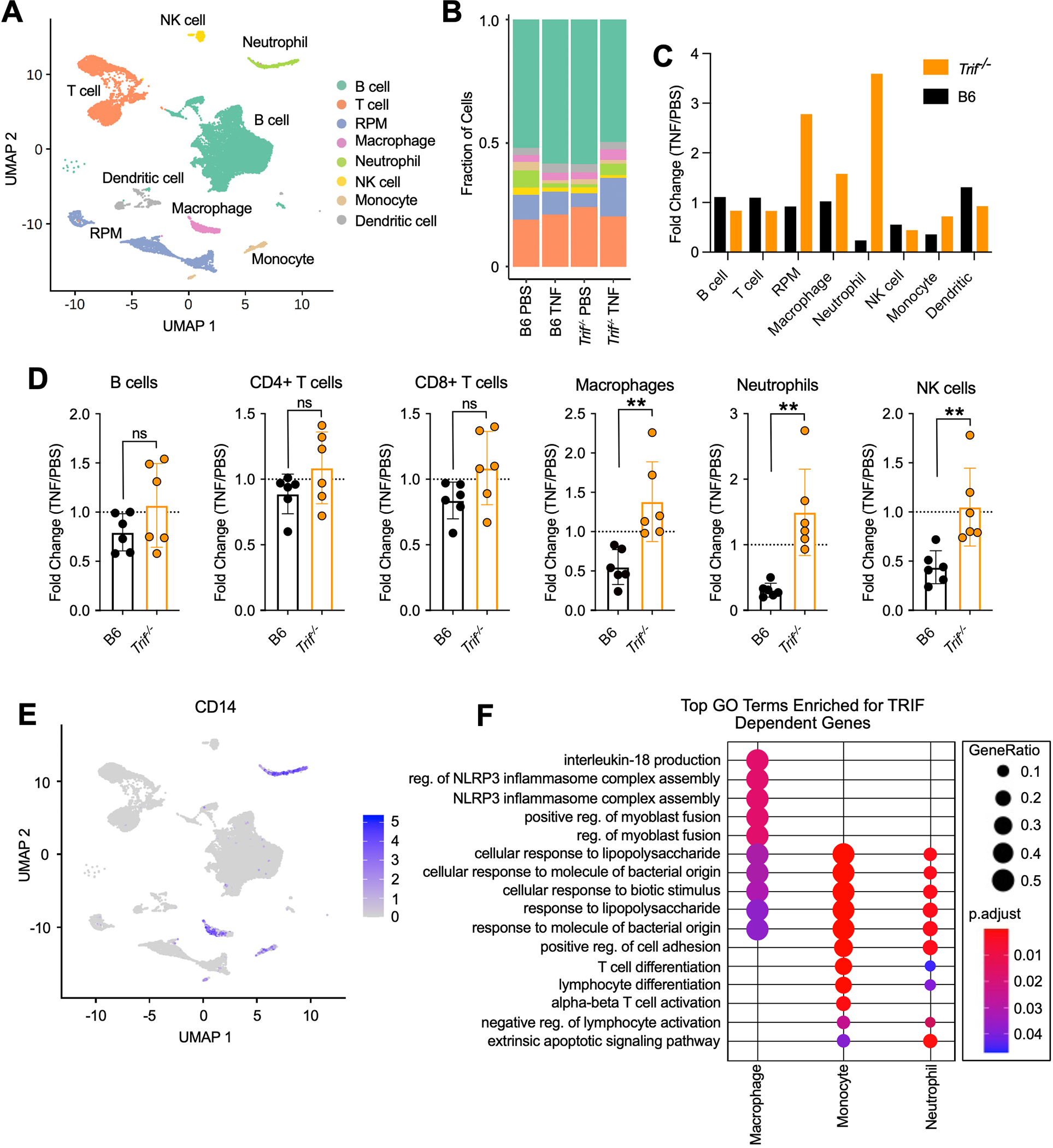
(A) UMAP of splenocytes from B6 and Trif−/− mice 4 hours after intravenous injection with 9ug mTNF or PBS. Colors represent cell type annotations. (B) Fraction of cells corresponding to each cell type annotation in B6 and Trif−/− mice 4 hours after intravenous injection with 9ug mTNF or PBS. (C) Fold change (TNF/PBS) in proportion of cells corresponding to each cell type annotation in B6 and Trif−/− mice 4 hours after intravenous injection. (D) Fold change in the number of immune cells in the spleen 4 hours after injection with TNF compared to PBS in B6 and Trif−/− mice. Data points indicate individual mice tested (B6 TNF, n=6; Trif−/− TNF, n=6). Bars represent the mean ± SD. Analysis of variance (ANOVA) was used for comparison between groups: ns, nonsignificant (P>0.05); *P<0.05; **P<0.01; ***P<0.001; ****P<0.0001. (E) UMAP showing CD14 expression in splenocytes from B6 and Trif−/− mice. (F) Top Gene Ontology (GO) Biological Process categories enriched for genes that were significantly upregulated in B6 mice in response to TNF, but significantly less upregulated in Trif−/− mice in indicated cell types. GeneRatio: The ratio of the number of genes in the query set that are annotated by the GO ID and the number of genes in the query set that are annotated in the database of all GO IDs. p.adjust calculated using the Benjamini-Hochberg method.
In confirmation of our SCS data, flow cytometry-analysis of mouse spleens 4-hours after injection with TNF revealed that the number of macrophages and neutrophils were dramatically decreased in B6 spleens, and increased in the spleens of Trif−/− mice in response to TNF (Fig. 4D, S4B, S5A). In contrast, the number of B and T cells remained largely consistent across genotypes in response to TNF (Fig. 4D, S4B, S5A). Although our SCS data suggested that NK cells respond to TNF in a TRIF-independent manner, flow cytometry-analysis suggested that like macrophages and neutrophils, responses in NK cells were also dependent on TRIF (Fig. 4D, S4B, S5A).
In further confirmation that TRIF-dependent responses to TNF correlated with the expression of TRIF and CD14, CD14 was highly expressed specifically in macrophage and neutrophil clusters (Fig. 4E), suggesting that dependency of TNF-responses on TRIF was observed in cells with high levels of CD14. To identify pathways that specifically relied on TRIF in response to TNF, we compared the top Gene Ontology (GO) Biological Process terms that were dependent on TRIF for each cluster (Fig. 4F, S5B). Of particular interest were the GO terms identified for the macrophage, monocyte and neutrophil populations, including “NLRP3 inflammasome complex assembly”, “cellular response to lipopolysaccharide” and “extrinsic apoptotic signaling pathway” (Fig. 4F, S5B). These data indicated that TRIF, which so far was believed to contribute exclusively to TLR-mediated responses, promoted TNF-induced lethality in vivo by driving inflammatory cytokine production and subsequent cell death, likely of specific cell populations. Additionally, these findings highlighted the importance of a balanced immune response, as the low levels of cytokine production observed in Trif−/− mice (Fig. 3C–E) were sufficient to promote immune cell recruitment and/or proliferation in the spleen (Fig. 4B–D), without driving massive cell death (Fig. 3F–K) and animal lethality (Fig. 3A, B).
Hematopoietic cells are the effectors of TRIF-dependent responses to TNF
To identify the subsets of cells that respond to TNF in a TRIF-dependent manner, we reconstituted lethally irradiated CD45.1 B6 mice with bone marrow from CD45.2 B6 or Trif−/− donors (Fig. S6A, B). Monitoring mouse body temperature (Fig. 5A) revealed that hematopoietic cells conferred the TRIF-dependent TNF-lethality phenotype. That is, when mice were reconstituted with B6 bone marrow (B6 BM), they succumbed to TNF-lethality, while mice reconstituted with Trif−/− bone marrow (Trif−/− BM) were highly protected (Fig. 5A). Interestingly, all recipient mice regardless of the bone marrow genotype exhibited an early drop in temperature reminiscent of the temperature drop observed in all mice regardless of genotype (aside from Tnfr1−/− mice), suggesting early cellular responses to TNF did not rely on TRIF (Fig. 3A, 5A). In further support of the importance of TRIF in hematopoietic cells, serum cytokine, LDH and ALT levels were attenuated in Trif−/− BM compared to B6 BM mice, suggesting that TRIF activity in hematopoietic cells was essential for the inflammatory and cell death responses to TNF (Fig. 5B–D).
Fig. 5. Hematopoietic cells are the effectors of TRIF-dependent responses to TNF.
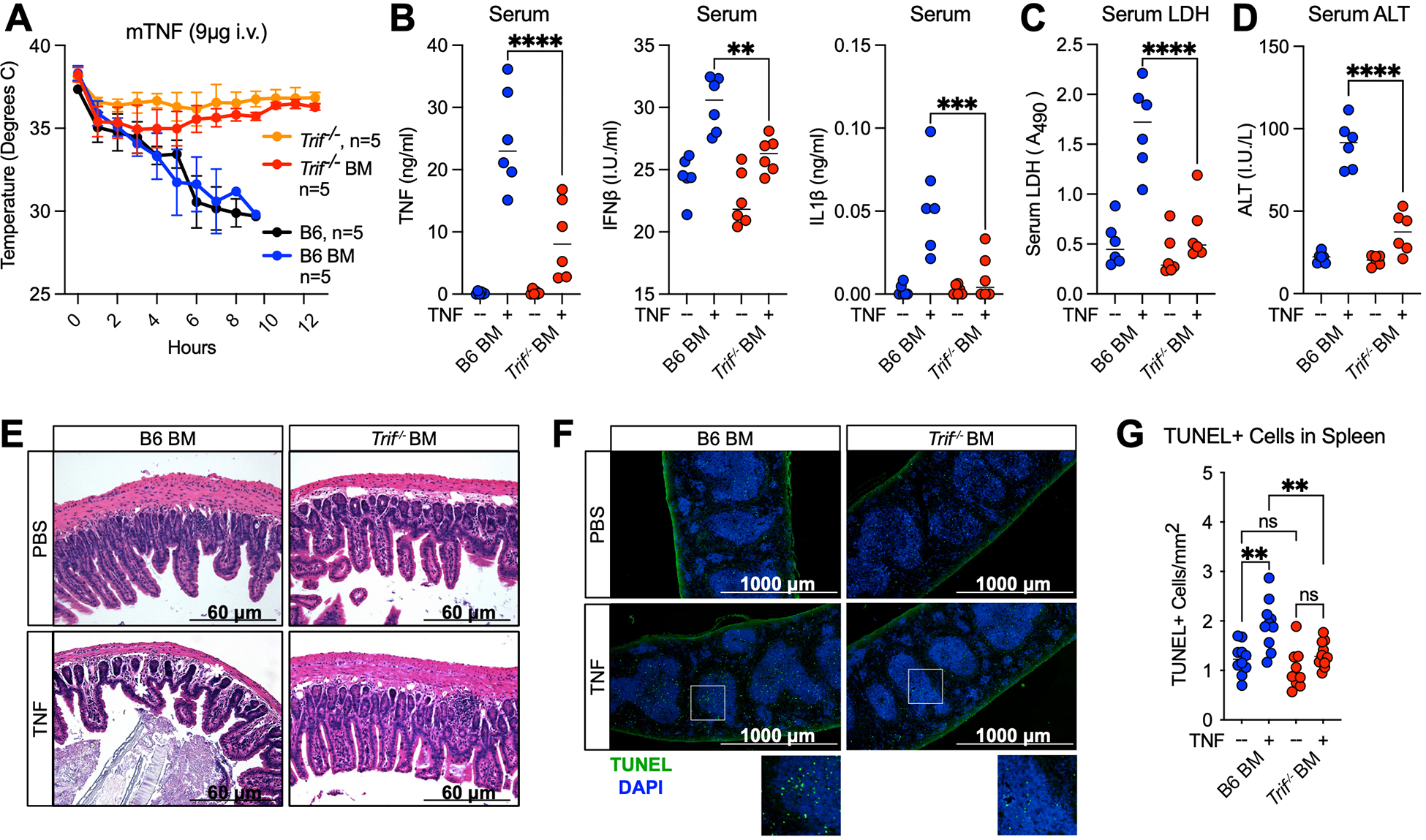
(A) Mouse body temperature over time in B6, Trif−/−, and lethally irradiated B6 mice reconstituted with B6 (B6 BM) or Trif−/− (Trif−/− BM) bone marrow followed by intravenous injection with 9ug mTNF. (B-D) Serum (B) TNF, IFNβ and IL-1β protein levels (C) relative lactate dehydrogenase (LDH) levels and (D) Alanine Aminotransferase levels (ALT) in indicated mice 4 hours after injection with 9ug mTNF. Data points indicate individual mice tested, n=6 (E) H&E staining of ileum sections from B6 BM and Trif−/− BM mice 4 hours after injection with 9ug mTNF. *indicate areas of villus blunting and edema. (F) TUNEL staining and (G) quantification of TUNEL+cells/mm2 in the spleen of B6 BM and Trif−/− BM mice 4 hours after injection with 9ug mTNF. Each point represents 1 field of view across 3 biological replicates. Analysis of variance (ANOVA) was used for comparison between groups: ns, nonsignificant (p>0.05); *p<0.05; **p<0.01; ***p<0.001; ****p<0.0001.
Finally, villus blunting and edema of the lamina propria observed in the ileum of B6 BM mice was abrogated in Trif−/− BM mice, suggesting that hematopoietic cells contributed to intestinal damage in the context of TNF-lethality in a TRIF-dependent manner (Fig. 5E, S6C). Intestinal damage was likely caused by the increase in neutrophil recruitment in B6 but not Trif−/− intestines, increasing local inflammation and driving cell death and necrosis (Fig. 3H, S3A). Histology of B6 BM spleens displayed decreased lymphoid regions and evidence of red pulp congestion that was lessened in the absence of TRIF in hematopoietic cells (Fig. S6D, E). Additionally, TNF-induced splenic cell death was markedly decreased in Trif−/− BM compared to B6 BM mice (Fig. 5F, G). These findings demonstrated that hematopoietic cells conferred TRIF-dependent cell death and inflammatory responses to TNF, and significantly contributed to the pathology observed in the intestine and spleen in the context of TNF-lethality.
In the context of TNF-lethality, neutrophils control TRIF-dependent inflammatory responses while macrophages mediate cell death
To identify which hematopoietic cells mediate TNF-toxicity in a TRIF-dependent manner, we observed no resistance to TNF-toxicity in Rag−/− mice, suggesting that B and T cells did not confer susceptibility to TNF (Fig. S7A). In fact, Rag−/− mice exhibited exacerbated hypothermia and elevated serum cytokine levels compared to B6 (Fig. S7B). These results are unsurprising given previous reports that adaptive immune cells play a role in dampening TNF-dependent innate immune responses (35). Conversely, neutrophil depletion with the monoclonal Ly6G antibody 1A8 (Fig. S7C, D) delayed and decreased lethality in B6 mice (Fig. 6A, B), suggesting a role for neutrophils in TNF-lethality. To determine if neutrophils were responsible for TRIF-dependent responses to TNF specifically, we performed a gain of function (GOF) experiment in which TNF-resistant Trif−/− mice were reconstituted with B6 or Trif−/− neutrophils. Strikingly, mice reconstituted with B6 but not Trif−/− neutrophils were susceptible to TNF-lethality, further suggesting that neutrophils likely contributed to TNF-lethality in a TRIF-dependent manner (Fig. 6C). Since macrophages and neutrophils exhibited similar population changes and TRIF-dependent pathways in our SCS analysis, we performed a similar GOF experiment in which Trif−/− mice were reconstituted with B6 or Trif−/− macrophages (Fig. 6C). Similar to neutrophils, mice reconstituted with B6 but not Trif−/− macrophages gained susceptibility to TNF (Fig. 6C). Interestingly, while addition of B6 neutrophils enhanced serum TNF and IFNβ levels in response to TNF, it had little to no impact on serum IL1b, LDH or ALT levels (Fig. 6D–F). Instead, accumulation of IL1β, LDH and ALT in the serum appeared to depend on macrophages in a TRIF-dependent manner (Fig. 6D–F). This finding was supported by our SCS data in which genes associated with “interleukin-1 production” and “NLRP3 inflammasome complex assembly” were enriched specifically in the macrophage population (Fig. 4F). These data further suggest that neutrophils were responsible for TRIF-dependent inflammatory responses to TNF, while cell death responses to TNF were regulated at the level of macrophages.
Fig. 6. In the context of TNF-lethality, neutrophils control TRIF-dependent inflammatory responses while macrophages mediate cell death.
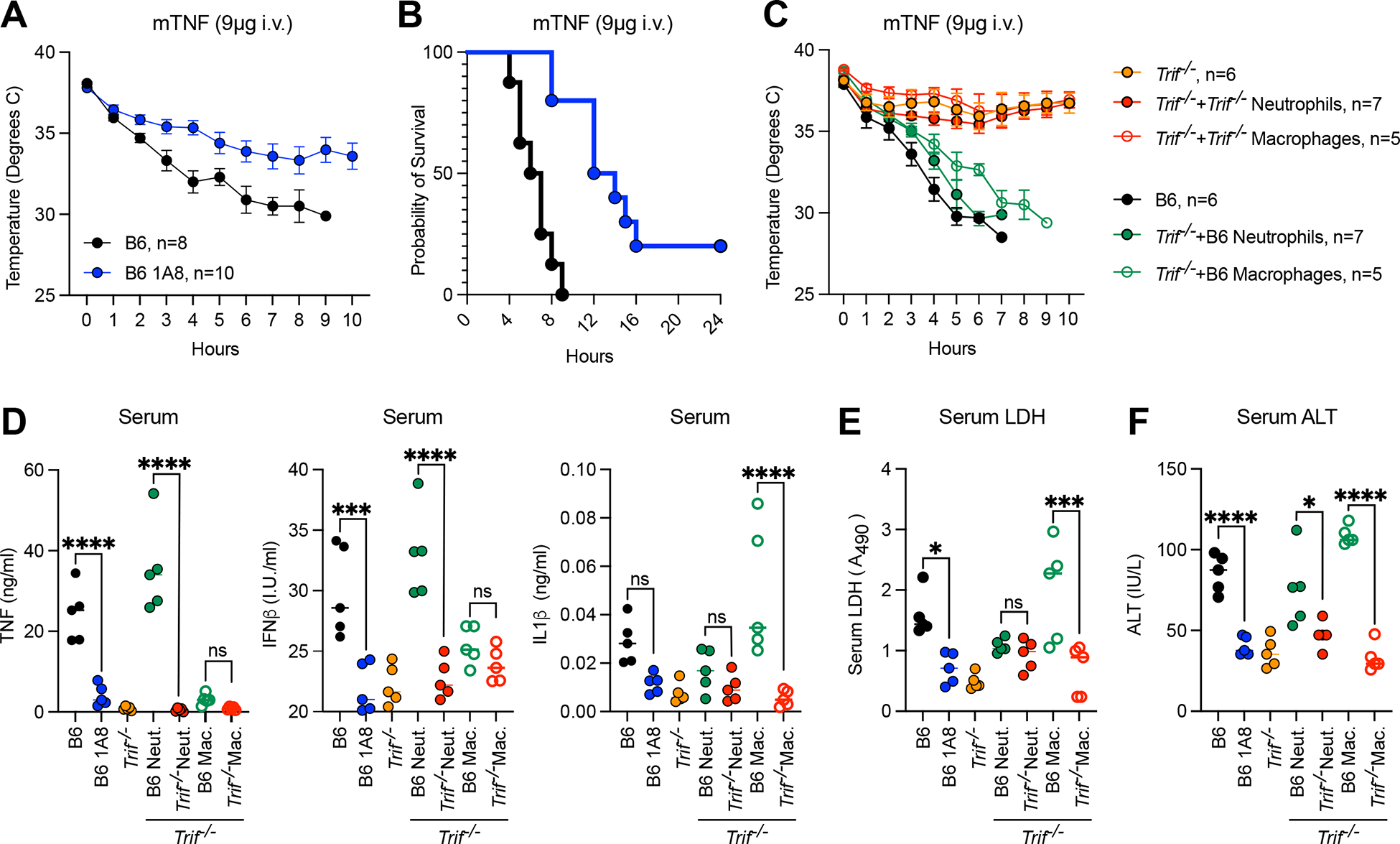
(A, B) Mouse body temperature (A) and survival (B) over time in neutrophil depleted (Ly6G antibody, 1A8, 24 hours) and isotype control treated B6 mice after injection with 9ug mTNF. (C) Mouse body temperature over time and serum (D) TNF, IFNβ and IL-1β protein levels (E) relative lactate dehydrogenase (LDH) levels and (F) Alanine Aminotransferase levels (ALT) 4 hours after injection with 9ug mTNF in Trif−/− mice reconstituted with 1×106 macrophages or neutrophils from B6 or Trif−/− donors. Data points indicate individual mice tested, n=5. Analysis of variance (ANOVA) was used for comparison between groups: ns, nonsignificant (p>0.05); *p<0.05; **p<0.01; ***p<0.001; ****p<0.0001.
DISCUSSION
The present study advances the model of TNF-induced toxicity in mice, which is poorly characterized compared to toxicity induced by LPS. Specifically, it has been reported that intestinal epithelial cells are the main targets of TNF (36), leading to “leaky gut”. Our data support the occurrence of epithelial injury but suggest that this injury is mediated by hematopoietic cells, perhaps via neutrophil inflammatory responses and NET formation and macrophage death. This study also establishes that TRIF and CD14 are critical mediators of TNF activity in vitro and in vivo. That is, TRIF− and CD14− deficient mice exhibited exquisite protection from TNF-induced lethality, which models the deleterious hyperinflammatory “cytokine storm” associated with systemic inflammatory response syndrome (SIRS) and sepsis. Not only were serum cytokine levels dampened by TRIF-deficiency, but intestinal epithelial cell death and villus blunting, often associated with TNF-induced lethality in mice (9), were ameliorated in the absence of TRIF. These finding allude to a similar role for TRIF and CD14 in other contexts such as Fas-mediated cell death.
At the molecular level, the kinase activity of RIPK1 drives vascular permeability and endothelial cell activation, conferring TNF-induced lethality (37, 38). In contrast, our results demonstrated that the TRIF-dependent response to TNF is conferred, at least in part by hematopoietic cells, specifically macrophages and neutrophils. However, a role for TRIF in nonhematopoietic cells such as endothelial cells cannot be excluded, particularly given the early drop in temperature observed in Trif−/− BM compared to TRIF whole body knockout mice. The possibility that TRIF contributes to TNF-responses across cell lineages is perhaps unsurprising given that TRIF acts upstream of RIPK1 to mediate both inflammation and cell death, while the role of the kinase activity of RIPK1 is confined mostly to the induction of cell death pathways. Furthermore, while treatment for sepsis remains difficult, simultaneous inhibition of inflammation and endothelial damage have shown encouraging results (39). Our results implicate TRIF and CD14 as potential therapeutic targets capable of simultaneously modulating both inflammatory and cell death responses in the context of SIRS or sepsis. To this end, clinical trials with monoclonal antibodies against CD14 have shown decreased systemic inflammation and organ damage in sepsis and COVID-19 patients (40, 41).
Mechanistically, we showed that TRIFosome formation underlies TNF-induced lethality, identifying an unexplored role for TRIF in the context of inflammatory diseases. Similar to its function in the context of TLR4 ligation, CD14 appears to regulate TNFR-1 endocytosis, placing TRAM in the proximity of TRIF. In this way, the TRIF-mediated pathway promotes the sustained activation of inflammatory signaling and cell death pathways in response to TNF. This function of TRIF is strikingly similar to the role of TRIF in the context of TLR4 signaling, in which MyD88 mediates early inflammatory signaling at the plasma membrane while TRIF regulates sustained inflammatory signaling and cell death within the endosomal compartment. In doing so, TRIF becomes a universal adaptor with dual roles in myeloid cells by mediating TNF-synthesis in response to LPS as well as responses to TNF. These findings point to the possibility of unexplored crosstalk between innate immune pathways, and imply that TRIF, functioning as a universal adaptor may function in signal transduction downstream of additional diverse receptors of immune relevance. Furthermore, these results suggest that the endocytosis pathway mediated via CD14 may engage additional receptors present at the plasma membrane such as TLRs or members of the TNF superfamily, stressing the need to study CD14 in various pathogenic contexts. For instance, in neurodegenerative conditions such as Alzheimer’s disease, the role of TLR4-activation in response to the “danger molecules” has been suggested (42), especially in CD14+ microglia. However, the possibility of CD14 mediating responses to TNF has not been considered. Conversely, the role of TNF as a mediator of acute brain injury has been established (43), but not in the context of CD14+ microglia. In this regard, our data provide rationale for further investigation of the role of CD14 in TNF-induced brain injury and in other neurodegeneration pathologies where the role of CD14+ cells has been established.
MATERIALS AND METHODS
Study Design
The aim of this study was to determine the role of key components of the TLR4 signaling pathway, TRIF and CD14 in the cell death and inflammation induced by TNF. We performed cellular viability assays, qPCR, immunoblot and immunofluorescence colocalization-based studies in bone marrow derived macrophages from various genetic knockout mouse lines to determine the effect and mechanism of TRIF and CD14-mediated interactions in the context of TNF-stimulation. To determine the physiological relevance of TRIF and CD14-deficiency, we induced TNF-lethality in various genetic knockout mice and bone marrow chimeras and assessed mouse survival, the levels of tissue and serum cytokines and cell death markers and histopathological changes in the ileum, liver, and spleen. To identify TRIF-dependent changes in immune cell populations as a result of proliferation, recruitment or cell death we performed single cell sequencing and flow cytometry on splenocytes from TNF-injected wildtype and TRIF-deficient mice.
Mice and Primary cells
C57BL/6 (B6), Tlr4−/−, Tlr3−/−, Ticam1−/− (Trif−/−), Cd14−/−, Rag−/−, Tnfr1−/− and Tnf−/− mice were obtained from The Jackson Laboratory. Trif−/−Cd14−/− and Tlr3−/−Tlr4−/− mice were generated by in-house breeding. Ripk1K45A/K45A (RIPK1 Kinase Inactive) and Ripk3K51A/K51A (RIPK3 Kinase Inactive) mice were provided by Dr. A. Degterev. Mlkl−/− mice were generated by Dr. W. Alexander and were a gift from Dr. M. Kelliher. Mice were housed according to protocols approved by the Tufts University Medical School Animal Care and Use Committees. Femurs from Ticam2−/− (Tram−/−) mice were generously donated by Dr. L. Li. Femurs from Gsdmd−/− mice were donated by Dr. K. Fitzgerald and were generated by Dr. V. Dixit. Femurs from Ripk3−/−Casp8−/− mice were donated by Dr. K. Fitzgerald and were originally generated by Dr. D. Green. Femurs from Casp3−/−Casp7−/− were generated and provided by Dr. Anthony Rongvaux. To generate the bone marrow derived macrophages (BMDMs) used in this study, bone marrow was isolated from the long bones of mice, propagated in RPMI containing 20% FBS, 2% Pen-Strep and 30% L cell supernatant on non-tissue culture treated Petri dishes for 7 days. Once differentiated, bone marrow derived macrophages were plated for experiments at a density of 1 × 106 cm2 in RPMI containing 20% FBS and 2% Pen-Strep. To isolate neutrophils, mice were primed with 1 ml thioglycolate for 8 hours. Peritoneal cells enriched for neutrophils were isolated in cold PBS and plated for experiments at a density of 1 × 106 cm2 in RPMI containing 20% FBS and 2% Pen-Strep.
TNF-induced lethality mouse model
Prior to induction of the TNF-induced lethality model, mice were co-housed for at least two weeks. 11-week-old male and female mice were injected intravenously with 9ug of recombinant murine TNF in sterile PBS (200 ul), or equal volume of sterile PBS for control mice. Temperature was monitored hourly by rectal thermometer. At indicated timepoints after administration of TNF, or when mouse body temperature reached < 30°C, mice were euthanized by CO2 asphyxiation, and spleen, liver and blood were harvested for quantification of cytokine levels by ELISA, serum lactate dehydrogenase levels using the CyQuant LDH Cytotoxicity Assay kit (C20300) and Alanine Aminotransferase (ALT) levels using the Pointe ALT kit according to the manufacturer’s instructions.
Murine TNF (DY410) and IL1β (DY401) DuoSet ELISA Kits were used according to the manufacturer’s instructions to quantify serum and tissue cytokine levels. For IFNβ ELISAs, 384-well ELISA plates were coated overnight at 4°C with monoclonal rat anti-mouse IFNβ antibody (Santa Cruz sc-57201, 1:500 dilution in 0.1M carbonate buffer) and blocked with 10% FBS in PBS for 2hrs at 37°C. Samples were incubated on plates overnight at 4°C, before washing with 0.005% TWEEN in PBS, and adding polyclonal rabbit anti-mouse IFNβ antibody (R&D Systems 32400–1, 1:2000 dilution in 10% FBS in PBS) overnight at 4°C. After washing, goat anti-rabbit-HRP antibody (Cell Signaling Technologies, 7074, 1:2000 dilution in 10% FBS in PBS) was added for 2–3 hrs at room temperature. TMB substrate was added and reaction was stopped with 2N H2SO4.
To quantify changes in cell populations within the spleen in response to TNF by flow cytometry, spleens were harvested from B6 and Trif−/− mice 4 hours after intravenous TNF-α or PBS injection. Single cell suspensions were generated via mechanical digestion and mixed with ACK lysis buffer for 3 min to lyse RBCs. Cells were mixed with CD16/CD32 Fc block for 20 min on ice. Cells were then stained with the following monoclonal antibodies for flow cytometry: CD19 (45-0193-80), F4/80 (12-4801-80), GR-1 (48-5931-80), and NK1.1 (63-5941-80) from Thermofisher Scientific; B220 (103221) and CD11c (11736) from Biolegend; Cd11b (25-112-U100) from Tonobo Biosciences. Cells were gated on live single-cell populations followed by separation of specific cell populations based on expression of the following cell surface markers: macrophages (Cd11b+ F4/80+), neutrophils (Cd11b+, GR-1+), natural killer cells (Cd11b+, NK1.1+) and B cells (CD19+, B220+). For histology and TUNEL staining, spleens were isolated from TNF or PBS injected mice after 4 hours, fixed in 10% formalin for 48-hours and transferred to 75% ethanol. H&E, TUNEL staining were performed by iHisto. TUNEL+ cells/mm2 were calculated on the BioTek Lionheart Automated microscope. Blinded histopathology assessment was performed independently by two veterinary pathologists. All experiments were performed in accordance with regulations and approval of the Tufts University Institutional Animal Care and Use Committee.
Bone Marrow Transfer and Gain of Function Experiments
Bone marrow (BM) cells were isolated from CD45.2 C57BL/6 (B6) and Ticam1−/− (Trif−/−) mice. BM cells were stained with biotin-conjugated anti-CD4, anti-CD8 and anti-TER-110 antibodies, and erythrocytes and T cells were depleted from the BM using streptavidin microbeads and LS columns (Miltenyi). Lethally irradiated CD54.1 C57BL/6 (B6) mice were reconstituted with 5×106 donor B6 or Trif−/− cells. After 8-weeks mice were injected with PBS or 9ug TNF and survival, cell death and inflammatory responses were quantified as described above in the TNF-induced lethality mouse model protocol. For neutrophil depletion experiments 11-week-old male and female B6 mice were injected intraperitoneally with anti-Ly6G antibody 1A8 (250ug) 24 hours prior to induction of TNF-induced lethality as described above. For neutrophil and macrophage gain of function experiments 11-week-old male and female Trif−/− mice were injected intravenously with 1×106 thioglycolate elicited neutrophils or bone marrow derived macrophages 1 hour prior to induction of TNF-induced lethality as described in the TNF-induced lethality mouse model protocol.
Reagents
Lipopolysaccharide (LPS) Escherichia coli 011:B4 (10 ng/ml, L4391), 5Z-7-Oxozeaenol (5z7, 125 nM), Necrostatin-1 (Nec-1, 10 uM, N9037) and cycloheximide (CHX, 10ug/ml) were purchased from Sigma. zVAD.fmk was purchased from Millipore (2109007, 50uM). Caspase-3/7 inhibitor I was purchased from Cayman Chemical. Recombinant human and murine TNF (50ng/ml) was purchased from PeproTech. SMAC mimetic SM-164 (1uM) was purchased from ApexBio.
Yersinia Growth Conditions
Yersinia pseudotuberculosis bacterial strains were generously provided by Dr. Ralph Isberg. Bacteria were grown from frozen glycerol stocks on LB plates containing Irgasan (Sigma). Cultures were grown overnight at 26°C for 2 additional hours prior to a shift to 37°C for 2 hours. Macrophages were infected at a multiplicity of infection (MOI) of 7.5 CFU/cell.
Kinetic Microscopy
The Cytation3 automated microscope was used for kinetic macrophage and neutrophil imaging assays, and built in environmental control maintained 37°C, 5% CO2 for the duration of the assay. Cells were seeded at a density of 1×106 cm2 in RPMI on 1.17 mm thick glass bottom imaging plates. To generate kinetic cell death curves, cells were imaged at 30-minute intervals at 4x magnification to capture approximately 5,000 cells/field of view. Propidium iodide (PI) incorporation was detected at 617 nm and PI+ nuclei were counted. Wells treated with 0.1% Triton X-100 were used as controls for 100% cell death.
RIPK1 and TRAM and FADD Immunoprecipitations
Cells were plated on 10 cm dishes, stimulated as indicated and harvested in immunoprecipitation lysis buffer (0.5% Triton X, 50 mM Tris Base (pH 7.4), 150 mM NaCl, 2 mM EDTA, 2 mM EGTA, 1X protease inhibitor cocktail). Lysed cells were rotated for 60 min at 4 degrees C with intermittent vortexing, centrifuged at 5,000 × g for 5 min, and the supernatant was incubated with α-RIPK1, α-TRAM or α-FADD antibody-conjugated Protein G agarose beads (Cell Signaling Technology 37478). Samples were washed three times in immunoprecipitation lysis buffer, and protein complexes were eluted with 1X Laemmli buffer containing 5% β-mercaptoethanol at 90 °C for 15 min.
Immunoblotting
After indicated treatments, cells were lysed in 1X Laemmli Buffer containing 5% β-mercaptoethanol, boiled for 15 min and incubated on ice for 15 min. Primary antibodies against p38 (9212), p-p38 (4511), ERK1/2 (4696), p-ERK1/2 (4370), IκBα (4814), RIPK1 (3493), TRAF3 (4729), TBK1 (3504), NEMO (2685), CASP8 (8592), CASP3 (9665), CASP7 (9492), CASP1 (24232), and GAPDH (2118) were purchased from Cell Signaling Technologies. TRAM antibody was purchased from Santa Cruz (sc-376076). FADD antibody (05–486) was purchased from Millipore Sigma. GSDMD antibody (ab209845). Secondary antibodies, anti-rabbit IgG (H+L) (DyLight™ 800 4X PEG Conjugate) (5151) and anti-mouse IgG (H+L) (DyLight™ 800 4X PEG Conjugate) (5257) were purchased from Cell Signaling Technologies.
RNA Isolation and Analysis
5×105 BMDMs or neutrophils were plated on 24-well tissue culture treated plates. Cells were lysed with TRIzol (Invitrogen) and RNA extraction was carried out according to the manufacturer’s instructions. Reverse transcription was performed using M-MuLV reverse transcriptase, RNase inhibitor, random primers 9, and dNTP mix (New England BioLabs) to synthesize cDNA. cDNa was analyzed for relative mRNA levels using SYBR Green (Applied Biosystems) and intron spanning primers. GAPDH was used to normalize mRNA levels. Post-amplification melting curve analysis was performed to confirm primer specificity.
Quantitative PCR Primers
TNF: (F) 5’-CTGTAGCCCACGTCGTAGC-3’,
(R) 5’-TTGAGATCCATGCCGTTG-3’
TNFR1: (F) 5’- GCCTCCCGCGATAAAGCCAACC-3’,
(R) 5’- CTTTGCCCACTTTCACCCACAGG-3’
IFNβ: (F) 5’-CAGCTCCAAGAAAGGACGAAC-3’,
(R) 5’-GGCAGTGTAACTCTTCTGCAT-3’
GAPDH: (F) 5’-GGAGAGTGTTTCCTCGTCCC-3’,
(R) 5’-TTCCCATTCTCGGCCTTGAC-3’.
Analysis of single cell RNA sequencing data
For single cell sequencing, spleens were isolated from B6 and Trif−/− mice 4 hours after injection with 9ug TNF or PBS. Single cell suspensions were generated, RBCs were lysed and MACs Dead Cell Removal Kit was be used to isolate viable cells. Libraries were generated from ~10,000 cells/sample to ensure representation of rare cell types using the 10x genomics Chromium Controller and associated equipment. Sequencing was performed on the NextSeq 550, using the High Output 150 Cycle Kit with paired-end reads. Paired-end reads were aligned with 10x Genomics Cell Ranger (v3.0) (44) against the mm10 Mouse reference genome. Putative doublet cells were removed using predictions generated from DoubletFinder (v2.0.3) (45). All samples were integrated to remove batch effects using the Seurat Single Cell Transform workflow (46) using the top 3000 variable features, followed by clustering using the Leiden algorithm on the shared nearest neighbor graph, as implemented in the Seurat function FindClusters function with resolution 0.8, and visualization by UMAP using the first 50 principal components. Cell clusters were annotated using R package SingleR (v1.2.4) with celldex (v.0.99.1) (47). MouseRNAseq reference database and an annotation was confirmed by manual inspection of top marker genes. Cell proportion bar graphs were produced with dittoSeq R package (48). Differential expression analysis was performed with MAST (49). TRIF-depedent genes were identified using a model with an interaction term, as described above for bulk-RNAseq analysis.
High Magnification Imaging
To observe TRIF, RIPK1, RAB5A, CD14 and TNF-R1 localization, macrophages were seeded at a density of 1×106 cm2 in RPMI on 1.17-mm-thick glass bottom imaging plates. Cells were stimulated with TNF or TNF/5z7 for indicated times, fixed in 4% paraformaldehyde for 15 minutes, blocked in 1X PBS (5% FBS, 0.2% Triton X-100) and incubated overnight with anti-TRIF (abcam ab13810), anti-RIPK1 (BD Biosciences 610459), anti-RAB5A (abcam, ab66746), anti-CD14 (abcam, ab221678) or anti-TNF-R1 (abcam, ab223352) antibody followed by a 2-hour incubation in the presence of Alexa Fluor 555 conjugated goat anti-mouse (ab150114) and Alexa Fluor 405 conjugated goat anti-rabbit (ab175652) antibodies. The Lionheart automated microscope was used to image cells at 60X magnification to capture approximately 25 cells/field of view in quadruplicate. Gen5 3.10 imaging software was used to quantify positive staining for each protein, and a mask was generated to calculate puncta/cell.
Quantification and Statistical Analysis
All statistical analyses were performed using GraphPad Prism 9 software. Error bars in qPCR experiments represent the standard deviation of three or more independent experiments. Data from imaging experiments are representative of three or more independent experiments. Immunoblots are representative of three or more independent experiments. For in vivo data, points represent individual mice. Significance was determined using a one-way or two-way ANOVA as appropriate: ns (non-significant) p > 0.05; *p < 0.05; **p <0.01; ***p < 0.001; ****p < 0.0001).
Supplementary Material
Acknowledgments:
We thank Dr. K. Fitzgerald, Dr. A. Degterev, Dr. M. Kelliher, Dr. L. Li and Dr. A. Rongvaux for sharing various mouse strains for this study.
Funding:
This work was supported by NIH grants AI135369 and AI056234 to A.P.
Footnotes
Competing interests: Authors declare no competing interests.
Data and materials availability:
All data needed to evaluate the conclusions in the paper are present in the paper or the Supplementary Materials. RNA-sequencing data have been deposited into the Gene Expression Omnibus public dataset with accession numbers GSM6475221, GSM6475222, GSM6475223 and GSM6475224.
References and Notes
- 1.Wajant H, Siegmund D, TNFR1 and TNFR2 in the Control of the Life and Death Balance of Macrophages. Front. Cell Dev. Biol. 7, 1–14 (2019). [DOI] [PMC free article] [PubMed] [Google Scholar]
- 2.Mcdermott MF, Aksentijevich I, Mcdermott EM, Ogunkolade BW, Centola M, Mansfield E, Gadina M, Karenko L, Pettersson T, Mccarthy J, Frucht DM, Aringer M, Torosyan Y, Teppo A, Wilson M, Karaarslan HM, Wan Y, Todd I, Wood G, Schlimgen R, Kumarajeewa TR, Cooper SM, Vella JP, Amos CI, Mulley J, Quane KA, Molloy MG, Ranki A, Powell RJ, Hitman GA, Shea JJO, Kastner DL, Germline Mutations in the Extracellular Domains of the 55 kDa TNF Receptor, TNFR1, Define a Family of Dominantly Inherited Autoinflammatory Syndromes. Cell 97, 133–144 (1999). [DOI] [PubMed] [Google Scholar]
- 3.Seymour HE, Worsley A, Smith JM, Thomas SHL, Anti-TNF agents for rheumatoid arthritis. Br J Clin Pharmacol 51, 201–208 (2001). [DOI] [PMC free article] [PubMed] [Google Scholar]
- 4.Garcia-carbonell R, Wong J, Kim JY, Close LA, Boland BS, Wong TL, Harris PA, Ho SB, Das S, Ernst PB, Sasik R, Sandborn WJ, Bertin J, Gough PJ, Chang JT, Kelliher MA, Boone D, Guma M, Karin M, Elevated A20 promotes TNF-induced and RIPK1- dependent intestinal epithelial cell death. PNAS 115, 9192–9200 (2018). [DOI] [PMC free article] [PubMed] [Google Scholar]
- 5.Del Valle DM, Kim-schulze S, Huang H, Beckmann ND, Nirenberg S, Wang B, Lavin Y, Swartz TH, Madduri D, Stock A, Marron TU, Xie H, Patel M, Tuballes K, Van Oekelen O, Rahman A, Kovatch P, Aberg JA, Schadt E, Jagannath S, Mazumdar M, Charney AW, Firpo-Betancourt A, Mendu DR, Jhang J, Reich D, Sigel K, Cordon-Cardo C, Feldmann M, Parekh S, Merad M, Gnjatic S, An inflammatory cytokine signature predicts COVID-19 severity and survival. Nat. Med. 26, 1636–1643 (2020). [DOI] [PMC free article] [PubMed] [Google Scholar]
- 6.Rowe RK, Harrison JL, Zhang H, Bachstetter AD, Hesson DP, O’Hara BF, Greene MI, Lifshitz J, Novel TNF receptor-1 inhibitors identified as potential therapeutic candidates for traumatic brain injury. J. Neuroinflammation 15, 1–14 (2018). [DOI] [PMC free article] [PubMed] [Google Scholar]
- 7.Marino MW, Dunn A, Grail D, Inglese M, Noguchi Y, Richards E, Jungbluth A, Wada H, Moore M, Williamson B, Basu S, Old LJ, Characterization of tumor necrosis factor-deficient mice. PNAS 94, 8093–8098 (1997). [DOI] [PMC free article] [PubMed] [Google Scholar]
- 8.Mandal P, Feng Y, Lyons JD, Berger SB, Otani S, Delaney A, Tharp GK, Maner-smith K, Burd EM, Hoffman S, Capriotti C, Roback L, Young CB, Ortlund EA, Dipaolo NC, Bosinger S, Bertin J, Gough PJ, Brodsky IE, Coopersmith CM, Shayakhmetov DM, Mocarski ES, Caspase-8 Collaborates with Caspase-11 to Drive Tissue Damage and Execution of Edotoxic Shock. Immunity 49, 42–55 (2018). [DOI] [PMC free article] [PubMed] [Google Scholar]
- 9.Van Hauwermeiren F, Vandenbroucke RE, Grine L, Lodens S, Van Wonterghem E, De Rycke R, De Geest N, Hassan B, Libert C, TNFR1-induced lethal inflammation is mediated by goblet and Paneth cell dysfunction. Mucosal Immunol. 8, 828–840 (2015). [DOI] [PubMed] [Google Scholar]
- 10.Tartaglia LA, Ayres MT, Wong GHW, Goeddel DV, A novel domain within the 55 kd TNf receptor signals cell death. Cell 74, 845–853 (1993). [DOI] [PubMed] [Google Scholar]
- 11.Park HH, Lo Y-C, Lin S-C, Wang L, Yang JK, Wu H, The Death Domain Superfamily in Intracellular Signaling of Apoptosis and Inflammation. Ann. Rev. Immunol. 25, 561–586 (2007). [DOI] [PMC free article] [PubMed] [Google Scholar]
- 12.Wajant H, Scheurich P, TNFR1-induced activation of the classical NF-kB pathway. FEBS J. 278, 862–876 (2011). [DOI] [PubMed] [Google Scholar]
- 13.Chen N, Chio IIC, Lin W, Duncan G, Chau H, Katz D, Huang H, Pike KA, Hao Z, Su Y, Yamamoto K, de Pooter RF, Zuniga-Pflucker JC, Wakeham A, Yeh W, Mak TW, Beyond tumor necrosis factor receptor: TRADD signaling in toll-like receptors. PNAS 105, 12429–12434 (2008). [DOI] [PMC free article] [PubMed] [Google Scholar]
- 14.Ermolaeva MA, Michallet M-C, Papadopoulou N, Utermohlen O, Kranidioti K, Kollias G, Tschopp J, Pasparakis M, Function of TRADD in tumor necrosis factor receptor 1 signaling and in TRIF-dependent inflammatory responses. Nat. Immunol. 9, 1037–1046 (2008). [DOI] [PubMed] [Google Scholar]
- 15.Pobezinskaya YL, Kim Y-S, Choksi S, Morgan MJ, Li T, Liu C, Liu Z, The function of TRADD in signaling via tumor necrosis factor receptor 1 and TRIF-dependent Toll-like receptors. Nat. Immunol. 9, 1047–1054 (2008). [DOI] [PMC free article] [PubMed] [Google Scholar]
- 16.Kondylis V, Kumari S, Vlantis K, Pasparakis M, The interplay of IKK, NF-kB and RIPK1 signaling in the regulation of cell death, tissue homeostasis and inflammation. Immunol. Rev. 277, 113–127 (2017). [DOI] [PubMed] [Google Scholar]
- 17.Feoktistova M, Geserick P, Kellert B, Dimitrova DP, Langlais C, Hupe M, Cain K, Macfarlane M, Hacker G, Leverkus M, cIAPs Block Ripoptosome Formation, a RIP1/Caspase-8 Containing Intracellular Cell Death Complex Differentially Regulated by cFLIP Isoforms. Mol. Cell 43, 449–463 (2011). [DOI] [PMC free article] [PubMed] [Google Scholar]
- 18.Micheau O, Tschopp J, Induction of TNF Receptor I-Mediated Apoptosis via Two Sequential Signaling Complexes. Cell 114, 181–190 (2003). [DOI] [PubMed] [Google Scholar]
- 19.Sarhan J, Liu BC, Muendlein HI, Li P, Nilson R, Tang AY, Rongvaux A, Bunnell SC, Shao F, Green DR, Poltorak A, Caspase-8 induces cleavage of gasdermin D to elicit pyroptosis during Yersinia infection. PNAS 115 (2018). [DOI] [PMC free article] [PubMed] [Google Scholar]
- 20.Orning P, Weng D, Starheim K, Ratner D, Best Z, Lee B, Brooks A, Pathogen blockade of TAK1 triggers caspase-8 – dependent cleavage of gasdermin D and cell death. Science (80-. ). 362, 1064–1069 (2018). [DOI] [PMC free article] [PubMed] [Google Scholar]
- 21.Chen KW, Demarco B, Heilig R, Shkarina K, Boettcher A, Farady CJ, Pelczar P, Broz P, Extrinsic and intrinsic apoptosis activate pannexin - 1 to drive NLRP3 inflammasome assembly. EMBO 38 (2019), doi: 10.15252/embj.2019101638. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 22.Muendlein HI, Jetton D, Connolly WM, Eidell K, Magri Z, Smirnova I, Poltorak A, cFLIPL Protects Macrophages From LPS Induced Pyroptosis via Inhibition of Complex II Formation. Science (80-. )., 1379–1384 (2020). [DOI] [PMC free article] [PubMed] [Google Scholar]
- 23.Muendlein HI, Connolly WM, Magri Z, Smirnova I, Ilyukha V, Gautam A, Degterev A, Poltorak A, ZBP1 promotes LPS-induced cell death and IL-1b release via RHIM-mediated interactions with RIPK1. Nat. Commun. 12, 1–13 (2021). [DOI] [PMC free article] [PubMed] [Google Scholar]
- 24.Peterson LW, Philip NH, Dillon CP, Bertin J, Gough PJ, Green DR, Brodsky IE, Cell-Extrinsic TNF Collaborates with TRIF Signaling To Promote Yersinia-Induced Apoptosis. J. Immunol. 197, 4110–4117 (2016). [DOI] [PMC free article] [PubMed] [Google Scholar]
- 25.Sarhan J, Liu BC, Muendlein HI, Li P, Nilson R, Tang AY, Caspase-8 induces cleavage of gasdermin D to elicit pyroptosis during Yersinia infection. PNAS 115, E10888–E10897 (2018). [DOI] [PMC free article] [PubMed] [Google Scholar]
- 26.Kagan JC, Su T, Horng T, Chow A, Akira S, Medzhitov R, TRAM couples endocytosis of Toll-like receptor 4 to the induction of interferon-Beta. Nat. Immunol. 9, 361–368 (2008). [DOI] [PMC free article] [PubMed] [Google Scholar]
- 27.Tanimura N, Saitoh S, Matsumoto F, Akashi-Takamura S, Miyake K, Roles for LPS-dependent interaction and relocation of TLR4 and TRAM in TRIF-signaling. Biochem. Biophys. Res. Commun. 368, 94–99 (2008). [DOI] [PubMed] [Google Scholar]
- 28.Polykratis A, Hermance N, Zelic M, Roderick J, Kim C, Van T-M, Thomas L, Chan FKM, Pasparakis M, Kelliher MA, Cutting Edge: RIPK1 Kinase Inactive Mice Are Viable and Protected from TNF-Induced Necroptosis In Vivo. J. Immunol. 193, 1539–1543 (2014). [DOI] [PMC free article] [PubMed] [Google Scholar]
- 29.Demarco B, Grayczyk JP, Bjanes E, Le Roy D, Tonnus W, Assenmacher C-A, Radaelli E, Fettrelet T, Mack V, Linkermann A, Roger T, Brodsky IE, Chen KW, Broz P, Caspase-8 – dependent gasdermin D cleavage promotes antimicrobial defense but confers susceptibility to TNF-induced lethality. Sci. Adv. 1, 1–16 (2020). [DOI] [PMC free article] [PubMed] [Google Scholar]
- 30.Kaufmann T, Jost PJ, Pellegrini M, Puthalakath H, Gugasyan R, Gerondakis S, Cretney E, Smyth MJ, Silke J, Hakem R, Bouillet P, Mak TW, Dixit VM, Strasser A, Fatal Hepatitis Mediated by Tumor Necrosis Factor TNFa Requires Caspase-8 and Involves the BH3-Only Proteins Bid and Bim. Immunity 30, 56–66 (2009). [DOI] [PMC free article] [PubMed] [Google Scholar]
- 31.Duprez L, Takahashi N, Van Hauwermeiren F, Vandendriessche B, Goossens V, Vanden Berghe T, Declercq W, Libert C, Cauwels A, Vandenabeele P, RIP Kinase-Dependent Necrosis Drives Lethal Systemic Inflammatory Response Syndrome. Immunity 35, 908–918 (2011). [DOI] [PubMed] [Google Scholar]
- 32.Kayagaki N, Stowe IB, Lee BL, O’Rourke K, Anderson K, Warming S, Cuellar T, Haley B, Roose-Girma M, Phung QT, Liu PS, Lill JR, Li H, Wu J, Kummerfeld S, Zhang J, Lee WP, Snipas SJ, Salvesen GS, Morris LX, Fitzgerald L, Zhang Y, Bertram EM, Goodnow CC, Dixit VM, Caspase-11 cleaves gasdermin D for non-canonical inflammasome signalling. Nature 526, 666–671 (2015). [DOI] [PubMed] [Google Scholar]
- 33.Piguet PF, Vesin C, Guo J, Donati Y, Barazzone C, TNF-induced enterocyte apoptosis in mice is mediated by the TNF receptor 1 and does not require p53. Eur. J. Immunol. 28, 3499–3505 (1998). [DOI] [PubMed] [Google Scholar]
- 34.Kurotaki D, Uede T, Tamura T, Functions and development of red pulp macrophages. Microbiol. Immunol. 59, 55–62 (2015). [DOI] [PubMed] [Google Scholar]
- 35.Dong Kim K, Zhao J, Auh S, Yang X, Du P, Tang H, Fu YX, Adaptive immune cells temper initial innate responses. Nat. Med. 13, 1248–1252 (2007). [DOI] [PMC free article] [PubMed] [Google Scholar]
- 36.Dannappel M, Vlantis K, Kumari S, Polykratis A, Kim C, Wachsmuth L, Eftychi C, Lin J, Corona T, Hermance N, Zelic M, Kirsch P, Basic M, Bleich A, Kelliher M, Pasparakis M, RIPK1 maintains epithelial homeostasis by inhibiting apoptosis and necroptosis. Nature 513, 90–94 (2014). [DOI] [PMC free article] [PubMed] [Google Scholar]
- 37.Zelic M, Roderick JE, O’Donnell JA, Lehman J, Lim SE, Janardhan HP, Trivedi CM, Pasparakis M, Kelliher MA, RIP kinase 1-dependent endothelial necroptosis underlies systemic inflammatory response syndrome. J. Clin. Invest. 128, 2064–2075 (2018). [DOI] [PMC free article] [PubMed] [Google Scholar]
- 38.Aird WC, The role of the endothelium in severe sepsis and multiple organ dysfunction syndrome. Blood 101, 3765–3777 (2003). [DOI] [PubMed] [Google Scholar]
- 39.Levi M, van der Poll T, Coagulation and sepsis. Thromb. Res. 149, 38–44 (2017). [DOI] [PubMed] [Google Scholar]
- 40.Axtelle T, Pribble J, An overview of clinical studies in healthy subjects and patients with severe sepsis with IC14, a CD14-specific chimeric monoclonal antibodyJ. Endotoxin Res. 9, 385–389 (2003). [DOI] [PubMed] [Google Scholar]
- 41.Martin TR, Wurfel MM, Zanoni I, Ulevitch R , Targeting innate immunity by blocking CD14: Novel approach to control inflammation and organ dysfunction in COVID-19 illness. EBioMedicine 57, 102836 (2020). [DOI] [PMC free article] [PubMed] [Google Scholar]
- 42.Yu Y, Ye RD, Microglial Aβ Receptors in Alzheimer’s Disease. Cell. Mol. Neurobiol. 35, 71–83 (2015). [DOI] [PMC free article] [PubMed] [Google Scholar]
- 43.Hennessy E, Griffin EW, Cunningham C, Astrocytes are primed by chronic neurodegeneration to produce exaggerated chemokine and cell infiltration responses to acute stimulation with the cytokines IL-1β and TNF-α. J. Neurosci. 35, 8411–8422 (2015). [DOI] [PMC free article] [PubMed] [Google Scholar]
- 44.Zheng GXY, Terry JM, Belgrader P, Ryvkin P, Bent ZW, Wilson R, Ziraldo SB, Wheeler TD, McDermott GP, Zhu J, Gregory MT, Shuga J, Montesclaros L, Underwood JG, Masquelier DA, Nishimura SY, Schnall-Levin M, Wyatt PW, Hindson CM, Bharadwaj R, Wong A, Ness KD, Beppu LW, Deeg HJ, McFarland C, Loeb KR, Valente WJ, Ericson NG, Stevens EA, Radich JP, Mikkelsen TS, Hindson BJ, Bielas JH, Massively parallel digital transcriptional profiling of single cells. Nat. Commun. 8 (2017), doi: 10.1038/ncomms14049. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 45.McGinnis CS, Murrow LM, Gartner ZJ, DoubletFinder: Doublet Detection in Single-Cell RNA Sequencing Data Using Artificial Nearest Neighbors. Cell Syst. 8, 329–337.e4 (2019). [DOI] [PMC free article] [PubMed] [Google Scholar]
- 46.Hafemeister C, Satija R, Normalization and variance stabilization of single-cell RNA-seq data using regularized negative binomial regression. Genome Biol. 20, 1–15 (2019). [DOI] [PMC free article] [PubMed] [Google Scholar]
- 47.Aran D, Looney AP, Liu L, Wu E, Fong V, Hsu A, Chak S, Naikawadi RP, Wolters PJ, Abate AR, Butte AJ, Bhattacharya M, Reference-based analysis of lung single-cell sequencing reveals a transitional profibrotic macrophage. Nat. Immunol. 20, 163–172 (2019). [DOI] [PMC free article] [PubMed] [Google Scholar]
- 48.Bunis DG, Andrews J, Fragiadakis GK, Burt TD, Sirota M, DittoSeq: Universal user-friendly single-cell and bulk RNA sequencing visualization toolkit. Bioinformatics 36, 5535–5536 (2020). [DOI] [PMC free article] [PubMed] [Google Scholar]
- 49.McDavid A, Finak G, Yajima M, MAST: Model-based Analysis of Single Cell Transcriptomics. R package version 1.20.0 (2021). [Google Scholar]
Associated Data
This section collects any data citations, data availability statements, or supplementary materials included in this article.
Supplementary Materials
Data Availability Statement
All data needed to evaluate the conclusions in the paper are present in the paper or the Supplementary Materials. RNA-sequencing data have been deposited into the Gene Expression Omnibus public dataset with accession numbers GSM6475221, GSM6475222, GSM6475223 and GSM6475224.