

Abstract
Methods for in vitro DNA cleavage and molecular cloning remain unable to precisely cleave DNA directly adjacent to bases of interest. Restriction enzymes (REs) must bind specific motifs, whereas wild-type CRISPR-Cas9 or -Cas12 nucleases require protospacer-adjacent motifs (PAMs). Here we explore the utility of our previously reported near-PAMless SpCas9 variant, named SpRY, to serve as a universal DNA cleavage tool for various cloning applications. By performing SpRY DNA digests (SpRYgests) using more than 130 gRNAs sampling a wide diversity of PAMs, we discover that SpRY is PAMless in vitro and can cleave DNA at practically any sequence, including sites refractory to cleavage with wild-type SpCas9. We illustrate the versatility and effectiveness of SpRYgests to improve the precision of several cloning workflows, including those not possible with REs or canonical CRISPR nucleases. We also optimize a rapid and simple one-pot gRNA synthesis protocol to streamline SpRYgest implementation. Together, SpRYgests can improve various DNA engineering applications that benefit from precise DNA breaks.
Ed summary:
A PAMless CRISPR nuclease is applied for precision DNA cleavage and cloning.
CRISPR-Cas nucleases cleave DNA in a programmable manner, offering advantages over restriction enzymes1 (REs) for in vitro molecular applications since the DNA specificities of REs impose challenges for precision cloning methods (i.e. isothermal assembly2; Sup. Figs. 1a–1c, Sup. Notes 1 and 2). Most of the DNA-targeting specificity of Cas enzymes is provided by a guide RNA (gRNA) that can be programmed to bind to nearly any sequence3 (Sup. Fig. 1d). However, canonical Cas9 and Cas12 nucleases are also encumbered by the requirement to recognize a short DNA motif adjacent to the target site, known as the protospacer adjacent motif4 (PAM; Sup. Fig. 1d). The dependence on the availability of a PAM proximal to the target site prohibits precise cleavage of DNA substrates (Sup. Fig. 1e). Indeed, prior approaches have attempted to utilize Cas9 or Cas12 enzymes for in vitro DNA digests, but these efforts have lacked precision targeting and flexibility due to the PAM requirements of Cas enzymes5–10, or unpredictability of Cas12a DSB ends8. Towards overcoming the PAM constraint, our group engineered a nearly PAMless CRISPR-Cas variant named SpRY11 that can target DNA sites with NNN PAMs in human cells, with a preference for NRN PAMs over NYN PAMs (where R is A or G and Y is C or T; Fig. 1a and Sup. Fig. 1f). Given that SpRY is less dependent on a PAM compared to wild-type (WT) SpCas9, we investigated whether SpRY could act as a fully programmable DNA endonuclease to cleave at any DNA base in vitro, simplifying the design and execution of molecular cloning protocols.
Figure 1: Characterization of SpRYgest in vitro cleavage efficiencies.

(a) Comparison of restriction enzymes (REs) that require fixed 4-8 nt motifs and the near-PAMless Cas9 variant, named SpRY, that can target and cleave DNA substrates without sequence constraints. (b) Illustration of SpRYgest in vitro cleavage reaction workflow. Categorization of substrate cleavage is determined at the final timepoint, judged as near-complete (>80%), partial (40-80%), or incomplete (<40%) digests. (c, d) Initial SpRYgest experiments assessing the DNA cleavage efficiencies of WT SpCas9 and SpRY against a linearized plasmid substrate by targeting either a specific region at 1 bp intervals using 12 gRNAs (panel c), or 8 gRNAs designed to be distributed across the substrate (panels d). (e) Comparison of the in vitro cleavage efficiencies of WT, SpG, and SpRY across 64 target sites representing all 2nd/3rd/4th position combinations of an NNNN PAM. Sites with a shifted NNGG PAM are indicated with an asterisk. (f, g) SpRYgest results using additional secondary gRNAs for primary sites in panels 1c and 1e for which partial or incomplete cleavage was observed. Secondary gRNAs were designed to target the opposite DNA strand placing the DSB at the same position as the primary gRNA (panel f), or to target different sites (spacers) but bearing the same PAMs as the primary gRNA (panel g). (h) Summary of the proportion of gRNAs that led to near-complete, partial, or incomplete substrate cleavage when using WT, SpG and SpRY. For panels c-g, cleavage of DNA substrates was quantified by capillary electrophoresis; mean shown for n = 3.
Here we discover that SpRY is essentially PAMless in vitro, enabling highly precise digestion of DNA molecules. The implementation of SpRY DNA digests (SpRYgests) overcomes the targeting constraints of REs, canonical CRISPR-Cas enzymes, and other nucleases, permitting DNA cleavage with single nucleotide resolution. We optimize various aspects of SpRYgests, including gRNA production, to enable a rapid, cost effective, and streamlined workflow. The flexibility to precisely SpRYgest DNA has advantages for several molecular DNA applications.
Results
Optimization of DNA digests with SpRY
We first investigated the abilities of WT SpCas9 or SpRY to generate DSBs along a DNA substrate at various locations harboring different PAMs. In our initial assays (Fig. 1b), we performed in vitro digests utilizing overexpressed Cas9 protein from human cell lysates (Sup. Figs. 2a and 2b) along with gRNAs produced using optimized and rapid in vitro transcription (IVT) conditions (Sup Figs. 3a–3c and Sup. Note 3). We assessed WT SpCas9 and SpRY activity in vitro by performing digests against 20 different target sites sampling NRN and NYN PAMs across a linearized plasmid substrate (Figs. 1c and 1d). We utilized 12 gRNAs targeting a specific region at 1 bp intervals and 8 gRNAs distributed across the substrate (Sup Fig. 4). Between the two experiments, WT SpCas9 digested only 4 of these 20 sites to near-completion (>80% digestion; Figs. 1c, 1d). In comparison, when using SpRY for the same DNA digests (SpRYgests) we observed near-complete cleavage of the substrate for 19 of 20 gRNAs, with the lone gRNA resulting in approximately 50% substrate digestion (Figs. 1c, 1d). These results provided evidence that SpRY could act as a potent PAM-agnostic endonuclease in vitro.
To further assess the PAMless nature of SpRYgests, we performed a large comparison of WT SpCas9, SpG (an SpCas9 variant previously engineered to target sites with NGN PAMs11), and SpRY using 64 additional gRNAs targeting a range of sites bearing all 2nd/3rd/4th position combinations of an NNNN PAM (Fig. 1e and Sup. Figs. 5a–c). Similar to previous reports12,13, WT SpCas9 efficiently digested substrates when programmed with gRNAs targeting sites harboring NGG PAMs, and sometimes exhibited activity against sites with NAG, NGA, or shifted NNGG PAMs (Fig. 1e and Sup. Fig. 5a). SpG recapitulated its preference to edit substrates with NGN PAMs (Fig. 1e and Sup. Fig. 5b). Finally, although an NRN PAM preference was observed with SpRY in mammalian cells11, SpRY digested the substrate to near-completion when using 59 of 64 gRNAs, partially digested the substrate with 2 gRNAs, and exhibited low-to-no activity with the remaining 3 (Fig. 1e and Sup. Fig. 5c). Together, 93% (78/84) of the gRNAs initially used in these two sets of SpRYgests led to extensive cleavage.
Next, we investigated potential causes for incomplete substrate digestion with SpRY. First, for sites where the primary gRNA exhibited partial or incomplete cleavage, we tested the ability of a secondary gRNA targeted to the opposite strand to generate a DSB at the exact same location (Fig. 1f, Sup. Figs. 6a and 6b). This strategy generates the same DSB but via a different target site and gRNA. We assessed the opposite-strand secondary gRNA approach for 6 primary gRNAs that did not reach >80% completion. We observed >80% cleavage for 5 of 6 new secondary gRNAs targeted to the opposite strand and 78% cleavage for the 6th gRNA (Fig. 1f and Sup. Fig. 6b), identifying a strategy to overcome low-activity gRNAs. Next, we explored whether low-activity sites could be attributed primarily to a spacer- or PAM-specific source. We tested additional gRNAs targeted to new sites/spacers bearing some of the PAMs that initially resulted in incomplete cleavage (Fig. 1g and Sup. Fig. 6c). For all 13 new gRNAs, we observed near-complete substrate cleavage, suggesting that the PAM is not a primary determinant for incompletely digested sites and that SpRY is generally PAMless in vitro. Collectively, our combined results using 103 gRNAs reveal the flexibility and effectiveness of SpRY for in vitro digests, with 93.2% of SpRYgests achieving near-complete substrate digestion, an efficiency dramatically higher than for WT SpCas9 or SpG11 (Fig. 1h).
To explore various putative sequence features that might predict SpRYgest efficiency, we calculated the rate constants for substrate cleavage for all WT, SpG, and SpRY reactions (Sup. Table 1 and Sup. Note 4). When classifying targetable sites based on rate constant, the PAM emerged as a major determinant of activity for WT SpCas9 and SpG, but had little impact on SpRY efficiency (Sup. Figs. 7a and 7b). Interestingly, for the target sites that either failed to reach complete cleavage with SpRY or had slow rate constants (< 0.01), all either encoded an NCN or NTN PAM (consistent with the NRN>NYN preference of SpRY11) or a cytosine in the most PAM-proximal position of the spacer (consistent with previously described sequence features that can negatively impact SpCas9 editing14). Of those sites, half encoded both an NYN PAM and a 1st spacer C (including the two sites refractory to cleavage), although other sites with both sequence features were efficiency cleaved (Sup. Table 1). Comparison of the experimental SpRYgest rate constants versus our previously described HT-PAMDA data for SpRY11 revealed little predictive power of the PAM alone (Sup. Fig. 7c), consistent with our conclusion that the spacer sequence is a larger determinant of SpRYgest success versus the PAM. We then trained an XGBoost15 regressor model for WT SpCas9 and SpRY and also performed SHAP analysis16 (Sup. Figs. 8a–8e and Sup. Note 4). The NGG PAM remained a clear determinant of efficiency for WT SpCas9 (Sup Figs. 8a and 8d) but the model was less predictive for SpRY (Sup. Figs. 8b, 8c, and 8e). Despite the poorer correlation, the NYN PAM and 1st spacer C once again emerged as weakly inhibitory features for SpRYgests (Sup. Fig. 8e). Finally, intramolecular gRNA interactions17,18 did not convincingly impact SpRYgest efficiency (Sup. Note 4).
SpRYgest applications
We compared SpRY to another alternative to REs, the DNA-guided prokaryotic Argonaute (Ago) proteins19–21. We initially examined the ability of commercially available Thermus thermophilus Argonaute (TtAgo) to generate custom DNA breaks when programmed with pairs of ssDNA guides (Sup. Figs. 1g and 9a). In experiments adhering to the restrictive target site design considerations for TtAgo (Sup. Fig. 9b and Sup. Note 5) and despite performing metal ion and enzyme dose optimizations (Sup. Figs. 9c and 9d, respectively), TtAgo was only able to cleave 2 of 5 substrates to near-completion (Fig. 2a and Sup. Fig. 9e). By comparison, all ten SpRYgests targeting either strand for each of the five TtAgo sites reached near-complete cleavage (Fig. 2a and Sup. Fig. 9e). We then tested TtAgo against two positive control sites and the 20 sites that we initially examined with SpRY (where 19/20 resulted in near-complete digestion; Figs. 1c and 1d). None of these 20 sites accommodated the restrictive TtAgo design requirements and we did not observe evidence of DNA cleavage at any of the 20 sites (Fig. 2b and Sup. Fig. 9f). Given that TtAgo fully digested the DNA substrate for only 8% of sites examined, at least under our current optimized conditions, TtAgo cannot generate DSBs in vitro as effectively as SpRY (Fig. 2b).
Figure 2: Molecular cloning via SpRYgest.

(a) Comparison of the DNA cleavage efficiencies of TtAgo and SpRY across 5 sites designed to adhere to TtAgo guide requirements. TtAgo reactions were performed with pairs of 5’P-ssDNA guides. TtAgo and SpRYgest reactions were performed for 60 and 216 minutes, respectively. Individual datapoints, mean, and s.e.m. shown for n = 3. (b) Proportion of TtAgo and SpRY guides that led to nearly complete, partial, or incomplete cleavage on the 20 target sites from Figs. 1c and 1d (see TtAgo results in Supplementary Fig. 9f) and the 5 sites from Fig. 2a. For panels a and b, cleavage of DNA substrates was quantified by capillary electrophoresis; mean shown for n = 3. (c, d) Schematics of the SpRYgest strategies to add P2A-EGFP sequences to SaCas9-ABE8e and PE2 via single and double SpRYgests, panels c and d, respectively. (e, f) Proportion of clones for which correct addition of the P2A-EGFP sequence was confirmed by Sanger sequencing for SaCas9-ABE8e and PE2 strategies, panels e and f, respectively. (g) Schematic of the SpRYgest strategy to add an NLS to the N-terminal end of SpCas9. (h) Proportion of clones for which correct addition of the N-terminal NLS was confirmed by Sanger sequencing.
To evaluate the practical utility of SpRYgests for molecular cloning applications, we first scaled up our cleavage reactions from ng to μg quantities of DNA substrate. Because these conditions necessitated the use of large quantities of SpRY, we overexpressed and purified SpRY from E. coli (Sup. Fig. 10). We initially tested purified SpRY in vitro using 3 different gRNAs targeting sites with NAT, NCA, and NGG PAMs across 9 different temperatures. Our data revealed that, consistent with prior reports for WT SpCas922, reactions at 37 °C were optimal for SpRY (Sup. Fig. 11a). The cleavage efficiencies of these 3 gRNAs with purified SpRY were consistent with our previous results using SpRY from a human cell lysate (Sup. Fig. 11b). For sites that exhibited lower activities using the SpRY from lysate, SpRY protein often improved editing efficiency (Sup. Fig. 11c). Upon scaling up the cleavage reactions to μg amounts of DNA substrate, we sometimes observed non-specific degradation of the digestion products (Sup. Fig. 12a). Reactions with gRNA-only conditions led to non-specific nicking of the supercoiled plasmid substrate, suggesting carry-forward of DNase (utilized for template removal during the IVT reaction) even following a magnetic bead-based clean-up step (Sup. Figs. 12a and 12b; Sup. Note 3). Omission of the of the DNase treatment during gRNA synthesis, or use of chemically synthesized gRNAs, eliminated the non-specific degradation of the SpRYgested DNA products (Sup. Fig. 12c), together indicating that the degradation was not due to SpRY.
We then explored the potential of SpRYgests to perform routine cloning applications where unique restriction sites were not available. First, we sought to precisely insert long ~1kb P2A-EGFP sequences into plasmids encoding two different genome editors, SaCas9-ABE8e23 and a prime editor24 (PE2) (Figs. 2c and 2d, respectively). To do so, we performed single and double gRNA SpRYgests using 4 μg of supercoiled plasmid substrate (Sup. Figs. 13a–13d). For both reactions, complete SpRYgestion was observed. Conversely, no cleavage was observed when using TtAgo targeted to these sites (Sup. Fig. 13e). We then generated a PCR product encoding the P2A-EGFP sequence and cloned it into the digested plasmids via isothermal assembly2 (Sup. Figs. 13a and 13b). Given the high precision of SpRYgests, we were able to utilize single short PCR products for inserts in the isothermal assembly reactions (Sup. Fig. 1c), which should increase assembly efficiency (whereas with restriction enzymes to open up the backbone, 3 PCR fragments would have been required for assembly, and thus be less efficient25; Sup. Fig. 1b). Of the 30 resulting clones that we sequenced for each assembly, 26 and 27 were correct (Figs. 2e and 2f). In another molecular cloning reaction, we performed a single SpRYgest of the N-terminus of a Cas9 expression plasmid to add a nuclear localization sequence (NLS) (Fig. 2g and Sup. Fig. 14). Isothermal assembly using a short PCR product was extremely efficient, leading to 23 out of 24 clones assembling correctly (Fig. 2h). If instead using REs, this approach would have utilized two distal RE sites and 3 PCR fragments. Together, SpRYgests simplified the complexity of these three practical and exemplary cloning experiments, resulting in high assembly efficiencies exceeding or comparable to typical RE-based cloning approaches.
SpRYgest cleavage specificity
Like genome editing experiments, off-target cleavage of closely related sequences could manifest in SpRYgests. When cloning the SaCas9-ABE8e-P2A-EGFP plasmid, we observed evidence of a very low-level secondary set of products likely caused by an off-target DSB (Sup. Fig. 13c). The weak off-target cleavage had only a minor impact on the intended digest band, being unlikely to impact cloning with the purified linear fragment. Closer inspection of the target site revealed that the off-target cleavage was the result of utilizing a gRNA that overlapped an NLS, for which there was a second NLS with high sequence similarity elsewhere in the plasmid, bearing 3 mismatches (Sup. Figs. 15a and 15b). We were able to completely mitigate off-target cleavage by utilizing a secondary gRNA targeted to the more unique sequence on the opposite strand (Sup. Figs. 15c and 15d). Another potential method to eliminate off-target editing is to utilize a high-fidelity version of SpRY, SpRY-HF111,26 (Sup. Fig. 15e), that has previously been shown to be more sensitive to mismatches. While SpRY-HF1 did not completely prevent off-target editing when utilizing the NLS-targeted gRNA at the final digestion timepoint (Sup. Fig. 15f), a comparison of SpRY and SpRY-HF1 when programmed with mismatched gRNAs revealed that in certain cases, SpRY-HF1 can reduce off-target cleavage in vitro, especially at earlier reaction timepoints (Sup. Fig. 15g).
Saturation mutagenesis using SpRYgests
Another potential application of SpRYgests is to generate plasmid libraries bearing customized regions at any location rapidly and cost-effectively. We sought to investigate biological properties of SpCas9 by generating two saturation mutagenesis libraries with randomized nucleotides in regions of SpCas9 that are critical for either the catalytic activity27 (Fig. 3a and Sup. Fig. 16a) or the PAM preference13,28 of SpCas9 (Fig. 3b and Sup. Fig. 16b). In two separate reactions, linearized an SpCas9-encoding plasmid via double-SpRYgest adjacent to HNH domain catalytic residues D839 and H840, or adjacent to PAM-interacting residues R1333, R13335, and T1337. Notably, due to a lack of nearby NGG PAMs, neither approach is possible with WT SpCas9 (Sup. Figs. 16a and 16b). Due to the proximal positioning of our SpRYgest sites, we were able to perform isothermal assembly reactions with short and inexpensive ssDNA oligonucleotides encoding degenerate NNS codons (where ‘N’ is any nucleotide and ‘S’ is G or C). Via both Sanger and next-generation sequencing, we observed balanced representation of nucleotides in the modified positions (Figs. 3c and 3d). The libraries also contained intentionally coded silent substitutions to enable assessment of library construction efficiency, which were introduced at > 99.8% suggesting highly effective synthesis with minimal background (Figs. 3c and 3d).
Figure 3: Rapid generation of saturation mutagenesis libraries via SpRYgest.

(a,b) Schematics of SpRYgest strategies to generate saturation mutagenesis libraries of SpCas9 residues important for catalytic activity (panel a) and NGG PAM preference (panel b). (c,d) Sanger sequencing traces and next-generation sequencing results from the libraries, illustrating the nucleotide diversity at mutated residues for the HNH-catalytic and PAM-interacting (PI) domain libraries, panels c and d, respectively. Recoded silent substitutions were intentionally included in the library to assess construction efficiency (highlighted in purple and indicated with a triangle). (e) Schematic of the bacterial positive selection assay13,29,30, which permits selection of cleavage competent SpCas9 enzymes from saturation mutagenesis libraries. Colonies survive on selective media only when SpCas9 and a gRNA cleave a target site on the toxic plasmid. Mutated regions of SpCas9 can be sequenced from the plasmids harbored within surviving colonies. (f,g) Post-selection results for cleavage competent SpCas9 variants from the catalytic domain HNH residue library (panel f), or from the PI domain library selected against toxic plasmids harboring target sites with NGG and NGAG PAMs (left and right sides of panels g, respectively). Pie charts illustrate the distribution of amino acids at each position in the pre-selection libraries (via NGS) and post-selection libraries (via Sanger sequencing of individual clones) in the top and bottom panels, respectively.
We then subjected these SpRYgest-constructed libraries to a previously described bacterial positive-selection13,29,30. Survival of colonies harboring transformed plasmids is dependent on the ability of SpCas9 to cleave a target site encoded within a selection plasmid that expresses a toxic gene (Fig. 3e). First, we performed experiments using the catalytic domain library that was varied at conserved HNH nuclease positions D839 and H840 (Figs. 3a and 3c). Selection for cleavage-competent clones revealed that, as expected31, only SpCas9 variants encoding D839 and H840 were cleavage competent (Fig. 3f). We also evaluated the SpCas9 PAM-interacting (PI) domain library that was varied at amino acids critical for PAM recognition including R1333, R1335, and T1337 (Fig. 3b and 3d). Selection of the library against a toxic plasmid encoding a target site with an NGG PAM led mostly to clones with R1333 and R1335, the two amino acid sidechains known to be important for specifying the guanines of the PAM28 (Fig. 3g). Notably, a variety of amino acids were observed at position 1337, though the native T1337 was enriched relative to the others. An additional selection with the same library against a target encoding an NGAG PAM led to enrichment of variants with R1333, R1335Q, and T1337R/K (Fig. 3g), consistent with expectations for amino acids that facilitate recognition of this non-canonical PAM32,33 (and previous results with the engineered variants SpCas9-VQR and SpCas9-VRQR13,26).
Simplification of gRNA production
To facilitate implementation of SpRYgests, we sought to simplify and expedite the gRNA synthesis protocol (Sup. Fig. 17a). To do so, we experimented with various one-pot gRNA synthesis conditions and methods that require minimal hands-on time by combining the template generation and IVT steps (Sup. Figs. 17a–17d and Sup. Note 6). We identified conditions with shortened IVT reaction times (<4 hours) that generate high gRNA yields (Sup. Figs. 17b and 17c). These optimized one-pot reaction conditions reduce SpRYgest hands-on times by approximately 3.5-fold, leading to workflows that are more similar to RE digests (Fig. 4a). One-pot gRNA IVT reactions performed at two different scales for four different gRNAs all supported complete digestion of a plasmid substrate (Fig. 4b), reducing the enzymatic cost of gRNA synthesis (Sup. Note 6). The optimization of one-pot gRNA synthesis methods dramatically minimizes hands-on time, minimizes cost by scaling-down gRNA reactions, and makes the SpRYgest workflow more similar to other molecular cloning experiments (Fig. 4a and Sup. Fig. 18).
Figure 4: Optimization of rapid, efficient, and specific SpRYgest reactions.
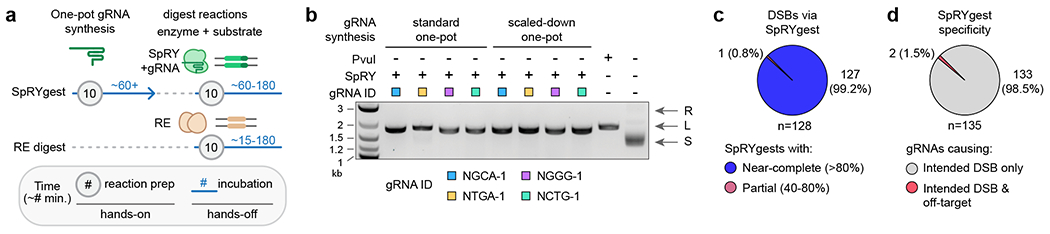
(a) Comparison of the hands-on and hands-off times of optimized SpRYgests (with gRNAs generated via one-pot reactions) versus RE digests. Approximate times in minutes are shown; lines not drawn to scale. The incubation times can vary for one-pot gRNA generation (see Sup. Fig. 17b), SpRYgest, or restriction enzyme digests. (b) Agarose gel of SpRYgest reactions performed with gRNAs taken directly from standard or scaled-down one-pot gRNA generation reactions (20 μL or 5 μL one-pot reactions, respectively). Plasmid conformations are: R, relaxed; L, linear; S, supercoiled; 1 kb Plus DNA Ladder (N.E.B.); n = 1. (c) Proportion of unique DSBs that were successfully generated via SpRYgest in all experiments of this study. (d) Proportion of gRNAs for which off-target cleavage products were detected during SpRYgests with gRNAs used in all experiments of this study.
Discussion:
Here we establish a method to manipulate DNA in vitro with unprecedented flexibility. We show that our previously reported near-PAMless SpRY nuclease11 is PAMless in vitro, providing a versatile tool for highly precise DNA digestion. This approach is not possible with WT SpCas9 or Cas12a enzymes, due to their NGG and T-rich PAM requirements, respectively5–10,34,35. With SpRYgests, standard and more complex cloning reactions are simplified, including the generation of saturation mutagenesis libraries to interrogate biological properties of impactful proteins (as demonstrated by our analysis of two SpCas9 domains). Our results suggest that the NRN>NYN PAM preference we previously observed in human cells11 is less prominent under defined in vitro conditions. This is likely due to a difference in absolute SpRY concentration in vitro versus in cells, or the fact that the ‘genome’ size of a plasmid is dramatically different than that of a eukaryotic organism, which influences SpRY target search time and how the preferred PAMs of a Cas variant are encountered and engaged36–38.
Across all experiments, we were able to generate a DNA break at every position that we sought to (Fig. 4c). For cases where the primary gRNA failed or was inefficient, reactions with the secondary gRNA that positions the DSB at the same position cleaved the substrate to near-completion (>80%) in all but one case (where this single gRNA reached ~78% substrate digestion; Fig. 4c). We anticipate that the digestion efficiency can be further improved and expedited through an improved understanding of gRNA-activity relationships, or by simply increasing the amount of SpRY-gRNA complex included in the SpRYgest reaction (including for weaker gRNAs that don’t initially lead to complete cleavage, though some gRNA/target site pairs might remain refractory to cleavage). Our results also indicate that the substrate preferences of Ago proteins19–21 limit their applicability and versatility for DNA digests.
The unconstrained targeting range of SpRY eliminates the need for a large repertoire of REs. Even with a comprehensive catalog of REs, rarely does a single cutter RE site exist at the intended location of the DNA modification. SpRYgests require only a single source of the SpRY protein (versus dozens of REs) and a gRNA, which can be generated by IVT (Fig. 4a and Sup. Fig. 17). To streamline the IVT process, we optimized a rapid, simple, and affordable one-pot gRNA generation method, the product of which can be added directly to a SpRYgest reaction (Fig. 4b and Sup. Note 6). Despite the already minimal cost and rapid workflow of SpRYgest reactions, further optimization is possible (e.g. of the IVT reaction39). SpRYgest timelines are not substantially different than traditional cloning workflows, which are similarly dependent on the design and receipt of custom oligonucleotides (Sup. Fig. 18). We provide additional guidance on performing SpRYgests and considerations for experimental design (e.g. omitting the DNase treatment during IVT of gRNAs; see Sup. Note 7). Since Cas9 enzymes predominantly leave blunt DNA breaks, SpRYgests do not leave overhangs typical of most REs (a property largely obviated by the use of isothermal assembly2). However, paired SpRY nickases could in principle be utilized to generate cohesive ‘sticky’ ends as needed.
Over the course of this study when performing 136 separate SpRYgests, we observed only very low level off-target cleavage for 2 gRNAs (both of which were attributable to related sequences in the plasmids; Fig. 4d and Sup Figs. 15b and 15h). These results indicate that achieving a single intended digestion product via SpRYgest is possible when considering other closely related sequences in the plasmid, and that simple methods can be used to mitigate off-target cleavage (by identifying when targeting the opposite strand would have fewer predicted off-target sites or by using SpRY-HF1, both of which can reduce or eliminate off-target cleavage). To prospectively identify gRNAs with closely matched off-target sites in DNA substrates, we have developed a web-based tool called SpOT-check (SpRYgest Off-Target checker; see Sup. Note 8).
We have identified some putative target site sequence features that in some cases can negatively impact SpRYgest efficiency, including the combination of an NYN PAM and a 1st spacer C, which can be detrimental to cleavage but is not absolutely inhibitory (see Sup. Note 4). We recommend that users ab initio design a second strand targeting gRNA when the initial gRNA and target site pair contains both features. Future efforts may further elucidate the causes of rare occurrences of incomplete or failed SpRYgests, either through more thorough spacer profiling, or gRNA engineering17,18,40 should intramolecular gRNA interactions be problematic.
The flexibility of SpRYgests to generate DSBs at specific positions within DNA substrates holds promise to simplify, accelerate, and improve the precision of molecular applications, many of which not previously possible when using REs. Beyond a general usefulness for standard cloning, SpRYgests can streamline and simplify various applications. For example, SpRYgests improve the efficiency of isothermal assembly reactions by reducing the number of input molecules25 (Sup. Fig. 1c), could improve the construction of saturation mutagenesis libraries (by enabling precise delineation of the region of interest and reducing oligonucleotide library cost), can enable general molecular cloning strategies when MCSs are not available or rendered unfit due to RE-site redundancy in the cloned CDS, targeted domain minimization or shuffling41 (via accurate sequence targeting), depleting unwanted sequences from mutagenic or sequencing libraries42 (due to increased targeting flexibility), for target enrichment in sequencing protocols43 (by increasing resolution to cleave at particular nucleotides), obviating the need to domesticate plasmid backbones via elimination of repeated RE sites, to generate more precisely terminated IVT templates, for cloning large plasmids44 or large genetic fragments6,7,34 (where unique RE sites no longer exist), to avoid plasmid backbone PCRs during isothermal assembly reactions2, to screen plasmids prior to sequencing, for DNA profiling20,45 to detect or diagnose genetic or infectious diseases, and many other uses. Nearly any molecular application that currently requires a DNA break should benefit from the precision and simplicity of SpRYgests. With SpRYgests, the design of molecular cloning approaches can now commence by simply focusing on the cloning objective, rather than anchoring the approach based on which unique restriction sites are available.
Online Methods:
Plasmids and oligonucleotides
Descriptions and Addgene IDs for all plasmids used in this study are available in Sup. Table 3; new plasmids have been deposited with Addgene (https://www.addgene.org/Benjamin_Kleinstiver/). A list of all SpRYgest target sites is provided in Sup. Table 4 that includes spacer sequences, PAMs, and gRNA generation methods. Oligonucleotide sequences and descriptions are available in Sup. Table 5; all oligonucleotides were ordered from Integrated DNA Technologies (IDT). Target sites for TtAgo are listed in Sup. Table 6. Additional details for plasmids and oligonucleotides (oligos) are provided below in the respective sections. The SpOT-check computed off-target profiles for all gRNAs used in this study are available in Sup. Table 7.
Human cell culture
Human HEK 293T cells (ATCC) were cultured in Dulbecco’s Modified Eagle Medium (DMEM) supplemented with 10% heat-inactivated FBS (HI-FBS) and 1% penicillin/streptomycin. The supernatant media from cell cultures was analyzed monthly for the presence of mycoplasma using MycoAlert PLUS (Lonza).
Expression of and normalization of SpCas9-containing human cell lysates
Expression plasmids encoding WT SpCas9, SpG, and SpRY each with a -P2A-EGFP signal (RTW3027, RTW4177 and RTW4830, respectively) were used to generate human cell lysates containing SpCas9 proteins. Approximately 20-24 hours prior to transfection, 1.5x105 HEK 293T cells were seeded in 24-well plates. Transfections containing 500 ng of human codon optimized nuclease expression plasmid and 1.5 μL TransIT-X2 were mixed in a total volume of 50 μL of Opti-MEM, incubated at room temperature for 15 minutes, and added to the cells. The lysate was harvested after 48 hours by discarding the media and resuspending the cells in 100 μL of gentle lysis buffer (containing 1X SIGMAFAST Protease Inhibitor Cocktail, EDTA-Free (Sigma), 20 mM Hepes pH 7.5, 100 mM KCl, 5 mM MgCl2, 5% glycerol, 1 mM DTT, and 0.1% Triton X-100). The amount of SpCas9 protein was approximated from the whole-cell lysate based on EGFP fluorescence. SpCas9 lysates were normalized to 180 nM fluorescien (Sigma) based on a standard curve. Fluorescence was measured in 384-well plates on a DTX 880 Multimode Plate Reader (Beckman Coulter) with λex = 485 nm and λem= 535 nm.
Production of gRNAs
The DNA substrates required to transcribe gRNAs were generated via two methods. First, plasmids for IVT of SpCas9 gRNAs were generated by annealing and ligating duplexed oligos (see Sup. Table 5) corresponding to spacer sequences into BsaI-digested MSP3485 for T7 promoter-driven transcription of gRNAs. The derivative pT7-spacer-gRNA plasmids were digested with HindIII (NEB) to permit run-off transcription near the 3’ end of the SpCas9 gRNA. Secondly, oligo-derived DNA templates for IVT were generated by combining a target specific oligo (encoding a T7 promoter, spacer sequence, and partial sequence of the SpCas9 crRNA) and a common SpCas9 gRNA scaffold oligo (oKAC682), and then incubating with either Klenow Fragment (3’→5’ exo-) (New England Biolabs (NEB), M0212L) in 1x NEBuffer 2 at 37 °C for 30 minutes, or Q5 polymerase (NEB) using the following program: 2 minutes 98 °C; 5 cycles of (10 seconds 98 °C, 10 seconds 65 °C , 30 seconds 72 °C); 5 minutes 72 °C. Plasmid or oligo-derived transcription templates were cleaned up using a MinElute PCR Purification Kit (Qiagen). SpCas9 gRNAs were transcribed at 37 °C for 16 hours using the T7 RiboMAX Express Large Scale RNA Production Kit (Promega). For gRNAs utilized in in vitro cleavage reactions containing SpRY from human cell lysates, the 37 °C incubation was followed by the addition of 1 μL RQ1 DNase at 37 °C for 15 minutes to degrade the DNA template. The DNase treatment step was omitted when preparing most gRNAs utilized for scaled-up SpRYgest reactions with purified SpRY protein. Following transcription and optional DNase treatment, gRNAs were purified using paramagnetic beads (prepared as previously described46; GE Healthcare Sera-Mag SpeedBeads (Fisher Scientific), washed in 0.1X TE and suspended in 20% PEG-8000 (w/v), 1.5 M NaCl, 10 mM Tris-HCl pH 8, 1 mM EDTA pH 8 and 0.05% Tween20). The gRNAs were purified by binding the transcription reaction to the beads, performing three washes with 70% EtOH, eluting in nuclease-free H2O, and then gRNAs were refolded by heating to 90 °C for 5 minutes and then cooling to room temperature at 1 °C every 2 seconds. Synthetic gRNAs were purchased from Synthego.
For one-pot gRNA IVT reactions, we utilized two general methods. First, gRNAs were generated using the EnGen sgRNA Synthesis Kit (NEB, E3322S) according to the manufacturer recommended protocol, or the EnGen sgRNA Synthesis Kit with increased oligo concentrations (final concentrations of 0.75 μM target-specific oligo and 0.75 μM common SpCas9 gRNA scaffold oligo (oKAC682)). The DNase step was omitted. Second, we also optimized a separate one-pot gRNA synthesis method using other commercial reagents. In this second method, 20 μL one-pot reactions were assembled containing final amounts or concentrations of 2.5 U Klenow Fragment (3’→5’ exo-), target-specific oligo at 0.5 or 1.5 μM (for standard or scaled-up reactions, respectively), common SpCas9 gRNA scaffold oligonucleotide (oKAC682) at 0.25 or 0.75 μM (for standard or scaled-up reactions, respectively), 125 μM dNTPs, 1x RiboMAX Express T7 Buffer (Promega, P1320), and 2 μL T7 Express Enzyme Mix (Promega, P1320) and incubated at 37 °C for 4 hours unless otherwise indicated. Appropriately scaled 5 μL reactions were assembled for smaller-scale one-pot reactions. For one-pot gRNAs used in SpRYgest reactions, the Promega recommended RQ1 DNase treatment was omitted and no clean-up of the gRNA was performed. To quantify gRNA yield, separate IVT reactions were performed that included the RQ1 DNase step and were purified using paramagnetic beads. Note that gRNA yield will vary based on incubation time.
Expression and purification of SpRY and SpRY-HF1 proteins
E. coli codon optimized SpRY and SpRY-HF1 coding sequences including an N-terminal MKIEE tag and C-terminal SV40 NLS and 6x histidine tag were synthesized (GenScript, NJ, USA) and cloned into pET28 expression vectors. The SpRY and SpRY-HF1 expression constructs and were used to express and purify the proteins as described previously47. Briefly, E. coli strain NiCo21(DE3) (C2529H from NEB) harbouring the recombinant construct was grown in 1-2 L of LB medium with 40 μg/mL Kanamycin at 30°C until mid-log phase. Overexpression of the target protein was induced by adding IPTG to a final concentration of 0.4mM with shaking overnight at 18°C. Cells were harvested and target protein expression was assessed by SDS PAGE prior to purification. Cells were disrupted by sonication in breakage buffer (50mM Tris-HCl (pH8.0), 300mM NaCl, 1mM EDTA, 1mM DTT, 2% (v/v) glycerol) supplemented with PMSF. The supernatant was passed through HiTrap DEAE Sepharose (Cytiva, MA, USA) in column buffer (20mM Tris(pH7.5) and 250mM NaCl) followed by subsequent purification on a HisTrap HP column (Cytiva). After 16x column volume wash in buffer containing 20mM Tris pH7.5, 250mM NaCl, 40mM imidazole, target proteins were eluted using a 40mM to 750mM imidazole gradient in the same buffer. Pooled fractions containing the proteins were further purified by loading onto HiTrap heparin HP columns (Cytiva), washed with 6 column volumes of a buffer containing 20mM Tris (pH8.0), 1mM EDTA, and 1mM DTT, and eluted using a 0.25 to 2M NaCl gradient in the same buffer. Pooled fractions were dialyzed in SEC column buffer (20mM HEPES (pH8.0), 250mM KCl, and 1mM DTT) and concentrated using an Amicon® Ultra-15 Centrifugal Filter Unit with 100 kDa molecular weight cut-off. Concentrated fractions were loaded on to a HiLoad 16/600 Superdex 200 pg column (Cytiva) using a 1 mL sample loop. Size exclusion chromatography was performed in SEC column buffer with a flow rate of 0.5mL/min. Eluted fractions were assessed by SDS-PAGE, pooled, dialyzed in storage buffer (20mM Tris (pH7.5), 300mM NaCl, 0.1mM EDTA, 1mM DTT and 50% (v/v) glycerol), and stored at −20°C. Protein concentration was determined by Bradford assay using BSA for standards.
In vitro cleavage reactions using SpCas9 from lysates
Plasmid KAC833 linearized with HindIII (NEB) was used as the DNA substrate for most in vitro cleavage reactions unless otherwise stated. SpCas9 ribonucleoprotein (RNP) complexes were formed by mixing 9 μL of SpCas9-containing normalized whole-cell lysate (normalized to 180 nM Fluorescein) with 11.25 pmol of transcribed or synthetic gRNA, and incubating for 5 minutes at 37 °C. Cleavage reactions were initiated by the addition of 34.82 fmol of linearized plasmid (digested with HindIII (NEB)) and buffer to bring the total reaction volume to 22.5 μL with a final composition of 10 mM HEPES pH 7.5, 150 mM NaCl, and 5 mM MgCl2. Reactions were performed at 37 °C and aliquots were terminated at timepoints of 1, 6, 36 and 216 minutes by removing 5 μL aliquots, mixing with 5 μL of stop buffer (50 mM EDTA and 2 mg/ml Proteinase K (NEB)), and incubating at room temperature for 10-minutes. Cleavage fragments were purified using paramagnetic beads and quantified via QIAxcel capillary electrophoresis (Qiagen). The relative abundances of substrate and products were analyzed using QIAxcel ScreenGel Software (v1.5.0.16, Qiagen) and plotted using GraphPad Prism 9 (v9.2.0). The rate constants for cleavage reactions were calculated using a custom script (Sup. Note 9), by fitting the substrate cleavage to an exponential decay model y(t) = Ae-kt, where y(t) is the percentage of uncleaved substrate over time, t is the time in minutes, k is the rate constant, and A is a constant, as previously described11,38.
In vitro cleavage reactions using purified SpRY
Small-scale in vitro cleavage reactions were performed as described above, except using 0.6-1 μM purified SpRY per reaction pool instead of 9 μL of SpCas9-containing normalized whole-cell lysate (0.6 μM in Sup. Figs. 11a and 11b and 1 μM in 11c). For scaled-up digests, 4 μg of supercoiled plasmid DNA was incubated at 37 °C for 3 hours with purified SpRY protein at a final concentration of 1 μM and IVT gRNA (prepared without DNase treatment) at a final concentration of 2 μM in Buffer 3.1 (NEB). Reactions were stopped by the addition of 1 μL of Proteinase K (NEB) and incubated at room temperature for 15 minutes. Cleavage fragments were resolved by 0.8% agarose gel electrophoresis with 1 μL of 1 kb Plus DNA Ladder (NEB) and visualized by ethidium bromide staining. Digestion products were purified using a QIAquick Gel Extraction Kit (Qiagen).
Molecular cloning reactions using purified SpRY
The C-terminal P2A-EGFP sequence was added to SaABE8e or pCMV-PE2 (Addgene IDs 138500 and 132775, respectively), and the N-terminal BPNLS was added to an SpCas9 plasmid similar to pCMV-T7-SpCas9 (Addgene plasmid ID 139987) via isothermal assembly. Reactions contained approximately 5 μL of isothermal assembly mix (prepared similar to as previously described2; 5x isothermal assembly buffer prepared with 3 mL 1M Tris-HCl pH 7.5, 300 μL 1M MgCl2, 600 μL of 10 mM each dNTP, 300 μL 1M DTT, 1.5 g PEG-8000, 20 mg NAD, H2O to 6 mL. Isothermal assembly mix (2x) prepared using 320 μL isothermal assembly buffer, 1.2 μL T5 Exonuclease (NEB), 20 μL Phusion Polymerase (NEB), and 160 μL Taq ligase (NEB), along with 700 μL H2O. Aliquots stored at −20 °C) or NEBuilder HiFi (NEB), 0.01 pmol of plasmid linearized via SpRYgest, and 0.03 pmol of PCR product insert in a final volume of 10 μL, and incubated at 50°C for 60 minutes. Cloning reactions were transformed into chemically competent XL1-Blue E. coli cells and grown at 37 °C for approximately 16 hours. Individual colonies were grown overnight at 37 °C, miniprepped (Qiagen), and fidelity of cloning was verified via Sanger sequencing. Saturation mutagenesis plasmid libraries for were constructed by incubating 0.02 pmol of BPK848 (linearized via SpRYgest) with 0.1 pmol of ~60bp ssDNA oligo (either oBK9102 or oBK9104) with 10 μL NEBuilder HiFi DNA Assembly Master Mix (NEB) in a final volume of 20 μL, and incubated at 50 °C for 15 minutes. NEBuilder reactions were cleaned up via MinElute (Qiagen) and eluted in 10 μL water, transformed into 100 μL of electrocompetent XL1-Blue E. coli, and recovered in 3 mL of SOC for 1 h at 37 °C. Next, 2 μL of the transformation recovery media was plated on LB + chloramphenicol, where the number of colonies following overnight at 37 °C were used to estimate library complexity. The remaining recovery was grown overnight in 150 mL LB + chloramphenicol and plasmid DNA was isolated by MaxiPrep (Qiagen). The complexity of the SpCas9 catalytic and PAM domain libraries were estimated to be 77,400 and 292,600 respectively. Plasmid libraries were sequenced via Sanger sequencing and NGS. For NGS, PCR amplicons were generated from the plasmids using primer pairs oKAC1589/oKAC1590 (for the catalytic domain) or oKAC1591/oKAC1592 (for the PI domain) and sequenced on a MiSeq (Illumina) to a depth of 12,378 and 409,221 reads for the catalytic and PI domain libraries, respectively. The resulting data was analyzed using CRISPResso2 (v2.0.30)48 to generate allele tables.
In vitro cleavage reactions using TtAgo
For TtAgo reactions, 5’-phosphorylated DNA guides were either purchased from Integrated DNA technologies or generated by incubating an unmodified oligonucleotide with T4 Polynucleotide Kinase (NEB) at 37 °C for 30 minutes, followed by heat-activation at 65°C for 20 min. Complexes of TtAgo programmed with ssDNA guides were prepared by combining final concentrations of 1 pmol TtAgo (NEB) and 2 pmol 5’-phosphorylated ssDNA guides and incubating at 70 °C for 20 minutes. Cleavage reactions were performed by combining TtAgo complexes with either 79.85 fmol of linearized KAC833 plasmid substrate (digested with PvuI, NEB) or 79.85 fmol supercoiled plasmid DNA (KAC1151 or MNW95) in ThermoPol buffer (NEB) with a final concentration of 10mM MgSO4. Reactions were performed at 80 °C for 60 minutes and terminated by the addition of 1 μL of Proteinase K (NEB). Cleavage fragments from pre-linearized substrates were purified using paramagnetic beads and quantified and analyzed as described above. Cleavage fragments from scaled-up plasmid DNA digests were resolved by 0.8% agarose gel electrophoresis and visualized by ethidium bromide staining.
Bacterial-based positive selection assay
Target plasmids for the selection assays were generated by cloning duplexed oligonucleotides into XbaI and SphI-digested p11-lacY-wtx1 (Addgene ID 69056)29 as previously described13, which contains an arabinose-inducible ccdB toxin gene. The derivative toxin-expressing plasmids contain target sites harboring either NGG or NGA PAMs (BPK740 and BPK754, respectively). To perform the selections, electrocompetent E. coli BW25141(λDE3)30 containing a toxin-expressing plasmid were transformed with BPK848-derived plasmids that express the SpCas9 variant libraries (encoding randomized codons in specified positions) in addition to a gRNA, both from separate T7 promoters. Following a 60-minute recovery in SOC media, transformations were spread on LB plates containing either chloramphenicol and 10 mM dextrose (non-selective) or chloramphenicol + 10 mM arabinose (selective). Transformation efficiency was assessed based on colony count from non-selective plates. The catalytic library selection resulted in approximately 9e4 colonies (from sampling approximately 87x library coverage). The PI domain library selections for NGG PAMs and NGA PAMs resulted in approximately 6e5 and 6.4e4 colonies (from sampling approximately 18x and 2x coverage of the libraries, respectively). Surviving colonies from selective plates were picked as single colonies for miniprep (Qiagen) followed by Sanger sequencing to verify the identities of the mutated amino acids.
Prediction of cleavage rates based on sequence information
To predict substrate cleavage rate constants for WT SpCas9 and SpRY, nucleotides within the 24 nt target sites (20 nt spacer and 4 nt PAM) were one-hot encoded and we trained an XGBoost regressor15. To obtain the optimal model for predicting WT SpCas9 and SpRY cleavage rates, we assessed 108 XGBoost models chosen from the following hyperparameter settings: the number of base trees (chosen from [50, 100, 200]), the maximum depth of the individual regression trees (chosen from [3, 4, 5]), the L1 regularization term (alpha) on weights (chosen from [0, 1, 3, 5]) and the subsample ratio of columns for constructing each tree (chosen from [0.3, 0.6, 1.0]). We performed 3-fold nested cross-validation to evaluate models using mean square error on WT SpCas9 and SpRY. The optimal WT SpCas9 and SpRY models (with the least mean square error) were evaluated in a leave-one-out cross-validation procedure using Pearson’s Correlation coefficients. We also performed SHAP (SHapley Additive exPlanations16) analysis on the trained XGBoost data to reveal the nucleotide importance across the 24-nt target sequence. Nucleotides with high absolute SHAP values have a strong impact on rate prediction, and the sign of the SHAP value indicates impact direction.
Supplementary Material
Acknowledgements:
We thank D.R. Edgell for helpful suggestions and S. Mahendraker for assistance developing the web version of SpOT-check. K.A.C. is supported by a Massachusetts General Hospital (MGH) Fund for Medical Discovery (FMD) Fundamental Research Fellowship Award. B.P.K. acknowledges support from an MGH Executive Committee on Research Howard M. Goodman Fellowship and NIH National Institutes of Health (NIH) P01 HL142494. R.T.W. is supported by the National Science Foundation Graduate Research Fellowship Program under Grant No. 1745302. L.P. is partially supported by NIH R35 HG010717.
Footnotes
Competing Interests Statement:
K.A.C., R.T.W., and B.P.K are inventors on patents and/or patent applications filed by Mass General Brigham that describe genome engineering technologies, including for the development of SpRY (R.T.W. and B.P.K.). B.P.K. is a consultant for EcoR1 capital and is an advisor to Acrigen Biosciences, Life Edit Therapeutics, and Prime Medicine. L.P. has financial interests in Edilytics and SeQure Dx, Inc. L.P.’s interests were reviewed and are managed by Massachusetts General Hospital and Partners HealthCare in accordance with their conflict-of-interest policies. M.M. and G.B.R. are employees of the Research Department at New England Biolabs Inc. (NEB). NEB is a commercial supplier of molecular biology reagents including some that have been used in this work. The remaining authors declare no competing interests.
Code Availability:
The script utilized to determine the SpRYgest rate constants is provided as Sup. Note 9.
Data Availability:
Primary datasets are available in Sup. Table 2; any other data that support this study are available from the corresponding author upon request.
References:
- 1.Loenen WAM, Dryden DTF, Raleigh EA, Wilson GG & Murray NE Highlights of the DNA cutters: a short history of the restriction enzymes. Nucleic Acids Research 42, 3–19 (2014). [DOI] [PMC free article] [PubMed] [Google Scholar]
- 2.Gibson DG et al. Enzymatic assembly of DNA molecules up to several hundred kilobases. Nat Methods 6, 343–345 (2009). [DOI] [PubMed] [Google Scholar]
- 3.Jinek M et al. A Programmable Dual-RNA–Guided DNA Endonuclease in Adaptive Bacterial Immunity. Science 337, 816–821 (2012). [DOI] [PMC free article] [PubMed] [Google Scholar]
- 4.Collias D & Beisel CL CRISPR technologies and the search for the PAM-free nuclease. Nature Communications 12, 555 (2021). [DOI] [PMC free article] [PubMed] [Google Scholar]
- 5.Wang J-W et al. CRISPR/Cas9 nuclease cleavage combined with Gibson assembly for seamless cloning. BioTechniques 58, 161–170 (2015). [DOI] [PubMed] [Google Scholar]
- 6.Jiang W et al. Cas9-Assisted Targeting of CHromosome segments CATCH enables one-step targeted cloning of large gene clusters. Nat Commun 6, 8101 (2015). [DOI] [PMC free article] [PubMed] [Google Scholar]
- 7.Jiang W & Zhu TF Targeted isolation and cloning of 100-kb microbial genomic sequences by Cas9-assisted targeting of chromosome segments. Nat Protoc 11, 960–975 (2016). [DOI] [PubMed] [Google Scholar]
- 8.Li S-Y, Zhao G-P & Wang J C-Brick: A New Standard for Assembly of Biological Parts Using Cpf1. ACS Synth. Biol 5, 1383–1388 (2016). [DOI] [PubMed] [Google Scholar]
- 9.Jeong YK, Yu J & Bae S Construction of non-canonical PAM-targeting adenosine base editors by restriction enzyme-free DNA cloning using CRISPR-Cas9. Sci Rep 9, 4939 (2019). [DOI] [PMC free article] [PubMed] [Google Scholar]
- 10.Shola DTN, Yang C, Kewaldar V-S, Kar P & Bustos V New Additions to the CRISPR Toolbox: CRISPR-CLONInG and CRISPR-CLIP for Donor Construction in Genome Editing. The CRISPR Journal 3, 109–122 (2020). [DOI] [PMC free article] [PubMed] [Google Scholar]
- 11.Walton RT, Christie KA, Whittaker MN & Kleinstiver BP Unconstrained genome targeting with near-PAMless engineered CRISPR-Cas9 variants. Science 368, 290–296 (2020). [DOI] [PMC free article] [PubMed] [Google Scholar]
- 12.Jiang W, Bikard D, Cox D, Zhang F & Marraffini LA RNA-guided editing of bacterial genomes using CRISPR-Cas systems. Nat Biotechnol 31, 233–239 (2013). [DOI] [PMC free article] [PubMed] [Google Scholar]
- 13.Kleinstiver BP et al. Engineered CRISPR-Cas9 nucleases with altered PAM specificities. Nature 523, 481–485 (2015). [DOI] [PMC free article] [PubMed] [Google Scholar]
- 14.Doench JG et al. Rational design of highly active sgRNAs for CRISPR-Cas9–mediated gene inactivation. Nat Biotechnol 32, 1262–1267 (2014). [DOI] [PMC free article] [PubMed] [Google Scholar]
- 15.Chen T & Guestrin C XGBoost: A Scalable Tree Boosting System. in Proceedings of the 22nd ACM SIGKDD International Conference on Knowledge Discovery and Data Mining 785–794 (Association for Computing Machinery, 2016). doi: 10.1145/2939672.2939785. [DOI] [Google Scholar]
- 16.Lundberg SM et al. From local explanations to global understanding with explainable AI for trees. Nat Mach Intell 2, 56–67 (2020). [DOI] [PMC free article] [PubMed] [Google Scholar]
- 17.Thyme SB, Akhmetova L, Montague TG, Valen E & Schier AF Internal guide RNA interactions interfere with Cas9-mediated cleavage. Nat Commun 7, 11750 (2016). [DOI] [PMC free article] [PubMed] [Google Scholar]
- 18.Moreb EA & Lynch MD A Meta-Analysis of gRNA Library Screens Enables an Improved Understanding of the Impact of gRNA Folding and Structural Stability on CRISPR-Cas9 Activity. The CRISPR Journal 5, 146–154 (2022). [DOI] [PubMed] [Google Scholar]
- 19.Swarts DC et al. DNA-guided DNA interference by a prokaryotic Argonaute. Nature 507, 258–261 (2014). [DOI] [PMC free article] [PubMed] [Google Scholar]
- 20.Enghiad B & Zhao H Programmable DNA-Guided Artificial Restriction Enzymes. ACS Synth. Biol 6, 752–757 (2017). [DOI] [PubMed] [Google Scholar]
- 21.Enghiad B et al. PlasmidMaker is a versatile, automated, and high throughput end-to-end platform for plasmid construction. Nat Commun 13, 2697 (2022). [DOI] [PMC free article] [PubMed] [Google Scholar]
- 22.Harrington LB et al. A thermostable Cas9 with increased lifetime in human plasma. Nat Commun 8, 1424 (2017). [DOI] [PMC free article] [PubMed] [Google Scholar]
- 23.Richter MF et al. Phage-assisted evolution of an adenine base editor with improved Cas domain compatibility and activity. Nature Biotechnology 38, 883–891 (2020). [DOI] [PMC free article] [PubMed] [Google Scholar]
- 24.Anzalone AV et al. Search-and-replace genome editing without double-strand breaks or donor DNA. Nature 576, 149–157 (2019). [DOI] [PMC free article] [PubMed] [Google Scholar]
- 25.Xia Y et al. T5 exonuclease-dependent assembly offers a low-cost method for efficient cloning and site-directed mutagenesis. Nucleic Acids Research 47, e15 (2019). [DOI] [PMC free article] [PubMed] [Google Scholar]
- 26.Kleinstiver BP et al. High-fidelity CRISPR–Cas9 nucleases with no detectable genome-wide off-target effects. Nature 529, 490–495 (2016). [DOI] [PMC free article] [PubMed] [Google Scholar]
- 27.Nishimasu H et al. Crystal Structure of Cas9 in Complex with Guide RNA and Target DNA. Cell 156, 935–949 (2014). [DOI] [PMC free article] [PubMed] [Google Scholar]
- 28.Anders C, Niewoehner O, Duerst A & Jinek M Structural basis of PAM-dependent target DNA recognition by the Cas9 endonuclease. Nature 513, 569–573 (2014). [DOI] [PMC free article] [PubMed] [Google Scholar]
- 29.Chen Z & Zhao H A highly sensitive selection method for directed evolution of homing endonucleases. Nucleic Acids Research 33, e154–e154 (2005). [DOI] [PMC free article] [PubMed] [Google Scholar]
- 30.Kleinstiver BP, Fernandes AD, Gloor GB & Edgell DR A unified genetic, computational and experimental framework identifies functionally relevant residues of the homing endonuclease I-BmoI. Nucleic Acids Research 38, 2411–2427 (2010). [DOI] [PMC free article] [PubMed] [Google Scholar]
- 31.Zuo Z et al. Structural and functional insights into the bona fide catalytic state of Streptococcus pyogenes Cas9 HNH nuclease domain. eLife 8, e46500 (2019). [DOI] [PMC free article] [PubMed] [Google Scholar]
- 32.Hirano S, Nishimasu H, Ishitani R & Nureki O Structural Basis for the Altered PAM Specificities of Engineered CRISPR-Cas9. Molecular Cell 61, 886–894 (2016). [DOI] [PubMed] [Google Scholar]
- 33.Anders C, Bargsten K & Jinek M Structural Plasticity of PAM Recognition by Engineered Variants of the RNA-Guided Endonuclease Cas9. Molecular Cell 61, 895–902 (2016). [DOI] [PMC free article] [PubMed] [Google Scholar]
- 34.Kudo K et al. In vitro Cas9-assisted editing of modular polyketide synthase genes to produce desired natural product derivatives. Nat Commun 11, 4022 (2020). [DOI] [PMC free article] [PubMed] [Google Scholar]
- 35.Enghiad B et al. Cas12a-assisted precise targeted cloning using in vivo Cre-lox recombination. Nat Commun 12, 1171 (2021). [DOI] [PMC free article] [PubMed] [Google Scholar]
- 36.Moreb EA, Hutmacher M & Lynch MD CRISPR-Cas “Non-Target” Sites Inhibit On-Target Cutting Rates. The CRISPR Journal 3, 550–561 (2020). [DOI] [PubMed] [Google Scholar]
- 37.Moreb EA & Lynch MD Genome dependent Cas9/gRNA search time underlies sequence dependent gRNA activity. Nat Commun 12, 5034 (2021). [DOI] [PMC free article] [PubMed] [Google Scholar]
- 38.Walton RT, Hsu JY, Joung JK & Kleinstiver BP Scalable characterization of the PAM requirements of CRISPR–Cas enzymes using HT-PAMDA. Nat Protoc 16, 1511–1547 (2021). [DOI] [PMC free article] [PubMed] [Google Scholar]
- 39.Gurevich VV, Pokrovskaya ID, Obukhova TA & Zozulya SA Preparative in vitro mRNA synthesis using SP6 and T7 RNA polymerases. Analytical Biochemistry 195, 207–213 (1991). [DOI] [PubMed] [Google Scholar]
- 40.Riesenberg S, Helmbrecht N, Kanis P, Maricic T & Pääbo S Improved gRNA secondary structures allow editing of target sites resistant to CRISPR-Cas9 cleavage. Nat Commun 13, 489 (2022). [DOI] [PMC free article] [PubMed] [Google Scholar]
- 41.Shams A et al. Comprehensive deletion landscape of CRISPR-Cas9 identifies minimal RNA-guided DNA-binding modules. Nat Commun 12, 5664 (2021). [DOI] [PMC free article] [PubMed] [Google Scholar]
- 42.Gu W et al. Depletion of Abundant Sequences by Hybridization (DASH): using Cas9 to remove unwanted high-abundance species in sequencing libraries and molecular counting applications. Genome Biology 17, 41 (2016). [DOI] [PMC free article] [PubMed] [Google Scholar]
- 43.Gilpatrick T et al. Targeted nanopore sequencing with Cas9-guided adapter ligation. Nat Biotechnol 38, 433–438 (2020). [DOI] [PMC free article] [PubMed] [Google Scholar]
- 44.Kim GB et al. Rapid Generation of Somatic Mouse Mosaics with Locus-Specific, Stably Integrated Transgenic Elements. Cell 179, 251–267.e24 (2019). [DOI] [PMC free article] [PubMed] [Google Scholar]
- 45.Roewer L DNA fingerprinting in forensics: past, present, future. Investigative Genetics 4, 22 (2013). [DOI] [PMC free article] [PubMed] [Google Scholar]
Methods-only references:
- 46.Rohland N & Reich D Cost-effective, high-throughput DNA sequencing libraries for multiplexed target capture. Genome Res. 22, 939–946 (2012). [DOI] [PMC free article] [PubMed] [Google Scholar]
- 47.Karvelis T et al. Rapid characterization of CRISPR-Cas9 protospacer adjacent motif sequence elements. Genome Biology 16, 253 (2015). [DOI] [PMC free article] [PubMed] [Google Scholar]
- 48.Clement K et al. CRISPResso2 provides accurate and rapid genome editing sequence analysis. Nature Biotechnology 37, 224–226 (2019). [DOI] [PMC free article] [PubMed] [Google Scholar]
Associated Data
This section collects any data citations, data availability statements, or supplementary materials included in this article.
Supplementary Materials
Data Availability Statement
Primary datasets are available in Sup. Table 2; any other data that support this study are available from the corresponding author upon request.