

Abstract
Background
Polygenic hazard scores (PHS) estimate age-dependent genetic risk of late-onset Alzheimer’s disease (AD), but there is limited information about the performance of PHS on real-world data where the population of interest differs from the model development population and part of the model genotypes are missing or need to be imputed.
Objective
We use Desikan PHS model developed based on a North American population, to obtain age-dependent risk of developing late-onset AD Nordic populations.
Methods
We used Desikan PHS model, based on Cox proportional hazards assumption, to obtain age-dependent hazard scores for AD from individual genotypes in the Norwegian DemGene cohort (n=2,772). We assessed the risk discrimination and calibration of Desikan model and extended it by adding new genotype markers (the Desikan Nordic model). Finally, we evaluated both Desikan and Desikan Nordic models in 2 independent Danish cohorts: The Copenhagen City Heart Study (CCHS) cohort (n=7,643) and The Copenhagen General Population Study (CGPS) cohort (n=10,886).
Results
We showed a robust prediction efficiency of Desikan model in stratifying AD risk groups in Nordic populations, even when some of the model SNPs were missing or imputed. We attempted to improve Desikan PHS model by adding new SNPs to it, but we still achieved similar risk discrimination and calibration with the extended model.
Conclusion
PHS modeling has the potential to guide the timing of treatment initiation based on individual risk profiles, and can help enrich clinical trials with people at high risk to AD in Nordic populations.
Keywords: Alzheimer’s Disease, Polygenic hazard score, Age at onset, Nordic ancestry
INTRODUCTION
Late-onset Alzheimer’s disease (AD) is the most common type of dementia. It affects nearly 50 million people worldwide, and this number is expected to increase to 152 million by 2050[1]. Since AD is an irreversibly progressive neurodegenerative disease, detecting individuals at high risk during the preclinical or pre-symptomatic stage could benefit early intervention strategies to slow down the progression of the disease[2, 3].
Genetic factors play an important role in the development of AD, evidenced by an estimated heritability (h2) of 60% to 80% in twin studies[4, 5]. This prominent genetic component has been attributed to a mixture of rare and common genetic risk variants.[6–9]. In particular, one of the most well-established genetic determinants for AD is the polymorphism in the apolipoprotein E (APOE) gene, determined by haplotypes including 2 single nucleotide polymorphisms (SNPs), rs7412 and rs429358, on chromosome 19[10]. Within this gene, the common risk variant ε4 increases the risk of AD 3~4-fold and the less common neuroprotective variant ε2 decreases the risk by half [10–12]. The age at onset of AD has also been reported as heritable (h2=42%)[13] and partially determined by genetic variants that also determine the risk of occurrence[14–17], such as the APOE ε4/ε2 alleles[17–19]. Previous studies have shown that the age at onset of AD can be predicted using a polygenic hazard score (PHS) which extends beyond the APOE region[17, 20, 21]. The PHS is built on a Cox proportional hazards model that estimates the risk of late-onset AD for an individual over time in longitudinal studies based on the additive effects of the individual’s genotypes over a set of polymorphic markers[17, 20]. The decreasing cost and the increasing quality of genotyping technologies, as well as the stability of additive genetic effects over generations, makes PHS modeling a powerful tool to assess the risk of developing AD before symptoms appear.
However, the accuracy of genetic tests trained in specific populations is often reduced when applied to other populations due to differences in genetic architecture across ancestral groups. Given the lack of genetic data from ancestrally diverse populations, this is currently a major barrier to future clinical applications of genetic testing and precision medicine[22]. Furthermore, compared to standard genetic tests such as polygenic risk scores which are trained on cross-sectional data, PHS models require longitudinal datasets with reliable phenotyping of symptom onset, making cross-ancestral validation even more challenging. Updating pre-existing PHS models with novel risk variants detected by new studies may also improve the predictive accuracy of PHS models[23, 24].
In the current study, we first examined the prediction efficiency of the PHS model developed using North American participants in the Alzheimer’s Disease Genetics Consortium (ADGC) cohort by Desikan et al. [17], henceforth called the Desikan model, in a Norwegian population from the longitudinal multi-center Norwegian Dementia Genetics Consortium (DemGene) study[25, 26]. Using the Desikan model, we stratified individuals in the DemGene population for the risk of late-onset AD based on the PHS quantiles[27]. In our attempt to improve the accuracy of the Desikan model, especially to compensate for the accuracy reduction due to the presence of missing or imputed SNPs, we extended the model with additional SNPs to generate a PHS model adjusted to Nordic populations, which we named the Desikan Nordic model. Finally, we evaluated both Desikan and Desikan Nordic models in 2 independent Danish populations: The Copenhagen City Heart Study (CCHS) and The Copenhagen General Population Study (CGPS).
MATERIAL AND METHODS
Participants and diagnosis
Norwegian population: DemGene cohort
The DemGene cohort consists of a network of clinical sites that prospectively collect data based on standardized examinations of cognitive, functional and behavioral measures of the participants[26]. Within this study, we followed 2772 individuals of Nordic ancestry (mean age = 72.25, 95% CI: (47.03, 90)) among whom 1350 had been diagnosed with AD, from 10 study centers including: the Norwegian Register of persons with Cognitive Symptoms (NorCog), the Progression of Alzheimer’s Disease and Resource use (PADR), the Dementia Study of Western Norway (DemVest), the Dementia Study in Rural Northern Norway (Nord Norge), Nord-Trøndelag Health Study, TrønderBrain study, the Dementia Disease Initiation study (DDI), Nursing home study, Karolinska Institute (KI) in Stockholm and Oslo PD study. For a complete description of the cohorts, please refer to Supplementary Table 1.
More than 80% of participants in DemGene cohort were older than 65 years old (Supplementary Table 1). As early-onset AD has shown distinct susceptibility genes from late-onset AD[28], we first selected participants, who were cognitively healthy and older than 65 years at enrollment. Next, we excluded participants who were diagnosed as other types of neurodegenerative disorder or mild cognitive impaired, leaving 1198 AD cases and 1026 participants cognitively healthy at the end of the study. AD cases were defined according to diagnostic guidelines from the National Institute on Aging–Alzheimer’s Association (NIA/AA) (AHUS), the NINCDS-ADRDA criteria (DemVest and TrønderBrain) or the ICD-10 diagnosis criteria (NorCog, PADR, Nord Norge and Nord-Trøndelag). The age of diagnosis for those developing AD was confirmed by a physician based on presenting signs and symptoms. Healthy subjects were also enrolled for follow-up and were screened with the same standardized interviews and cognitive tests applied to the AD population.
Of the 10 included cohorts, we used 9 for testing the stratification accuracy of the Desikan model and developing the Desikan Nordic PHS model. Those 9 cohorts included 1878 individuals of whom 1048 were diagnosed with AD and 830 were censored at the end of the follow-up period. We kept one cohort, Nord Norge, for validation and comparing of the predictive performance of the two PHS models. The Nord Norge contained 346 subjects of whom 150 were diagnosed with AD and 196 were right-censored. The inclusion and exclusion criteria of the participants and their number in each validation or development set are shown in Figure 1.
Figure 1.
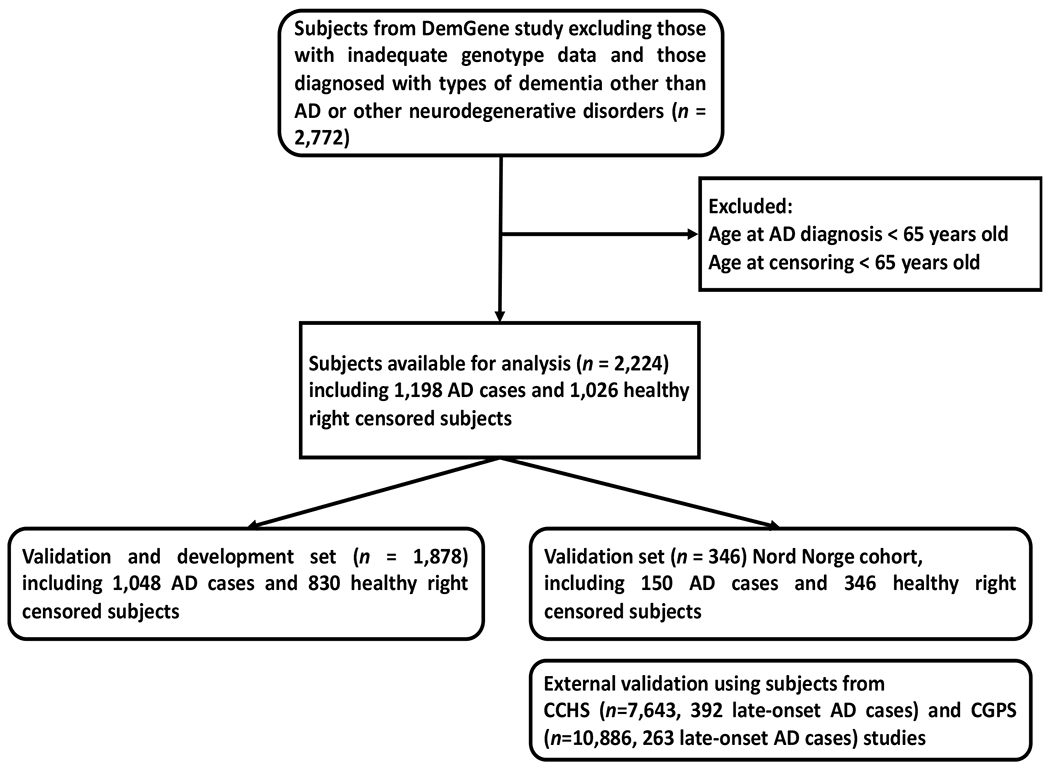
Flow chart illustrating the inclusion and exclusion criteria of the Norwegian DemGene participants in the validation and development set (used to validate the Desikan model and develop the Desikan Nordic model) and the validation set (used to validate both models). Both models were also externally evaluated using two Danish studies CGPS and CCHS.
Danish populations: CCHS and CGPS studies
The CCHS study is a prospective study of the Danish general population initiated in 1976–78 with follow-up examinations in 1981–1983, 1991–1994, and 2001–2003[29–31]. For this study, data was collected using questionnaires and physical examinations, as well as using blood sampling for biochemical and DNA analyses. We included 7643 unrelated individuals of Danish descent without any dementia at baseline who gave blood for biochemical and DNA analyses at the 1991–1994 examination, among whom 392 developed late-onset AD during follow-up (end of 2018).
The CGPS study is also a prospective study of the Danish General population, with its first enrolments between 2003-2015[29–31]. Individuals aged 20-100 years were randomly selected from the national Danish Civil Registration System and their data were obtained from a self-administered questionnaire on lifestyle and health, as well as from physical examinations and blood samples. The first 10886 unrelated participants of Danish descent with no dementia at baseline were used for genotyping, of whom 263 developed late-onset AD disease during follow-up (end of 2018).
Both CCHS and CGPS studies used ICD-10 codes, G30 and F00, from the national Danish Patient Registry (hospital diagnoses) for AD diagnosis[30, 31].
Genotyping and imputation
The genotypes for the DemGene cohort were obtained using Human Omni Express-24 v.1.1 (Illumina Inc., San Diego, CA, USA) chip at deCODE Genetics (Reykjavik, Iceland). Quality control (QC) steps were performed using PLINK[32], including removal of SNPs with genotype call rate lower than 0.95, minor allele frequency (MAF) lower than 0.01, less than 1e-06 probability of being in Hardy-Weinberg equilibrium, and ambiguous strand assignment (A/T, C/G SNPs). Next, we applied quality control on participants by removing participants with ambiguous sex information, a call rate < 0.95, an estimated relatedness of >0.1, as well as outliers based on population structure using principal components analysis (PCA)[26].
Illumina Exome Chip was used to obtain genotypes for the Danish cohorts. The minimum call-rate threshold was 0.98 for keeping a SNP (sample call-rate) and for keeping an individual (genotyping call-rate). The ethnicity of the participants was also checked using PCA only individuals of Nordic ancestry were included in the analysis.
For both Norwegian and Danish cohorts, Minimac software[33] (based on MaCH algorithm[34]) was used to impute genotypes using the reference haplotypes derived from samples of European ancestry in the haplotype reference consortium (HRC) v1.1[35]. We required at least 50% confidence for keeping an imputed genotype in the analysis. Post imputation QC steps were the same as the steps explained above.
Polygenic hazard scoring (PHS) model
The Desikan PHS model is composed of the APOE ε2 and ε4 variants as well as 31 other SNPs, i.e. the polygenic component of the model (Supplementary Table 2)[17]. The SNP array data of DemGene included the APOE ε2/ε4 variants and 17 out of the 31 polygenic SNPs. The SNP array of CCHS and CGPS contained the APOE ε2/ε4 variants and only 9 out of the 31 polygenic SNPs. After imputation, 2 SNPs were still missing in the DemGene population due to low imputation quality, while 10 SNPs remained missing in CCHS and CGPS populations (Supplementary Table 2). These SNPs were left-out from PHS calculations, i.e. their effects were set to zero. To estimate PHS scores, undetermined individual alleles of the APOE haplotypes and the other remaining SNPs were set to the population average after numerically coding of the genotypes according to the dosages of the reference allele.
The Desikan log proportional hazard score of each individual was obtained according to:
| (1) |
where the first two terms of the right-side take the APOE ε2/ε4 alleles into account and represents the genotypes of 31 SNPs, , in the polygenic part of the model. The genetic effects β in (1) were provided by Desikan et al[17], and the corresponding effect was left out of the summation in (1) if was excluded due to low imputation quality.
We examined the utility of the Desikan model in stratifying the Nordic population based on liability to AD, by defining risk groups based on the PHS quantiles and calculating the between group hazard ratios. The median incidence rate within each risk group was considered as its hazard score and the between group hazard ratio was determined according to:
| (2) |
where was obtained by fitting a Cox model to the age at onset using the PHS alone, as described elsewhere[27]. Specifically, we calculated the hazard ratios between the top 20% and bottom 20%, as well as between the top 2% and bottom 50% and between the top 50% and bottom 20% percentile groups of the proportional hazard scores. These ratios namely represent high to low risk, extremely high to medium low risk and medium high to low risk ratios. In calculating the PHS quantiles, we used those individuals who had been right-censored under the age of 70, as well as healthy subjects younger than 65 years old. We considered these PHS quantiles as references for defining the risk groups, in order to avoid inflation of the between group hazard ratios due to selection bias[17].
To compare the survival curves of AD over time using the Desikan PHS model with the non-parametric Kaplan-Meier estimates for each risk group, we used Breslow’s method[36] for obtaining the baseline population hazard from the DemGene population, with the baseline hazard function smoothed according to previously established procedures[17].
To extend the Desikan model using DemGene population, candidate SNPs (excluding those used in the Desikan model) were incrementally added as predictors to a Cox proportional hazards model including the Desikan log proportional hazard score. We also included sex and the first 4 principal components of the population genotype matrix (which explained around 25% of the total variance of the genotypes) as covariates in the Cox model to adjust for non-genetic and population structure effects. The candidate SNPs were obtained from a group of SNPs associated with the risk of AD according to a large meta-analysis from the International Genomics of Alzheimer’s Project (IGAP)[37]. The meta-analysis contained 69,814 AD cases and 375,741 controls of European ancestry, with samples from DemGene excluded. Using a significance threshold of p < 10−5, we included 4566 AD-associated SNPs and extracted corresponding genotypes from DemGene cohort. The set of the candidate SNPs obtained from IGAP was complemented using the results of a recent GWAS study of late onset AD by the Psychiatric Genomics Consortium, combining 90,338 AD cases and 1,036,225 controls from 13 cohorts after exclusion of DemGene subject[38]. Having determined the SNP effect estimates on the updated sample, we used a selection threshold of p < 10−5 and identified 985 additional SNPs, summing up in total to 5551 candidate SNPs for Desikan Nordic model. To ensure numerical stability of the statistical procedures and avoid spurious associations, we only considered those SNPs from the candidate set who had an imputation rate of at least 0.9 and a minimum allele frequency (MAF) of at least 0.05 in the training population. After quality control, 5060 candidate SNPs remained in the PHS analyses to obtain the Desikan Nordic model.
In developing the Cox regression model of Desikan Nordic, a subset of the candidate SNPs was preselected by testing the association of each SNP with the diagnosis of AD in the training population using univariate trend tests (α=0.05). The p-values of these trend tests were also used to sort the order of addition of the SNPs to the model, i.e., from the most strongly to the most weakly associated SNP. SNPs in the preselected subset were retained in the Desikan Nordic model if their addition significantly increased the Cox partial likelihood at α=0.05.
The predictive value of this modeling approach was evaluated by repeated 10-fold cross-validation using 100 random seeds. Accordingly, we selected a final set of predictive SNPs for the Desikan Nordic PHS model by considering those that appeared on at least 50 percent of the repetitions[24]. We also only allowed those SNPs in the final model whose linkage disequilibrium (LD) with the APOE ε2 and ε4 variants were r2 < 0.1, as APOE variants are already included in the model.
In the same manner as described for the Desikan model, we defined risk groups by PHS quantiles of the Desikan Nordic model and obtained the between group hazard ratios and confidence intervals according to Equation (2). To calculate the Desikan Nordic log proportional hazard scores, we used the Desikan PHS scores obtained by Equation (1) as well as the effect estimates of the m additional Desikan Nordic SNPs from each training set according to:
| (3) |
where shows the genotype dosage for SNP among the m SNPs additional to the 31 SNPs in Equation (1). Computation of the hazard ratios between the risk groups was performed using the same approach as the Desikan PHS model, obtaining the in Equation (3) from the training split and the in Equation (2) from the test split.
We validated the Desikan Nordic model and compared it with the original Desikan model by investigating the relationship between the resulting hazard scores and age at onset, as well as the risk of AD diagnosis, in the Nord Norge cohort, which was left out in the estimation of Desikan Nordic model. In particular, we compared 0.2, 0.5, 0.8 and 0.98 quantiles of the PHS scores among the right-censored individuals to define 5 risk groups that represent very low risk, low risk, medium risk, high risk and very high-risk groups, respectively. Using the 5 risk groups, we checked the association of the risk groups with the observed risk of AD using generalized Cochran-Mantel-Haenszel test stratified by gender. We also calculated Harrell’s C-index[39] to evaluate the discriminatory power and predictive accuracy of the two PHS models.
Finally, both Desikan and Desikan Nordic models were independently evaluated in the Danish CCHS and CGPS populations. In addition to obtaining the hazard ratios between the risk groups defined above, we also calculated the intra-class correlation coefficient (ICC) of the ages at onset within the defined risk groups for these populations.
RESULTS
Age of onset prediction in the Norwegian population
The average call-rate of the APOE ε2/ε4 alleles and 29 imputed SNPs from Desikan model was 0.98 (SD=0.02, minimum = 0.91) in the Nordic population, except for SNPs rs115124923 and rs155675626 on chromosome 6 which were missing (Supplementary Table 2). Based on the available predictive markers, the median hazard ratio between the top 20% risk percentile versus the bottom 20% was while the corresponding hazard ratio was for the top 2% risk group versus the bottom 50% and for the top 50% versus the bottom 20% (Figure 2). Figure 3-a depicts the obtained survival curves using the Desikan model along with the Kaplan-Meier estimates of survival for the bottom and top 20% percentiles, as well as the mid 40% percentiles, i.e. between 30% and 70%, and the top 2% percentile.
Figure 2.
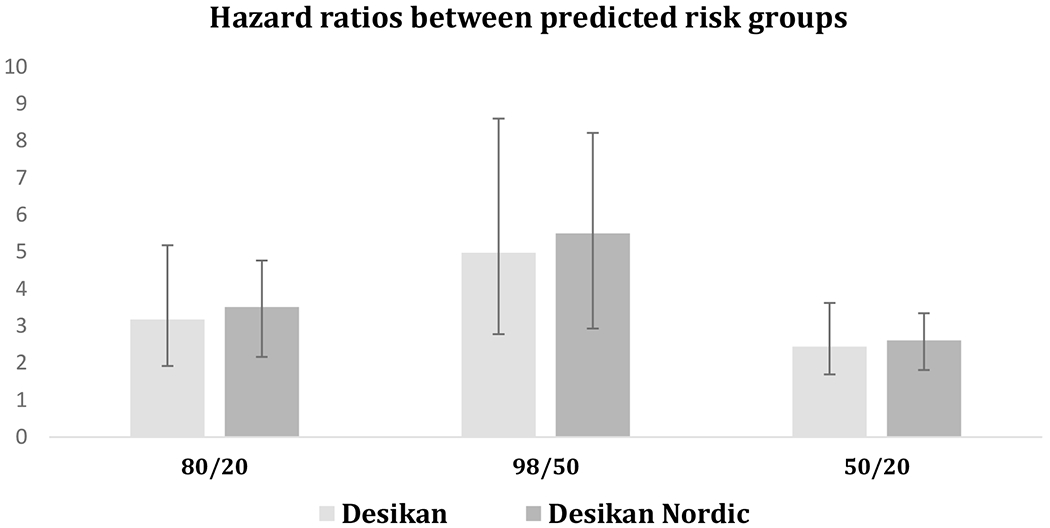
Hazard ratios between different risk groups obtained by Desikan model (light gray) and Desikan Nordic model (dark gray) in DemGene population. Ratios represent high to low risk (the top 20% to bottom 20% percentile) (left), extremely high to medium low risk (the top 2% to bottom 50% percentile) (middle) and medium high to low risk (the top 50% to bottom 20% percentile) (right).
Figure 3.
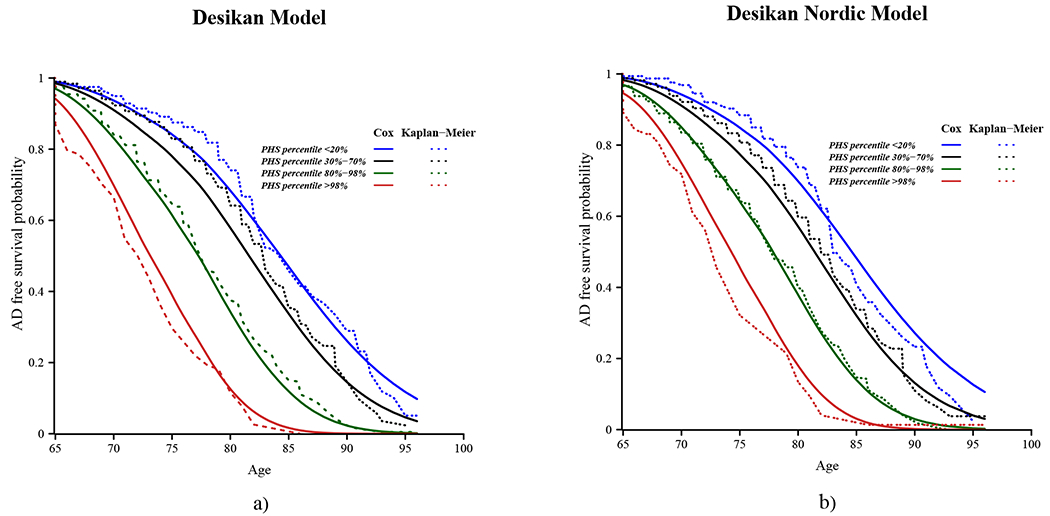
Cumulative survival with age in the Nordic population used to develop the Desikan Nordic model (11 out of 12 cohorts of the DemGene cohort, excluding the Nord Norge cohort) for different polygenic hazard score percentiles predicted by a) Desikan and b) Desikan Nordic PHS model. The solid lines show the survival estimated by Cox proportional hazards assumption, with 95% confidence intervals shown shaded, and the dashed lines show Kaplan-Meier estimates. The risk groups are defined by below 0.2 PHS percentile (blue), mid 40% PHS percentiles, i.e. between 0.3-0.7 (black), between 0.8-0.98 (green) and above 0.98 (red) PHS percentiles.
The list of the Desikan Nordic specific SNPs and their effects on the log proportional hazard of AD are provided in Table 1. A total of m=5 additional SNPs were added to the Desikan Nordic model in Equation (3), including one located over each of chromosomes 2, 8, 11 and 16 and one other on chromosome 19 downstream the APOE gene. Among these 5 SNPs (Table 1), rs7581787 on chromosome 2 was in considerable LD with rs10202748 from the original Desikan model (), and rs480781 on chromosome 11 was in considerable LD with two of the SNPs from the original Desikan model ( with rs526904 and with rs543293) in the DemGene cohort. All other LD values were negligible between these Nordic SNPs and the original Desikan SNPs or APOE alleles ().
Table 1.
Predictive SNPs in the Desikan Nordic model and their effects on the log proportional hazard of developing Alzheimer’s disease. The given call rate and MAF are for the DemGene model development population.
| rsID | Chromosome | Position (bp) | Reference Allele | Alternative Allele | Call rate | MAF | SNP Effect | 95%CI-L | 95%CI-U |
|---|---|---|---|---|---|---|---|---|---|
| rs7581787 | 2 | 234,077,240 | A | C | 0.998 | 0.481 | 0.16 | 0.13 | 0.18 |
| rs35112405 | 8 | 27,511,807 | A | T | 0.999 | 0.05 | −0.48 | −0.53 | −0.42 |
| rs480781 | 11 | 85,795,950 | A | G | 0.994 | 0.29 | −0.14 | −0.15 | −0.1 |
| rs1549299 | 16 | 31,154,146 | G | A | 0.993 | 0.287 | −0.09 | −0.16 | −0.05 |
| rs57355367 | 19 | 45,698,085 | A | G | 0.964 | 0.122 | 0.19 | 0.16 | 0.21 |
Compared with the Desikan PHS model, the Desikan Nordic model resulted in similar risk stratification, with a slight increase in the group hazard ratios: (Figure 2). The Kaplan-Meier estimates of survival with age are depicted together with the survival curves obtained by the Desikan Nordic model for each the bottom and top 20% percentiles, as well as the mid 40% and top 2% percentiles in Figure 3-b.
We validated the obtained results using the Nord Norge cohort, which we left out from the training. The association of the 5 risk groups defined by 0.2, 0.5, 0.8 and 0.98 PHS quantiles with the risk of AD was significant for Desikan Nordic model in this cohort. This association was weaker and not significant for the Desikan model . Harrell’s C-index was also slightly better with Desikan Nordic model () compared with Desikan model (). The mean difference of in the C-index was small, but significant ().
Age of onset prediction in the Danish population
The average imputation or call-rate of the available SNPs in the two Danish populations was 0.86 (SD=0.15). 10 SNPs were removed from the Desikan model due to low imputation quality (Supplementary Table 2), while all the 5 additional SNPs in the Desikan Nordic model where covered. Using Desikan model, we achieved in the CCHS population and in the CGPS population. Using Desikan Nordic model, similar results were obtained: and for the CCHS and CGPS populations, respectively. The Kaplan-Meier and Cox survival estimates in these populations are shown in Supplementary Figures 1–2, respectively. The intraclass correlation coefficient (ICC) of the ages at onset within the risk groups defined by 0.2, 0.5, 0.8 and 0.98 quantiles was and with the Desikan model, and and with the Desikan Nordic model, in CGPS and CCHS populations, respectively.
Among the Nordic SNPs (Table 1), the same 2 SNPs as in the DemGene cohort were in considerable LD with SNPs from the original Desikan model also in the Danish cohorts: rs7581787 on chromosome 2 was in considerable LD with rs10202748 from the original Desikan model (), and rs480781 on chromosome 11 was in considerable LD with two of the SNPs from the original Desikan model ( with rs526904 and with rs543293). All other LD values between the Nordic SNPs and the original Desikan SNPs or APOE alleles were in the Danish cohorts.
DISCUSSION
We showed that the Desikan PHS model for age-dependent risk of late-onset AD based on a North-American cohort [17], provided satisfactory prediction efficiency in Nordic cohorts. By extending the Desikan PHS model using AD-associated SNPs, we also constructed a Desikan Nordic model, which led to marginal improvement of prediction efficiency.
The PHS model, as a personalized genetic assessment of an individual’s age-dependent risk for a disorder, has potential to risk-stratify individuals who would otherwise be missed by traditional screening approaches, thereby identifying those at high risk at an early stage and helping prioritize preventive and therapeutic interventions [40]. The PHS model can also be extended by relevant environmental and biological information, such as sex [41], amyloid and tau protein disposition [20, 42], and healthy lifestyle scores [43] to more accurately predict the age-dependent risk of AD and its underlying mechanisms in individuals. With the introduction of new AD drugs, identifying high-risk groups prior to neurodegeneration and onset of cognitive-impairment or clinical symptoms may be the best strategy to reduce the incidence and disability-adjusted life year of AD. Using an appropriate PHS model, prospective clinical trials to investigate risk-lowering strategies could be enriched with individuals with homogenous age-dependent genetic liability, to improve trial efficiency and reduce the number of participants needed.
The Desikan PHS model has shown a ranging from 3.4 to 3.8 for North American participants with ages from 60 to 95 years [17]. In comparison, the same model resulted in values from 3.2 to 5.2 in Nordic populations. The comparison of the Cox survival curves of AD with the corresponding Kaplan-Meier curves for different risk groups (Figure 3, Supplementary Figures 1–3) showed that the assumption of proportional polygenic hazard was reasonably valid. The robustness of the Desikan model was further confirmed by the marginal improvement of the Desikan Nordic model. There are two factors that may account for the cross-ancestral prediction efficiency shown in the present study. First of all, the Desikan model was developed based on the North American participants in the ADGC cohort [17, 44], which is composed of various immigrant populations including Scandinavians [45]. Secondly, although there are genetic differences between the Nordic population and the North American population, the two populations still share genetic similarities. For example, according to a recent study by Han et al. (2017) [45] within a typical population gathered across the United States, 96% of the individuals assigned to the Scandinavian cluster had at least 20% of their alleles descended from the postulated cluster (individual admixture proportion ), while the same was still true for 14% of the individuals outside this cluster [45]. However, as we did not have access to the individual level genotype data of the ADGC cohort, we could not estimate its exact genetic distance to the Nordic cohorts. Future research is therefore required to validate Desikan PHS model in genetically more distant populations, and to boost power to discover larger proportions of the polygenic component of AD risk.
One out of the 5 additional SNPs in the Desikan Nordic model was located near the APOE locus on chromosome 19. Similarly, 3 out of the 4 other additional SNPs were close to the loci already included in the Desikan model while two SNPs where in noticeable LD () with the neighboring Desikan SNPs. The added value of the Nordic SNPs could therefore be in part due to their covering for missing Desikan genotypes, but also due to genetic heterogeneity of the disease [46].
Among the Nordic SNPs, rs480781 detected on chromosome 11 is not associated with any coding region but may have intergenic or intronic effects. SNP rs7581787 on chromosome 2 is located on INPP5D gene reported to be associated with late onset AD through modulating the inflammatory process and immune response in Caucasian populations[47, 48]. Interestingly, the SNP rs35112405 on chromosome 8 is located on SCARA3 gene locus, which encodes a macrophage scavenger protein that plays an important role in protecting cells from oxidative stress. Variations in this gene are shown to be associated with the progression of various types of malignant tumors[49, 50] as well as with the virally induced hand-foot-and-mouth disease (HFMD)[51]. The only novel locus in the Desikan Nordic model was detected by SNP rs1549299 on chromosome 16, located on PRSS36 gene whose expression in the brain is shown to be associated with AD and Parkinson’s disease [52].
In the current study, different follow-up and examination protocols were applied across the recruitment centers. Due to the discrete follow-up examination times, the age at diagnosis could differ up to a few months from the actual age at onset. The former was also influenced by the different diagnostic procedures employed at different centers. Similarly, different genotyping platforms were used across cohorts. As a result, different SNPs were missing in the applied PHS models and the accuracy of the included genotypes also varied. Unless the genotyping specifically targets the SNPs in the applied PHS model, the PHS models should therefore inevitably be modified to be used in practice, as was the case in our study. These limitations affect the obtained hazard scores and hazard ratios and make cross-population comparisons more difficult. However, these limitations are most likely introducing random noise to the model. Therefore, it is expected that the prediction performance will improve with more stringent genotyping and clinical assessment protocols.
CONCLUSION
In the current study, we applied the North American Desikan PHS model [17] to Nordic cohorts, including the Norwegian DemGene cohort and the Danish CGPS and CCHS cohorts. We extended this model by including additional genetic variants to identify risk groups of late-onset AD. We showed that the Desikan PHS model performs well, and its performance is only slightly improved by extension with additional SNPs in the Nordic populations. In summary, the Desikan PHS model showed potential to detect Nordic individuals who are at high age-dependent risk of AD. This further supports the potential use of PHS in precision medicine approaches to design and evaluate preventive interventions in the preclinical stage more efficiently.
Supplementary Material
ACKNOWLEDGEMENT
The authors would like to thank the participants of the Norwegian Dementia Genetics Network (DemGene) and the clinical units involved in data collection, as well as the participants and staff of the Copenhagen City Heart Study (CCHS) and the Copenhagen General Population Study (CGPS) for their important contributions. EM and WC received funding from the European Union’s Horizon 2020 research and innovation program under the Marie Skłodowska-Curie grant agreement No 801133. The other authors were funded by the Research Council of Norway (OAA: 213837, 223273, 229129, 248778, 248980, 262656, 273291, 283798, 311993), the South-Eastern Norway Regional Health Authority (OAA: 2013-123, 2017-112, 2019-108, 2020-034), National Institute on Aging (R01 AG08724, R01 AG17561, R01 AG028555, R01 AG060470), EU JPND: PMI-AD RCN 311993 and the EU’s Horizon2020 (grant #847776 CoMorMent). We gratefully acknowledge support from the American National Institute of Health (NS057198, EB00790), KG Jebsen Stiftelsen, and the Norwegian Health Association (22731). The CCHS and CGPS studies were funded by the Danish Heart Association, the Danish Lung Association, the Velux Foundation, and the Lundbeck Foundation.
CONFLICTS OF INTEREST
Dr. Andreassen has received speaker’s honorarium from Lundbeck and Sunovion, and is a consultant to HealthLytix. Dr. Dale is a Founder of and holds equity in CorTechs Labs, Inc, and serves on its Scientific Advisory Board. He is a member of the Scientific Advisory Board of Human Longevity, Inc. and receives funding through research agreements with General Electric Healthcare and Medtronic, Inc. The terms of these arrangements have been reviewed and approved by UCSD in accordance with its conflict-of-interest policies. The other authors declare no competing interests.
Footnotes
DATA STATEMENT
The study was approved by the institutional review board of each participating center and written informed consent was obtained from each patient or a legal representative.
References
- [1].Livingston G, Huntley J, Sommerlad A, Ames D, Ballard C, Banerjee S, Brayne C, Burns A, Cohen-Mansfield J, Cooper C (2020) Dementia prevention, intervention, and care: 2020 report of the Lancet Commission. The Lancet 396, 413–446. [DOI] [PMC free article] [PubMed] [Google Scholar]
- [2].Sperling RA, Aisen PS, Beckett LA, Bennett DA, Craft S, Fagan AM, Iwatsubo T, Jack CR Jr, Kaye J, Montine TJ (2011) Toward defining the preclinical stages of Alzheimer’s disease: recommendations from the National Institute on Aging-Alzheimer’s Association workgroups on diagnostic guidelines for Alzheimer’s disease. Alzheimer’s & Dementia 7, 280–292. [DOI] [PMC free article] [PubMed] [Google Scholar]
- [3].Sperling RA, Karlawish J, Johnson KA (2013) Preclinical Alzheimer disease—the challenges ahead. Nature Reviews Neurology 9, 54. [DOI] [PMC free article] [PubMed] [Google Scholar]
- [4].Gatz M, Pedersen NL, Berg S, Johansson B, Johansson K, Mortimer JA, Posner SF, Viitanen M, Winblad B, Ahlbom A (1997) Heritability for Alzheimer’s disease: the study of dementia in Swedish twins. The Journals of Gerontology Series A: Biological Sciences and Medical Sciences 52, M117–M125. [DOI] [PubMed] [Google Scholar]
- [5].Ertekin-Taner N (2007) Genetics of Alzheimer’s disease: a centennial review. Neurologic Clinics 25, 611–667. [DOI] [PMC free article] [PubMed] [Google Scholar]
- [6].Cruchaga C, Chakraverty S, Mayo K, Vallania FL, Mitra RD, Faber K, Williamson J, Bird T, Diaz-Arrastia R, Foroud TM (2012) Rare variants in APP, PSEN1 and PSEN2 increase risk for AD in late-onset Alzheimer’s disease families. PLoS One 7, e31039. [DOI] [PMC free article] [PubMed] [Google Scholar]
- [7].Long T, Hicks M, Yu H-C, Biggs WH, Kirkness EF, Menni C, Zierer J, Small KS, Mangino M, Messier H (2017) Whole-genome sequencing identifies common-to-rare variants associated with human blood metabolites. Nature Genetics 49, 568–578. [DOI] [PubMed] [Google Scholar]
- [8].Naj AC, Schellenberg GD, Consortium AsDG (2017) Genomic variants, genes, and pathways of Alzheimer’s disease: an overview. American Journal of Medical Genetics Part B: Neuropsychiatric Genetics 174, 5–26. [DOI] [PMC free article] [PubMed] [Google Scholar]
- [9].Gatz M, Reynolds CA, Fratiglioni L, Johansson B, Mortimer JA, Berg S, Fiske A, Pedersen NL (2006) Role of genes and environments for explaining Alzheimer disease. Archives of General Psychiatry 63, 168–174. [DOI] [PubMed] [Google Scholar]
- [10].Yamazaki Y, Zhao N, Caulfield TR, Liu C-C, Bu G (2019) Apolipoprotein E and Alzheimer disease: pathobiology and targeting strategies. Nature Reviews Neurology 15, 501–518. [DOI] [PMC free article] [PubMed] [Google Scholar]
- [11].Neu SC, Pa J, Kukull W, Beekly D, Kuzma A, Gangadharan P, Wang LS, Romero K, Arneric SP, Redolfi A, Orlandi D, Frisoni GB, Au R, Devine S, Auerbach S, Espinosa A, Boada M, Ruiz A, Johnson SC, Koscik R, Wang JJ, Hsu WC, Chen YL, Toga AW (2017) Apolipoprotein E Genotype and Sex Risk Factors for Alzheimer Disease: A Meta-analysis. JAMA Neurology 74, 1178–1189. [DOI] [PMC free article] [PubMed] [Google Scholar]
- [12].Goldberg TE, Huey ED, Devanand D (2020) Association of APOE e2 genotype with Alzheimer’s and non-Alzheimer’s neurodegenerative pathologies. Nature Communications 11, 1–8. [DOI] [PMC free article] [PubMed] [Google Scholar]
- [13].Li YJ, Scott WK, Hedges DJ, Zhang F, Gaskell PC, Nance MA, Watts RL, Hubble JP, Koller WC, Pahwa R, Stern MB, Hiner BC, Jankovic J, Allen FA Jr., Goetz CG, Mastaglia F, Stajich JM, Gibson RA, Middleton LT, Saunders AM, Scott BL, Small GW, Nicodemus KK, Reed AD, Schmechel DE, Welsh-Bohmer KA, Conneally PM, Roses AD, Gilbert JR, Vance JM, Haines JL, Pericak-Vance MA (2002) Age at onset in two common neurodegenerative diseases is genetically controlled. American Journal of Human Genetics 70, 985–993. [DOI] [PMC free article] [PubMed] [Google Scholar]
- [14].Zhang Q, Sidorenko J, Couvy-Duchesne B, Marioni RE, Wright MJ, Goate AM, Marcora E, Huang K-l, Porter T, Laws SM, Masters CL, Bush AI, Fowler C, Darby D, Pertile K, Restrepo C, Roberts B, Robertson J, Rumble R, Ryan T, Collins S, Thai C, Trounson B, Lennon K, Li Q-X, Ugarte FY, Volitakis I, Vovos M, Williams R, Baker J, Russell A, Peretti M, Milicic L, Lim L, Rodrigues M, Taddei K, Taddei T, Hone E, Lim F, Fernandez S, Rainey-Smith S, Pedrini S, Martins R, Doecke J, Bourgeat P, Fripp J, Gibson S, Leroux H, Hanson D, Dore V, Zhang P, Burnham S, Rowe CC, Villemagne VL, Yates P, Pejoska SB, Jones G, Ames D, Cyarto E, Lautenschlager N, Barnham K, Cheng L, Hill A, Killeen N, Maruff P, Silbert B, Brown B, Sohrabi H, Savage G, Vacher M, Sachdev PS, Mather KA, Armstrong NJ, Thalamuthu A, Brodaty H, Yengo L, Yang J, Wray NR, McRae AF, Visscher PM, Australian Imaging B, Lifestyle S (2020) Risk prediction of late-onset Alzheimer’s disease implies an oligogenic architecture. Nature Communications 11, 4799. [DOI] [PMC free article] [PubMed] [Google Scholar]
- [15].Escott-Price V, Shoai M, Pither R, Williams J, Hardy J (2017) Polygenic score prediction captures nearly all common genetic risk for Alzheimer’s disease. Neurobiology of Aging 49, 214. e217–214. e211. [DOI] [PubMed] [Google Scholar]
- [16].Escott-Price V, Sims R, Bannister C, Harold D, Vronskaya M, Majounie E, Badarinarayan N, GERAD/PERADES, consortia I, Morgan K (2015) Common polygenic variation enhances risk prediction for Alzheimer’s disease. Brain 138, 3673–3684. [DOI] [PMC free article] [PubMed] [Google Scholar]
- [17].Desikan RS, Fan CC, Wang Y, Schork AJ, Cabral HJ, Cupples LA, Thompson WK, Besser L, Kukull WA, Holland D (2017) Genetic assessment of age-associated Alzheimer disease risk: Development and validation of a polygenic hazard score. PLoS Medicine 14, e1002258. [DOI] [PMC free article] [PubMed] [Google Scholar]
- [18].Corder EH, Saunders AM, Strittmatter WJ, Schmechel DE, Gaskell PC, Small G, Roses A, Haines J, Pericak-Vance MA (1993) Gene dose of apolipoprotein E type 4 allele and the risk of Alzheimer’s disease in late onset families. Science 261, 921–923. [DOI] [PubMed] [Google Scholar]
- [19].Strittmatter WJ, Saunders AM, Schmechel D, Pericak-Vance M, Enghild J, Salvesen GS, Roses AD (1993) Apolipoprotein E: high-avidity binding to beta-amyloid and increased frequency of type 4 allele in late-onset familial Alzheimer disease. Proceedings of the National Academy of Sciences 90, 1977–1981. [DOI] [PMC free article] [PubMed] [Google Scholar]
- [20].Tan CH, Bonham LW, Fan CC, Mormino EC, Sugrue LP, Broce IJ, Hess CP, Yokoyama JS, Rabinovici GD, Miller BL (2019) Polygenic hazard score, amyloid deposition and Alzheimer’s neurodegeneration. Brain 142, 460–470. [DOI] [PMC free article] [PubMed] [Google Scholar]
- [21].Holland D, Frei O, Desikan R, Fan C-C, Shadrin AA, Smeland OB, Sundar V, Thompson P, Andreassen OA, Dale AM (2020) Beyond SNP heritability: Polygenicity and discoverability of phenotypes estimated with a univariate Gaussian mixture model. PLoS Genetics 16, e1008612. [DOI] [PMC free article] [PubMed] [Google Scholar]
- [22].Peterson RE, Kuchenbaecker K, Walters RK, Chen C-Y, Popejoy AB, Periyasamy S, Lam M, Iyegbe C, Strawbridge RJ, Brick L (2019) Genome-wide association studies in ancestrally diverse populations: opportunities, methods, pitfalls, and recommendations. Cell 179, 589–603. [DOI] [PMC free article] [PubMed] [Google Scholar]
- [23].Karunamuni RA, Huynh-Le M-P, Fan CC, Thompson W, Eeles RA, Kote-Jarai Z, Muir K, Lophatananon A, Schleutker J, Pashayan N (2021) Additional SNPs improve risk stratification of a polygenic hazard score for prostate cancer. Prostate Cancer and Prostatic Diseases, 1–10. [DOI] [PMC free article] [PubMed] [Google Scholar]
- [24].Karunamuni RA, Huynh-Le MP, Fan CC, Thompson W, Eeles RA, Kote-Jarai Z, Muir K, collaborators U, Lophatananon A, Tangen CM (2021) African-specific improvement of a polygenic hazard score for age at diagnosis of prostate cancer. International Journal of Cancer 148, 99–105. [DOI] [PMC free article] [PubMed] [Google Scholar]
- [25].Steinberg S, Stefansson H, Jonsson T, Johannsdottir H, Ingason A, Helgason H, Sulem P, Magnusson OT, Gudjonsson SA, Unnsteinsdottir U (2015) Loss-of-function variants in ABCA7 confer risk of Alzheimer’s disease. Nature Genetics 47, 445–447. [DOI] [PubMed] [Google Scholar]
- [26].Witoelar A, Rongve A, Almdahl IS, Ulstein ID, Engvig A, White LR, Selbæk G, Stordal E, Andersen F, Brækhus A (2018) Meta-analysis of Alzheimer’s disease on 9,751 samples from Norway and IGAP study identifies four risk loci. Scientific Reports 8, 1–8. [DOI] [PMC free article] [PubMed] [Google Scholar]
- [27].Seibert TM, Fan CC, Wang Y, Zuber V, Karunamuni R, Parsons JK, Eeles RA, Easton DF, Kote-Jarai Z, Al Olama AA (2018) Polygenic hazard score to guide screening for aggressive prostate cancer: development and validation in large scale cohorts. BMJ 360. [DOI] [PMC free article] [PubMed] [Google Scholar]
- [28].Van Giau V, Bagyinszky E, Yang YS, Youn YC, An SSA, Kim SY (2019) Genetic analyses of early-onset Alzheimer’s disease using next generation sequencing. Scientific Reports 9, 1–10. [DOI] [PMC free article] [PubMed] [Google Scholar]
- [29].Frikke-Schmidt R, Nordestgaard BG, Stene MC, Sethi AA, Remaley AT, Schnohr P, Grande P, Tybjærg-Hansen A (2008) Association of loss-of-function mutations in the ABCA1 gene with high-density lipoprotein cholesterol levels and risk of ischemic heart disease. JAMA 299, 2524–2532. [DOI] [PubMed] [Google Scholar]
- [30].Juul Rasmussen I, Rasmussen KL, Nordestgaard BG, Tybjærg-Hansen A, Frikke-Schmidt R (2020) Impact of cardiovascular risk factors and genetics on 10-year absolute risk of dementia: risk charts for targeted prevention. European Heart Journal 41, 4024–4033. [DOI] [PMC free article] [PubMed] [Google Scholar]
- [31].Rasmussen KL, Tybjærg-Hansen A, Nordestgaard BG, Frikke-Schmidt R (2020) APOE and dementia–resequencing and genotyping in 105,597 individuals. Alzheimer’s & Dementia 16, 1624–1637. [DOI] [PMC free article] [PubMed] [Google Scholar]
- [32].Purcell S, Neale B, Todd-Brown K, Thomas L, Ferreira MA, Bender D, Maller J, Sklar P, De Bakker PI, Daly MJ (2007) PLINK: a tool set for whole-genome association and population-based linkage analyses. The American Journal of Human Genetics 81, 559–575. [DOI] [PMC free article] [PubMed] [Google Scholar]
- [33].Das S, Forer L, Schönherr S, Sidore C, Locke AE, Kwong A, Vrieze SI, Chew EY, Levy S, McGue M (2016) Next-generation genotype imputation service and methods. Nature Genetics 48, 1284–1287. [DOI] [PMC free article] [PubMed] [Google Scholar]
- [34].Li Y, Willer CJ, Ding J, Scheet P, Abecasis GR (2010) MaCH: using sequence and genotype data to estimate haplotypes and unobserved genotypes. Genetic Epidemiology 34, 816–834. [DOI] [PMC free article] [PubMed] [Google Scholar]
- [35].McCarthy S, Das S, Kretzschmar W, Delaneau O, Wood AR, Teumer A, Kang HM, Fuchsberger C, Danecek P, Sharp K (2016) A reference panel of 64,976 haplotypes for genotype imputation. Nature Genetics 48, 1279. [DOI] [PMC free article] [PubMed] [Google Scholar]
- [36].Breslow N (1974) Covariance analysis of censored survival data. Biometrics, 89–99. [PubMed] [Google Scholar]
- [37].Lambert J-C, Ibrahim-Verbaas CA, Harold D, Naj AC, Sims R, Bellenguez C, Jun G, DeStefano AL, Bis JC, Beecham GW (2013) Meta-analysis of 74,046 individuals identifies 11 new susceptibility loci for Alzheimer’s disease. Nature Genetics 45, 1452–1458. [DOI] [PMC free article] [PubMed] [Google Scholar]
- [38].Wightman DP, Jansen IE, Savage JE, Shadrin AA, Bahrami S, Holland D, Rongve A, Børte S, Winsvold BS, Drange OK (2021) A genome-wide association study with 1,126,563 individuals identifies new risk loci for Alzheimer’s disease. Nature Genetics 53, 1276–1282. [DOI] [PMC free article] [PubMed] [Google Scholar]
- [39].Harrell FE Jr, Lee KL, Califf RM, Pryor DB, Rosati RA (1984) Regression modelling strategies for improved prognostic prediction. Statistics in Medicine 3, 143–152. [DOI] [PubMed] [Google Scholar]
- [40].Denny JC, Collins FS (2021) Precision medicine in 2030—seven ways to transform healthcare. Cell 184, 1415–1419. [DOI] [PMC free article] [PubMed] [Google Scholar]
- [41].Fan CC, Banks SJ, Thompson WK, Chen C-H, McEvoy LK, Tan CH, Kukull W, Bennett DA, Farrer LA, Mayeux R (2020) Sex-dependent autosomal effects on clinical progression of Alzheimer’s disease. Brain 143, 2272–2280. [DOI] [PMC free article] [PubMed] [Google Scholar]
- [42].Tan CH, Fan CC, Mormino EC, Sugrue LP, Broce IJ, Hess CP, Dillon WP, Bonham LW, Yokoyama JS, Karch CM (2018) Polygenic hazard score: an enrichment marker for Alzheimer’s associated amyloid and tau deposition. Acta Neuropathologica 135, 85–93. [DOI] [PMC free article] [PubMed] [Google Scholar]
- [43].Lourida I, Hannon E, Littlejohns TJ, Langa KM, Hyppönen E, Kuźma E, Llewellyn DJ (2019) Association of lifestyle and genetic risk with incidence of dementia. JAMA 322, 430–437. [DOI] [PMC free article] [PubMed] [Google Scholar]
- [44].Naj AC, Jun G, Beecham GW, Wang L-S, Vardarajan BN, Buros J, Gallins PJ, Buxbaum JD, Jarvik GP, Crane PK (2011) Common variants at MS4A4/MS4A6E, CD2AP, CD33 and EPHA1 are associated with late-onset Alzheimer’s disease. Nature Genetics 43, 436–441. [DOI] [PMC free article] [PubMed] [Google Scholar]
- [45].Han E, Carbonetto P, Curtis RE, Wang Y, Granka JM, Byrnes J, Noto K, Kermany AR, Myres NM, Barber MJ (2017) Clustering of 770,000 genomes reveals post-colonial population structure of North America. Nature Communications 8, 1–12. [DOI] [PMC free article] [PubMed] [Google Scholar]
- [46].George-Hyslop S, Haines J, Farrer L, Polinsky R, Broeckhoven CV, Goate A, McLachlan D, Orr H, Bruni A, Sorbi S (1990) Genetic linkage studies suggest that Alzheimer’s disease is not a single homogeneous disorder. Nature 347, 194–197. [DOI] [PubMed] [Google Scholar]
- [47].Jing H, Zhu J-X, Wang H-F, Zhang W, Zheng Z-J, Kong L-L, Tan C-C, Wang Z-X, Tan L, Tan L (2016) INPP5D rs35349669 polymorphism with late-onset Alzheimer’s disease: a replication study and meta-analysis. Oncotarget 7, 69225. [DOI] [PMC free article] [PubMed] [Google Scholar]
- [48].Rosenthal SL, Kamboh MI (2014) Late-onset Alzheimer’s disease genes and the potentially implicated pathways. Current Genetic Medicine Reports 2, 85–101. [DOI] [PMC free article] [PubMed] [Google Scholar]
- [49].Jiang L, Hu G, Chen F, Du X, Liu B, Liu C (2017) CSR1 suppresses tumor growth and metastasis of human hepatocellular carcinoma via inhibition of HPIP. Eur Rev Med Pharmacol Sci 21, 3813–3820. [PubMed] [Google Scholar]
- [50].Brown CO, Schibler J, Fitzgerald MP, Singh N, Salem K, Zhan F, Goel A (2013) Scavenger receptor class A member 3 (SCARA3) in disease progression and therapy resistance in multiple myeloma. Leukemia Research 37, 963–969. [DOI] [PMC free article] [PubMed] [Google Scholar]
- [51].Tian Y, Zhou K, Hu J, Shan M-F, Chen H-J, Cheng S, Liu L-F, Mei X-L (2019) Scavenger receptor class a, member 3 is associated with severity of hand, foot, and mouth disease in a case-control study. Medicine 98. [DOI] [PMC free article] [PubMed] [Google Scholar]
- [52].Baird DA, Liu JZ, Zheng J, Sieberts SK, Perumal T, Elsworth B, Richardson TG, Chen C-Y, Carrasquillo MM, Allen M (2021) Identifying drug targets for neurological and psychiatric disease via genetics and the brain transcriptome. PLoS Genetics 17, e1009224. [DOI] [PMC free article] [PubMed] [Google Scholar]
Associated Data
This section collects any data citations, data availability statements, or supplementary materials included in this article.