

Abstract
The CRISPR prime editor PE2 consists of a Streptococcus pyogenes Cas9 nickase (nSpCas9) fused at its C-terminus to a Moloney Murine Leukemia Virus reverse transcriptase (MMLV-RT). Here, we show that separated nSpCas9 and MMLV-RT proteins function as efficiently as intact PE2 in human cells. We use this Split-PE system to rapidly identify and engineer more compact prime editor architectures that also broaden the types of RTs used for prime editing.
Prime editing uses CRISPR-guided reverse transcription to enable the programmable introduction of any desired base substitution or small insertion/deletion1. Mutations are induced by a prime editing (PE) protein (e.g., PE2) together with a PE gRNA (pegRNA) (Supplementary Fig. 1). For PE2, the pegRNA directs nSpCas9 activity to create an R-loop with a nicked DNA strand, which anneals to a primer binding sequence (PBS) at the 3’ end of the pegRNA (Supplementary Figs. 1a, 1b). The RT part of the PE protein then reverse transcribes the reverse transcription template (RTT) that is adjacent to the PBS into DNA encoding the desired edit of interest (Supplementary Fig. 1c). This DNA template then mediates introduction of the edit into the genomic locus by a mechanism that is not yet fully defined. Editing efficiency can be further enhanced with the PE3 system in which an additional secondary nick mediated by a nicking gRNA (ngRNA) is introduced either up- or down-stream of the desired edit site and on the strand opposite the one nicked by the PE protein/pegRNA complex (Supplementary Fig. 1c)1. Recent work has shown that concomitant overexpression of a dominant negative mutant of human MLH1 (termed hMLH1dn), a protein involved in DNA mismatch repair, can further enhance prime editing efficiencies in human cells2. Moreover, recent optimization of both PE and pegRNA designs has been shown to enable improved prime editing outcomes across different cell types and organisms3, 4. One challenge for use of all PE systems is the large size of the required PE2 protein (2117 aa encoded by 6351 bps), a difficulty that is exacerbated if one also needs to encode an additional ngRNA and/or the hMLH1dn protein (a 753 aa protein encoded by 2259 bps).
In the process of attempting to modify the architecture of the PE2 protein, we inadvertently discovered that the pentamutant MMLV-RT (hereafter referred to as MMLV-RT) is separable from nSpCas9. In initial experiments, we found that alternative configurations of the components that make up PE2, including fusion of MMLV-RT to the N-terminus of nSpCas9 and certain inlaid fusions of MMLV-RT within the Cas9 nickase5, showed frequencies of pure prime edits (PPEs – alleles with the precise desired edit) that were similar to or only moderately reduced relative to the original PE2 fusion when tested with 11 pegRNA/ngRNA combinations in HEK293T cells (Fig. 1 and Supplementary Figs. 2a, b). In addition, the frequencies of impure prime edits (IPEs - alleles with the desired edit together with an additional mutation) and byproducts (alleles with indels and/or substitutions but not the desired edit) we observed with the 11 pegRNA/ngRNA pairs and these alternative PE2 architectures did not appear to differ from those observed with PE2. Note that for pegRNAs designed to introduce insertion and deletion edits, it is not always possible to distinguish IPE and byproduct alleles; in these cases, we group IPE and byproduct frequencies together and show them as combined outcome frequencies as we have done previously6.
Fig. 1. Split and intact prime editors function with similar efficiencies in human HEK293T cells.

Schematics at the top illustrate the location of MMLV-RT (pink box) with respect to nSpCas9-H840A (white box) for three intact variants (C-terminal, N-terminal, and inlaid fusion at G1247) and the separate expression of nSpCas9 and the MMLV-RT pentamutant for Split-PE (not drawn to scale). Dot and bar plots represent the frequencies of prime editing induced at 11 genomic loci targeted with prime editing gRNAs (pegRNAs) and nicking gRNAs (ngRNAs) using the PE3 approach. The types of desired edits induced are grouped as substitutions, insertions (ins.), or deletions (del.). For substitution edits, frequencies of pure prime edits (PPE), impure prime edits (IPE), and byproducts are shown separately. For insertion and deletion edits, IPE and byproduct frequencies are added together and shown as a single bar next to their respective PPE frequencies6. Bar graphs represent the mean, error bars show standard deviation (s.d.), and dots represent values of replicates (n=3; independent replicates). bp, base pairs. FLAG, Flag tag (DYKDDDDK)33 with an SGS-linker).
These unexpected findings suggested to us that MMLV-RT, rather than functioning in cis on the same protein molecule with the nSpCas9 protein, might be acting in trans from another PE2 molecule not tethered to the target site. This in turn suggested that a split PE2 architecture with the nSpCas9 and the MMLV-RT expressed as wholly separate proteins from different plasmids might also function comparably to intact PE2 protein. Indeed, we found that a Split-PE2 architecture was comparably efficient to the original intact PE2 when tested with the same 11 pegRNA/ngRNA pairs in HEK293T cells (Fig. 1; Supplementary Fig. 2c). In addition, we observed similar results in U2OS cells with Split-PE2 showing similar or higher activities than intact PE2 with seven out of eight pegRNA/ngRNA pairs we tested (Supplementary Fig. 3a). We also tested whether a split version of another prime editor based on a Staphylococcus aureus Cas9 KKH PAM recognition variant nickase (nSaCas9-KKH)7 might function similarly to its intact counterpart (Supplementary Fig. 2d), and again found this to be true with six different pegRNA/ngRNA pairs targeting various endogenous gene sites in human HEK293T cells (Supplementary Fig. 2d).
We next explored whether the splitting of PE2 into separate RT and nickase components might alter the off-target effects of prime editing. To do this, we assessed editing frequencies at 18 genomic sites using six pegRNA/ngRNA combinations. These genomic sites had previously been found to exhibit off-target editing with either intact PE2 and/or SpCas9 nuclease in human cells ((Supplementary Fig. 4)1, 8, 9. In our experiments, intact PE2 and Split-PE2 showed similar on-target editing efficiencies with all six pegRNA/ngRNA combinations. We also observed similar editing frequencies with intact PE2 and Split-PE2 at an off-target site that had been previously reported for two different pegRNA/ngRNA combinations at HEK site 4 (Supplementary Fig. 4)1. Notably, we did not observe any evidence of new editing with Split-PE2 at any of the 17 other potential off-target sites that did not previously show evidence of editing with intact PE2 (Supplementary Fig. 4).
An important implication of our findings with split PE proteins is that alternative RT enzymes or CRISPR-Cas nickases could potentially be rapidly tested as separate domains, without the need to optimize linker lengths or relative positions within a fusion protein. To test this idea, we assessed six truncation mutants of MMLV-RT in the Split-PE2 configuration with three pegRNA/ngRNA pairs targeting different endogenous human gene target sites (Fig. 2a). This included a previously described N-terminal truncation variant (truncation 2, lacking 23 residues)10, 11 as well as C-terminal truncation variants that included truncations of the connection (truncations 1, 3, and 4) and/or RNAse H domains (truncation 5)11–14. From these experiments, we observed that a MMLV-RT variant (truncation 5) lacking the RNase H domain (MMLV-RTΔRH) exhibited PE activities equivalent to full-length MMLV-RT (Fig. 2a, Supplementary Fig. 5a). This truncated RT is encoded by 1488 bps and is therefore 26.7% smaller than the parental MMLV-RT. A recent study published by others while this work was in progress has also described a PE variant with a MMLV-RT truncation of the RNase H domain15.
Fig. 2. Rapid screening of variant RT domains using the Split-PE platform in HEK293T cells.
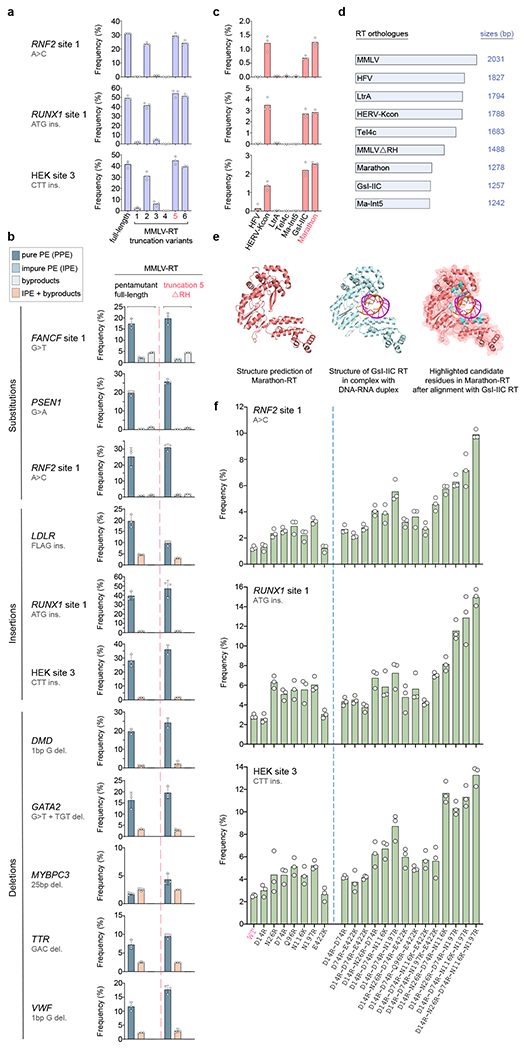
a, Dot and bar plots showing PPE frequencies with Split-PE using full-length MMLV-RT or six truncation variants tested with three pegRNA/ngRNA combinations (ΔRH in pink). Experiments were performed as technical replicates and so no error bars are shown (also applies to c and f). n=3, technical replicates. b, Dot and bar plots comparing PPE, IPE, and byproduct or combined IPE and byproduct frequencies observed with Split-PE using MMLV-RT-ΔRH or the full-length MMLV-RT together with 11 pegRNA/ngRNA combinations. Data shown for full-length MMLV-RT (left of the red dashed line) are the same as those shown for Split-PE in Fig. 1. Bar graphs represent the mean, error bars show standard deviation (s.d.), and dots represent values of replicates (n=3; independent replicates), c, Dot and bar plots showing PPE frequencies of seven non-MMLV RTs tested (Split-PE) with three pegRNA/ngRNA combinations (Marathon-RT in pink). Human codon-optimized non-MMLV RTs tested were from human foamy virus (HFV), human endogenous retrovirus K (HERV-Kcon), lactococcal group II intron Ll.ltrB (LtrA), Thermosynechococcus elongatus group II intron (TeI4c), Methanosarcina aromaticovorans intron 5 (Ma-Int5), Geobacillus stearothermophilus GsI-IIC intron (GsI-IIC), and Eubacterium rectale (Eu.re.I2) group II intron (Marathon). n=3, technical replicates d, Schematic showing the lengths of all non-MMLV RTs tested in c in comparison to MMLV-RT (without counting start codons), e, Structural representation of Marathon-RT (salmon; Phyre2 structure prediction), GsI-IIC RT (blue) in complex with an RNA template-DNA primer duplex (PDB 6AR1), and Marathon-RT (cartoon inside of mesh) with highlighted candidate residues (cyan) that are located within the modeled DNA/RNA binding pocket, based on the alignment with GsI-IIC RT (generated with PyMol; Methods), f, Dot and bar plots showing PPE frequencies (Split-PE) of seven Marathon-RT single residue mutants (left of dashed blue line) that were used to generate the 14 most efficient Marathon-RT combination variants (right of blue line). The data for wild-type (WT) Marathon-RT are the same as those shown in c. n=3, technical replicates. PPE frequencies induced by all 30 single and 18 combinatorial variants (inclusive of those shown here) are presented in Supplementary Fig. 6.
To further assess the activity of the MMLV-RTΔRH truncation, we tested it with eight additional pegRNA/ngRNA pairs and found it functioned as efficiently or better than full-length MMLV-RT in the Split-PE2 configuration with 10 out of 11 pegRNA/ngRNA pairs in HEK293T cells (Fig. 2b; Supplementary Fig. 5b). We obtained similar results in U2OS cells, with Split-PE2 using truncated MMLV-RTΔRH performing similarly to or better than Split-PE2 using the full-length MMLV-RT for seven out of the eight pegRNA/ngRNA pairs we tested (Supplementary Fig. 3a). Furthermore, we also observed similar editing efficiencies when the truncated MMLV-RTΔRH was expressed as a cleavable P2A translational fusion with the nSpCas9 from a single plasmid (and promoter) with the same 11 pegRNA/ngRNA pairs in HEK293T cells (Supplementary Fig. 5c). In addition, we tested whether the MMLV-RTΔRH truncation could mediate PE with different nickases, and found it worked as efficiently as full-length MMLV-RT when co-expressed with nSaCas9 or the nSaCas9-KKH variant and as a fusion with nSaCas9-KKH in HEK293T cells (Supplementary Figs. 6a and b). Finally, to test the MMLV-RTΔRH in a more disease-relevant, non-cancer cell line, we transfected human induced pluripotent stem cell (hiPSC)-derived cardiomyocytes with constructs expressing intact and Split-PE prime editor architectures using MMLV-RTΔRH together with 4 pegRNA/ngRNA combinations. We observed PE at all four sites with both intact and split PE2ΔRH (range of mean PPE frequencies across all four sites of 1.4 to 16.7%) (Supplementary Fig. 3b). At all 4 sites in hiPSC-derived cardiomyocytes, the editing activities of intact and split PE2-ΔRH variants were also similar as expected (Supplementary Fig. 3b).
We additionally leveraged the simplified screening enabled by the split PE framework to test a set of seven different RT enzymes, all smaller in size than MMLV-RT. The coding sequences for these enzymes (without start codons) ranged in length from 1242 to 1827 bps relative to 2031 bp for MMLV-RT (Figs. 2c - 2d; Supplementary Fig. 7). Two of the seven RTs we tested were of viral (human foamy virus; HFV)16, 17 or human endogenous retroviral (HERV)18, 19 origin, and the remaining five were group II intron RT domains (Fig. 2c)20–26. Testing of these RTs co-expressed with nSpCas9 and using three different pegRNA/ngRNA pairs revealed low PE frequencies in HEK293T cells (Fig. 2c). The best performing RTs among the seven we tested were the HERV-Kcon RT (frequencies ranging from 1.2 - 3.5%) and the bacterial group II intron RTs GsI-IIC and Marathon (0.7 - 2.8%). Because of its small size and consistent activity across the three different pegRNA/ngRNA pairs tested, we selected the Marathon-RT (a maturase RT from Eubacterium rectale that is also commonly used for in vitro laboratory applications26) to carry forward for additional optimization.
To further improve the activity of Marathon-RT for PE, we created a series of rationally designed mutants and tested each of these with co-expressed nSpCas9 in human cells. To guide the choice of the mutations we created, we initially used Phyre227 to generate a predicted structural model of Marathon-RT and also used published high-resolution structures of Marathon-RT in isolation (PDB 5HHL25) and of the homologous GsI-IIC group II intron maturase RT (commercially available as TGIRT-III) complexed with an RNA template-DNA primer duplex (PDB 6AR124) (Fig. 2e; Online Methods). By aligning our Marathon-RT structure prediction with the structure of GsI-IIC RT in complex with the RNA-DNA duplex, we identified 15 negatively charged or polar uncharged amino acid residues in Marathon-RT that were predicted to lie within the modeled DNA/RNA binding pocket of the enzyme (Fig. 2e). We hypothesized that changing each of these 15 positions to positively charged residues might potentially increase binding of the RT domain to the pegRNA and/or the nicked DNA exposed in the R-loop generated by a nickase Cas9. Based on this reasoning, we screened 30 different Marathon-RT variants harboring mutations at each of these positions with nSpCas9 and identified 15 that showed increased PE efficiencies relative to wild-type Marathon-RT when co-expressed with three different pegRNA/ngRNA pairs in HEK293T cells (Supplementary. Fig. 8). We also tested 18 additional Marathon-RT variants harboring various combinations of the seven most promising mutations (again with nSpCas9 and three pegRNA/ngRNA pairs) in HEK293T cells, and found several of these variants showed further improved activity. Notably, one Marathon-RT variant harboring five amino acid substitutions (D14R-N26R-D74R-N116K-N197R) showed 5.2- to 7.9-fold (mean of 6.1-fold) higher editing activity relative to the original Marathon-RT, and achieved absolute prime editing frequencies ranging from 9.9 - 15% (Fig. 2f and Supplementary Fig. 8).
To further validate our findings, we tested MMLV-RTΔRH and Marathon-RT in both intact and split PE configurations with 11 pegRNA/ngRNA combinations. These experiments in HEK293T cells showed that intact and split PEs with MMLV-RTΔRH exhibited similar editing between intact and split architectures at 5 out of 11 sites, and somewhat reduced editing with the split configuration at the remaining six sites (Supplementary Figs. 9; Supplementary Table 1). Overall, the intact and split PE2ΔRH editors showed similar PPE frequencies ranging from 7.4 – 53% and 2.3 – 46.6%, respectively (Supplementary Figure 10a; Supplementary Table 1). For intact and split PE architectures made with the engineered tetramutant and pentamutant Marathon-RTs, the split versions outperformed the intact ones at 5 out of 11 sites (tetramutant) and 9 out of 11 sites (pentamutant), respectively, with PPE frequencies ranging from 0.4 – 26.2% (tetramutant, split) and 0.4 – 22.7% (pentamutant, split) (Supplementary Fig. 9 and Supplementary Table 1). The relative efficiencies of each of our Split-PE architectures using the MMLV-RTΔRH and pentamutant Marathon-RT differed substantially across the 11 different pegRNA/ngRNA pairs tested (Supplementary Fig. 9 and Supplementary Fig. 10b), but we did not observe any obvious correlations between activities observed and the various lengths of the PBS and RTT regions of the pegRNAs tested (Supplementary Table 2).
Finally, we sought to compare our most active Split-PE2 architecture (using MMLV-RTΔRH) with an alternative split-intein PE2 protein that was published during the course of our experiments28. As noted above, the large size of the intact PE2 protein precludes its delivery using viral vectors such as adeno-associated virus (AAV) or lentiviral vectors. However, it has been shown that PE2 can be divided into two parts in the middle of the SpCas9 nickase, and then reconstituted into intact functional PE2 if trans splicing inteins are placed at the location of the split (Fig. 3a)28. The components of this split-intein PE2 can be delivered into cells in vivo using dual AAV vectors to mediate prime editing events28. To compare this system with ours, we transfected HEK293T cells with plasmids encoding 11 pegRNA/ngRNA combinations and either our most efficient minimized Split-PE architecture (Split-PE2ΔRH) or the previously described split-intein PE2 architecture. For all 11 sites, we observed higher PPE frequencies with Split-PE2ΔRH compared with the split-intein PE2 (Fig. 3b), perhaps at least partly reflecting the additional requirement for a bimolecular fusion reaction necessary to generate functional PE2 in the latter system. We additionally tested whether our split PE system could be delivered using two AAV vectors. For this initial experiment, we encoded the entire SpCas9 nickase in one AAV vector and the pegRNA/ngRNA combination for HEK site 3 (CTT insertion) together with the MMLV-RTΔRH-P2A-eGFP construct in the other (Fig. 3c). Following sorting for GFP-positive cells (Methods), delivery of both vectors to U2OS cells yielded a mean PPE frequency of nearly 4% while delivery of only the pegRNA/ngRNA/RT vector did not yield detectable PPEs (Fig. 3d). This initial experiment establishes the feasibility of using AAV vectors to deliver our Split-PE2 components even without extensive optimization of experimental parameters such as number and ratios of viral particles.
Fig. 3. Comparison of Split-PEΔRH with a split-intein PE system in HEK293T cells and dual AAV delivery of Split-PEΔRH to U2OS cells.
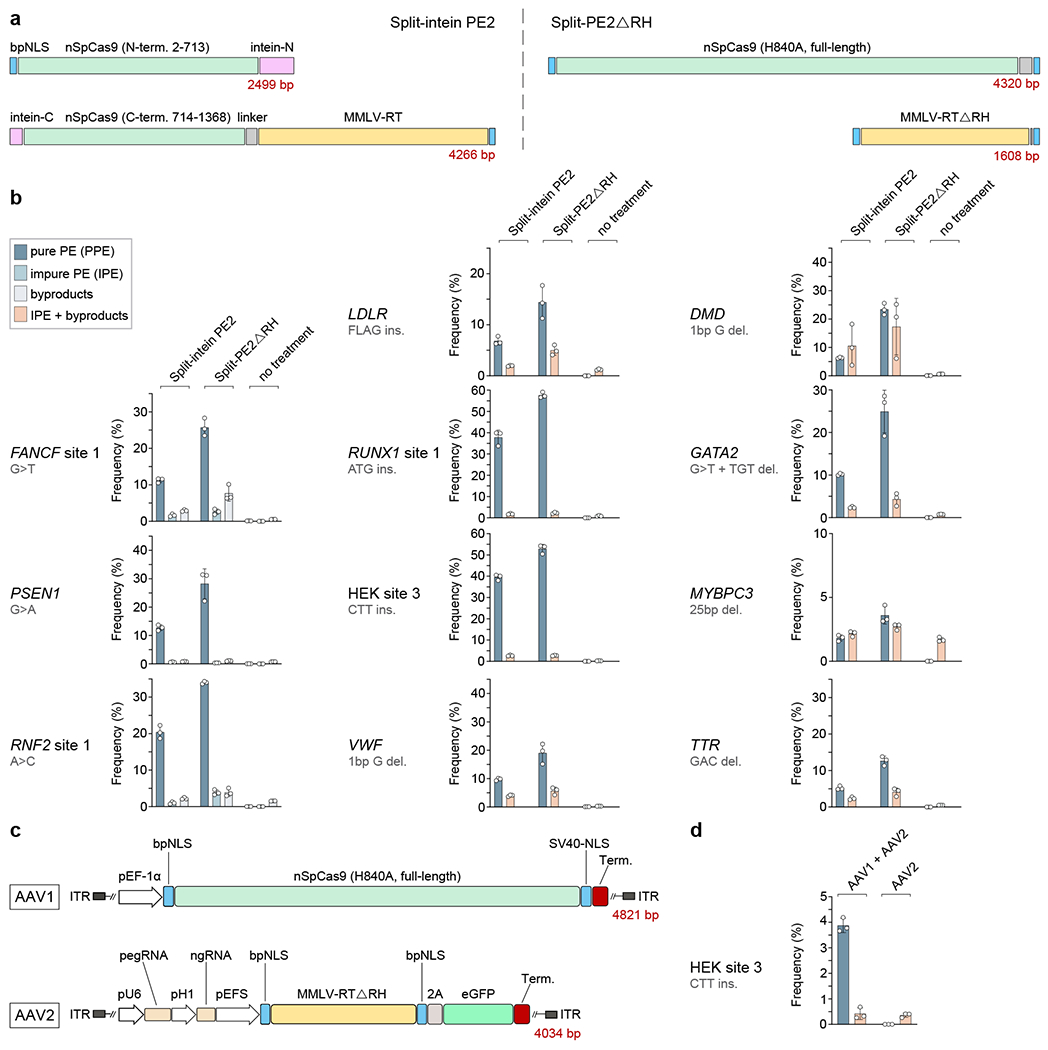
a, Schematic of Split-intein PE2 and Split-PE2ΔRH architectures, based on the nSpCas9-H840A variant and MMLV-RT. Both components of both systems were expressed from a CMV promoter. PegRNA and ngRNA plasmids were co-transfected separately and both gRNAs were expressed from a human U6 promoter. Red numbers indicate the length of the respective component in base pairs (bp), b, Dot and bar plots showing PPE, IPE and byproduct frequencies or combined IPE and byproducts induced by Split-intein PE2 and Split-PE2ΔRH as well as a no treatment control using 11 peg/ngRNA combinations in HEK293T cells. Bar graphs represent the mean, error bars show standard deviation (s.d.), and dots represent values of replicates (n=3; independent replicates), c, Schematic of the Split-PE2ΔRH architecture for dual AAV delivery, d, Dot and bar plots showing PPE and combined IPE and byproducts induced at HEK site 3 (desired edit: CTT insertion) in U2OS cells by Split-PE2-ΔRH (AAV1+AAV2) and a control (AAV2 only). Split-PE2-ΔRH was delivered via dual-AAV transduction. The AAV expressing the RT and peg/ngRNAs also co-translationally expressed eGFP. One week post-transduction, cells were sorted for the top ~10-20% GFP MFI and cultured for another 72h before cell harvest and gDNA extraction (Methods). Bar graphs represent the mean, error bars show standard deviation (s.d.), and dots represent values of replicates (n=3; independent replicates).
Our mechanistic finding that the RT and nCas9 components of PE proteins function efficiently even when separate has important implications for improving prime editing and for better understanding its other potential effects on cells. Our results strongly suggest that with existing intact PE proteins, the RT activity is likely provided by a second PE molecule that is presumably not bound to the target DNA site, possibly from solution. This in turn implies that the efficiency of PE might be further increased by creating different next-generation fusions in which the RT actually must function in cis to the nCas9 (i.e., a configuration in which RT activity is dependent on being tethered to the on-target site). Our findings also raise the possibility that with existing prime editors a RT may be able to act from solution on other off-target genomic sites in which a nicked DNA-RNA hybrid might be present, although it is not clear whether such an intermediate does in fact occur or if it would have any biological consequence in human or other cells. While this work was in progress, others have also demonstrated that efficient prime editing is possible when nSpCas9 and the RT domain are expressed separately29. This same study also showed that efficient prime editing is possible even with the components of the pegRNA are split into two RNAs: one a standard guide RNA and the other a linear or circular RNA that encodes the PBS and RTT29.
The Split-PEs and reduced size RTs we describe here provide reagents and architectures that should enhance and better enable the delivery of PE components and accelerate further improvements to the platform. Split-PEs address a limitation imposed by size-constrained AAV vectors – namely that the full-length PE2 protein is currently too large to fit into a single AAV vector. By leveraging the Split-PE architecture, one can encode the nSpCas9 protein in one AAV and the pegRNA/gRNA and RT in another, thereby creating a configuration in which only cells that are transduced by both vectors will undergo editing without the need for additional components such as split intein sequences used previously with CRISPR nucleases, base editors, and prime editors1, 30, 31. In our direct comparisons, we also found the additional benefit that our split architecture is more efficient than the previously described split-intein system, most likely because there is no need for the additional step of reconstituting a necessary protein component in our split configuration. We also note that our split-PE system would be expected to enhance and simplify both RNA and ribonucleoprotein delivery methods due to more efficient expression of shorter-length nCas9 and RT components instead of a full-length fusion of these two components. Our studies demonstrate that the split architecture can facilitate more rapid screening of new PE variants with improved properties. Rather than cloning and sequencing a new lengthy fusion for each RT variant and determining where and how to fuse each of these to a nicking Cas9, we were able to rapidly construct and then screen a large series of different viral, non-viral, and engineered RTs to identify those with desired activities. Similarly, this modularity should also permit the rapid screening of alternative nicking Cas9 or other nickases for PE.
Lastly, to our knowledge, the studies described here provide the first demonstration that non-viral, bacterial group II intron reverse transcriptases (G2I-RTs) can be engineered to function efficiently as intact or split prime editors in human cells. This finding opens up the possibility of mining the very large number of G2I-RTs that exist in diverse bacteria and archaea as starting points to engineer prime editors that are not only smaller but that may also provide improved function such as higher fidelity or greater processivity. The enormous size of this treasure trove of RTs that might be used for PE is evident from a recent computational search that yielded over 4,000 G2I-RTs32. In sum, our results and reagents should help to accelerate the optimization and application of prime editing for research and therapeutic uses.
Methods
Molecular cloning.
Prime editor (PE), Cas9 nuclease, and reverse transcriptase (RT) constructs used in this study (Supplementary Table 3) were cloned into a pCMV-T7 mammalian expression vector backbone obtained by AgeI-HF and NotI-HF (New England Biolabs, NEB) restriction digest of Addgene plasmid no. 112101 or 132775 as described below. All constructs that express PE2, SpCas9(H840A), MMLV-RT and its variants, XTEN linkers, and/or bipartite NLSs were cloned using Addgene plasmid no. 132775 as the PCR template. SaCas9-KKH based constructs were cloned using Addgene plasmid no. 70708 as a template. WT SaCas9 based constructs were cloned using Addgene plasmid no. 61594 as a template. Some constructs were cloned as P2A-eGFP fusions to obtain co-translational expression of enhanced GFP (eGFP; P2A-eGFP generated using Addgene no. 112101 as template). DNA encoding alternative RTs were purchased from IDT as synthetic dsDNA products (IDT gblocks) with codon optimization for expression in human cells (GenScript GenSmart codon optimization tool). The split-intein PE2 constructs were obtained from Addgene (plasmid no. 164909 and 164908) and subcloned into the abovementioned pCMV-T7 backbone with bpNLS on both termini of Split-intein PE2. The C-terminal portion had a silent G>T mutation at L1144. Gibson fragments with complementary overhangs were generated by PCR using Phusion high-fidelity DNA polymerase (NEB), which were then directly purified using paramagnetic beads34 or purified after agarose gel electrophoresis and extraction using Qiaquick gel extraction kit (Qiagen). The purified DNA fragments were then assembled with a pCMV backbone at 50 °C for 1 h using Gibson mix35 and used to transform chemically competent Escherichia coli XL1-Blue (Agilent). The prime editing gRNAs (pegRNAs) used in this study (Supplementary Table 4) were cloned based on the protocol described by Anzalone et al1. First, the oligos for the spacer, 5’ phosphorylated scaffold, and 3’ extension for each guide were annealed to form dsDNA fragments (95 °C for 5 min, then cooled to 10 °C at a rate of −5 °C/min) with compatible overhangs for ligation to each other and to the Bsal-digested pUC19-based hU6-pegRNA-gg-acceptor entry vector (Addgene no. 132777). Subsequently, the vector backbone and the DNA duplexes were ligated using T4 ligase (NEB). Construction of SpCas9 and SaCas9 pegRNAs required different scaffolds (Supplementary Table 4). Nicking gRNAs (ngRNAs) were generated in a similar fashion using only spacer oligos along with the BsmBI-digested pUC19-based hU6 gRNA entry vector BPK152036 (Addgene no. 65777) for SpCas9 ngRNAs and BPK26607 (Addgene no. 70709) for SaCas9 ngRNAs. All the plasmids used in this study were purified using Qiagen Mini/Midi Plus kits.
Cell culture.
We used STR-authenticated HEK293T cells (CRL-3216, ATCC) and U2OS cells (similar match to HTB-96; gain of no. 8 allele at the D5S818 locus), both cultured in Dulbecco’s modified Eagle medium supplemented with 10% FBS and 50 units/ml penicillin and 50 μg/ml streptomycin (all from Gibco). U2OS cells were supplemented with an additional 1% GlutaMAX (Gibco). Cells were grown at 37 °C with 5% CO2 and passaged every 2-3 days when cells reached approximately 80% confluency. For experiments with iCell Cardiomyocytes (obtained from Cellular Dynamics/Fujifilm, item 11713), plating medium (Cellular Dynamics) was thawed overnight at 4°C before thawing the cells according to the manufacturer’s recommendations. After resuspension and counting, 2.5 x 104 cells were seeded in 100μL plating medium per well of a 96-well plate that had previously been coated with 0.1% gelatin for 4 hours. Maintenance medium (Cellular Dynamics) was thawed overnight at 4°C 24h before use, followed by equilibration at 37°C. Cells were carefully washed with maintenance medium 48h post-seeding and plating medium was replaced with 90μL maintenance medium per well, which was replaced every other day. Cells were maintained at 37°C under 5% CO2. Every 4 weeks, cell cultures were tested for mycoplasma contamination using the MycoAlert PLUS mycoplasma detection kit (Lonza) and all the results were negative for the duration of this study.
Transfections and Nucleofections.
For transfections, HEK293T cells were seeded at 1.25 x 104 cells in 92 μL growth medium/well in 96-well flat-bottom cell culture plates (Corning). After 18-24 h of growth, the cells were transfected with 43.3 ng of plasmid DNA in total (30 ng PE, 10 ng pegRNA, 3.3 ng ngRNA for intact PE variants; 15 ng nCas9, 15 ng RT, 10 ng pegRNA, 3.3 ng ngRNA for split PE variants, using 0.3 μL of lipofection reagent TransIT-X2 (Mirus) and 9 μL of Opti-MEM (Gibco) per well. For off-target experiments, HEK293T cells were seeded into a 24-well plate flat-bottom format (Corning) (6.25 x 104 cells/well). After 18-24 h of growth, the cells were transfected with 216.5 ng of plasmid DNA in total (150 ng PE, 50 ng pegRNA, 16.5 ng ngRNA for intact PE variants; 75 ng nCas9, 75 ng RT, 50 ng pegRNA, 16.5 ng ngRNA for split variants). For experiments with U2OS cells, 4 x 106 cells were seeded into a 15-cm dish (Corning) in 25 ml growth medium. After 18-24 h of incubation, 2 x 105 cells/sample were electroporated with 1083.3 ng of total plasmid DNA (800 ng PE, 200 ng pegRNA, 83.3 ng ngRNA for intact PE variants; 400 ng nCas9, 400 ng RT, 200 ng pegRNA, 83.3 ng ngRNA for split variants) using the SE cell Line Nucleofector X Kit (Lonza) according to the manufacturer’s protocol. Subsequently, the electroporated cells were plated in 500 μL growth media in 24-well flat-bottom plates (Corning). iCell cardiomyocytes were transfected using Transit-LT1 transfection reagent37 (Mirus) on days 5, 6, and 7 post-thawing, using 150 ng PE, 50 ng pegRNA, and 17 ng ngRNA for intact PE variants or 75 ng nCas9, 75 ng RT, 50 ng pegRNA, and 17 ng ngRNA for split PE variants as well as 9μL Opti-MEM (Gibco) and 0.6μL Transit-LT1 per well. Maintenance medium was replaced 3h pre-transfection and 24h post-transfection. Transfected and electroporated cells were incubated at 37°C under 5% CO2 for 72 h, followed by genomic DNA (gDNA) extraction.
AAV experiments.
AAVs were produced in HEK293T cells by PEI triple transfection of ΔF6 helper plasmid (Addgene no. 112867), AAV2/2 package plasmid (Addgene no. 104963), and an AAV2 ITR-flanked transgene containing plasmid. AAVs were purified and concentrated by sucrose density gradient ultracentrifugation to a final titer between 1012 and 1013 genome copies/ml. The viruses were packaged at the MGH Vector Core Facility, Massachusetts General Hospital Neuroscience Center, Charlestown, MA. Transductions were carried out in 96-well format, where 10μl of each of the two AAVs (or of one only for the negative control), encoding either nSpCas9 or MMLV-RTΔRH-P2A-eGFP and the two guide RNAs were applied to 1.5 x 104 U2OS cells per well which were cultured in 50μl of DMEM. One week post-transduction, we sorted the top 21.8 - 22.4% FITC signal for the single-AAV negative controls (gRNA and RT-P2A-eGFP only) and the top 10.7 – 12.4% FITC signal for the dual-AAV Split-PE treatment samples (Supplementary Note 1). These cells were then seeded and cultured for another 72 hours before gDNA extraction.
DNA extraction.
After an initial wash step with 1x PBS, cells in 96-well format experiments were lysed with 43.5 μL gDNA lysis buffer (100 mM Tris-HCl (pH 8), 200 mM NaCl, 5 mM EDTA, 0.05% SDS), 1.25 μL 1 M DTT (Sigma), and 5.25 μL Proteinase K (800 U/ml, NEB) per well. Cells transfected or electroporated in a 24-well plate were lysed with the same components as listed but with 4x the amount, totaling 200 μL/well. Cells were lysed overnight in a shaker (HT Infors Multitron) at 500 rpm, at 55 °C and the gDNA was extracted with 2x paramagnetic beads as described previously 34. DNA bound to beads was washed with 70% ethanol three times using a Biomek FXp Laboratory Automation Workstation (Beckman Coulter) and eluted in 35-75 μL 0.1x Buffer EB (Qiagen).
Library preparation for targeted amplicon sequencing.
Concentrations of gDNA were determined using the Qubit4 fluorometer with the dsDNA HS Assay Kit (Thermo Fisher). Amplicons for sequencing were produced using a 2-PCR process to first amplify the specific target sequence and add Illumina adapter sequences (PCR1), and to subsequently add Illumina barcodes (PCR2). In PCR1, the target sequence was amplified from approximately 5-20 ng of gDNA using primers carrying Illumina-compatible adapter sequences (Supplementary Table 4) with Phusion DNA polymerase (NEB) under the following reaction conditions: 98 °C for 2 min, followed by 30-35 cycles of 98 °C for 10 s, 68 °C for 12 s, and 72 °C for 12 s, and a final 72 °C extension for 10 min. For off-target amplicons, the same gDNA was used to amplify both the on-target and off target sites using different PCR1 primers with Illumina-compatible adapter sequences (Supplementary Table 4). The PCR products were purified with 0.7x paramagnetic beads, eluted in 30 μL EB buffer and quantified using the Quantifluor dsDNA quantification system (Promega) on a Synergy HT microplate reader (BioTek; set to 485/528 nm). In PCR2, unique Illumina-compatible barcodes were added to each PCR1 amplicon (based on NEBnext E7600 barcodes, as well as custom barcodes; Supplementary Table 4) using approximately 50-200 ng of the clean PCR1 product per sample (or per pool), and Phusion DNA polymerase (NEB). The reaction conditions were as follows: 98 °C for 2 min, 5-10 cycles of 98 °C for 10 s, 65 °C for 30 s, and 72 °C for 30 s, followed by a 72 °C extension for 10 min. In some cases, when PCR1 products stemmed from non-overlapping genomic sites, they were quantified using the Quantiflour system (Promega) and pooled before barcoding to allow sequencing of more samples per run. PCR2 products were cleaned with 0.7x paramagnetic beads, quantified with the Quantifluor system (Promega), and pooled to ensure equal representation of samples in the final library. The pooled PCR2 products were subjected to a final cleanup using 0.6x paramagnetic beads to reduce residual primers and primer-dimers. The resulting amplicons were sequenced using Illumina Miseq kits or Miseq micro kits (Miseq Reagent Kit v2; 300 cycles, 2 × 150 bp, paired-end). Demultiplexed sequencing data were downloaded in the form of FASTQ files via BaseSpace (Illumina).
Deep sequencing analysis.
Sequencing files were analyzed using CRISPResso238 in HDR (homology directed repair) mode using standard parameters (unless otherwise indicated below). CRISPResso2 HDR categorizes sequencing reads into three distinct groups including ‘HDR’, ‘reference’ and ‘ambiguous’. Reads in the HDR group have a higher degree of sequence homology to the edited than to the unedited amplicons. The reads in the reference group have a higher degree of sequence homology to the unedited amplicons than to the edited amplicons. Reads in the ambiguous group are equally homologous to the edited and unedited amplicons (this can for example occur if the locus of the intended edit is deleted). The HDR group contained all reads harboring hallmarks of PE activity including pure PE containing only the intended edits and impure PE containing both the intended and unintended edits. To distinguish pure PE from impure PE, two editing windows were defined: One editing window spans from one bp before the predicted PE2 nicking location to one bp after the end of the DNA sequence that is homologous to the pegRNA RT template. The second HDR window spans from one bp before to one bp after the putative nicking site of the ngRNA. If apart from the intended edit, other mutations were detected within the editing window, reads were categorized as impure PE, otherwise as pure PE. The reference group contained all reads with neither the intended edit nor other mutations in the editing window. CRISPResso2 HDR categorizes reads without the intended edit but with additional mutations as ambiguous (if the locus of the intended edit was deleted) or as NHEJ (if the locus of the intended edit was intact but an edit was observed within the editing window). The reads of both groups (“ambiguous” and “NHEJ”) were interpreted as representing undesired PE byproducts. CRISPResso2 HDR was run with quality filtering (only reads with an average quality score >= 30 were considered). Read depth (number of reads aligned per experiment) is reported in Supplementary Table 4.
Analysis of editing frequencies at off-target sites.
Sequencing files were analyzed with CRISPResso2. An editing window was defined for every pegRNA which ranged from the first base before the putative Cas9 induced nick to one base after the end of the pegRNA RTT at the on-target site. The size of this editing window is defined as A. For every off-target candidate of a particular pegRNA, an editing window of size A was defined starting from the first base before the putative Cas9 nick. Sequencing reads with basepair insertions or deletions overlapping with the editing window were defined as edited; the remaining reads were defined as unedited. The fraction of edited reads is reported as the editing frequency.
PyMOL analysis.
The structure of the E. rectale RT (Marathon-RT; PDB 5HHL25) and of the GsI-IIC group II intron maturase RT (commercially available as TGIRT-III) complexed with an RNA template-DNA primer duplex (PDB 6AR124) were downloaded from the PDB and visualized with PyMOL v.2.3.4 and 2.5 (Schrodinger). A structure prediction of full-length Marathon-RT was generated using Phyre 227 and was subsequently aligned with the structure of GsI-IIC RT in complex with an RNA-DNA duplex (PDB 6AR1) using the ‘align’ command (‘align structurel, structure2, object=alnobj’). All illustrations (Fig. 2e) were generated with PyMOL 2.5.
Statistics and data reporting.
All bar graphs show the mean and error bars represent the standard deviation (s.d.). Error bars are shown when three independent replicates were performed (i.e. not in screening conditions, e.g. Figs. 2a, c, f). All sequencing data were processed using CRISPResso 2.1.3 (Python 3.8). Microsoft Excel for Mac 16.19 (181109) was used to perform the unpaired, two-tailed t-tests (homoscedastic, i.e. assuming the two samples have equal or similar variance) that were used to calculate the p-values shown in Supplementary Table 1. GraphPad Prism 9.2.0 was used for final data analyses and generation of graphs. For the scatter plots in Fig. 2c and Supplementary Fig. 10, we used simple linear regression via GraphPad Prism 9.2.0. We did not predetermine sample sizes based on statistical methods. Investigators were not blinded to experimental conditions or assessment of experimental outcomes.
Supplementary Material
Supplementary Table 1: Editing frequencies in comparison experiments between intact and split PE architectures.
Supplementary Table 2: Editing frequencies and PBS/RTT lengths for a subset of comparison experiments between intact and split PE architectures.
Supplementary Table 3: Constructs used in this study.
Supplementary Table 4: Guide RNA and amplicon sequences, NGS barcodes and read depth of individual experiments.
Acknowledgements
Support for this work was provided by the National Institutes of Health (RM1 HG009490 and R35 GM118158 to J.K.J.). J.K.J. is additionally supported by the Desmond and Ann Heathwood MGH Research Scholar Award and the Robert B. Colvin, M. D. Endowed Chair in Pathology. J.G. was funded by the Deutsche Forschungsgemeinschaft (DFG, German Research Foundation) – Projektnummer 416375182. The project described was supported by a Career Development Award from the American Society of Gene & Cell Therapy (awarded to J.G.). The content is solely the responsibility of the authors and does not necessarily represent the official views of the American Society of Gene & Cell Therapy. K.P. was funded by the Deutsche Forschungsgemeinschaft (DFG, German Research Foundation) – Projektnummer 417577129. We thank M. K. Clement for technical advice, R. Zhou, J.Y. Hsu, A. Schmidts, J.F. Angstman, and P. Exconde for discussions and technical advice and L. Paul Pottenplackel for assistance with editing the manuscript.
Competing interests
J.K.J. and two other investigators who work on the NIH award supporting this research, but are not authors on this publication, are co-founders of and have a financial interest in SeQure, Dx, Inc., a company developing technologies for gene editing target profiling. J.K.J. also has, or had during the course of this research, financial interests in several companies developing gene editing technology: Beam Therapeutics, Blink Therapeutics, Chroma Medicine, Editas Medicine, EpiLogic Therapeutics, Excelsior Genomics, Hera Biolabs, Monitor Biotechnologies, Nvelop Therapeutics (f/k/a ETx, Inc.), Pairwise Plants, Poseida Therapeutics, and Verve Therapeutics. J.K.J.’s interests were reviewed and are managed by Massachusetts General Hospital and Mass General Brigham in accordance with their conflict of interest policies. J.K.J. is a co-inventor on various patents and patent applications that describe gene editing and epigenetic editing technologies. K.P. has a financial interest in SeQure Dx, Inc. K.P.’s interests and relationships have been disclosed to Massachusetts General Hospital and Mass General Brigham in accordance with their conflict of interest policies. P.K.C. is a paid consultant to and has financial interests in Nvelop Therapeutics (f/k/a ETx, Inc.). J.G., B.R.M, and J.K.J. are co-inventors on a patent application that has been filed by Mass General Brigham/Massachusetts General Hospital on engineered bipartite PE architectures, reduced size PEs, and non-MMLV-RT PE architectures.
Footnotes
Reporting Summary. Additional information on experimental designs, procedures, and analyses can be found in the Nature Research Reporting Summary.
Code availability
Custom python scripts that were generated for CRISPResso analyses are provided in the Supplementary Information.
Data availability
Plasmids encoding constructs used in this study have been deposited at Addgene and will be available at: https://www.addgene.org/Keith_Joung (Addgene No. 190104 – 190112). All targeted amplicon sequencing data have been deposited at the NCBI’s Sequence Read Archive (SRA) and can be accessed via http://www.ncbi.nlm.nih.gov/bioproject/86123739.
References
- 1.Anzalone AV et al. Nature 576, 149–157 (2019). [DOI] [PMC free article] [PubMed] [Google Scholar]
- 2.Chen PJ et al. Cell 184, 5635–5652 e5629 (2021). [DOI] [PMC free article] [PubMed] [Google Scholar]
- 3.Li X et al. Nat Commun 13, 1669 (2022). [DOI] [PMC free article] [PubMed] [Google Scholar]
- 4.Xu W et al. Nat Plants 8, 45–52 (2022). [DOI] [PubMed] [Google Scholar]
- 5.Wang Y et al. Signal Transduct Target Ther 4, 36 (2019). [DOI] [PMC free article] [PubMed] [Google Scholar]
- 6.Petri K et al. Nat Biotechnol 40, 189–193 (2022). [DOI] [PMC free article] [PubMed] [Google Scholar]
- 7.Kleinstiver BP et al. Nat Biotechnol 33, 1293–1298 (2015). [DOI] [PMC free article] [PubMed] [Google Scholar]
- 8.Tsai SQ et al. Nat Biotechnol 33, 187–197 (2015). [DOI] [PMC free article] [PubMed] [Google Scholar]
- 9.Kleinstiver BP et al. Nature 529, 490–495 (2016). [DOI] [PMC free article] [PubMed] [Google Scholar]
- 10.Gu J et al. J Mol Biol 305, 341–359 (2001). [DOI] [PubMed] [Google Scholar]
- 11.Das D et al. Protein Sci 10, 1936–1941 (2001). [DOI] [PMC free article] [PubMed] [Google Scholar]
- 12.Katano Y et al. Biosci Biotechnol Biochem 81, 2339–2345 (2017). [DOI] [PubMed] [Google Scholar]
- 13.Cote ML et al. Virus Res 134, 186–202 (2008). [DOI] [PMC free article] [PubMed] [Google Scholar]
- 14.Das D et al. Structure 12, 819–829 (2004). [DOI] [PubMed] [Google Scholar]
- 15.Bock D et al. Sci Transl Med 14, eabl9238 (2022). [DOI] [PMC free article] [PubMed] [Google Scholar]
- 16.Yu SF et al. Science 271, 1579–1582 (1996). [DOI] [PubMed] [Google Scholar]
- 17.Wohrl BM Viruses 11 (2019). [DOI] [PMC free article] [PubMed] [Google Scholar]
- 18.Berkhout B et al. J Virol 73, 2365–2375 (1999). [DOI] [PMC free article] [PubMed] [Google Scholar]
- 19.Lee YN et al. PLoS Pathog 3, e10 (2007). [DOI] [PMC free article] [PubMed] [Google Scholar]
- 20.Mills DA et al. J Bacteriol 178, 3531–3538 (1996). [DOI] [PMC free article] [PubMed] [Google Scholar]
- 21.Mohr S et al. RNA 19, 958–970 (2013). [DOI] [PMC free article] [PubMed] [Google Scholar]
- 22.Dai L et al. RNA 9, 14–19 (2003). [DOI] [PMC free article] [PubMed] [Google Scholar]
- 23.Blocker FJ et al. RNA 11, 14–28 (2005). [DOI] [PMC free article] [PubMed] [Google Scholar]
- 24.Stamos JL et al. Mol Cell 68, 926–939 e924 (2017). [DOI] [PMC free article] [PubMed] [Google Scholar]
- 25.Zhao C et al. Nat Struct Mol Biol 23, 558–565 (2016). [DOI] [PMC free article] [PubMed] [Google Scholar]
- 26.Zhao C et al. RNA 24, 183–195 (2018). [DOI] [PMC free article] [PubMed] [Google Scholar]
- 27.Kelley LA et al. Nat Protoc 10, 845–858 (2015). [DOI] [PMC free article] [PubMed] [Google Scholar]
- 28.Liu P et al. Nat Commun 12, 2121 (2021). [DOI] [PMC free article] [PubMed] [Google Scholar]
- 29.Liu B et al. Nat Biotechnol (2022). [Google Scholar]
- 30.Truong DJ et al. Nucleic Acids Res 43, 6450–6458 (2015). [DOI] [PMC free article] [PubMed] [Google Scholar]
- 31.Levy JM et al. Nat Biomed Eng 4, 97–110 (2020). [DOI] [PMC free article] [PubMed] [Google Scholar]
- 32.Toro N et al. RNA Biol 16, 1486–1493 (2019). [DOI] [PMC free article] [PubMed] [Google Scholar]
- 33.Hopp TP et al. Bio/Technology 6, 1204–1210 (1988). [Google Scholar]
Methods-only References
- 34.Rohland N et al. Genome Res 22, 939–946 (2012). [DOI] [PMC free article] [PubMed] [Google Scholar]
- 35.Gibson DG et al. Nat Methods 6, 343–345 (2009). [DOI] [PubMed] [Google Scholar]
- 36.Kleinstiver BP et al. Nature 523, 481–485 (2015). [DOI] [PMC free article] [PubMed] [Google Scholar]
- 37.Gramlich M et al. EMBO Mol Med 7, 562–576 (2015). [DOI] [PMC free article] [PubMed] [Google Scholar]
- 38.Clement K et al. Nat Biotechnol 37, 224–226 (2019). [DOI] [PMC free article] [PubMed] [Google Scholar]
- 39.Grünewald J et al. Datasets. Sequence Read Archive (SRA). http://www.ncbi.nlm.nih.gov/bioproject/861237 (2022).
Associated Data
This section collects any data citations, data availability statements, or supplementary materials included in this article.
Supplementary Materials
Supplementary Table 1: Editing frequencies in comparison experiments between intact and split PE architectures.
Supplementary Table 2: Editing frequencies and PBS/RTT lengths for a subset of comparison experiments between intact and split PE architectures.
Supplementary Table 3: Constructs used in this study.
Supplementary Table 4: Guide RNA and amplicon sequences, NGS barcodes and read depth of individual experiments.
Data Availability Statement
Plasmids encoding constructs used in this study have been deposited at Addgene and will be available at: https://www.addgene.org/Keith_Joung (Addgene No. 190104 – 190112). All targeted amplicon sequencing data have been deposited at the NCBI’s Sequence Read Archive (SRA) and can be accessed via http://www.ncbi.nlm.nih.gov/bioproject/86123739.