

Abstract
Lysosomes coordinate cellular metabolism and growth upon sensing of essential nutrients, including cholesterol. Through bioinformatic analysis of lysosomal proteomes, we identified LYsosomal CHOlesterol Signaling (LYCHOS, previously annotated as G-protein coupled receptor 155), a multidomain transmembrane protein that enables cholesterol-dependent activation of the master growth regulator, the protein kinase mechanistic Target of Rapamycin Complex 1 (mTORC1). Cholesterol bound to the N-terminal permease-like region of LYCHOS, and mutating this site impaired mTORC1 activation. At high cholesterol concentrations, LYCHOS bound to the GATOR1 complex, a GTPase-activating protein for the Rag guanosine triphosphatases, through a conserved cytoplasm-facing loop. By sequestering GATOR1, LYCHOS promotes cholesterol- and Rag-dependent recruitment of mTORC1 to lysosomes. Thus, LYCHOS functions in a lysosomal pathway for cholesterol sensing, and couples cholesterol concentrations to mTORC1-dependent anabolic signaling.
One Sentence Summary: LYCHOS senses cholesterol to activate mTORC1
Cholesterol, an essential building block for membrane biogenesis, is also a signaling molecule that regulates embryonic development and numerous physiological processes by acting either as a ligand or as a precursor for oxysterols and steroid hormones. Aberrant amounts and activity of cholesterol are associated with pathological conditions such as obesity, atherosclerosis, infertility, and cancer, making its accurate sensing essential (1, 2). For example, dedicated machinery on the endoplasmic reticulum senses local cholesterol concentrations and, in response, fine-tunes the rate of cholesterol synthesis and uptake (3, 4).
In actively proliferating cells, including cancer cells, the phosphatidylinositol 3-kinase (PI3K)-AKT-mechanistic Target of Rapamycin Complex 1 (mTORC1) pathway stimulates both de novo cholesterol synthesis and uptake to meet the increased demand for both cholesterol itself and its growth-promoting biosynthetic intermediates (5–7). However, where in the cell and how cholesterol abundance is signaled to growth-regulating pathways is not well understood.
The lysosome is a key nutrient-sensing center for the cell (6). At the lysosome, the master growth regulator protein kinase, mTORC1, integrates many environmental signals, including nutrients, growth factors, energy, oxygen and stress and, in response, triggers downstream anabolic programs that increase cell mass (8). When intracellular concentrations of nutrients, including cholesterol, are increased, mTORC1 localizes to the lysosomal membrane, where it contacts the small guanosine triphosphatase (GTPase) Rheb, which activates mTORC1 kinase function (8–10). Conversely, when nutrient abundance is low, mTORC1 relocalizes to the cytosol, where it remains inactive until nutrient concentrations are restored.
Multiple proteins control the nutrient-dependent localization of mTORC1 to the lysosome. The Rag GTPases are heterodimers composed of either RagA or RagB in complex with either RagC or RagD (11, 12). A key event when nutrients are plentiful is the loading of RagA or B with GTP, which enables it to physically bind to mTORC1 and anchor it to the lysosomal membrane (8, 13, 14). In turn, the nucleotide state of RagA or B is controlled by other lysosome-associated proteins. The GATOR1 complex is a dedicated GTPase activating protein (GAP) for RagA. When nutrient concentrations are low, GATOR1 triggers GTP hydrolysis on RagA, thereby promoting the inactive state of the Rag GTPase complex that leads to cytoplasmic relocalization and inactivation of mTORC1 (15, 16). This negative regulatory activity of GATOR1 is countered by the amino acids leucine, arginine, and methionine, through dedicated sensors that inhibit the GAP activity of GATOR1 either directly or via a second complex known as GATOR2 (8). Whether cholesterol regulates the GAP activity of GATOR1 toward RagA is unknown but, consistent with this possibility, genetic inactivation of GATOR1 renders mTORC1 constitutively active even if cholesterol concentrations are low (17).
In cells and in vitro, cholesterol on the lysosomal limiting membrane directly participates in the recruitment and activation of mTORC1. This cholesterol pool is highly regulated: the sterol carrier, oxysterol binding protein (OSBP), localizes at ER-lysosome membrane contact sites, where it transfers cholesterol from the ER to the lysosome to enable mTORC1 activation (1, 6, 17). Conversely, the cholesterol transporter Niemann-Pick C1 (NPC1) promotes export of cholesterol from the lysosomal surface, thereby inhibiting mTORC1 signaling (10, 17).
How cholesterol interacts with the mTORC1-scaffolding machinery is not well understood. Various cholesterol pools co-exist at the lysosomal membrane, one derived from low-density lipoprotein (LDL), another deposited across membrane contact sites (17). Moreover, the concentration of cholesterol likely varies across different regions of the lysosomal membrane and as a function of metabolic states (18). Thus, like sensing of amino acids, cholesterol sensing may rely on multiple cholesterol-sensing factors with distinct localization, affinity for the sterol ligand and upstream regulatory mechanisms. One important player is the lysosomal transmembrane protein, SLC38A9 (19, 20), which participates in cholesterol-dependent activation of mTORC1 through conserved sterol-interacting motifs within its transmembrane domains (10). However, SLC38A9 primarily relays arginine abundance to mTORC1, whereas a dedicated sensor for cholesterol remains to be identified.
More generally, it is likely that the lysosome has as yet undiscovered nutrient sensors that could regulate cellular metabolism through mTORC1-dependent or independent pathways. Identifying putative nutrient sensors can be challenging, because they generally have weak interactions with their cognate metabolites and have diverse domain composition and topologies (21). Building on recent advances in immunoisolation and proteomic profiling of lysosomes combined with a robust bioinformatic pipeline that identifies and prioritizes putative signaling proteins, we identified GPR155, which we rename Lysosomal Cholesterol Sensing (LYCHOS) protein, as a candidate lysosomal cholesterol sensor that controls signaling functions of this organelle.
LYCHOS is required for cholesterol-mediated mTORC1 activation
To identify candidate nutrient-sensing factors that reside at the lysosome, we devised a bioinformatic method to analyze a 611-protein ‘master list’ that combines published and unpublished lysosomal proteomic datasets (17, 22–24) (Fig. 1A and S1A and Table S1). Gene ontology (GO) analysis of this list showed the expected enrichment of biological processes associated with the lysosome (Fig. S1B).
Fig. 1. Lysosomal transmembrane protein LYCHOS is required for cholesterol-mediated mTORC1 activation.
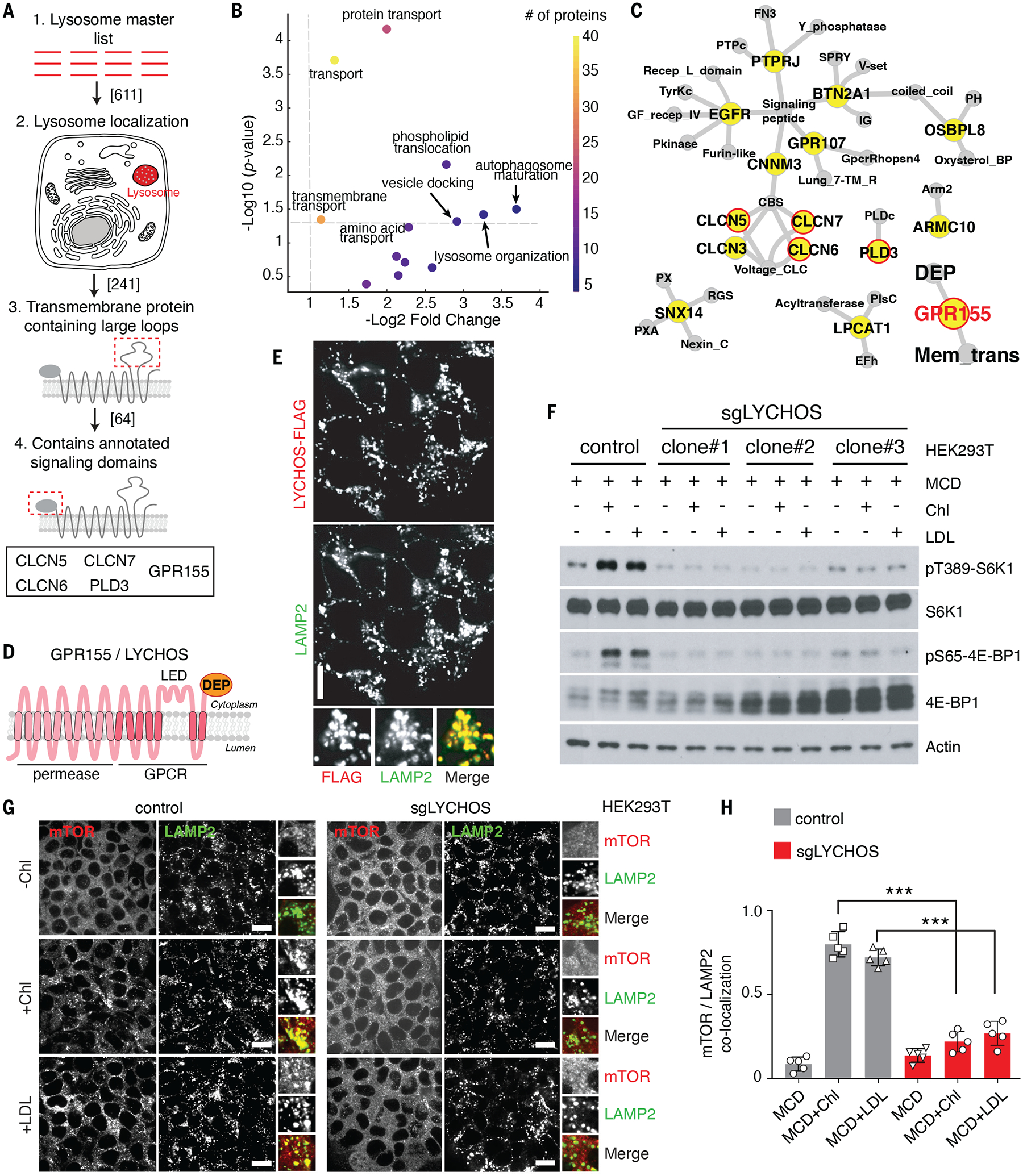
(A) Summary chart of the workflow for the identification of lysosomal transmembrane signaling proteins..
(B) Volcano plots of ‘biological process’ GO terms enriched in lysosome-resident transmembrane proteins relative to all transmembrane proteins.
(C) Network representation of lysosome-associated transmembrane proteins with large loops (blue). Gray nodes show annotated signaling domains. Predicted lysosome-resident transmembrane proteins are circled in red.
(D) Schematic of the predicted GPR155/LYCHOS topology and domain organization.
(E) LYCHOS is a lysosomal protein. HEK293T cells stably expressing LYCHOS-FLAG was fixed and stained with antibodies targeting FLAG and LAMP2. Scale bar, 10 μm.
(F) LYCHOS is required for mTORC1 activation by cholesterol. Control HEK293T cells or LYCHOS-deleted cells (sgLYCHOS) were depleted of sterols using methyl-β-cyclodextrin (MCD, 0.75% w/v) for 2 hours, followed by re-feeding for 2 hours with 50 μM cholesterol (chl) in complex with 0.1% MCD or with 50 μg/ml LDL. Cell lysates were blotted with the indicated antibodies.
(G) LYCHOS is required for cholesterol-dependent mTORC1 recruitment to lysosomes. LYCHOS-deleted HEK293T cells were subject to cholesterol depletion and restimulation, followed by immunofluorescence of endogenous mTOR and LAMP2. Representative images are shown. Scale bars, 10 μm.
(H) Quantification of co-localization of mTOR with LAMP2-positive lysosomes in the indicated genotypes and conditions. Data are mean ±s.d. Statistical analysis was performed using ANOVA with Dunnett’s multiple comparison test; ***p<0.001
From this we selected proteins that satisfied all the following criteria: i) transmembrane topology, commonly found in metabolite receptors and ‘transceptors’ (21) ii) presence of relatively large loops that could facilitate interaction with cytoplasmic effectors iii) presence of structural domains and architecture associated with signal transduction iv) absence of reported localization to other membrane compartments, which may indicate pleiotropic roles (Fig. 1A). Consistent with the role of the lysosome in releasing the products of macromolecular breakdown to the cytosol, the 127 lysosome-specific transmembrane proteins were highly enriched for metabolite transport and translocation compared to the 5208 transmembrane proteins annotated in UniProt (Fig. 1B, S1C and S1D).
Five transmembrane proteins satisfied all of the above criteria: chloride voltage-gated channel (CLCN) 5, 6 and 7, phospholipase D family member 3 (PLD3) and G-protein couple receptor 155 (GPR155) (Fig. 1C). Of these, GPR155 (also known as DEP domain containing 3) stood out due to its topological features, consisting of a 10 transmembrane (TM) domain N-terminal portion with similarity to solute carriers (SLCs), a 7-TM central portion with similarity to class B GPCRs (25), which contains a large (118 aa) insertion between TM helices 15 and 16, and a C-terminal region containing a Dishevelled, Egl-10, and Pleckstrin (DEP) domain. Based on UniProt and Alphafold predictions, we predict a 17-TM topology for GPR155 (Fig. 1D and Fig. S1E).
We confirmed the lysosomal localization of GPR155 by immunoblotting of immunoprecipitated lysosomal samples with an antibody to the endogenous protein (Fig. S2A). Moreover, double immunofluorescence of endogenous LAMP2 and FLAG-labeled GPR155, stably expressed in human embryonic kidney (HEK)-293T cells, showed that GPR155 specifically localized to lysosomes, with no detectable signal in vesicular structures that lack LAMP2, in the Golgi, or in the plasma membrane (Fig. 1E and S2, B and C). Through immunostaining with antibodies directed against the TM15–16 loop and the DEP domain under semi-permeabilized conditions, we experimentally verified that both domains face the cytoplasm, not the lysosomal lumen (Fig. S2, D and E). For reasons described below, we hereafter refer to GPR155 as LYsosomal CHOlesterol Signaling protein (LYCHOS), and its TM15–16 loop as the LYCHOS Effector Domain (LED).
Because the domain architecture of LYCHOS is consistent with a signaling function, we tested whether LYCHOS regulated mTORC1 activation. Depletion of LYCHOS by CRISPR-Cas9 or shRNA in HEK-293T cells decreased mTORC1 signaling under full nutrient conditions, as shown by loss of phosphorylation of canonical substrates p70 S6-kinase 1 (S6K1) and 4E-Binding Protein 1 (4E-BP1) (11, 12) (Fig. S3, A and B). Conversely, transient overexpression of LYCHOS boosted phosphorylation of mTORC1 substrates in a dose-dependent manner (Fig. S3C).
Consistent with decreased mTORC1 signaling, LYCHOS depletion resulted in significant suppression of cell proliferation, which was rescued by reconstituting LYCHOS-deleted cells with LYCHOS WT (Fig. S3, D and E). Moreover, RNAseq analysis of LYCHOS-depleted cells showed a decreased expression of several genes involved in glycolysis, the pentose phosphate pathway, and lipid biosynthesis, a gene signature similar to that observed upon pharmacological mTORC1 inhibition (26) (Fig. S3, F through H). These gene expression changes were completely reversed by re-expressing an shRNA-resistant isoform of LYCHOS (Fig. S3H).
A bioinformatic query of the 611-gene lysosomal master list in the NIH GEO Profiles database identified LYCHOS as one of 8 lysosomal genes that have decreased expression upon fasting in liver, muscle and adipose tissue of mice (Fig. S4, A and B and Table S2). We confirmed these in silico results by quantitative PCR (qPCR) from livers of mice subjected to 6-h and 24-h fasting, which showed a time-dependent decrease in Lychos expression (Fig. S4C). Conversely Npc1, a negative regulator of cholesterol-dependent mTORC1 activation (10, 17), was one of 14 lysosomal genes with increased expression in fasting animals (Fig. S4D).
We tested whether LYCHOS enables mTORC1 activation by specific nutrient stimuli. In LYCHOS knockout (KO) cells, mTORC1 activation by acute starvation-refeeding with amino acids (Fig. S5A) or glucose (Fig. S5B) were unperturbed. In contrast, LYCHOS was required for mTORC1 stimulation by cholesterol, delivered to cells in complex with methyl-beta cyclodextrin (MCD), or in LDL particles, following its depletion with MCD plus the cholesterol synthesis inhibitor, mevastatin (10, 17) (Fig. 1F and Fig. S5, C and D).
Like amino acids and glucose, cholesterol induces mTORC1 relocalization from the cytosol to the lysosomal membrane (8, 10, 17). In LYCHOS KO cells, mTORC1 failed to localize to lysosomes upon stimulation with either MCD:cholesterol or LDL (Fig. 1, G and H and Fig. S5, E and F). In contrast, LYCHOS deletion did not affect lysosomal localization of the Rag GTPases or their membrane anchor, the Ragulator complex. Thus, LYCHOS appears to be required for cholesterol-dependent activation of the mTORC1 lysosomal scaffolding complex, not for its physical integrity (Fig. S5, G through J).
LYCHOS binds to cholesterol through its N-terminal domain.
The presence of both an N-terminal permease-like domain and two signaling modules (GPCR-like and DEP) indicates that LYCHOS may be either a cholesterol transporter or an effector. Lipidomic analysis of immuno-isolated lysosomes from control cells and cells lacking LYCHOS did not reveal a significant difference in total lysosomal cholesterol content following loss of LYCHOS (Fig. S6, A and B).
We also ablated LYCHOS in cells lacking NPC1, loss of which causes cholesterol to accumulate both within the lysosomal lumen and on the limiting membrane (17, 27, 28). The two pools were visualized, respectively, with filipin and with mCherry-D4H, a recombinant, fluorescently-tagged cholesterol probe based on the fourth domain of Clostridium perfringens theta-toxin (17, 29, 30). Depleting LYCHOS in cells lacking NPC1 did not reveal significant changes in the filipin or mCherry-D4H signal, whereas inactivating OSBP, which transfers cholesterol from the ER to the lysosomal limiting membrane, ablated mCherry-D4H but not filipin staining in cells lacking NPC1 (Fig. S6, C through E).
Loss of NPC1, and the resulting cholesterol accumulation on the lysosomal membrane renders mTORC1 constitutively and aberrantly active (10, 17, 31). Depleting LYCHOS from cells lacking NPC1 completely suppressed constitutive mTORC1 signaling (Fig. S6F). Thus, LYCHOS does not control the concentrations of lysosomal cholesterol, but is required for lysosomal cholesterol to activate mTORC1.
To determine whether LYCHOS directly binds to cholesterol, we expressed LYCHOS recombinantly, verified its purity (Fig. S7, A and B) and incubated it with increasing concentrations of [3H]-cholesterol under detergent conditions that preserve protein stability (Fig. S7C). LYCHOS exhibited saturable binding to [3H]-cholesterol with an apparent Kd between 100 and 200 nM, which was competed by unlabeled cholesterol but not by its 3-OH epimer, epicholesterol (Fig. 2, A and B and Table S3). Cholesterol appeared to bind to LYCHOS in a stereo-specific manner, because binding of [3H]-cholesterol was competed by 25- and 19-hydroxycholesterol but not by 4β-hydroxycholesterol, indicating that binding is sensitive to modifications in specific positions of the cholesterol structure (Fig. 2C and Table S3).
Fig. 2. Cholesterol binding at the N-terminal region of LYCHOS is essential for mTORC1 activation.
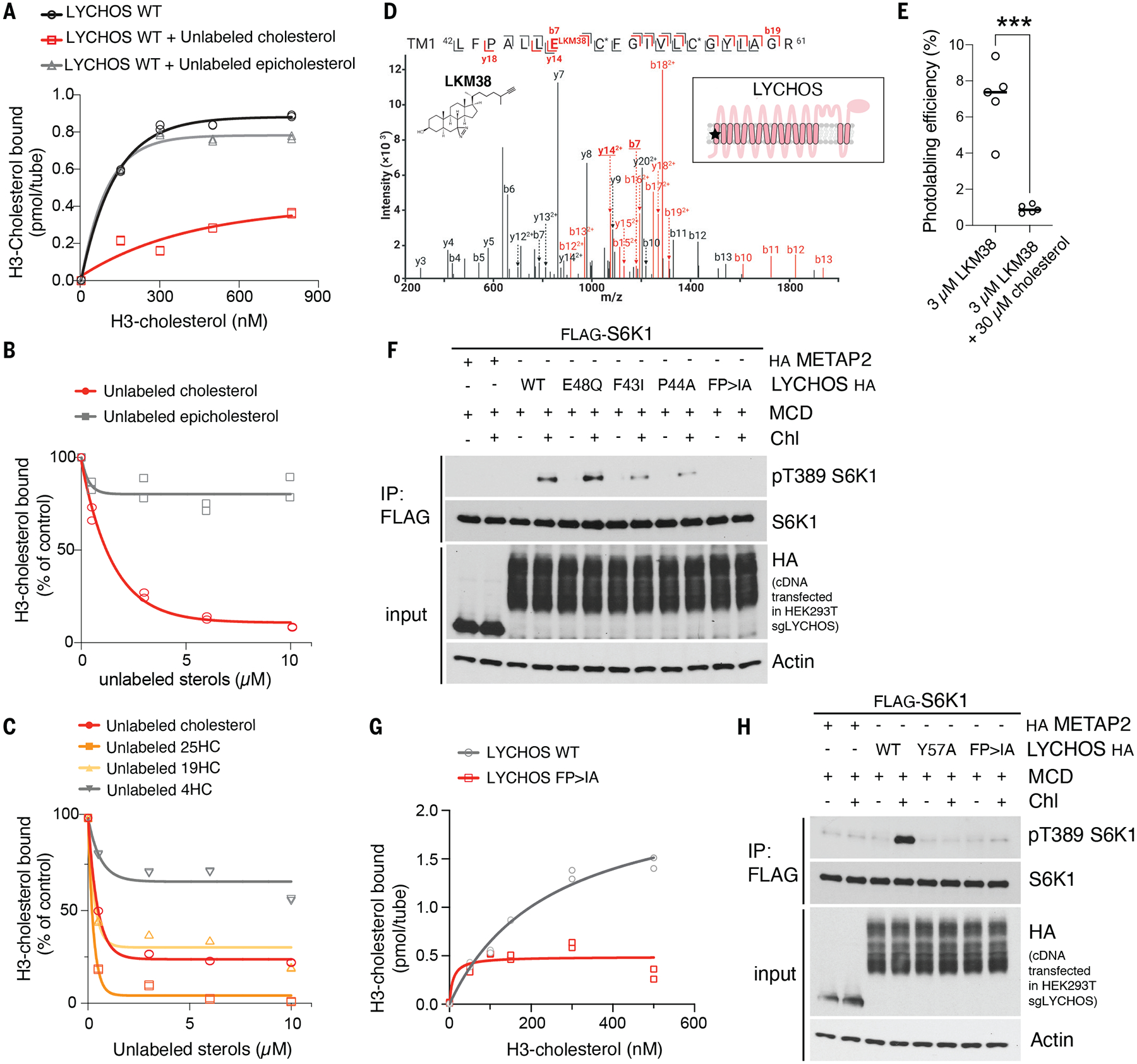
(A) [3H]-cholesterol binding to LYCHOS WT. 150ng of purified LYCHOS was incubated with indicated concentration of [3H]-cholesterol, in the presence or absence of 10μM cold cholesterol or epicholesterol. Bound [3H]-cholesterol was measured by scintillation counting. The assay was performed in duplicates and each data point is shown.
(B,C) Competitive binding of unlabeled sterols to LYCHOS. 150ng LYCHOS was incubated with 500nM [3H]-cholesterol along with increasing concentrations of the indicated unlabeled sterol. Bound [3H]-cholesterol was measured by scintillation counting. The assay was performed in duplicates and each data point is shown.
(D) CID product ion spectrum of the TM1 tryptic peptide photolabeled with 3μM LKM38. TM1 peptide (m/z=934.53, z=3) is photolabeled by LKM38 at E48. Red and black indicate product ions that do or do not contain LKM38 adduct, respectively. The C* indicates that the cysteine is alkylated by NEM. The inset schematic of GPR155 in the panel indicates the approximate location of the residues labeled by LKM38 (red star). The numerical data are included in Table S3. NEM, N-ethylmaleimide; TM, transmembrane helix.
(E) Photolabeling efficiency of recombinant LYCHOS by LKM38 in the absence or presence of excess unlabeled cholesterol. Data are mean of ±s.d of n=5. Statistical analysis was performed using student t-test; ***p<0.001
(F) LYCHOS TM1 is required for cholesterol-mediated mTORC1 activation. HEK293T/sgLYCHOS were transfected with FLAG-S6K1 along with HA-tagged METAP2 (neg. control), LYCHOS WT and TM1 mutants. Cells were cholesterol-starved, or starved and restimulated as indicated in the presence of 50μM mevalonate and mevastatin, followed by FLAG immunoprecipitation (IP) and immunoblotting for the phosphorylation state and levels of the indicated proteins
(G) [3H]-cholesterol binding to LYCHOS WT and LYCHOS FP>IA mutant. 150ng of purified LYCHOS were incubated with indicated concentration of [3H]-cholesterol, and bound radioactive cholesterol was measured by scintillation counting. The assay was performed in duplicates and each data point is shown. (H) LYCHOS TM1 mutants FP>IA and Y57A blunt cholesterol-mediated mTORC1 activation. HEK293T/ sgLYCHOS cells were transfected with FLAG-S6K1 along with HA-METAP2 (neg. control) or LYCHOS WT, Y57A or FP>IA-HA and analyzed as in (F).
To map the site(s) of cholesterol binding, we labeled recombinantly expressed LYCHOS in vitro with photo-crosslinkable cholesterol analogs that combine a UV-activated diazirine group to create a peptide-steroid adduct, and with an alkyne group for identification of the labeled site via copper-catalyzed cycloaddition and mass spectrometry (10, 32). Bulk photolabeling of LYCHOS was competed by excess cholesterol or cholesterol hemisuccinate, indicative of a specific binding reaction (Fig. S8, A and B). Mass spectrometry profiling of peptide adducts with the analog LKM38, which bears the photoreactive diazirine group in ring 2, identified two sites of labeling, one corresponding to transmembrane helix 1 (TM1) in the permease-like N-terminal region (42LFPALLECFGIVLCGYIAGR61), the second in the DEP domain (809 LVQGGVIQHITNEYEFRDEYLFYR832) (Fig. 2D, S8C and Table S4 and S5). Both labeling sites were competed by excess free cholesterol (Fig. 2E and Fig. S8, D and E). Moreover, the TM1 site was independently labeled by a second cholesterol analog, KK231, which bears the photoreactive diazirine group in the aliphatic tail (Fig. S8F and Table S6).
To determine whether the TM1 or DEP represent a cholesterol-binding regulatory site for mTORC1 activation, we reconstituted LYCHOS-deleted cells with isoforms lacking the TM1-containing permease-like domain, the DEP domain, or the LED. All three modified proteins were expressed in amounts comparable to those of the the full-length protein and localized to the lysosome (Fig. S9, A through C). However, whereas deleting the DEP had no effect on cholesterol-dependent mTORC1 activation, removing the permease-like domain completely abolished it (Fig. S9C). Removing the LED also ablated cholesterol-dependent mTORC1 activation; however, recombinantly expressed LED, held in a loop configuration by a leucine zipper, did not show appreciable binding to [3H]-cholesterol (Fig. S9D). Thus, we further pursued TM1 as a cholesterol-dependent regulatory site.
Aromatic amino acids are often found in cholesterol-binding pockets (28, 33, 34). The TM1 domain has two conserved aromatic residues, Phe43 and Tyr57, located near the sites of adduct formation by LKM38 and KK231 (Glu48 and Cys55, respectively) (Fig. 2D and Fig.S8F). In LYCHOS-deleted cells reconstituted with a Phe43>Ile mutant, cholesterol-dependent mTORC1 activation was blunted (Fig. 2F). Nearby Pro44 may help establish the conformation of the cholesterol-binding pocket. Consistent with this possibility, binding of [3H]-cholesterol to the F43I-P44A double mutant LYCHOS was nearly abolished, and this double mutant had a stronger mTORC1 activation defect than either the F31I or P44A single mutants (Fig. 2, F and G). In contrast, mutating Glu48 to Gln had no effect on mTORC1 signaling, indicating that this residue, which is labeled by the photoreactive diazirine of LKM38, is not involved in binding to native cholesterol (Fig. 2F). Finally, mutating the second aromatic residue, Tyr57 to Ala also blunted cholesterol-dependent mTORC1 activation (Fig. 2H). All of these mutants localized correctly to lysosomes and were expressed at near-identical amounts (Fig. 2F and H and Fig. S9E).
LYCHOS promotes mTORC1 signaling via cholesterol-regulated interaction with GATOR1.
To delineate the mechanisms by which LYCHOS communicates cholesterol abundance to mTORC1, we used proximity biotinylation coupled with proteomics (35). We C-terminally fused LYCHOS to the TurboID biotin ligase (LYCHOS-TiD, which correctly localized to lysosomes, Fig. S10A), along with NPC1-TiD as a control.
Although our lysosomal ‘master list’ includes several trimeric G-proteins known to transduce signals downstream of canonical GPCRs (Table S1), none scored as LYCHOS interactors in TiD experiments (Table S7) or by co-immunoprecipitation (Fig. S10, B and C). Instead, LYCHOS-TiD specifically biotinylated the GATOR1 subunits NPRL2, NPRL3 and DEPDC5, and the SZT2 subunit of the GATOR1-associated KPTN, ITFG2, C12orf66, and SZT2-containing regulator of mTORC1 (KICSTOR) complex (36) (Fig. 3, A and B and Table S7). Confirming the proximity biotinylation results, endogenous LYCHOS, tagged with a triple FLAG epitope by CRISPR-Cas9, bound to endogenous NPRL2 and NPRL3 in pull-down experiments, albeit to a smaller extent than DEPDC5 (also endogenously tagged with FLAG) (Fig. S10D). No binding of LYCHOS to the KICSTOR subunit KPTN was observed, indicating that LYCHOS primarily interacts with GATOR1, not KICSTOR (Fig. S10D).
Fig. 3. LYCHOS promotes mTORC1 signaling via cholesterol-regulated interaction with GATOR1.
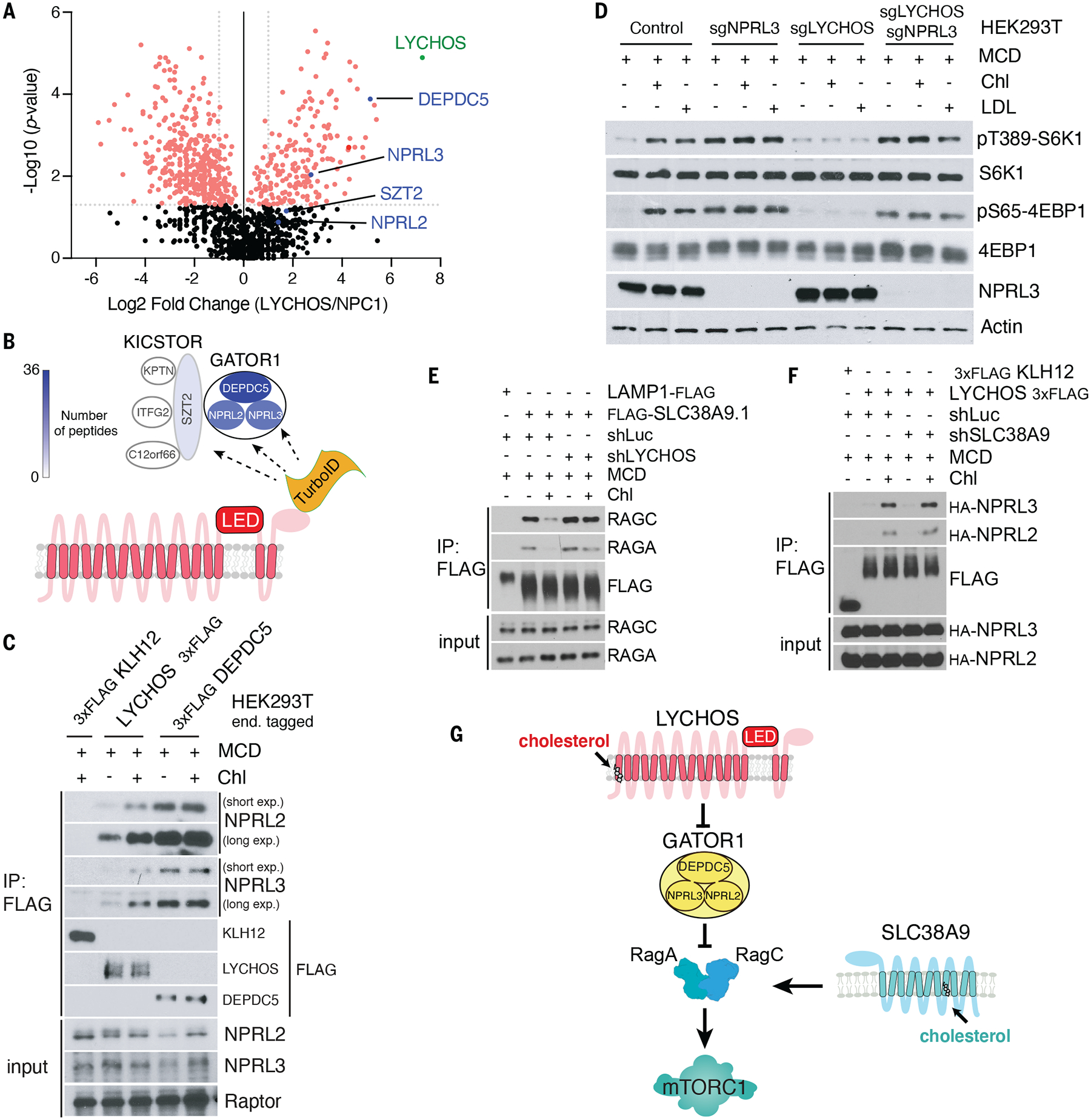
(A) TurboID-based proximity labeling combined with LC-MS/MS identified GATOR1 complex components (DEPDC5, NPRL2, and NPRL3) as interactors of LYCHOS. Volcano plot of the ratio of LYCHOS-FLAG-TurboID (LYCHOS) to NPC1-FLAG-TurboID (NPC1) is shown. Proteins with statistically significant (p value≥0.05, two-tailed unpaired t test) with fold change LYCHOS/NPC1≥2 are displayed as red circles.
(B) Cartoon summarizing the TurboID proteomic analysis in (A). GATOR1 and KICSTOR subunits are color-coded according to their peptide counts.
(C) Cholesterol strengthens the LYCHOS-GATOR1 interaction. HEK293T cells bearing endogenously 3xFLAG-tagged KLH12, LYCHOS and DEPDC5 were depleted of sterols then re-fed with 50 μM cholesterol, followed by FLAG immunoprecipitation and immunoblotting for the indicated proteins.
(D) LYCHOS regulates cholesterol-dependent mTORC1 signaling via GATOR1. HEK293T cells lacking GATOR1 (sgNPRL3), LYCHOS (sgLYCHOS) or both (sgNPRL3/sgLYCHOS) were cholesterol-starved, or starved and refed with 50 μM cholesterol or 50 μg/ml LDL, followed by immunoblotting for the indicated proteins.
(E) LYCHOS functions at upstream of RagA GTP loading. HEK293T cells stably expressing LAMP1-FLAG or FLAG-SLC38A9.1 were infected with shRNA targeting luciferase or LYCHOS for FLAG immunoprecipitation to assess SLC38A9.1-RAG A/C interaction.
(F) SLC38A9.1 functions at downstream of RagA GTP loading. SLC38A9.1 was knockdown in HEK293T cells endogenously 3xFLAG-tagged KLH12 and LYCHOS cells, followed by FLAG immunoprecipitation and immunoblotting for the indicated proteins.
(G)) LYCHOS and SLC38A9 mediate distinct cholesterol-sensing pathways, converging on mTORC1.
We tested whether cholesterol controls the LYCHOS-GATOR1 interaction. In co-immunoprecipitation (co-IP) experiments, LYCHOS-FLAG bound more GATOR1 subunits when isolated from cells treated with cholesterol, than from cholesterol-depleted cells (Fig. 3C). In contrast, cholesterol did not affect the interaction of DEPDC5 with its partner GATOR1 subunits (Fig. 3C). Also, amino acids had no effect on the LYCHOS-GATOR1 interaction (Fig. S10E).
Stimulation of LYCHOS-GATOR1 interaction by cholesterol was dose-dependent, with an EC50 of 31.5uM that matched that of cholesterol-induced activation of mTORC1 signaling (Fig. S11, A and B). Based on measurements of lysosomal lipid content by lyso-IP and mass spectrometry, this value corresponded to a 33% molar ratio of cholesterol to phospholipids, although what fraction of this cholesterol is unbound by sphingomyelin and accessible by LYCHOS is unknown (37) (Fig. S11C).
These data suggest that LYCHOS promotes mTORC1 signaling by interacting with and inhibiting GATOR1. Consistent with this hypothesis, deleting the essential GATOR1 subunit NPRL3 resulted in constitutively active mTORC1 signaling regardless of cholesterol abundance or LYCHOS deletion (Fig. 3D). Further supporting LYCHOS action through the GATOR1-Rag GTPase axis, stable expression of the GTP-locked RagBQ99L mutant, which is refractory to GATOR1-mediated inhibition (15, 16), induced constitutive mTORC1 signaling that bypassed loss of LYCHOS (Fig. S11D).
SLC38A9 is also required for cholesterol-dependent activation of mTORC1 (10). However, SLC38A9-dependent and LYCHOS-dependent signaling are mechanistically distinct. SLC38A9 binds to the Rag GTPases and LAMTOR complex, but not to GATOR1, whereas LYCHOS binds to GATOR1 but not Rag GTPases or LAMTOR (Fig. S12, A and B). Treatment of cells with cholesterol weakens SLC38A9 interaction with the Rag GTPases, likely because RagA becomes GTP-loaded in cells with high cholesterol concentrations (10, 19, 38). However, in LYCHOS-deleted cells the SLC38A9-Rag GTPase interaction became unresponsive to cholesterol (Fig. 3E). On the contrary, cholesterol-dependent strengthening of the LYCHOS-GATOR1 interaction occurred irrespective of SLC38A9 status (Fig. 3F). Thus, through its interaction with GATOR1, LYCHOS functions at a step upstream of GTP loading of RagA, whereas SLC38A9 functions downstream of it (Fig. 3G). Whereas deleting the GATOR1 subunit NPRL3 fully rescued loss of LYCHOS, depleting SLC38A9 in NPRL3-deleted cells blunted cholesterol-dependent mTORC1 activation, further supporting the function of LYCHOS and SLC38A9 on distinct but converging pathways (Fig. S12C).
Cholesterol disrupts the GATOR1-KICSTOR interaction through the LYCHOS LED
Because of our previous results indicating that the LED does not bind to cholesterol but is nonetheless required for cholesterol-dependent mTORC1 activation, we tested whether the LED might function in binding to GATOR1. Consistent with this possibility, deleting the LED abolished cholesterol-dependent interaction between LYCHOS and the GATOR1 subunits NPRL2 and NPRL3 (Fig. S13A).
We sought to identify conserved amino acids within the LED that are essential for LYCHOS-GATOR1 interaction and mTORC1 activation. The LED contains a cluster of highly conserved Cys residues (Fig. 4A). Cys-rich motifs participate in signal transduction by promoting protein-protein as well as protein-lipid interactions (39). Mutating 4 of the conserved cysteines to alanine (C595>A, C604>A, C629>A, C638>A, collectively termed 4CA) abolished the ability of LYCHOS to support cholesterol-mediated mTORC1 activation, although the 4CA mutant of LYCHOS was expressed in amounts comparable to the wild-type protein and had normal lysosomal localization (Fig. 4B and S13B). We also tested Tyr551, which is conserved from human to zebrafish (Fig. 4A). Similar to the 4-cysteine cluster, Tyr551 also appeared to be essential for LYCHOS signaling activity, because a LYCHOSY551A mutant failed to restore cholesterol-dependent mTORC1 activation in LYCHOS-deleted cells (Fig. 4B).
Fig. 4. Cholesterol disrupts the GATOR1-KICSTOR interaction via the LYCHOS LED.
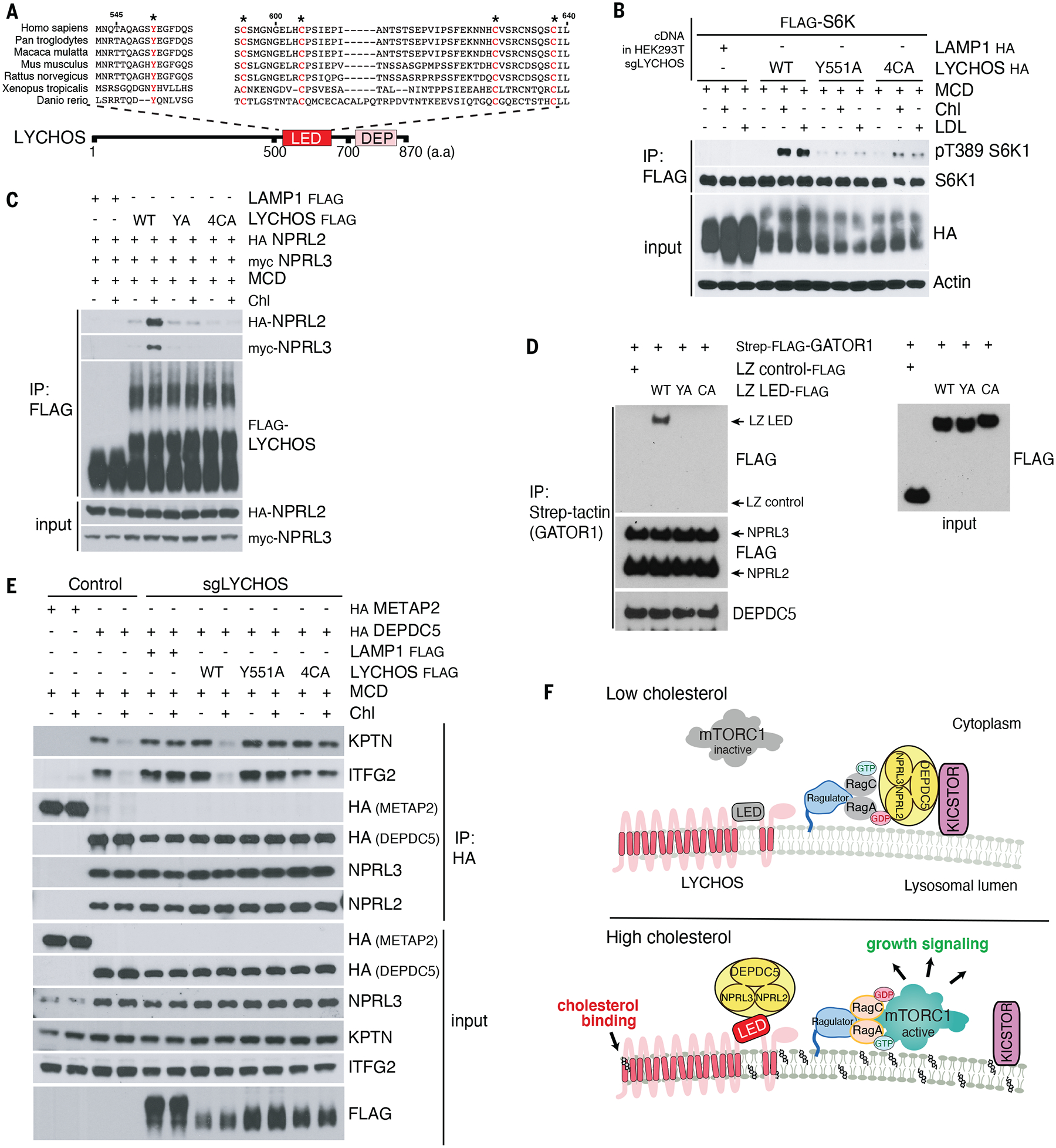
(A) Sequence alignment of LYCHOS LED domain. Highly conserved residues selected for mutagenesis are highlighted in red.
(B) HEK293T/ sgLYCHOS cells were transfected with FLAG-S6K1 along with LAMP1-HA (neg. control) or LYCHOS WT, Y551A or 4CA-HA, followed by FLAG immunoprecipitation and immunoblotting for the phosphorylation state and levels of the indicated proteins.
(C) LYCHOS LED mutations disrupt cholesterol-dependent LYCHOS interaction with GATOR1. HEK293T/sgLYCHOS cells were reconstituted with the indicated WT and LED-mutant LYCHOS constructs along with GATOR1. FLAG immunoprecipitates were analyzed by immunoblotting.
(D) LYCHOS LED is sufficient for GATOR1 interaction: in vitro binding assay between purified GATOR1 and recombinant, wild-type or mutant LYCHOS LED fused to a leucine zipper (LZ). LZ alone was used as negative control.
(E) In high cholesterol, LYCHOS disrupts the GATOR1-KICSTOR interaction via its LED. Control HEK293T cells, or HEK293T/sgLYCHOS cells reconstituted with the indicated WT and mutant FLAG-LYCHOS constructs, and co-expressing HA-METAP2 or HA-DEPDC5 as indicated, were cholesterol-starved, or starved and restimulated, followed by HA immunoprecipitation. Lysates were analyzed by immunoblotting
(F) Molecular mechanisms of LYCHOS-dependent regulation of cholesterol-mTORC1 signaling. Under low cholesterol, stable GATOR1-KICSTOR complex promotes GATOR1-dependent GTP hydrolysis of RagA/B, which maintains mTORC1 inactive in the cytosol. In high cholesterol, a conformational change in the LED stimulates LYCHOS interaction with GATOR1 while displacing KICSTOR, thus favoring the GTP-loaded state of RagA/B that leads to lysosomal recruitment and activation of mTORC1.
In contrast to wild-type LYCHOS, and consistent with their inability to support cholesterol-dependent mTORC1 activation, the Y551A and 4CA LED mutants showed barely detectable interaction with GATOR1 subunits NPRL2 and NPRL3, which was not strengthened by treating cells with cholesterol (Fig. 4C). Consistent with the LED as the GATOR1-interacting domain in LYCHOS, recombinantly expressed wild-type LED, held in a loop configuration with a leucine zipper, was sufficient for binding to GATOR1, whereas the interaction was abolished by the Y551A and 4CA mutations (Fig. 4D).
To gain mechanistic insight into how the cholesterol-regulated LYCHOS-GATOR1 interaction promotes mTORC1 signaling, we tested whether the LED can inhibit the RagA-GAP activity of GATOR1. However, neither recombinantly expressed LED, nor full-length LYCHOS affected GATOR1-dependent GTP-to-GDP conversion on RagA, when added in molar excess to GATOR1 in vitro (40) (Fig. S13, C through E). Based on lack of direct inhibition of GATOR1 GAP activity, and given that LYCHOS displayed a weak interaction with the KICSTOR subunit SZT2 in proximity biotinylation experiments, we considered the alternative possibility that LYCHOS may regulate the association between GATOR1 and the KICSTOR complex, which is essential for the GAP activity of GATOR1 toward RagA or B in cells (36).
Under full nutrient conditions, the interaction between the GATOR1 subunit DEPDC5 and the KICSTOR subunit, KPTN, was strengthened in LYCHOS-depleted cells compared to that in LYCHOS WT cells, suggesting that LYCHOS inhibits GATOR1-KICSTOR binding (Fig. S14, A and B). Cholesterol inhibited the GATOR1-KICSTOR interaction in a LYCHOS-dependent manner. In LYCHOS WT cells depleted of cholesterol, DEPDC5 interacted with the KICSTOR subunits, KPTN and ITFG2, whereas cholesterol refeeding inhibited this interaction (Fig. 4E). In contrast, in cells lacking LYCHOS, DEPDC5 and KICSTOR interacted strongly both in the absence and presence of cholesterol (Fig. 4E). Re-expression of wild-type LYCHOS restored cholesterol-dependent inhibition of GATOR1-KICSTOR binding, whereas both the 4CA and Y551A LED mutants (expressed at comparable or higher amounts than wild type) failed to do so (Fig. 4E). Consistent with a key role for the LED in regulating GATOR1-KICSTOR binding, recombinantly expressed wild-type LED was sufficient to disrupt a preformed GATOR1-KICSTOR complex in vitro in a dose-dependent manner. In contrast the Y551 and 4CA mutants, added in equal amounts to the wild-type, were largely ineffective (Fig. S14, C and D). Unlike cholesterol, neither amino acids nor glucose stimulation affected the strength of GATOR1-KICSTOR binding (Fig. S14E), further supporting a specific role of LYCHOS-dependent GATOR1-KICSTOR modulation for cholesterol sensing upstream of mTORC1.
Collectively, these results support a model in which, upon binding of cholesterol to the permease-like region of LYCHOS, the LED engages GATOR1, antagonizes its interaction with KICSTOR, and thus interferes with the ability of GATOR1 to act as a GAP for RagA/B. Conversely, when cholesterol concentrations are low LYCHOS is unable to perturb the KICSTOR-GATOR1 complex, favoring GATOR1-dependent GTP hydrolysis on RagA/B and leading to inhibition of mTORC1 signaling (Fig. 4F). Consistent with this model, deleting the essential KICSTOR subunit KPTN completely bypassed loss of LYCHOS, as did GATOR1 inactivation (Fig. S14F).
Discussion
We identified LYCHOS as a component of a lysosome-based pathway that transduces cholesterol levels into activation of mTORC1 signaling. Unlike SLC38A9, which also participates in regulation of mTORC1 by cholesterol (as well as arginine) (10, 19, 20), LYCHOS does not directly interact with the Rag GTPases but rather regulates their nucleotide state through cholesterol-dependent interaction with GATOR1 (Fig. 4F). Thus, LYCHOS functions in a manner analogous to amino acid sensors that modulate the GAP activity of GATOR1, either directly (e.g. SAMTOR) or via the GATOR2 complex (e.g. Sestrin, CASTOR) (8).
Despite its GPCR-like core, LYCHOS appears not to function like a classical GPCR. The specific localization of LYCHOS at the lysosomal membrane makes it unlikely that this protein senses extracellular ligands. Based on our mapping of a cholesterol-binding site to TM1 within the permease-like domain, we propose that cholesterol within the lysosomal membrane is in fact the main ligand for LYCHOS.
Cholesterol binding to TM1 may initiate a reorientation of specific helices within LYCHOS that enables the interaction of the LED with GATOR1. The LED is located in a position analogous to the intracellular loop 3 (ICL3) in canonical GPCRs, but is larger (~110 residues) compared to ICL3 in most GPCRs (25–50 residues) (41). Rather than binding to a trimeric G-protein as canonical ICL3s do, the LYCHOS LED appears to directly bind to GATOR1 and disrupt its interaction with KICSTOR, thereby inhibiting the RagA-GAP function of GATOR1.
The identification of a GPCR-like protein as a putative cholesterol sensor upstream of mTORC1 suggests intriguing parallels with the Hedgehog (Hh) pathway. In the Hh pathway, the Smoothened GPCR protein binds to cholesterol (and hydroxysterols) and, in response, triggers a signaling cascade that culminates in activation of transcriptional programs for differentiation and morphogenesis (42–47). The Hh pathway also includes Patched, a NPC1-related protein that moves cholesterol away from Smo to block its activation, a similar function to that of NPC1 in inhibiting cholesterol-dependent mTORC1 activation (10, 48–50).
Regulation of LYCHOS expression by fasting or feeding points to how cholesterol-mTORC1 signaling may be integrated with the metabolic state of the organism. Higher LYCHOS levels in fed states may promote cholesterol-dependent mTORC1 signaling when metabolic building blocks are abundant, a possibility consistent with the LYCHOS-dependent anabolic gene signature we identified (6, 26). Conversely, during starvation, simultaneous decreased expression of LYCHOS and increased expression of NPC1 may help shut down cholesterol-mTORC1 signaling, thus favoring conservation of cellular resources.
Supplementary Material
Acknowledgements
We thank R. Irannejad, J. Thorner and all the members of the Zoncu lab for critical reading of the manuscript. We thank M. Rape for providing endogenously tagged 3xFLAG KLH12 HEK293T cells, R. Irannejad for the Gi/Gs/Gq cDNA constructs, the Stroud lab for the anti-GFP nanobody Sepharose, and Taia Wu in the J.Gestwicki lab for assistance with the DSF experiment.
Funding:
This work was supported by NIH R01GM127763, a University of Notre Dame/APMRF grant and an Edward Mallinckrodt, Jr. Foundation Scholar Award to R.Z., a 2019 AACR-Amgen Fellowship in Clinical/Translational Cancer Research (19-40-11-SHIN) and a 2021 AFTD-Holloway postdoctoral fellowship (2021-002) to H.R.S., NIH R01MH110550, NIH R01HL067773 to D.F.C., NIH R01GM108799 to A.S.E., and 1P50MH122379 to D.F.C and A.S.E.
Footnotes
Publisher's Disclaimer: This manuscript has been accepted for publication in Science. This version has not undergone final editing. Please refer to the complete version of record at http://www.sciencemag.org/. The manuscript may not be reproduced or used in any manner that does not fall within the fair use provisions of the Copyright Act without the prior, written permission of AAAS
Competing interests: R.Z. is a co-founder, stockholder, and scientific advisor for Frontier Medicines Corp., and a scientific advisor for Nine Square Therapeutics and Apertor Pharmaceuticals. D.K. N. is a co-founder, shareholder, and adviser for Frontier Medicines and Vicinitas Therapeutics. D.K.N. is also on the scientific advisory board of The Mark Foundation for Cancer Research, Photys Therapeutics, and Apertor Pharmaceuticals, and is a consultant for MPM Capital and Droia Ventures. The other authors declare that they have no competing interests.
Data and materials availability:
The RNA-sequencing data discussed in this manuscript and performed in this study are deposited in the NCBI Gene Expression Omnibus (NCBI-GEO) Accession number GSE46495 and GSE196384 respectively. All other data are available in the main text or the supplementary materials.
References
- 1.Luo J, Yang H, Song B-L, Nat Rev Mol Cell Biol. 21, 225–245 (2020). [DOI] [PubMed] [Google Scholar]
- 2.Riscal R, Skuli N, Simon MC, Mol Cell. 76, 220–231 (2019). [DOI] [PMC free article] [PubMed] [Google Scholar]
- 3.Radhakrishnan A, Goldstein JL, McDonald JG, Brown MS, Cell Metab. 8, 512–521 (2008). [DOI] [PMC free article] [PubMed] [Google Scholar]
- 4.Yan R et al. , Science (2021), doi: 10.1126/science.abb2224. [DOI] [Google Scholar]
- 5.Düvel K et al. , Mol. Cell 39, 171–183 (2010). [DOI] [PMC free article] [PubMed] [Google Scholar]
- 6.Shin HR, Zoncu R, Dev. Cell (2020), doi: 10.1016/j.devcel.2020.06.010. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 7.Lu X-Y et al. , Nature (2020), doi: 10.1038/s41586-020-2928-y. [DOI] [Google Scholar]
- 8.Liu GY, Sabatini DM, Nat. Rev. Mol. Cell Biol (2020), doi: 10.1038/s41580-019-0199-y. [DOI] [Google Scholar]
- 9.Sancak Y et al. , Cell. 141, 290–303 (2010). [DOI] [PMC free article] [PubMed] [Google Scholar]
- 10.Castellano BM et al. , Science. 355, 1306–1311 (2017). [DOI] [PMC free article] [PubMed] [Google Scholar]
- 11.Sancak Y et al. , Science. 320, 1496–1501 (2008). [DOI] [PMC free article] [PubMed] [Google Scholar]
- 12.Kim E, Goraksha-Hicks P, Li L, Neufeld TP, Guan K-L, Nat. Cell Biol 10, 935–945 (2008). [DOI] [PMC free article] [PubMed] [Google Scholar]
- 13.Rogala KB et al. , Science. 366, 468–475 (2019). [DOI] [PMC free article] [PubMed] [Google Scholar]
- 14.Anandapadamanaban M et al. , Science. 366, 203–210 (2019). [DOI] [PMC free article] [PubMed] [Google Scholar]
- 15.Bar-Peled L et al. , Science. 340, 1100–1106 (2013). [DOI] [PMC free article] [PubMed] [Google Scholar]
- 16.Panchaud N, Péli-Gulli M-P, De Virgilio C, Sci Signal. 6, ra42 (2013). [DOI] [PubMed] [Google Scholar]
- 17.Lim C-Y et al. , Nat. Cell Biol 21, 1206–1218 (2019). [DOI] [PMC free article] [PubMed] [Google Scholar]
- 18.Toulmay A, Prinz WA, J. Cell Biol 202, 35–44 (2013). [DOI] [PMC free article] [PubMed] [Google Scholar]
- 19.Wang S et al. , Science. 347, 188–194 (2015). [DOI] [PMC free article] [PubMed] [Google Scholar]
- 20.Rebsamen M et al. , Nature. 519, 477–481 (2015). [DOI] [PMC free article] [PubMed] [Google Scholar]
- 21.Chantranupong L, Wolfson RL, Sabatini DM, Cell. 161, 67–83 (2015). [DOI] [PMC free article] [PubMed] [Google Scholar]
- 22.Wyant GA et al. , Science. 360, 751–758 (2018). [DOI] [PMC free article] [PubMed] [Google Scholar]
- 23.Schröder B et al. , Traffic. 8, 1676–1686 (2007). [DOI] [PubMed] [Google Scholar]
- 24.Chapel A et al. , Mol Cell Proteomics. 12, 1572–1588 (2013). [DOI] [PMC free article] [PubMed] [Google Scholar]
- 25.Vassilatis DK et al. , Proc Natl Acad Sci U S A. 100, 4903–4908 (2003). [DOI] [PMC free article] [PubMed] [Google Scholar]
- 26.Düvel K et al. , Mol. Cell 39, 171–183 (2010). [DOI] [PMC free article] [PubMed] [Google Scholar]
- 27.Saha P et al. , Elife. 9, e57089 (2020).32410728 [Google Scholar]
- 28.Kwon HJ et al. , Cell. 137, 1213–1224 (2009). [DOI] [PMC free article] [PubMed] [Google Scholar]
- 29.Maekawa M, Fairn GD, J. Cell. Sci 128, 1422–1433 (2015). [DOI] [PubMed] [Google Scholar]
- 30.Li J, Lee PL, Pfeffer SR, Methods Mol Biol. 1583, 85–95 (2017). [DOI] [PubMed] [Google Scholar]
- 31.Davis OB et al. , Dev Cell (2020), doi: 10.1016/j.devcel.2020.11.016. [DOI] [Google Scholar]
- 32.Budelier MM et al. , J Biol Chem. 292, 9294–9304 (2017). [DOI] [PMC free article] [PubMed] [Google Scholar]
- 33.Bukiya AN, Dopico AM, J Lipid Res. 58, 1044–1054 (2017). [DOI] [PMC free article] [PubMed] [Google Scholar]
- 34.Hanson MA et al. , Structure. 16, 897–905 (2008). [DOI] [PMC free article] [PubMed] [Google Scholar]
- 35.Cho KF et al. , Nat Protoc. 15, 3971–3999 (2020). [DOI] [PubMed] [Google Scholar]
- 36.Wolfson RL et al. , Nature. 543, 438–442 (2017). [DOI] [PMC free article] [PubMed] [Google Scholar]
- 37.Das A, Brown MS, Anderson DD, Goldstein JL, Radhakrishnan A, Elife. 3 (2014), doi: 10.7554/eLife.02882. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 38.Fromm SA, Lawrence RE, Hurley JH, Nat Struct Mol Biol. 27, 1017–1023 (2020). [DOI] [PMC free article] [PubMed] [Google Scholar]
- 39.Huang P et al. , Cell. 174, 312–324.e16 (2018). [DOI] [PMC free article] [PubMed] [Google Scholar]
- 40.Lawrence RE et al. , Science (2019), doi: 10.1126/science.aax0364. [DOI] [Google Scholar]
- 41.Kobilka BK et al. , Science. 240, 1310–1316 (1988). [DOI] [PubMed] [Google Scholar]
- 42.Cooper MK et al. , Nat Genet. 33, 508–513 (2003). [DOI] [PubMed] [Google Scholar]
- 43.Huang P et al. , Cell. 166, 1176–1187.e14 (2016). [DOI] [PMC free article] [PubMed] [Google Scholar]
- 44.Deshpande I et al. , Nature. 571, 284–288 (2019). [DOI] [PMC free article] [PubMed] [Google Scholar]
- 45.Qi X et al. , Nature. 571, 279–283 (2019). [DOI] [PMC free article] [PubMed] [Google Scholar]
- 46.Xiao X et al. , Mol Cell. 66, 154–162.e10 (2017). [DOI] [PubMed] [Google Scholar]
- 47.Qi X, Friedberg L, De Bose-Boyd R, Long T, Li X, Nat Chem Biol. 16, 1368–1375 (2020). [DOI] [PMC free article] [PubMed] [Google Scholar]
- 48.Qian H et al. , Cell. 182, 98–111.e18 (2020). [DOI] [PubMed] [Google Scholar]
- 49.Qi X, Schmiege P, Coutavas E, Li X, Science. 362 (2018), doi: 10.1126/science.aas8843. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 50.Zhang Y et al. , Cell. 175, 1352–1364.e14 (2018). [DOI] [PMC free article] [PubMed] [Google Scholar]
Associated Data
This section collects any data citations, data availability statements, or supplementary materials included in this article.
Supplementary Materials
Data Availability Statement
The RNA-sequencing data discussed in this manuscript and performed in this study are deposited in the NCBI Gene Expression Omnibus (NCBI-GEO) Accession number GSE46495 and GSE196384 respectively. All other data are available in the main text or the supplementary materials.