

Abstract
The protective efficacy of serum antibody results from the interplay of antigen-specific B cell clones of different affinities and specificities. These cellular dynamics underlie serum-level phenomena such as “Original Antigenic Sin” (OAS), a proposed propensity of the immune system to rely repeatedly on the first cohort of B cells engaged by an antigenic stimulus when encountering related antigens, in detriment of inducing de novo responses1–5. OAS-type suppression of new, variant-specific antibodies may pose a barrier to vaccination against rapidly evolving viruses such as influenza and SARS-CoV-26,7. Precise measurement of OAS-type suppression is challenging because cellular and temporal origins cannot readily be ascribed to antibodies in circulation; thus, its impact on subsequent antibody responses remains unclear5,8. Here, we introduce a molecular fate-mapping approach in which serum antibodies derived from specific cohorts of B cells can be differentially detected. We show that serum responses to sequential homologous boosting derive overwhelmingly from primary cohort B cells, while later induction of new antibody responses from naïve B cells is strongly suppressed. Such “primary addiction” decreases sharply as a function of antigenic distance, allowing reimmunization with divergent viral glycoproteins to produce de novo antibody responses targeting epitopes absent from the priming variant. Our findings have implications for the understanding of OAS and for the design and testing of vaccines against evolving pathogens.
The ability of serum antibody to protect against infection is an emergent property of the complex mixture of immunoglobulins secreted over time by B cell clones of various specificities and a range of affinities. The plasma cells that produce this antibody arise through multiple parallel pathways, ranging from direct differentiation from naïve B cell precursors upon primary infection or immunization to elaborate routes involving one or more rounds of affinity maturation in germinal centers (GCs) and intercalating memory B cell phases. The complexity of these cellular pathways compounds markedly with repeated antigenic exposure9–12, and their ultimate contribution to the serum antibody pool has been difficult to deconvolute. On one hand, molecular analyses of immunoglobulin genes obtained from memory or GC B cells do not directly assess the composition of antibodies in serum13–16; on the other, direct studies of the clonal composition of serum antibody cannot readily assign a cellular or temporal origin to antibodies of different specificities17,18. The clonal dynamics of immune phenomena that take place at the serum level thus remain poorly understood.
A serum-level phenomenon that has been particularly difficult to unravel is OAS, described in the 1950s as a tendency of individuals exposed to a given strain of influenza to respond with antibodies that react more strongly to the first strain of influenza they had met in early childhood than to the exposure strain itself1,19. OAS was originally attributed to a propensity of the immune system to repeatedly reuse the first cohort of B cells that respond to an antigen, whose reactivity will necessarily be biased towards the strain that originally elicited it. However, unlike related concepts such as “antigenic seniority,” 4,20 OAS (as defined herein) requires active suppression of the de novo recruitment of new B cell clones from the naïve repertoire upon boosting2–4, thus restricting the ability of the immune system to mount specific antibody responses to escape epitopes. The extent to which this active suppression exists and influences subsequent responses has been debated for decades5,8. More recently, B cell fate-mapping experiments in mice have shown that, in apparent contrast the predictions of OAS, GCs that form in response to boosting consist almost exclusively of naïve rather than memory-derived B cells21–23. This later addition implies that either the effects of OAS in mice are negligible, or that OAS is a phenomenon observed exclusively at the serum level.
Molecular fate-mapping of serum antibody
Resolving this issue—as well as generally understanding the effect of OAS on the response to repeated antigen exposure—would require the ability to transpose such cellular fate-mapping experiments to serum antibody itself. To achieve this, we adapted the classic fate-mapping strategy to enable detection of the cellular and temporal origin of antibodies in serum, an approach we call “molecular fate-mapping.” We engineered mice in which the C-terminus of the immunoglobulin kappa (Igκ) light chain gene (Igk) is extended to encode for a LoxP-flanked FLAG-tag, followed by a downstream Strep-tag (Fig. 1a and Extended Data Fig. 1). B cells bearing this “K-tag” allele produce immunoglobulins that are FLAG-tagged unless exposed to Cre recombinase, upon which they permanently switch the FLAG-tag for a Strep-tag. Cre-mediated recombination thus fate-maps the antibodies these B cells and their plasma cell descendants express on their surfaces and/or secrete into serum. This allows for differential detection of pre- and post-fate-mapping Igκ+ antibody using secondary reagents specific for each tag.
Figure 1 |. The K-tag system for molecular fate-mapping of serum antibodies.
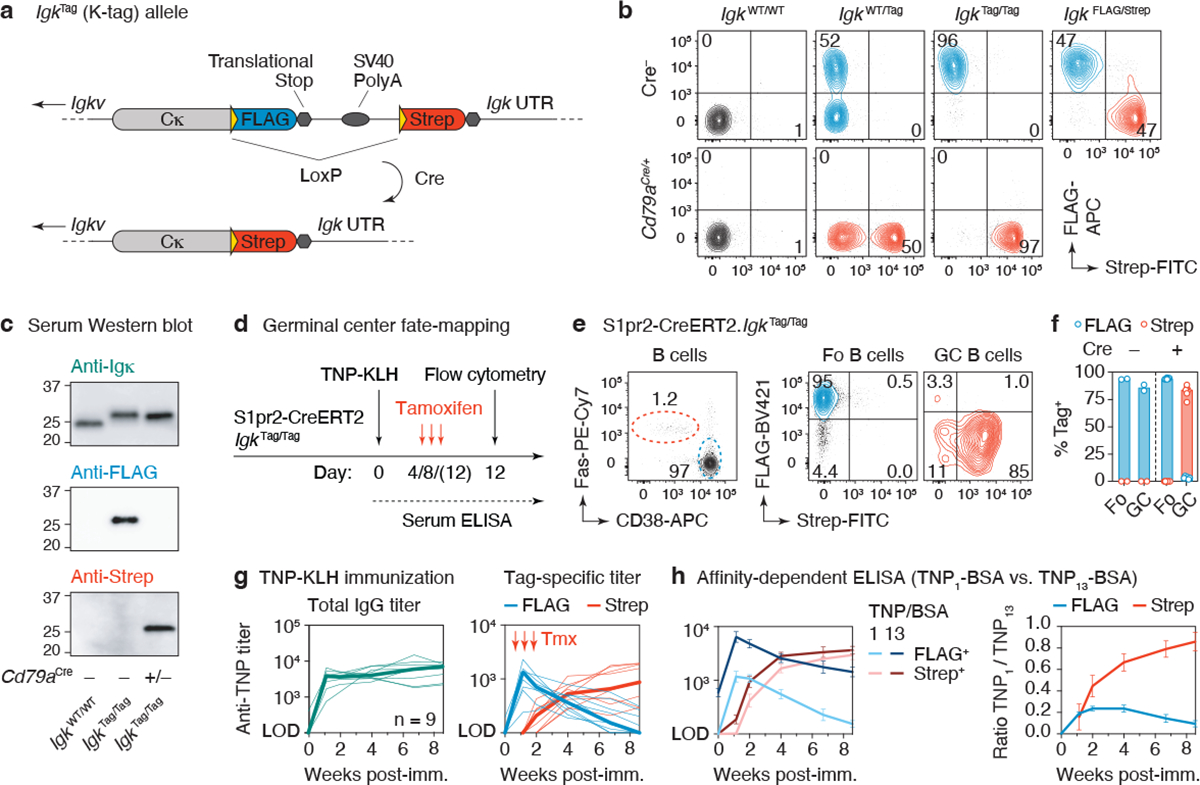
(a) Schematic representation of the IgkTag allele prior to and following cre-mediated recombination. (b) Flow cytometry of blood B cells showing expression of FLAG- and Strep-tagged B cell receptors on mice of the indicated genotype. (c) Western blotting of serum obtained from mice of the indicated genotype, stained for Igκ light chain or FLAG/Strep-tags. Representative of 2 experiments. (d) Schematic representation of the immunization strategy used to fate-map GC B cells and their antibody output. (e) Flow cytometry of popliteal lymph node 12 days after footpad immunization with TNP-KLH in alum adjuvant. Left panel shows B cells (B220+, CD4− CD8− CD138−) stained for GC (FAS+ CD38−) and follicular (Fo) B cell (FAS− CD38+) markers. (f) Quantification of data in (e). Each dot represents an individual lymph node, bars represent the median. (g) Anti-TNP total IgG and tag-specific endpoint titers as determined by TNP4 -BSA ELISA in mice immunized i.p. with TNP-KLH in alhydrogel adjuvant. Thin lines represent individual mice, thick lines link medians of log transformed titer values at each time point. Results are from 9 mice from 2 independent experiments. (h) Relative affinity of anti-TNP FLAG+ and Strep+ antibodies of the same samples shown in (g) as estimated by ELISA using TNP1-BSA or TNP13-BSA as capture reagents. The means of the log transformed titer values are shown with error bars representing SEM. The ratio between anti-TNP1-BSA and anti-TNP13-BSA titers was calculated per sample, shown in the right panel.
To verify the functionality of the K-tag allele, we first determined that B cells in IgkTag mice expressed tagged B cell receptors on their surface. Following the rules of allelic exclusion24, roughly 50% and 95% of B cells from heterozygous IgkWT/Tag (WT, wild-type) or homozygous IgkTag/Tag mice were FLAG+ (as expected, about 5% of B cells in homozygous mice carried an Igλ light chain24; Extended Data Fig. 2a). K-tag mice in which all B cells constitutively expressed Cre recombinase (IgkTag/Tag.Cd79aCre/+) replaced FLAG- with Strep-tag in virtually all Igκ+ B cells (Fig. 1b). Importantly, K-tag mice appropriately secreted FLAG- or Strep-tagged antibodies into serum in the absence or presence of Cre recombinase, respectively (Fig. 1c), without affecting steady state serum antibody levels (Extended Data Fig. 2b). Generation and maturation of K-tag B cells was unimpaired, as indicated by the equal proportion of tagged and untagged circulating B cells in IgkWT/Tag mice (Fig. 1b). The same was true of circulating B cells and bone marrow plasma cells expressing FLAG- and Strep-tags in IgkFLAG/Strep mice, in which one of the two K-tag alleles was pre-recombined by Cre expression in the germline (Fig. 1b and Extended Data Fig. 2c).
To ensure that FLAG+ and Strep+ B cells were equally competitive throughout the course of B cell activation, affinity maturation, and plasma cell differentiation, we primed and boosted IgkFLAG/Strep mice with the model antigen 2,4,6-trinitrophenyl-keyhole limpet hemocyanin (TNP-KLH) in alum adjuvant and followed the serum titers of antibodies bearing each tag over time. To enable direct comparison of titers of anti-TNP antibodies bearing each tag, we diluted secondary (anti-FLAG or anti-Strep) antibodies to achieve similar detection of standard curves generated using recombinant FLAG- or Strep-tagged monoclonal antibodies (Extended Data Fig. 2d). Endpoint ELISA titers normalized using these curves showed a similar range of anti-TNP reactivity between FLAG+ and Strep+ fractions (Extended Data Fig. 2e), indicative of equal competitiveness of the differently tagged B cells.
Following the serum antibody produced by B cells engaged at different stages of the immune response requires temporally restricted fate-mapping of activated B cell clones. To enable this, we crossed IgkTag mice to the GC-specific, tamoxifen-inducible S1pr2-CreERT2 BAC-transgenic allele25 (to generate S1pr2-IgkTag mice). Tamoxifen treatment of mice on days 4 and 8 after TNP-KLH immunization led to efficient recombination (96.1% +/− 0.50 SEM ((Strep+/Tag+)*100) ) of the K-tag allele in GC B cells but not in non-GC B cells in the same lymph node at 12 days post-immunization (d.p.i.) (Fig. 1d–f). Again, tagged B cells were found at similar proportions to untagged B cells in heterozygous S1pr2-IgkWT/Tag mice (on average 41% +/− SD 9.0 Tag+), indicating that expression of the tag does not impair B cell competitiveness in the GC (Extended Data Fig. 2f, g). GC B cells in Cre− animals (Fig. 1f) or S1pr2-IgkWT/Tag mice not treated with tamoxifen remained FLAG+, with only minimal spontaneous recombination (1.3% ± 1.0 SD Strep+ at day 12 d.p.i.) in the latter (Extended Data Fig. 2g).
Total anti-TNP IgG antibodies in S1pr2-IgkTag mice immunized intraperitoneally (i.p.) with TNP-KLH in alhydrogel and treated with tamoxifen on days 4, 8 and 12 were first detected in serum at 8 d.p.i. and increased progressively through 60 d.p.i. (Fig. 1g). Deconvolution of GC-derived (Strep+) and non-GC derived (FLAG+) antibodies showed that an initial wave of extrafollicular FLAG+ antibodies that peaked at 8 d.p.i. was progressively replaced by GC-derived Strep+ antibodies that were first detected in serum at 14 d.p.i. (Fig. 1g). FLAG+ anti-TNP antibodies regressed to near baseline levels between 47–60 d.p.i., as expected from their extrafollicular origin. An affinity-dependent anti-TNP ELISA showed detectable affinity maturation only in the GC-derived Strep+ antibody fraction (Fig. 1h), confirming the efficient fate-mapping of GC-derived antibody. Background signal in control animals not given tamoxifen remained below the limit of detection (LOD) throughout the primary response (Extended Data Fig. 2h). Thus, the S1pr2-IgkTag mouse model enables us to discern antibodies derived from the first wave of B cells that entered a GC reaction in response to immunization. Our data also show that extrafollicular responses are of relatively short duration in these settings and that virtually all antibody detectable after the first few weeks of immunization, and especially antibody with high affinity, is derived from plasma cells of GC origin.
Primary addiction in homologous boosting
With this system in hand, we sought to measure the extent to which OAS-type suppression affects the development of de novo antibody responses to homologous boosting (we refer to this suppression generically as “primary addiction,” to encompass the homologous regimen). To this end, we took advantage of the ability to trigger Cre-mediated recombination of the K-tag allele in a time-resolved manner by administration of tamoxifen to mark serum antibodies produced by B cells that formed GCs in response to primary immunization (the “primary cohort”). In addition to labeling primary-cohort B cells, this approach also “reverse fate-maps” with a FLAG-tag any antibodies that arose from clones engaged de novo by subsequent booster doses. This property allows us to distinguish between two models of recall antibody response: (i) a simple sequential contribution model, in which de novo responses, although smaller than memory-derived ones, are nevertheless allowed to progress with similar kinetics to a new primary response, adding up as more doses of antigen are provided (related to antigenic seniority); and (ii) a primary addiction model (related to OAS), where the primary response actively suppresses the emergence of subsequent de novo antibody responses even after several boosts (Fig. 2a).
Figure 2 |. Primary addiction upon homologous boosting.
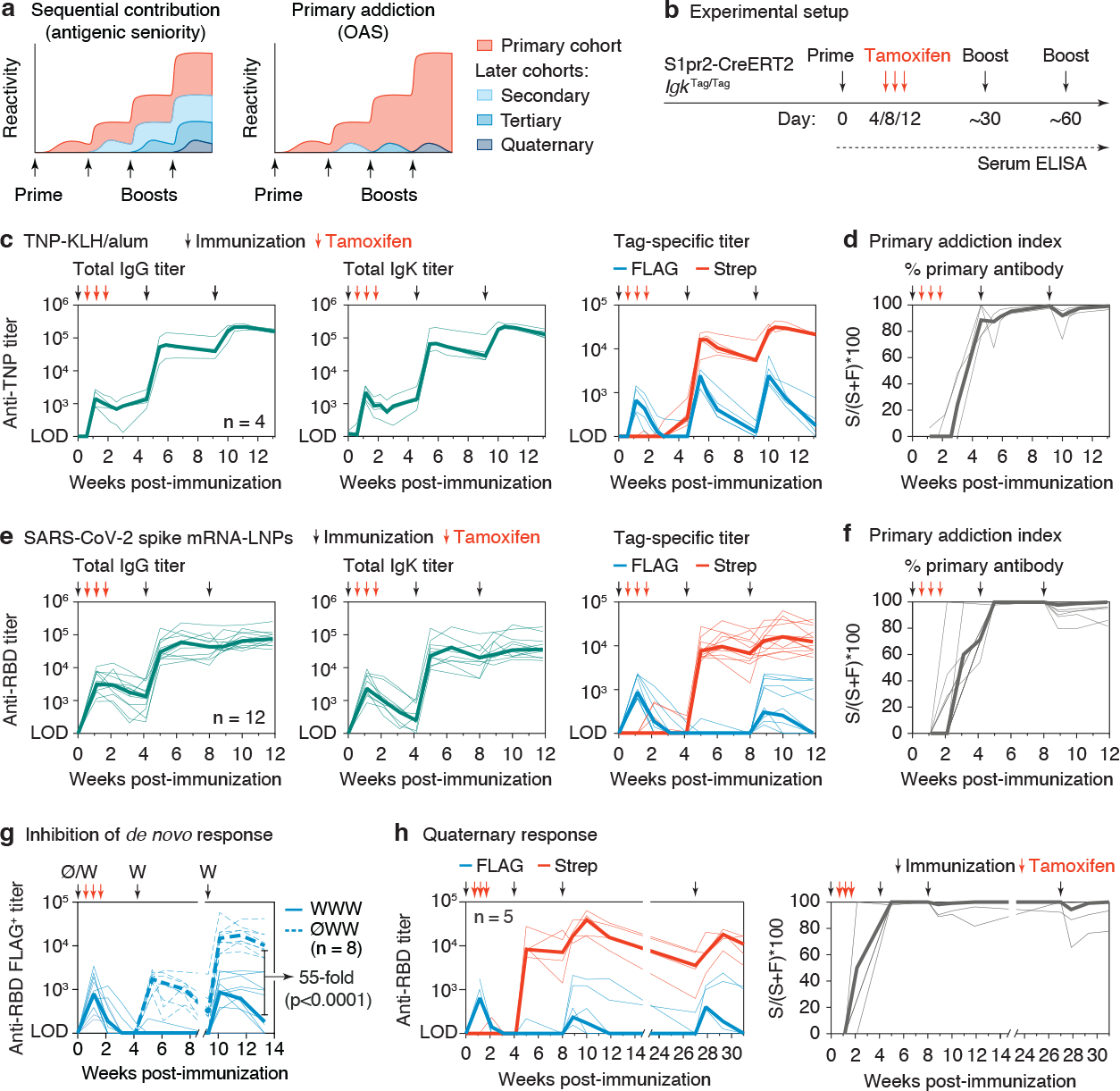
(a) Schematic representation of the “sequential contribution” and “primary addiction” models of antigenic imprinting. (b) General immunization strategy used to measure primary addiction. S1pr2-IgkTag/Tag mice were immunized on the days indicated by black arrows with TNP-KLH in alum (c-d) or WH1 mRNA-LNP (e-g) and treated with tamoxifen at 4, 8 and 12 d.p.i. as indicated by the red arrows. (c) Anti-TNP serum IgG (left panel), Igκ (middle panel) and tag-specific titers (right panel) as measured using TNP4 -BSA by ELISA. Results are from 4 mice from 2 independent experiments. Thin lines represent individual mice, thick lines link medians of log transformed titer values at each time point. (d) The percentage of the TNP-titer derived from the primary cohort of B cells (the “primary addiction index”) was calculated by dividing the Strep+ titer of each individual sample by its total titer (Strep+ + FLAG+), multiplied by 100 (S/(S+F)*100). (e) Anti-WH1 RBD IgG (left panel), Igκ (middle panel), and tag-specific titers (right panel) as measured by ELISA. Results are from 12 mice from 3 independent experiments. (f) Primary addiction index calculated as in (d). (g) Comparison of de novo FLAG+ antibody responses in the presence or absence of primary immunization. WWW, three doses of WH1 S mRNA; ØWW, first dose and fate-mapping omitted. WWW data are for the same samples as in panel (e), re-measured in the same assay as ØWW. (h) Anti-WH1 RBD response (left) and primary addiction index (right) in mice receiving a 4th dose of mRNA-LNP at 133 days after the previous dose, for one of the cohorts shown in (e). Two of five mice were not sampled at day 0.
We primed S1pr2-IgkTag mice i.p. with alum-adjuvanted TNP-KLH and administered tamoxifen at 4, 8, and 12 d.p.i. to fate-map primary cohort GC B cells and their memory and plasma cell progeny (Fig. 2b). In this setting, all primary cohort-derived antibodies are Strep+, whereas any antibody produced by B cell clones expanded de novo by secondary or higher-order boosting (including by their memory or plasma cell progeny) will be reverse fate-mapped as FLAG+. Importantly, since we do not rely on differences between antigen variants to distinguish primary from secondary or later antibodies, this approach allows us to measure primary addiction at “zero-antigenic distance”—i.e., when priming and boosting with the exact same antigen. Because suppression of de novo antibody responses is likely to decrease as antigenic distance increases26,27, this approach allows us to estimate the strength of primary addiction when it is at its strongest.
Homologous boosting one and two months after the primary immunization resulted in the expected increases in recall TNP titers (Fig. 2c) and the formation of recall GCs that were dominated by naïve-derived B cells21 (Extended Data Fig. 3a). Deconvolution of these responses using tag-specific ELISA revealed that both secondary and tertiary titers were strongly dominated by fate-mapped (Strep+) antibodies derived from primary cohort B cells and. Whereas FLAG+ TNP-specific antibodies also appeared after each boost, their titers peaked at much lower levels and decayed markedly with time. (Fig. 2c). Importantly, counter to the expectations of the sequential contribution model (Fig. 2a), FLAG+ titers did not increase progressively between the second and third antigen doses, when any FLAG+ memory B cells would have been reactivated. To synthesize both measures, we created a “primary addiction index,” computed by dividing Strep+ by total Strep+ + FLAG+ titers (S/(S+F)*100). This showed that almost all detectable recall antibody (mean 95% and 97% of serum reactivity at 14 days after the first and second boosts, respectively) was derived from the B cell cohort engaged in the primary GC response (Fig. 2d). Depletion of IgM from post-boost serum samples resulted in a sharp reduction in FLAG+ but not Strep+ recall TNP titers, supporting the notion that naïve-derived B cells engaged by recall generate primarily an extrafollicular-like (IgM-dominated) B cell response (Extended Data Fig. 3b,c).
To extend these findings to a clinically relevant setting, we immunized and boosted mice as in Fig. 2b but using a lipid nanoparticle (LNP)-formulated nucleoside-modified mRNA vaccine encoding the prefusion-stabilized (2P) form of the SARS-CoV-2 Wuhan-Hu-1 (WH1) spike (S) protein, similar to available SARS-CoV-2 mRNA vaccines28. Secondary and tertiary anti-S protein receptor binding domain (RBD) antibodies were again almost entirely derived from primary cohort (Strep+) B cells (Fig. 2e,f). As with GC B cells (Extended Data Fig. 2g), low-level spontaneous recombination to Strep+ antibody was detected in recall responses in control mice not given tamoxifen. This resulted in a slight underestimation of FLAG+ antibody titers (median 2.1 and 2.8 % two weeks after second and third immunizations respectively; Extended Data Fig. 3d). Although primary addiction was more pronounced than for the SARS-CoV-2 RBD than for TNP-KLH after the first boost (no new (FLAG+) antibody was detected at this time point), 5/12 mice developed low but stable titers of FLAG+ anti-RBD antibody after the third dose. This bimodality was independent of the experimental cohort and of whether boosting was done ipsilaterally or contralaterally to the site of the primary dose (Extended Data Fig. 3e) and is therefore likely ascribable to stochastic variability inherent to highly oligoclonal recall responses21. To quantify the extent to which de novo antibody responses to the boost were suppressed by previous priming (i.e., the magnitude of the primary addiction suppressive effect), we compared FLAG+ antibodies in mice that were given three doses of WH1 mRNA-LNPs (WWW) to an additional group in which the priming and fate-mapping steps were omitted (ØWW). FLAG+ responses were 55-fold lower in WWW compared to ØWW mice at 4 weeks after the final dose (Fig. 2g), indicating strong suppression of new B cell responses in primed animals compared to what they would have been in the absence of priming. Finally, primary addiction was long-lasting, as even a fourth immunization of a subset of mice (>133 days after the previous boost) was dominated by Strep-tagged antibodies (Fig. 2h and Extended Data Fig. 3f), again failing to demonstrate the progressive increase in FLAG+ antibody titers predicted by a simple sequential contribution model (Fig. 2a). We conclude that primary addiction can be remarkably strong when measured at zero antigenic distance, evidence of OAS-type suppression of de novo B cell responses by preexisting immunity.
Antigenic drift limits primary addiction
To measure how primary addiction responds to increases in antigenic distance between priming and boosting antigens, we employed historical series of drifted influenza virus hemagglutinin (HA) variants as models. We first used an influenza infection/immunization model (Fig. 3a) based on two of the strains for which OAS was originally described—A/Puerto Rico/8/1934 (PR8) and A/Fort Monmouth/1/1947 (FM1) 1,19—whose HAs share 90% identity at the amino acid level (Fig. 3b). As with hapten and mRNA immunization, the primary response to HAPR8 was characterized by high extrafollicular (FLAG+) titers that peaked between 8 and 16 days post-infection and were subsequently replaced by GC-derived (Strep+) titers (Fig. 3c). Homologous boosting with recombinant HAPR8 protein subcutaneously at 3 and 4 months post-infection resulted in a 1-log increase in Strep+ HAPR8 binding titers after the first boost and a less pronounced increase after the second boost. As with protein immunization, the contribution of non-primary (FLAG+) antibodies to total titers was minor: even though it increased progressively between the first and second boosts, its peak median value was roughly 10% of HA reactivity (Fig. 3c). Heterologous boosting with HAFM1 led to only slight back-boosting of primary Strep+ HAPR8 titers and had virtually no effect on FLAG+ HAPR8 reactivity (Fig. 3d), indicative of substantial antigenic distance between these variants. Accordingly, crossreactive primary titers towards HAFM1 were completely absent from the primary extrafollicular response to PR8 infection and began to emerge only at approximately 4 weeks in the Strep+ antibody fraction (Fig. 3d), likely as a side-effect of affinity maturation towards HAPR8. Heterologous boosting not only increased these crossreactive (Strep+) titers by close to 1 log, but, importantly, also induced substantial responses from de novo clones elicited by the boost, in that roughly half of all serum reactivity to HAFM1 was derived from the FLAG+ fraction after the second boost (Fig. 3d). Comparing these levels to those achieved by two doses of HAFM1 in the absence of prior infection showed that primary addiction suppressed new responses by 3.8-fold (Extended Data Fig. 4a), much less than the 55-fold suppression achieved in homologous mRNA-vaccination (Fig. 2g). Thus, heterologous boosting partly circumvents primary addiction, allowing improved expansion and serum contribution of variant-specific B cell clones not involved in the primary response.
Figure 3 |. Primary addiction decreases with antigenic distance.
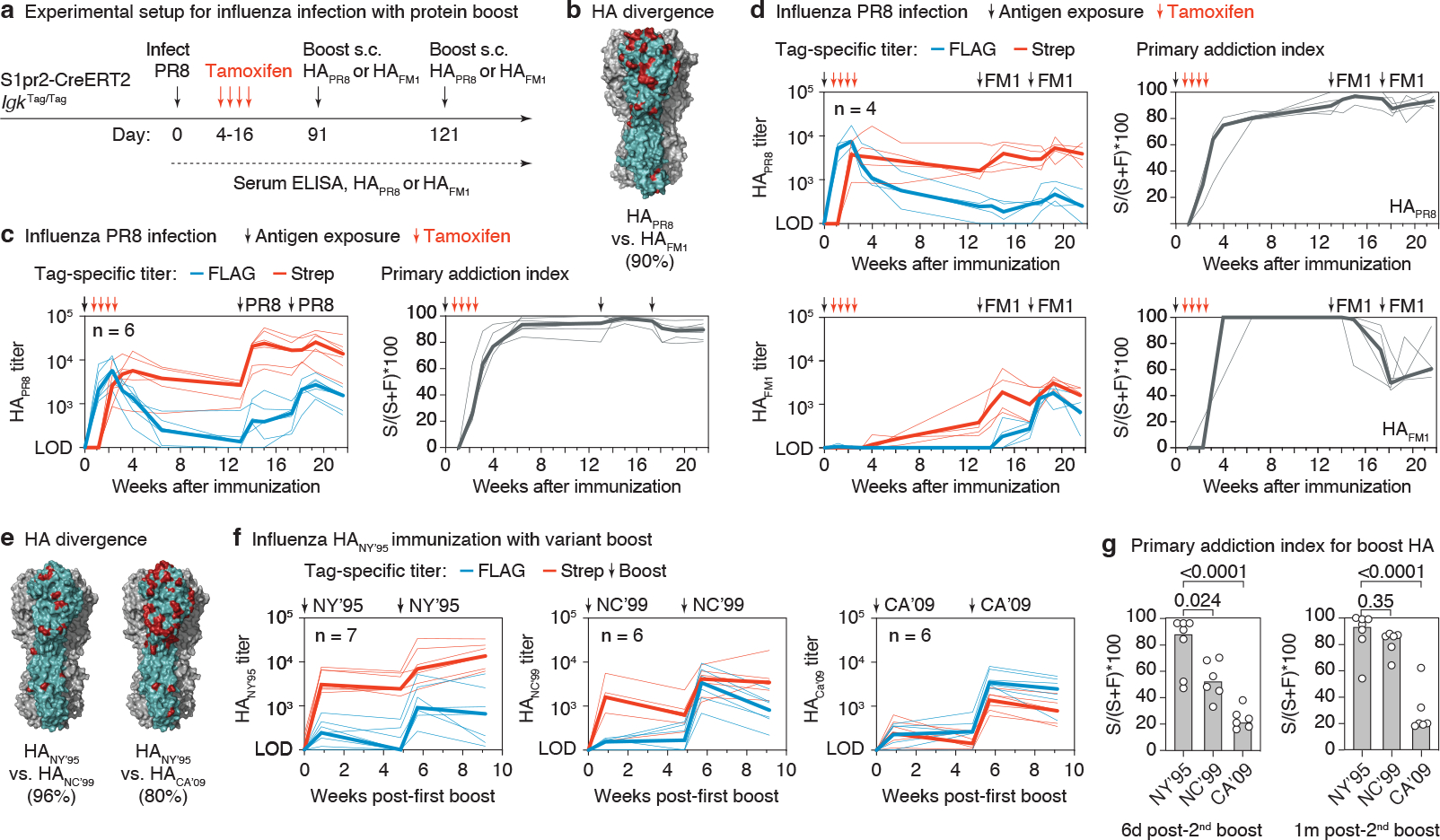
(a) Schematic representation of influenza infection and HA boosting strategy. S1pr2-IgkTag/Tag mice were infected intranasally with PR8 influenza and boosted subcutaneously (s.c.) with HAPR8 or HAFM1 in alhydrogel at the indicated time points. (b) Rendering of the HAPR8 trimer structure (PDB: 1RU7) with one monomer highlighted in teal and the amino acids that diverge between HAPR8 and HAFM1 in red. Percent amino acid identity is given in parentheses. (c) Anti-HAPR8 FLAG+ and Strep+ titers in S1pr2-IgkTag/Tag mice homologously boosted with HAPR8 (left) and quantification of the primary addiction index score (right). (d) Anti-HAPR8 (top) and anti-HAFM1 (bottom) ELISA reactivity in mice heterologously boosted with HAFM1. Tag-specific titers are shown in the left panels, and quantification of the primary addiction index is shown on the right. (e) Divergence between HANC’95 and HANC’99 or HACA’09, colored as in (b). Modeled on the structure of HACA’09 (PDB: 3LZG). (f) Anti-HA tag-specific titers in mice primed with HANY’95 protein, shown after the first boost with HANY’95 (homologous) or with variants HANC’99 or HACA’09 (heterologous), as outlined in Extended Data Fig. 4b. Antibody reactivity against the boosting antigen is shown. The full time-course and reactivities against all three HAs as measured by ELISA for the top serum dilution are shown in Extended Data Fig. 4c. (g) Primary addiction index for the boosting HA, 6 days (left) and 1 month (right) after the second boost (third dose). P-values are for two-tailed Student’s T test comparing the primary addiction index of the homologous to each heterologous boost. Thin lines represent individual mice, thick lines link medians of log transformed titer values at each time point. All results are from 2 independent experiments, with the number of mice in each group indicated in the graphs.
To verify this notion over a wider range of antigenic distances, we immunized mice i.p. with recombinant H1 from strain A/New York/614/1995 (HANY’95) in alhydrogel adjuvant, then boosted these mice twice, either homologously with HANY’95 or heterologously with H1s from strains A/New Caledonia/20/1999 (HANC’99; a slightly drifted strain with 96% amino acid identity to HANY’95) or pandemic A/California/07/2009 (HACA’09; an “antigenic shift” strain, with 80% amino acid identity, Fig. 3e and Extended Data Fig. 4b). Generally, primary addiction was weaker and more variable in this setting even with homologous boosting, possibly due to the overall weak primary response elicited by recombinant HAPR8 protein (Extended Data Fig. 4c). Nevertheless, we observed a progressive decrease in primary addiction as the antigenic distance between the primary and boost antigens increased, so that up to 80% of total serum responses to HACA’09 were FLAG-tagged (corresponding to 20% primary addiction) upon boosting with this variant (Fig. 3f,g). Pooling data for the infection and immunization experiments according to the similarity between priming and boosting HAs showed a highly significant linear decrease in primary addiction as antigenic distance increased (Extended Data Fig. 4d). We conclude that increased antigenic distance between priming and boosting antigens counteracts primary addiction, thus enabling the generation of new, variant-specific antibody responses.
Heterologous SARS-CoV-2 spike boosting
A setting in which subversion of primary addiction by antigenic distance is clinically important is the response to Omicron strains of SARS-CoV-2 in individuals previously exposed to antigens from the WH1 strain. We used the K-tag system to estimate the degree to which boosting with mRNA-LNP encoding the S protein from the Omicron BA.1 strain was capable of overcoming primary addiction generated by priming with WH1 S-encoding mRNA-LNP (WH1 and BA.1 strains have 98% and 92% amino acid identity in the full S protein and RBD domains, respectively). We primed S1pr2-IgkTag mice with WH1 mRNA-LNP in the right leg, then boosted these mice one and two months later with either BA.1 or WH1 mRNA-LNP distally in the left leg (Fig. 4a). Boosting induced similar total IgG responses to WH1 and BA.1 RBDs in both groups (Fig. 4b). By contrast, whereas sera from both groups neutralized a WH1 pseudovirus29 equally, BA.1-boosted serum was on average 15-fold more potent against BA.1 pseudovirus, indicating a strong qualitative difference between the heterologous and homologous boosting regimens. Deconvolution of these effects by tag-specific ELISA revealed responses to the WH1 RBD that were indistinguishable between homologously and heterologously boosted animals, in that primary (Strep+) antibodies were strongly dominant in both settings, with no substantial FLAG+ response after the initial extrafollicular response (Fig. 4c,d). Whereas little to no Strep+ antibodies to the BA.1 RBD were observed after primary immunization, recall Strep+ reactivity to the BA.1 RBD was equally strong regardless of which variant was used for boosting (Fig. 4c,d). This observation agrees with previous reports documenting the evolution of crossreactivity to other strains as a consequence of affinity maturation towards WH1 vaccination in humans30. Importantly, however, heterologous boosting resulted in a pronounced increase in BA.1 RBD titers generated by newly recruited (FLAG+) clones not crossreactive with the WH1 strain, a reactivity otherwise absent from mice boosted homologously (Fig. 4c,d). At their peak (2 weeks post 2nd boost), Strep+ antibodies accounted for an average of 73% (± 19% SD) of total anti-BA.1 reactivity across heterologously boosted mice (Fig. 4d). The induction of new (FLAG+) antibodies upon double BA.1 boost was even more pronounced when assaying for reactivity against the full-length WH1 and BA.1 S proteins (Fig. 4e). Two doses of BA.1 in the absence of prior WH1 immunization (ØBB) generated responses that were only 3.6-fold higher than the WBB group (a difference that did not reach statistical significance; Fig. 4f), again showing that the effect of primary addiction in this setting is greatly reduced compared to that observed for homologous boosting (Fig. 2g). We conclude that BA.1 is sufficiently divergent from WH1 to induce substantial de novo antibody responses, even if not able to entirely overcome primary addiction. Moreover, the key difference between boosting homologously and heterologously is that only the latter can elicit a robust de novo response to the drifted strain.
Figure 4 |. Subversion of primary addiction by heterologous SARS-CoV-2 boosting.

(a) Schematic representation of immunization strategies. (b) Anti-WH1 (left) and BA.1 RBD IgG titers (middle) and NT50 of BA.1 pseudovirus (right) 2 weeks after indicated dose in homologously (WWW) or heterologously (WBB) immunized S1pr2-IgkTag/Tag mice. P-value: 2-tailed T test. FC, fold-change. (c) Evolution of anti-WH1 and BA.1 RBD tag-specific titers. Thin lines represent individual mice, thick lines link medians of log transformed titer values. (d) Comparison of FLAG and Strep anti-RBD titers shown in (c) at 2 weeks after 3rd immunization (left) along with primary addiction index (right). P-values: 2-tailed T test. (e) Anti-full S tag-specific titers and primary addiction index for the same samples as in (d). Results in (b-d) are from 2 independent experiments, with the number of mice in each group indicated. (f) Comparison of de novo FLAG+ antibody responses in WBB mice versus mice in which the 1st dose and fate-mapping were omitted (ØBB). ØBB data are for 10 mice from 2 independent experiments. WBB data are from (c), remeasured in the same assay as ØBB. (g) Schematic representation of antibody fractionation. (h) BA.1 NT50 titers for WBB mice at the time point as in (d). Post-depletion NT50s normalized as described in Methods. P-values: 1-tailed paired T test. (i) Potency of anti-RBD BA.1 Strep+ (FLAG-depleted) and FLAG+ (Strep-depleted) fractions. Raw NT50 values for each fraction were divided by their respective RBD ELISA titers. P-values: 2-tailed paired T test. (j) WH1 RBD structure (PDB: 6MOJ) with amino acid changes in BA.1 highlighted in red. (k) DMS of serum samples obtained from 3 WBB mice 2 weeks after 3rd immunization (d). Antibody binding sites on RBD are shaded according to escape fraction. The positions most highly targeted by each serum fraction are indicated (BA.1-specific residues in parentheses).
To determine whether the enhanced neutralization of BA.1 observed upon heterologous boosting (Fig. 4b) was due to induction of new BA.1-specific antibody in this setting, we fractionated WBB serum samples taken two weeks after the third immunization into FLAG-depleted (Strep+, primary) and Strep-depleted (FLAG+, new) preparations (Fig. 4g and Extended Data Fig. 5a) and measured their neutralizing potency against WH1 and BA.1 pseudovirus. As expected, depletion of FLAG+ antibodies had minimal effect on WH1 neutralization, whereas Strep+ depletion resulted in a much larger decrease (1.7 vs 8.5-fold, respectively; Extended Data Fig. 5b,c). By contrast, removing new (FLAG+) antibodies from WBB sera resulted in a greater reduction in BA.1 neutralization than removing primary (Strep+) antibodies (4.9 vs 1.9-fold decrease; Fig. 4h) even though Strep+ antibodies bound more avidly to the BA.1 RBD by ELISA (Fig. 4d). To estimate the neutralizing potency per unit of specific antibody, we divided the 50% neutralization titer (NT50) derived from the BA.1 pseudovirus assay by the endpoint binding titer obtained by BA.1 RBD-specific ELISA. When normalized to reactivity in this manner, new (FLAG+) antibody was on average 7.0-fold more potent at neutralizing BA.1 than the Strep+ antibody produced by primary-cohort B cell clones in response to WH1 (Fig. 4i). As calculated in Extended Data Fig. 5d, roughly 80% of the excess BA.1 neutralization by WBB compared to WWW samples could be attributed to de novo generation of BA.1 specific antibodies from naïve cells rather than to secondary affinity maturation or preferential selection of primary-cohort memory B cells. Thus, the antibodies that escape primary addiction upon BA.1 boosting are optimized to neutralize the variant strain.
Finally, to gain mechanistic insight into how antigenic drift leads to attenuation of primary addiction, we carried out deep mutational scanning31,32 to define the WH1 and BA.1 RBD epitopes targeted by FLAG+ and Strep+ antibodies in heterologously boosted mice (Fig. 4j and Extended Data Fig. 6). This approach allowed us to separately determine the epitopes targeted by primary and new antibody in the same mouse. We measured the antibody escape patterns of four mice against BA.1 (both Strep+ and FLAG+ antibodies) and WH1 RBD (Strep+ antibodies only), three of which showed interpretable dominance peaks (Extended Data Fig. 7). In these three mice, there was clear segregation of the epitopes targeted by primary Strep+ and new FLAG+ antibodies (Fig. 4k and Extended Data Fig. 7). In two mice, Strep+ antibodies targeted residues of the “class 3” epitope located on the outer face of the RBD (R346, R357, I468), which, as expected, were conserved between WH1 and BA.1 (Fig. 4j,k). By contrast, FLAG+ antibodies in both mice were focused on the BA.1-specific residue R493 (Q493 in WH1), located on the top of the RBD in the “class 2” region of the ACE2-binding surface. The third mouse showed analogous segregation between primary and new antibodies but targeted to different epitopes. Whereas Strep+ antibodies were heavily focused on the conserved class 1/2 epitope that includes G485/F486 (on the “top” of the RBD at the ACE2 interface), FLAG+ antibodies, bound primarily to an epitope that includes the BA.1-specific K440 residue (N440 in WH1) on the side face of the RBD distal to G485/F486 (class 3). Notably, both N440K and Q493R have been reported to lead to escape from neutralization by various monoclonal antibodies33–35. Thus, all three mice followed a logic in which new antibodies elicited by heterologous immunization preferentially targeted epitopes that contained BA.1-specific escape mutations and that did not overlap with epitopes bound by crossreactive primary antibodies. We conclude that primary addiction, by acting in an epitope-specific manner, suppresses the de novo generation of antibodies to conserved epitopes, while allowing the induction of new antibodies targeted specifically to drifted epitopes.
Discussion
Taken together, our findings using the K-tag system make two major points. First, suppression of de novo antibody responses by existing immunity, a necessary feature of OAS as we define it, is extremely potent when measured at zero antigenic distance. These findings support the “primary addiction/OAS2” model (Fig. 2a) in which existing responses prevent the emergence of new serum antibodies to the same antigen, over a “sequential contribution/seniority20” model in which first-cohort responses are larger simply because they were established first and thus boosted a greater number of times. Second, primary addiction weakens markedly as antigenic distance between priming and boosting strains increases. This observation suggests an explanation for why OAS has been so difficult to document experimentally in a consistent manner5: traditional measurements, which rely on differences between drifted antigens to assign antibodies to primary or de novo cohorts, may only be able to distinguish these cohorts reliably when antigenic distance is too great to allow for clear detection of primary addiction. A case in point is that our model estimates that primary addiction between PR8 and FM1, the influenza virus strains for which OAS was initially described1,19, is relatively weak (equivalent to a 3.8-fold suppression of the de novo response), and may thus be difficult to ascertain without the precision afforded by the K-tag model.
Our data are in line with observations in humans showing that, after seasonal influenza vaccination, a large proportion of serum antibody clonotypes are recalled from pre-existing pools, although these studies remained agnostic to whether vaccine-induced antibody clonotypes arose from de novo responses or from recall of undetected memory B cells17,36. Humans, even with their rich influenza antigen exposure history, are able to mount significant de novo responses in recall GCs, while relying on memory B cells for the immediate plasma cell response37. Therefore, we expect the general mechanics of primary addiction reported here to apply to humans as well, at least in general terms. In the SARS-CoV-2 setting, OAS-type suppression may explain the small and variable differences in the preferential induction of Omicron neutralization by variant-specific or bivalent compared to homologous vaccination38–42. Our data suggest that a second dose of an Omicron-containing vaccine may be required to reveal the full extent of de novo antibody induction by Omicron boosting. However, in practice, the repeated exposure of most individuals to WH1 antigens prior to Omicron boosting, as well as the potential greater propensity of human GCs to allow entry of reactivated memory B cells37 may lead to stronger OAS-type suppression of Omicron-specific antibodies in real-life scenarios.
Mechanistically, our measurements at zero antigenic distance indicate a functional divide between recall GC and serum responses in mice: whereas the former consist almost exclusively of naïve-derived B cell clones21–23, the latter are dominated by the effects of primary addiction. A potential explanation for this divergence is that the naïve B cells that contribute to secondary GCs are not of sufficient affinity either to exit the GC as plasma cells or to secrete antibody that is detectable by direct ELISA. Low antigen binding among naïve-derived secondary GC B cells has been reported previously by others22, which is consistent with the latter hypothesis. Because antigenic drift for both influenza virus and SARS-CoV-2 is primarily driven by immune escape, the loosening of OAS in heterologous settings should in principle focus newly recruited B cell clones on drifted neutralizing epitopes. This view is supported by the results of our neutralization and epitope mapping experiments. Moreover, RBD-binding antibodies that escaped primary addiction tended to focus on novel residues located within epitopes that were not dominant targets of the crossreactive first-cohort response. This pattern suggests antibody-mediated epitope masking—whereby serum antibody either present prior to boosting or produced acutely by boosted memory B cells competes with naïve B cells for binding a specific epitope—as a potential mechanism for primary addiction. Similar effects have been observed previously by infusion of monoclonal antibodies prior to induction of an immune response in mice and humans and are predicted to affect the fine-specificity of recall responses43–46. These findings not only indicate that the suppression of de novo antibody responses afforded by primary addiction is epitope- rather than antigen-specific (epitope masking, rather than antigen trapping47), but also suggest a teleological explanation for why it might be advantageous for memory B cells to avoid re-entering secondary GCs21,48, since competition by memory B cells could inhibit the ability of naïve cells to generate antibodies tailored to novel epitopes in viral escape variants. In such a framework, the primary role of secondary GCs would be to circumvent the worst effects of OAS.
METHODS
Mice
Wild type C57BL/6J and B6.C(Cg)-Cd79atm1(cre)Reth/EhobJ (“Cd79aCre/+”, also known as Mb1-Cre49) mice were obtained from The Jackson Laboratory. S1pr2-CreERT2 BAC-transgenic mice25 were a kind gift from T. Kurosaki and T. Okada (U. Osaka, RIKEN-Yokohama). IgkTag mice were generated at the Rockefeller University. We designed the allele as indicated in Fig. 1a and Extended Data Fig. 1, with a FLAG-tag (DYKDDDDK) and Strep-II tag (WSHPQFEK) separated by stop codons and the SV40 poly-A transcriptional terminator. The 522 nucleotide single-stranded DNA template (including 5’ and 3’ homology arms, each 100 nucleotides long) and the CRISPR guide-RNA (“GGAGCTGGTGGTGGCGTCTC”) were purchased from IDT and prepared according to the Easi-CRISPR gene targeting method50 by the Rockefeller University Gene Targeting Resource Center and injected into zygotes obtained from C57BL/6 mice by the Rockefeller University Transgenic Services core facility. We verified that the allele was correctly inserted by Sanger sequencing across the entire locus using genomic primers located outside of the homology arms. To decrease the risk of potential CRISPR off-targets, one founder mouse was back-crossed for at least 5 generations onto C57BL/6J mice prior to use in experiments. To generate IgkFLAG/Strep mice, germline-excised (Cre-negative mice displaying Strep+ B cell surface staining) mice, obtained from occasional spontaneous germline recombination in Cd79aCre/+ IgkTag breedings, were crossed to the parental IgkTag strain. All mice were held at the immunocore clean facility at the Rockefeller University under specific pathogen-free conditions. All mouse procedures were approved by the Rockefeller University’s Institutional Animal Care and Use Committee.
Immunizations, infections, and treatments
Immune responses were induced in 7- to 12-week-old mice by either: subcutaneous footpad immunization with 10 μg TNP17 -KLH (Biosearch, #T-5060) supplemented with 1/3 volume of Imject™ alum (ThermoScientific) or i.p. immunization with 50 μg TNP-KLH prepared with 1/3 volume of either alum or aluminum hydroxide gel (alhydrogel, Invivogen); 20 μg recombinantly produced trimer-stabilized HA (see below) in alhydrogel; intramuscular immunization of quadricep muscles with 3 μg WH151 or BA.1 spike mRNA encapsulated in lipid nanoparticles (mRNA-LNP), generated as described below; or by intranasal infection with mouse-adapted PR8 influenza virus produced in embryonated chicken eggs (~33 PFU, kindly provided by M. Carroll, Harvard University Medical School). In S1pr2-IgkTag mice, the primary immune response was fate-mapped by oral gavage of 200 μl tamoxifen (Sigma) dissolved in corn oil at 50 mg/ml, on days 4 and 8 for the day 12 flow cytometry experiment (Fig. 1), on days 4, 8 and 12 for all other immunization experiments, and on days 4, 8, 12, and 16 for the influenza infection experiment (Fig. 3). In TNP recall experiments (Fig. 2c), boosting was performed identically as the primary immunization, on the indicated time points. For homologous mRNA-LNP experiments (Fig. 2e), boosting was performed in the same way as priming, except that some mice the left quadricep muscle was boosted (contralaterally, stratified in Extended Data Fig. 3e). For heterologous mRNA-LNP recall experiments (Fig. 4) boosting was contralateral in all cases. For HA immunization experiments (Fig. 3), homologous or heterologous HA protein boosting was performed ipsilaterally, exactly as the primary immunization. For infection experiments, mice were boosted after 3 months by subcutaneous immunization of the footpad with 5 μg HAPR8 or HAFM1 prepared with 1/3 volume alhydrogel, as described previously21. In all cases, additional booster immunizations were performed identically to the first boost. Blood samples were collected via cheek puncture into microtubes prepared with clotting activator serum gel (Sarstedt, #41.1378.005).
Generation of recombinant and hapten-conjugated proteins
Recombinant HAs used for immunizations and ELISAs were produced in-house using a CHO cell protein expression system, as described previouosly21. Cysteine residues were introduced into the HA sequence to create trimer-stabilizing disulfide bonds, as originally described for H1/A/California/07/200952. We described the production of HAPR8 and HACa’09 before21. For HAFM1 and HANC’99, the same procedure was followed, including the introduction of trimer-stabilizing mutations. For immunizations, C-terminal domains not native to HA (foldon, Avi-tag, His-tag) were removed by thrombin cleavage and HAs were subsequently FPLC-purified prior to storage in phosphate-buffered saline (PBS). For ELISA, non-thrombin treated FPLC-purified proteins were used. A high affinity IgY-specific mAb obtained from CGG-immunized mice (clone 2.153) was modified for use as a standard for FLAG/Strep ELISA detection. Heavy and light chain constant regions in the original human mAb plasmids54 were replaced with mouse IgG1 and Igκ constant regions and the C-terminus of Cκ was modified to encode a LoxP site and Ser-Gly-Gly linker followed by either a FLAG or Strep-tag, yielding Cκ chains identical to those produced by IgkTag mice prior to and after recombination, respectively. The mAb-FLAG or mAb-Strep light chain plasmids were transfected together with the heavy chain plasmid into 293F cells (ThermoScientific #R79007) and purified using protein-G affinity chromatography as described53. Cell lines tested negative for mycoplasma and were not authenticated besides testing the protein they produced. To compare affinity maturation between FLAG+ and Strep+ anti-TNP antibody titers (Fig. 1h), custom low and high hapten-Bovine serum Albumin (BSA) conjugations were made in-house. BSA (in PBS; ThermoScientific #77110) at 2.5 mg/ml was incubated with TNP-ε-Aminocaproyl-OSu (Biosearch Technologies #T-1030) in PBS with 20% dimethyl sulfoxide at a molar ratio of either 1:2 or 1:20 for 2 hours at room temperature while rotating. Unconjugated TNP-ε-Aminocaproyl-OSu was removed by dialysis in PBS. Final TNP: BSA conjugation ratios were estimated to be ~1:1 and ~1:13 by measuring absorbance at 280 and 348 nm, these reagents are referred to as TNP1 -BSA and TNP13 -BSA. The BSA concentration was corrected by determining the extinction coefficient for TNP-ε-Aminocaproyl-OSu at 280 nm. Besides in Fig. 1h, commercial TNP4 -BSA (Biosearch Technologies #T-5050) was used for all other TNP ELISAs, see description below.
Production of mRNA-LNP
The WH1 S mRNA vaccine was designed based on the SARS-CoV-2 S protein sequence (Wuhan-Hu-1, GenBank: MN908947.3) where the lysine (K) and valine (V) amino acids in positions 986–987 are modified to prolines (P) to obtain a prefusion-stabilized mRNA-encoded immunogen. The BA.1 S amino acid sequence was obtained from the WH1 S by introducing BA.1-specific modifications. Coding sequences of the WH1 and BA.1 S were codon-optimized, synthesized and cloned into an mRNA production plasmid (GenScript) as described55. mRNA production and LNP encapsulation was performed as described55. Briefly, mRNAs were transcribed to contain 101 nucleotide-long poly(A) tails. m1Ψ−5′-triphosphate (TriLink) instead of UTP was used to generate modified nucleoside-containing mRNAs. Capping of the in vitro transcribed mRNAs was performed co-transcriptionally using the trinucleotide cap1 analog, CleanCap (TriLink). mRNA was purified by cellulose (Sigma) purification, as described 56. All mRNAs were analyzed by agarose gel electrophoresis and were stored frozen at −20°C. Cellulose-purified m1Ψ-containing RNAs were encapsulated in LNP using a self-assembly process as previously described wherein an ethanolic lipid mixture of ionizable cationic lipid, phosphatidylcholine, cholesterol and polyethylene glycol-lipid was rapidly mixed with an aqueous solution containing mRNA at acidic pH57. The ionizable cationic lipid and LNP composition are described in the patent application WO 2017/004143. The RNA-loaded particles were characterized and subsequently stored at −80°C at a concentration of 1 μg μl−1. The mean hydrodynamic diameter of these mRNA-LNP was ~80 nm with a polydispersity index of 0.02–0.06 and an encapsulation efficiency of ~95%.
Flow cytometry
For flow cytometry of peripheral B cells, blood was collected in microtubes with EDTA to prevent coagulation and treated with ACK buffer (Lonza) to lyse red-blood cells. For lymph node samples, cell suspensions were obtained by mechanical disassociation with disposable micropestles (Axygen). Spleens were homogenized by filtering through a 70-μm cell strainer and treated with ACK buffer. Bone-marrow cells were extracted by centrifugation of punctured tibiae and femurs at up to 10,000 xG for 10 s, then treated with ACK buffer. Cells from each tissue were resuspended in PBS supplemented with 0.5% BSA and 1 mM EDTA and incubated first with FC-block (rat anti-mouse CD16/32, clone 2.4G2, Bio X Cell) for 30 min on ice and subsequently with various fluorescently-labeled antibodies (see Table S1) for 30 min. Cells were filtered and washed with the same buffer before analysis on a BD FACS Symphony cytometer. Data were analyzed using FlowJo v.10 software.
Western blotting
To determine the presence of epitope-tagged antibodies in IgkTag mice, serum samples from steady state adult mice and precision plus dual color protein standards (Biorad) were run in triplicate on SDS-PAGE mini-protean TGX protein gels (Biorad) under denaturing conditions, to separate heavy and light antibody chains. Samples were transferred to a PVDF membrane using the Iblot gel transfer system (Invitrogen). Membranes were blocked for 2 hours at room temperature while gently shaking with 5% nonfat dry milk in PBS-Tween (0.05%), prior to overnight incubation in the same buffer with 1:2000 anti-FLAG-HRP (clone D6W5B, CellSignallingTechnology #86861S) or anti-Strep (clone Strep-tag II StrepMAB-Classic, Biorad #MCA2489P) or goat anti-mouse Igκ-HRP (SouthernBiotech #1050-05). Membranes were extensively washed with PBS-Tween and subsequently incubated with western blotting ECL substrate (Amersham) prior to chemiluminescence detection on an Azure c300 gel imager (Azure Biosystems).
ELISA
ELISAs were performed as described21, with specific modifications to allow for direct FLAG/Strep comparison. FLAG/Strep ELISAs were performed side by side and with internal standards on each 96-well plate. To detect antigen-specific serum antibody titers, plates were coated overnight at 4°C with antigen in PBS (10 μg/ml for TNP4 -BSA, 2 μg/ml for in-house conjugated TNP1/13-BSA (see above), 1 μg/ml for HAs and SARS-CoV-2 Spike or RBD proteins (Sinobiological #40592-V08H, #40592-V08H121, #40589-V08H26; WH1 S and RBD proteins were a kind gift from P. Wilson)). For FLAG/Strep standard curves, wells were coated with 10 μg/ml purified IgY (Gallus Immunotech). After washing with PBS-Tween (PBS + 0.05% Tween20, Sigma), plates were blocked for 2 hours at room temperature with 2.5% BSA in PBS. Serum samples were diluted 1:100 in PBS and serially titrated in 3-fold dilutions. Mouse anti-IgY mAb-FLAG or mAb-Strep were also serially titrated in 3-fold dilutions (Extended Data Fig. 2d). Samples were incubated for 2 hours and then washed with PBS-Tween, before adding one of the following HRP-detection Abs: goat anti-mouse IgG (Jackson Immunoresearch #15-035-071), rat anti-mouse Igκ (abcam #ab99632), goat anti-mouse IgM (Southern Biotech #1020-05), rabbit anti-FLAG-HRP (clone D6W5B) or mouse anti-Strep (clone Strep-tag II StrepMAB-Classic) for 30–45 minutes. Dilutions of anti-FLAG and anti-Strep antibodies were defined so that the curves generated by titration of FLAG- and Strep-tagged mAbs were equivalent (Extended Data Fig. 2d). After washing with PBS-Tween, samples were incubated with 3,3′,5,5′-Tetramethylbenzidine substrate (slow kinetic form, Sigma) and the reaction was stopped with 1N HCl. Optical Density (OD) absorbance was measured at 450 nm on a Fisher Scientific accuSkan FC plate reader. To normalize FLAG and Strep endpoint titers, the serum titer dilution was calculated at which each sample passed the threshold OD value of its respective mAb at a fixed concentration of either 20 or 6.67 ng/μl. Titers were calculated by logarithmic interpolation of the dilutions with readings immediately above and immediately below the mAb OD used58.
For total serum IgG ELISAs, plates were coated with anti-mouse IgG. Standard curves were generated using unlabeled mouse IgG (Southern Biotech #0107-01), and detection was performed with anti-mouse IgG-HRP (Southern Biotech). To deplete IgM from serum samples, anti-mouse IgM agarose beads (Sigma #A4540) were used according to instructions from the manufacturer. Beads were washed with PBS and samples were incubated at a ratio of 1:20 sample to beads overnight at 4°C with rotation. The bead-bound IgM fraction was removed by centrifugation for 3 minutes at 10,000 G, and the unbound supernatant fraction was used for subsequent ELISAs. To confirm the efficiency of IgM depletion, total IgM levels were measured as described above for total IgG, with goat anti-mouse IgM, unlabeled IgM and anti-mouse IgM-HRP (Southern Biotech).
Serum fractionation and SARS-Cov2 pseudoneutralization assay
Virus neutralization titers were assessed in FLAG+ versus Strep+ serum fractions of samples collected from S mRNA-LNP immunized S1pr2-IgkTag mice. To separate fractions, immunoprecipitation with anti-FLAG M2 magnetic beads (Sigma #M8823–5ML) and MagStrep “type3” XT beads (IBA #2-4090-010) was performed as per the manufacturer’s instructions. In brief, magnetic beads were washed with sample buffer (Tris buffered saline for FLAG beads, and 1x buffer W (IBA # 2-1003-100) for MagStrep beads) and samples were incubated at a ratio of 20:1 sample to bead resin overnight at 4°C with rotation. Bead-bound fractions were separated using a magnetic separator and discarded, while the unbound fraction was collected. Fractionated samples were concentrated by centrifugation to half the input concentration and heat inactivated. The degree to which the total and fractionated serum samples neutralized WH1 and BA.1 SARS-CoV-2 was approximated using SARS-CoV-2 spike pseudotyped HIV-1 based NanoLuc luciferase reporter assay described previously29. Briefly, serum samples were five-fold serially diluted with a final top dilution of 1:00 serum and incubated for 1 h at 37°C with SARS-CoV-2 WH1 or BA.1 spike pseudotyped HIV-1 reporter virus and then transferred to HT1080/ACE2.cl14 cells59. At 48 h, the cells were washed, lysed and luciferase activity was measured using the Nano-Glo Luciferase Assay System (Promega) and the Glomax Navigator luminometer (Promega). The relative luminescence units were normalized using cells infected in the absence of serum and then plotted in GraphPad Prism. NT50 values were calculated using four-parameter non-linear regression (least squares regression method without weighting) of the curves shown in Extended Data Fig. 5b. The mean of two technical duplicates is shown, outlier points were excluded. For NT50 comparisons between input and fractions (Fig. 4g and Extended Data Fig. 5c), the NT50 of the fractionated samples was adjusted to equalize the BA.1 RBD ELISA titer of the un-depleted tag compared to its corresponding ELISA titer in the input fraction.
Deep Mutational Scanning
Construction of yeast-displayed deep mutational scanning libraries of Omicron BA.1 RBD
Duplicate single-mutant site-saturation variant libraries were designed in the background of the SARS-CoV-2 Omicron BA.1 spike RBD and produced by Twist Bioscience, essentially the same as has been done previously for other SARS-CoV-2 variants60,61. The Genbank map of the plasmid encoding the unmutated Omicron BA.1 RBD in the yeast-display vector is available at https://github.com/jbloomlab/SARS-CoV-2-RBD_DMS_Omicron/blob/main/data/3294_pETcon-SARS2-RBD_Omicron-BA1.gb. The site-saturation variant libraries were delivered as double-stranded DNA fragments by Twist Bioscience and were barcoded and cloned in bulk into the yeast-display vector backbone. The barcoded mutant library plasmid DNA was electroporated into E. coli (NEB 10-beta electrocompetent cells, New England BioLabs #C3020K), and bottlenecked to ~1 × 105 cfus (an average of >25 barcodes per single-mutant). Plasmid DNA was purified and transformed into the AWY101 yeast strain. 16-nucleotide barcodes were associated with their BA.1 variants by PacBio sequencing, and the effects of mutations of RBD expression and ACE2 binding were measured, essentially as described61. These experiments are described and analyzed at https://github.com/jbloomlab/SARS-CoV-2-RBD_DMS_Omicron.
FACS sorting of yeast libraries to select mutations with reduced binding by polyclonal sera from immunized mice
Experiments mapping mutations that reduce RBD binding of sera from immunized mice were performed in biological duplicate with independent mutant WH1 or BA.1 RBD libraries, similarly to as previously described for monoclonal antibodies62, human polyclonal plasma samples63. First, 75 μL of each of the sera was twice-depleted of nonspecific yeast-binding antibodies by incubating for 2 hours at room temperature or overnight at 4°C with 37.5 OD units of AWY101 yeast containing an empty vector, as described63. WH1 and BA.1 mutant RBD yeast libraries61 were induced with galactose-containing, low-dextrose synthetic defined medium with casamino acids (SD-CAA, 6.7g/L Yeast Nitrogen Base, 5.0g/L Casamino acids, 1.065 g/L MES acid, and 2% w/v galactose + 0.1% w/v dextrose) to express RBD, then washed and incubated with diluted serum for 1 hour at room temperature with gentle agitation. Each tested combination of mouse serum against each WH1 or BA.1 RBD mutant library for loss of binding of Strep or FLAG-tag antibodies was performed independently. For each serum, a sub-saturating dilution was used such that the amount of fluorescent signal due to serum antibody binding to RBD was approximately equal across samples (1:1000 for mapping of Strep antibodies against the WH1 libraries, 1:200 for mapping of Strep antibodies against the BA.1 libraries, and 1:50 for mapping of the FLAG antibodies against the BA.1 libraries). The yeast libraries were then secondarily labeled for 1 hour with 1:100 FITC-conjugated anti-MYC antibody (Immunology Consultants Lab, #CYMC-45F) to label for RBD expression and either 1:200 APC-conjugated Streptavidin (Invitrogen S-868) to label for bound Strep antibodies or APC-conjugated rat anti-FLAG (BioLegend #637308) to label for bound FLAG-tagged antibodies. A flow cytometric selection gate was drawn to capture RBD mutants with reduced antibody binding for their degree of RBD expression. For each sample, ~4 × 106 cells were processed on the BD FACSAria II cell sorter. Serum-escaped cells were grown overnight in SD-CAA as defined above with 2% w/v dextrose, no galactose, and 100 U/mL penicillin + 100 μg/mL streptomycin to expand cells prior to plasmid extraction.
DNA extraction and Illumina sequencing
Plasmid DNA was extracted from 30 OD units (1.6 × 108 colony forming units (cfus)) of pre-selection yeast populations and approximately 5 OD units (~3.2 × 107 cfus) of overnight cultures of serum-escaped cells (Zymoprep Yeast Plasmid Miniprep II) as previously described60,62. The 16-nucleotide barcodes identifying each WH1 or BA.1 RBD variant were amplified by polymerase chain reaction (PCR) and prepared for Illumina sequencing as described previously60,62. Barcodes were sequenced on an Illumina NextSeq 2000 with 50 bp single-end reads.
Analysis of deep sequencing data to compute each mutation’s escape fraction
Escape fractions were computed essentially as described62. We used the dms_variants package (https://jbloomlab.github.io/dms_variants/, version 1.4.0) to count each barcoded RBD variant in each pre-selection and serum-escape population. For each selection, we computed the escape fraction for each barcoded variant via the formula provided in Greaney et al. 62. These escape fractions represent the estimated fraction of cells expressing that specific variant that falls in the escape bin, such that a value of 0 means the variant is always bound by serum and a value of 1 means that it always escapes serum binding. We then applied a computational filter to remove variants with >1 amino-acid mutation, low sequencing counts (< 50 in the pre-selection condition), or highly deleterious mutations that might cause antibody escape simply by leading to poor expression of properly folded RBD on the yeast cell surface (an ACE2 binding score of < −2 or an RBD expression score of < −1.25 or −0.83361 for the WH1 and BA.1 mutant libraries, respectively, reflecting the different baseline expression levels of the two wild-type RBDs). The reported antibody-escape scores throughout the paper are the average across duplicate libraries; these scores are also in Supplemental Spreadsheet 1. Correlations in final single-mutant escape scores are shown in Extended Data Fig. 6c. Full documentation of the computational analysis is at https://github.com/jbloomlab/SARS-CoV-2-RBD_MAP_OAS.
Data visualization
The serum-escape map logo and line plots were created using the dmslogo package (https://jbloomlab.github.io/dmslogo, version 0.6.2). The height of each letter indicates the escape fraction for that amino-acid mutation. For each serum, the logo plots feature any site where for >=1 library/antibody tag condition, the site-total antibody escape was >10x the median across all sites and at least 10% the maximum of any site. For each sample, the y-axis is scaled to be the greatest of (a) the maximum site-wise escape metric observed for that sample, or (b) 20x the median site-wise escape fraction observed across all sites for that plasma. The code that generates these logo plot visualizations is available at https://github.com/jbloomlab/SARS-CoV-2-RBD_MAP_OAS/blob/main/results/summary/escape_profiles.md. To visualize serum escape on the RBD structure, the WH1 RBD surface (PDB: 6M0J) was colored by the site-wise escape metric at each site, with white indicating no escape and red indicating the site with the most escape.
Statistical analysis and software
Statistical tests used to compare conditions are indicated in figure legends. No statistical methods were used to determine sample size. Statistical analysis was carried out using GrahPad Prism v.9. Flow cytometry analysis was carried out using FlowJo v.10 software. Graphs were plotted using Prism v.9, and edited for appearance using Adobe Illustrator CS. For data plotted on logarithmic scales (e.g., serum antibody titers), statistical analysis was performed on the log-transformed data. Samples with reactivities below the limit of detection were assigned a value of 100, as the top dilution was 1:100.
Extended Data
Extended Data Figure 1 |. Design of the K-tag allele.

Nucleotide sequence of the 522 bp DNA template used to generate the IgkTag allele, with amino acid translations given for all coding sequences (bold font). Nucleotide numbers are given for each line. Amino acid translation is positioned below the center nucleotide of each codon.
Extended Data Figure 2 |. Characterization of the K-tag system.
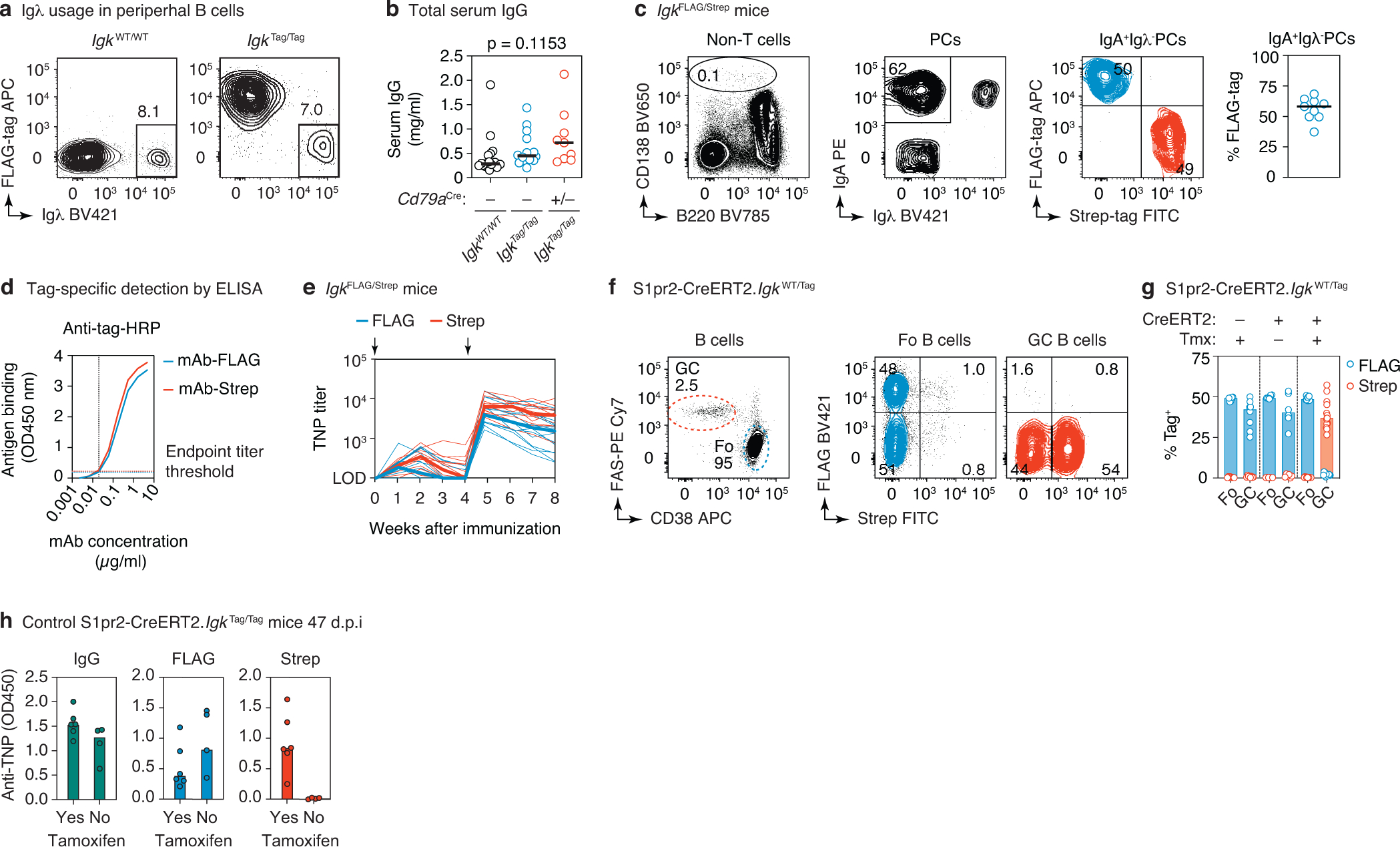
(a) Representative flow cytometry plots of peripheral B cells (gated: B220+CD4−CD8−CD138−) obtained from the blood of WT and IgkTag/Tag mice, stained for surface expression of Igλ and FLAG-tagged immunoglobulins. (b) Total IgG concentration in the serum of WT, IgkTag/Tag, and Cd79aCre/+.IgkTag/Tag mice as determined by ELISA. Differences were not significant by one-way ANOVA (p<0.05). (c) Flow cytometry of steady-state bone marrow plasma cells (PCs) obtained from adult (6-week old) IgkFLAG/Strep mice. Gating strategy for PCs is shown in the left panel (pre-gated on non-CD4/CD8 T cells), for surface IgA+ and Igλ− PCs in the middle panel, and for FLAG/Strep in the right panel. Quantification across 9 mice from 3 independent experiments is shown in the rightmost panel (each dot represents an individual IgkFLAG/Strep mouse, line represents the median). (d) ELISA standard curves with monoclonal antibody (mAb)-FLAG and mAb-Strep detected at dilutions of the respective HRP antibodies where the curves overlap. The mAb concentration at which the curves crossed the absorbance background threshold (indicated by the dotted lines) was used to calculate the endpoint titer, as described in the methods section. (e) Tag-specific anti-TNP titers in IgkFLAG/Strep mice immunized and boosted i.p. with TNP-KLH/alum at the timepoints indicated by black arrows. Results are from 14 mice from 2 independent experiments. The day 7 timepoint was not collected for the first cohort. Thin lines represent individual mice, thick lines link medians of log transformed titer values at each time point. (f) Flow cytometry of S1pr2-IgkWT/Tag mice as in Fig. 1d, e. with quantification in (g), cre− and no tamoxifen control groups are included. Data points are from 6–13 popliteal lymph nodes per group from at least 2 independent experiments. (h) Anti-TNP ELISA reactivity for S1pr2-IgkTag/Tag mice immunized as in Fig. 1d. Serum was obtained at 47 d.p.i from 6 mice that received tamoxifen and 4 that did not receive tamoxifen. IgG (left), FLAG (middle) and Strep (right) ELISA absorbance is shown. Samples were diluted 1:100.
Extended Data Figure 3 |. Recall antibodies and GCs.
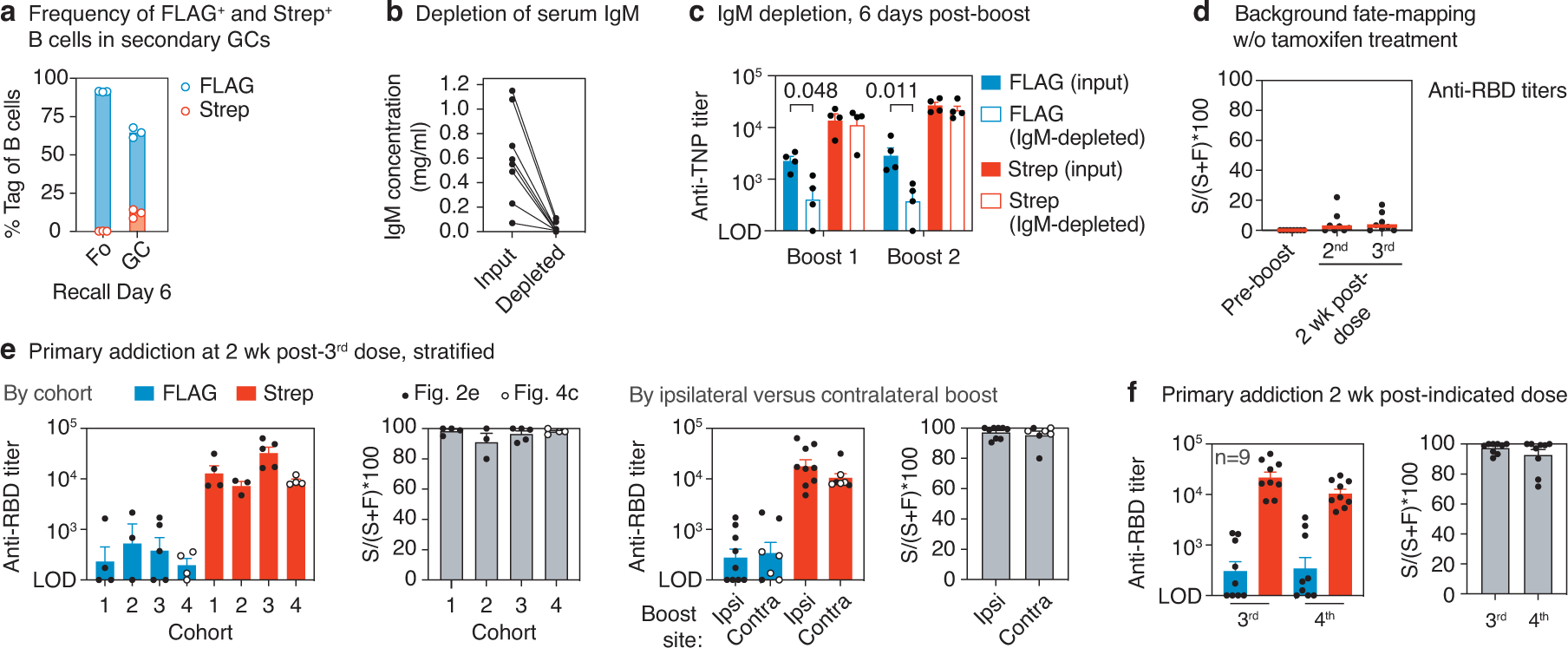
(a) Flow cytometry of secondary GCs in the spleen of 3 TNP-KLH i.p. primed and boosted S1pr2-IgkTag/Tag mice, with tamoxifen labeling at 4, 8, and 12 d.p.i.. Gated on GC B cells (FAS+CD38−B220+CD4−CD8−CD138−) expressing FLAG or Strep-tag as in Fig. 1e. (b) Total IgM concentrations of serum samples pre and post IgM-depletion via immunoprecipitation, measured by ELISA. Data are from 8 serum samples, from the same 4 mice as in Fig. 2c. (c) Tag-specific anti-TNP titers before and after IgM depletion for samples collected 6 days after the first and second boost with TNP-KLH (same samples as in (b) and Fig. 2c). Bars represent the means of log transformed titers and the error bars are SEM. P-values are for two-tailed, paired T-test, only statistically significant (p < 0.05) values are shown. (d) Background Strep+ anti-RBD titers in S1pr2-IgkTag/Tag control mice immunized as in Fig. 2b,e, but not treated with tamoxifen. Graphs show median percentage of the anti-RBD titer that is Strep+ ((S/(S+F)*100)) in the absence of tamoxifen at the pre-boost time point and two weeks after the second and third immunizations. This represents the median percentage by which FLAG+ titers in recall responses are likely to be underestimated by spontaneous recombination by the S1pr2-CreERT2 driver. Data are from 8 mice from 2 independent experiments. (e) Comparison of primary addiction data shown in Fig. 2e,f, stratified by cohort and ipsilateral versus contralateral boost. The 4th cohort of mice is the homologously boosted group shown in Fig. 4b–e, depicted here by open circles. Bars represent the mean of log transformed titers, error bars are SEM. Data are from 16 mice from 4 independent cohorts. (f) Comparison of primary addiction between 3rd and 4th responses in 9 mice from 2 cohorts, based on data from Fig. 2e,f,h. Bars represent the mean of log transformed titers, error bars are SEM.
Extended Data Figure 4 |. Primary cohort recall upon heterologous HA protein boosting.
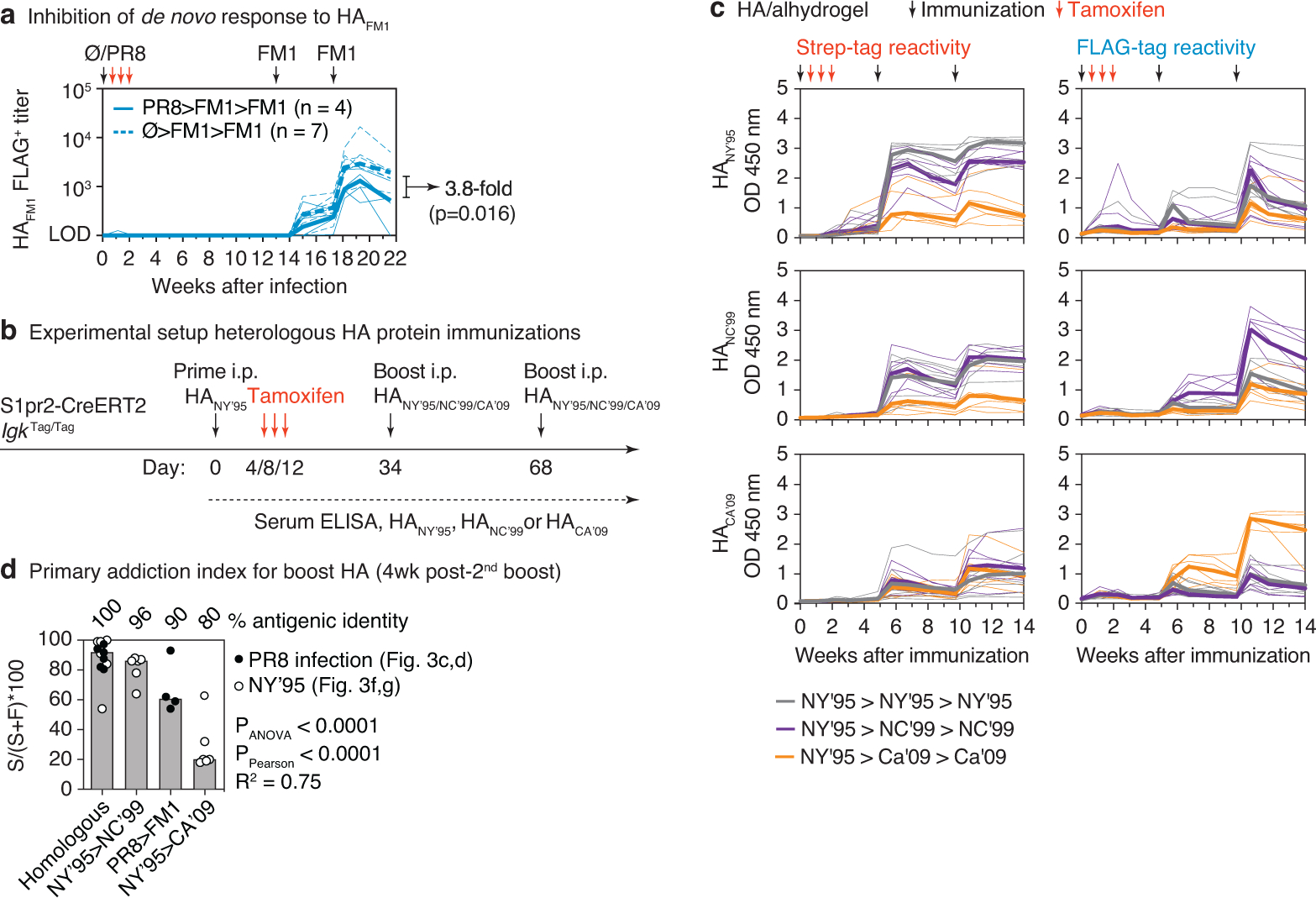
(a) Comparison of de novo FLAG+ antibody responses to HAFM1 in the presence or absence of primary infection with influenza virus PR8. PR8>FM1>FM1 data are for the same samples as in Fig. 3d, re-measured in the same assay as Ø>FM1>FM1. (b) Schematic representation of HA prime-boost strategy. S1pr2-IgkTag/Tag mice were primed i.p. with HANY’95 in alhydrogel and boosted homologously or heterologously with HANC’99 or HACA’09 in alhydrogel as indicated. (c) Full time course of anti-HA tag-specific ELISA reactivity (optical density at 1:100 dilution) of the same mice shown in Fig. 3f. Anti-HANY’95 (top), HANC’99 (middle) and HACA’09 (bottom) ELISAs are shown, with Strep (left) and FLAG (right) detection. (d) Relationship between primary addiction and antigenic distance for infection and immunization experiments. Graph compiles primary addiction indices from Figs. 3d and 3g. Bars are ordered by amino acid identity between the priming and boosting HAs. P-values are for one-way ANOVA, with % identity as a categorical variable, or Pearson correlation, with (100 – % identity) as a linear variable.
Extended Data Figure 5 |. Neutralization of WH1 and BA.1 pseudoviruses by serum antibody fractions from BA.1 boosted mice.
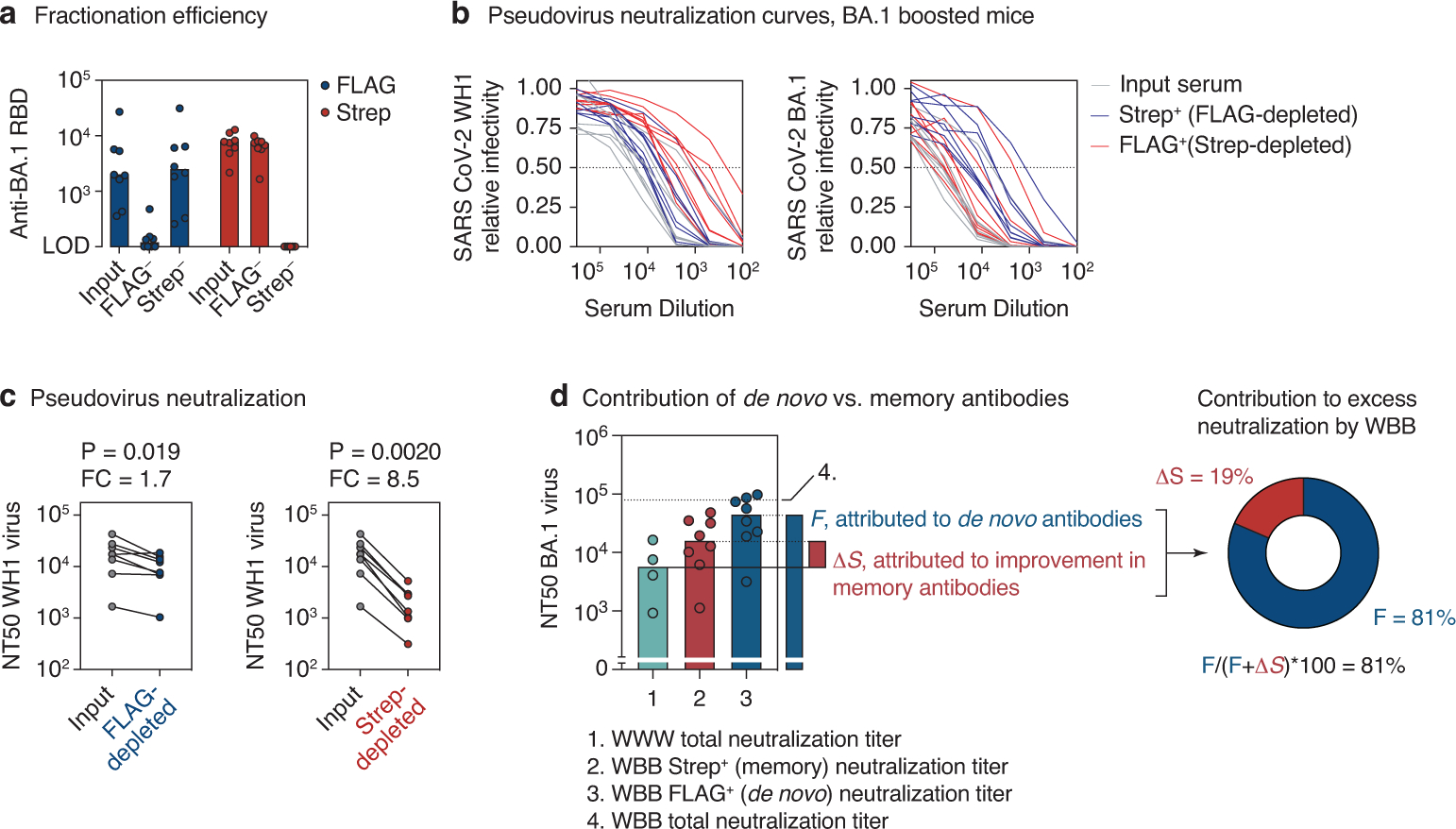
(a) Efficiency of serum fractionation into FLAG and Strep-depleted fractions, as measured by anti-RBD ELISA of input vs. post-depletion samples. Serum was obtained from heterologously immunized mice two weeks after the 3rd immunization, Fig. 4d (8 mice from 2 independent experiments). (b) Neutralization of WH1 (left) and BA.1 (right) SARS-CoV-2 S-expressing pseudotyped HIV-1 virus by serum fractions shown in (a). Mean values of technical duplicates are shown. (c) WH1 S pseudovirus NT50 titers for samples in (b). Post-depletion NT50s were normalized to input serum based on the BA.1 RBD ELISA titers (a), by applying a correction equalizing the anti-RBD Strep-titer of the FLAG-depleted fraction to the input, and the anti-RBD FLAG-titer of the Strep-depleted fraction to the input. P-values are for one-tailed paired T test. FC, fold-change. (d) Estimating the contribution of de novo vs. memory-derived antibodies to excess BA.1 neutralization by the WBB regimen. The contribution of secondary affinity maturation or preferential selection of crossreactive memory B cells by BA.1 boosting, μS, is calculated as the difference between Strep+ WBB and total WWW BA.1 neutralization titers (the latter are assumed to be all FLAG+). The contribution of BA.1-specific antibodies induced de novo by BA.1 boosting, F, is given as the FLAG+ WBB titer. Percent contribution of F to the improved BA.1 neutralization in WBB is calculated as (F/(F+μS))*100. Bars and dotted lines represent the medians for each condition, which were used to calculate F and μS. The upper dotted line (4.) represents the median neutralization of total WBB antibodies and is shown for reference purposes only. Data in groups 1 and 4 are reproduced from Fig. 4b, data in group 2 and 3 from Fig. 4h.
Extended Data Figure 6 |. Yeast-displayed deep mutational scanning to map mutations that reduce binding of immunized mouse serum.

(a) Top: Representative plots of nested FACS gating strategy used for all experiments to select for single yeast cells. Bottom: Gating strategy to select for RBD-expressing single cells (FITC-A vs. FSC-A). (b) FACS gating strategy for one of two independent libraries to select cells expressing BA.1 or WH1 RBD mutants with reduced Strep or FLAG antibody binding (cells in blue), as measured by secondary staining with APC-conjugated streptavidin or APC-conjugated anti-FLAG antibody, respectively. Gates were set manually for each sample to capture cells that have a reduced amount of tagged antibody binding for their degree of RBD expression. FACS scatter plots were qualitatively similar between the two libraries. The mouse identifier (#1–4), DMS target library (WH1 or BA.1), and antibody tag (Strep or FLAG) are indicated above each plot. (c) Mutation (left)- and site (right)-level correlations of escape scores between two independent biological replicate libraries.
Extended Data Figure 7 |. RBD serum-escape logo plots.

Deep mutational scanning results of serum collected from 4 heterologously immunized mice, 2 weeks after the 3rd dose (Fig. 4d). Each mutation’s “escape fraction” was measured, which ranges from 0 (no cells effect on antibody binding) to 1 (all cells with the mutation have decreased antibody binding). Mouse 4 is not shown in the main text as there were no interpretable peaks in antibody binding to BA.1 libraries for either antibody fraction. Logo plots show the antibody-escape fractions for individual amino-acid mutations at key sites of strong escape. Sites in which BA.1 differs from the WH1 sequence are shown in purple font. All escape scores are shown in Supplemental Spreadsheet 1 and are available online at https://github.com/jbloomlab/SARS-CoV-2-RBD_MAP_OAS/blob/main/results/supp_data/all_raw_data.csv.
Supplementary Material
Acknowledgments
We would like to thank the Rockefeller University Transgenics and Gene Targeting facilities for generating the K-tag mouse strain and Comparative Biosciences Center for mouse housing. We thank P. Wilson for recombinant SARS-CoV-2 WH1 Spike and RBD protein, J.T. Jacobsen for technical assistance, and all Rockefeller University staff for their continuous support. This study was funded by NIH/NIAID grants R01AI119006 and R01AI139117 to G.D.V., P01AI165075 to P.D.B., R01AI146101 and R01AI153064 to N.P., and R01AI141707 to J.D.B. Work in the Victora laboratory is additionally supported by NIH grant DP1AI144248 (Pioneer award) and the Robertson Foundation. A.S. was supported by a Boehringer-Ingelheim Fonds PhD fellowship. P.D.B. and J.D.B. are HHMI investigators. G.D.V. is a Burroughs-Wellcome Investigator in the Pathogenesis of Infectious Disease, a Pew-Stewart Scholar, and a MacArthur Fellow.
Footnotes
Competing interests
N.P. is named on a patent describing the use of nucleoside-modified mRNA in lipid nanoparticles as a vaccine platform. He has disclosed those interests fully to the University of Pennsylvania and has an approved plan in place for managing any potential conflicts arising from the licensing of that patent. Paulo J.C. Lin and Ying K. Tam are employees of Acuitas Therapeutics, a company involved in the development of mRNA-LNP therapeutics. Ying K. Tam is named on patents that describe lipid nanoparticles for the delivery of nucleic acid therapeutics, including mRNA, and the use of modified mRNA in lipid nanoparticles as a vaccine platform. P.D.B. has done consulting work in the area of COVID vaccines for Pfizer Inc.. J.D.B. consults or has recently consulted for Apriori Bio, Oncorus, Merck, and Moderna on topics related to viruses, vaccines, and viral evolution. J.D.B, T.N.S., and A.J.G. are inventors on Fred Hutch licensed patents related to viral deep mutational scanning. G.D.V. and J.D.B. are advisors for the Vaccine Company, Inc..
Code availability
The full code that analyzes the deep mutational scanning experiments is available at https://github.com/jbloomlab/SARS-CoV-2-RBD_MAP_OAS. The code that generates the logo plot visualizations is available at https://github.com/jbloomlab/SARS-CoV-2-RBD_MAP_OAS/blob/main/results/summary/escape_profiles.md.
Data Availability
The raw Illumina reads of the 16-nucleotide variant barcodes from the deep mutational scanning experiments are available on the NCBI SRA under BioProject PRJNA770094, BioSample SAMN30086726. All escape scores are shown in Supplemental Spreadsheet 1 and are available online at https://github.com/jbloomlab/SARS-CoV-2-RBD_MAP_OAS/blob/main/results/supp_data/all_raw_data.csv. Renderings of Spike RBD and hemagglutinin structures were obtained from the Protein Data Bank (PDB), with accession codes PDB: 6MOJ (https://www.rcsb.org/structure/6m0j), PDB: 1RU7 (https://www.rcsb.org/structure/1RU7), PDB: 3LZG (https://www.rcsb.org/structure/3LZG).
MAIN TEXT REFERENCES
- 1.Francis T On the Doctrine of Original Antigenic Sin. Proceedings of the American Philosophical Society 104, 572–578 (1960). [Google Scholar]
- 2.Fazekas de St. Groth S & Webster RG Disquisitions on Original Antigenic Sin. II. Proof in lower creatures. J Exp Med 124, 347–361 (1966). 10.1084/jem.124.3.347 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 3.Cobey S & Hensley SE Immune history and influenza virus susceptibility. Curr Opin Virol 22, 105–111 (2017). 10.1016/j.coviro.2016.12.004 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 4.Henry C, Palm AE, Krammer F & Wilson PC From Original Antigenic Sin to the Universal Influenza Virus Vaccine. Trends Immunol 39, 70–79 (2018). 10.1016/j.it.2017.08.003 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 5.Yewdell JW & Santos JJS Original Antigenic Sin: How Original? How Sinful? Cold Spring Harb Perspect Med 11 (2021). 10.1101/cshperspect.a038786 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 6.Krammer F The human antibody response to influenza A virus infection and vaccination. Nat Rev Immunol 19, 383–397 (2019). 10.1038/s41577-019-0143-6 [DOI] [PubMed] [Google Scholar]
- 7.Wheatley AK et al. Immune imprinting and SARS-CoV-2 vaccine design. Trends Immunol 42, 956–959 (2021). 10.1016/j.it.2021.09.001 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 8.Monto AS, Malosh RE, Petrie JG & Martin ET The Doctrine of Original Antigenic Sin: Separating Good From Evil. J Infect Dis 215, 1782–1788 (2017). 10.1093/infdis/jix173 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 9.Victora GD & Nussenzweig MC Germinal Centers. Annu Rev Immunol (2022). 10.1146/annurev-immunol-120419-022408 [DOI] [PubMed] [Google Scholar]
- 10.Baumgarth N The Shaping of a B Cell Pool Maximally Responsive to Infections. Annu Rev Immunol 39, 103–129 (2021). 10.1146/annurev-immunol-042718-041238 [DOI] [PubMed] [Google Scholar]
- 11.Bhattacharya D Instructing durable humoral immunity for COVID-19 and other vaccinable diseases. Immunity 55, 945–964 (2022). 10.1016/j.immuni.2022.05.004 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 12.Pulendran B & Ahmed R Immunological mechanisms of vaccination. Nat Immunol 12, 509–517 (2011). 10.1038/ni.2039 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 13.Gaebler C et al. Evolution of antibody immunity to SARS-CoV-2. Nature 591, 639–644 (2021). 10.1038/s41586-021-03207-w [DOI] [PMC free article] [PubMed] [Google Scholar]
- 14.Tan J et al. A LAIR1 insertion generates broadly reactive antibodies against malaria variant antigens. Nature 529, 105–109 (2016). 10.1038/nature16450 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 15.Davis CW et al. Longitudinal Analysis of the Human B Cell Response to Ebola Virus Infection. Cell 177, 1566–1582 e1517 (2019). 10.1016/j.cell.2019.04.036 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 16.Turner JS et al. SARS-CoV-2 mRNA vaccines induce persistent human germinal centre responses. Nature 596, 109–113 (2021). 10.1038/s41586-021-03738-2 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 17.Lee J et al. Molecular-level analysis of the serum antibody repertoire in young adults before and after seasonal influenza vaccination. Nat Med 22, 1456–1464 (2016). 10.1038/nm.4224 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 18.Han J et al. Polyclonal epitope mapping reveals temporal dynamics and diversity of human antibody responses to H5N1 vaccination. Cell Rep 34, 108682 (2021). 10.1016/j.celrep.2020.108682 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 19.Francis T Jr. Influenza: the new acquayantance. Ann Intern Med 39, 203–221 (1953). 10.7326/0003-4819-39-2-203 [DOI] [PubMed] [Google Scholar]
- 20.Lessler J et al. Evidence for antigenic seniority in influenza A (H3N2) antibody responses in southern China. PLoS Pathog 8, e1002802 (2012). 10.1371/journal.ppat.1002802 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 21.Mesin L et al. Restricted Clonality and Limited Germinal Center Reentry Characterize Memory B Cell Reactivation by Boosting. Cell 180, 92–106 e111 (2020). 10.1016/j.cell.2019.11.032 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 22.Viant C et al. Antibody Affinity Shapes the Choice between Memory and Germinal Center B Cell Fates. Cell 183, 1298–1311 e1211 (2020). 10.1016/j.cell.2020.09.063 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 23.Kuraoka M et al. Recall of B cell memory depends on relative locations of prime and boost immunization. Sci Immunol 7, eabn5311 (2022). 10.1126/sciimmunol.abn5311 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 24.Mostoslavsky R, Alt FW & Rajewsky K The lingering enigma of the allelic exclusion mechanism. Cell 118, 539–544 (2004). 10.1016/j.cell.2004.08.023 [DOI] [PubMed] [Google Scholar]
- 25.Shinnakasu R et al. Regulated selection of germinal-center cells into the memory B cell compartment. Nat Immunol 17, 861–869 (2016). 10.1038/ni.3460 [DOI] [PubMed] [Google Scholar]
- 26.Smith DJ, Forrest S, Ackley DH & Perelson AS Variable efficacy of repeated annual influenza vaccination. Proc Natl Acad Sci U S A 96, 14001–14006 (1999). 10.1073/pnas.96.24.14001 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 27.Fonville JM et al. Antibody landscapes after influenza virus infection or vaccination. Science 346, 996–1000 (2014). 10.1126/science.1256427 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 28.Hogan MJ & Pardi N mRNA Vaccines in the COVID-19 Pandemic and Beyond. Annu Rev Med 73, 17–39 (2022). 10.1146/annurev-med-042420-112725 [DOI] [PubMed] [Google Scholar]
- 29.Schmidt F et al. Measuring SARS-CoV-2 neutralizing antibody activity using pseudotyped and chimeric viruses. J Exp Med 217 (2020). 10.1084/jem.20201181 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 30.Muecksch F et al. Affinity maturation of SARS-CoV-2 neutralizing antibodies confers potency, breadth, and resilience to viral escape mutations. Immunity 54, 1853–1868 e1857 (2021). 10.1016/j.immuni.2021.07.008 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 31.Greaney AJ et al. Comprehensive mapping of mutations in the SARS-CoV-2 receptor-binding domain that affect recognition by polyclonal human plasma antibodies. Cell Host Microbe 29, 463–476 e466 (2021). 10.1016/j.chom.2021.02.003 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 32.Starr TN et al. Prospective mapping of viral mutations that escape antibodies used to treat COVID-19. Science 371, 850–854 (2021). 10.1126/science.abf9302 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 33.Liu L et al. Striking antibody evasion manifested by the Omicron variant of SARS-CoV-2. Nature 602, 676–681 (2022). 10.1038/s41586-021-04388-0 [DOI] [PubMed] [Google Scholar]
- 34.McCallum M et al. Structural basis of SARS-CoV-2 Omicron immune evasion and receptor engagement. Science 375, 864–868 (2022). 10.1126/science.abn8652 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 35.Weisblum Y et al. Escape from neutralizing antibodies by SARS-CoV-2 spike protein variants. Elife 9 (2020). 10.7554/eLife.61312 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 36.Lee J et al. Persistent Antibody Clonotypes Dominate the Serum Response to Influenza over Multiple Years and Repeated Vaccinations. Cell Host Microbe 25, 367–376 e365 (2019). 10.1016/j.chom.2019.01.010 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 37.Turner JS et al. Human germinal centres engage memory and naive B cells after influenza vaccination. Nature 586, 127–132 (2020). 10.1038/s41586-020-2711-0 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 38.Alsoussi WB et al. SARS-CoV-2 Omicron boosting induces de novo B cell response in humans. bioRxiv (2022). 10.1101/2022.09.22.509040 [DOI] [PubMed] [Google Scholar]
- 39.Collier AY et al. Immunogenicity of the BA.5 Bivalent mRNA Vaccine Boosters. bioRxiv (2022). 10.1101/2022.10.24.513619 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 40.Wang Q et al. Antibody responses to Omicron BA.4/BA.5 bivalent mRNA vaccine booster shot. bioRxiv, 2022.2010.2022.513349 (2022). 10.1101/2022.10.22.513349 [DOI] [Google Scholar]
- 41.Davis-Gardner ME et al. mRNA bivalent booster enhances neutralization against BA.2.75.2 and BQ.1.1. bioRxiv, 2022.2010.2031.514636 (2022). 10.1101/2022.10.31.514636 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 42.Kurhade C et al. Low neutralization of SARS-CoV-2 Omicron BA.2.75.2, BQ.1.1, and XBB.1 by 4 doses of parental mRNA vaccine or a BA.5-bivalent booster. bioRxiv, 2022.2010.2031.514580 (2022). 10.1101/2022.10.31.514580 [DOI] [PubMed] [Google Scholar]
- 43.McNamara HA et al. Antibody Feedback Limits the Expansion of B Cell Responses to Malaria Vaccination but Drives Diversification of the Humoral Response. Cell Host Microbe 28, 572–585 e577 (2020). 10.1016/j.chom.2020.07.001 [DOI] [PubMed] [Google Scholar]
- 44.Meyer-Hermann M Injection of Antibodies against Immunodominant Epitopes Tunes Germinal Centers to Generate Broadly Neutralizing Antibodies. Cell Rep 29, 1066–1073 e1065 (2019). 10.1016/j.celrep.2019.09.058 [DOI] [PubMed] [Google Scholar]
- 45.Tas JMJ et al. Antibodies from primary humoral responses modulate recruitment of naive B cells during secondary responses. Immunity (2022). 10.1016/j.immuni.2022.07.020 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 46.Schaefer-Babajew D et al. Antibody feedback regulation of memory B cell development in SARS-CoV-2 mRNA vaccination. medRxiv (2022). 10.1101/2022.08.05.22278483 [DOI] [Google Scholar]
- 47.Kok A, Fouchier RAM & Richard M Cross-Reactivity Conferred by Homologous and Heterologous Prime-Boost A/H5 Influenza Vaccination Strategies in Humans: A Literature Review. Vaccines (Basel) 9 (2021). 10.3390/vaccines9121465 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 48.Pape KA, Taylor JJ, Maul RW, Gearhart PJ & Jenkins MK Different B cell populations mediate early and late memory during an endogenous immune response. Science 331, 1203–1207 (2011). 10.1126/science.1201730 [DOI] [PMC free article] [PubMed] [Google Scholar]
ADDITIONAL REFERENCES
- 49.Hobeika E et al. Testing gene function early in the B cell lineage in mb1-cre mice. Proc Natl Acad Sci U S A 103, 13789–13794 (2006). 10.1073/pnas.0605944103 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 50.Quadros RM et al. Easi-CRISPR: a robust method for one-step generation of mice carrying conditional and insertion alleles using long ssDNA donors and CRISPR ribonucleoproteins. Genome biology 18, 92 (2017). 10.1186/s13059-017-1220-4 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 51.Laczko D et al. A Single Immunization with Nucleoside-Modified mRNA Vaccines Elicits Strong Cellular and Humoral Immune Responses against SARS-CoV-2 in Mice. Immunity 53, 724–732 e727 (2020). 10.1016/j.immuni.2020.07.019 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 52.Lee PS, Zhu X, Yu W & Wilson IA Design and Structure of an Engineered Disulfide-Stabilized Influenza Virus Hemagglutinin Trimer. J Virol 89, 7417–7420 (2015). 10.1128/JVI.00808-15 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 53.Tas JM et al. Visualizing antibody affinity maturation in germinal centers. Science 351, 1048–1054 (2016). 10.1126/science.aad3439 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 54.Wardemann H et al. Predominant autoantibody production by early human B cell precursors. Science 301, 1374–1377 (2003). 10.1126/science.1086907 [DOI] [PubMed] [Google Scholar]
- 55.Freyn AW et al. A Multi-Targeting, Nucleoside-Modified mRNA Influenza Virus Vaccine Provides Broad Protection in Mice. Mol Ther 28, 1569–1584 (2020). 10.1016/j.ymthe.2020.04.018 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 56.Baiersdorfer M et al. A Facile Method for the Removal of dsRNA Contaminant from In Vitro-Transcribed mRNA. Mol Ther Nucleic Acids 15, 26–35 (2019). 10.1016/j.omtn.2019.02.018 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 57.Maier MA et al. Biodegradable lipids enabling rapidly eliminated lipid nanoparticles for systemic delivery of RNAi therapeutics. Mol Ther 21, 1570–1578 (2013). 10.1038/mt.2013.124 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 58.Ersching J et al. Germinal Center Selection and Affinity Maturation Require Dynamic Regulation of mTORC1 Kinase. Immunity 46, 1045–1058 e1046 (2017). 10.1016/j.immuni.2017.06.005 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 59.Schmidt F et al. Plasma Neutralization of the SARS-CoV-2 Omicron Variant. N Engl J Med 386, 599–601 (2022). 10.1056/NEJMc2119641 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 60.Starr TN et al. Deep Mutational Scanning of SARS-CoV-2 Receptor Binding Domain Reveals Constraints on Folding and ACE2 Binding. Cell 182, 1295–1310 e1220 (2020). 10.1016/j.cell.2020.08.012 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 61.Starr TN et al. Shifting mutational constraints in the SARS-CoV-2 receptor-binding domain during viral evolution. Science 377, 420–424 (2022). 10.1126/science.abo7896 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 62.Greaney AJ et al. Complete Mapping of Mutations to the SARS-CoV-2 Spike Receptor-Binding Domain that Escape Antibody Recognition. Cell Host Microbe 29, 44–57 e49 (2021). 10.1016/j.chom.2020.11.007 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 63.Greaney AJ et al. A SARS-CoV-2 variant elicits an antibody response with a shifted immunodominance hierarchy. PLoS Pathog 18, e1010248 (2022). 10.1371/journal.ppat.1010248 [DOI] [PMC free article] [PubMed] [Google Scholar]
Associated Data
This section collects any data citations, data availability statements, or supplementary materials included in this article.
Supplementary Materials
Data Availability Statement
The raw Illumina reads of the 16-nucleotide variant barcodes from the deep mutational scanning experiments are available on the NCBI SRA under BioProject PRJNA770094, BioSample SAMN30086726. All escape scores are shown in Supplemental Spreadsheet 1 and are available online at https://github.com/jbloomlab/SARS-CoV-2-RBD_MAP_OAS/blob/main/results/supp_data/all_raw_data.csv. Renderings of Spike RBD and hemagglutinin structures were obtained from the Protein Data Bank (PDB), with accession codes PDB: 6MOJ (https://www.rcsb.org/structure/6m0j), PDB: 1RU7 (https://www.rcsb.org/structure/1RU7), PDB: 3LZG (https://www.rcsb.org/structure/3LZG).