

Abstract
The fundamental biological importance and complexity of allosterically regulated proteins stem from their central role in signal transduction and cellular processes. Recently, machine learning approaches have been developed and actively deployed to facilitate theoretical and experimental studies of protein dynamics and allosteric mechanisms. We surveyed recent developments in applications of machine learning methods for studies of allosteric mechanisms, prediction of allosteric effects and allostery-related physicochemical properties, and allosteric protein engineering. We also reviewed the applications of machine learning strategies for characterization of allosteric mechanisms and drug design targeting SARS-CoV-2 virus. Continuous development and task-specific adaptation of machine learning methods for protein allosteric mechanisms will play an increasingly important role in bridging a wide spectrum of data-intensive experimental and theoretical technologies.
Keywords: Allostery, machine learning, mechanism, prediction, protein design, drug discovery
Protein allostery at the intersection of modern molecular biology and data science
Allosteric regulation serves as an efficient strategy for molecular communication and is a common mechanism employed by proteins for regulation of activity and adaptability [1–3]. Allosteric effects occur when a certain perturbation occurs at a distal site of a protein that is topographically distinct from the protein’s orthosteric function site and consequently modulates the protein’s activity [1–3]. Since the term “allosteric” was introduced in 1961 [4], protein allostery has been one of the focuses of structural biology and is often referred as the “second secret of life”, second only to the genetic code [5,6]. A quantitative elucidation of such fundamental and elusive phenomenon is critical to understanding life process and disease therapy [7–9]. It has been further proposed that all proteins are allosteric: even if the protein is not known to be allosteric, under given conditions such as the presence of appropriate allosteric effectors or mutations, the protein could be observed to be allosteric [10,11].
The remarkable progress and recent breakthroughs in X-ray crystallography (see Glossary), nuclear magnetic resonance (NMR) spectroscopy, fluorescence resonance energy transfer (FRET), and hydrogen–deuterium exchange mass spectrometry (HDXMS) experimental technologies have enabled structural and dynamic studies of large biomolecules at atomic resolution and are often employed as diagnostic tools of allosteric interactions and communications in signaling proteins [12]. Recent advances in single-particle cryogenic electron microscopy (cryo-EM) have enabled the determination of near-atomic resolution structures for well-ordered proteins and large macromolecular assemblies, breaking resolution barriers for studies of allosteric events and allosteric drug discovery [13–15].
Computational approaches have complemented experimental methods and provided detailed molecular insights into allosteric transformations and regulatory mechanisms. Molecular dynamics (MD) simulations-based and elastic network models (ENM)-based approaches represent two main flavors of computational methods to interrogate allosteric mechanisms based on protein dynamics [16–19]. Many other computational approaches correlate protein structural information at various levels with their allosteric functions. Allosteric molecular events involve a complex interplay of thermodynamic and dynamic changes that are difficult to observe, simulate and interpret. The quantitative elucidation of these highly dynamic processes continues to present formidable technical and conceptual challenges [20].
Due to its universal importance, protein allostery has been studied through wide range of aspects (Figure 1). The past decade has witnessed the rapid development of machine learning and deep learning (DL) techniques and their applications to model increasingly complex chemical and biological phenomena [21–23]. In this review, we surveyed recent developments and applications of machine learning to protein allostery along three main themes: prediction and analysis of allosteric mechanisms, properties prediction, and allosteric protein design. We also provide a perspective to the future development of computational and machine learning approaches for studies of protein allostery.
Figure 1.
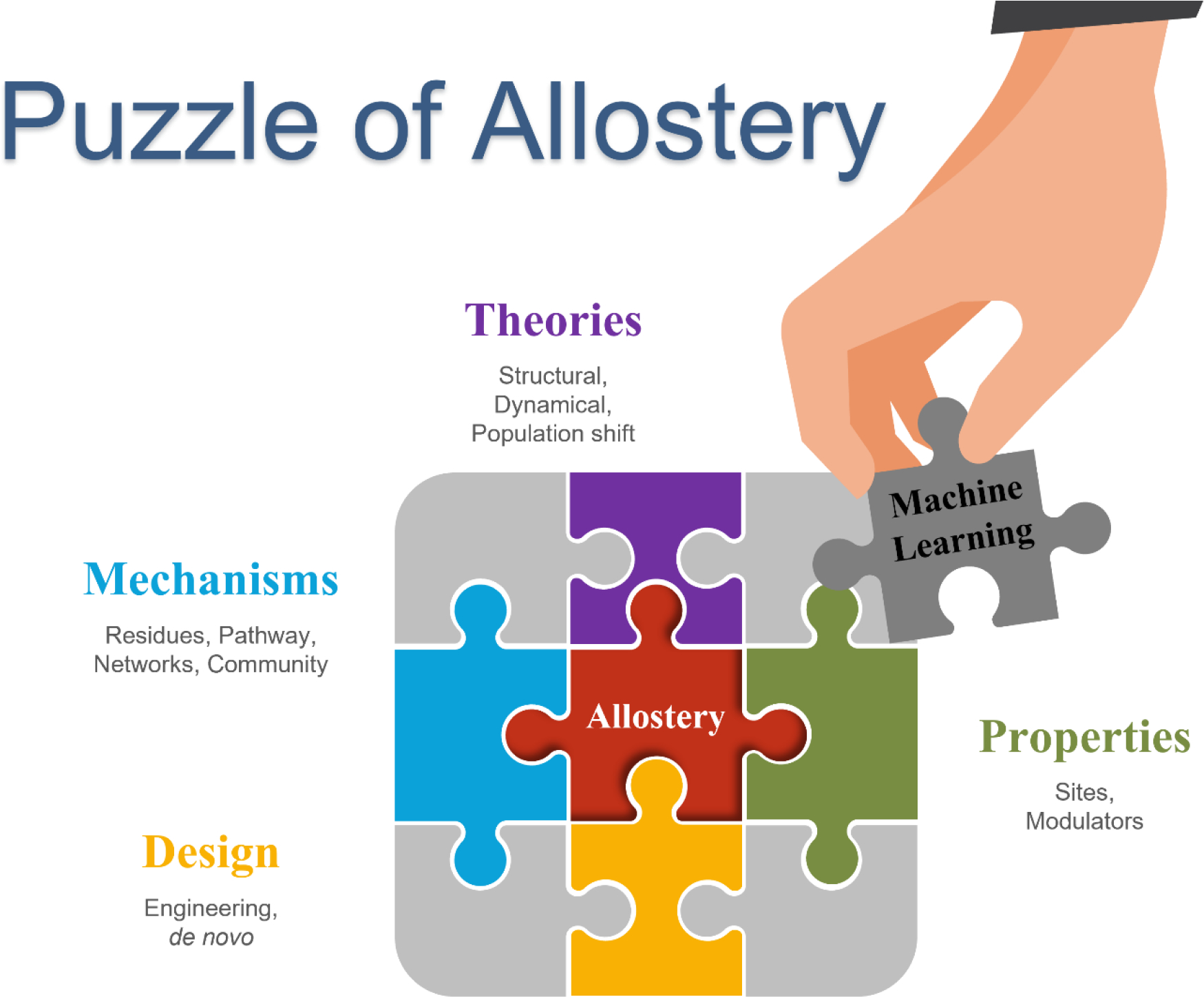
Solving the puzzle of allostery with machine learning. Protein allostery has been investigated from multiple aspects, including fundamental theories, allostery mechanisms, allostery related properties, and allosteric protein design. With increasing amounts of information and data related to allostery available, machine learning methods add another piece to the puzzle and have been employed more widely to study protein allostery in various areas.
Machine learning studies of protein allostery
Dynamics-driven allosteric models have described protein allosteric mechanisms as signal propagation through dynamically modulated functional motions that can occur in the absence of visible structural changes. The current view recognizes that allostery can often involve an equilibrium shift of the pre-existing conformational ensembles due to an effector binding [11,24]. In some perturbation-based simulation methods, mechanical forces are exerted on the allosteric proteins during simulations to probe protein dynamical and allosteric responses [25–29].
Combined with network models, these approaches can provide insight into mechanistic details of signaling pathways, predict the response to various perturbations and guide the identification of regulatory sites. Despite the established view that many proteins function as dynamic and versatile allosteric regulatory machines, our atomistic understanding of allosteric mechanisms is still at a rudimentary level, and our knowledge of allosteric functional states and allosteric communication networks that govern diverse protein functions is surprisingly limited. Due to the lack of a universal theory, current studies aim to interpret protein allostery at various protein structural levels (Figure 2). A substantial challenge in investigating the allosteric mechanisms for large multi-domain protein systems is the inherent difficulty of adapting experimental and computational methodologies to capture the intrinsic flexibility of these structures essential for functionality. The fundamental biological importance and complexity of these processes require a multi-faceted platform of integrated approaches for characterization of allosteric functional states and atomistic reconstruction of allosteric regulatory mechanisms. In this review, we detail how machine learning methods can be productively utilized to capitalize on the rapidly growing information and rich multidimensional data on protein dynamics and allosteric protein landscapes. We suggest that machine learning approaches have the potential to become a unifying data-centric research tool for synthesizing advances in theory and experimental technologies, ultimately leading to the development of robust and efficient computational models and expert systems for prediction of diverse allosteric effects in protein systems.
Figure 2.
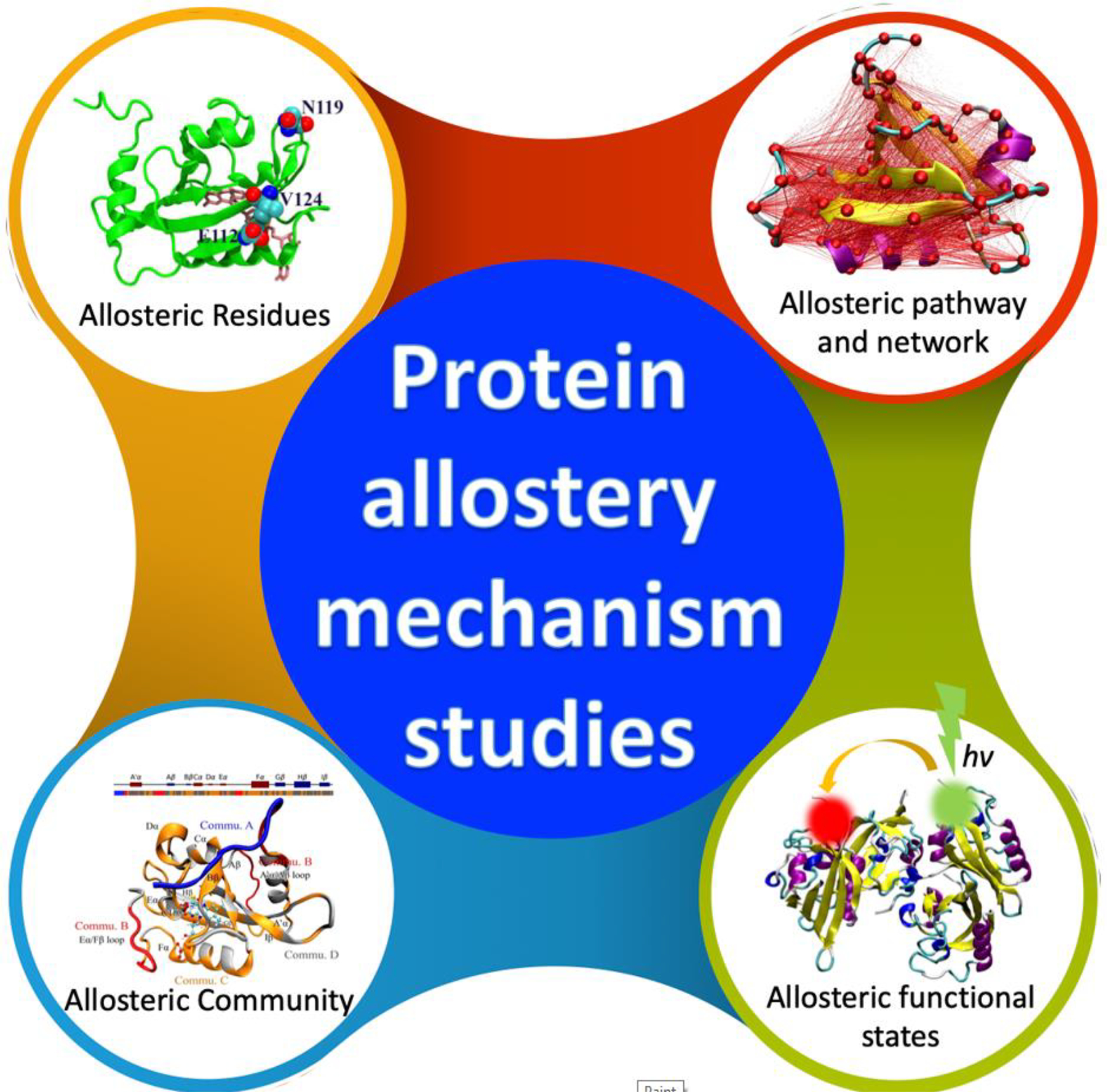
Allostery study facilitated by machine learning. Due to the lack of a universal theoretical framework for protein allostery, the mechanisms of allostery have been elucidated in multiple levels. At the residue level, key individual residues are identified as important for functions of the target allosteric proteins. At the pathway level, allosteric pathways consisting of multiple residues are identified as main communication channels between the allosteric site and the main functional site. In some cases, multiple pathways could form networks to enable allosteric signal transduction within the protein structure. The allosteric community comprises a group of closely related residues associated with allostery. Allosteric protein structure could be divided into several communities which interact with each other synergistically to carry out allosteric function. From dynamical point of view, proteins need to transition between different functional states when fulfilling their allosteric functions. These allosteric functional states could be identified through both computational and experimental studies.
Machine learning approaches for molecular simulations and characterization of allosteric functional states
Without a universally accepted fundamental theory, experimental observations remain as the foundation of protein allostery. New advances in experimental techniques often provide new insight into this ubiquitous phenomenon. For example, the recent breakthroughs in single molecule fluorescence resonance energy transfer (smFRET) technologies have enabled dynamic studies of large biomolecules. These advances provided semi-direct experimental observation of allostery related protein dynamics. Combined with MD simulations at microsecond scale, smFRET experiments could directly probe transitions among allosteric states with significant conformational changes [30–32]. Recently, the DeepFRET method was developed using a DL model to bridge experimental data and protein dynamics [33]. These emerging experimental advances provide a solid foundation for computational and theoretical studies of protein allostery seen in recent years.
Machine learning approaches have been widely employed to facilitate conformational sampling with MD simulations via optimal selection of reaction coordinates [34–37], enhanced conformational sampling by active learning [38–40] and even autonomous generation of equilibrium ensembles without performing MD simulations [41]. With help from machine learning methods, time-dependent structural changes can be quantitatively analyzed to provide insight into underlying allosteric mechanisms. Takami et al. [42] applied three time-series clustering methods, the unsupervised machine learning technique for time-series data, to analyze multiple Tight-Relaxed state transition trajectories of Human adult hemoglobin (HbA). These trajectories were classified by time-series clustering methods and analyzed to investigate the effect of oxygen molecules on the structural change of HbA.
In many other cases, the allostery related structural changes could not be easily recognized or characterized. Therefore, a number of machine learning models have been developed to identify structural features that can properly describe the slowest dynamics underlying conformational changes. These features could be used to model protein kinetics that underly allosteric processes [43–45].
Identifying key protein allosteric residues
As fundamental building blocks of protein, individual residues are the focus of many protein allosteric mechanism studies. Machine learning methods provide quantitative means to correlate global protein allostery with individual residues. Many studies aim to identify key residues for protein allostery through informative and insightful analysis of protein dynamics data using various machine learning methods. Zhou et al. [46] applied supervised learning methods, decision tree and neural networks, to build classification models for allosteric states based on the simulation data of the second PDZ domain from human PTP1E protein (PDZ2). These accurate classification models provide numerical measurement of importance of each residue for overall allostery. The key allosteric residues identified based on this importance has excellent agreement with experimental and computational studies. Similarly, Hayatshahi et al. [47] applied deep neural networks (DNNs) to build a classification model of the PDZ3 domain from the adaptor protein PSD95 to distinguish otherwise similar allosteric states using MD simulation data. Their classification model, a residue response map as a 2D property-residue map, could be constructed to represent allosteric effects as residue-specific properties. More recently, Do et al. [48] introduced a Gaussian-accelerated molecular dynamics (GaMD), DL, and free energy profiling workflow (GLOW), to characterize both activation and allosteric modulation of a G protein-coupled receptor (GPCR). A convolutional neural network (CNN) model was employed in GLOW to classify the residue contact maps, from which important residues could be identified.
Mapping of allosteric networks and communication pathways using machine learning
The nature and atomistic details of the allosteric communication between the allosteric site and the functional site are often difficult to dissect. The experimental approaches could reveal allosteric hotspots and potential communication pathways in protein structures. Using a combination of mutagenesis, mass spectrometry, amide HDXMS, and FRET studies, the atomistic details and allosteric pathways of the Hsp70 chaperone regulation mechanisms have been mapped, revealing the previously unrecognized dichotomy of allosteric control in the chaperone [49–51].
There are many machine-learning-based methods to identify allosteric pathway or networks. Graph theory-based methods are among the most widely used approaches. By mapping dynamic fluctuations onto a graph, network-based approaches can describe signal transmission via cascades of coupled residue fluctuations and characterize allosteric communication pathways in proteins. Zhu et al. [52] applied a graph neural network (GNN)-based neural relational inference (NRI) model, which adopts an encoder-decoder architecture to simultaneously infer latent interactions for probing protein allosteric processes as dynamic networks of interacting residues. From the MD trajectories, this model successfully learned the long-range interactions and pathways that can mediate the allosteric communications between distant sites. Machine learning methods could also be applied to develop various dynamic network models of allosteric interactions to decrypt the underlying mechanisms driving allosteric effects in proteins [53].
Other machine-learning-based methods use various correlation relations among residues to identify potential allosteric pathways. Zhou et al. [54] used relative entropy concept from information theory to develop the relative entropy-based dynamical allosteric network (REDAN) model. The relative entropy is used to measure the response of each residue pair to external perturbation. The potential allosteric pathways are identified as a series of short-range residue pairs with the most significant response. Botlani et al. [55] extended the underlying mechanism of allostery by exploring correlation between ensembles of protein in different allosteric states. They applied support vector machine (SVM) to quantitively evaluate these correlations using simulations representing different allosteric states of the same protein. Undirected weighted graph theory was also employed to identify the shortest pathway possible for allosteric signaling mechanisms. Yan et al. [56] proposed the node-weighted amino acid contact energy network (NACEN) to characterize and predict three types of functional residues, namely, hot spots, catalytic residues, and allosteric residues. These studies demonstrate the viability and diversity, as well as uncertainty, of using machine learning methods to evaluate allosteric contribution from individual residues.
Allosteric community models divide different residues into different groups, referred to as communities. These allosteric communities are not necessarily correlated with protein secondary or tertiary structural components and could provide a higher level of information than pathway and network models. Zhou et al. [57] and Ibrahim et al. [58] developed a community analysis algorithm based on their machine learning based classification model for protein allostery. The allosteric communities are built in a way such that the impacts of external perturbations on the distribution differences are maximum across different communities and minimum within the same community. This algorithm was applied to reveal allosteric mechanism of fungal circadian clock photoreceptor Vivid (VVD), as one member of light-oxygen-voltage (LOV) domain, upon photo activation. Interestingly, two distal loop regions were identified in the same community. This means that despite the distance between these two secondary structures, residue pairs across these two loop regions in VVD carry minimal allosteric significance. On the other hand, these two loops together carry significant contribution to overall allosteric effects.
Stetz and Verkhivker [59] applied a graph-based model on Hsp70 chaperones to construct residue interaction networks. The allosteric communities in Hsp70 were constructed as stable clusters of residues along the simulations. Verkhivker and co-workers [60,61] developed allosteric community models for Hsp90 through residue interaction networks analysis and noted that different allosteric communities were correlated through intermodular pathways for long-range communications. They also applied the community models to characterize functional mechanisms of Hsp90 allosteric modulation through binding with various allosteric modulators as well as other protein domains for its regulation [62,63]. Chen et al. [64] applied dynamic network analysis to build community model to reveal the regulatory effect on GPCR through binding with G-protein-mimicking Nanobody80 (Nb80). Both supervised (neural network) and unsupervised (principal component analysis) learning methods were used for feature extraction and key residues identification for dynamical response to binding event. In comparison to pathway and network models, allosteric community models do not target certain sites in protein and could provide a more comprehensive view of underlying protein allosteric mechanisms. Based on protein dynamics, these community models provide alternative views of protein structure related to allostery other than conventional primary, secondary, and tertiary structures. The communities within protein structures identified in these allosteric community models provide functional information regarding protein allostery in addition to convention secondary and tertiary structural information.
Machine learning approaches for prediction of allosteric binding sites, hotspots, and phenotypes, and applications in allosteric drug design
Allosteric drug development is among the most promising fields based on allostery for many reasons: the allosteric drugs could be more selective and less toxic with fewer side effects; they can either activate or inhibit proteins; they can be used in conjunction with orthosteric drugs. Discovery of allosteric drugs presents challenges beyond those encountered in orthosteric drug discovery. To address this challenge, Zhang and coworkers constructed AlloSteric Database (ASD) [65], which is a platform providing comprehensive information of allosteric proteins and their modulators. The database now contains a total of 1949 entries of allosteric proteins. ASBench [66], an optimized selection of ASD data, includes a core set with 235 unique allosteric sites and a core-diversity set with 147 structurally diverse allosteric sites. However, in many cases the location of allosteric sites is unknown. It is also difficult to accurately predict whether the drug will activate or inhibit the protein strength of the allosteric regulation [67,68]. Machine learning and deep learning, leveraging existing sample data to make predictions or decisions, can help predict the allosteric components.
Predicting allosteric sites
Several methods have been developed to detect and predict allosteric sites in proteins. These studies can be classified as sequence-based, structure-based, dynamics-based, normal mode analysis (NMA)-based, or combined prediction approaches [69]. Machine learning can help with the detection task since it can deal with numerous input features, including local or static features of pockets and delocalized or dynamic features of proteins (Table 1).
Table 1.
Representative allosteric site prediction methods
| Features | Methods | Datasetsa | Refs |
|---|---|---|---|
|
| |||
| Static pocket features | Naïve Bayes and neural networks | ASD and ASBench | [70] |
| Static pocket features | GCNN with XGBoost | ASD | [71] |
| Static pocket features | Automated machine learning | ASD and ASBench | [72] |
| Static pocket features | Random forest | ASD b | [73] |
| Static pocket features | Support vector machine | ASD | [74] |
| Pocket features with NMA perturbation | Support vector machine | ASBench | [75] |
| Pocket features with NMA perturbation | Logistic regression | ASBench | [76] |
| Features at residue level | Random forest | ASBench | [77] |
| Crystal additive location | DBSCAN | ASD and ASBenchc | [78] |
The original datasets used to obtain allosteric site data. The data was filtered for high-resolution and non-redundant structures, individually.
The PDBbind database was used to obtain information on orthosteric sites.
The RCSB protein data bank was used to obtain protein-crystallographic additives complexes.
The static features, such as pocket volume, pocket flexibility, and pocket hydrophobicity, characterize the conformation of protein pockets, and further provide information for classifiers to identify allosteric sites. Akbar et al. [70] characterized allosteric pockets using a set of physicochemical descriptors and trained a predictive model based on Naïve Bayes and artificial neural networks. The predictive models were capable of prioritizing allosteric pockets in a set of pockets found on a given protein and were encapsulated in publicly accessible program ALLO. Tian and Xiao et al. adapted an ensemble learning method combining eXtreme gradient boosting (XGBoost) and graph convolutional neural networks (GCNNs), and an automated machine learning method (AutoGluon and AutoKeras) to predict plausible allosteric sites. They deployed both models to Protein Allosteric Site Server (passer.smu.edu) [71,72]. Chen et al. [73] used the structures of the sites and the co-crystallized ligands to calculate 43 structural descriptors. These structural descriptors were used to build a three-way predictive model based on random forest to characterize protein-ligand binding sites as allosteric, regular or orthosteric. Huang et al. applied SVM for prediction of allosteric sites using static pocket features as well, leading to a web server Allosite [74].
Dynamic features were also used for allosteric site prediction because allostery is a dynamic behavior of the whole protein. Greener et al. utilized perturbed NMA and pocket descriptors in SVM to sort pockets in proteins and developed the AlloPred web server to predict allosteric pockets [75]. Song et al. [76] combined pocket features with NMA-based perturbation analysis to build a logistic regression model, AllositePro, to predicts allosteric sites in proteins.
Other features were also explored for allosteric site prediction. Mishra et al. [77] used various features at residue level, including amino acid physicochemical properties, rate of residue evolution, and features for protein geometry and dynamics, to build the Active and Regulatory site Prediction (AR-Pred) model. Fogha et al. [78] found that crystal additives (CA), which stabilize proteins during the crystallization process, tend to aggregate in protein hotspots, especially near the binding cavities, so that it can be a criterion to make site-type decisions. They proposed an efficient and easy way to use the structural information of CA to identify allosteric sites.
With comparable accuracy but using different methods, these prediction models for allosteric sites provide ample choice for users. One could also apply methods using different strategies in the same study and use the consensus results for better outcome. The workflow of an allosteric site analysis web-server AlloFinder is illustrated in Figure 3.
Figure 3.
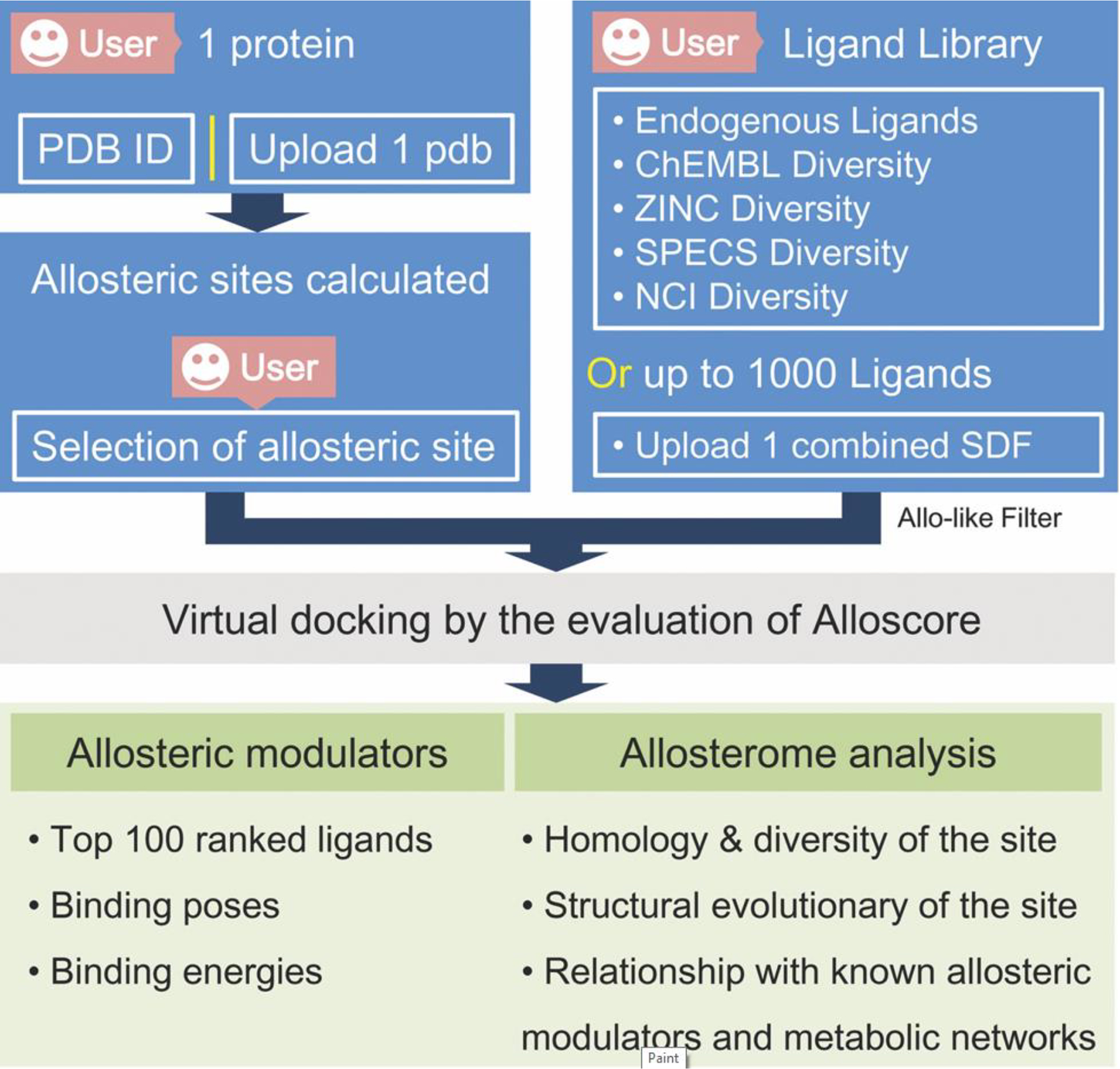
The workflow of AlloFinder. After the user uploads a query protein to AlloFinder, all putative allosteric sites on the protein are predicted. The user can choose one allosteric site to screen a predefined ligand library virtually. The pocket-generated pharmacophore model for the selected allosteric site is generated for quickly ruling out unbound compounds in the library. Conformational sampling of an ensemble of docked conformations are performed for each compound. The most favorable binding energy of each compound is evaluated and ranked. The top 100 compounds are provided by the AlloFinder server. Finally, the predicted allosteric sites and modulators are harnessed to perform allosterome mapping analyses in the human proteome. [94]
The reversed allosteric communication theory has been proposed [79] and achieved several successful studies. It is based on the premise that allosteric signaling in proteins is bidirectional and can propagate from an allosteric to an orthosteric site and vice versa [80,81]. Some of the reversed allosteric communication approaches are rooted in the dynamic network-based models of inter-residue interaction [82,83]. An integrated computational and experimental strategy exploited the reversed allosteric communication concepts to combine MD simulations with Markov state model (MSM) for characterization of binding shifts in the protein ensembles and identification of cryptic allosteric sites [84]. In MSM, the dimensionality reduction techniques are employed to generate suitable collective variables to characterize protein conformational space. The simulation of allosteric protein could be projected into the space using these collective variables as the distribution in the conformational space. Clustering methods are generally applied to cluster these distribution into metastable states. Accordingly, transition probability among these metastable states could be estimated based on the simulation data.
Using the reversed allosteric communication concept, machine learning methods enable reconstruction and analysis of the comparative perturbed ensembles of the allosteric states and characterize redistribution of dynamic states in the inhibitor-bound versus inhibitor-free systems following allosteric binding [85]. It should be pointed out that these machine-learning based models either as classification or regression models cannot account for the signal transduction between the distal sites and function related active sites as no such information is included in the training data to develop these models.
As the predicted allosteric sites could potentially be used directly for allosteric drug development and due to the recent breakthrough of protein structure prediction including AlphaFold2 and many others, the allosteric sites prediction methods possess huge potential and significance related to protein allostery.
Machine learning models based on deep mutational scanning
Currently, experimental data remain as the primary foundation for the development of allostery related computational models for understanding, predicting, and engineering biophysical properties of allosteric proteins. Emerging deep mutational scanning (DMS) experiments combine saturation mutagenesis of a protein with a high-throughput functional test and deep sequencing and provide unbiased and systematic single mutational information of target proteins. Such large and quantitative datasets enabled machine learning approaches to predict allosteric properties from sequence. Leander et al. [86] carried out DMS of four homologous bacterial allosteric transcription factors (aTF). They further developed prediction models using neural network model and genetic algorithms to identify hotspots of homolog proteins and to predict the structural and molecular properties of allosteric hotspots. Faure et al. [87] generated mutagenesis libraries of the C-terminal SH3 domain of the human growth factor receptor-bound protein 2 (GRB2-SH3) and third PDZ domain from the adaptor protein PSD95 (PSD95-PDZ3) domains containing both single and double amino acid substitutions. Neural network model was developed using DMS data to predict the binding free energy change upon single amino acid substitution on both systems. These prediction models were used to map the energetic and allosteric landscapes of the target domains.
These recent studies demonstrated the potential of DMS data to facilitate the development of machine learning based methods for protein allostery related properties at residue level and even theoretical landscaping models for protein allostery.
Evaluating allosteric effectors
Binding with allosteric modulators is the main allosteric perturbation in many cases. Some studies aimed to distinguish allosteric modulators from non-allosteric modulators. Several physically relevant compound descriptors of molecules were computed, and the feature differences were then connected into chemical property differences. Wang et al. [88] and Smith et al. [89] concluded that allosteric modulators are generally more aromatic, structural rigid, and more hydrophobic. This general idea can help with preliminary screening of allosteric modulators.
Similar to using machine learning models to identify allosteric sites for proteins, machine learning models could be developed to classify modulators as allosteric or non-allosteric. For example, Hou et al. [90] trained six types of machine learning models using different combinations of features for a an 11-class classification task with 10 GPCR subtype classes and a random compounds class. It is the first work on the multi-class classification of GPCR allosteric modulators.
Other studies focus on developing generative models to build and evaluate allosteric inhibitors targeting various receptors. Different methods and training data were used to develop various machine learning based models with comparable performance. Bian et al. [91] first established a general molecule generation model (g-DeepMGM) with a half million compounds collected from the ZINC database, and then constructed a target-specific molecule generation model (t-DeepMGM) based on the transfer learning process of reported cannabinoid receptor 2 (CB2) ligands. Yang et al. [92] first trained a Transformer-encoder-based generator on ChEMBL’s 1.6 million data sets to learn the grammatical rules of known drug molecules. Transfer Learning is used to introduce the prior knowledge of drugs with known activities against particular targets into the generative model to construct new molecules similar to the known ligands. Reinforcement Learning is used to combine the generative model and the predictive model to generate molecules with drug-like properties that are expected to bind well with the target.
Vennila et al. [93] utilized the voxelized representation of five different conformational states of PDK1 allosteric site, PIF pocket, to predict 1D SMILES imparted in LiGANN pipeline in playmolecule platform, in which, for a given protein shape, a generative adversarial neural network (GANN) produces complementary ligand shapes in a multimodal fashion. Huang et al. [94] built AlloFinder that identifies potential endogenous or exogenous allosteric modulators and their involvement in human allosterome. AlloFinder automatically amalgamates allosteric site identification, allosteric screening, and allosteric scoring evaluation of modulator–protein complexes to identify allosteric modulators, followed by allosterome mapping analyses of predicted allosteric sites and modulators in the human proteome. More recently, Miljkovic et al. [95] applied random forest, SVM, and DNN models to predict different classes of kinase inhibitors targeting different allosteric sites. Compounds were represented using molecular fingerprints without other structural information being considered. As the authors were struck by consistently good performance across different methods used in this study, this demonstrated that machine learning methods in general could extract key chemical features for certain properties using appropriate features.
Identifying receptors for allosteric inhibitors
In some scenarios, potential receptors need to be identified for known substrates with significant pharmaceutical effects. These substrates may include allosteric effectors interacting with pharmacology networks. Rodrigues et al. [96] developed a novel strategy to identify potential targets of known allosteric effectors using self-organizing map–based prediction of drug equivalence relationships (SPiDER) model. The SPiDER model uses a consensus of unsupervised self-organizing maps, consensus scoring, and statistical analysis to identify potential targets for known active substrates. Using this approach, the authors identified 5-lipoxygenase as an allosteric inhibiting target for β-lapachone as a clinical-stage, natural product with thorough validation. As an emerging field of computer-aided molecule design, there are many potential directions that machine learning methods could be applied specifically for allosteric modulator development.
Machine learning studies for allosteric protein design
One of the goals of studying protein allostery is developing novel proteins which carry improved or novel allosteric functions. As this is a new area, large amounts of data related to the allostery of different proteins have yet to be employed in the developing process. In an early study, Zayner et al. [97] studied over 100 mutations of Avena sativa light-oxygen-voltage domain 2 (AsLOV2) as a light-activated protein. In this experimental study, the authors used various experimental methods to characterize the target mutations of AsLOV2. The biggest lesson learned through this study was that most mutations, which were expected to be highly disruptive substitutions, turned out to be modest or had no effect on function, even with many mutations displaying enhanced photoactivity. These counterintuitive results signify the importance of deeper and more comprehensive understanding of protein allostery in the effort to design an enhanced or novel allosteric molecular apparatus.
Weinkam et al. [98] used simulation data of a set of ten proteins and their mutations to build prediction models for allostery. They built a decision-based machine learning model with a wide range of features, including geometric- and energy-based features, to predict mutational effect on protein allosteric activity. This prediction function no doubt will help with the efforts of protein engineering to develop modified protein allosteric activities and functions. Xiao et al. [99] employed systematic machine learning approaches to analyze allostery of thrombin as a multifunctional serine protease at conformational ensemble level. Their study provided mechanistic insight into allostery of one key thrombin mutant with ample intramolecular interaction details.
Currently, successful cases of allosteric protein design are still mainly based on special expertise and experience of researchers. For example, García-Fernández et al. [100] developed a novel biosensor by fusing two ion channels, a tetrameric viral Kcv channel and the dimeric mouse TREK-1 channel, to a physiologically unrelated membrane protein, GPCR. The GPCR displayed regulatory effort toward both fused ion channels. A great deal of effort was spent to fine tune the length of linkers connecting GPCR with two ion channels. The success of the fusion between two physiologically unrelated allosteric proteins to design novel biosensors indicates a direction for computational studies based on structural and simulation data and machine learning modeling to identify potential candidates and appropriate design for linkers. In a more traditional study, D’Amico et al. [101] developed enhanced tryptophan synthases through mutations at a distant, surface-exposed network residue. It is expected that data-driven strategies using machine learning methods could catalyze the breakthrough in allosteric proteins designs in the near future.
There is at least one study using a machine learning method to model evolutionary relations among allosteric proteins. Astl and Verkhivker [102] used a systematic approach and carried out ENM analysis of 235 unique allosteric protein entries from ASBench. Using residue interaction networks models of the target proteins, they evaluated the coevolutionary of key residues for different allosteric proteins and identified unifying molecular signatures shared by allosteric systems. The application of their models on protein kinases revealed molecular signatures of known regulatory allosteric residues. Allostery related protein evolution is relatively uncharted area, mainly due to the lack of unified theoretical models for protein allostery. The applications of suitable machine learning models to correlate protein allosteric mechanism with evolution point to a new direction of deciphering protein allostery.
Concluding remarks and future perspectives
Allostery is an intrinsic but elusive ubiquitous phenomenon in proteins. We have reviewed research progress in protein allostery using machine learning methods in various frontiers. Although many theories and models were developed to interpret this phenomenon, there is no simple equation to quantify allostery. Machine learning helps explain the mechanism in different dimensions, residues, pathways, networks, and communities. As there is no universal theory for all allosteric regulations, it could be the case that protein allostery theory or mechanisms cannot be unified given the diversity of proteins structures and dynamical behaviors.
Important implementations of protein allostery include prediction of various protein allostery related properties. Suitable for processing large amounts of data and developing reliable prediction models in general, data-driven machine learning methods have been applied to develop computational models to predict protein allosteric binding sites and modulators. Those prediction models have been made available with easy access to the research community and have been widely used in many studies related to protein allostery. The biggest impact made to protein allostery studies using machine learning methods is mainly in applications. This was demonstrated by the emphasis on machine learning method-based approaches that focused on allosteric mechanisms of SARS-CoV-2 (Box 1) and modulators as ligands to target various receptors in SARS-CoV-2 (Box 2).
Box 1. Allosteric mechanisms for SARS-CoV-2 viral spike protein.
COVID-19, caused by severe acute respiratory syndrome coronavirus 2 (SARS-CoV-2), emerged in 2019 and then quickly spread around the globe. The infection involves the attachment of the receptor-binding domain (RBD) of the SARS-CoV-2 viral spike (S) protein to the angiotensin-converting enzyme 2 (ACE2) receptors on the peripheral membrane of host cells [103]. The open and closed conformations of ACE2 differ from each other by the degree of opening of the catalytic site cleft of the peptidase domain. These structural insights identified ACE2 as a viable target to block S1 recognition through allosteric control of open–closed transitions necessary for S1 recognition [104,105].
Extensive studies have revealed that SARS-CoV-2 shares many biological features with but has higher infectivity than SARS-CoV [106]. Delgado et al. [107] aimed to understand the host receptor recognition mechanism of SARS-CoV-2 to explain this. Affinity propagation algorithm, an unsupervised machine learning algorithm was employed for clustering analysis of CoV and CoV-2 spike-ACE2 systems. Trozzi et al. [108] developed a Collective Variable-guided Convolutional Neural Network (CV-CNN) model as a novel scheme to capture the functional and structural differences of ACE2 extracellular N-terminal peptidase domain (PD). The REDAN model was employed to obtain the pathway information of residue-residue interactions that characterize ACE2 PD functional dynamics. Uyar et al. [109] distinguished several all-atom molecular dynamics simulations by linear discriminant analysis (LDA) method to show persistent differences in the ACE2 structure upon binding. This allows the prediction for which compounds lead to free versus bound states and to pinpoint long-range ligand-induced allosteric changes in the ACE2 structure. Ray et al. [103] focused on the correlations between the RBD and residues in distant, allosteric sites. These computational studies provided insight at the atomistic level into the infection process of SARS-CoV-2 virus and paved the way for allosteric drug design to cope with COVID-19.
Box 2. Allosteric drug development against SARS-CoV-2 virus.
During the COVID-19 pandemic, developing drugs based on an allosteric mechanism of recognition between SARS-CoV-2 spike protein and ACE2 proteins was an important strategy. Iyengar [110] used the machine learning method Partial Order Optimum Likelihood (POOL) to predict allosteric binding sites in protein structures from SARS-CoV-2. Some other studies focused on identifying allosteric modulators for either SARS-CoV-2 spike proteins or ACE2 as potential drug. Karki et al. [104] introduced an application of a deep neural network-based drug screening method, validating it using a docking algorithm on approved drugs for drug repurposing efforts, and extending the screen to a library of 750,000 compounds. Jain et al. [111] built predictive models, using both machine learning and pharmacophore-based modeling, with the screening data from the SARS-CoV-2 cytopathic effect reduction assay. Experimental testing with live virus provided 100 active compounds out of the predicted hits from the screening result of optimized models. SARS-CoV-2 main protease (Mpro) is required for maturation of the virus and infection of host cells, so the key question is how to block the activity of Mpro. Kaptan et al. [112] combined atomistic simulations with machine learning methods, the Gaussian Mixture Model (GMM) and the Partial Least Squares based Functional Mode Analysis (PLS-FMA) model, and found that the enzyme regulates its own activity by a collective allosteric mechanism that involves dimerization and binding of a single substrate. Their results suggest that dimerization of main proteases is a general mechanism to foster coronavirus proliferation and propose a strategy that does not depend on the frequently mutating spike proteins at the viral envelope. Verkhivker and co-workers [113–119] have done a series of computational work to explore allosteric mechanisms and potential regulatory effects of SARS-CoV-2 spike proteins for different strains (Figure I). They applied different allosteric models using various machine learning methods to formulate allosteric interaction pathway and networks model for the binding of the SARS-CoV-2 spike proteins. These studies demonstrate the positive impact that advanced and mature computational modeling of protein allostery using machine learning methods could exert on real global public health emergency.
Figure I.
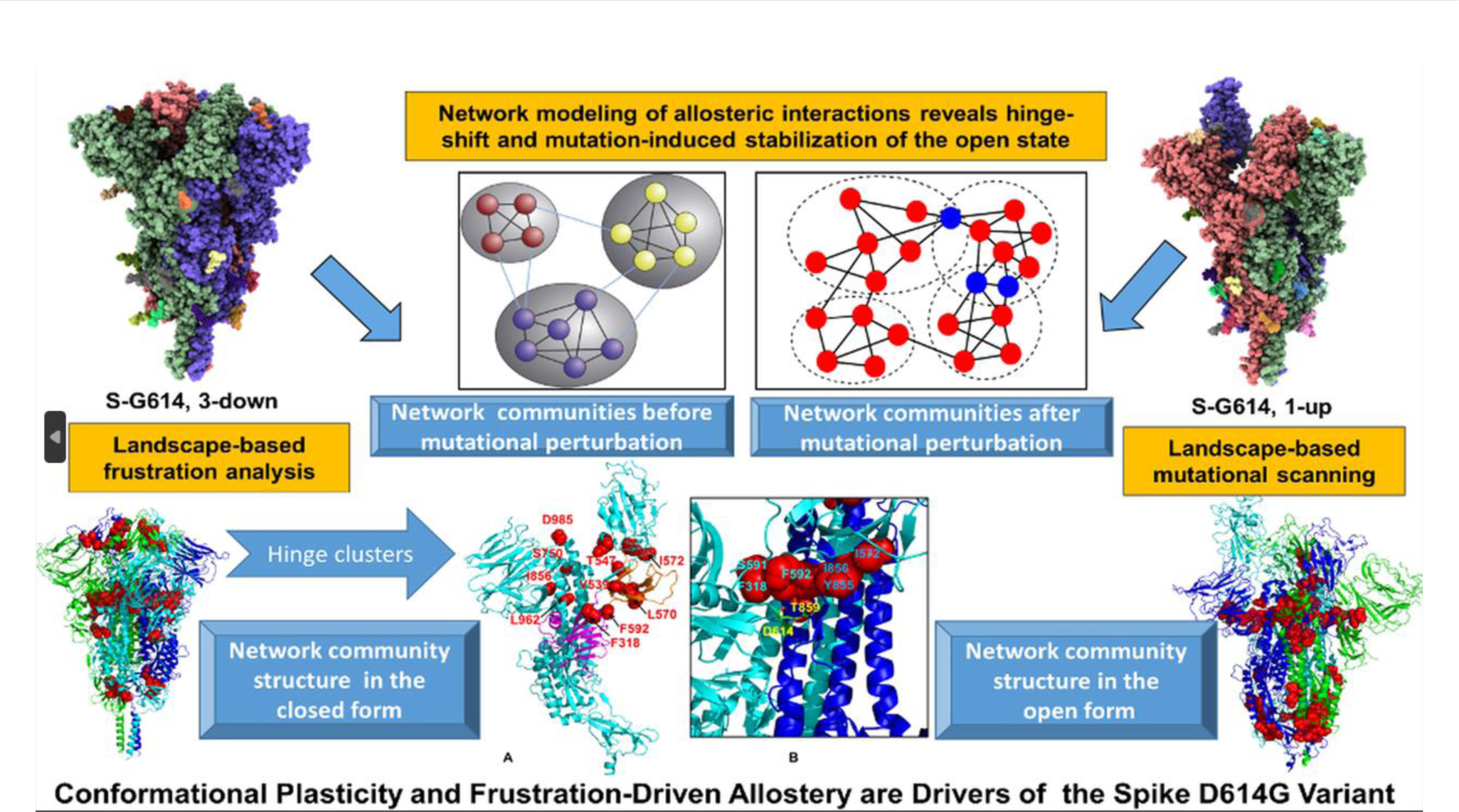
Landscape-based protein stability analysis and network modeling of multiple conformational states of the SARS-CoV-2 spike D614G mutant. Multiple computational methods and models were employed in this study of SARS-CoV-2 spike protein allostery focusing in on its D614G mutant. Coarse-grained simulations were carried out for trimers of this protein. Residue interaction networks were identified based on both dynamic correlations and coevolutionary residue couplings. A community model was built based on a graph theory representation of protein structure. The impact on protein allostery through mutational perturbation was revealed through both network and community models. The ensemble-based analysis characterizes the dynamic signatures of the conformational landscapes for the target protein. The combination of multiple allosteric models reveals a hinge-shift mechanism leading to the increased stability of the open form in the mutant. [119]
Despite the promising developments presented in this review, the readers should also be aware of the limitation of the machine learning based methods for protein allostery study. In general, the usage of a machine learning model is restricted by the training data source and model construction. Machine learning based models may not lead to a universal theory to explain general allosteric events.
Nevertheless, given the success in numerous studies of protein allostery using machine learning methods, we expect to see the current trend to continue with more applications using machine learning methods suitable for protein systems, especially dynamical processes. Due to the uniqueness of protein systems, there is a need to develop machine learning methods for different purposes, including dimensionality reduction methods with accurate decoding functionality, time-dependent data series analysis, and features suitable for chemical structures, protein structures and protein assembly structures. With more data available and deeper insight into protein allosteric mechanisms, we expect to see systematic development in allosteric protein engineering and even de novo allosteric protein design. With continuous accumulation of more data and information in chemical and biological sciences related to protein allostery, there is increasing room and opportunities for advanced and specific machine learning methods to be integrated into this interdisciplinary field (see Outstanding questions).
Outstanding questions.
How the underlying mechanisms for protein allostery could be formulated at different structural levels, including individual residues, allosteric pathways, and networks?
How could the advanced experimental technics, such as single molecule FRET, be used to characterize protein allostery at microscopic level?
Could protein ensembles generated from simulations be used directly to shed light on underlying allosteric mechanisms?
With their strength in analyzing large amount of data to build highly performing prediction models, how could machine learning methods be used to develop prediction models for allosteric sites?
How could potential modulators targeting allosteric proteins with desired properties be effectively developed using machine learning based approaches?
Is it feasible to engineer or even develop novel allosteric proteins with desired properties? If so, how could machine learning methods be used to facilitate these developments?
How could machine learning-based computational analysis and prediction methods related to protein allostery be used to address the pharmaceutical challenges caused by COVID-19 pandemic?
Highlights.
Machine learning methods provide unprecedented opportunities for the studies in understanding and exploiting protein allostery.
Large amount of data including simulations related to protein allostery were subjected to various types of machine learning methods to provide deeper insight into underlying allosteric mechanisms at levels of allosteric residues, pathways and networks, communities, and protein ensembles.
Machine learning methods have done exceptionally well to develop prediction models for protein allosteric properties, including allosteric sites and effectors.
Allosteric protein engineering and design are emerging fields with accumulating information for further applications of machine learning methods.
Protein allostery played a key role in many computational studies using machine learning methods targeting SARS-CoV-2 virus, aiming to mitigate the COVID-19 pandemic.
Acknowledgements
This work was supported by the National Institute of General Medical Sciences of the National Institutes of Health under Award No. R15GM122013.
Glossary
- Allosterome
is systematic identification of protein allosteric interactions. It provides entire allosteric landscapes for related proteins of interest.
- Angiotensin-converting enzyme 2 (ACE2)
a vital element in the renin-angiotensin-aldosterone system (RAAS) pathway that is critical to the regulation of processes such as blood pressure, wound healing, and inflammation. ACE2 helps modulate the many activities of angiotensin II (ANG II). When the SARS-CoV-2 virus binds to ACE2, it prevents ACE2 from performing its normal function to regulate ANG II signaling.
- Cryogenic electron microscopy (Cryo-EM):
a microscopy technique applied to samples cooled to cryogenic temperatures. It can be used to provide 3D structural information on biological molecules and assemblies by imaging non-crystalline specimens. The structures of the samples are preserved by embedding in an environment of low temperature.
- Elastic network model (ENM)
a computational model used to describe proteins as structured elastic objects at a coarse-grained level. In ENM, proteins are treated as points in space with mass and connected by springs. ENMs can provide essential vibrational dynamics associated with the given structure and have been widely used to study protein dynamics, function, and conformational changes.
- Fluorescence resonance energy transfer (FRET)
a distance-dependent physical process by which energy is transferred nonradiatively from an excited molecular fluorophore (the donor) to another fluorophore (the acceptor) by means of intramolecular long-range dipole–dipole coupling. FRET can be an accurate measurement of molecular proximity at distances between 10 and 100 Å and highly efficient if the donor and acceptor are positioned within the Förster radius (the distance at which half the excitation energy of the donor is transferred to the acceptor, typically 3–6 nm).
- Hydrogen–deuterium exchange mass spectrometry (HDXMS)
protein is exposed to D2O and induces rapid amide H → D exchange in disordered regions that lack stable hydrogen-bonding. Tightly folded elements are much more protected from HDX, resulting in slow isotope exchange that is mediated by the structural dynamics (“breathing motions”) of the protein.
- Machine learning
part of artificial intelligence, which leverages data to improve performance on sets of tasks. It builds a model based on sample data, known as training data, to make predictions or decisions without being explicitly programmed to do so.
- Markov state model (MSM)
a theoretical model employed to study the allosteric regulatory events. The first step is using robust dimensionality reduction techniques to identify suitable collective variables. Simulation data can be projected and represented by these collective variables. Clustering methods are applied to divide the projection of simulation into metastable states. Transition probabilities among these metastable states could be estimated based on the simulation data.
- Molecular dynamics (MD) simulation
a computational method for analyzing the movements of atoms and molecules in space. The MD trajectories of atoms and molecules are determined by numerically solving Newton’s equations of motion for a system of interacting particles. The forces between the particles and their potential energies are calculated using molecular mechanics force fields. These simulations can capture a wide variety of important biomolecular processes, including conformational change, ligand binding, and protein folding.
- Normal mode analysis (NMA)
provides vibrational modes accessible to a system in an equilibrium state, approximating the system in harmonic potentials. This computational model has been applied to identify and characterize the slow and global motions in a macromolecular system.
- Nuclear magnetic resonance (NMR) spectroscopy
used to obtain information about the structure and dynamics of proteins, nucleic acids, and their complexes. The sample is placed inside a powerful magnet to measure the absorption of radio frequency signals. Types of nuclei and distances between adjacent nuclei can be determined from absorption information and can be used to determine the overall structure of the protein. NMR spectroscopy can monitor both conformations and dynamics and can be applied to partially unfolded proteins.
- X-ray crystallography
an experimental technique to determine the 3D structure of a compound in crystal. The crystallized sample is exposed to an X ray beam to obtain diffraction patterns. These patterns can be processed to yield information about the crystal packing symmetry, the size of the repeating unit, and a map of the electron density. The molecular structure can be built and refined based on electron density information from diffraction patterns.
Footnotes
Declaration of interests
There are no conflicts to declare.
Publisher's Disclaimer: This is a PDF file of an unedited manuscript that has been accepted for publication. As a service to our customers we are providing this early version of the manuscript. The manuscript will undergo copyediting, typesetting, and review of the resulting proof before it is published in its final form. Please note that during the production process errors may be discovered which could affect the content, and all legal disclaimers that apply to the journal pertain.
References
- 1.Zha J et al. (2022) Explaining and Predicting Allostery with Allosteric Database and Modern Analytical Techniques. J. Mol. Biol. 434, 167481. [DOI] [PubMed] [Google Scholar]
- 2.Lu S et al. (2019) Allosteric Methods and Their Applications: Facilitating the Discovery of Allosteric Drugs and the Investigation of Allosteric Mechanisms. Acc. Chem. Res. 52, 492–500 [DOI] [PubMed] [Google Scholar]
- 3.Liu J and Nussinov R (2016) Allostery: An Overview of Its History, Concepts, Methods, and Applications. PLOS Comput. Biol. 12, e1004966. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 4.Monod J and Jacob F (1961) General Conclusions: Teleonomic Mechanisms in Cellular Metabolism, Growth, and Differentiation. Cold Spring Harb. Symp. Quant. Biol. 26, 389–401 [DOI] [PubMed] [Google Scholar]
- 5.Fenton AW (2008) Allostery: an illustrated definition for the ‘second secret of life.’ Trends Biochem. Sci. 33, 420–425 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 6.Monod J (1971) Chance and Necessity: An Essay on the Natural Philosophy of Modern Biology, New York: Vintage Books [Google Scholar]
- 7.Motlagh HN et al. (2014) The ensemble nature of allostery. Nature 508, 331–339 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 8.Freiburger LA et al. (2011) Competing allosteric mechanisms modulate substrate binding in a dimeric enzyme. Nat. Struct. Mol. Biol. 18, 288–294 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 9.Nussinov R et al. (2013) The (still) underappreciated role of allostery in the cellular network. Annu. Rev. Biophys. 42, 169–189 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 10.Gunasekaran K et al. (2004) Is allostery an intrinsic property of all dynamic proteins? Proteins Struct. Funct. Bioinforma. 57, 433–443 [DOI] [PubMed] [Google Scholar]
- 11.Tsai C-J et al. (2008) Allostery: Absence of a Change in Shape Does Not Imply that Allostery Is Not at Play. J. Mol. Biol. 378, 1–11 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 12.Grutsch S et al. (2016) NMR Methods to Study Dynamic Allostery. PLOS Comput. Biol. 12, e1004620. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 13.Merk A et al. (2016) Breaking Cryo-EM Resolution Barriers to Facilitate Drug Discovery. Cell 165, 1698–1707 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 14.Coffino P and Cheng Y (2022) Allostery Modulates Interactions between Proteasome Core Particles and Regulatory Particles. Biomolecules 12, 764. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 15.Gulati S et al. (2019) Cryo-EM structure of phosphodiesterase 6 reveals insights into the allosteric regulation of type I phosphodiesterases. Sci. Adv. 5, eaav4322. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 16.Raman S (2018) Systems Approaches to Understanding and Designing Allosteric Proteins. Biochemistry 57, 376–382 [DOI] [PubMed] [Google Scholar]
- 17.Yamato T and Laprévote O (2019) Normal mode analysis and beyond. Biophys. Physicobiology 16, 322–327 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 18.Hollingsworth SA and Dror RO (2018) Molecular Dynamics Simulation for All. Neuron 99, 1129–1143 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 19.Na H and Song G (2014) Bridging between normal mode analysis and elastic network models. Proteins Struct. Funct. Bioinforma. 82, 2157–2168 [DOI] [PubMed] [Google Scholar]
- 20.Wodak SJ et al. (2019) Allostery in Its Many Disguises: From Theory to Applications. Structure 27, 566–578 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 21.Yang KK et al. (2019) Machine-learning-guided directed evolution for protein engineering. Nat. Methods 16, 687–694 [DOI] [PubMed] [Google Scholar]
- 22.Butler KT et al. (2018) Machine learning for molecular and materials science. Nature 559, 547–555 [DOI] [PubMed] [Google Scholar]
- 23.Mater AC and Coote ML (2019) Deep Learning in Chemistry. J. Chem. Inf. Model. 59, 2545–2559 [DOI] [PubMed] [Google Scholar]
- 24.Guzel P and Kurkcuoglu O (2017) Identification of potential allosteric communication pathways between functional sites of the bacterial ribosome by graph and elastic network models. Biochim. Biophys. Acta BBA - Gen. Subj. 1861, 3131–3141 [DOI] [PubMed] [Google Scholar]
- 25.Lu H-M and Liang J (2009) Perturbation-based Markovian Transmission Model for Probing Allosteric Dynamics of Large Macromolecular Assembling: A Study of GroEL-GroES. PLOS Comput. Biol. 5, e1000526. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 26.Verkhivker GM (2020) Molecular Simulations and Network Modeling Reveal an Allosteric Signaling in the SARS-CoV-2 Spike Proteins. J. Proteome Res. 19, 4587–4608 [DOI] [PubMed] [Google Scholar]
- 27.Villani G (2020) A Time-Dependent Quantum Approach to Allostery and a Comparison With Light-Harvesting in Photosynthetic Phenomenon. Front. Mol. Biosci. 7, 156. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 28.Hakhverdyan Z et al. (2021) Dissecting the Structural Dynamics of the Nuclear Pore Complex. Mol. Cell 81, 153–165.e7 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 29.Brotzakis ZF et al. (2021) A method of incorporating rate constants as kinetic constraints in molecular dynamics simulations. Proc. Natl. Acad. Sci. 118, e2012423118. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 30.Matsunaga Y and Sugita Y (2018) Linking time-series of single-molecule experiments with molecular dynamics simulations by machine learning. eLife 7, e32668. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 31.Matsunaga Y and Sugita Y (2020) Use of single-molecule time-series data for refining conformational dynamics in molecular simulations. Curr. Opin. Struct. Biol. 61, 153–159 [DOI] [PubMed] [Google Scholar]
- 32.Zheng Y and Cui Q (2018) Multiple Pathways and Time Scales for Conformational Transitions in apo-Adenylate Kinase. J. Chem. Theory Comput. 14, 1716–1726 [DOI] [PubMed] [Google Scholar]
- 33.Thomsen J et al. (2020) DeepFRET, a software for rapid and automated single-molecule FRET data classification using deep learning. eLife 9, e60404. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 34.Wehmeyer C and Noé F (2018) Time-lagged autoencoders: Deep learning of slow collective variables for molecular kinetics. J. Chem. Phys. 148, 241703. [DOI] [PubMed] [Google Scholar]
- 35.Hernández CX et al. (2018) Variational encoding of complex dynamics. Phys. Rev. E 97, 062412. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 36.Mardt A et al. (2018) VAMPnets for deep learning of molecular kinetics. Nat. Commun. 9, 5. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 37.Ribeiro JML et al. (2018) Reweighted autoencoded variational Bayes for enhanced sampling (RAVE). J. Chem. Phys. 149, 072301. [DOI] [PubMed] [Google Scholar]
- 38.M. Sultan M and Pande VS (2017) tICA-Metadynamics: Accelerating Metadynamics by Using Kinetically Selected Collective Variables. J. Chem. Theory Comput. 13, 2440–2447 [DOI] [PubMed] [Google Scholar]
- 39.Doerr S and De Fabritiis G (2014) On-the-Fly Learning and Sampling of Ligand Binding by High-Throughput Molecular Simulations. J. Chem. Theory Comput. 10, 2064–2069 [DOI] [PubMed] [Google Scholar]
- 40.Zimmerman MI and Bowman GR (2015) FAST Conformational Searches by Balancing Exploration/Exploitation Trade-Offs. J. Chem. Theory Comput. 11, 5747–5757 [DOI] [PubMed] [Google Scholar]
- 41.Noé F et al. (2019) Boltzmann generators: Sampling equilibrium states of many-body systems with deep learning. Science 365, eaaw1147. [DOI] [PubMed] [Google Scholar]
- 42.Takami K et al. (2020) Performance Research of Clustering Methods for Detecting State Transition Trajectories in Hemoglobin. J. Comput. Chem. Jpn. 19, 154–157 [Google Scholar]
- 43.Konovalov KA et al. (2021) Markov State Models to Study the Functional Dynamics of Proteins in the Wake of Machine Learning. JACS Au 1, 1330–1341 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 44.Bonati L et al. (2021) Deep learning the slow modes for rare events sampling. Proc. Natl. Acad. Sci. 118, e2113533118. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 45.Brandt S et al. (2018) Machine Learning of Biomolecular Reaction Coordinates. J. Phys. Chem. Lett. 9, 2144–2150 [DOI] [PubMed] [Google Scholar]
- 46.Zhou H et al. (2018) Recognition of protein allosteric states and residues: Machine learning approaches. J. Comput. Chem. 39, 1481–1490 [DOI] [PubMed] [Google Scholar]
- 47.Hayatshahi HS et al. (2019) Probing Protein Allostery as a Residue-Specific Concept via Residue Response Maps. J. Chem. Inf. Model. 59, 4691–4705 [DOI] [PubMed] [Google Scholar]
- 48.Do HN et al. (2022) GLOW: A Workflow Integrating Gaussian-Accelerated Molecular Dynamics and Deep Learning for Free Energy Profiling. J. Chem. Theory Comput. 18, 1423–1436 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 49.Kityk R et al. (2015) Pathways of allosteric regulation in Hsp70 chaperones. Nat. Commun. 6, 8308. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 50.Mashaghi A et al. (2016) Alternative modes of client binding enable functional plasticity of Hsp70. Nature 539, 448–451 [DOI] [PubMed] [Google Scholar]
- 51.Kityk R et al. (2018) Molecular Mechanism of J-Domain-Triggered ATP Hydrolysis by Hsp70 Chaperones. Mol. Cell 69, 227–237.e4 [DOI] [PubMed] [Google Scholar]
- 52.Zhu J et al. (2022) Neural relational inference to learn long-range allosteric interactions in proteins from molecular dynamics simulations. Nat. Commun. 13, 1661. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 53.Arantes PR et al. (2022) Emerging Methods and Applications to Decrypt Allostery in Proteins and Nucleic Acids. J. Mol. Biol. 434, 167518. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 54.Zhou H and Tao P (2019) REDAN: relative entropy-based dynamical allosteric network model. Mol. Phys. 117, 1334–1343 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 55.Botlani M et al. (2018) Machine learning approaches to evaluate correlation patterns in allosteric signaling: A case study of the PDZ2 domain. J. Chem. Phys. 148, 241726. [DOI] [PubMed] [Google Scholar]
- 56.Yan W et al. (2018) Node-Weighted Amino Acid Network Strategy for Characterization and Identification of Protein Functional Residues. J. Chem. Inf. Model. 58, 2024–2032 [DOI] [PubMed] [Google Scholar]
- 57.Zhou H et al. (2019) Allosteric mechanism of the circadian protein Vivid resolved through Markov state model and machine learning analysis. PLOS Comput. Biol. 15, e1006801. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 58.Ibrahim MT et al. (2022) Dynamics of hydrogen bonds in the secondary structures of allosteric protein Avena Sativa phototropin 1. Comput. Struct. Biotechnol. J. 20, 50–64 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 59.Stetz G and Verkhivker GM (2017) Computational Analysis of Residue Interaction Networks and Coevolutionary Relationships in the Hsp70 Chaperones: A Community-Hopping Model of Allosteric Regulation and Communication. PLOS Comput. Biol. 13, e1005299. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 60.Astl L et al. (2020) Allosteric Mechanism of the Hsp90 Chaperone Interactions with Cochaperones and Client Proteins by Modulating Communication Spines of Coupled Regulatory Switches: Integrative Atomistic Modeling of Hsp90 Signaling in Dynamic Interaction Networks. J. Chem. Inf. Model. 60, 3616–3631 [DOI] [PubMed] [Google Scholar]
- 61.Stetz G et al. (2020) Exploring Mechanisms of Communication Switching in the Hsp90-Cdc37 Regulatory Complexes with Client Kinases through Allosteric Coupling of Phosphorylation Sites: Perturbation-Based Modeling and Hierarchical Community Analysis of Residue Interaction Networks. J. Chem. Theory Comput. 16, 4706–4725 [DOI] [PubMed] [Google Scholar]
- 62.Astl L et al. (2020) Dissecting Molecular Principles of the Hsp90 Chaperone Regulation by Allosteric Modulators Using a Hierarchical Simulation Approach and Network Modeling of Allosteric Interactions: Conformational Selection Dictates the Diversity of Protein Responses and Ligand-Specific Functional Mechanisms. J. Chem. Theory Comput. 16, 6656–6677 [DOI] [PubMed] [Google Scholar]
- 63.Verkhivker GM (2022) Exploring Mechanisms of Allosteric Regulation and Communication Switching in the Multiprotein Regulatory Complexes of the Hsp90 Chaperone with Cochaperones and Client Proteins: Atomistic Insights from Integrative Biophysical Modeling and Network Analysis of Conformational Landscapes. J. Mol. Biol. 434, 167506. [DOI] [PubMed] [Google Scholar]
- 64.Chen Y et al. (2021) Allosteric Effect of Nanobody Binding on Ligand-Specific Active States of the β2 Adrenergic Receptor. J. Chem. Inf. Model. 61, 6024–6037 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 65.Liu X et al. (2020) Unraveling allosteric landscapes of allosterome with ASD. Nucleic Acids Res. 48, D394–D401 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 66.Huang W et al. (2015) ASBench: benchmarking sets for allosteric discovery. Bioinformatics 31, 2598–2600 [DOI] [PubMed] [Google Scholar]
- 67.Greener JG et al. (2017) Predicting Protein Dynamics and Allostery Using Multi-Protein Atomic Distance Constraints. Structure 25, 546–558 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 68.Xie J et al. (2022) Uncovering the Dominant Motion Modes of Allosteric Regulation Improves Allosteric Site Prediction. J. Chem. Inf. Model. 62, 187–195 [DOI] [PubMed] [Google Scholar]
- 69.Lu S et al. (2014) Recent computational advances in the identification of allosteric sites in proteins. Drug Discov. Today 19, 1595–1600 [DOI] [PubMed] [Google Scholar]
- 70.Akbar R and Helms V (2018) ALLO: A tool to discriminate and prioritize allosteric pockets. Chem. Biol. Drug Des. 91, 845–853 [DOI] [PubMed] [Google Scholar]
- 71.Tian H et al. (2021) PASSer: prediction of allosteric sites server. Mach. Learn. Sci. Technol. 2, 035015. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 72.Xiao S et al. (2022) PASSer2.0: Accurate Prediction of Protein Allosteric Sites Through Automated Machine Learning. Front. Mol. Biosci. 9, 879251. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 73.Chen AS-Y et al. (2016) A Random Forest Model for Predicting Allosteric and Functional Sites on Proteins. Mol. Inform. 35, 125–135 [DOI] [PubMed] [Google Scholar]
- 74.Huang W et al. (2013) Allosite: a method for predicting allosteric sites. Bioinformatics 29, 2357–2359 [DOI] [PubMed] [Google Scholar]
- 75.Greener JG and Sternberg MJ (2015) AlloPred: prediction of allosteric pockets on proteins using normal mode perturbation analysis. BMC Bioinformatics 16, 335. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 76.Song K et al. (2017) Improved Method for the Identification and Validation of Allosteric Sites. J. Chem. Inf. Model. 57, 2358–2363 [DOI] [PubMed] [Google Scholar]
- 77.Mishra SK et al. (2019) Coupling dynamics and evolutionary information with structure to identify protein regulatory and functional binding sites. Proteins Struct. Funct. Bioinforma. 87, 850–868 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 78.Fogha J et al. (2020) Computational Analysis of Crystallization Additives for the Identification of New Allosteric Sites. ACS Omega 5, 2114–2122 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 79.Tsai C-J and Nussinov R (2014) A Unified View of “How Allostery Works.” PLOS Comput. Biol. 10, e1003394. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 80.Zhang W et al. (2019) Correlation Between Allosteric and Orthosteric Sites. In Protein Allostery in Drug Discovery (Zhang J and Nussinov R, eds), pp. 89–105, Springer; [DOI] [PubMed] [Google Scholar]
- 81.Leroux AE and Biondi RM (2020) Renaissance of Allostery to Disrupt Protein Kinase Interactions. Trends Biochem. Sci. 45, 27–41 [DOI] [PubMed] [Google Scholar]
- 82.Tee W-V et al. (2018) Reversing allosteric communication: From detecting allosteric sites to inducing and tuning targeted allosteric response. PLOS Comput. Biol. 14, e1006228. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 83.Fan J et al. (2021) Harnessing Reversed Allosteric Communication: A Novel Strategy for Allosteric Drug Discovery. J. Med. Chem. 64, 17728–17743 [DOI] [PubMed] [Google Scholar]
- 84.Ni D et al. (2021) Discovery of cryptic allosteric sites using reversed allosteric communication by a combined computational and experimental strategy. Chem. Sci. 12, 464–476 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 85.Ferraro M et al. (2021) Machine Learning of Allosteric Effects: The Analysis of Ligand-Induced Dynamics to Predict Functional Effects in TRAP1. J. Phys. Chem. B 125, 101–114 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 86.Leander M et al. (2022) Deep mutational scanning and machine learning reveal structural and molecular rules governing allosteric hotspots in homologous proteins. eLife 11, e79932. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 87.Faure AJ et al. (2022) Mapping the energetic and allosteric landscapes of protein binding domains. Nature 604, 175–183 [DOI] [PubMed] [Google Scholar]
- 88.Wang Q et al. (2012) Toward understanding the molecular basis for chemical allosteric modulator design. J. Mol. Graph. Model. 38, 324–333 [DOI] [PubMed] [Google Scholar]
- 89.Smith RD et al. (2017) Are there physicochemical differences between allosteric and competitive ligands? PLOS Comput. Biol. 13, e1005813. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 90.Hou T et al. (2021) Integrated Multi-Class Classification and Prediction of GPCR Allosteric Modulators by Machine Learning Intelligence. Biomolecules 11, 870. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 91.Bian Y and Xie X-Q (2022) Artificial Intelligent Deep Learning Molecular Generative Modeling of Scaffold-Focused and Cannabinoid CB2 Target-Specific Small-Molecule Sublibraries. Cells 11, 915. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 92.Yang L et al. (2021) Transformer-Based Generative Model Accelerating the Development of Novel BRAF Inhibitors. ACS Omega 6, 33864–33873 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 93.N. Vennila K and P. Elango K (2022) Multimodal generative neural networks and molecular dynamics based identification of PDK1 PIF-pocket modulators. Mol. Syst. Des. Eng. 7, 1085–1092 [Google Scholar]
- 94.Huang M et al. (2018) AlloFinder: a strategy for allosteric modulator discovery and allosterome analyses. Nucleic Acids Res. 46, W451–W458 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 95.Miljković F et al. (2020) Machine Learning Models for Accurate Prediction of Kinase Inhibitors with Different Binding Modes. J. Med. Chem. 63, 8738–8748 [DOI] [PubMed] [Google Scholar]
- 96.Rodrigues T et al. (2018) Machine intelligence decrypts β-lapachone as an allosteric 5-lipoxygenase inhibitor. Chem. Sci. 9, 6899–6903 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 97.Zayner JP et al. (2013) Investigating Models of Protein Function and Allostery With a Widespread Mutational Analysis of a Light-Activated Protein. Biophys. J. 105, 1027–1036 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 98.Weinkam P et al. (2013) Impact of Mutations on the Allosteric Conformational Equilibrium. J. Mol. Biol. 425, 647–661 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 99.Xiao J et al. (2019) Probing light chain mutation effects on thrombin via molecular dynamics simulations and machine learning. J. Biomol. Struct. Dyn. 37, 982–999 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 100.García-Fernández MD et al. (2021) Distinct classes of potassium channels fused to GPCRs as electrical signaling biosensors. Cell Rep. Methods 1, 100119. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 101.D’Amico RN et al. (2021) Substitution of a Surface-Exposed Residue Involved in an Allosteric Network Enhances Tryptophan Synthase Function in Cells. Front. Mol. Biosci 8 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 102.Astl L and Verkhivker GM (2019) Data-driven computational analysis of allosteric proteins by exploring protein dynamics, residue coevolution and residue interaction networks. Biochim. Biophys. Acta BBA - Gen. Subj. DOI: 10.1016/j.bbagen.2019.07.008 [DOI] [PubMed] [Google Scholar]
- 103.Ray D et al. (2021) Distant residues modulate conformational opening in SARS-CoV-2 spike protein. Proc. Natl. Acad. Sci. 118, e2100943118. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 104.Karki N et al. (2021) Predicting Potential SARS-COV-2 Drugs—In Depth Drug Database Screening Using Deep Neural Network Framework SSnet, Classical Virtual Screening and Docking. Int. J. Mol. Sci. 22, 1573. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 105.Bhattarai A et al. (2021) Mechanism and Pathways of Inhibitor Binding to the Human ACE2 Receptor for SARS-CoV1/2. Biophys. J. 120, 204a [Google Scholar]
- 106.Nishiga M et al. (2020) COVID-19 and cardiovascular disease: from basic mechanisms to clinical perspectives. Nat. Rev. Cardiol. 17, 543–558 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 107.Delgado JM et al. (2021) Molecular basis for higher affinity of SARS-CoV-2 spike RBD for human ACE2 receptor. Proteins Struct. Funct. Bioinforma. 89, 1134–1144 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 108.Trozzi F et al. (2022) Allosteric control of ACE2 peptidase domain dynamics. Org. Biomol. Chem. 20, 3605–3618 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 109.Uyar A and Dickson A (2021) Perturbation of ACE2 Structural Ensembles by SARS-CoV-2 Spike Protein Binding. J. Chem. Theory Comput. 17, 5896–5906 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 110.Iyengar SM et al. (2021) Prediction and Analysis of Multiple Sites and Inhibitors of SARS-CoV-2 Proteins. Biophys. J. 120, 204a [Google Scholar]
- 111.Jain S et al. (2021) Hybrid In Silico Approach Reveals Novel Inhibitors of Multiple SARS-CoV-2 Variants. ACS Pharmacol. Transl. Sci. 4, 1675–1688 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 112.Kaptan S et al. (2022) Maturation of the SARS-CoV-2 virus is regulated by dimerization of its main protease. Comput. Struct. Biotechnol. J. 20, 3336–3346 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 113.Verkhivker GM et al. (2021) Atomistic Simulations and In Silico Mutational Profiling of Protein Stability and Binding in the SARS-CoV-2 Spike Protein Complexes with Nanobodies: Molecular Determinants of Mutational Escape Mechanisms. ACS Omega 6, 26354–26371 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 114.Verkhivker GM et al. (2021) Computational analysis of protein stability and allosteric interaction networks in distinct conformational forms of the SARS-CoV-2 spike D614G mutant: reconciling functional mechanisms through allosteric model of spike regulation. J. Biomol. Struct. Dyn. DOI: 10.1080/07391102.2021.1933594 [DOI] [PubMed] [Google Scholar]
- 115.Verkhivker GM et al. (2021) Allosteric Control of Structural Mimicry and Mutational Escape in the SARS-CoV-2 Spike Protein Complexes with the ACE2 Decoys and Miniprotein Inhibitors: A Network-Based Approach for Mutational Profiling of Binding and Signaling. J. Chem. Inf. Model. 61, 5172–5191 [DOI] [PubMed] [Google Scholar]
- 116.Verkhivker GM and Di Paola L (2021) Dynamic Network Modeling of Allosteric Interactions and Communication Pathways in the SARS-CoV-2 Spike Trimer Mutants: Differential Modulation of Conformational Landscapes and Signal Transmission via Cascades of Regulatory Switches. J. Phys. Chem. B 125, 850–873 [DOI] [PubMed] [Google Scholar]
- 117.Verkhivker G (2022) Allosteric Determinants of the SARS-CoV-2 Spike Protein Binding with Nanobodies: Examining Mechanisms of Mutational Escape and Sensitivity of the Omicron Variant. Int. J. Mol. Sci. 23, 2172. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 118.Verkhivker G et al. (2022) Computer Simulations and Network-Based Profiling of Binding and Allosteric Interactions of SARS-CoV-2 Spike Variant Complexes and the Host Receptor: Dissecting the Mechanistic Effects of the Delta and Omicron Mutations. Int. J. Mol. Sci. 23, 4376. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 119.Verkhivker GM et al. (2022) Landscape-Based Protein Stability Analysis and Network Modeling of Multiple Conformational States of the SARS-CoV-2 Spike D614G Mutant: Conformational Plasticity and Frustration-Induced Allostery as Energetic Drivers of Highly Transmissible Spike Variants. J. Chem. Inf. Model. 62, 1956–1978 [DOI] [PubMed] [Google Scholar]