

Abstract
Nearly 1/3 of lung adenocarcinomas have loss of STK11 (LKB1) function. Herein, a bioinformatics approach was used to determine how accurately preclinical model systems reflect the in vivo biology of STK11 loss in human patients. Hierarchical and K-mean clustering, principle component, and gene set enrichment analyses were employed to model gene expression due to STK11 loss in patient cohorts representing nearly 1000 lung adenocarcinoma patients. K-means clustering classified STK11 loss patient tumors into three distinct sub-groups; positive (54%), neuroendocrine (NE) (35%) and negative (11%). The positive and NE groups are both defined by the expression of NKX2-1. In addition to NKX2-1, NE patients express neuroendocrine markers such as ASCL1 and CALCA. In contrast, the negative group does not express NKX2-1 (or neuroendocrine markers) and is characterized by significantly reduced survival relative to the two other groups. Two gene expression signatures were derived to explain both neuroendocrine features and differentiation (NKX2-1 loss) and were validated through two public datasets involving chemical differentiation (DCI) and NKX2-1 reconstitution. Patients results were then compared with established cell lines, transgenic mice, and patient-derived xenograft models of STK11 loss. Interestingly, all cell line and PDX models cluster and show expression patterns similar with the NKX2-1 negative subset of STK11-loss human tumors. Surprisingly, even mouse models of STK11 loss do not resemble patient tumors based on gene expression patterns. Results suggest pre-clinical models of STK11 loss are pronounced by marked elimination of type II pneumocyte identity, opposite of most in vivo human tumors.
Keywords: Type II pneumocyte, STK11 (LKB1), NKX2-1 (TTF-1), Immunotherapy
Introduction
As of 2020, lung malignancies remain the leading cause of cancer death with adenocarcinoma being the most common histological subtype(1). Expression of the STK11 (LKB1) protein, encoded by the tumor suppressor gene STK11, is lost in over 30% of these tumors(2–4), but is rare in other common tumors. Currently, there are no drugs used clinically to target STK11 loss cancers. Furthermore, STK11 loss tumors have reduced infiltrates of cytotoxic T-cells and respond poorly to anti-PD1 or anti-PDL-1 immunotherapies regardless of the PDL-1 status(5–11). For these reasons, many patients will benefit from a better understanding of the biology of STK11 loss.
Biologically, STK11 plays roles in cell polarity, metabolic regulation, inflammation, DNA repair, apoptosis, and relevant to this manuscript, it is also involved in cell lineage switching(12–14). The type II pneumocyte (ATII) is the cell lineage that is thought to most commonly give rise to adenocarcinoma. ATII cells are characterized by expression of NKX2-1 (commonly known as TTF1 and used as a clinical biomarker of the adenocarcinoma histology)and pulmonary surfactants which are responsible for regulating gas exchange and injury/inflammatory response in the lung(15–17). NKX2-1 is a master homeobox transcriptional regulator expressed in the thyroid, the lung, and the diencephalon of the brain which links the nervous system and the endocrine system.
Neuroendocrine cells of the pulmonary vasculature act as sensors, providing feedback and transmitting signals pertaining to the lung environment(18–20). These cells exist in isolation or cluster into neuroepithelial bodies and express the lineage biomarkers ASCL1 (Achaete-Scute Family BHLH Transcription Factor) and CALCA (Calcitonin related polypeptide alpha)(21, 22). Serving as sentinels, these cells are capable of sensing hypoxia among other environmental stimuli and can both communicate to other tissues in the body as well as influence the local microenvironment(23). In addition to their role in homeostatic maintenance and signal transduction, neuroendocrine cells produce neurotransmitters such as γ-aminobutyric acid (GABA) and are thought to play a vital role in allergen response(19).
One hurdle in understanding complex biological systems is the development of models capable of accurately recapturing patient biology. In the current manuscript, we explore how well current preclinical models of STK11 loss reflect the predominant adenocarcinoma phenotypes observed in patients. The results suggest that nearly all STK11-loss model systems reflect a single and rare classification of lung adenocarcinoma that is characterized by loss of NKX2-1 expression and thus ATII cell identity. These findings demonstrate the current limitations of surrogate in vivo research regarding STK11 loss and highlight the importance of studying STK11 loss primarily in human patients.
Materials and Methods
Public datasets
Series matrix files were downloaded through public NCBI GEO (https://www.ncbi.nlm.nih.gov/gds/). Datasets that contained linear gene expression were log2 transformed. TCGA data was downloaded from the Xena browser (https://xenabrowser.net/hub/) for gene expression (RNA-seq), protein expression (RPPA), copy number variants and somatic mutation. MLOS (Moffitt Lung Adenocarcinoma with Overall Survival) has been previously described as GSE72094(6). Additional resources include cell line datasets; GSE36133 (Cancer Cell Line Encyclopedia or CCLE(24)), GSE21581 (mouse model 1, MM1(25)) GSE118246 (mouse model 2, MM2(14)), GSE78806 (patient derived xenograft, PDX(26)), and two human datasets mentioned in our previous study MLOS(2, 6, 27) and TCGA. The CCLE contains 44 annotated lung adenocarcinoma samples, 15 are classified as STK11 mutant and 29 as wildtype. MM1 contains 18 primary lung cancer samples, 9 KRAS mutants and 9 KRAS/STK11 mutants. MM2 contains 8 KRAS/TP53 mutants, 9 STK11 mutant SOX2 cre, 6 STK11/PTEN mutants, 8 SOX2 overexpressed STK11 mutants, and 3 normal lung tissues. The PDX study contains 46 adenocarcinoma samples determined by clustering and of these 10 were STK11 mutant and 29 STK11 wildtype. MLOS contains 442 patient tumors, 145 predicted STK11 mutants and 297 wildtypes. TCGA contains 515 lung adenocarcinoma tumors, 178 predicted STK11 mutants and 337 wildtypes. In addition to these datasets, we also explored two separate characteristics of ATII cellular identity through viral transduction of NKX2-1 into A549 cells (STK11 mutant, NKX2-1 null; GSE40584, described herein as cohort NKX) and combination dexamethasone and cyclic AMP treatment of fetal lung epithelial cells to differentiate them into ATII cells (GSE3306, described herein as cohort, DCI). Supplemental Files 1–11 provide data and test statistics that were generated in various analyses.
Clustering
Hierarchical Clustering was performed using the Python programming language. The gene expression matrix was first Z-score transformed for each gene. Distance was calculated through scipy.spacial.distance.pdist using the Euclidean distance metric. Linkage was then calculated through scipy.cluster.hierarchy.linkage using Ward linkage. Dendrograms were created for the clustering of both samples and genes and represented using scipy.cluster.hierarchy.dendrogram. Heatmaps were displayed on a color bar scale by Z-score from −3 to 3. K-means clustering classified the three STK11-loss subtypes through the Biopython library and the Bio.Cluster.kcluster function generated (k=3) clusters. The average method was used and Euclidean distance with 100 permutations.
Principal component analysis was performed through the Biopython library (https://biopython.org/) using the Bio.Cluster pca function. The data matrix was refined to the genes of interest prior to calculating component scores of each gene. Signature scores were generated by first calculating the number of standard deviations from the mean (z-score) for each gene for each patient. The averaged principal component loading coefficients were multiplied by the z-score for that same gene. This cumulative score was then divided by the number of genes in the signature to get an average score per sample.
Gene Set Enrichment Analysis, Venn Diagrams and Kaplan-Meier Curves
Gene Set Enrichment Analysis was performed with the Broad Institute’s Hallmark gene-sets (http://software.broadinstitute.org/gsea/msigdb/annotate.jsp) with a FDR cutoff of 0.05. Venn diagrams were generated using (http://www.interactivenn.net/) as referenced (28). Kaplan Meier survival curves were generated using Python’s lifelines library (https://lifelines.readthedocs.io/en/latest/). The KaplanMeierFitter was called and passed vital status information as (alive = 0) and (dead = 1) and respective survival duration. Log-rank assessment was performed using the logrank_test in the lifelines library.
Statistical Tests
Statistical analysis was performed using the Scipy python package and the function scipy.stats.ttest_ind for p-value between arrays. Pearson correlation was calculated through the scipy.stats.pearsonr function. The scipy.stats.fisher_exact function was used for the statistical test determining the significance between mutations occurring in each STK11-loss/Mut subset compared to STK11 WT.
Results
Optimization of a patient-derived gene expression classifier for STK11 loss.
Previous studies, including our own, have developed classifiers to identify STK11-loss tumors based on gene expression changes(2–4). Universally, these studies concluded that the identification of STK11 loss in patients is significantly under-estimated by genomic sequencing. While these signatures are nonetheless good classifiers, the genes that they contain, in retrospect, do not fully represent the in vivo biology of STK11 loss since the signatures relied heavily on cell lines and/or mouse models in their derivation. To correct for this deficiency, we two very large well-annotated lung adenocarcinoma datasets: TCGA (N=515)(29) and our previously published MLOS cohort (N=442)(2, 6, 30). Each of these datasets contains both DNA sequencing and RNA expression data on most patients. First, we used known STK11 mutation data derived from genomic sequencing to derive an initial 29-gene expression classifier. Next, we used the initial gene expression classifier to reclassify the patients as STK11 loss or STK11 wildtype (WT). Finally, we used the reclassified patients to define the full spectrum of gene expression changes associated with STK11 loss. This process generated a novel patient derived 137-gene signature to classify patients by STK11 loss of function. The entire process is described in more detail in Supplementary Materials, including Supplementary Table 1 and Figures 1 and 2. This signature was used, when appropriate, to classify patient tumors as STK11 WT or STK11 loss in the studies that follow.
Ex vivo models of STK11 loss lose ATII lineage marker NKX2-1
It became apparent that a subset of patients with predicted STK11 loss also lost expression of a subset of genes in the derived signature. This same gene network was not found in our previous in vitro signature and contained NKX2-1, a critical regulator of the ATII cell lineage and the principle biomarker of lung adenocarcinoma clinically. To explore NKX2-1 expression in pre-clinical models of lung adenocarcinoma, we characterized the expression changes of NKX2-1 in six model systems with respect to STK11 status. Surprisingly, in three of the four pre-clinical model systems, STK11 mutant samples had statistically significant reductions in their expression of NKX2-1 (Fig 1 A–D) relative to samples with wildtype STK11. The one exception was our first public mouse model (MM1). While the MM1 result is not statistically significant, the STK11 mutant tumors in MM1 trend toward reduced NKX2-1 expression (Fig 1 C). In contrast, when we explored NKX2-1 expression in the MLOS and TCGA patient cohorts, we find only a small number of patients with reduced NKX2-1 in both the STK11-loss and wildtype subsets. Notably, unlike the pre-clinical models, there was no change in frequency of NKX2-1 loss with respect to STK11 status in MLOS (p = 0.246) or TCGA (p=0.212) (Fig 1 E–F). Thus, in human lung adenocarcinoma tumors, STK11 loss is not associated with loss of NKX2-1; however, in pre-clinical models STK11 loss is strongly associated with a loss of NKX2-1 expression (and thereby ATII lineage differentiation).
Figure 1 |. STK11 loss results in loss of type II pneumocyte identity ex vivo.
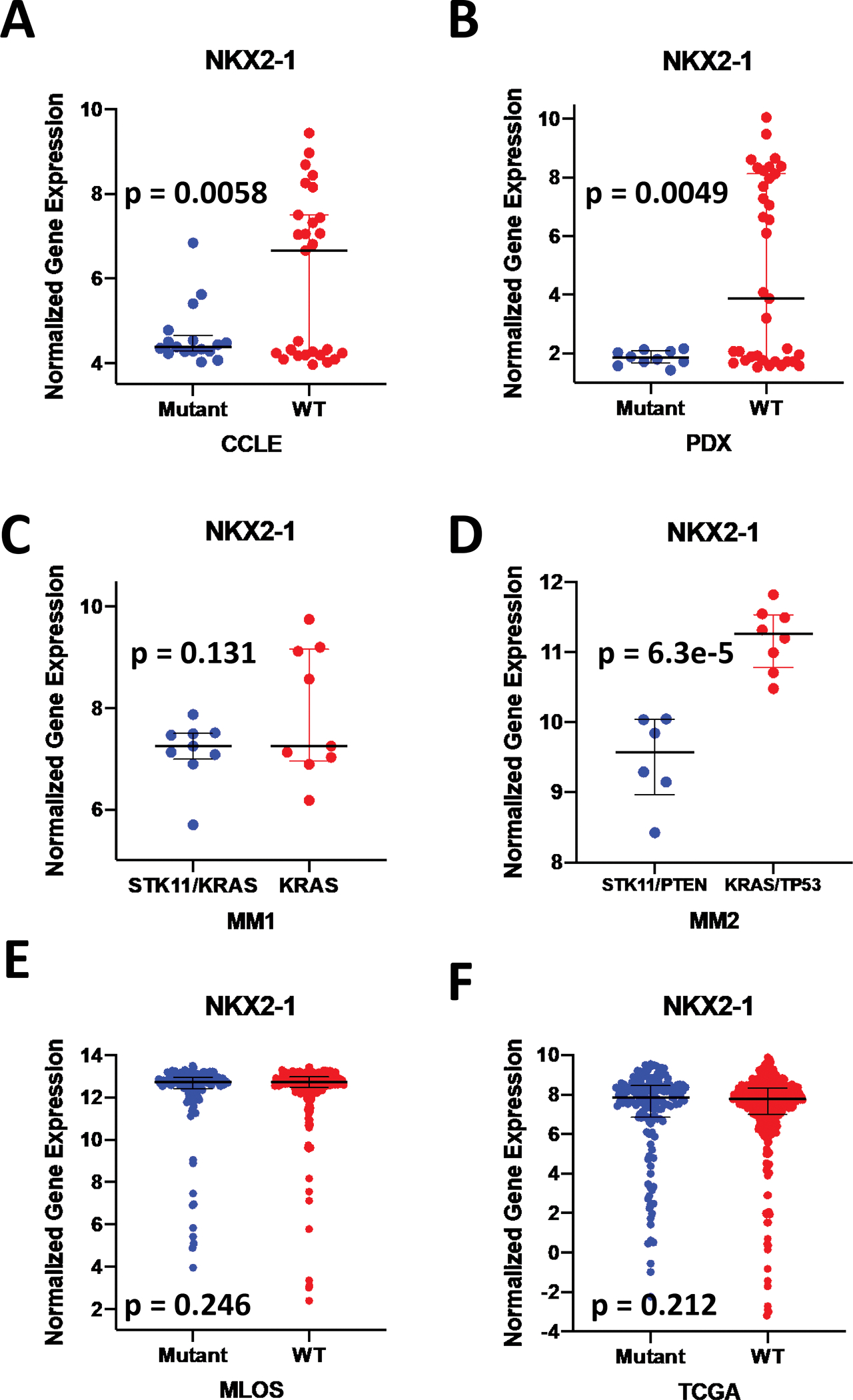
(a-f) Log2 transformed expression of NKX2-1 mRNA. (a) Cancer Cell Line Encyclopedia (CCLE) lung adenocarcinoma cell lines predicted with the STK11 loss signature cutoff WT (n = 29) < 0 > Mutant (n = 15). (b) Patient derived xenograft (PDX) models predicted with the STK11 loss signature with a cutoff of WT (n=29) < 0, Mutant (n =10) > 0.1. (c) Mouse model 1 (MM1) of n=9 KRAS tumors vs n=9 KRAS/STK11 tumors. (d) Mouse model 2 (MM2) of n=8 KRAS/TP53 tumors and n=6 STK11/PTEN tumors. (e) MLOS lung adenocarcinoma patient tumors predicted for STK11 status with WT (n=297) and Mutant (n=145). (f) TCGA lung adenocarcinoma patient tumors predicted for STK11 status with WT (n=337) and Mutant (n=178).
Classification of STK11 loss tumors into three distinct biological subtypes.
The fact that a subset of patient samples with predicted STK11 loss show reduced NKX2-1 expression (Fig 1 E–F) provides an opportunity to assess the impact of STK11 loss in the context of cellular lineage identity both within patient tumors and ex vivo models. Previous work has shown that a subset of lung adenocarcinomas expresses neuroendocrine markers (31, 32). Interestingly, we noticed that a large fraction of patient tumors with STK11 loss upregulated at least two of these markers; Achaete-Scute Family BHLH Transcription Factor 1, or ASCL1 (TCGA p = 8.38e-31; MLOS p = 2.19e-16) and Calcitonin Related Polypeptide Alpha or CALCA (TCGA p = 1.13e-54; MLOS p = 3.78e-41) (Fig 2 A and B). In contrast, Fig 2 A and B reveals that high expression of these neuroendocrine markers are rare in STK11 WT tumors. This suggests that a large fraction of pathologically classified lung adenocarcinoma with STK11 loss may undergo partial lineage switching to a neuroendocrine-like phenotype.
Figure 2 |. Classification of STK11 loss (mutant) patient tumors into positive, NE, and negative subsets.
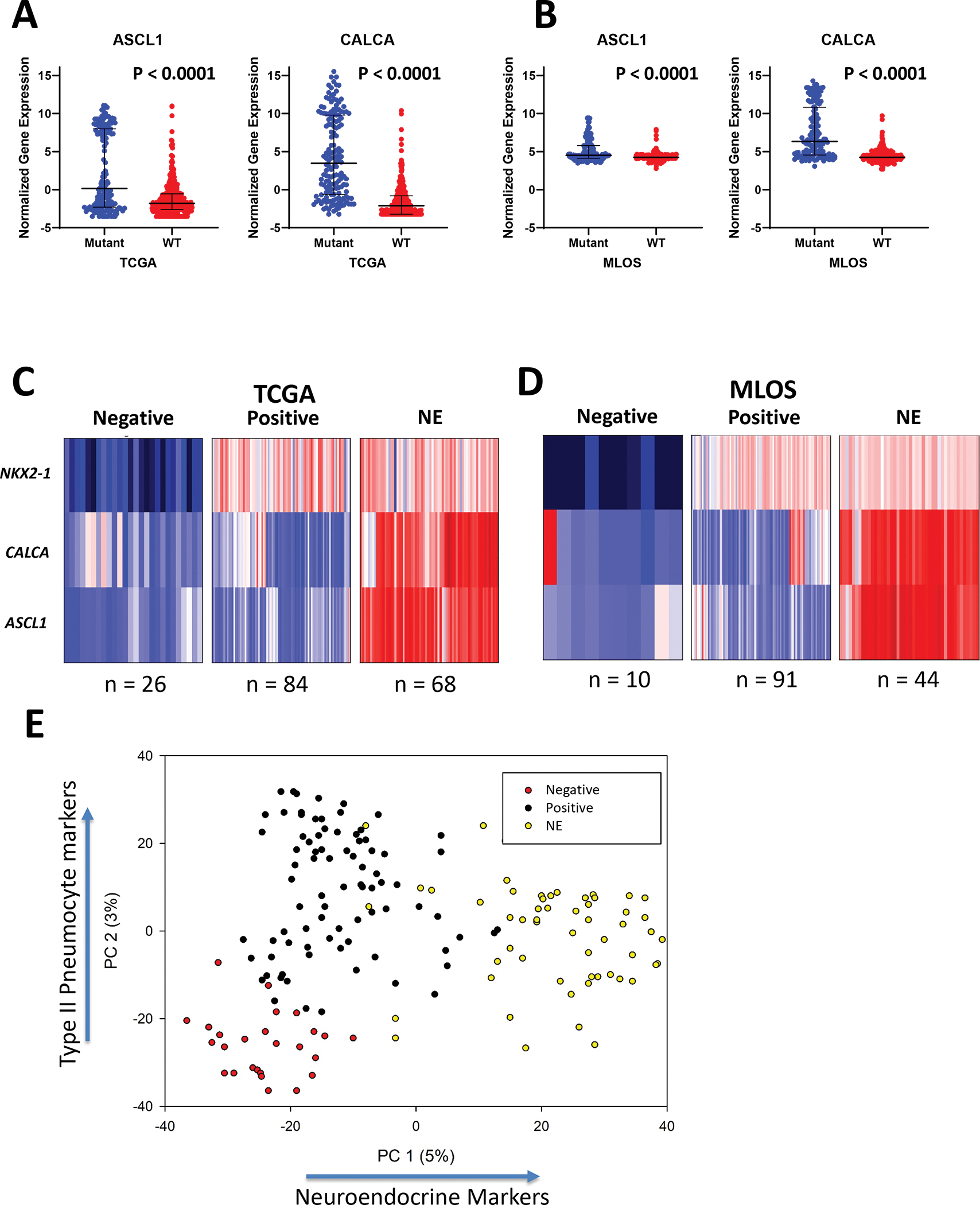
(a) Gene expression of neuroendocrine markers ASCL1 and CALCA in STK11 normal and STK11 loss in TCGA (left) and MLOS (right). (b-c) K-means clustering of STK11 loss patients in TCGA (b) and MLOS (c) into negative, positive, and NE subsets based on the markers NKX2-1, ASCL1, and CALCA. (d) Scatter plot of PC-1(neuroendocrine signature) and PC-2 (differentiation signature) of principal component analysis performed on 544 genes describing variance of STK11-loss subtypes.
To assess this hypothesis, K-means clustering was used to further classify STK11-loss patient tumors based on their expression of neuroendocrine markers, ASCL1 and CALCA, in addition to the ATII marker, NKX2-1. We annotated tumors in MLOS and TCGA with high expression of ASCL1, CALCA, and NKX2-1 as “neuroendocrine (NE)”, high expression of NKX2-1 and low ASCL1/CALCA as “positive”, and “negative” as low expression of all these markers. In TCGA, patients clustered into these 3 subtypes with 84 positive tumors, 68 NE, and 26 negative tumors (Fig 2 C). MLOS contained 91 positive, 44 NE, and 10 negative tumors (Fig 2 D). The overall percentage of patients with each subtype was 54% positive, 35% NE and 11% negative.
Further, we performed principal component analysis on the most significantly altered genes between these three clinical subtypes to describe lineage associated expression patterns. We find that two principal components sufficiently explain this variance with PC-1 describing neuroendocrine features and PC-2 describing features of NKX2-1 loss, or de-differentiation (Fig 2 E). Loading coefficients of genes most significantly contributing to PC-1 include well-characterized NE markers (ASCL1, CALCA, RET, KLK12, PCP4, CTNND2, and NEUROD1). (Supplemental File 3). Top coefficients of PC-2 include NKX2-1, PCSK2, GKN2, C16ORF89 (LINC00473), SLC14A2, NAPSA, LPL, and HOPX.
The three STK11-loss subtypes have distinct biology and clinical outcome
Lung adenocarcinoma subtypes have been related to survival and other clinical outcomes in several published studies (5, 9, 33–35). We further characterized our STK11-loss subtypes by defining their molecular characteristics to understand their clinical and biological ramifications. We analyzed gene expression, protein expression, DNA mutations, and copy number alterations using data from the TCGA cohort and gene expression, survival, and clinical data from both TCGA and MLOS (Fig 3 A–D). We briefly summarize the findings here.
Figure 3 |. Molecular and clinical characterization of STK11 loss subsets.
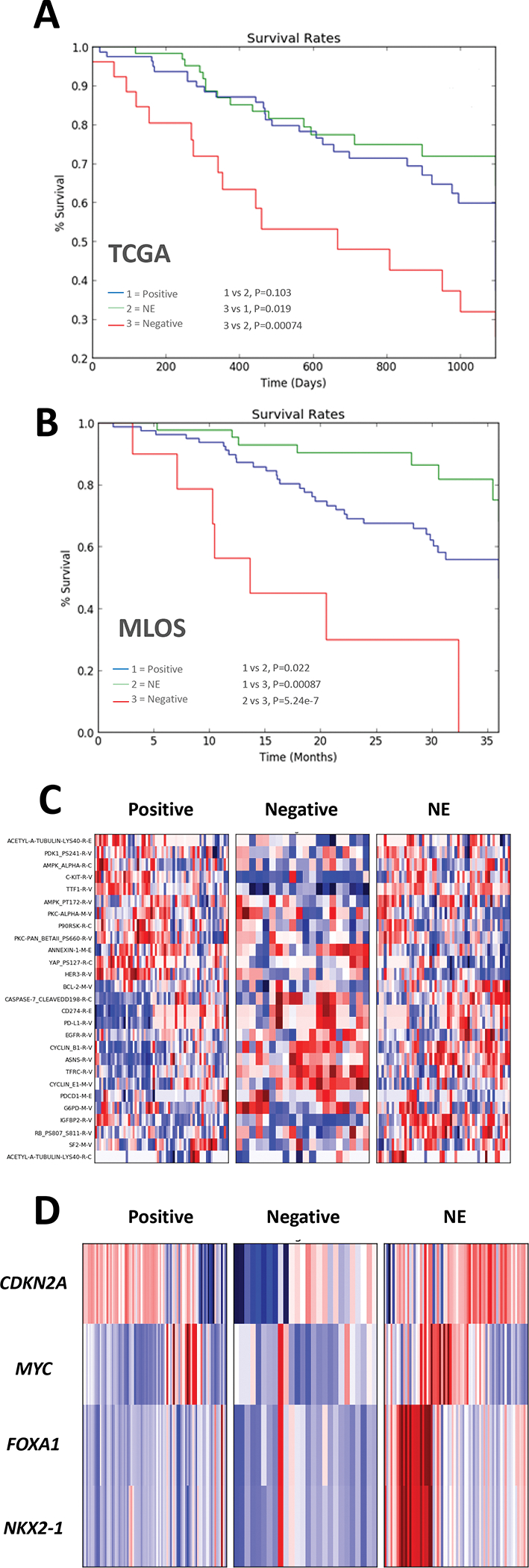
(a-b) Kaplan Meier curves of 3-year survival in TCGA (a) and MLOS (b) based on the STK11-loss subtypes. (c) Heatmap of Z-score (scale −3 to 3) transformed protein expression from TCGA RPPA (d) Heatmap of Z-score transformed copy number changes (scale −3 to 3) between the three STK11-loss subtypes.
Survival
Overall survival was assessed at 3 years (1095 days or 36 months) in the 3 subtypes through Kaplan-Meyer analysis and the log-rank test (Fig 3 A–B). The negative cohort displayed the worst survival compared to both the positive (TCGA p = 0.019, MLOS p = 0.00087) and NE (TCGA p = 0.00074, MLOS p = 5.24e-7) cohorts with a median survival of less than 700 days. There was no significant difference between the positive and NE cohort in TCGA (p = 0.103) but the NE subset had a statistically improved prognosis in MLOS (p = 0.022). These data suggest that de-differentiation and loss of cellular identity is coincident with poor patient prognosis.
Protein Expression
Through use of TCGA’s RPPA protein expression data, we profiled the three STK11-loss subtypes for defining characteristics (Fig 3C). In the positive subgroup, there was a significant reduction in cyclin B1 (CCNB1), glutamine dependent asparagine synthetase (ASNS), caspase 7 cleavage, and immunosuppressive programmed cell death 1 ligand 1 (PD-L1/CD274). There was a significant increase in napsin A (involved in the processing of surfactants), HER3, and YAP pS127 which lies downstream of AMPK activation (downstream of STK11) and is activated during high cellular density. The negative cohort had significant increases in glucose-6-phosphate dehydrogenase G6PD a hallmark of NRF2 activation, TFRC, cyclinE1 and had the highest levels of both PD-1 and PD-L1 expression. There was a reduction in NKX2-1 (as one would predict), c-KIT, and IGFBP2. The NE cohort was characterized by elevated IGFBP2, XRCC1, BCL2, acetyl-a-tubulin lys40 with decreases in YAP pS127. Through both gene and protein expression there is a remarkable correlation of KIT to NKX2-1 levels (KIT is highly mutated in gastrointestinal cancers (36) and may be related to latent gastrointestinal signaling mediated in lung adenocarcinoma through NKX2-1 (37)).
Gene Copy number
Gene copy number profiles showed very interesting lineage specific amplifications (Fig 3D). The NE cohort displayed copy number amplifications of the NKX2-1/FOXA1 locus, suggesting that amplification of NKX2-1 may have an oncogenic role in this subset. NKX2-1 and FOXA1 have been shown to have opposite roles in the regulation of differentiation and lung cancer progression (37, 38). CDKN2A (p16), had an increased rate of deletion in the negative cohort, followed by the positive, and much less so in the NE subset. While MYC was amplified in each cohort, it was most commonly amplified in the NE subset.
NKX2-1 addition and ATII differentiation in ex vivo models, mimics patient phenopes
To distinguish the changes that take place as a result of type II pneumocyte differentiation, we explored a public dataset (called DCI in our studies) in which human fetal lung epithelial cells were differentiated into type II pneumocytes through treatment with dexamethasone and cyclic AMP (Supplemental File 4) (39). A student’s t-test was performed on the dataset comparing the 5 treated samples to untreated and changing genes were displayed through volcano plot (Fig 4A). In the treated cohort, significant upregulation was seen in ALDH3A2, SFTPC, SFTPB, CIT, CALCA, HOPX, NR4A2, LPL, PGC, and MAOA; all of which are genes shown to associate with NKX2-1 expression in TCGA (Fig 4C). Many of these genes upregulated during ATII differentiation play critical roles in ATII cell biology and are seen in neither ex vivo models nor the negative cohort of patients with STK11 loss.
Figure 4 |. ATII differentiation and NKX2-1 addition share transcriptional changes associated with STK11 signaling.
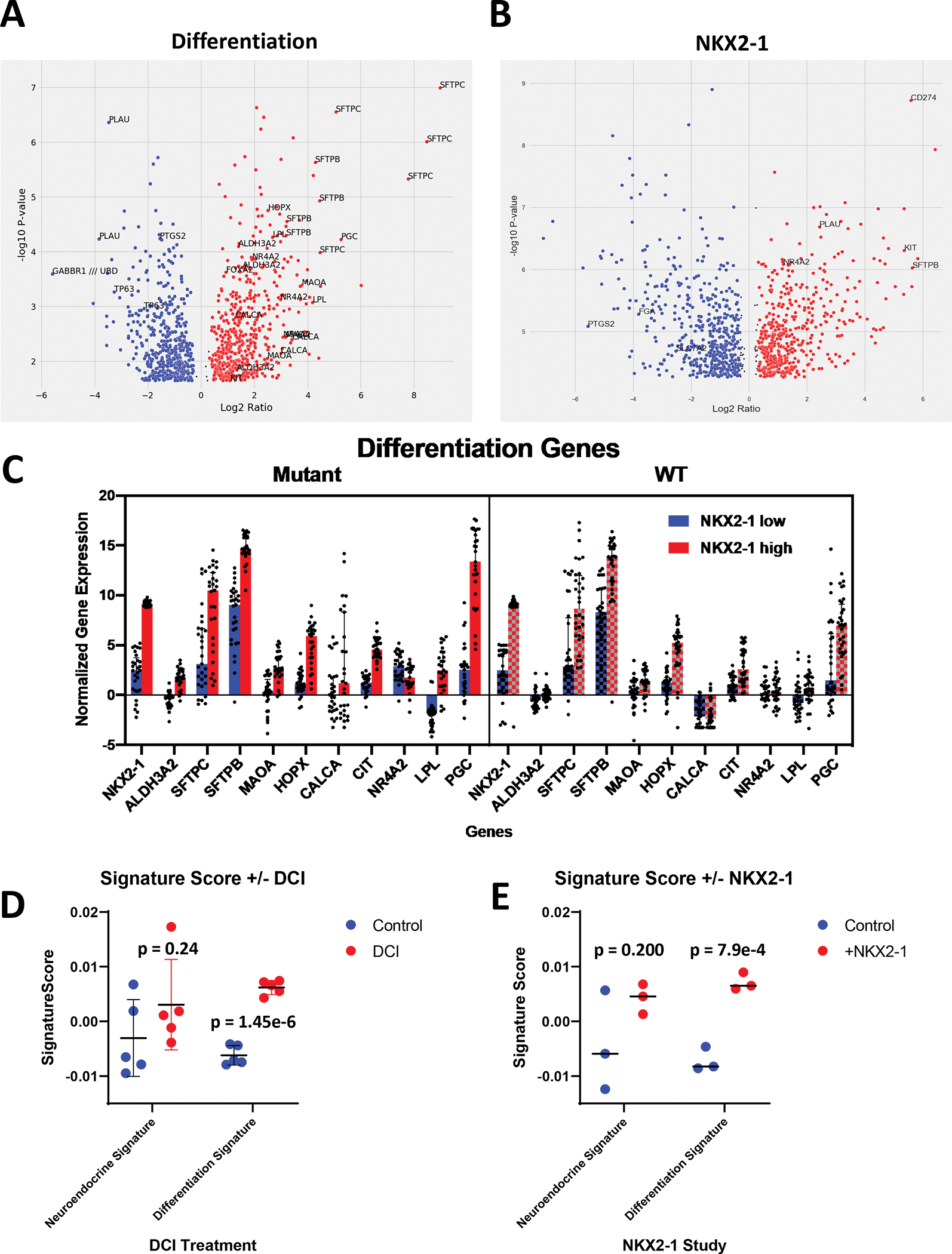
(a) Volcano plot of gene expression changes as a result of type II pneumocyte differentiation. (b) Volcano plot of gene expression changes as a result of NKX2-1 addition to A549 cells. (c) Expression of genes pertaining to ATII differentiation in the top and bottom 10% of patients based on NKX2-1 expression in both STK11 WT and loss patients (d) Swarm plot of PC-1 (left) and PC-2 (right) in ATII differentiation study. (e) Swarm plot of the neuroendocrine signature (left) and the differentiation signature (right) in NKX2-1 addition study.
As a second test, we analyzed data from a previous publication that used a lentivirus to overexpress NKX2-1 in A549 lung adenocarcinoma cells which lack expression of both NKX2-1 and STK11 (Supplemental File 5) (40). Student’s t-test was performed on the gene expression of this dataset and the results are summarized in a volcano plot (Fig 4B). Notable increases in gene expression include PD-L1, KIT, and SFTPB (Supplemental Fig 1E). Decreases in PTGS2 (COX2), FGA, and HLA-DMB were also observed. The complementation of NKX2-1 to A549 cells restored a subset of gene expression seen in patients with STK11 loss, including increases in KIT and decreases in class II HLA’s. This dataset highlights the role that the NKX2-1 transcription factors plays in the context of STK11 loss and further displays its importance in regulating the expression of genes lost in ex vivo studies.
We applied the principal component derived signatures from our classification of in vivo STK11 loss tumors (From 3.3 reference). These signatures explain the gene expression changes associated with neuroendocrine features and NKX2-1 loss respectively and were applied to studies of both NKX2-1 addition to A549 cells and ATII differentiation. In both the NKX2-1 study (p=0.244) and ATII differentiation study (DCI) (p=0.200) the neuroendocrine signature was not significantly altered. However, the differentiation signature was significantly elevated through both reconstitution of NKX2-1 (p=0.00079) and DCI treatment (p=1.45e-6) (Fig 4C–D). Thereby providing evidence for the aforementioned observations: 1) That in vitro STK11 loss models can partially regain in vivo expression patterns when NKX2-1 expression is restored 2) that in vivo loss of NKX2-1 expression mimics gene expression patterns of genes altered during ATII differentiation 3) that in vitro models of STK11 loss are representing only the fraction of STK11 loss patients with NKX2-1 loss.
Ex vivo models cluster with the NKX2-1 and ASCL1/CALCA negative subgroup of STK11-loss adenocarcinomas
We characterized gene expression patterns conserved between studies, represented by the 5-way Venn diagram displaying overlaps of genes changing by either +/− 1.5-fold (Fig 5A). Using the log2 gene expression changes in each public dataset used in this study (Supplemental Files 6–11), a matrix was created for every gene that had expression data. In both MLOS and TCGA, we added the relative fold-change from the positive, NE, and negative cohort compared to STK11 WT patients. In addition, we added the averaged fold change between all STK11 mutants and WT, labeled as overall. Each study was hierarchically clustered based on the genes that were used in the neuroendocrine signature and differentiation signature (Supplemental File 3). The NE, overall, and positive cohorts all cluster together from each study while the negative cohorts cluster with both PDX and CCLE models of STK11 loss (Fig 5B). More surprising is that mouse models of STK11 loss exhibit the least resemblance to STK11 loss patient tumors and cluster alongside the experimental studies (named DCI and NKX).
Figure 5 |. Ex vivo models of STK11 loss cluster with and mimic the negative subset of STK11 mutant patients lacking inflammatory signaling.
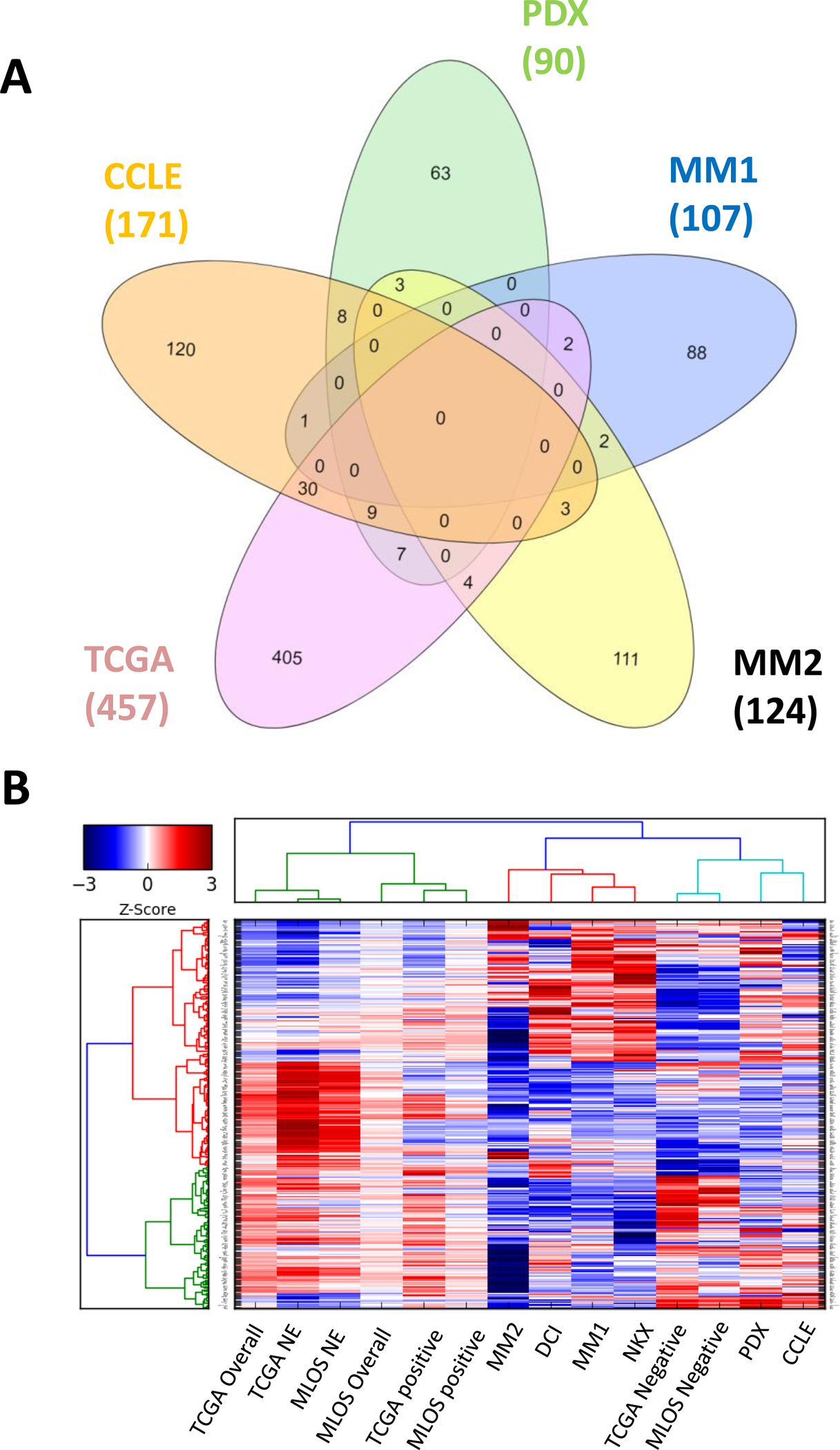
(a) 5-way Venn Diagram of average STK11 loss vs WT, cell lines (CCLE), patient-derived xenografts (PDX), and mouse models (MM1 and MM2). (b) Hierarchical clustering of gene expression fold changes of each study. TCGA and MLOS were further divided into fold changes of subtype vs WT and average vs WT. Genes selected were used for PCA signature generation.
Genes lost in ex vivo models are markers of both neuroepithelial cells and type II pneumocytes. Clustering of cell line and PDX models to the subset of patients with loss of NKX2-1 suggests that ex vivo models of STK11 loss fail to retain cellular lineage identity. This contrasts with roughly 90% of in vivo tumors of STK11 loss. Gene Set Enrichment Analysis (GSEA) on each of the datasets described here demonstrate how different pre-clinical models of STK11 loss impact biology. (See section on GSEA analysis and Figure 3 in Supplemental Materials)
Discussion
Herein, we show that STK11 loss results in changes in cell lineage markers unanimously in non-patient model systems. We first profiled several known in vivo and ex vivo datasets in the context of STK11 loss. We showed the specific loss of cellular identity in ex vivo models of STK11 loss through NKX2-1 expression, while revealing that most patient tumors retained this identity. In addition, we observed the enrichment of predominant neuroendocrine characteristics in a subset of in vivo STK11 loss tumors through expression of ASCL1 and CALCA. We clustered patients into 3 subsets dependent upon their expression of lineage markers into positive, NE, and negative. These cohorts were then analyzed for recurrent changes in copy number alterations, protein expression, mRNA expression, DNA mutation profiles, and overall survival. Negative cohorts displayed loss of ATII identity and subsequently other hallmarks of STK11 loss via urea cycle utilization, polyamine metabolism, and an increase in inflammatory response. This same negative cohort most closely related to ex vivo models of STK11 loss, which lose NKX2-1 expression.
It is important to understand the differences in each of these models. Cell lines, mouse models, and PDX models are much more homogenous than most human tumors with a much quicker initiation in the case of PDX and mouse models. Being a mediator of energy sensing and metabolic stress, STK11 loss in environments with vastly altered resource and nutrient availability likely contribute to the significant alterations in both cellular differentiation and gene expression seen in in vitro culture. Culture conditions most commonly provide an excess of amino acids and sugar and are grown at normoxia (20% O2), all of which are significantly different from in vivo models. Most surprising is how far mouse models miss the mark of recapturing patterns of human gene expression changes. Previous genetically engineered mouse models have shown the impact of STK11 loss on an adenocarcinoma to squamous cell carcinoma lineage switch through both SOX2 mediated suppression(14) and epigenetic changes driven by the EZH2 polycomb recombination complex(13). While, these observations are relevant in mice, we notice that this same lineage switch does not occur in patient tumors of STK11 loss, which maintain the histological identity of adenocarcinoma. In this setting, most environmental stressors such as oxygen availability, immune response, and lung specific signaling should remain intact. It is possible that mouse models that do not specifically target NKX2-1 positive cells with the induction of STK11 loss could result in a tumor with mixed histology given the low frequency of ATII cells to other cell types. Mouse models differ significantly in tumor initiation from that of patient tumors, which undergo a much more stringent and elaborate adaptive process in disease progression. Further, another explanation of the discrepancy of cell line and PDX models is a selection for the most aggressive phenotype of STK11 loss, which happens to be cells lacking ATII identity.
Previous studies of STK11 loss noted that neutrophils modulate the immune microenvironment and result in lack of immunotherapy efficacy through an increase in proinflammatory cytokines CXCL7, G-CSF, and IL-6 (41). While these observations are true of mouse models, these same markers are not seen to be elevated in patient studies nor do we observe any significant changes in myeloid cell populations through gene expression patterns. Flow cytometry of patient tumors with STK11 loss revealed that of all basic immune cell populations, T-cells were the most significantly reduced cell population (9). This study was carried out in mice with genetic backgrounds of KRAS/TP53 and KRAS/TP53/STK11. We find that TP53 mutations occur more frequently in the negative cohort of patients (58% occurrence) than in the positive cohort (32%, p=0.02) and previous publications have shown the dichotomy of NKX2-1 signaling dependent on TP53 status and STK11 loss (42, 43). Additionally, it has been shown that TP53 and p16 alterations are exclusive for subsets of large cell neuroendocrine carcinoma with dual KRAS/STK11 mutation (44). TP53 mutations are exclusively associated with both KRAS and STK11 mutations in lung adenocarcinoma and this could also play a role in the maintenance of both cell state and subsequently inflammatory signaling.
Model systems are created with the purpose of deepening our understanding of patient biology and thus providing a mechanism towards discovering therapies to be used in our fight against cancer. Given the implications of STK11 loss on both the metabolism and immune response make this exceedingly difficult. This study highlights the importance of having model systems that are not only capable of manipulation, but also ones that accurately recapitulate patient biology, especially in the context of immunotherapy. Understanding the limitations of such systems will help to not only keep an open mind to previous failed treatments but also to improve our current models in a way that accurate depicts the disease we are attempting to eliminate. Tissue specificity influences the frequency of genomic aberrations, as confirmed from the diverse landscape of genomic alterations in specific cancer types. With respect to lung cancer and STK11 loss, we hope that this consideration will drastically alter the ways in which we study this unique genetic abnormality and provide insight into the lack of immune response in this cohort.
Supplementary Material
Acknowledgments
We thank Gabriela Wright and Edna Gordián for critical discussion of this work and specific comments on this manuscript. This work was funded by the James and Esther King Biomedical Research Program Grant (5JK06) from the Florida Department of Health (WDC) which had no role in directing this work or writing the manuscript. Our study also received valuable assistance from the Moffitt’s Lung Cancer Center of Excellence program, and the Cancer Informatics and Genomics Core Facilities at the H. Lee Moffitt Cancer Center & Research Institute, an NCI designated Comprehensive Cancer Center, supported under NIH grant P30-CA76292. These funding sources did not play a role in study design or in the decision to submit this work for publication.
Funding Support:
This work was funded by the James and Esther King Biomedical Research Program Grant (5JK06) from the Florida Department of Health (WDC) which had no role in directing this work or writing the manuscript. Our study also received valuable assistance from the Moffitt’s Lung Cancer Center of Excellence program, and the Cancer Informatics and Genomics Core Facilities at the H. Lee Moffitt Cancer Center & Research Institute, an NCI designated Comprehensive Cancer Center, supported under NIH grant P30-CA76292. These funding sources did not play a role in study design or in the decision to submit this work for publication.
Footnotes
Conflict of interest statement: The listed authors have any NO conflicts of interest relative to this publication. Specifically, we have no affiliations with or involvement in any organization or entity with any financial interest (such as honoraria; educational grants; participation in speakers’ bureaus; membership, employment, consultancies, stock ownership, or other equity interest; and expert testimony or patent-licensing arrangements), or non-financial interest (such as personal or professional relationships, affiliations, knowledge or beliefs) in the subject matter or materials discussed in this manuscript.
References
- 1.ACS. Cancer Facts and Figures 2018. Atlanta, GA: American Cancer Society; 2018. p. 1–76. [Google Scholar]
- 2.Chen L, Engel BE, Welsh EA, Yoder SJ, Brantley SG, Chen DT, et al. A sensitive NanoString-based assay to score STK11 (LKB1) pathway disruption in lung adenocarcinoma. J Thorac Oncol. 2016;11(16):838–49. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 3.Kaufman JM, Yamada T, Park K, Timmers CD, Amann JM, Carbone DP. A Transcriptional Signature Identifies LKB1 Functional Status as a Novel Determinant of MEK Sensitivity in Lung Adenocarcinoma. Cancer Res. 2017;77(1):153–63. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 4.He N, Kim N, Song M, Park C, Kim S, Park EY, et al. Integrated analysis of transcriptomes of cancer cell lines and patient samples reveals STK11/LKB1-driven regulation of cAMP phosphodiesterase-4D. Mol Cancer Ther. 2014;13(10):2463–73. [DOI] [PubMed] [Google Scholar]
- 5.Skoulidis F, Byers LA, Diao L, Papadimitrakopoulou VA, Tong P, Izzo J, et al. Co-occurring genomic alterations define major subsets of KRAS - mutant lung adenocarcinoma with distinct biology, immune profiles, and therapeutic vulnerabilities. Cancer Discov. 2015. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 6.Schabath MB, Welsh EA, Fulp WJ, Chen L, Teer JK, Thompson ZJ, et al. Differential association of STK11 and TP53 with KRAS mutation-associated gene expression, proliferation and immune surveillance in lung adenocarcinoma. Oncogene. 2016;35(24):3209–16. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 7.Biton J, Mansuet-Lupo A, Pecuchet N, Alifano M, Ouakrim H, Arrondeau J, et al. TP53, STK11 and EGFR Mutations Predict Tumor Immune Profile and the Response to anti-PD-1 in Lung Adenocarcinoma. Clin Cancer Res. 2018. [DOI] [PubMed] [Google Scholar]
- 8.Piton N, Lamy A, Guisier F, Marguet F, Sabourin JC. STK11 Mutations Are Associated With Lower PDL1 Expression in Lung Adenocarcinoma. Modern Pathol. 2018;31:746-. [Google Scholar]
- 9.Skoulidis F, Goldberg ME, Greenawalt DM, Hellmann MD, Awad MM, Gainor JF, et al. STK11/LKB1 Mutations and PD-1 Inhibitor Resistance in KRAS-Mutant Lung Adenocarcinoma. Cancer Discov. 2018. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 10.Skoulidis F, Arbour KC, Hellmann MD, Patil PD, Marmarelis ME, Awad MM, et al. Association of STK11/LKB1 genomic alterations with lack of benefit from the addition of pembrolizumab to platinum doublet chemotherapy in non-squamous non-small cell lung cancer. Journal of Clinical Oncology. 2019;37(15_suppl):102-. [Google Scholar]
- 11.Aggarwal C, Thompson JC, Chien AL, Quinn KJ, Hwang WT, Black TA, et al. Baseline plasma tumor mutation burden predicts response to pembrolizumab-based therapy in patients with metastatic non-small cell lung cancer. Clin Cancer Res. 2020. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 12.Udd L, Makela TP. LKB1 signaling in advancing cell differentiation. Fam Cancer. 2011;10(3):425–35. [DOI] [PubMed] [Google Scholar]
- 13.Zhang H, Fillmore Brainson C, Koyama S, Redig AJ, Chen T, Li S, et al. Lkb1 inactivation drives lung cancer lineage switching governed by Polycomb Repressive Complex 2. Nat Commun. 2017;8:14922. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 14.Mollaoglu G, Jones A, Wait SJ, Mukhopadhyay A, Jeong S, Arya R, et al. The Lineage-Defining Transcription Factors SOX2 and NKX2-1 Determine Lung Cancer Cell Fate and Shape the Tumor Immune Microenvironment. Immunity. 2018;49(4):764–79 e9. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 15.Beers MF, Moodley Y. When Is an Alveolar Type 2 Cell an Alveolar Type 2 Cell? A Conundrum for Lung Stem Cell Biology and Regenerative Medicine. Am J Respir Cell Mol Biol. 2017;57(1):18–27. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 16.Mason RJ. Biology of alveolar type II cells. Respirology. 2006;11 Suppl:S12–5. [DOI] [PubMed] [Google Scholar]
- 17.Yamaguchi T, Hosono Y, Yanagisawa K, Takahashi T. NKX2-1/TTF-1: an enigmatic oncogene that functions as a double-edged sword for cancer cell survival and progression. Cancer Cell. 2013;23(6):718–23. [DOI] [PubMed] [Google Scholar]
- 18.Klein Wolterink RGJ, Pirzgalska RM, Veiga-Fernandes H. Neuroendocrine Cells Take Your Breath Away. Immunity. 2018;49(1):9–11. [DOI] [PubMed] [Google Scholar]
- 19.Branchfield K, Nantie L, Verheyden JM, Sui P, Wienhold MD, Sun X. Pulmonary neuroendocrine cells function as airway sensors to control lung immune response. Science. 2016;351(6274):707–10. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 20.Bordon Y Mucosal immunology: Neuroendocrine cells regulate lung inflammation. Nat Rev Immunol. 2016;16(2):76–7. [DOI] [PubMed] [Google Scholar]
- 21.George J, Walter V, Peifer M, Alexandrov LB, Seidel D, Leenders F, et al. Integrative genomic profiling of large-cell neuroendocrine carcinomas reveals distinct subtypes of high-grade neuroendocrine lung tumors. Nat Commun. 2018;9(1):1048. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 22.Augustyn A, Borromeo M, Wang T, Fujimoto J, Shao C, Dospoy PD, et al. ASCL1 is a lineage oncogene providing therapeutic targets for high-grade neuroendocrine lung cancers. Proc Natl Acad Sci U S A. 2014;111(41):14788–93. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 23.Kobayashi Y, Tata PR. Pulmonary Neuroendocrine Cells: Sensors and Sentinels of the Lung. Dev Cell. 2018;45(4):425–6. [DOI] [PubMed] [Google Scholar]
- 24.Barretina J, Caponigro G, Stransky N, Venkatesan K, Margolin AA, Kim S, et al. The Cancer Cell Line Encyclopedia enables predictive modelling of anticancer drug sensitivity. Nature. 2012;483(7391):603–7. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 25.Carretero J, Shimamura T, Rikova K, Jackson AL, Wilkerson MD, Borgman CL, et al. Integrative genomic and proteomic analyses identify targets for Lkb1-deficient metastatic lung tumors. Cancer Cell. 2010;17(6):547–59. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 26.Gao H, Korn JM, Ferretti S, Monahan JE, Wang Y, Singh M, et al. High-throughput screening using patient-derived tumor xenografts to predict clinical trial drug response. Nat Med. 2015;21(11):1318–25. [DOI] [PubMed] [Google Scholar]
- 27.Chen L, Kurtyka CA, Welsh EA, Rivera JI, Engel BE, Munoz-Antonia T, et al. Early2 factor (E2F) deregulation is a prognostic and predictive biomarker in lung adenocarcinoma. Oncotarget. 2016;7(50):82254–65. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 28.Heberle H, Meirelles GV, da Silva FR, Telles GP, Minghim R. InteractiVenn: a web-based tool for the analysis of sets through Venn diagrams. BMC Bioinformatics. 2015;16:169. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 29.TCGA. Comprehensive molecular profiling of lung adenocarcinoma. Nature. 2014;511(7511):543–50. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 30.Chen Z, Li JL, Lin S, Cao C, Gimbrone NT, Yang R, et al. cAMP/CREB-regulated LINC00473 marks LKB1-inactivated lung cancer and mediates tumor growth. The Journal of clinical investigation. 2016;126(6):2267–79. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 31.Bhinge K, Yang L, Terra S, Nasir A, Muppa P, Aubry MC, et al. EGFR mediates activation of RET in lung adenocarcinoma with neuroendocrine differentiation characterized by ASCL1 expression. Oncotarget. 2017;8(16):27155–65. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 32.Kosari F, Ida CM, Aubry MC, Yang L, Kovtun IV, Klein JL, et al. ASCL1 and RET expression defines a clinically relevant subgroup of lung adenocarcinoma characterized by neuroendocrine differentiation. Oncogene. 2014;33(29):3776–83. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 33.Pecuchet N, Laurent-Puig P, Mansuet-Lupo A, Legras A, Alifano M, Pallier K, et al. Different prognostic impact of STK11 mutations in non-squamous non-small-cell lung cancer. Oncotarget. 2017;8(14):23831–40. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 34.Biton J, Mansuet-Lupo A, Pecuchet N, Alifano M, Ouakrim H, Arrondeau J, et al. TP53, STK11, and EGFR Mutations Predict Tumor Immune Profile and the Response to Anti-PD-1 in Lung Adenocarcinoma. Clin Cancer Res. 2018;24(22):5710–23. [DOI] [PubMed] [Google Scholar]
- 35.Wilkerson MD, Yin X, Walter V, Zhao N, Cabanski CR, Hayward MC, et al. Differential pathogenesis of lung adenocarcinoma subtypes involving sequence mutations, copy number, chromosomal instability, and methylation. PLoS ONE. 2012;7(5):e36530. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 36.Ke H, Kazi JU, Zhao H, Sun J. Germline mutations of KIT in gastrointestinal stromal tumor (GIST) and mastocytosis. Cell Biosci. 2016;6:55. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 37.Snyder EL, Watanabe H, Magendantz M, Hoersch S, Chen TA, Wang DG, et al. NKX2-1 represses a latent gastric differentiation program in lung adenocarcinoma. Mol Cell. 2013;50(2):185–99. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 38.Deutsch L, Wrage M, Koops S, Glatzel M, Uzunoglu FG, Kutup A, et al. Opposite roles of FOXA1 and NKX2-1 in lung cancer progression. Genes Chromosomes Cancer. 2012;51(6):618–29. [DOI] [PubMed] [Google Scholar]
- 39.Wade KC, Guttentag SH, Gonzales LW, Maschhoff KL, Gonzales J, Kolla V, et al. Gene induction during differentiation of human pulmonary type II cells in vitro. Am J Respir Cell Mol Biol. 2006;34(6):727–37. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 40.Maeda Y, Tsuchiya T, Hao H, Tompkins DH, Xu Y, Mucenski ML, et al. Kras(G12D) and NKX2-1 haploinsufficiency induce mucinous adenocarcinoma of the lung. The Journal of clinical investigation. 2012;122(12):4388–400. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 41.Koyama S, Akbay EA, Li YY, Aref AR, Skoulidis F, Herter-Sprie GS, et al. STK11/LKB1 deficiency promotes neutrophil recruitment and proinflammatory cytokine production to suppress T cell activity in the lung tumor microenvironment. Cancer Res. 2016. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 42.Tsai LH, Chen PM, Cheng YW, Chen CY, Sheu GT, Wu TC, et al. LKB1 loss by alteration of the NKX2-1/p53 pathway promotes tumor malignancy and predicts poor survival and relapse in lung adenocarcinomas. Oncogene. 2014;33(29):3851–60. [DOI] [PubMed] [Google Scholar]
- 43.Chen PM, Wu TC, Cheng YW, Chen CY, Lee H. NKX2-1-mediated p53 expression modulates lung adenocarcinoma progression via modulating IKKbeta/NF-kappaB activation. Oncotarget. 2015;6(16):14274–89. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 44.Rekhtman N, Pietanza MC, Hellmann MD, Naidoo J, Arora A, Won H, et al. Next-Generation Sequencing of Pulmonary Large Cell Neuroendocrine Carcinoma Reveals Small Cell Carcinoma-like and Non-Small Cell Carcinoma-like Subsets. Clin Cancer Res. 2016;22(14):3618–29. [DOI] [PMC free article] [PubMed] [Google Scholar]
Associated Data
This section collects any data citations, data availability statements, or supplementary materials included in this article.