

SUMMARY
Aryl diazonium salts are ubiquitous building blocks in chemistry, as they are useful radical precursors in organic synthesis as well as for the functionalization of solid materials. They can be reduced electrochemically or through a photo-induced electron transfer reaction. Here we provide a detailed picture of the ground and excited-state reactivity of a series of 9 rare and earth abundant photosensitizers with 13 aryl diazonium salts, which also included 3 macrocyclic calix[4]arene tetradiazonium salts. Nanosecond transient absorption spectroscopy confirmed the occurrence of excited-state electron transfer and was used to quantify cage-escape yields, i.e. the efficiency with which the formed radicals separate and escape the solvent cage. Cage-escape yields were large; increased when the driving force for photo-induced electron transfer increased and also tracked with the C-N2+ bond cleavage propensity, amongst others. A photo-induced borylation reaction was then investigated with all the photosensitizers and proceeded with yields between 9 and 74%.
Keywords: Photoredox, Catalysis, Electron Transfer, Diazonium, Mechanism, Photo-induced, Hammett, Transient, Cage Escape
Graphical Abstract
eTOC blurb:
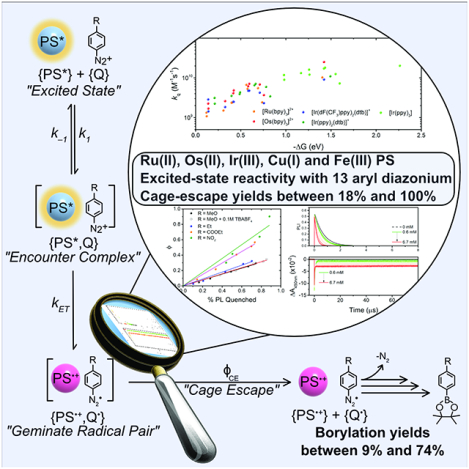
The excited-state reactivity of 9 rare and earth abundant photosensitizers with a series of aryl diazonium derivatives was investigated by means of steady-state and time-resolved spectroscopic techniques. The efficiency with which the oxidized and reduced species separate after electron transfer, i.e. the cage-escape yields, was quantified with several photosensitizers and ranged from 18% to 100%. Iron(III) photosensitizers exhibited both light and dark reactivity with aryl diazonium salts. A novel kinetic model was developed to account for these observations.
INTRODUCTION
Diazonium salts, first prepared by Peter Griess in the late 1850’s,1 represent a well-known class of reactive molecules that has always attracted much attention because of their interesting chemistry as well as ease of preparation.2 Indeed, diazotization is simply performed by reacting the parent aniline with a nitroso group, either formed in situ via the reaction of NaNO2 with HCl for example, or via commercial derivatives such as tBuONO or NOBF4. The main drawback of aryl diazonium derivatives is their lack of thermal stability, which can result in more challenging handling. Stability is usually increased when diazonium are isolated as tetrafluoroborate salts.
In spite of this, diazonium salts have found numerous applications, and probably the most widespread one is in the dyeing industry, as the azo-coupling reactions often yield intensely colored pigments. Organic chemists have taken an interest in the ease with which the N2 group can be displaced, and this displacement was historically used in the Sandmeyer, Pschorr, Gomberg-Bachmann, Meerwein or Balz-Schiemann reactions and are nowadays commonly used in (transition-metal catalyzed) aryl C–C and aryl C–Heteroatom bond formation.2 With the exception of the Balz-Schiemann reaction that occurs via thermal or light activation, these historic reactions proceed via the exogenous reduction of aryl diazonium salts using a (sacrificial) electron donor, where the chemical reduction generates the corresponding reactive aryl radical and the inert N2 gas. The resulting radical can then be used to drive a plethora of organic chemistry transformation.2–7 Alternatively, the radicals can also be used for surface modification of conducting, semi-conducting and insulating surfaces as well as nanoparticles, as pioneered by Pinson et al. in the 1990s.6,8–11
Aryl diazonium salts are also very versatile reagents in photoredox catalysis, where they can be reduced via excited-state electron transfer from a photosensitizer. In 1984, Cano-Yelo and Deronzier reported one of the first visible light activated Pschorr reactions using the prototypical [Ru(bpy)3]2+ photosensitizer.12,13 In this example, the photochemical approach led to intramolecular cyclization with 100% yield, whereas the thermal dark pathway led to lower yields and isomeric side-products. This reaction sparked a renewed interest in photo-redox catalysis and its use toward the activation of aryl diazonium derivatives.
The photophysical scheme governing the photoreaction, including the different steps relevant for the present study, is presented in Scheme 1. Photon absorption by the photosensitizer (PS) leads to the formation of an excited state (PS*) that can diffuse to generate an encounter complex with the quencher, 4-MeO-benzene diazonium in this case. A geminate radical pair composed of the oxidized photosensitizer (PS•+) and the reduced diazonium moiety is then formed if the driving force for electron transfer is favorable. This geminate radical pair can either undergo geminate charge recombination (kGCR) to form the initial reactants or, more interestingly for the field of photoredox catalysis, undergo cage-escape (ϕCE) to generate the separated, charged species (Scheme 1). The solvent separated radicals can either undergo back-electron transfer (kBET) or bond dissociation of the reduced diazonium can occur, freeing the inert N2 gas and the reactive aryl radical for further reactivity. Often mentioned in the literature is the regeneration of the ground-state photosensitizer via oxidation of an organic intermediate at a given stage within the reaction scheme (PS•+ to PS in Scheme 1).
Scheme 1.
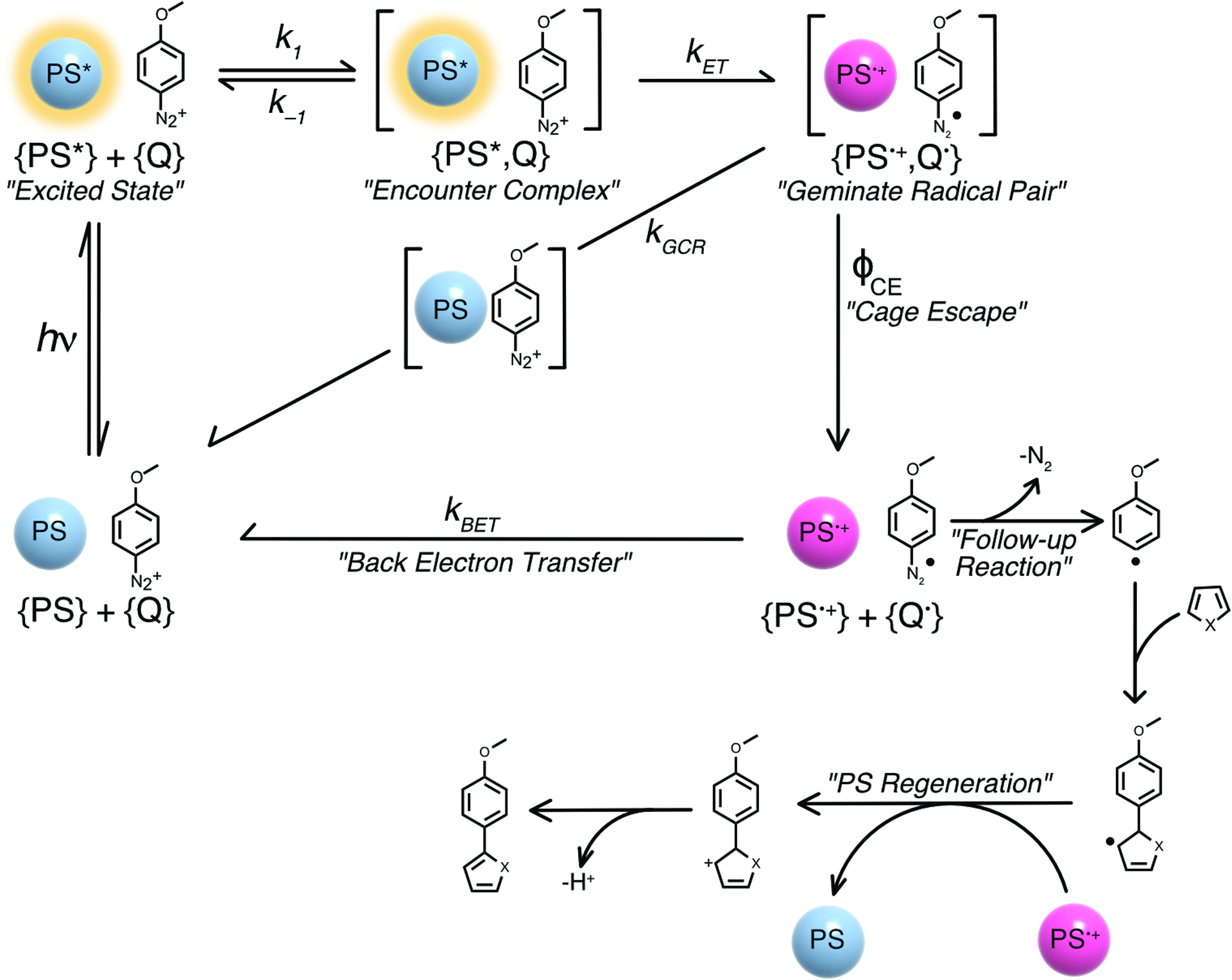
Photophysical and photochemical scheme describing the reaction between a photosensitizer (PS) and 4-MeO-benzene diazonium (Q) under irradiation. The successive reaction is a representative example reported by König et al..14
Numerous examples of the use of aryl diazonium in photoredox catalysis have since emerged. König et al. used these substrates for the direct C-H arylation of furan, thiophene and Boc-protected pyrrole scaffolds.14 Tlili et al. also recently reported the conversion of aryl diazonium salts into arylsulfonyl fluorides using DABSO, KHF2 and a cyanoarene photosensitizer (3DPAFIPN).15 Sandford et al. reported the ligand-directed C-H arylation reaction using aryl diazonium salts through the merger of palladium-catalyzed C-H functionalization and visible-light photoredox catalysis with a [Ru(bpy)3]2+ photosensitizer.16 Finally, Yan et al. used Eosin Y as photosensitizer for the visible light-induced borylation of aryldiazonium salts.17 Several reviews are dedicated to the (photo)chemical reduction of aryl diazoniums for organic chemistry and surface modification.2–7
In light of the widespread applicability of photosensitized aryl diazonium activation, we became interested in understanding key factors that govern their excited-state reactivity, product separation after electron transfer and N2 dissociation (Scheme 1). To reach that goal, a series of 9 rare and earth abundant transition metal photosensitizers were selected. This permitted tuning of ground and excited-state redox properties, nature of excited-state transitions, as well as modulation of the visible absorption range. We investigated their excited-state reactivity with a series of 13 aryl diazonium salts, which included 3 macrocyclic calix[4]arene tetradiazonium derivatives,4,11 using a variety of steady-state and time-resolved spectroscopic techniques. The emphasis was placed on determining mechanistically relevant information that would be useful for the future design of catalytic photoredox transformations.
RESULTS AND DISCUSSION
Synthesis
Figure 1 displays the structure of the 9 photosensitizers as well as the 10 aryl-diazonium and 3 calix[4]arene tetradiazonium derivatives included in this study. Most of the photosensitizers were either commercially available or were synthesized via reported procedures.18–20 [Fe(Br-phtmeimb)2]+, where Br-phtmeimb is tris(3-methylimidazolium-1-yl)(4-Br-phenyl)borate, was synthesized by reaction between FeBr2 and Br-phtmeimb in anhydrous THF at room temperature for 48 hours. Wärnmark et al. very recently reported the synthesis of [Fe(Br-phtmeimb)2]+ which occurred with similar yields but in much shorter reaction times in anhydrous DMF.21 Br-phtmeimb was synthesized in two steps starting from (4-bromophenyl)-trimethylsilane that first underwent reaction with BBr3 in dichloromethane at −78 °C following a related procedure reported by Kaufmann in 1987.22 The resulting 4-bromo-dibromophenylborane was directly reacted with imidazole in toluene in the presence of TMSCl, similarly to the procedure reported by Smith et al.23 The use of TMSCl allowed for easier recovery of tris(3-methylimidazolium-1-yl)(4-Br-phenyl)borate in 80% yield over two steps after ion metathesis using ammonium hexafluorophosphate in water. The different diazonium derivatives were either commercially available or were synthesized by reaction of the parent aniline with NOBF4 in acetonitrile at −40 °C for 2 hours.10,24 The diazonium salts were obtained in almost quantitative yields following several washing steps. As depicted in Figure 1, the aryl diazonium salts were para substituted with several electron donating/withdrawing groups that permitted tuning of the Hammett parameter (σ) from – 0.83 for 4-(N,N)-diethylamine benzene diazonium tetrafluoroborate to 0.78 for the 4-nitrobenzene diazonium tetrafluoroborate analogue.25 The calix[4]arene tetradiazonium derivatives are particularly interesting substrates as they represent polydiazonium compounds which have not been investigated thus far for excited-state quenching applications. In addition, tetrasubstituted calix[4]arenes represent a very important class of macrocyclic compounds and developing strategies to poly-functionalize these macrocycle is therefore essential.4,26 The possibility to trigger light activated processes using these compounds could in the future lead to a new class of synthetic approaches towards these macrocyclic derivatives.
Figure 1.
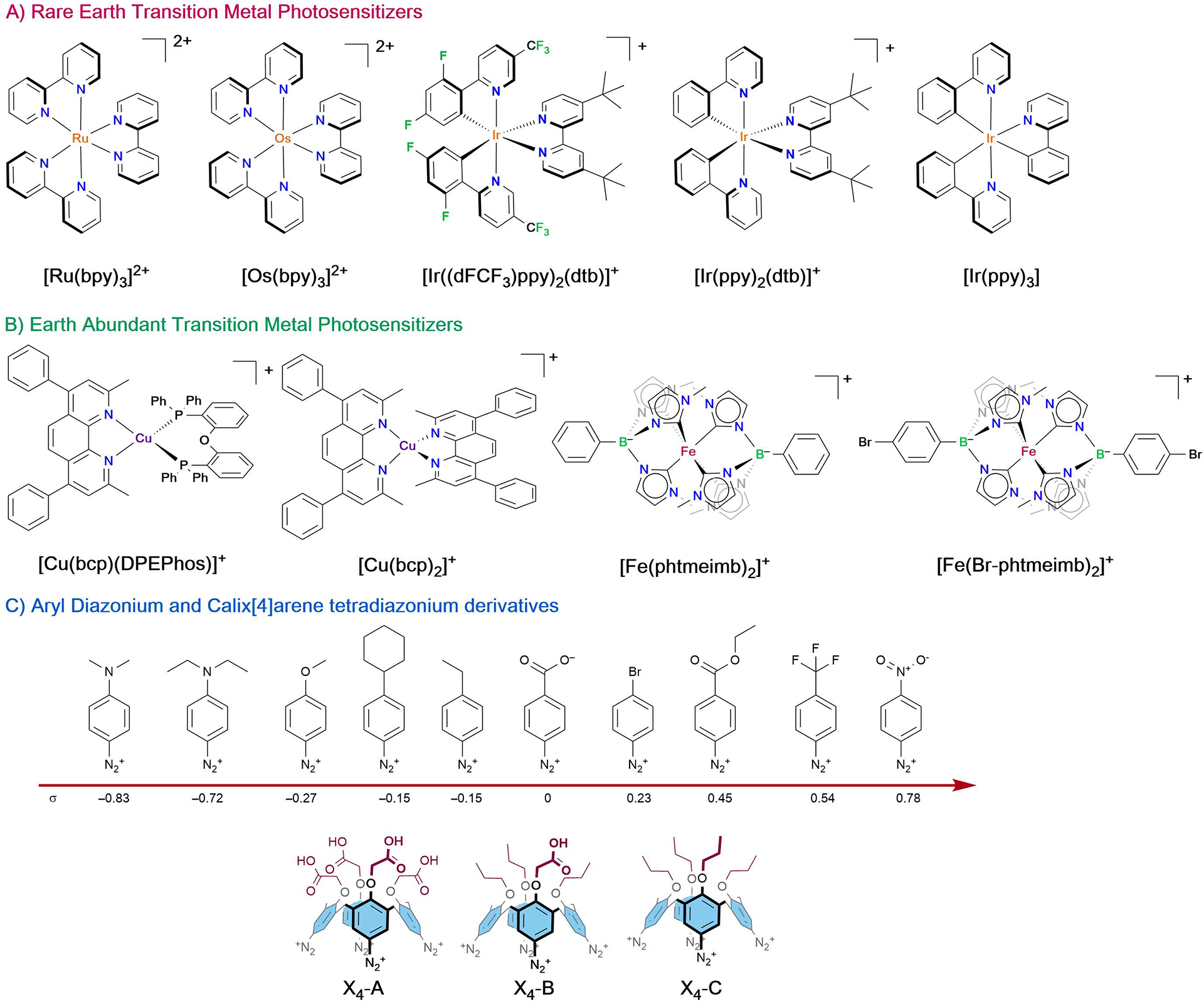
Structures of the different rare (A) and earth abundant (B) photosensitizers isolated as hexafluorophosphate salts (when charged) as well as (C) the calix[4]arene tetradiazonium and aryl diazonium salts investigated herein, including their respective Hammett parameter (σ). All overall positively charged diazonium derivatives had tetrafluoroborate counter ion(s).
Ground and Excited-State Properties
The ground-state absorption spectra of the different photosensitizers were recorded in argon purged acetonitrile at room temperature (Figure 2). The three Ir(III) photosensitizers exhibited moderate absorption features at wavelengths greater than 400 nm. [Ru(bpy)3]2+ exhibited the classical singlet metal-to-ligand charge transfer (1MLCT) band around 450 nm with molar absorption coefficient of 12000 M−1cm−1.27–29 Similar 1MLCT transitions were observed for [Os(bpy)3]2+ with a slightly larger molar absorption coefficient. In addition, direct 3MLCT excitation at wavelengths greater than 550 nm were also observed due to the larger spin-orbit coupling conferred by the heavier Os(II) center.27,30 The heteroleptic [Cu(bcp)(DPEPhos)]+ absorbed very weakly in the visible region whereas [Cu(bcp)2]+ exhibited intense 1MLCT transitions with molar absorption coefficients around 13000 M−1cm−1 at 480 nm.31–34 Finally, both iron(III) photosensitizers exhibited a doublet ligand-to-metal charge transfer band at 500 nm with a molar absorption coefficient of 2950 M−1cm−1.20,21 The introduction of Br groups had no significant impact on the UV-Visible absorption features.
Figure 2.
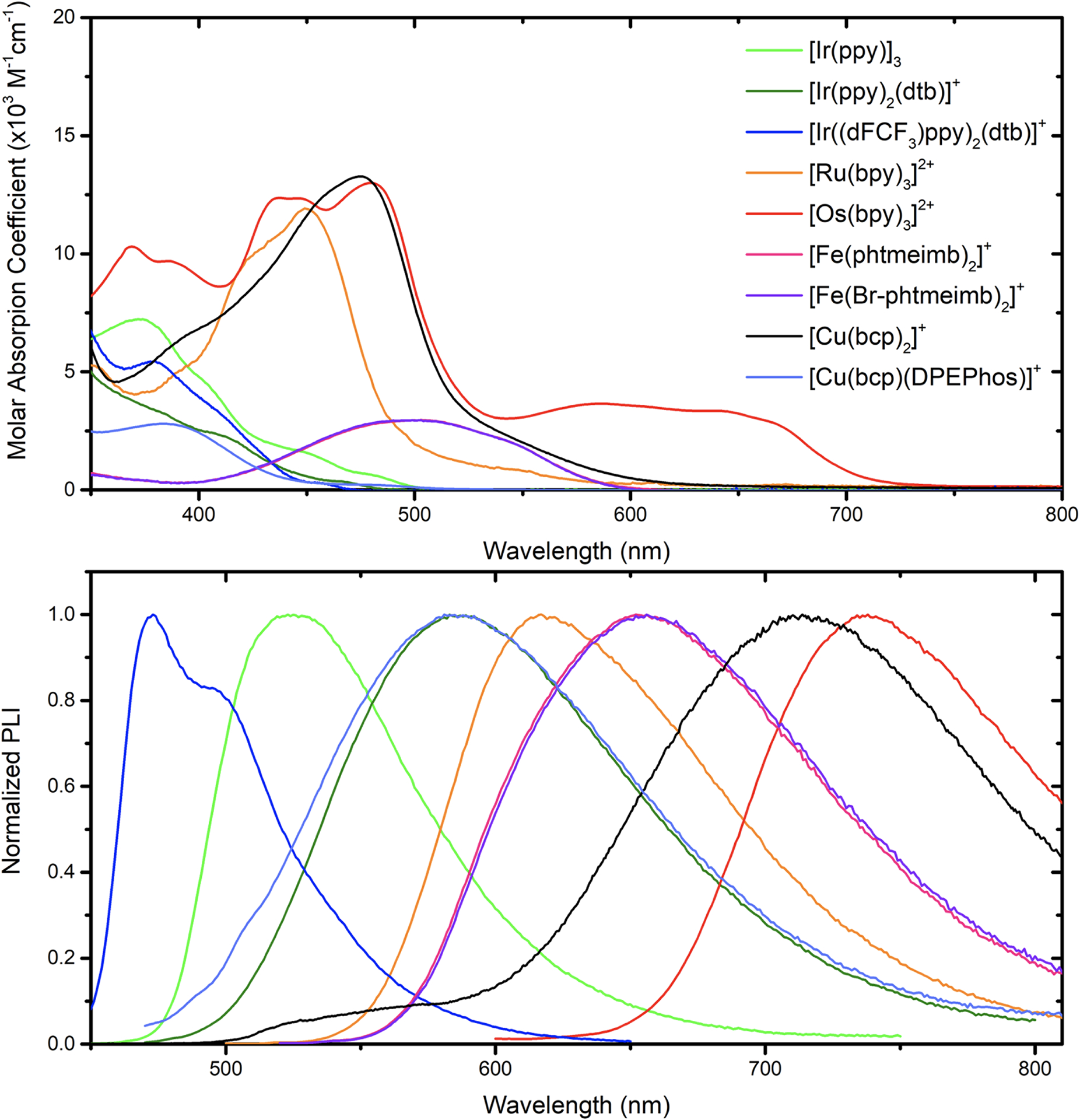
Absorption (top) and photoluminescence (bottom) spectra of [Ir(ppy)3] (green), [Ir(ppy)2(dtb)]+ (dark green), [Ir((dFCF3)ppy)2(dtb)]+ (blue), [Ru(bpy)3]2+ (orange), [Os(bpy)3]2+ (red), [Fe(phtmeimb)2]+ (pink), [Fe(Br-phtmeimb)2]+ (violet), [Cu(bcp)2]+ (black, recorded in dichloromethane), and [Cu(bcp)(DPEPhos)]+ (light blue) recorded at room temperature in argon purged acetonitrile.
Excitation of the different photosensitizers in acetonitrile within the visible range of the solar spectrum led to room temperature photoluminescence that ranged from 472 nm for [Ir((dFCF3)ppy)2(dtb)]+, to 737 nm for [Os(bpy)3]2+, respectively. Note that the emission of [Cu(bcp)2]+ was extremely weak in acetonitrile under argon and was thus recorded in dichloromethane. Time-resolved measurements of the photoluminescence in argon purged acetonitrile containing 0.1M TBABF4 were well described by first-order kinetics which yielded excited-state lifetimes ranging from 2 to 2010 ns (Table 1). The decision to perform measurements in 0.1M TBABF4 were motivated by the desire to work at fixed ionic strength to avoid further salt effects that could have originated upon the titration of aryl diazonium tetrafluoroborate derivatives (vide infra). For [Fe(phtmeimb)]+ and [Fe(Br-phtmeimb)]+, the excited-state lifetimes were obtained through reconvolution of the instrument response function (IRF) and a simulated single-exponential function to model the measured decay in signal (Figures S88–S95). This yielded an excited-state lifetime of 2 ns for [Fe(phtmeimb)]+ as well as for [Fe(Br-phtmeimb)]+, in agreement with literature values.20,21
Table 1.
Excited-state lifetimes of the 9 photosensitizers in different media
| PS | CH3CN | CH3CN + 0.1M TBABF4 | ||
|---|---|---|---|---|
| τ (ns)a | τ (ns)b | τ (ns)a | τ (ns)b | |
| [Ru(bpy)3]2+ | 193 | 850 | 165 | 890 |
| [Os(bpy)3]2+ | 39 | 57 | 37 | 55 |
| [Ir((dFCF3)ppy)2(dtb)]+ | 157 | 2010 | 142 | 1890 |
| [Ir(ppy)2(dtb)]+ | 63 | 554 | 64 | 543 |
| [Cu(bcp)(DPEPhos)]+ | 120 | 1100 | 153 | 1050 |
| [Cu(bcp)2]+c | 50 | 58 | 34 | 35 |
| [Fe(Br-phtmeimb)2]+ | 2 | 2 | 2 | 2 |
| [Fe(phtmeimb)2]+ | 2 | 2 | 2 | 2 |
| [Ir(ppy)3] | 45 | 1400 | 24 | 1420 |
Recorded under air.
Recorded under argon.
Recorded in dichloromethane
Cyclic voltammetry and differential pulse voltammetry (DPV) allowed determination of the reduction potentials of the different aryl diazoniums and photosensitizers. In Table 2 the photosensitizers are sorted according to their excited-state oxidation potential Eox*, i.e., according to their ability to release one electron from their excited state. The energy stored in the excited state (E00) was estimated using the generalizable method of extrapolating a tangent line on the blue edge of the corrected PL spectrum to the emission baseline.35–37 The excited-state redox potentials were then determined using E00 and the following equations:
| Equation 1a |
| Equation 1b |
The values obtained from these equations matched those reported in the literature.20,21,31,38–40 [Ru(bpy)3]2+, [Os(bpy)3]2+, [Ir((dFCF3)ppy)2(dtb)]+, [Ir(ppy)2(dtb)]+ and [Cu(bcp)(DPEPhos)]+ exhibit similar excited-state oxidation potentials ranging from −0.63V to −0.78V vs. NHE. The largest difference between those photosensitizers stems from the ground-state oxidation potentials, which range from 1.02 V vs. NHE for [Os(bpy)3]2+ to +1.93 V vs. NHE for [Ir((dFCF3)ppy)2(dtb)]+. [Ir(ppy)3], [Fe(phtmeimb)2]+ and [Fe(Br-phtmeimb)2]+ are the strongest reductants in their excited state, with Eox* of −1.49, −1.29 and −1.23 V vs. NHE, respectively. They exhibit ground-state oxidation potentials of +1.01, +0.85 and +0.91 V vs. NHE for [Ir(ppy)3], [Fe(phtmeimb)2]+ and [Fe(Br-phtmeimb)2]+, respectively.
Table 2.
Electrochemical properties of the 9 photosensitizers
| PS | Ground State | Excited State | ||
|---|---|---|---|---|
| Eoxa,b | Ereda,b | Eox*b | Ered*b | |
| [Ru(bpy)3]2+ | +1.50 | −1.11 | −0.63 | +1.02 |
| [Os(bpy)3]2+ | +1.02 | −1.06 | −0.65 | +0.61 |
| [Ir((dFCF3)ppy)2(dtb)]+ | +1.93 | −1.13 | −0.65 | +1.45 |
| [Ir(ppy)2(dtb)]+ | +1.45 | −1.27 | −0.72 | +0.90 |
| [Cu(bcp)(DPEPhos)]+ | +1.49 | −1.40 | −0.78 | +0.87 |
| [Cu(bcp)2]+ | +0.92 | −1.38 | −1.05 | +0.59 |
| [Fe(Br-phtmeimb)2]+ | +0.91 | −0.51 | −1.23 | +1.58 |
| [Fe(phtmeimb)2]+ | +0.85 | −0.56 | −1.29 | +1.63 |
| [Ir(ppy)3] | +1.01 | −1.95 | −1.49 | +0.55 |
Recorded in CH3CN containing 0.1M TBAPF6
In V vs. NHE.
The aryl diazonium reduction waves were irreversible, and the corresponding potentials were estimated by DPV. Reduction potentials ranged from −0.51 to 0.77 V vs. NHE and trended linearly with their Hammett parameters (Figure 3a).
Figure 3.
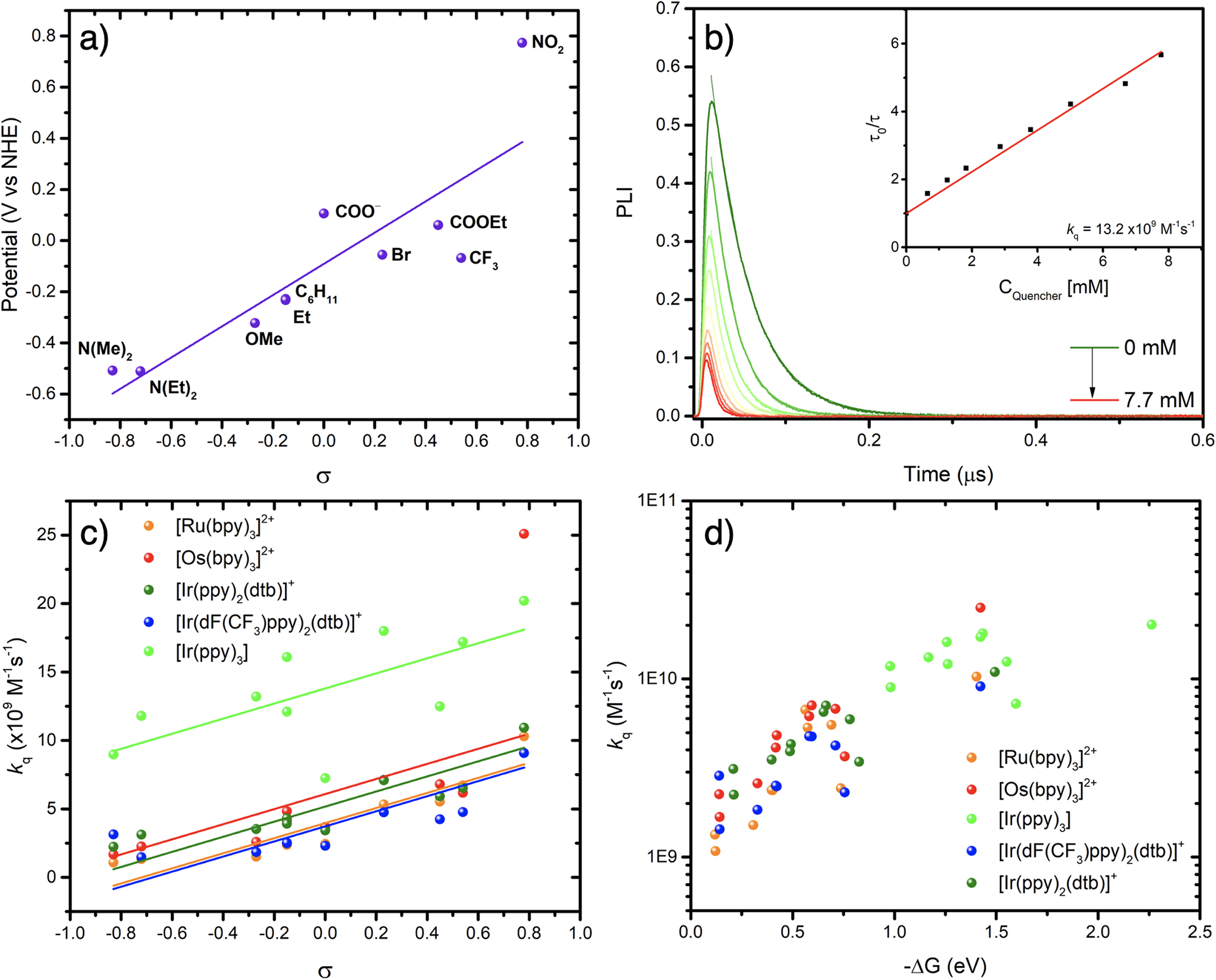
Electrochemical and excited-state characterization.
(a) Reduction potential of the aryl diazonium as a function of Hammett parameters (σ) recorded in 0.1M TBAPF6 CH3CN electrolyte at room temperature. (b) Time-resolved photoluminescence quenching experiments and Stern-Volmer plot (inset) of [Ir(ppy)]3 with increased concentration of 4-MeO-benzene diazonium. Experiments were carried out under argon in 0.1 M TBABF4 CH3CN solutions. (c) Bimolecular quenching rate constants (kq) as a function of Hammett parameters (σ) and (d) as a function of driving force (ΔG), for [Ir(ppy)3] (green), [Ir(ppy)2(dtb)]+ (dark green), [Ir((dFCF3)ppy)2(dtb)]+ (blue), [Ru(bpy)3]2+ (orange), and [Os(bpy)3]2+ (red).
Excited-State Quenching
The electrochemical data and the excited-state redox properties suggested that excited-state electron transfer from the photosensitizers to the aryl diazonium derivatives would be in all cases thermodynamically favorable as diazonium derivatives possess reduction potentials in the −0.5 to +0.8V vs. NHE range. Hence, time-resolved Stern-Volmer titration were carried out with all photosensitizers, with the exception of both iron(III) photosensitizers that were investigated via steady-state photoluminescence quenching experiments. Both copper complexes were also studied but showed lack of stability and are hence not presented herein. All steady-state and time-resolved quenching experiments were carried out in argon purged acetonitrile containing 0.1M TBABF4. The lifetime of the photosensitizers in acetonitrile and acetonitrile containing 0.1M TBABF4 are reported in Table 1. We chose to work at fixed ionic strength to avoid salt effects that could have originated upon the titration of aryl diazonium tetrafluoroborate derivatives. A representative example of the excited-state quenching of [Ir(ppy)3] with 4-MeO-benzene diazonium is presented in Figure 3b. In almost all cases, the gradual increase of quencher concentration led to appreciable quenching of the photosensitizer’s excited-state lifetime (Figures S1–S66). In some cases, additional static quenching was observed by a decrease of the initial time-resolved photoluminescence amplitude. The quenching rate constants (kq) obtained with [Ru(bpy)3]2+, [Os(bpy)3]2+, [Ir((dFCF3)ppy)2(dtb)]+, [Ir(ppy)2(dtb)]+ and [Ir(ppy)3] with all the diazonium derivatives, were in the (1.08–25.1) ×109 M−1s−1 range, i.e., close to the solvent diffusion limit. These values are compiled in Table 3. These quenching rate constants increased with the driving force for electron transfer (Figure 3d), before reaching a plateau when [Ir(ppy)3], the photosensitizer with the highest excited-state oxidation potential, was used. This plateau was reached at a driving force of around −ΔGET = ~1.2 eV which is in line or slightly larger than the ~1eV commonly observed in the literature for other systems.41–45 Interestingly, the quenching rate constants also trended with Hammett parameters (Figure 3c). In all cases, a similar slope afforded reasonable fits of the data. The calix[4]arene tetradiazonium derivatives exhibited quenching rate constant that were slighlty larger than the other ether derivatives, i.e. 4-MeO-benzene diazonium. This probably originates from the fact that the concentration of quencher, i.e. calix[4]arene is taken into consideration when determining the quenching rate constant, and not the concentration of diazonium unit. In addition, most Stern-Volmer plots with calix[4]arenre tetradiazonium moiety exhibited downward curvature at higher concentrations. The reasons for such downward curvature are still unknown but could stem from changes in concentration of quencher during the experiments or from possible intramolecular side-reactions upon reduction.
Table 3.
Quenching rate constant (kq) and numerical value of Hammett parameters (σ) for the indicated photosensitizer/aryl diazonium combinations.
| Diazonium (σ) | [Ru(bpy)3]2+a | [Os(bpy)3]2+a | [Ir(ppy)3]a | [Ir((dFCF3)ppy)2(dtb)]+ a | [Ir(ppy)2(dtb)]+ a |
|---|---|---|---|---|---|
| NMe2 (−0.83) | 1.08 | 1.67 | 8.97 | 1.43 | 2.25 |
| NEt2 (−0.72) | 1.34 | 2.25 | 11.8 | 2.86 | 3.11 |
| MeO (−0.27) | 1.51 | 2.59 | 13.2 | 1.84 | 3.52 |
| C6H11 (−0.15) | 2.37 | 4.83 | 12.1 | 2.49 | 4.32 |
| Et (−0.15) | 2.37 | 4.12 | 16.1 | 2.52 | 3.93 |
| COO− (0) | 2.43 | 3.67 | 7.24 | 2.30 | 3.42 |
| Br (0.23) | 5.33 | 7.11 | 17.9 | 4.74 | 7.06 |
| COOEt (0.45) | 5.52 | 6.08 | 12.5 | 4.24 | 5.92 |
| CF3 (0.54) | 6.71 | 6.17 | 17.2 | 4.78 | 6.53 |
| NO2 (0.78) | 10.3 | 25.1 | 20.2 | 9.07 | 10.9 |
| X4-A | 4.37 | 10.8 | 20.5 | 5.59 | 10.3 |
| X4-B | 4.37 | 7.30 | 11.5 | 3.18 | 8.68 |
| X4-C | 5.70 | 10.2 | 16.7 | 5.12 | 10.0 |
kq (×109 M−1s−1). Experiments were carried out at room temperature in argon purged acetonitrile containing 0.1M TBABF4 electrolyte.
Unfortunately, neither copper complexes remained stable enough in 0.1M TBABF4 CH3CN solution during the experiment to afford unambiguous quenching rate constants. This was in line with the well-established heteroleptic to homoleptic conversion46,47 via a ligand association-dissociation mechanism that occurs more rapidly in coordinating solvents, such as acetonitrile48, than in non-coordinating ones such as dichloromethane.49 This process was also shown to occur upon oxidation of Cu(I) to Cu(II), either electrochemically50 or under illumination.51,52 Surprisingly, despite a large driving force for electron transfer, the iron (III) complexes did not exhibit appreciable excited-state quenching (Figures S65–S66). The possibility to perform excited-state oxidative quenching of [Fe(phtmeimb)2]+ was already realized by Wärnmark and coworkers using methylviologen with concentrations ranging from 10 mM to 500 mM.20 We observed similar results when methylviologen was used with [Fe(phtmeimb)2]+ or [Fe(Br-phtmeimb)2]+, with kq of 1.8 ×109 and 1.6 ×109 M−1s−1, respectively. We were unable to increase the concentration range of 4-MeO-benzenediazonium to reach those used for the quenching by methylviologen, as dark reactivity was observed concomitantly, as will be further described (vide infra).
Photolysis
The quenching rate constants highlight that the 5 rare earth transition metal complexes were able to photo-react, supposedly via excited-state electron transfer, with all the aryl diazonium derivatives. Thermodynamically, all earth abundant photosensitizers should also be able to undergo excited-state reactivity with the diazonium. In an attempt to corroborate the excited-state reactivity, or lack thereof, and to further investigate the issue of photostability noted for the copper complexes, we performed photolysis experiments.53 These were carried out with the different photosensitizers in the presence of 30 mM of 4-MeO-benzene diazonium in argon-purged acetonitrile. Three main pieces of information were expected from these photolysis experiments. The first one was to determine whether excited-state electron transfer can occur, even for the photosensitizers for which excited-state quenching could not be monitored. This investigation is crucial for future transient absorption measurements as well as for photoredox catalytic measurements. The second one was the photostability of the photosensitizer in the ground and oxidized form in the presence of 30 mM of 4-MeO-benzene diazonium. The third one was to obtain the molar absorption coefficient of the oxidized and reduced photosensitizers. This is important for measurements of cage-escape yields (vide infra) that require Δε values.
A typical photolysis experiment is presented in Figure 4 with [Os(bpy)3]2+ and with [Fe(phtmeimb)2]+. Illumination of a solution of [Os(bpy)3]2+ with 30 mM of 4-MeO-benzene diazonium led to the gradual bleach of the MLCT transitions within 10 minutes of continuous irradiation, indicative of the formation of Os(III). Ascorbic acid was then added to test reversibility which allowed for the immediate recovery of the authentic [Os(bpy)3]2+ spectra. The difference observed between the original spectra and the one after the addition of ascorbic acid stems from a combination of the absorbance of reduced aromatic compounds that tails into the visible range as well as some scattering induced by the moderately soluble ascorbic acid. When [Fe(phtmeimb)2]+ was used, in conditions where no dark reactivity occurs (vide infra), illumination for 60 minutes led to the gradual bleach of the 2LMCT transition accompanied by the concomitant growth of a broad absorbance centered around 700 nm. This band was previously attributed to the formation of Fe(IV) species.20 The longer irradiation time probably stems from smaller quenching rate constant, lower cage-escape yields or the fact that the excited-state electron transfer from the iron-localized excited state to the diazonium could be kinetically slow. Nevertheless, the formation of Fe(IV) was also reversible, as observed by the recovery of the initial 2LMCT transition upon the addition of ascorbic acid. Similar reactivity was observed for [Fe(Br-phtmeimb)2]+ which exhibited a molar absorption coefficient for the Fe(IV) form of 7030 M−1cm−1 (Figure S75). Reversibility was also observed with [Ru(bpy)3]2+ (Figure S79). [Ir(ppy)3] seemed to exhibit some degree of irreversibility under these conditions (Figure S78), whereas for the two other iridium complexes, the changes in absorption spectra upon photolysis were too small to unambiguously discuss reversibility (Figures S76–S77). We also encountered partial reversibility for [Cu(bcp)2]+ (Figure S73) and observed the growth of a peak around 480 nm when [Cu(bcp)(DPEPhos)]+ was used, consistent with the in situ formation of [Cu(bcp)2]+ upon illumination in these conditions, as already known in the literature for similar complexes (Figure S74).48,54
Figure 4.
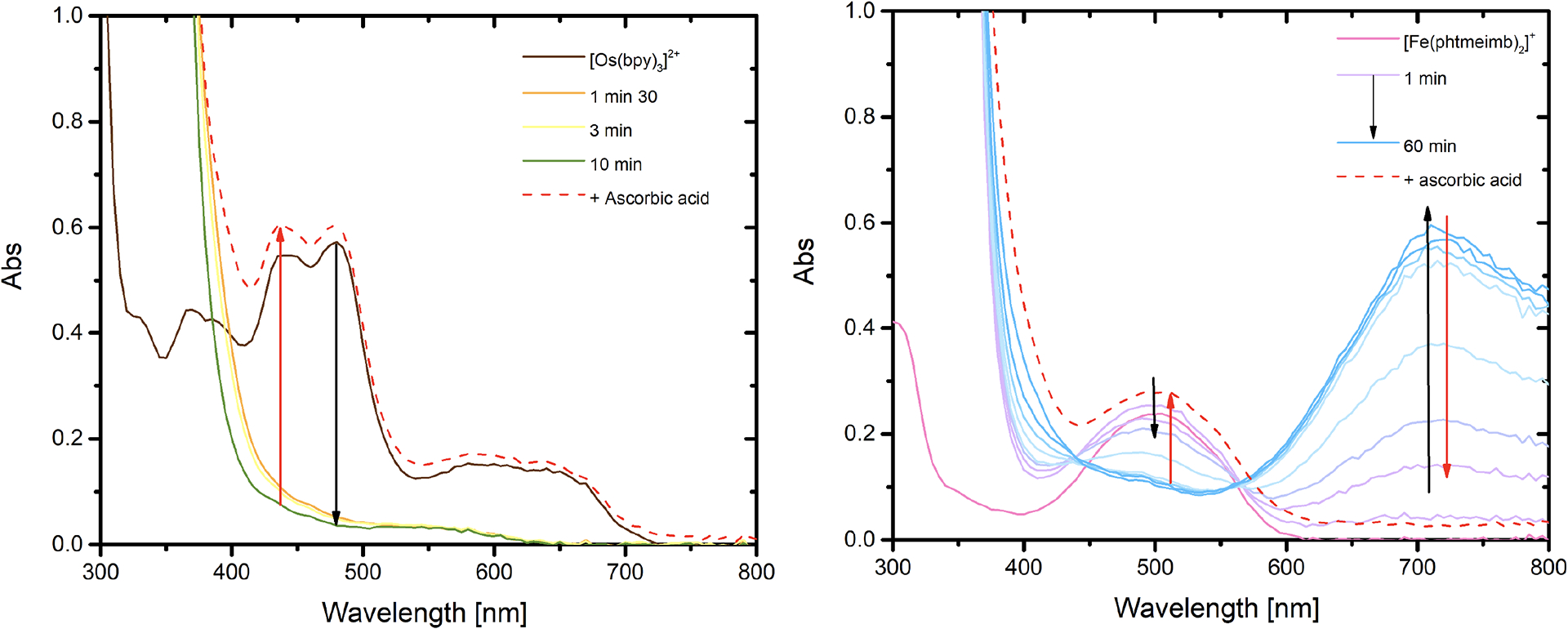
Photolysis experiments of [Os(bpy)3]2+ (left) and [Fe(phtmeimb)2]+ (right) in argon purged acetonitrile in the presence of 30 mM MeO-benzene diazonium. Reversibility was assessed through the addition of ascorbic acid at the end of the photolysis experiment (red dashed lines).
Dark Reactivity
As mentioned before, reactivity in the dark was observed when [Fe(phtmeimb)2]+ or [Fe(Br-phtmeimb)2]+ was mixed with selected diazonium salts. For example, in a solution mixture containing 4-Br-benzene diazonium, the bright red color of [Fe(phtmeimb)2]+ gradually changed to a persistent blue, which was unambiguously attributed to the formation of Fe(IV). Also, when the more electron deficient 4-NO2-benzene diazonium was used as the oxidant, the Fe(IV/III) color change was almost instantaneous to the unaided eye. These qualitative observations were fully consistent with a ground-state thermal electron transfer, despite thermodynamic predictions suggesting a highly endergonic ΔG of 0.80 eV for electron transfer based on the one-electron reduction potentials (Fe(IV/III) = 0.85 V vs. NHE and 4-Br-benzene diazonium reduction (Br-N2(+/•) ≈ 0.05 V vs. NHE)). When, instead, the more electron rich 4-MeO-benzene diazonium was used, the solution mixture maintained the Fe(III) red color for several hours, indicating that the thermal electron transfer was significantly inhibited in these experimental conditions.
The reaction sequence for the net activation of the diazonium substrates in the dark is described here by a consecutive irreversible chemical reaction with a reversible electron transfer step according to equation 2.
| Equation 2 |
Quantitatively, equation 2 is expressed by a set of coupled differential equations that describe the time-dependent concentration changes of all reactants and products,
| Equation 2.1 |
| Equation 2.2 |
| Equation 2.3 |
To test the proposed reaction pathway described by equation 2, solution mixtures of 230 μM of [Fe(phtmeimb)2]+ were prepared at various concentrations of Br-benzene diazonium, and the changes in absorption were monitored as a function of time. Kinetic traces monitored at 700 nm, Figure 5, or at 500 nm, which were representative of Fe(IV) formation, were analyzed by two different – but complementary – methods. First, by reasonably assuming k2 ≫ k1, the kinetics of equation 2 reduces to a single first order kinetic reaction under the steady-state approximation, with k1 being the rate determining step. A linear fit of the observed rates plotted against the concentration of diazonium provided a rate constant k1 of 0.115 M−1s−1 (Figure 5)
Figure 5.
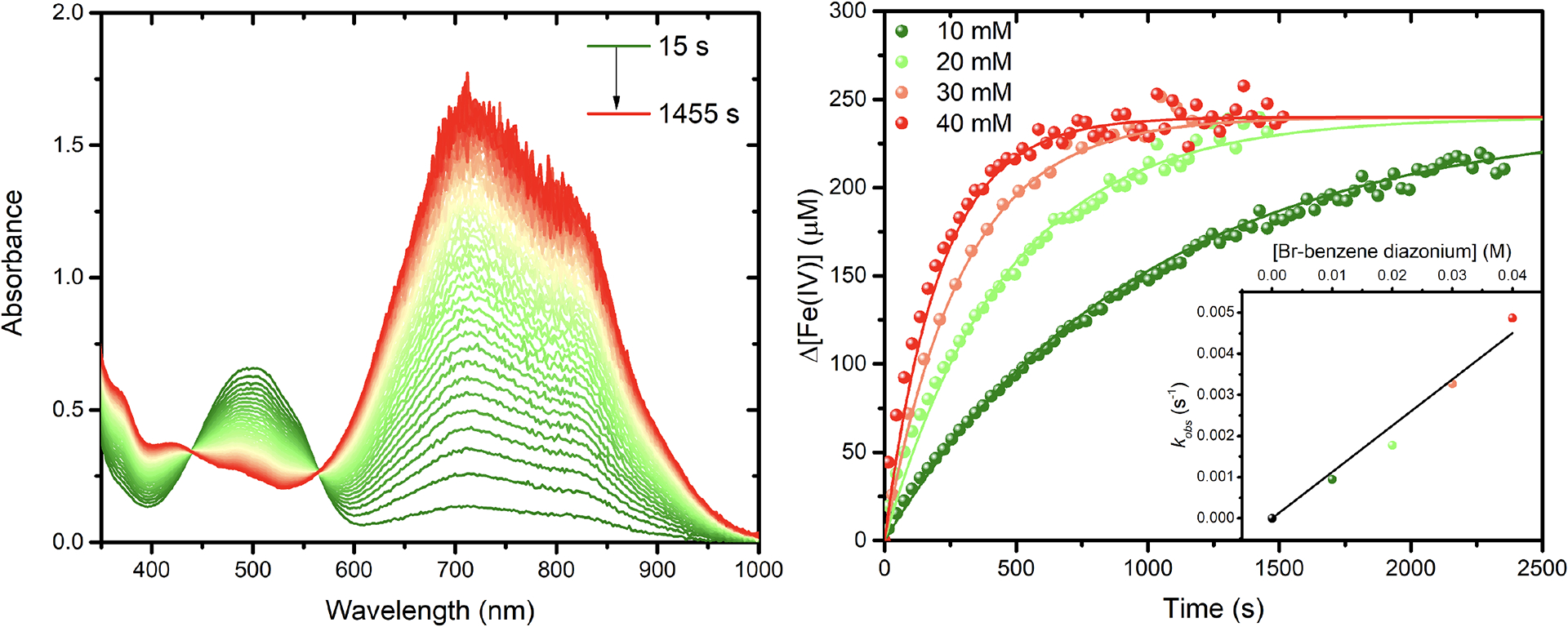
Dark reactivity of [Fe(phtmeimb)2]+ with 4-Br-benzene diazonium. Absorption changes (left) recorded in acetonitrile at selected time intervals for a solution of [Fe(phtmeimb)2]+ (230 μM) in the presence of 20 mM 4-Br-benzene diazonium. Absorption changes recorded at 700 nm (right) at different concentration of 4-Br-benzene diazonium. The inset shows the observed rate at each concentration that was used to determine the rate constant for electron transfer. This rate constant was used, in concert with equations 2.1–2.3, to fit the absorption changes at 700 nm.
The kinetic traces in Figure 5 were then analyzed by numerically fitting the data to the set of differential equations using k1 = 0.115 M−1s−1 as a fixed input parameter. The reverse rate constant k−1 was substituted by the relationship k−1 = k1/Keq, where Keq = exp(−ΔG/RT) with ΔG = 0.80 eV (at 298K) as the initial guess. A global analysis of the kinetic traces in Figure 5 produced the best fits when ΔG = 0.66 eV (Keq = 8 × 10−12) and k2 ≤ 1.0 × 108 s−1. Values for the dissociation constant of R•N2 faster than 10 ns did not significantly improve the quality of the fit; in contrast, slower values of k2 allow the reversible electron transfer, controlled by k−1, to compete kinetically with the irreversible R•N2 dissociation step. Using the value of ΔG = 0.66 eV, and the Fe(IV/III) redox potential, the one electron reduction potential for 4-Br-benzene diazonium was estimated at 0.19 V vs. NHE. This value is about 0.14 V more positive than that estimated by DPV measurements, and naturally raised questions as to whether DPV or cyclic voltammetry methods provide accurate reduction potentials for irreversible reduction events, as observed for the series of benzene-substituted diazonium compounds.
In a pure reversible equilibrium, highly endergonic electron transfer reactions are often considered improbable when, in reality, a dynamic equilibrium exists where the concentrations of reactants and products are thermodynamically controlled by the balance of opposing forward and reverse rate constants, or the equilibrium constant. For reactions like the one described by equation 2, the presence of a fast and irreversible chemical step – the dissociation of R•N2 that frees N2 – creates a reaction sink for the reversible electron transfer that shifts the product concentrations towards the right-hand side of equation 2, in agreement with the Le Chatelier principle.
Transient Absorption Spectroscopy
Nanosecond transient absorption was then carried out to provide evidence that oxidative excited-state electron transfer was indeed occurring. First, the excited-state absorption spectra were recorded for the different photosensitizers. Representative examples for [Os(bpy)3]2+ and [Ir(ppy)3] are shown in Figure 6 whereas the transient absorption spectra of [Ru(bpy)3]2+, [Ir((dFCF3)ppy)2(dtb)]+ and, [Ir(ppy)2(dtb)]+ are gathered in the supporting information (Figures S67–S72). The excited-state absorption spectra of [Os(bpy)3]2+ was typical for this class of photosensitizer, i.e. a ground state bleach associated with the MLCT transitions, and positive absorption features below 410 nm and above 690 nm attributed to transition located on the reduced 2,2′-bipyridine following metal to ligand charge transfer. The transient absorption spectra, monitored at 490 nm, decayed with the same time constant as the one determined via time-resolved photoluminescence measurements. When similar experiments were carried out in the presence of 30 mM of 4-MeO-benzene diazonium tetrafluoroborate, bleached signals were observed in almost the entire spectral window. The absence of positive absorption features coupled with the extremely long (greater than 300 μs) lifetime of the transient species are clear indicators of excited-state electron transfer and formation of the corresponding Os(III) center.
Figure 6.
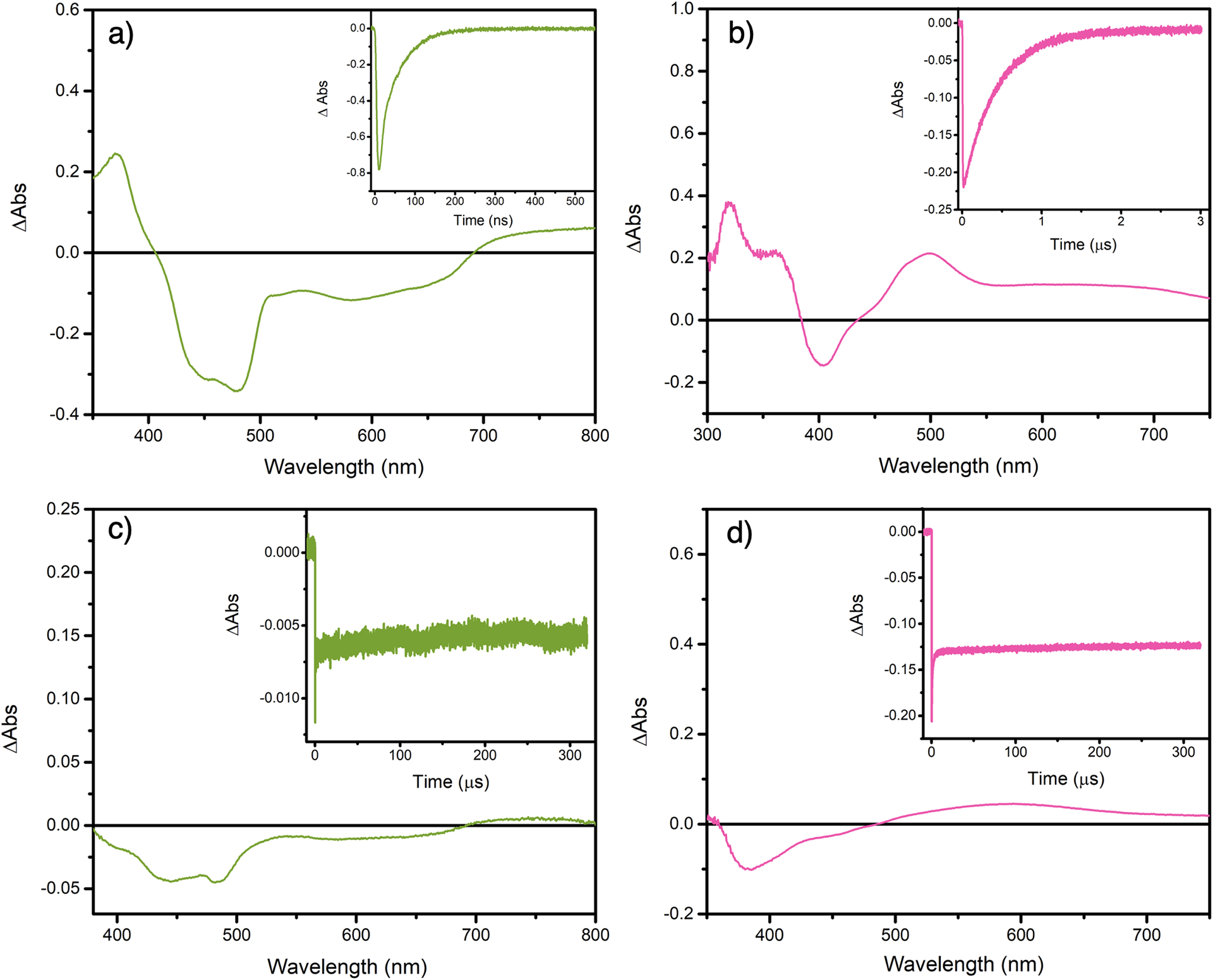
Excited-state characterization and reactivity of [Os(bpy)3]2+ and [Ir(ppy)3]. Nanosecond transient absorption spectra of [Os(bpy)3]2+ (a) and [Ir(ppy)3] (b) recorded 10 ns after the laser pulse (integrated for 50 ns) in argon purged acetonitrile under argon atmosphere. Changes in absorption of [Os(bpy)3]2+ (c) and [Ir(ppy)3] (d) recorded 100 ns after the laser pulse (integrated for 100 ns) in the presence of 30 mM MeO-benzene diazonium. Insets represent the single wavelength absorption changes recorded at 450 nm for [Os(bpy)3]2+ and 400 nm for [Ir(ppy)3], respectively. Experiments were carried out in argon purged acetonitrile at room temperature. The sample were excited at 420 nm with a laser fluence of 10 mJ/pulse.
Similar results were obtained for [Ir(ppy)3] in argon purged acetonitrile. The excited-state absorption spectra exhibited a bleach around 400 nm accompanied by positive absorption features below 370 nm and above 440 nm. Recovery of the ground-state bleach occurred with a similar rate as the one determined by time-resolved photoluminescence measurements. In the presence of 30 mM of 4-MeO-benzene diazonium tetrafluoroborate, bleached signals were observed below 490 nm accompanied by positive absorption features above 490 nm. These spectral changes as well as the greater than 300 μs lifetime were also consistent with excited-state oxidation of [Ir(ppy)3]. Similar observations were made with [Ru(bpy)3]2+, [Ir((dFCF3)ppy)2(dtb)]+ and [Ir(ppy)2(dtb)]+ (Figures S67–S72). Transient absorption measurements were not attempted with the different copper complexes due to their previously noted lack of stability.
The results presented so far are in line with the accepted excited-state reaction scheme in solution that follows the undermentioned sequence: the excited photosensitizer diffuses towards the quencher (diazonium in this case), forming a so-called “encounter complex” (Scheme 1). If thermodynamically favorable, the electron transfer event can occur, generating the corresponding geminate pair of radicals (oxidized and reduced species), {PS•+;Q•−}. The latter was evidenced by Stern-Volmer experiments, photolysis and nanosecond transient absorption spectroscopy. The geminate pair or radicals can either recombine via geminate charge recombination to regenerate the initial state or undergo a cage-escape process to generate the charge-separated products that can be further used to drive additional organic transformations (Scheme 1). To date, there is no clear theory that allow prediction of the efficiency with which these oxidized and reduced species separate, yet it is of paramount importance for further improvement of light-driven applications.19,41,55–58
Cage-Escape Yields
The literature of cage-escape yields (ϕCE) for inorganic PS is mostly centered around [Ru(bpy)3]2+ that has often been considered as the prototypical inorganic photosensitizer. In a seminal paper, Olmsted and Meyer sought to understand the factors influencing ϕCE. To do so, they used methylviologen (MV2+) as electron acceptor and a series of Ru(II) and Os(II) PS, as well as some anthracene derivatives as electron donors. Olmsted and Meyer observed that ϕCE for the {M3+,MV•+} pair varied only slightly within the series, with values that ranged from 14% to 27%.59 However, when using an organic triplet as the electron donor, such as 9-methylanthracene or acridine yellow, ϕCE reached almost 100%. The yields dropped to 30% when perturbation by heavy atoms was introduced, as in the case of 9-bromoanthracene in solution containing CH3I. The reported results highlighted that ϕCE were strongly affected by the rate of triplet-singlet interconversion of the triplet pair generated in the quenching event. In the absence of triplet-singlet mixing, as in the case of pure triplet organic donors, back-electron-transfer, which requires a spin change, was slow compared to diffusion out of the solvent cage which ultimately led to large ϕCE. When spin-orbit coupling was substantial, as with heavy-atom perturbators or in transition metal complexes, back-electron-transfer became competitive with diffusional cage escape and thus lead to lower ϕCE.
Kalyanasundaram and Neumann-Spallart later investigated the effects of added electrolyte on the ϕCE for the reaction between [Ru(bpy)3]2+* and MV2+. They found that ϕCE could vary by a factor of four upon changes in the nature of the medium by the addition of electrolytes. Experimentally, they determined that the quenching rate constant between the two doubly charged species increased as the ionic strength of the solution increased according to a “primary kinetic salt effect”, as can be explained in terms of the Bronsted-Debye equation.60 A ϕCE of 42% was determined in acetonitrile containing 0.1M tetrabutylammonium perchlorate as electrolyte. This value decreased to 35% when 5% water was added to acetonitrile and reached 25% in neat or in buffered water at pH 4.7, in line with the results of Olmsted and Meyer.
We have recently attempted to understand factors influencing ϕCE using iron(III) photosensitizer and to compare them with Ru(II) PS.19,58 When triethylamine was used as electron donor, ϕCE for Ru(II) PS increased with solvent polarity – lower yields of 15% were found for CH2Cl2, moderate yields of 39% were found in CH3CN, and much higher values were determined in DMF, ϕCE = 58%. This observation was in line with the expectation that polar solvents are better at stabilizing charged species. On the contrary, ϕCE values determined for the Fe(III) PS exhibited the opposite trend as those of Ru(II) PS, i.e. higher yields were measured in low polarity solvents (ϕCE = 21% in CH2Cl2) whereas lower ϕCE were measured in DMF (ϕCE = 9%) and CH3CN (negligible ϕCE).40
When iron(III) PS was quenched by aniline derivatives, i.e., N,N-dimethyl-p-toluidine (DMT), dimethylaniline (DMA) or tritolylamine (TTA), ϕCE measured by comparative actinometry in DMF and CH3CN were less than 7% but reached much higher values (between 36% and 63%) in CH2Cl2. This occurred despite very similar excited-state quenching rate constants in all three solvents. The low photo-product yields observed in CH3CN were consistent with a report by Wärnmark et al. who determined ϕCE = 5% for both a N-phenylaniline electron donor and a methylviologen electron acceptor in CH3CN.20
Our initial hypothesis that sparked this research project was that compounds like aryl diazonium, which are known to be very reactive and irreversibly generate the inert N2 gas upon one electron reduction would lead to increase cage-escape yields by impeding back-electron transfer within the solvent cage. The cage-escape yields (ϕCE) were determined for [Os(bpy)3]2+, [Ru(bpy)3]2+, [Ir((dFCF3)ppy)2(dtb)]+, [Ir(ppy)2(dtb)]+ and [Ir(ppy)3] with a series of diazonium. These cage-escape yields were determined in neat CH3CN as well as in the presence of 100 mM TBABF4 for [Ru(bpy)3]2+. The cage-escape yields were determined by comparative actinometry using equations 3 and 4 using [Ru(bpy)3]2+ as actinometer.61 The maximum absorption changes generated from the oxidized PS (PS+) was compared to the absorption maxima of the excited state of the reference [Ru(bpy)3]2+ (ESref) and normalized by their respective absorptances at the excitation wavelength (λexc). A Δε value of −11000 M−1cm−1 for the actinometer at 450 nm was used.62–64 Δε450nm = −9000 M−1cm−1 and Δε470nm = −12000 M−1cm−1 were determined through the photolysis experiments for [Ru(bpy)3]2+ and [Os(bpy)3]2+, respectively, while Δε450nm = 1600 M−1cm−1 for [Ir((dFCF3)ppy)2(dtb)]+ and [Ir(ppy)2(dtb)]+ was estimated following a modified reported procedure using transient absorption spectroscopy and MV2+ as electron acceptor.65,66 The Δε450nm = −9000 M−1cm−1 was in line with the value obtained via spectroelectrochemistry which enables to confirm that, in this case, the photolysis approach allowed to determine useful values of Δε. A Δε530nm = 1700 M−1cm−1 was used based on reported spectroelectrochemical measurements.67 Final cage-escape yields (ϕCE) values were obtained by comparing the relative yield of PS+ produced (ϕ) to the percentage of quenched PL (%PL).
| Equation 3 |
| Equation 4 |
A representative example for [Ru(bpy)3]2+ with 4-MeO-benzene diazonium is represented in Figure 7.
Figure 7.
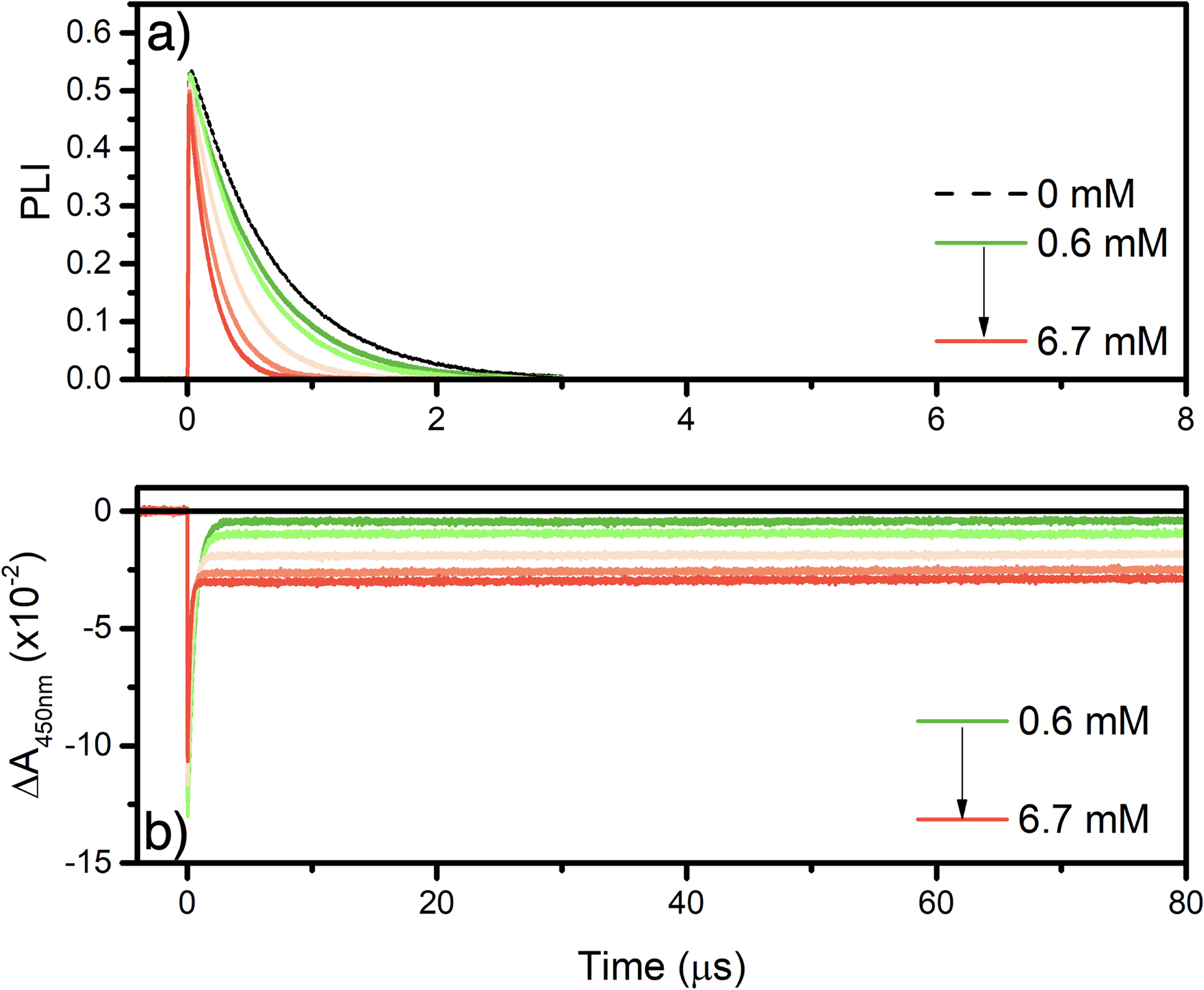
Representative data set for [Ru(bpy)3]2+ quenched by 4-MeO-benzene diazonium in argon purged acetonitrile at room temperature. a) Excited-state lifetime at different concentration of 4-MeO-benzene diazonium used to determine the percentage of PL quenched. b) Changes in absorption recorded at 450 nm following pulse light excitation at similar concentration of 4-MeO-benzene diazonium as in panel a).
The gradual Increase of 4-MeO-benzene diazonium led to increased excited-state quenching, which was used to determine the percentage of PL quenched used in equation 3. Each data point represents a freshly prepared sample with the indicated concentration of aryl diazonium. The percentage of quenching was determined either via time-resolved photoluminescence or via steady-state photoluminescence when static quenching was observed. In parallel, the changes in absorption following pulsed light excitation are recorded at 450 nm. The value of ΔA recorded 10 μs after the laser pulse was used in equation 4. Plots of ϕ versus the %PL quenched were all linear and the actual cage-escape values were extracted from the slope (Figure 8). The cage-escape yields are gathered in Table 4.
Figure 8.
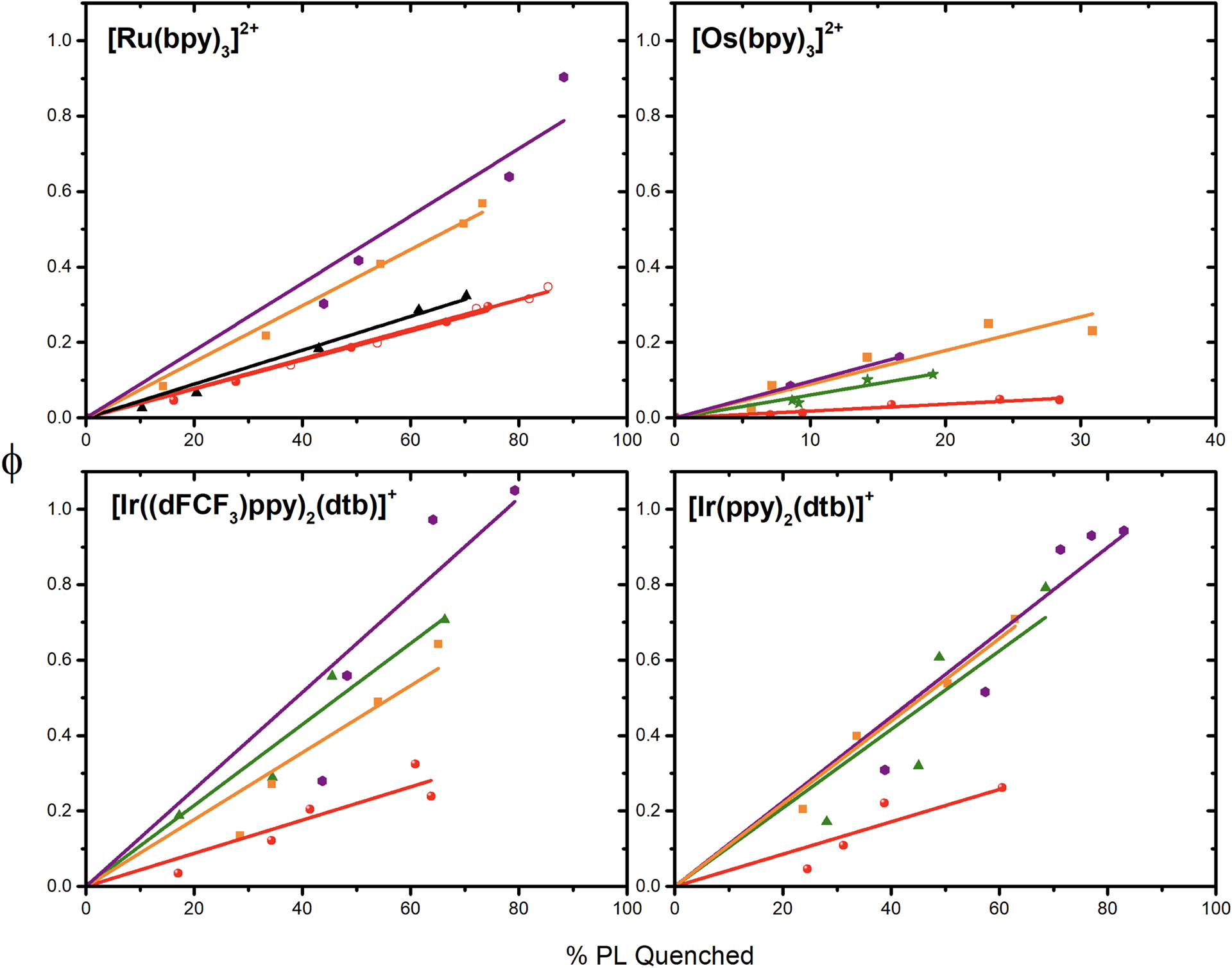
Plots of the relative yield of product (ϕ) versus the percentage of quenched photoluminescence (% PL quenched) for the indicated photosensitizers in the presence of R-benzene diazonium, with R = MeO (red), ethyl (black), COOEt (orange), NO2 (purple) and Br (green). Values recorded in 0.1M TBABF4 (for [Ru(bpy)3]2+) are represented with open red circles. The slope was used to extract the actual cage-escape yield which are tabulated in Table 4.
Table 4.
Cage-escape yields (ϕCE) for the indicated photosensitizer/aryl diazonium combinations recorded in argon-purged acetonitrile.
| ϕCE | [Ru(bpy)3]2+a | [Os(bpy)3]2+ | [Ir(ppy)3] | [Ir((dFCF3)ppy)2(dtb)]+ | [Ir(ppy)2(dtb)]+ |
|---|---|---|---|---|---|
| MeO | 38%b | 18% | 61% | 44% | 43% |
| Br | N.D | 61% | 100% | 100% | 100% |
| COOEt | 74% | 89% | N.D | 89% | 100% |
| NO2 | 98% | 97% | N.D | 100% | 100% |
a cage escape yield of 45% was determined for [Ru(bpy)3]2+ using 4-ethylbenzene diazonium tetrafluoroborate.
a value of 39% was determined in acetonitrile containing 0.1M TBABF4.
An error of 10% is assumed on each value. N.D = Not determined
The cases of [Ru(bpy)3]2+ and [Os(bpy)3]2+ are the most straightforward to discuss. Indeed, the cage escape yield increases from minimum values of 18% to maximum values of 98% when the driving force for electron transfer increases. The addition of 0.1M TBABF4 electrolyte did not impact the cage-escape yield when 4-MeO-benzene diazonium was used. In addition, these cage-escape yields also follow the C-N2+ bond cleavage propensity, as exemplified by the nuclear Fukui function (see below). For the iridium photosensitizers, the cage-escape yield for 4-MeO-benzene diazonium was between 43% and 61% and reached values of 100% when the driving force was increased. The origin for the different cage-escape yields observed with a series of photosensitizers using the same aryl diazonium is still unknown. Charge, electrostatic interactions or dipole moment changes upon electron transfer might be responsible for these changes.
Quantum Mechanical Calculations
Quantum Mechanical calculations were used to investigate if further correlations between the series of diazonium salts could be obtained. MP2 calculations68–72 were first carried out using the 6–311G(d,p) basis set73,74 with the Gaussian 16 program package75. An optimization of the geometry for the neutral species was performed in acetonitrile using the integral equation formalism variant of the PCM model76, and the minimal energy conformation was verified by a frequency calculation. Figure 9 presents the frontier orbital plots of the LUMO of the studied molecules. As can be observed, the LUMO is almost uniformly distributed along the molecule, with very limited effect from electron donating or withdrawing substituents.
Figure 9.
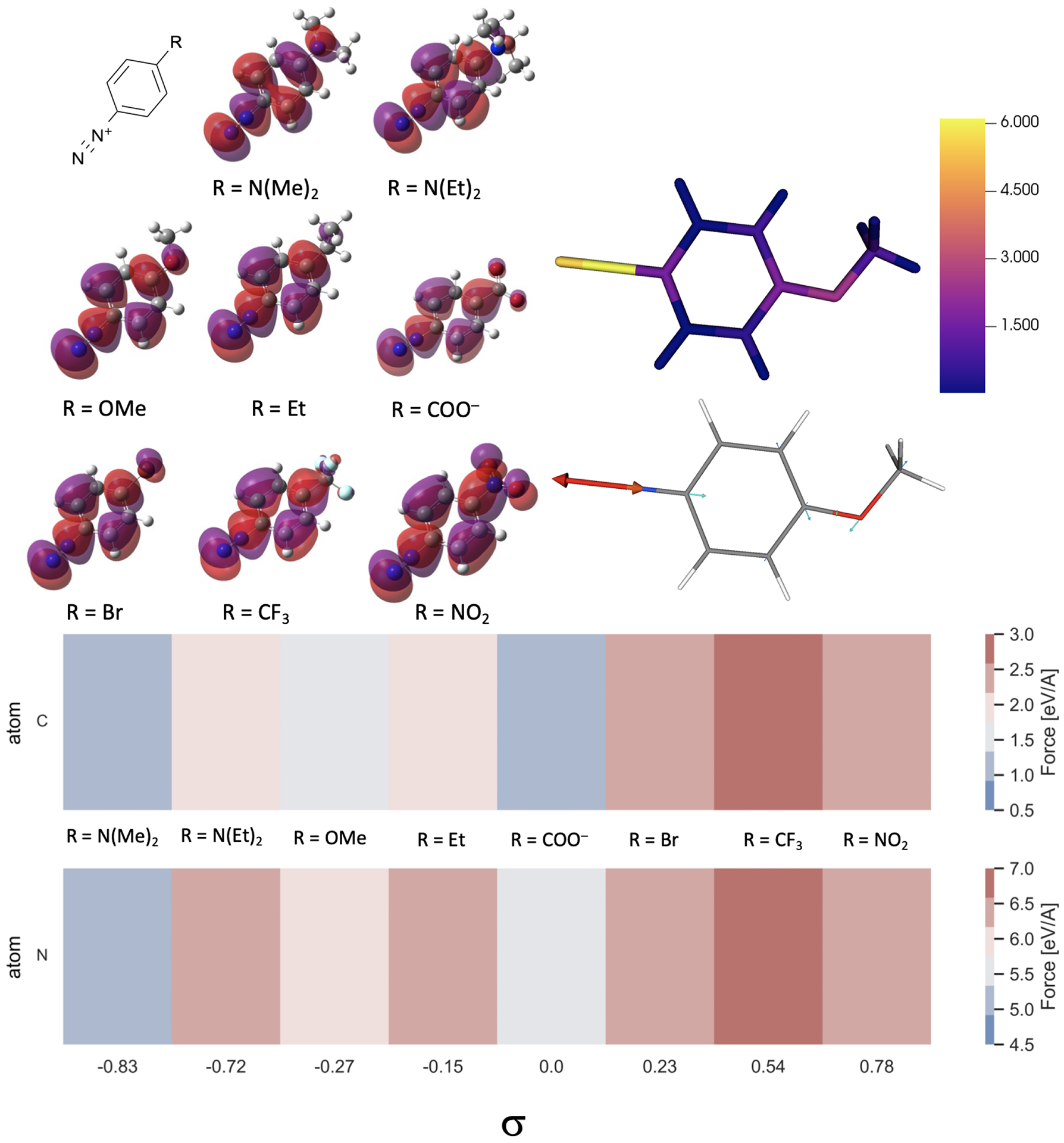
General structure of the aryl diazonium salt and the corresponding LUMO calculated at the MP2 level using the 6–311G(d, p) basis. Force (in eV/Å) on the indicated atom for selected aryl diazonium derivatives, classified according to their Hammett parameters (σ). The color map represents the force experienced by the indicated diazonium upon one-electron reduction.
Next, we sought to understand the response of the atomic nuclei due to the perturbation of the electron density upon reduction of the diazonium derivatives. To do so, a Conceptual Density Functional Theory (CDFT) reactivity descriptor, i.e. the nuclear Fukui function77–79 was used. This function allows to characterize the changes in the force and in positions that the different nuclei experience upon reduction by one electron. The nuclear Fukui function calculations were performed in a finite difference method77 which means that two separate calculations, performed on the neutral and reduced species, were carried out using the optimized geometry of the neutral species. These calculations were performed at the DFT/B3LYP80–82/6–311G(d,p) level of theory with the Northwest Computational Chemistry Package program package (NWChem 6.8)83 and enabled to estimate the magnitude of the change in force (projected vectors) acting on the different nuclei. The magnitudes of the CDFT reactivity descriptor that the atoms of aryl diazonium derivatives experience following the electron attachment is shown in Figure 9.
The nuclear forces indicate that, upon one-electron reduction, the C-N2+ exhibits the most drastic elongation. This is in line with the reported fragmentation process that occurs along the C-N2+ bond axis, freeing N2 and the corresponding aryl radical. It is interesting to note that the magnitude of the force on the nitrogen atom is between 5.18 and 6.88 eV/Å. This magnitude seems to be greater for diazonium with more positive Hammett parameters. These values are also much larger than those recently calculated on halogenated substrate that showed forces between 2.5 and 4.9 eV/Å for homolytic C–Cl or C–Br bond cleavage, respectively. Hence, these strong forces observed with the diazonium series are in line with very favorable bond dissociation upon reduction.
Photoredox Catalysis
With a relatively clear mechanistic picture of excited-state reactivity and charge separation following excited-state electron transfer, we sought to transpose these observations to actual photoredox catalysis. As described in the introduction, there are numerous examples in the literature that use diazonium as starting building block for organic transformation. We decided to use the series of photosensitizers for the visible light-induced borylation or aryldiazonium salts. This approach was reported by Yan et al. using Eosin Y in acetonitrile at room temperature using 25 W visible light lamps.17 This transformation seems therefore compatible with our choice of solvent and with the excited-state lifetime of the different photosensitizers. We focused our endeavor on 4-MeO-benzene diazonium tetrafluoroborate and bis(pinacolato)diboron (B2pin2) as borylating agent. For that reaction, 0.5 mmol of diazonium, 0.5 mol% of photosensitizer and 2 eq of B2pin2 were used. The solutions were irradiated using commercial blue, green or orange LED, depending on the absorption profile of the photosensitizer (Figure 2). The power was measured and set at 30 mW/cm2 by adjusting the distance of the LED lamp relative to the reaction round bottom flask. The spectral profiles of the LED are shown in Figure S80. Irradiation for 17 hours led to borylation yields of 4-MeO-benzene diazonium tetrafluoroborate that ranged from 9 to 74%, depending on the photosensitizer (Figure 10). The yields were determined by 1H NMR using 3,4,5-MeO-benzaldehyde as internal reference. The overall yields are to some extent not essential, but the trend observed between the photosensitizers in otherwise identical conditions is probably the best indicator of reaction success. The lowest yields were observed for the earth abundant photosensitizers. This is not surprising as both copper photosensitizers showed lack of stability under irradiation or during the course of Stern-Volmer titrations in the presence of diazonium derivatives. The two iron(III) photosensitizers also led to small yields. As both compounds are very photostable, it seems like the smaller yields are most probably related to the slow kinetics for excited-state electron transfer or to inefficient regeneration of the iron(III) after oxidation. Indeed, both complexes required much longer irradiation time during photolysis experiments than the other rare earth transition metal photosensitizers (vide supra). Yields of 22% with low conversions are obtained with [Os(bpy)3]2+ whereas higher conversions and yields (between 54% and 74%) are obtained with [Ru(bpy)3]2+ and the three iridium(III) photosensitizers. Note that for [Os(bpy)3]2+ a red side product was obtained after reaction, with no presence of photosensitizer left. At this stage we are unable to provide clear-cut explanation as to why those yields differ. Indeed, with the exception of [Ir((dFCF3)ppy)2(dtb)]+, the ground-state oxidation potential for most of the photosensitizers and quenching rate constant are all in the same range. The difference in yields could come from changes in cage-escape yields or from the relative stability of the different photosensitizers, as 2,2′-bipyridine derivatives could undergo side-reactions with the aryl radicals, but further experiments are needed to elucidate these aspects and remediate the overall moderate yields.
Figure 10.
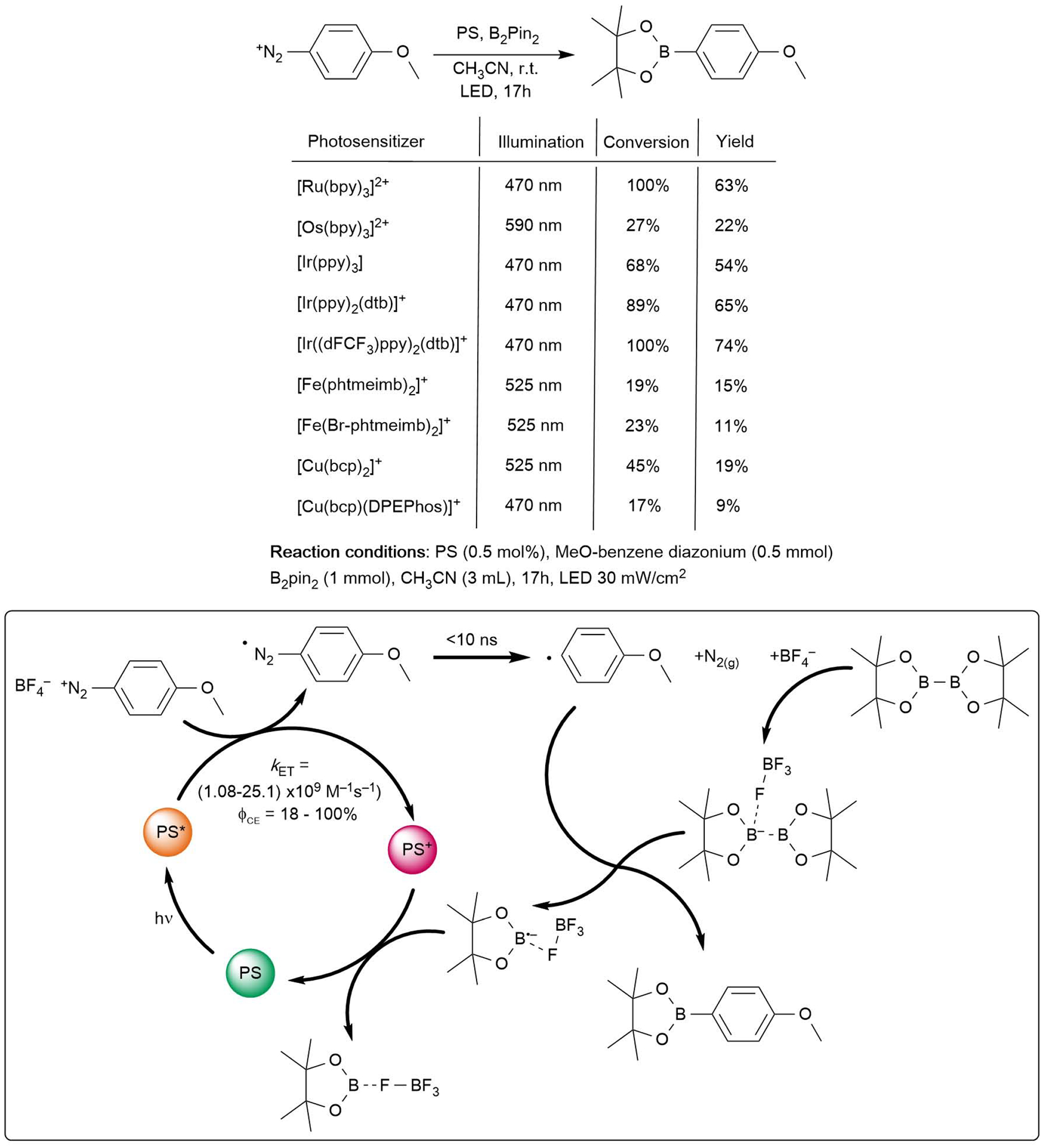
Photoredox catalysis results. Reaction conditions and 1H NMR yields for the indicated reaction using the series of earth abundant and rare earth transition metal photosensitizer (top). Proposed mechanism for the visible light mediated borylation of 4-MeO-benzene diazonium (bottom).
Conclusions
In conclusion, we have investigated the excited-state reactivity of 9 photosensitizers with 13 aryl diazonium derivatives. The quenching rate constant were determined and ranged from (1.08–25.1) ×109 M−1s−1. Nanosecond transient absorption and steady-state photolysis experiments confirmed that the excited-state quenching proceeded via oxidative electron transfer, efficiently transferring one electron from the excited-state to the diazonium. Reaction kinetics recorded in the dark using Iron(III) photosensitizers not only allowed to obtain kinetic rate constants for electron transfer in the dark, but also yielded precious information related to possible discrepancies between electrochemical potential of sacrificial electron donor/acceptors, and potentials determined by kinetic measurements. In addition, these kinetic measurements also informed that the electron transfer process to the diazonium moiety is reversible and that the C-N2• dissociation occurs on a timescale of 10 ns or shorter. This reversibility was also evidenced when cage-escape yields were lower than 100%, despite having favorable driving force. Nevertheless, aryl diazonium derivatives were shown to lead to very large cage-escape yields that are not often observed in the literature. Indeed, except for some triphenylamine derivatives,84 most of the cage-escape yields reported thus far for transition metal complexes are in the 10–40% range. Hence, highly reactive diazonium salts seem to offer new alternatives for more efficient visible-light mediated transformations. At this stage, we are unable to explain the difference observed in cage-escape yields observed with a series of photosensitizers using the same aryl diazonium. Future studies will focus on further expanding the effect of charge, electrostatic interactions or dipole moment changes upon electron transfer on these cage-escape yields using inorganic and organic photosensitizers. Such studies would help determine key parameters that control cage-escape yields and could lead to more efficient light-activated organic transformations with other substrates.
EXPERIMENTAL PROCEDURES
Resource availability
Lead contact
Further information and requests for resources should be directed to and will be fulfilled by the lead contact, Ludovic Troian-Gautier (Ludovic.Troian@uclouvain.be).
Materials availability
All other data supporting the findings of this study are available within the article and the supplemental information or from the lead contact upon reasonable request.
Data and code availability
This study did not generate any datasets.
Materials
Acetonitrile 99.9% (VWR), dry acetonitrile 99.8% (Sigma-Aldrich), dichloromethane 99% (VWR), diethyl ether 99% (VWR), dry diethyl ether 99.5% (Acros Organics), toluene 99.9% (VWR), tetrahydrofuran anhydrous (stabilized with BHT) >99.5% (TCI), methanol 99.9% (VWR), n-butyllithium solution (1.6 M in hexanes) (Acros Organics), Hexamethyldisilazane 98% (Acros Organics), ultra-dry Iron(II) bromide 99.995% (Alfa Aesar), 1-Methylimidazole 99% (BASF), NaOH pellet for analysis (VWR), NH4PF6 99% (Fluorochem), trimethylsilyl chloride 98% (Acros Organics), dichlorophenylborane 97% (Acros Organics), (4-bromophenyl)trimethylsilane 97% (Ambeed), boron tribromide (1M in dichloromethane) (ThermoFisher), nitrosonium tetrafluoroborate 97% (Acors Organics), methoxybenzenediazonium tetrafluoroborate 98% (ThermoFisher), bromobenzenediazonium tetrafluoroborate 96% (Alfa Aesar), nitrobenzenediazonium tetrafluoroborate 97% (Alfa Aesar), N,N-dimethyl-p-phenylenediamine 97% (Acros Organics), N,N-diethyl-p-phenylenediamine 97% (Acros Organics), 4-ethylaniline 99.48% (Ambeed), 4-(trifluoromethyl)aniline 99.88% (Ambeed), 4-cyclohexylaniline 97% (Ambeed), ethyl 4-aminobenzoate 99.9% (Ambeed), sodium 4-aminobenzoate 95% (Ambeed), bi(pinacolato)diboron (Fluorochem), tetrabutylammonium tetrafluoroborate 98% (Acros Organics), 3,4,5-trimethoxybenzaldehyde 99% (Acros Organics), SiO2 40–63 μm (Rocc) and neutral aluminum oxide Brockmann 50–200 μm 60Å (Acros Organics) were purchased from commercial suppliers and used as received.
Experimental Protocols
Synthesis of tris(3-methylimidazolium-1-yl)(4-Br-phenyl)borate bis(hexafluorophosphate).
A solution of 1M BBr3 in CH2Cl2 (4.8 mL, 4.8 mmol) was placed under argon and cooled to −78°C in a flame-dried round-bottom flask. (4-bromophenyl)-trimethylsilane (920 mg, 4 mmol) was added in 15 minutes to the cooled solution and the solution was stirred at −78°C for one hour then at room temperature for 3 hours. The volatiles were evaporated at 30°C under reduced pressure. The resulting 4-bromo-dibromophenylborane was used without further purification. It was first dissolved in 10 mL of toluene and was followed by the dropwise addition of 1-Me-imidazole (1.12 mL, 14 mmol). The solution was stirred for 15 minutes and TMSCl (1.27 mL, 10 mmol) was then added in one portion. The reaction was then heated for 24 hours at 80°C under argon. After reaction, the mixture was evaporated, and the residue was dissolved in water. A saturated solution of ammonium hexafluorophosphate was then added which induced precipitation of a beige solid that was collected by filtration, washed with water and dried under static vacuum. The desired product was then isolated in 80% yield after column chromatography on silica using CH3CN/MeOH mixtures.
1H NMR (300 MHz, CD3CN) δH (ppm) 8.00 (s, 3H), 7.60 (d, J = 8.3 Hz, 2H), 7.44 (t, J = 1.7 Hz, 3H), 7.24 − 6.94 (m, 5H), 3.82 (s, 9H). 13C NMR (75 MHz, CD3CN) δC (ppm) 140.21, 136.42, 135.96, 132.59, 131.50, 125.76, 124.85, 36.54. HRMS (ESI) m/z: [M]2+ for C18H22BBrN6: 206.05857; Found: 206.05891.
Synthesis of Bis(tris(3-methlimidazol-1-ylidene)(phenyl)borate) Fe(III) hexafluorophosphate [Fe(Br-phtmeimb)2]+.
A flame-dried and argon-purged 25 mL Schlenk flask equipped with a stirring bar, was charged with dry HMDS (0.73 mL, 0.565 g, 3.5 mmol) and cooled to 0 °C. A solution of n-BuLi (1.6 M in hexanes, 2.25 mL, 3.5 mmol) was added dropwise at 0 °C. The reaction mixture was then allowed to reach room temperature and stirred for an additional 30 minutes. Afterwards, the volatiles were slowly removed under reduced pressure to afford a white solid. The resulting product was then solubilized in 3.5 mL of dry THF, forming the corresponding LiHMDS solution.
A flame-dried and argon-purged 50 mL Schlenk flask equipped with a stirring bar was charged with tris(3-methylimidazolium-1-yl)(4-Br-phenyl)borate bis(hexafluorophosphate) (0.703 g, 1.0 mmol) and the solid was dried under vacuum for 2 hours before being purged under argon. Dry THF (20 mL) was then added under argon. The mixture was cooled to −78 °C and the previously obtained LiHMDS solution was added dropwise. The reaction mixture was stirred at this temperature for 30 minutes. A solution of anhydrous FeBr2 was prepared by adding FeBr2 (0.108 g, 0.5 mmol) into a flame-dried and argon-purged 10 mL Schlenk flask followed by 10 mL of dry THF. The mixture was sonicated for 20 minutes to ensure solubilization of the iron source. Afterwards, the cooling bath was removed and the solution of FeBr2 was added dropwise to the deprotonated ligand solution. The reaction mixture was allowed to slowly reach room temperature under argon and then was stirred at room temperature for 72h in the dark. Upon exposure to air, the solution became dark red and the solvent was removed under reduced pressure. The resulting residue was dissolved in 30 mL of CH2Cl2, filtered through a porosity 4 frit, washed with 2×15 mL of CH2Cl2 and the filtrate was evaporated under reduced pressure resulting in a dark red solid. The residue was then solubilized in a minimum amount of CH3CN and poured in approximately 200 mL of diethyl ether which induced precipitation of a red solid that was collected by filtration and washed with 3×30 mL of diethyl ether and dried under vacuum. The resulting solid was then washed with methanol, filtered, and the precipitate was washed with methanol before being dried under static vacuum. The desired compound was obtained as a red powder (153 mg, 30%).
1H NMR (300 MHz, CD3CN) δH (ppm) 14.77 (d, J = 7.3 Hz, 4H), 10.60 (d, J = 5.8 Hz, 4H), 4.97 (s, 18H), 1.37 (s, 6H), −12.46 (s, 6H). HRMS (ESI) m/z: [M]+ for C36H38B2Br2N12Fe: 876.12278; Found: 876.12226.
General procedure for the synthesis of aryl diazonium derivatives.
Aniline (0.71 mmol) was dissolved on 3 mL of argon-purged acetonitrile. The solution was kept under argon and cooled to −40°C. NOBF4 (261 mg, 1.77 mmol) was added in one portion and the medium was allowed to stir at −40°C for two hours. After reaction, the mixture was evaporated under reduced pressure and the residue was triturated three times with 10mL diethylether. The formed precipitate was isolated and dried under reduced pressure which afforded the corresponding diazonium with >95% yields. The diazonium derivatives were then stored in the freezer at −18°C.
Caution! Although we have not encountered any problem, it is noted that diazonium salts are potentially explosive and should be handled with appropriate precautions. Tetrafluoroborate counter-ions using increase stability of the resulting diazonium salts. Calix[4]arene derivatives have been shown to exhibit thermal stability up to 150°C.
4-trifluoromethyl-benzene diazonium tetrafluoroborate. 1H NMR (CD3CN, 300 MHz) δH (ppm) 8.69 (d, J = 8.7 Hz, 2H), 8.23 (d, J = 8.8 Hz, 2H).
4-cyclohexyl)benzene diazonium tetrafluoroborate. 1H NMR (CD3CN, 300 MHz) δH (ppm) 8.38 (d, J = 8.5 Hz, 2H), 7.78 (d, J = 8.5 Hz, 2H), 2.83 (m, 1H), 1.92 − 1.67 (m, 5H), 1.57 − 1.21 (m, 5H).
4-ethyl-benzene diazonium tetrafluoroborate. 1H NMR (CDCl3, 300 MHz) δH (ppm) 8.51 (d, J = 8.4 Hz, 2H), 7.62 (d, J = 8.3 Hz, 2H), 2.85 (q J = 7.6 Hz, 2H), 1.28 (t, J = 7.5 Hz, 3H).
4-carboxylate-benzene diazonium. 1H NMR (D2O, 300 MHz) δH (ppm) 8.65 (dt, J = 9.1, 2.0 Hz, 2H), 8.37 (dt, J = 8.9, 2.0 Hz, 2H).
4-ethylester-benzene diazonium tetrafluoroborate. 1H NMR (CD3CN, 300 MHz) δH (ppm) 8.58 (dt, J = 9.1, 1.9 Hz, 2H), 8.42 (dt, J = 9.1, 2.0 Hz, 2H), 4.44 (q, J = 7.1 Hz, 2H), 1.39 (t, J = 7.1 Hz, 2H).
4-N,N-dimethylamino-benzene diazonium tetrafluoroborate. 1H NMR (CD3CN, 300 MHz) δH (ppm) 7.98 (d, J = 9.8 Hz, 2H), 6.93 (d, J = 9.8 Hz, 2H), 3.26 (s, 6H).
4-N,N-diethylamino-benzene diazonium tetrafluoroborate. 1H NMR (CD3CN, 300 MHz) δH (ppm) 7.97 (d, J = 9.8 Hz, 2H), 6.94 (d, J = 9.8 Hz, 2H), 3.61 (q, J = 7.2 Hz, 4H), 1.23 (t, J = 7.2 Hz, 6H).
General procedure for the photoredox catalysis experiments.
4-MeO-benzene diazonium tetrafluoroborate (111 mg, 0.5 mmol), bis(pinacolato)diboron (254 mg, 1 mmol) and the photosensitizer (0.5 mol%) were place in 3 mL of argon-purged acetonitrile. The solution was kept under argon and was irradiated with the appropriate LED for 17 hours. After reaction, the mixture was evaporated to dryness and the residue was dried under static vacuum for 24 hours before being subjected to quantitative NMR using 3,4,5-trimethoxy benzaldehyde as internal reference. The final products can also be purified on silica gel using cyclohexane/ethyl acetate mixtures as eluent.
UV-Vis spectroscopy
UV–Visible Absorption. UV–vis absorption spectra were recorded on a Shimadzu UV-1700 with 1 cm path length quartz cell.
Time-Resolved Photoluminescence
Time-resolved photoluminescence spectra were recorded on a LP980-K spectrometer from Edinburgh Instruments (see nanosecond transient absorption for full description). The optically diluted samples were excited with pulsed light and the excited-state lifetime was measured on a PMT LP detector (Hamamatsu R928) which covers the spectral range from 185 to 870 nm. An average of 30 scans per measurement was used.
Electrochemistry
Cyclic voltammetry was performed with an Autolab PGSTAT 100 potentiostat using a standard three-electrode-cell, i.e. a glassy carbon disk working electrode (approximate area = 0.03 cm2), a platinum wire counter electrode and an aqueous Ag/AgCl reference electrode (salt bridge: 3M KCl/saturated AgCl). Experiments were performed in dry 0.1 M tetrabutylammonium perchlorate acetonitrile electrolyte, and the samples were purged with Ar before each measurement. For comparison purposes, the electrochemical potentials were converted to NHE by adding 0.197 V.
Steady-State Photoluminescence
Room temperature steady-state photoluminescence (PL) spectra were recorded on a Horiba Scientific-FL-1000 fluorimeter and were corrected by calibration of the instrument’s response with a standard tungsten-halogen lamp. The photoluminescence intensity was integrated for 0.1 s at 1 nm resolution and averaged over 3 scans. Alternatively, some steady-state PL spectra were recorded on a Varian Cary Eclipse spectrometer and were not corrected for the instrument’s response.
Stern-Volmer Experiments
A photosensitizer stock solution with an absorbance of ~ 0.1 to ~ 0.2 at the excitation wavelength was prepared in acetonitrile containing 0.1M TBABF4. The stock solution was purged with argon for 30 minutes. Aryl-diazonium tetrafluoroborate salts were then weighted in scintillation vials to reach a final desired concentration between 20 and 30 mM. 2 mL of the photosensitizer stock solution were added to the scintillation vial kept under argon, and 3 mL of the stock solution were transferred to a quartz cuvette equipped with a 24/40 joint, also kept under argon. The cuvette and the scintillation vials were purged with argon for another 5 minutes. The desired Aryl-diazonium tetrafluoroborate quencher was then gradually added to the cuvette. This allowed to increase the concentration of quencher while keeping the concentration of photosensitizer constant. The excited-state quenching was monitored by time-resolved spectroscopy using the LP980-K spectrometer from Edinburgh Instruments (see below) or by steady-state photoluminescence when [Fe(phtmeimb)2]+ or [Fe(Br-phtmeimb)2]+ were used. The decrease of excited-state lifetime or photoluminescence is directly related to the concentration of quencher and the respective Stern-Volmer plots were extrapolated using equation 5. The quenching rate constant (kq) was then determined by dividing the slope by the initial lifetime without any quencher, determined for each experiment.
| Equation 5 |
Transient Absorption Spectroscopy
Nanosecond transient absorption measurements were recorded on a LP980-K spectrometer from Edinburgh Instruments equipped with an iCCD detector from Andor (DH320T). The excitation source was a tunable Nd:YAG Laser NT342 Series from EKSPLA. The third harmonic (355 nm) at 150 mJ was directed into an optical parametric oscillator (OPO) to enable wavelength tuning starting from 410 nm. The laser power was then attenuated to reach appreciable signal/noise and the integrity of the samples was verified by UV–Vis measurements. The LP980-K is equipped with a symmetrical Czerny-Turner monochromator. For single wavelength absorption changes, a 1800 g mm−1 grating, blazed at 500 nm is used, which affords wavelength coverage from 200 to 900 nm. For spectral mode (iCCD), a 150 g mm−1 grating, blazed at 500 nm is used, offering a wavelength coverage of 540 nm over the full wavelength range extending from 250 to 900 nm. Single wavelength absorption changes were monitored using a PMT LP detector (Hamamatsu R928) which covers the spectral range from 185 to 870 nm. The probe was a 150 W ozone-free xenon short arc lamp (OSRAM XBO 150W/CR OFR) that was pulsed at the same frequency of the laser. The excitation wavelength depended on the photosensitizers but in all case the concentration at the excitation wavelength was adjusted to reach absorbance values between 0.3 and 0.5. All measurements were performed in argon-purged acetonitrile containing 0.1M TBABF4 at room temperature. An average of 30 to 90 scans per measurement was used.
Quantum Mechanical Calculations
MP2 calculations have been performed using the Gaussian16 program,75 applying default procedures, integration grids, algorithms and parameters. Selected diazonium salts were considered in this theoretical study. The geometry of the ground state species was optimized at the MP2/6–311G(d,p) level. MP2 with the same basis set was used to study the effect of electron correlation on the calculated density. Additional calculations were done with the NWChem program package83 in order to calculate the nuclear Fukui function.78,79 The nuclear Fukui function characterizes the changes in the forces (energy gradient) that nuclei experience when the total number of electrons of the system is changed and can be calculated using the finite difference method.77 For that, two separate B3LYP/6–311G(d,p) calculations, performed on the ground and reduced species, are carried out, both with the geometry of the ground state. The magnitude of the change in force acting on the nuclei was estimated as well as the projected forces along the bonds involving the nuclei. These were depicted as vectors with the origin located on the nuclei. Both quantities were visualized using VisIt 3.1.485 and can help to interpret some trends in nuclear reactivity as they pertain to the initial mechanistic steps of electron-induced attachment.86
Supplementary Material
Bigger Picture:
Factors controlling excited-state reactivity are of paramount importance as a better understanding of these would yield more efficient photo-induced processes. This is relevant, not only for photoredox catalysis, but for all fields that use light to trigger reactions, which include, but are not limited to, photochemotherapy and solar fuel formation such as water oxidation, halide oxidation, proton or carbon dioxide reduction. Here, we investigated reactions between a large series of Ru (II), Os(II), Ir(III), Cu(I) and Fe(III) photosensitizers and 13 diazonium salts, which included macrocyclic derivatives. Clear explanations of dark and excited-state reactivity are provided. The efficiency of formation of charge-separated products, also termed cage-escape yields, generating radical species of paramount importance for additional targeted transformation were quantified in different media and with varying driving force for electron transfer.
Highlights:
Excited-state reactivity of 9 photosensitizers with 13 aryl diazonium salts
Cage-escape yields quantified and ranged from 18% to 100%.
Kinetic model developed to explain dark reactivity of Iron(III) photosensitizers
Photo-induced borylation reaction with yields between 9% and 74%.
ACKNOWLEDGMENTS
Financial support was provided to A.R. by the F.R.S.-FNRS (FRIA fellowship). This work was supported by the Fonds de la Recherche Scientifique (F.R.S.-FNRS) under grant no. U.N021.21 (B.E.). L.T.-G. is a Chercheur Qualifié of the Fonds de la Recherche Scientifique – FNRS. S.D.K., B.E. and L.T.-G. gratefully acknowledge the UCLouvain for financial support. Computational resources have been provided by the Consortium des Équipements de Calcul Intensif (CÉCI), funded by the Fonds de la Recherche Scientifique de Belgique (F.R.S.-FNRS) under Grant No. 2.5020.11 and by the Walloon Region. Financial support was provided to U.K.T by W. W. Caruth, Jr. Endowed Scholarship, Welch Foundation (I-1748), National Institutes of Health (R01GM102604), American Chemical Society Petroleum Research Fund (59177-ND1), and Teva Pharmaceuticals Marc A. Goshko Memorial Grant (60011-TEV). Financial support was provided to C.A.V. by Sarah and Frank McKnight Fund Graduate Fellowship.
INCLUSION AND DIVERSITY
We support inclusive, diverse, and equitable conduct of research.
Footnotes
Publisher's Disclaimer: This is a PDF file of an unedited manuscript that has been accepted for publication. As a service to our customers we are providing this early version of the manuscript. The manuscript will undergo copyediting, typesetting, and review of the resulting proof before it is published in its final form. Please note that during the production process errors may be discovered which could affect the content, and all legal disclaimers that apply to the journal pertain.
SUPPLEMENTAL INFORMATION
Document S1. Excited-state quenching (Figures S1–S66), Nanosecond transient absorption spectroscopy (Figures S67–S72), Photostability (Figures S73–S79) LED profiles (Figure S80), Excited-state lifetime (Figures S81–S95), 1H NMR, 13C NMR and HRMS of relevant compounds (Figures S96–S108), Electrochemistry of [Fe(Br-phtmeimb)2]+ (Figure S109).
DECLARATION OF INTERESTS
The authors declare no competing interests.
REFERENCES*
- 1.Griess JP, and Hofmann AWV (1864). XVIII. On a new series of bodies in which nitrogen is substituted for hydrogen. Philosophical Transactions of the Royal Society of London 154, 667–731. doi: 10.1098/rstl.1864.0018. [DOI] [Google Scholar]
- 2.Mo F, Qiu D, Zhang L, and Wang J (2021). Recent Development of Aryl Diazonium Chemistry for the Derivatization of Aromatic Compounds. Chem. Rev 121, 5741–5829. 10.1021/acs.chemrev.0c01030. [DOI] [PubMed] [Google Scholar]
- 3.Mo F, Dong G, Zhang Y, and Wang J (2013). Recent applications of arene diazonium salts in organic synthesis. Org. Biomol. Chem 11, 1582–1593. 10.1039/C3OB27366K. [DOI] [PubMed] [Google Scholar]
- 4.Troian-Gautier L, Mattiuzzi A, Reinaud O, Lagrost C, and Jabin I (2020). Use of calixarenes bearing diazonium groups for the development of robust monolayers with unique tailored properties. Org. Biomol. Chem 18, 3624–3637. 10.1039/D0OB00070A. [DOI] [PubMed] [Google Scholar]
- 5.Hari DP, and König B (2013). The Photocatalyzed Meerwein Arylation: Classic Reaction of Aryl Diazonium Salts in a New Light. Angew. Chem. Int. Ed 52, 4734–4743. 10.1002/anie.201210276. [DOI] [PubMed] [Google Scholar]
- 6.Bélanger D, and Pinson J (2011). Electrografting: a powerful method for surface modification. Chem. Soc. Rev 40, 3995–4048. 10.1039/C0CS00149J. [DOI] [PubMed] [Google Scholar]
- 7.Babu SS, Muthuraja P, Yadav P, and Gopinath P (2021). Aryldiazonium Salts in Photoredox Catalysis – Recent Trends. Adv. Synth. Catal 363, 1782–1809. 10.1002/adsc.202100136. [DOI] [Google Scholar]
- 8.Delamar M, Hitmi R, Pinson J, and Saveant JM (1992). Covalent modification of carbon surfaces by grafting of functionalized aryl radicals produced from electrochemical reduction of diazonium salts. J. Am. Chem. Soc 114, 5883–5884. 10.1021/ja00040a074. [DOI] [Google Scholar]
- 9.Pinson J, and Podvorica F (2005). Attachment of organic layers to conductive or semiconductive surfaces by reduction of diazonium salts. Chem. Soc. Rev 34, 429–439. 10.1039/B406228K. [DOI] [PubMed] [Google Scholar]
- 10.Troian-Gautier L, Valkenier H, Mattiuzzi A, Jabin I, den Brande NV, Mele BV, Hubert J, Reniers F, Bruylants G, Lagrost C, and Leroux Y (2016). Extremely robust and post-functionalizable gold nanoparticles coated with calix[4]arenes via metal-carbon bonds. Chem. Commun 52, 10493–10496. 10.1039/C6CC04534K. [DOI] [PubMed] [Google Scholar]
- 11.Mattiuzzi A, Jabin I, Mangeney C, Roux C, Reinaud O, Santos L, Bergamini J-F, Hapiot P, and Lagrost C (2012). Electrografting of calix[4]arenediazonium salts to form versatile robust platforms for spatially controlled surface functionalization. Nature Comm. 3, 1130. 10.1038/ncomms2121. [DOI] [PubMed] [Google Scholar]
- 12.Cano-Yelo H, and Deronzier A (1984). Photo-oxidation of tris(2,2′-bipyridine)ruthenium(II) by para-substituted benzene diazonium salts in acetonitrile. Two-compartment photoelectrochemical cell applications. J. Chem. Soc., Faraday Trans 1 80, 3011–3019. 10.1039/F19848003011. [DOI] [Google Scholar]
- 13.Cano-Yelo H, and Deronzier A (1984). Photo-oxidation of some carbinols by the Ru(II) polypyridyl complex-aryl diazonium salt system. Tetrahedron Lett. 25, 5517–5520. 10.1016/S0040-4039(01)81614-2. [DOI] [Google Scholar]
- 14.Hari DP, Schroll P, and König B (2012). Metal-Free, Visible-Light-Mediated Direct C–H Arylation of Heteroarenes with Aryl Diazonium Salts. J. Am. Chem. Soc 134, 2958–2961. 10.1021/ja212099r. [DOI] [PubMed] [Google Scholar]
- 15.Louvel D, Chelagha A, Rouillon J, Payard P-A, Khrouz L, Monnereau C, and Tlili A (2021). Metal-Free Visible-Light Synthesis of Arylsulfonyl Fluorides: Scope and Mechanism. Chem-Eur. J 27, 8704–8708. 10.1002/chem.202101056. [DOI] [PubMed] [Google Scholar]
- 16.Kalyani D, McMurtrey KB, Neufeldt SR, and Sanford MS (2011). Room-Temperature C–H Arylation: Merger of Pd-Catalyzed C–H Functionalization and Visible-Light Photocatalysis. J. Am. Chem. Soc 133, 18566–18569. 10.1021/ja208068w. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 17.Yu J, Zhang L, and Yan G (2012). Metal-Free, Visible Light-Induced Borylation of Aryldiazonium Salts: A Simple and Green Synthetic Route to Arylboronates. Adv. Synth. Catal 354, 2625–2628. 10.1002/adsc.201200416. [DOI] [Google Scholar]
- 18.Lentz C, Marcélis L, Troian-Gautier L, Robeyns K, Cauët E, and Elias B (2021). Excited-state behavior and photoinduced electron transfer of pH-sensitive Ir(III) complexes with cyclometallation (C/N–) ratios between 0/6 and 3/3. J. Photoch. Photobio. A 405, 112957. 10.1016/j.jphotochem.2020.112957. [DOI] [Google Scholar]
- 19.Aydogan A, Bangle RE, Cadranel A, Turlington MD, Conroy DT, Cauët E, Singleton ML, Meyer GJ, Sampaio RN, Elias B, and Troian-Gautier L (2021). Accessing Photoredox Transformations with an Iron(III) Photosensitizer and Green Light. J. Am. Chem. Soc 143, 15661–15673. 10.1021/jacs.1c06081. [DOI] [PubMed] [Google Scholar]
- 20.Kjær KS, Kaul N, Prakash O, Chábera P, Rosemann NW, Honarfar A, Gordivska O, Fredin LA, Bergquist K-E, Häggström L, et al. (2019). Luminescence and reactivity of a charge-transfer excited iron complex with nanosecond lifetime. Science 363, 249–253. 10.1126/science.aau7160. [DOI] [PubMed] [Google Scholar]
- 21.Prakash O, Lindh L, Kaul N, Rosemann NW, Losada IB, Johnson C, Chábera P, Ilic A, Schwarz J, Gupta AK, et al. (2022). Photophysical Integrity of the Iron(III) Scorpionate Framework in Iron(III)–NHC Complexes with Long-Lived 2LMCT Excited States. Inorg. Chem 10.1021/acs.inorgchem.2c02410. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 22.Kaufmann D (1987). Borylierung von Arylsilanen, I Ein allgemeiner, einfacher und selektiver Zugang zu Phenyldihalogenboranen. Chemische Berichte 120, 853–854. 10.1002/cber.19871200527. [DOI] [Google Scholar]
- 23.Forshaw AP, Bontchev RP, and Smith JM (2007). Oxidation of the Tris(carbene)borate Complex PhB(MeIm)3MnI(CO)3 to MnIV[PhB(MeIm)3]2(OTf)2. Inorg. Chem 46, 3792–3794. 10.1021/ic070187w. [DOI] [PubMed] [Google Scholar]
- 24.Troian-Gautier L, Martínez-Tong DE, Hubert J, Reniers F, Sferrazza M, Mattiuzzi A, Lagrost C, and Jabin I (2016). Controlled Modification of Polymer Surfaces through Grafting of Calix[4]arene-Tetradiazoate Salts. J. Phys. Chem C 120, 22936–22945. 10.1021/acs.jpcc.6b06143. [DOI] [Google Scholar]
- 25.Hansch C, Leo A, and Taft RW (1991). A survey of Hammett substituent constants and resonance and field parameters. Chem. Rev 91, 165–195. 10.1021/cr00002a004. [DOI] [Google Scholar]
- 26.Sachdeva G, Vaya D, Srivastava CM, Kumar A, Rawat V, Singh M, Verma M, Rawat P, and Rao GK (2022). Calix[n]arenes and its derivatives as organocatalysts. Coord. Chem. Rev 472, 214791. 10.1016/j.ccr.2022.214791. [DOI] [Google Scholar]
- 27.Kober EM, and Meyer TJ (1982). Concerning the absorption spectra of the ions M(bpy)32+ (M = Fe, Ru, Os; bpy = 2,2′-bipyridine). Inorg. Chem 21, 3967–3977. 10.1021/ic00141a021. [DOI] [Google Scholar]
- 28.Cerfontaine S, Wehlin SAM, Elias B, and Troian-Gautier L (2020). Photostable Polynuclear Ruthenium(II) Photosensitizers Competent for Dehalogenation Photoredox Catalysis at 590 nm. J. Am. Chem. Soc 142, 5549–5555. 10.1021/jacs.0c01503. [DOI] [PubMed] [Google Scholar]
- 29.Juris A, Balzani V, Barigelletti F, Campagna S, Belser P, and von Zelewsky A (1988). Ru(II) polypyridine complexes: photophysics, photochemistry, eletrochemistry, and chemiluminescence. Coord. Chem. Rev 84, 85–277. 10.1016/0010-8545(88)80032-8. [DOI] [Google Scholar]
- 30.Maurer AB, and Meyer GJ (2020). Stark Spectroscopic Evidence that a Spin Change Accompanies Light Absorption in Transition Metal Polypyridyl Complexes. J. Am. Chem. Soc 142, 6847–6851. 10.1021/jacs.9b13602. [DOI] [PubMed] [Google Scholar]
- 31.Ruthkosky M, Castellano FN, and Meyer GJ (1996). Photodriven Electron and Energy Transfer from Copper Phenanthroline Excited States. Inorg. Chem 35, 6406–6412. 10.1021/ic960503z. [DOI] [PubMed] [Google Scholar]
- 32.Luo S-P, Mejía E, Friedrich A, Pazidis A, Junge H, Surkus A-E, Jackstell R, Denurra S, Gladiali S, Lochbrunner S, and Beller M (2013). Photocatalytic Water Reduction with Copper-Based Photosensitizers: A Noble-Metal-Free System. Angew. Chem. Int. Ed 52, 419–423. 10.1002/anie.201205915. [DOI] [PubMed] [Google Scholar]
- 33.Kirchhoff JR, Gamache RE Jr., Blaskie MW, Del Paggio AA, Lengel RK, and McMillin DR (1983). Temperature dependence of luminescence from Cu(NN)2+ systems in fluid solution. Evidence for the participation of two excited states. Inorg. Chem 22, 2380–2384. 10.1021/ic00159a008. [DOI] [Google Scholar]
- 34.Ichinaga AK, Kirchhoff JR, McMillin DR, Dietrich-Buchecker CO, Marnot PA, and Sauvage JP (1987). Charge-transfer absorption and emission of Cu(NN)2+ systems. Inorg. Chem 26, 4290–4292. 10.1021/ic00272a030. [DOI] [Google Scholar]
- 35.McCusker CE, Chakraborty A, and Castellano FN (2014). Excited State Equilibrium Induced Lifetime Extension in a Dinuclear Platinum(II) Complex. J. Phys. Chem. A 118, 10391–10399. 10.1021/jp503827e. [DOI] [PubMed] [Google Scholar]
- 36.Yarnell JE, McCusker CE, Leeds AJ, Breaux JM, and Castellano FN (2016). Exposing the Excited-State Equilibrium in an IrIII Bichromophore: A Combined Time Resolved Spectroscopy and Computational Study. Eur. J. Inorg. Chem 2016, 1808–1818. 10.1002/ejic.201600194. [DOI] [Google Scholar]
- 37.Ito A, and Meyer TJ (2012). The Golden Rule. Application for fun and profit in electron transfer, energy transfer, and excited-state decay. Phys. Chem. Chem. Phys 14, 13731–13745. 10.1039/C2CP41658A. [DOI] [PubMed] [Google Scholar]
- 38.Vlcek AA, Dodsworth ES, Pietro WJ, and Lever ABP (1995). Excited State Redox Potentials of Ruthenium Diimine Complexes; Correlations with Ground State Redox Potentials and Ligand Parameters. Inorg. Chem 34, 1906–1913. 10.1021/ic00111a043. [DOI] [Google Scholar]
- 39.Prier CK, Rankic DA, and MacMillan DWC (2013). Visible Light Photoredox Catalysis with Transition Metal Complexes: Applications in Organic Synthesis. Chem. Rev 113, 5322–5363. 10.1021/cr300503r. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 40.Rosemann NW, Chábera P, Prakash O, Kaufhold S, Wärnmark K, Yartsev A, and Persson P (2020). Tracing the Full Bimolecular Photocycle of Iron(III)–Carbene Light Harvesters in Electron-Donating Solvents. J. Am. Chem. Soc 142, 8565–8569. 10.1021/jacs.0c00755. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 41.Bürgin TH, Glaser F, and Wenger OS (2022). Shedding Light on the Oxidizing Properties of Spin-Flip Excited States in a CrIII Polypyridine Complex and Their Use in Photoredox Catalysis. J. Am. Chem. Soc 144, 14181–14194. 10.1021/jacs.2c04465. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 42.Bevernaegie R, Wehlin SAM, Piechota EJ, Abraham M, Philouze C, Meyer GJ, Elias B, and Troian-Gautier L (2020). Improved Visible Light Absorption of Potent Iridium(III) Photo-oxidants for Excited-State Electron Transfer Chemistry. J. Am. Chem. Soc 142, 2732–2737. 10.1021/jacs.9b12108. [DOI] [PubMed] [Google Scholar]
- 43.Rehm D, and Weller A (1970). Kinetics of Fluorescence Quenching by Electron and H-Atom Transfer. Isr. J. Chem 8, 259–271. doi: 10.1002/ijch.197000029. [DOI] [Google Scholar]
- 44.Deetz AM, Troian-Gautier L, Wehlin SAM, Piechota EJ, and Meyer GJ (2021). On the Determination of Halogen Atom Reduction Potentials with Photoredox Catalysts. J. Phys. Chem. A 125, 9355–9367. 10.1021/acs.jpca.1c06772. [DOI] [PubMed] [Google Scholar]
- 45.Miller JR, Calcaterra LT, and Closs GL (1984). Intramolecular long-distance electron transfer in radical anions. The effects of free energy and solvent on the reaction rates. J. Am. Chem. Soc 106, 3047–3049. 10.1021/ja00322a058. [DOI] [Google Scholar]
- 46.Xiao P, Dumur F, Zhang J, Fouassier JP, Gigmes D, and Lalevée J (2014). Copper Complexes in Radical Photoinitiating Systems: Applications to Free Radical and Cationic Polymerization upon Visible LEDs. Macromolecules 47, 3837–3844. 10.1021/ma5006793. [DOI] [Google Scholar]
- 47.Kuang S-M, Cuttell DG, McMillin DR, Fanwick PE, and Walton RA (2002). Synthesis and Structural Characterization of Cu(I) and Ni(II) Complexes that Contain the Bis[2-(diphenylphosphino)phenyl]ether Ligand. Novel Emission Properties for the Cu(I) Species. Inorg. Chem 41, 3313–3322. 10.1021/ic0201809. [DOI] [PubMed] [Google Scholar]
- 48.Scaltrito DV, Thompson DW, O’Callaghan JA, and Meyer GJ (2000). MLCT excited states of cuprous bis-phenanthroline coordination compounds. Coord. Chem. Rev 208, 243–266. 10.1016/S0010-8545(00)00309-X. [DOI] [Google Scholar]
- 49.Kaeser A, Mohankumar M, Mohanraj J, Monti F, Holler M, Cid J-J, Moudam O, Nierengarten I, Karmazin-Brelot L, Duhayon C, et al. (2013). Heteroleptic Copper(I) Complexes Prepared from Phenanthroline and Bis-Phosphine Ligands. Inorg. Chem 52, 12140–12151. 10.1021/ic4020042. [DOI] [PubMed] [Google Scholar]
- 50.Nishikawa M, Kakizoe D, Saito Y, Ohishi T, and Tsubomura T (2017). Redox Properties of Copper(I) Complex Bearing 4,7-Diphenyl-2,9-dimethyl-1,10-phenanthroline and 1,4-Bis(diphenylphosphino)butane Ligands and Effects of Light in the Presence of Chloroform. B. Chem. Soc. Jpn 90, 286–288. 10.1246/bcsj.20160339. [DOI] [Google Scholar]
- 51.Zhang Y, Heberle M, Wächtler M, Karnahl M, and Dietzek B (2016). Determination of side products in the photocatalytic generation of hydrogen with copper photosensitizers by resonance Raman pectroelectrochemistry. RSC Adv. 6, 105801–105805. 10.1039/C6RA21469J. [DOI] [Google Scholar]
- 52.Moonshiram D, Garrido-Barros P, Gimbert-Suriñach C, Picón A, Liu C, Zhang X, Karnahl M, and Llobet A (2018). Elucidating the Nature of the Excited State of a Heteroleptic Copper Photosensitizer by using Time-Resolved X-ray Absorption Spectroscopy. Chem-Eur. J 24, 6464–6472. 10.1002/chem.201800330. [DOI] [PubMed] [Google Scholar]
- 53.Wodon M, De Kreijger S, Sampaio RN, Elias B, and Troian-Gautier L (2022). Accumulation of mono-reduced [Ir(piq)2(LL)] photosensitizers relevant for solar fuels production. Photochem. Photobio. Sci 21, 1433–1444. 10.1007/s43630-022-00233-z. [DOI] [PubMed] [Google Scholar]
- 54.Shaw GB, Grant CD, Shirota H, Castner EW, Meyer GJ, and Chen LX (2007). Ultrafast Structural Rearrangements in the MLCT Excited State for Copper(I) bis-Phenanthrolines in Solution. J. Am. Chem. Soc 129, 2147–2160. 10.1021/ja067271f. [DOI] [PubMed] [Google Scholar]
- 55.Fajardo J Jr., Barth AT, Morales M, Takase MK, Winkler JR, and Gray HB (2021). Photoredox Catalysis Mediated by Tungsten(0) Arylisocyanides. J. Am. Chem. Soc 143, 19389–19398. 10.1021/jacs.1c07617. [DOI] [PubMed] [Google Scholar]
- 56.Glaser F, Kerzig C, and Wenger OS (2021). Sensitization-initiated electron transfer via upconversion: mechanism and photocatalytic applications. Chem. Sci 12, 9922–9933. 10.1039/D1SC02085D. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 57.Ilic A, Schwarz J, Johnson C, de Groot LHM, Kaufhold S, Lomoth R, and Wärnmark K (2022). Photoredox catalysis via consecutive 2LMCT- and 3MLCT-excitation of an Fe(iii/ii)–N-heterocyclic carbene complex. Chem. Sci 13, 9165–9175. 10.1039/D2SC02122F. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 58.Aydogan A, Bangle RE, De Kreijger S, Dickenson JC, Singleton ML, Cauët E, Cadranel A, Meyer GJ, Elias B, Sampaio RN, and Troian-Gautier L (2021). Mechanistic investigation of a visible light mediated dehalogenation/cyclisation reaction using iron(iii), iridium(iii) and ruthenium(ii) photosensitizers. Catalysis Science & Technology 11, 8037–8051. 10.1039/D1CY01771C. [DOI] [Google Scholar]
- 59.Olmsted J, and Meyer TJ (1987). Factors affecting cage escape yields following electron-transfer quenching. J. Phys. Chem 91, 1649–1655. 10.1021/j100290a071. [DOI] [Google Scholar]
- 60.Kalyanasundaram K, and Neumann-Spallart M (1982). Influence of added salts on the cage escape yields in the photoredox quenching of Ru(bpy)2+3 excited states. Chem. Phys. Lett 88, 7–12. 10.1016/0009-2614(82)80059-6. [DOI] [Google Scholar]
- 61.Mallouk TE, Krueger JS, Mayer JE, and Dymond CMG (1989). Reductive quenching of ruthenium polypyridyl sensitizers by cyanometalate complexes. Inorg. Chem 28, 3507–3510. 10.1021/ic00317a023. [DOI] [Google Scholar]
- 62.Yoshimura A, Hoffman MZ, and Sun H (1993). An evaluation of the excited state absorption spectrum of Ru(bpy)32+ in aqueous and acetonitrile solutions. J. Photoch. Photobio. A 70, 29–33. 10.1016/1010-6030(93)80005-T. [DOI] [Google Scholar]
- 63.Goez M, von Ramin-Marro D, Othman Musa MH, and Schiewek M (2004). Photoionization of [Ru(bpy)3]2+: A Catalytic Cycle with Water as Sacrificial Donor. J. Phys. Chem. A 108, 1090–1100. 10.1021/jp036790r. [DOI] [Google Scholar]
- 64.Müller P, and Brettel K (2012). [Ru(bpy)3]2+ as a reference in transient absorption spectroscopy: differential absorption coefficients for formation of the long-lived 3MLCT excited state. Photochem. Photobio. Sci 11, 632–636. 10.1039/C2PP05333K. [DOI] [PubMed] [Google Scholar]
- 65.DiMarco BN, O’Donnell RM, and Meyer GJ (2015). Cation-Dependent Charge Recombination to Organic Mediators in Dye-Sensitized Solar Cells. J. Phys. Chem. C 119, 21599–21604. 10.1021/acs.jpcc.5b07342. [DOI] [Google Scholar]
- 66.Wehlin SAM, Troian-Gautier L, Li G, and Meyer GJ (2017). Chloride Oxidation by Ruthenium Excited-States in Solution. J. Am. Chem. Soc 139, 12903–12906. 10.1021/jacs.7b06762. [DOI] [PubMed] [Google Scholar]
- 67.Kurtz DA, and Dempsey JL (2019). Proton-Coupled Electron Transfer Kinetics for the Photoinduced Generation of a Cobalt(III)-Hydride Complex. Inorg. Chem 58, 16510–16517. 10.1021/acs.inorgchem.9b02445. [DOI] [PubMed] [Google Scholar]
- 68.Frisch MJ, Head-Gordon M, and Pople JA (1990). A direct MP2 gradient method. Chemical Physics Letters 166, 275–280. 10.1016/0009-2614(90)80029-D. [DOI] [Google Scholar]
- 69.Frisch MJ, Head-Gordon M, and Pople JA (1990). Semi-direct algorithms for the MP2 energy and gradient. Chemical Physics Letters 166, 281–289. 10.1016/0009-2614(90)80030-H. [DOI] [Google Scholar]
- 70.Head-Gordon M, and Head-Gordon T (1994). Analytic MP2 frequencies without fifth-order storage. Theory and application to bifurcated hydrogen bonds in the water hexamer. Chemical Physics Letters 220, 122–128. 10.1016/0009-2614(94)00116-2. [DOI] [Google Scholar]
- 71.Head-Gordon M, Pople JA, and Frisch MJ (1988). MP2 energy evaluation by direct methods. Chemical Physics Letters 153, 503–506. 10.1016/0009-2614(88)85250-3. [DOI] [Google Scholar]
- 72.Sæbø S, and Almlöf J (1989). Avoiding the integral storage bottleneck in LCAO calculations of electron correlation. Chemical Physics Letters 154, 83–89. 10.1016/0009-2614(89)87442-1. [DOI] [Google Scholar]
- 73.Krishnan R, Binkley JS, Seeger R, and Pople JA (1980). Self‐consistent molecular orbital methods. XX. A basis set for correlated wave functions. J. Chem. Phys 72, 650–654. 10.1063/1.438955. [DOI] [Google Scholar]
- 74.McLean AD, and Chandler GS (1980). Contracted Gaussian basis sets for molecular calculations. I. Second row atoms, Z=11–18. The Journal of Chemical Physics 72, 5639–5648. 10.1063/1.438980. [DOI] [Google Scholar]
- 75.Frisch MJ, Trucks GW, Schlegel HB, Scuseria GE, Robb MA, Cheeseman JR, Scalmani G, Barone V, Petersson GA, Nakatsuji H, et al. (2016). Gaussian 16 Rev. C.01.
- 76.Tomasi J, Mennucci B, and Cancès E (1999). The IEF version of the PCM solvation method: an overview of a new method addressed to study molecular solutes at the QM ab initio level. J. Mol. Struc. Theochem 464, 211–226. 10.1016/S0166-1280(98)00553-3. [DOI] [Google Scholar]
- 77.Balawender R, and Geerlings P (2001). Nuclear Fukui function from coupled perturbed Hartree–Fock equations. The Journal of Chemical Physics 114, 682–691. 10.1063/1.1331359. [DOI] [Google Scholar]
- 78.Cohen MH, Ganduglia-Pirovano MV, and Kudrnovský J (1994). Electronic and nuclear chemical reactivity. The Journal of Chemical Physics 101, 8988–8997. 10.1063/1.468026. [DOI] [Google Scholar]
- 79.Cohen MH, Ganduglia-Pirovano MV, and Kudrnovský J (1995). Reactivity kernels, the normal modes of chemical reactivity, and the hardness and softness spectra. The Journal of Chemical Physics 103, 3543–3551. 10.1063/1.470238. [DOI] [Google Scholar]
- 80.Becke AD (1988). Density-functional exchange-energy approximation with correct asymptotic behavior. Phys. Rev. A 38, 3098–3100. 10.1103/PhysRevA.38.3098. [DOI] [PubMed] [Google Scholar]
- 81.Lee C, Yang W, and Parr RG (1988). Development of the Colle-Salvetti correlation-energy formula into a functional of the electron density. Phys. Rev. B 37, 785–789. 10.1103/PhysRevB.37.785. [DOI] [PubMed] [Google Scholar]
- 82.Miehlich B, Savin A, Stoll H, and Preuss H (1989). Results obtained with the correlation energy density functionals of becke and Lee, Yang and Parr. Chem. Phys. Lett 157, 200–206. 10.1016/0009-2614(89)87234-3. [DOI] [Google Scholar]
- 83.Apra E, Bylaska EJ, de Jong WA, Govind N, Kowalski K, Straatsma TP, Valiev M, van Dam HJJ, Alexeev Y, Anchell J, et al. (2020). NWChem: Past, present, and future. J Chem Phys 152, 184102. 10.1063/5.0004997. [DOI] [PubMed] [Google Scholar]
- 84.Shan B, and Schmehl R (2014). Photochemical Generation of Strong One-Electron Reductants via Light-Induced Electron Transfer with Reversible Donors Followed by Cross Reaction with Sacrificial Donors. J. Phys. Chem. A 118, 10400–10406. 10.1021/jp503901v. [DOI] [PubMed] [Google Scholar]
- 85.Childs H, Brugger E, Whitlock B, Meredith J, Ahern S, Pugmire D, Biagas K, Miller M, Harrison C, Weber G, et al. (2012). VisIt: An end-user tool for visualizing and analyzing very large data. High Performance Visualization-Enabling Extreme-Scale Scientific Insight, 357–372. [Google Scholar]
- 86.Cauët E, Bogatko S, Liévin J, De Proft F, and Geerlings P (2013). Electron-attachment-induced DNA damage: instantaneous strand breaks. J Phys Chem B 117, 9669–9676. 10.1021/jp406320g. [DOI] [PubMed] [Google Scholar]
Associated Data
This section collects any data citations, data availability statements, or supplementary materials included in this article.
Supplementary Materials
Data Availability Statement
This study did not generate any datasets.