

Summary
Previously, we demonstrated the efficacy of human pluripotent stem cell (hPSC)-derived GABAergic cortical interneuron (cIN) grafts in ameliorating seizures. However, a safe and reliable clinical translation requires a mechanistic understanding of graft function, as well as the assurance of their long-term efficacy and safety. By employing hPSC-derived chemically matured migratory cINs in two models of epilepsy, we demonstrate lasting efficacy in treating seizures and comorbid deficits, as well as safety without uncontrolled growth. Host inhibition do not increase with increasing grafted cIN densities, assuring their safety without the risk of over-inhibition. Furthermore, their closed-loop optogenetic activation abortes seizure activity, revealing mechanisms of graft-mediated seizure control, and allowing graft modulation for optimal translation. Monosynaptic tracing shows their extensive and specific synaptic connections with host neurons, resembling developmental connection specificity. These results offer confidence in stem-cell based therapy for epilepsy as a safe and reliable treatment for patients suffering from intractable epilepsy.
Keywords: Temporal lobe epilepsy, human pluripotent stem cells, cortical interneurons, seizure, transplantation, optogenetics, monosynaptic tracing
eTOC blurb
hPSC-derived migratory cINs show lasting efficacy to treat seizures and comorbid behavioral deficits, as well as safety without uncontrolled growth. Host-inhibition did not increase with increasing grafted cIN densities, and their closed-loop optogenetic activation aborted seizure activity. Monosynaptic tracing showed their extensive and specific synaptic connections with host neurons.
Introduction
More than 70 million people globally suffer from epilepsy, a complex brain disorder characterized by recurrent seizures resulting from abnormal and unpredictable neuronal firing in the brain1–6. The use of anti-seizure medication (ASM) as the first-line treatment for the management of epilepsy is mostly cost-effective7; however, about one-third of patients have intractable epilepsy, which does not respond to ASM8,9. While surgical resection is a possible treatment for drug-refractory seizures, it is not effective for all patients10. Neuromodulation devices offer patients new options, but pose various risks11. Therefore, it is imperative to develop alternative, and even restorative, therapeutic options for patients with limited treatment options.
In the brain, inhibitory GABAergic cortical interneurons (cINs) comprise less than 20% of the neuronal cell population, yet they play critical roles in regulating the function of cortical circuitry12,13. The loss of interneurons is consistently found in seizure foci14–16. A previous study showed the transplantation of human embryonic stem cell (hESC)-derived cINs in a preclinical mouse model of temporal lobe epilepsy (TLE)—the most common and most refractory type of epilepsy in adults17—and observed significant reductions of seizure frequency, in addition to the restoration of cognitive deficits in the transplanted mice18. More recently, similar anti-epileptic efficacy was also demonstrated using human induced pluripotent stem cell (iPSC)-derived cINs19. These studies have shown that human pluripotent stem cell (hPSC)-derived GABAergic cINs hold great promise for epilepsy treatment.
There are, however, a number of critical issues to be addressed before this promising experimental therapy can be introduced into the clinic. First, the use of hPSCs is associated with safety issues, such as tumor formation20. Our previous study developed a way to chemically mature hPSC-derived cIN progenitors to generate homogeneous populations of highly migratory, early postmitotic cINs21, with the potential to eliminate the risk of uncontrolled growth, and to increase host integration. Thus, in this study, we tested whether these well-defined populations of early postmitotic cINs are indeed safe without tumor formation, even after long-term engraftment. In addition, it is also important to assess their long-term in vivo viability in the inflammatory epileptic brain environment22, especially considering the more fragile nature of postmitotic neurons compared to proliferating progenitor cells. Furthermore, considering a cIN transplantation study which observed only transient anti-epileptic efficacy23, it is critical to investigate their anti-epileptic efficacy over an extended period of time. Lastly, the mechanisms of human cIN graft function requires clarification. Such mechanisms include synaptic connection specificity, the modality of graft-mediated seizure inhibition, and the relationship between graft density vs host inhibition.
Here, we addressed these key issues by transplanting chemically matured synchronous populations of cINs derived from hPSCs into the hippocampi of the pilocarpine-induced TLE model (PILO-TLE) and the intrahippocampal KA injection-induced TLE model (KA-TLE) in Nod Scid gamma (NSG) mice. Chemically matured human cINs extensively migrated and spread within the host hippocampus after grafting, and were integrated into the host circuitry. This was accompanied by the restoration of synaptic density deficits in TLE mice. Furthermore, in the two independent models of TLE, we observed robust graft survival, and the maintenance of efficacy to ameliorate seizures and comorbid behavioral deficits during an extended time period, that lasted up to nine months, without uncontrolled growth. Host inhibition did not linearly increase as the graft density increased, suggesting the presence of an auto-regulatory mechanism that prevents over-inhibition. This is in contrast to GABA receptor agonist usage, which is associated with over-inhibition-related side effects24. Closed-loop optogenetic stimulation of grafted human cINs aborted seizures, demonstrating action potential firing-dependent seizure control, and providing the potential for further optimization of cIN transplantation therapy to include graft modulation. Monosynaptic tracing revealed extensive host synaptic connections onto grafted human cINs, mostly from within the hippocampus, but also from long-range projections from the cortex, amygdala, and septum, consistent with their endogenous connection patterns25,26. These results demonstrate the lasting efficacy and safety of chemically matured human cIN grafts, and illuminate the mechanisms of their functional efficacy, bringing this novel experimental therapy one step closer to reality.
Results
Authentic phenotypes of chemically matured GABAergic cINs in vitro and in vivo
For extensive characterization of chemically matured human cINs for their potential for cell therapy use, we transplanted H9 hESC-derived cINs into the hippocampi of the PILO-TLE model NSG mice27, and analyzed them as summarized in Fig 1A. After three weeks of differentiation in vitro according to our published protocol21, 86±2.4% of cells displayed the telencephalon fate (FOXG1+), 79±2.7% of cells expressed MGE progenitor markers NKX2.1, 97±1.0% of cells expressed the neuronal progenitor marker NESTIN, and 90.5±1.5% of cells expressed the neuronal lineage marker TUJ1 (Fig. 1B and Fig S1A). Furthermore, 86±1.0% of cells expressed the longitudinal human MGE-lineage cIN marker SOX6 (Ma et al., 2013) (Fig 1B). In addition, 38±6.0% of the cells expressed the early ventral telencephalic marker OLIG2, and 55±2.0% of the cells expressed the cIN-lineage maker DLX (Fig 1B). 0.2±0.2% of cells expressed PAX6, a marker for dorsal telencephalic neural progenitors (Fig 1B). 2±0.7% of cells expressed the mature neuronal marker NeuN, and 1.3±0.4% of cells expressed the post-mitotic cIN marker GABA at this stage, with 44±3.0% of cells expressing the mitotic marker KI67 (Fig 1B). Many transplantation studies have shown that MGE progenitor cells are “phenotype-determined” progenitors that will spontaneously generate MGE-type cINs, regardless of the location of their transplantation28,29, which was also observed in our previous studies18,30. Overall, these results demonstrated efficient induction of MGE progenitors from hPSCs.
Fig. 1. Histological properties of hPSC-derived cINs in vitro and in vivo.
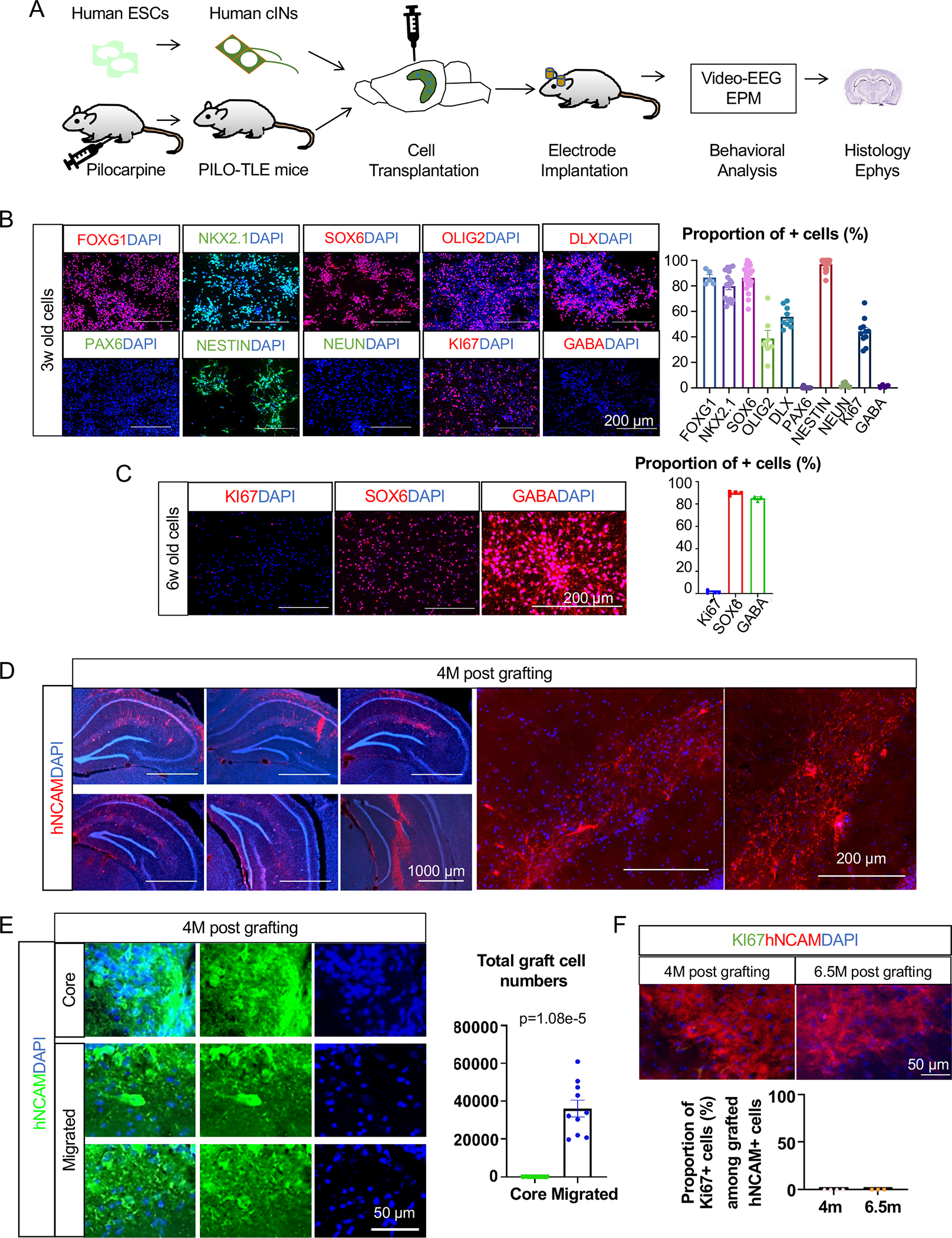
(A) Schematic for the experiments conducted in this study. Human cINs derived from H9 human ES cells were transplanted into the hippocampus of mice with TLE, and characterized by various analyses.
(B) Immunocytochemistry and cell counting analysis of H9-derived MGE cells after three weeks of differentiation (from three independent differentiation batches).
(C) Immunocytochemistry and cell counting analysis of H9-derived six-week-old cINs after one week CDP treatment starting from three weeks of age (from three independent differentiation batches).
(D) Migration and integration of H9-derived human cINs into the PILO-TLE mouse hippocampi analyzed with human-specific NCAM antibody four months after transplantation.
(E) Stereological analysis of grafted cINs in the graft cores vs cINs that migrated out of the graft cores four months after transplantation as analyzed from every 12th 40μm coronal brain sections.
(F) Immunostaining and cell counting analysis of KI67 expression on grafted cells at 4 months and 6.5 months post-transplant as analyzed from every 12th 40μm coronal brain section (4 mice for 4 months post-transplant, 3 mice for 6.5 months post-transplant).
Error bars are the standard error of the mean (SEM). See also Fig S1 and Fig S2.
Generated MGE progenitors were treated for one week with a chemical combination of CultureOne supplement, DAPT, and PD0332991 (CDP) to promote synchronous cell cycle exit into post-mitotic cINs21. Following treatment, 85±1.0% of cells expressed GABA, and 89±0.8% of cells expressed SOX6, while expression of KI67 (1.0±0.7%) and OCT4 (0.0±0.0%) was negligible (Fig 1C and Fig. S1B), demonstrating the generation of a homogeneous population of post-mitotic cINs. This is consistent with our previous study where we observed that CDP treatment results in a synchronized cell cycle exit without change in MGE lineage phenotype21. In our previous RNA-seq analysis on these MGE-type cINs derived from 28 different iPSC lines31, we observed significant expression of KCC2 and NKCC1 (Fig. S1C). We also observed robust KCC2 expression in grafted cINs (Fig. S1D), suggesting that these cells will normally respond to released GABA with hyperpolarization. These early post-mitotic neuronal populations are expected to be safe with minimal uncontrolled growth, yet more robust for passaging and handling compared to fully mature neurons, thus providing an optimal time window for cell transplantation during their differentiation. Furthermore, highly migratory properties of early post-mitotic cINs, just as during their extensive in vivo developmental migration, will ensure that they efficiently integrate into the host circuitry.
The characteristics of chemically matured human cINs were analyzed in vivo after transplantation into PILO-TLE mouse hippocampi. Four months post-transplantation, chemically matured human cINs extensively migrated within the hippocampi (Fig. 1D and Fig. S1E) with 0.066±0.066% of grafted cells left in the graft core area (the area with high grafted cell density, as defined in the STAR Methods section) (Fig 1E). Next, in vivo phenotypes of transplanted human cINs were characterized by immunohistochemistry analysis four months post-transplant. At this age, only about 29±2.0% of engrafted neurons expressed NKX2.1, exhibiting the loss of NKX2.1 expression as they further matured in vivo (Fig S2). 69±4.0% of cells expressed DLX, 82±3.0% of cells expressed GABA, 81±2.0% of cells expressed SOX6, and 100±0.0% expressed TUJ1, together demonstrating their phenotype as MGE-derived cINs (Fig S2). 28±3.0% of grafted human cINs also expressed LHX6, which is expressed in certain subgroups of MGE-derived cINs (Fig S2). Grafted cINs started to express specific cIN subtype markers such as calbindin (CB) (29±0.5%), somatostatin (SST) (8±0.7%), and parvalbumin (PV) (10±0.6%), as expected from their MGE origin. Human PSC differentiation well-recapitulates the endogenous timeline of human embryonic development32, where the protracted maturation of MGE-type cINs, including the expression of PV, occurs mostly postnatally and is not completed until adolescence33. Accordingly, we observed that the majority of human MGE-derived cINs at this time point expressed SOX6 but not yet PV or SST. 5±0.3% of cells expressed vasoactive peptide (VIP), a marker for caudal ganglionic eminence (CGE)-derived cINs (Fig S2). Together, these analyses indicate that transplanted hPSC-derived cINs mature into the proper phenotype in vivo after grafting into the mouse epileptic hippocampus, and extensively migrate and integrate into host neuronal networks.
One of the critical criteria for the safe clinical translation of cell therapy for epilepsy is the safety of cell sources, free from uncontrolled growth, which can lead to tumor formation in patient brains. In our previous study, one month after transplantation, we observed a significant number of KI67+ proliferating cells in the human cIN graft without CDP treatment, but not in the CDP-treated graft21. To further investigate this issue, we analyzed the expression of the proliferating cell marker KI67 at 4 months and 6.5 months post-transplant. Remarkably, there were no detectable KI67+ cells at either time, ensuring the in vivo safety of chemically matured human cINs (Fig 1F).
Maintained anti-seizure efficacy and long-term safety of chemically matured human cIN grafts in PILO-TLE mice
To characterize the seizure-limiting efficacy of chemically matured human cINs, the seizure activity of control PILO-TLE mice vs. PILO-TLE mice with chemically matured cIN grafts was analyzed by video-EEG four months post-engraftment. Seizure frequency was significantly decreased by chemically matured human cIN grafts without significant changes in the duration of each seizure event (Fig 2B), possibly due to the provision of extra inhibition in hyperexcitable epileptic circuitry. Considering that surface screw electrodes were used in this experiment, the absence of change in seizure duration could reflect the fact that grafted cINs can regulate seizure initiation within the hippocampus, where they reside, but have no role in abating seizures once they propagate outside the hippocampus. Given that anxiety is a common comorbidity of TLE34, here we analyzed the impact of human cIN grafts on anxiety. There was a significant increase in the time spent in the open arms of the Elevated Plus Maze (EPM) in mice with human cIN grafts, without significant changes in total distance traveled at five months post-engraftment (Fig 2C), suggesting a reduction of anxiety by human cIN grafts similar to the anxiolytic effects of GABA receptor agonists24.
Fig.2. Anti-epileptic efficacy and safety of chemically matured cINs.
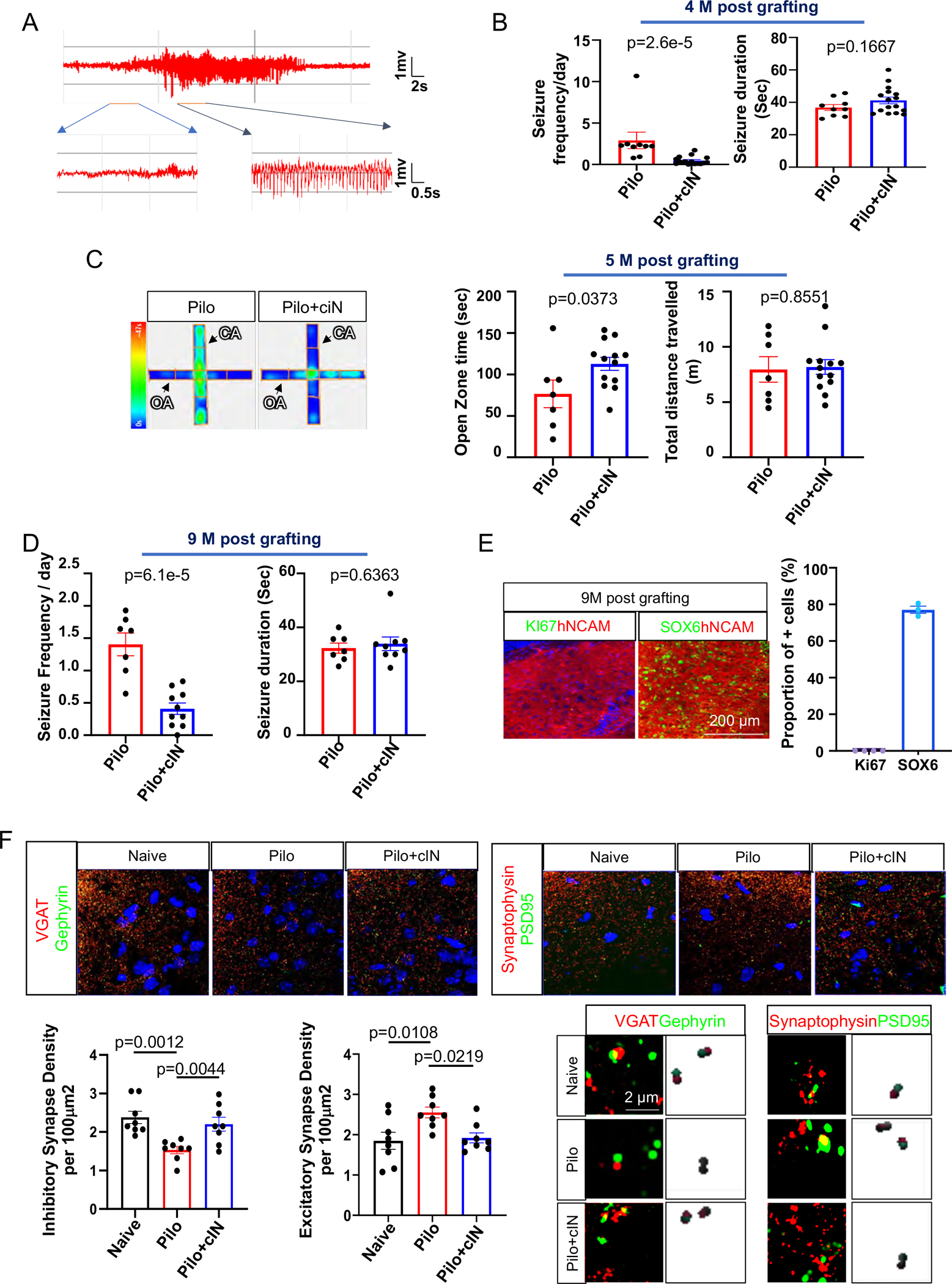
(A) Typical seizure EEG activity.
(B) Seizure frequency and duration in the PILO-TLE group vs the PILO-TLE + cIN group at four-month post-transplant.
(C) Open zone times and total distances between the PILO-TLE group vs the PILO-TLE + cIN group in EPM test at five months post-transplant.
(D) Seizure frequency and duration in the PILO-TLE group vs the PILO-TLE + cIN group at nine months post-transplant.
(E) Immunostaining and cell counting analysis of KI67 and SOX6 expression on grafted cINs at nine months post-transplant as analyzed from every 12th 40μm coronal brain section from four grafted mice per group.
(F) Abnormalities of inhibitory synapse densities and excitatory synapse densities in the dentate gyrus of PILO-TLE mice was restored by cIN transplantation.
Error bars are the SEM.
As previous human cIN grafting studies conducted the analysis of anti-seizure efficacy three to four months post-engraftment18,19, at which time point the human neurons start to display full efficacy, in this study we analyzed whether the observed efficacy of human cINs is maintained after long-term engraftment, even in the epileptic brain environment (Fig. S7C)22. This is important to ascertain, as a previous study observed only transient efficacy of cIN grafts23. Nine months after transplantation, seizure frequencies remained significantly decreased by human cIN grafts (Fig 2D and Fig. S3A–D). Importantly, the histological results of nine-month-old grafts showed that the majority of the cells expressed SOX6 without detectable KI67 expression, even after extended time for proliferation in vivo, confirming the safety of chemically matured human cIN grafts (Fig 2E). Stereological analysis showed no significant changes in total survived cell numbers between grafts after four and nine months (Fig. S3E); but significant increases in total graft volume at nine months were seen (Fig. S3F), suggesting continued cell migration during these time periods. No apoptotic grafted neurons or host neurons were observed at one week and four months post-transplant, as analyzed by the expression of the apoptotic marker cleaved caspase-3 (Fig. S3G), though necrosis of host neurons in the needle tract due to mechanical trauma is possible in the acute phase after grafting.
To gain mechanistic insight into anti-seizure efficacy, we analyzed changes in the densities of inhibitory and excitatory synapses in the dentate gyrus (DG), where synaptic density aberrations were observed in TLE mice in previous studies35,36. Compared to naïve mice, inhibitory synapse density in the DG was significantly decreased in TLE mice; whereas this deficit was reversed after the transplantation of chemically matured human cIN into the hippocampus (Fig. 2F and Fig. S3H). In addition, excitatory synapse density in the DG was significantly higher in TLE mice compared to naïve mice, but these differences were abolished in TLE mice with human cIN transplantation (Fig 2F and Fig. S3H). This suggests that the loss of inhibitory/excitatory balance in the DG of TLE mice can be restored by cIN grafting, providing a mechanistic basis of seizure attenuation.
Chemically matured human cINs regulate host inhibition without over-inhibition
We further characterized chemically matured human cINs within epileptic hippocampal circuitry using whole-cell patch-clamp recordings. Ten months after transplantation, grafted cINs were identified with green fluorescence in acute brain slices (GFP+ cells, Fig. 3A). Grafted cells displayed passive membrane properties of maturing cINs [resting membrane potential (RMP) = −66.3 ± 2.6 mV; membrane resistance (Rm) = 0.70 ± 0.15 GΩ; membrane capacitance (Cm) = 68 ± 10 pF; average ± SEM; n = 6 cells; Fig. 3A]. When voltage pulses were applied, grafted cells showed rapidly desensitizing inward currents and sustained outward currents (Fig. 3B–C), suggesting the expression of voltage-gated Na+ channels and K+ channels in grafted cells. In current-clamp mode, the injection of depolarizing currents induced action potential (AP) firings in all grafted cells examined with an AP threshold of −56.3 ± 2.3 mV and the afterhyperpolarization (AHP) of 17.4 ± 1.6 mV (6 cells; Fig. 3D–E). At the RMP, spontaneous AP firings were detected in two out of five grafted cells (Fig. 3F). Moreover, grafted cells displayed spontaneous excitatory postsynaptic currents (sEPSC) at −85 mV in voltage-clamp mode (Fig. 3G), indicating functional synaptic integration of grafted cells into host hippocampal circuits. These results demonstrate that the transplanted, chemically matured cINs manifest neuronal functions within host epileptic circuitry.
Fig.3. Grafted human interneurons functionally integrated into host hippocampus.
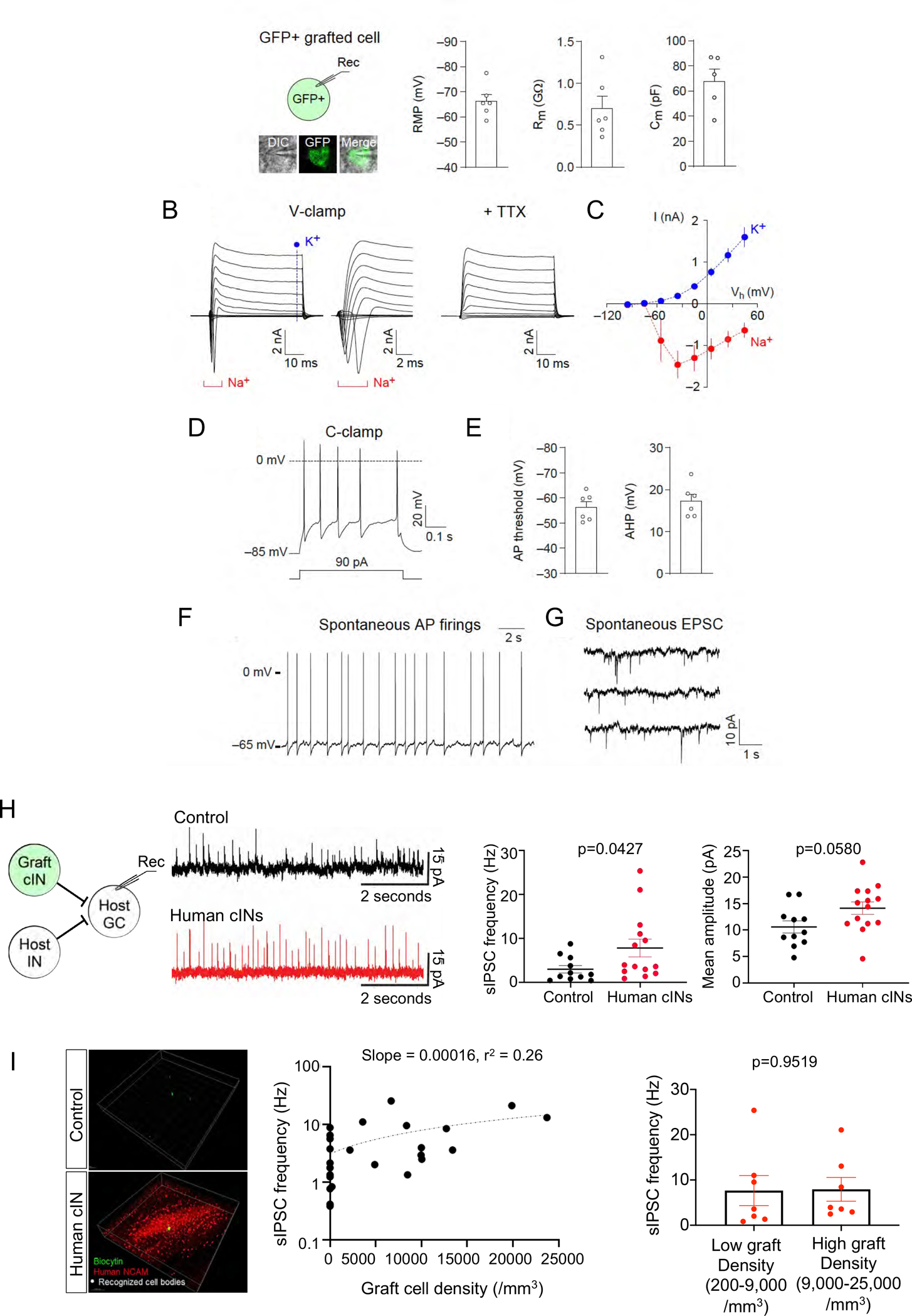
(A) Left panel: whole-cell patch-clamp recordings were performed with grafted cells, which were identified with green fluorescence in acute brain slices of the mouse hippocampus (inset). Right panel: quantification of the resting membrane potential (RMP), membrane resistance (Rm) and membrane capacitance (Cm) of grafted cells (5–6 cells) ten months after transplantation.
(B) Representative traces showing currents induced by voltage pulses in a grafted cell. Membrane potential was held at −85 mV in voltage-clamp mode (V-clamp). Left panel: square voltage pulses from −115 mV to 65 mV with increments of 20 mV (50 ms long) induced both transient inward (Na+, a bracket) and sustained outward currents (K+, a vertical dotted line), which are likely to be mediated by voltage-gated Na+ and K+ channels, respectively. Middle panel: traces zoomed in to visualize the transient inward currents mediated by voltage-gated Na+ channels. Right panel: the application of tetrodotoxin (TTX, 1 mM) blocked the transient inward currents mediated by voltage-gated Na+ channels.
(C) The current-voltage plots of currents mediated by voltage-gated Na+ and K+ channels as recorded in (B). Currents mediated by voltage-gated Na+ channels were quantified by measuring peak transient inward currents induced by depolarizing voltage pulses (red symbols, 5 cells). Currents mediated by voltage-gated K+ channels were quantified by measuring sustained outward currents induced by voltage pulses (blue symbols, 5 cells).
(D) A representative trace of action potentials (AP) induced by depolarizing current injection (90 pA, 500 ms long) and recorded in a graft cell in current-clamp mode (C-clamp) at approximately −85 mV.
(E) Quantification of AP threshold and afterhyperpolarization (AHP, 6 cells).
(F) A representative trace of spontaneous AP firings in a grafted cell. AP firings were recorded at the RMP in current-clamp mode without current injection or withdrawal.
(G) A representative trace of spontaneous excitatory postsynaptic currents (EPSC) recorded in a grafted cell at −85 mV in voltage-clamp mode.
(H) Left: host granule cells received GABAergic inputs from host interneurons and grafted cINs. Middle: the representative traces of sIPSCs from control group vs human cIN graft group. Right: the mean frequency and the mean amplitude of sIPSCs recorded in no graft control group vs human cIN graft group.
(I) Left: Representative images of 3D analysis of electrophysiologically analyzed brain slices. The green cell is the recorded biocytin-labelled host granule cell. Grafted cells are stained for human-specific NCAM. White dots are human cIN cell bodies recognized using IMARIS software spot analysis option. Middle: Correlation between grafted human cIN density and sIPSC frequency (slope = 0.00016, r2 = 0.26) of recorded host neurons. Right: sIPSC frequencies between low graft densities (200–9,000/mm3) vs high graft densities (9,000–25,000/mm3).
Error bars are the SEM.
GABA receptor agonists can cause various adverse side effects such as sedation, drowsiness, and motor weakness when given at high doses24 by over-inhibition of the circuitry. As such, we tested whether grafted human cINs may cause over-inhibition of circuitry at high densities. We analyzed spontaneous inhibitory postsynaptic currents (sIPSCs) of host granule cells at six months post-transplant. Compared to naïve mouse controls, sIPSC frequencies in host granule cells were significantly increased in the human cIN graft group, but not sIPSC amplitudes (Fig. 3H), suggesting an increase in pre-synaptic inhibitory inputs to these neurons. This is in line with our previous studies where we observed the efficient inhibition of host neurons by grafted human cINs using optogenetic activation of grafted neurons18,31. Next, to understand the relationship between grafted human cIN density vs. inhibition of host neurons, we determined grafted human cIN densities by 3D imaging analysis using IMARIS software, and correlated graft neuron densities within the recorded brain slices with sIPSC frequencies (Fig. 3I). Although human cIN grafts significantly increased host granule cell sIPSC frequencies (Fig. 3H), that increase reached a plateau at relatively low graft densities with minimal further elevations as graft densities increased over a large range (Fig. 3I). This observation suggests a possible auto-regulatory mechanism which limits total inhibition as grafted cIN density increases. These results further indicate that human cIN grafts are unlikely to cause over-inhibitory side effects in grafted subjects, supporting their in vivo safety.
Chemically matured human cIN grafts abort seizure upon closed-loop optogenetic activation
To further ascertain the long-term seizure-reducing efficacy of chemically matured cINs, we utilized another model of TLE by intrahippocampal injection of kainic acid (KA) (Fig. 4A). Intrahippocampal KA injection results in granule cell dispersion and glial activation in the hippocampus (Fig. S4), well-modeling the hippocampal sclerosis histopathology observed in human TLE patients37. These mice develop frequent spontaneous recurrent seizures (SRS) with >20 seizures per day (Fig. S5A), allowing for reliable seizure analysis. In addition, the KA-TLE model in mice consistently shows a loss of inhibitory interneurons as observed in TLE patients (Fig. S5B–C), allowing us to analyze the restoration of inhibition by human cIN transplantation. Since we have extensively characterized human iPSC-derived cINs both in vivo and in vitro and demonstrated their authenticity and functionality in our previous study31, we used iPSC-derived cINs for transplantation into the KA-TLE model, considering the therapeutic potential of iPSCs for autologous cell therapy. At one-month post-transplant, chemically matured human cIN grafts reduced seizure frequency mildly, yet significantly (Fig. 4B and Fig. S6A). We observed more robust seizure reduction at three months, six months and even nine months post-engraftment (Fig. 4B and Fig. S6A). Further reduction of seizure frequency up to six months post-transplant suggests the continued integration and maturation of grafted human cINs during this interval. Again, grafted neurons extensively migrated within the KA-TLE hippocampus (Fig. S6B). cIN grafts did not revert mossy fiber sprouting (Fig. S7A), suggesting that seizure suppression is independent of mossy fiber sprouting38; however, cIN grafts significantly restored granule cell dispersion (Fig. S7B). Microglial and astrocyte activation caused by the KA-TLE model generation was not suppressed by cIN grafting (Fig. S7C), suggesting that the reversal of microglial or glial activation is not a prerequisite for seizure control by human cIN grafts. These results, along with Fig. S3E, demonstrate the robust survival of chemically matured human cINs in neuroinflammatory epileptic brain environments for an extended time period. There was no significant difference in mortality among groups (Fig. S7D).
Fig. 4. Chemically matured human cIN grafts abort seizure upon closed-loop optogenetic activation in KA-TLE model mice.
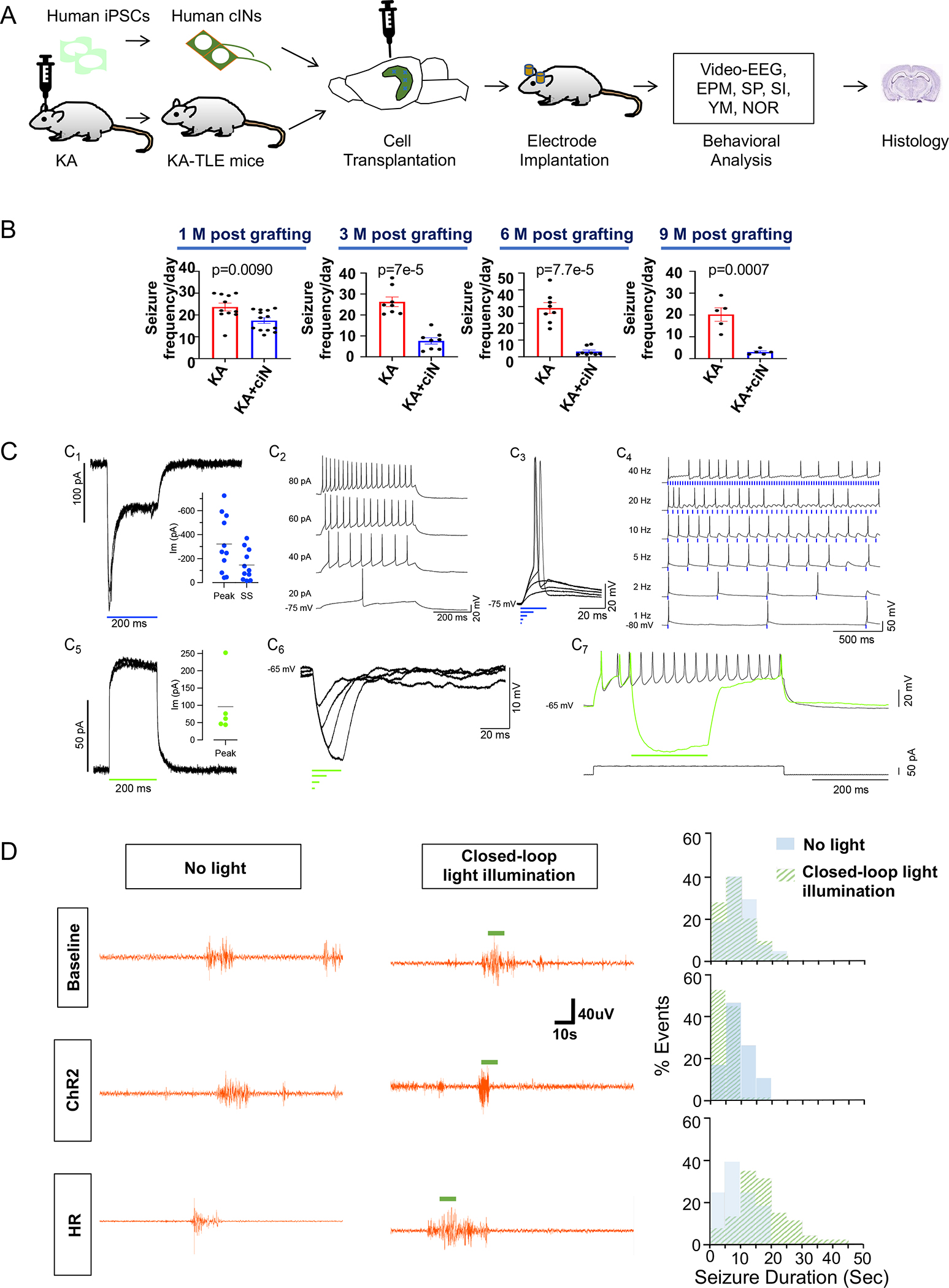
(A) Schematic representation of experimental design. KA-TLE model in NSG mice were transplanted with human cINs derived from iPSCs and analyzed for EEG, behavioral assays and histology.
(B) Seizure frequency analysis by EEG at one month, three months, six months and nine months post-transplant.
(C) ChR2 and HR expression in cINs resulted in large photocurrents. C1. ChR2 expression resulted in a characteristic inward ChR2 current in cINs evoked by 200 ms duration flashes of blue light (holding potential = −66 mV). Inset: distribution of maximal peak (Peak) and steady-state (SS) ChR2 currents evoked by 200 ms flashes (n = 11). C2. The cIN recorded in C1 had overshooting action potentials and sustained repetitive firing in response to injected current pulses. C3. Brief lights pulses of 20, 10 and 5 ms duration produced suprathreshold depolarizations, while shorter pulses (2 and 1 ms) did not. C4. Firing fidelity was assessed with trains of 10 ms light flashes delivered at the indicated frequencies. C5. HR expression resulted in characteristic outward currents evoked by 200 ms duration flashes of green light in cINs. Inset: distribution of maximal peak (Peak) HR currents evoked by 200 ms flashes (n = 5). C6. Brief light pulses (2–20 ms) produced strong membrane hyperpolarization. C7. HR current activated during repetitive firing was able to completely stop firing.
(D) Closed-loop optogenetic modulation of grafted human cINs in KA-TLE model in NSG mice. Left panel: Representative EEG tracing at baseline (one week post-transplant) or one month after grafting with ChR2-expressiong human cINs or HR-expressing human cINs, either without light illumination or with closed-loop light illumination. Right panel: histograms of seizure duration without light illumination (light blue) or with closed loop light illumination (light green).
Error bars are the SEM. See also Fig S4, Fig S5, Fig S6, Fig S7 and Fig S8.
Next, we explored the mechanisms of seizure control by chemically matured cINs using a closed-loop optogenetic approach. Using this approach, we can detect seizures in real-time by EEG and trigger optogenetic illumination upon seizure detection39,40. For this, chemically matured cINs were infected with a lentivirus that expresses Channelrhodopsin2 (ChR2)-YFP or Halorhodopsin (HR)-YFP prior to grafting, allowing us to selectively activate or inhibit grafted cINs, respectively, in the grafted brains. To confirm functional opsin expression, whole cell recordings were made from ChR2-expressing or HR-expressing human cINs. Neurons were selected for recording using IR DIC and were confirmed to express the opsin following recording by fluorescence imaging of the YFP tag. Recorded neurons expressing ChR2 (Cm = 25.2 ± 3.2 pF, Rm = 1.2 ± 0.3 GΩ, n = 11) had strong inward photocurrents that rapidly peaked and then relaxed to a steady-state. Peak values ranged from −724 to −41 pA (−322 ± 72 pA, n =11), and steady state values ranged from −370 to −15 pA (−146 ± 38 pA) when measured from a holding potential of −72.8 ± 1.6 mV (n =11; Fig. 4 C1 and inset). In current clamp mode, most cells fired one or more overshooting action potentials, and some showed robust repetitive firing (Fig. 4C2) in response to depolarizing current pulses. In cells with spikes, brief ChR2 currents of 5–20 ms in duration were adequate to drive spiking (Fig. 4 C3) and 10 ms light pulses could drive high fidelity spike following up to 5 Hz. At higher pulse frequencies, firing appeared to saturate at ~7 spikes per second (see 20 and 40 Hz pulse trains; Fig. 4 C4). Recorded neurons expressing HR (Cm = 34.4 ± 11.3 pF, Rm = 1.2 ± 0.6 GΩ, n = 5) showed strong outward currents following illumination (Fig. 4C5). The peak outward HR current ranged from 237 to 44 pA (96.0 ± 39.5 pA, n =5) when measured from a holding potential of −63 ± 4.1 mV (n = 5; Fig. 4 C5 inset). In current clamp mode, brief light flashes (2 – 20 ms) produced strong membrane hyperpolarization (Fig. 4C6) and these photocurrents could completely block repetitive firing (Fig. 4 C7). These findings confirm that the expression of ChR2 and HR can be used to powerfully excite and inhibit cINs with a high degree of temporal precision. When the mice grafted with ChR2-expressing cINs were analyzed one-week post-transplant using closed-loop optogenetics, we did not observe any impacts by light illumination, as shown in the baseline data (Fig. 4D and Fig. S8). In these experiments, light illumination was randomly triggered during 50% of seizure events, whereas the other random 50% seizure events did not trigger light illumination, the latter of which served as an internal negative control. We analyzed these mice again at one-month post-transplant—at which time, the point efficacy of cIN grafts started to emerge but was not fully established (Fig. 4B)—to address the question of whether optogenetic activation of grafted cINs will further enhance their anti-epileptic efficacy. At this time point, closed-loop activation of grafted human cINs by light illumination aborted seizures in mice grafted with ChR2-expressing human cINs (Fig. 4D and Fig. S8). These results demonstrate action potential firing-dependent seizure control mechanisms, as well as the sufficiency of modulation of grafted human cINs as a method for seizure control. On the contrary, closed-loop optogenetic inhibition of HR-expressing cIN grafts resulted in prolongation of seizure duration (Fig. 4D and Fig. S8), demonstrating the necessity of proper human cIN function in modulating seizure activity once they integrate into the host circuitry.
Chemically-matured human cIN grafts restore comorbid behavioral deficits
Since there are neuropsychiatric comorbidities of TLE, we analyzed whether such comorbid behavioral deficits can be restored by chemically matured cIN grafts in a KA-TLE model in mice. Anxiety is one such comorbidity of TLE34. TLE model animals have been shown to display anxiety-related behaviors41. KA-TLE mice spent significantly less time in the open arms of the elevated plus maze, demonstrating their anxiety phenotype, which was reverted by chemically matured cIN grafts both at three months and nine months post-transplant (Fig. 5A and Fig. S9). This result is consistent with the result observed in PILO-TLE mice (Fig. 2C). Depression is another comorbidity of epilepsy42. Thus, we analyzed the anhedonia phenotype in KA-TLE mice (Fig. 5B) using a sucrose preference test. KA-TLE mice showed significant decreases in sucrose preference compared to naïve mice, as reported previously19,43,44, which was reverted by human cIN grafting (Fig. 5B). Social interaction deficits also have been recognized as one of the comorbidities of epilepsy 45–47. When given the choice between an object and a mouse, TLE model animals spent less time interacting with other mice as compared to naïve mice48,49, suggesting deficits in social interaction. These deficits were reverted by human cIN grafting at three months and nine months post-transplant (Fig. 5C and Fig. S9).
Fig. 5. Chemically matured human cIN grafts restore comorbid behavioral deficits.
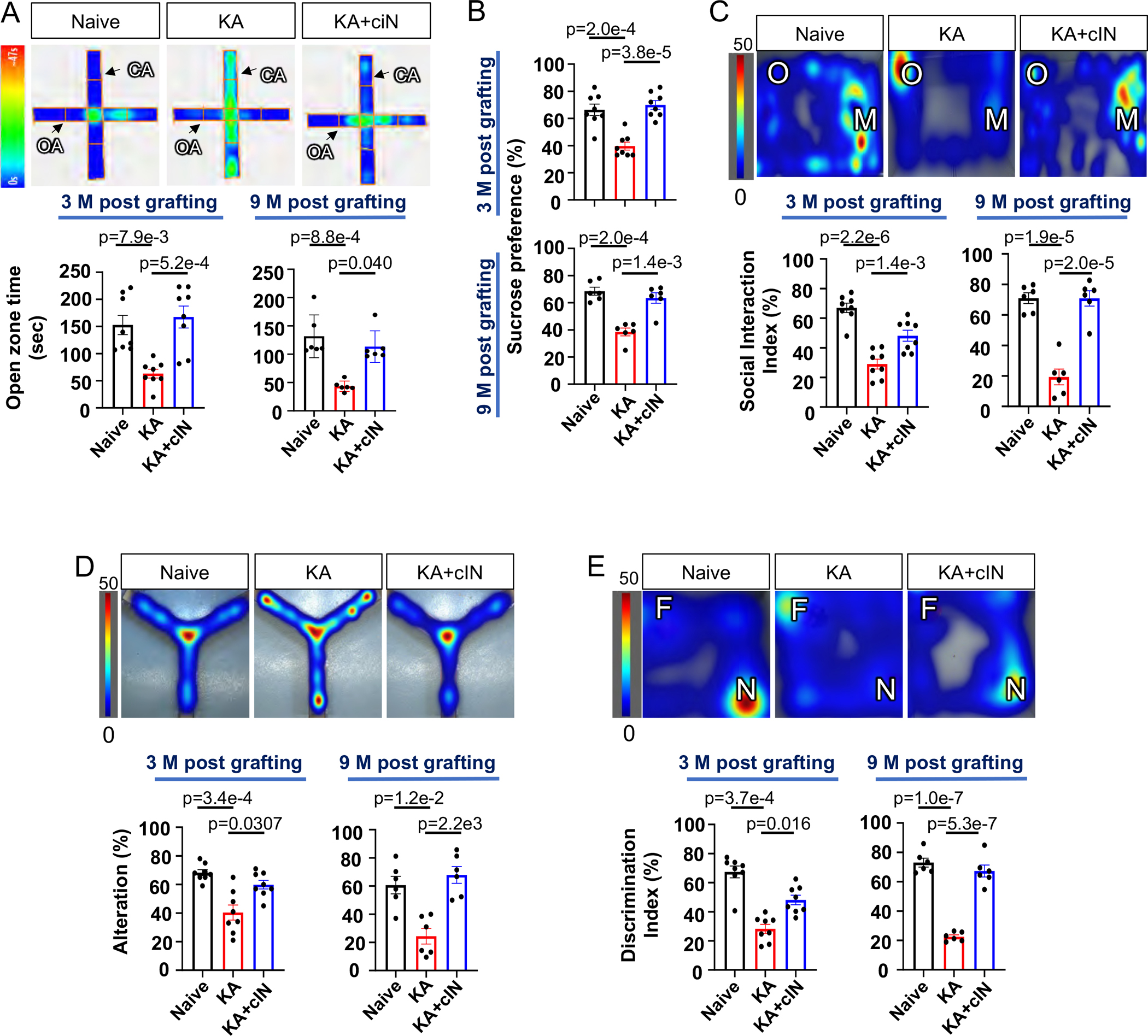
(A) EPM analysis of human cIN-grafted KA-TLE mice. Upper panel: representative heat map image. Lower panel: time spent in open arms by naïve, KA-TLE and KA-TLE + cIN groups at three months and nine months post-transplant.
(B) Sucrose preference test in naïve, KA-TLE and KA-TLE + cIN groups at three months and nine months post-transplant.
(C) Social interaction test. Upper panel: representative heat map image. Lower panel: Social interaction index in naïve, KA-TLE and KA-TLE + cIN groups at three months and nine months post-transplant.
(D) Y-maze analysis. Upper panel: representative heat map image. Lower panel: % of alteration of naïve, KA-TLE and KA-TLE + cIN groups at three months and nine months post-transplant.
(E) Novel object recognition test. Upper panel: representative heat map image. Lower panel: discrimination index of naïve, KA-TLE and KA-TLE + cIN groups at three months post-transplant and nine months post-transplant.
Heat map indicates total duration of stay (in seconds) in a given spot. Error bars are the SEM. See also Fig S9.
In addition to comorbidities of emotional faculties, cognitive defects are also well-known comorbidities of TLE50. Previous studies showed cognitive deficits in different TLE models that were reverted by human cIN grafts18,19. Here, we tested the impact of human cIN grafts on the cognitive deficits in KA-TLE mice, which show lesions closely resembling those seen in patients with hippocampal sclerosis. When analyzed by Y maze and Novel Object Recognition, KA-TLE mice showed significant cognitive deficits18,19,51–55, which were reverted by chemically matured cIN grafts both at three months and nine months post-transplant (Fig. 5D–E and Fig. S9).
Chemically matured human cINs receive extensive host-innervation in a manner similar to developmental integration
We addressed the question of synaptic connectivity of grafted cINs using rabies monosynaptic tracing56, which reveals the neurons that directly innervate the starter neurons that express EnvA receptor TVA and rabies G-protein (Fig. S10A–B). TVA expression on starter cells allowed the specific infection of these cells with EnvA-pseudotyped rabies that expressed mCherry, and were G-deleted to prevent further spreading of rabies beyond the directly innervated neurons (monosynaptic tracing; Fig. S10A–B). Rabies-infected starter neurons release rabies virions coated with G-protein, which will infect only directly innervated presynaptic neurons because of the restricted expression of G-receptors in presynaptic nerve terminals (Fig. S10C). Human cINs were infected with lentiviruses that expressed TVA-P2A-eGFP-2A-oG prior to transplantation. Three months after grafting, an EnvA-pseudotyped rabies vector expressing mCherry was injected into the grafted site, infecting only grafted cINs that expressed the TVA receptor. One week after this injection, cIN-engrafted mice were perfused for histological analysis. Immunohistochemistry analysis showed extensive innervation of GFP−mCherry+Human-specific NCAM (hNCAM)− host-traced neurons (Fig. 6Aii) onto GFP+mCherry+hNCAM+ grafted human neurons (Fig. 6Ai). Most of the innervation onto grafted human cINs comes from within the hippocampus including the hilus, CA1, and CA3 (Fig. 6B), but there were also long-range projections from the cortex, amygdala, and septum (Fig. 6C). Long-range projections from the cortex and septum to hippocampal interneurons were reported previously25,26,56. Our observations suggest that such developmental innervation specificity from cortical or septal long-range projection neurons to hippocampal interneurons is maintained even in an adult brain environment with grafted human cINs. To further analyze synaptic connection specificity, we quantified neuronal subtypes among traced host neurons. The contribution of MGE-derived cINs among traced host neurons are negligible for both SST+ cINs (0.00%; Fig. 6D) and PV+ cINs (1.85%; Fig. 6E), but the majority of traced host neurons in the hippocampus are CamKII+ principal neurons (66.02%; Fig. S10D). These results assure that grafted human cINs in an epileptic brain environment will integrate into the host circuitry in a manner that enables them to detect excitation of host principal neurons, and in turn, release GABA to counteract the hyperexcitable circuitry.
Fig. 6. Widespread host neuron innervation onto grafted cINs.
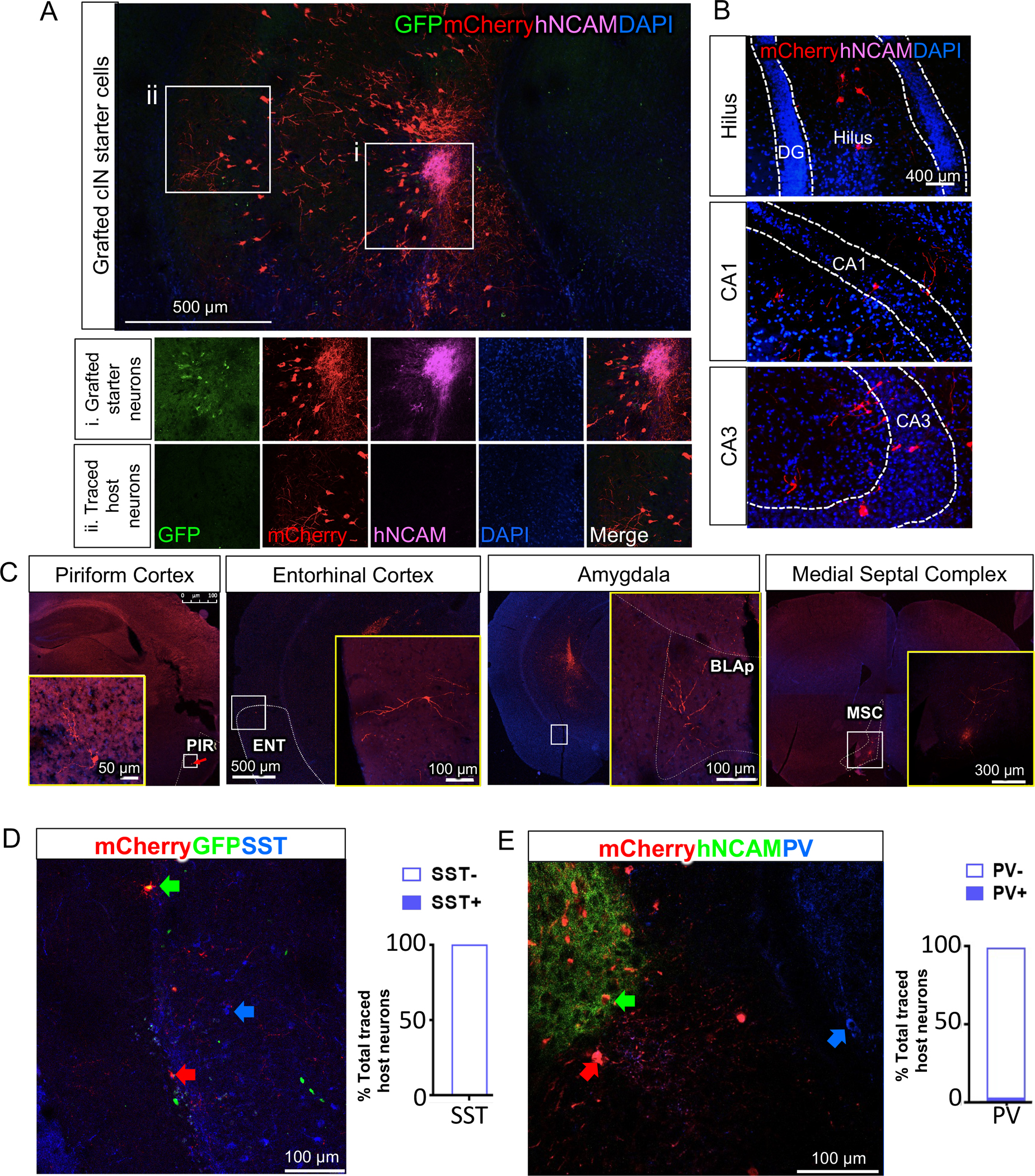
(A) Grafted hNCAM+ neurons receive extensive host synaptic connections demonstrated by large numbers of mCherry+GFP−hNCAM− host neurons that made synaptic connections onto grafted human cINs, as analyzed from six grafted mice.
(B) Traced mCherry+hNCAM− host neurons within hippocampus.
(C) Host neurons also make synaptic connections with grafted human cINs via long-range projections (PIR: piriform cortex, ENT: Entorhinal cortex, BLAp: anterior basolateral amygdala, MSC: medial septal complex).
(D) Host PV+ cINs seldom innervate onto grafted human cINs. Bar graph showing the proportion of PV+ vs PV− cells among traced host neurons (total 108 traced host neurons).
(E) Host SST+ cINs seldom innervate onto grafted human cINs. Bar graph showing proportion of SST+ and SST− cells among traced host neurons (total 115 traced host neurons).
See also Fig S10.
Discussion
In this study, we extensively characterized hPSC-derived chemically matured cINs and unraveled their mechanisms of functional efficacy. Chemically matured human cINs efficiently migrated and integrated into host epileptic circuitry as shown by the scarcity of the graft core area after integration into host circuitry, in contrast to the un-integrating graft core area observed in progenitor grafting strategies18. Grafted chemically matured human cINs maintained their antiepileptic efficacy during prolonged grafting periods of over nine months, as verified in two independent models of TLE. This time frame far exceeds the three to four month timeline of previous human cIN grafting studies18,19, which was just enough for human neurons to integrate into adult epileptic circuitry and start to show robust efficacy. A previous study observed a more transient efficacy of MGE cells23, and thus a future side by side analysis comparing each of the different factors between these studies (e.g. grafted cell types, graft location and immune environment etc.) would be necessary to identify factors contributing to transient vs long-term efficacy. Outstandingly, the observed anti-epileptic activity was accompanied by robust long-term survival of grafted chemically matured human cINs, even in the inflammatory epileptic brain environment (Fig. S7C). At the same time, chemically matured cIN grafts did not display any uncontrolled growth or risk of tumor formation following an extended engraftment period post-transplant. These results showcase homogeneous populations of early post-mitotic human cINs as optimal cell sources that are robust, unlike fully matured cINs, yet safe compared to proliferating progenitors. Furthermore, these cells exerted enhanced inhibitory control over host neurons without circuit over-inhibition, assuring the safety of the grafts with a minimal risk of over-inhibition-related side effects, as seen in the systemic use of GABA receptor agonists24. Overall, these results provide important answers to several critical issues for the safe and reliable translation of this promising novel therapeutic.
The optogenetic approach involves light-illumination-mediated fast activation or inhibition of specific cell populations, allowing for the analysis of specific neuronal function and activity in real-time in behaving mice. Here, by combining optogenetic modulation of grafted human cINs with real-time EEG seizure detection, the light illumination was triggered by seizure detections (closed loop optogenetic modulation)39,40. Specific activation of grafted human cINs upon seizure detection aborted seizure activity, whereas specific inhibition of grafted human cINs upon seizure detection showed the opposite effect. Chr2-mediated grafted cIN activation results demonstrated the sufficiency of grafted human cINs in modulating and aborting seizures. On the other hand, HR-mediated grafted cIN inhibition results suggest that grafted cINs are well-integrated into the host circuitry, and thus, are necessary to elicit full seizure control in the epileptic brain. Recently developed ultra-sensitive opsins could provide minimally invasive optogenetic modulation of neurons in the brain57. Thus, based on proven functional modulation of human cIN grafts by optogenetics, it may be possible, as technologies further develop, to fine-tune cell therapy for epilepsy to include non-invasive modulation of graft function.
As a mechanism underlying lasting epileptic efficacy, we observed a reversal of abnormal synapse formation in TLE mice by chemically matured cINs. Previous studies reported reduced inhibitory synaptic input35 and increased excitatory synaptic input36 onto granule cells in TLE models. Consistent with these studies, we also observed synaptic density abnormalities in the DG of TLE models in NSG mice, which was corrected by grafted human cINs. In line with this histological observation, we also observed increased sIPSCs onto granule cells by whole-cell patch clamp analysis, indicating that increased inhibition from grafted cINs provided restraint to the hyperexcitable epileptic circuitry. The robust integration of grafted cINs within host circuitry was further demonstrated by sEPSCs recorded in grafted neurons. Our observation that about 40% of the grafted neurons showed spontaneous AP firing at RMP suggests that they provide tonic inhibition to the host epileptic circuitry, in addition to activity-dependent inhibition upon activation from the host synaptic inputs. However, it is re-assuring that the extra inhibitions provided by grafted cINs by either mechanism do not over-inhibit the circuitry, possibly due to the presence of self-regulatory mechanisms. The absence of over-inhibition by grafted cINs will aid the efficacious yet safe translation of cell therapy for epilepsy.
Monosynaptic tracing of grafted human cINs using rabies vector showed extensive innervation from host neurons onto grafted human cINs. The majority of inputs were from within the hippocampus, but we also observed long-range projection neurons from the areas known to have long-range synaptic connections onto hippocampal cINs, such as the cortex and the septum25,26. These results suggest that human cINs grafted into the adult brain environment still maintain the specificity of developmentally established synaptic connections. The majority of host neurons that synapsed onto grafted human cINs were principal neurons, suggesting that grafted human cINs can be activated by host principal neurons and release GABA to hyperexcitable epileptic circuitry. Overall, these observations provide confidence in cell therapy for epilepsy, in that grafted neurons are restoring natural and needed connections.
Extensive behavioral analysis revealed the efficacy of grafted human cINs in restoring comorbid behavioral abnormalities of TLE, such as anxiety, depression and cognitive deficits. Anxiety and depression are frequent comorbidities of TLE34,42, and TLE model animals also display anxious behavior evidenced by EPM performance and depression-related anhedonic behavior seen in a sucrose preference test19,43,44,58. These emotional comorbidities were successfully reversed by grafted human cINs, demonstrating the wider efficacy of grafted human cINs in addition to their capacity to control seizures. This result is consistent with previous studies that linked abnormal hippocampal cIN function, especially ventral hippocampal cINs, with anxiety and depressive behaviors44,59. Cognitive deficits are another well-known comorbidity of TLE50, and animal models of TLE display cognitive deficits18,19,51–55. The reversal of cognitive deficits by human cIN grafts was reported in previous studies18,19, the results of which were corroborated here in an independent model of TLE using intrahippocampal KA injection.
NSG mice used in this study lack B-cells, T-Cells, and NK cells60; thus, only activated astrocytes and microglia mediate neuroinflammation in the NSG mouse brain (Fig. S7C), without infiltrating peripheral immune cells. Activated astrocytes and microglia release various cytokines and reactive oxygen species61. Given that NSG mice lack the common gamma chain comprising receptors for six different cytokines (e.g., IL-2, IL-4, IL-7, IL-19, IL-15, and IL-21), the downstream signaling pathways for these cytokines are inhibited, resulting in an overall dampening of inflammatory responses compared to naïve mice. Considering that most CNS cell therapies in patients employ allografts with immunosuppression62, it is reasonable to study preclinical grafting in immunodeficient animals.
Cell therapies for epilepsy could provide an advantage over current treatment strategies, in that they are truly restorative by providing enhanced inhibition in an activity-dependent manner, avoiding systemic adverse effects seen in ASM, or the risk of damage to healthy neural tissues seen in resection of seizure foci. The use of fetal tissues, as employed in Parkinson’s disease cell therapy63, comes with less risk of uncontrolled growth and tumor formation64,65, as seen in stem cell-based grafting66; however, it also comes with ethical and practical issues, The establishment of safe and efficacious cell sources of unlimited quantities from renewable hPSCs67,68 is a critical step towards the realization of cell therapy for epilepsy. This was demonstrated in this study by hPSC-derived chemically matured human cINs. hPSC-derived chemically matured cINs were prepared using methods employing xeno-free, feeder-free components that can be readily adapted to cGMP-compatible production. Furthermore, the homogeneity of the generated cell population, unlike many other hPSC progenies which are more heterogeneous, provides a unique opportunity to avoid side effects resulting from unwanted cell types in the cell preparation. Most hPSC-derived cell therapy clinical trials thus far are carried out using human ESC-derived allografts69 due to the prohibitively high cost of autologous iPSC generation70. hESC-derived allografts that are more cost effective, however, come with the risk of immunosuppression confounder and/or immune rejection in patients. To mitigate this issue, cell therapy strategies incorporating hypoimmunogenic iPSCs are being developed71 to enable off-the-shelf use of cryopreserved cINs with minimal need for immunosuppression. Another remaining issue standing in the way of cell therapy treatment in clinical settings could be how well the long-term efficacy observed in the lifespan of mice (months) will translate to humans (years). For this issue, one can consider the precedents from Parkinson’s disease (PD) cell therapy efforts, in which long-term graft survival and efficacy was observed for more than 10 years in inflammatory PD brain environments72–74. Considering that there are many other brain disorders associated with compromised inhibition of circuitry, such as neuropathic pain, amyotrophic lateral sclerosis, Alzheimer’s disease, Parkinson’s disease, autism spectrum disorders and schizophrenia, to name a few75, the establishment of safe and efficacious stem cell-derived cINs shown in this study may have far-reaching implications in treating multiple brain disorders.
This is the first study to demonstrate stem cell-derived human cINs with long-term antiepileptic efficacy and safety, free from tumor formation or over-inhibition. In addition to seizure suppression by enhanced inhibition, we also observed the reversal of emotional and cognitive comorbid behavioral abnormalities by grafted human cINs, highlighting the truly restorative nature of cell therapy for epilepsy. Furthermore, the novel mechanistic insights of human cIN grafts revealed in this study further bolster the confidence for the safe and reliable translation of cell therapy for epilepsy. Overall, the results presented here offer a promising future for the clinical application of human cINs for patients with intractable epilepsy.
STAR Methods
RESOURCE AVAILABILITY
Lead contact
Further information and requests for resources and reagents should be directed to and will be fulfilled by the lead contact, Dr. Sangmi Chung (schung8@nymc.edu).
Materials availability
This study did not generate new unique reagents.
Data and code availability
Any additional information required to reanalyze the data reported in this paper is available from the lead contact upon request. This paper does not report original code.
EXPERIMENTAL MODEL AND SUBJECT DETAILS
Human pluripotent stem cells
Human H9 ESCs (WiCell, Madison, WI, US, passage 30–50) or human iPSC 31731 were maintained on Geltrex (Thermo Fisher, Waltham, MA, USA) in Essential 8 Medium (Thermo Fisher, Waltham, MA, USA) with daily media change. ROCK inhibitor (Y27632, 10uM, ApexBio, Boston, MA, USA) was added to the culture for 24 hours after passaging to prevent single cell-induced cell death of hPSCs.
Nod Scid Gamma (NSG) mice
The Animal Care and Use Committee at New York Medical College approved all animal procedures. Mice were housed under a 12-hour light/dark cycle with water and food available ad libitum. For induction of PILO-TLE mice, five-week-old male and female NSG mice (The Jackson Laboratory stock number 005557) were used, to prevent xeno-graft rejection. For induction of KA-TLE mice, four-week-old male and female NSG mice (The Jackson Laboratory) were used, again to support human graft survival in mouse brains.
METHOD DETAILS
Human pluripotent stem cell culture and differentiation
For MGE differentiation, H9 ESCs or iPSCs were grown as spheres after trypsinization in low adherent flasks with SRM media (DMEM, 15% knockout serum replacement (KSR), 2 mM L-glutamine, and 10 μM β-mercaptoethanol (all from Thermo Fisher, Waltham, MA, USA)) from day 0 to day 14. Y27632 (10 μM) was added on the first day of differentiation. From day 0 to day 7, SRM media was supplemented with 0.1 μM LDN193189 (Selleck Chem, Houston, MA, USA), 10 μM SB431452 (Tocris Bioscience, Minneapolis, MN, US), 0.1 μM SAG (Selleck Chem, Houston, MA, USA) and 5 μM IWP2 (Selleck Chem, Houston, MA, USA). From day 8 to day 14, cells were cultured in SRM media supplemented with 0.1 μM LDN193189 and 0.1 μM SAG. From day 15 to day 21, cells were cultured in N2AA media [DMEM/F12 media with N2 supplement (1:200, Life Technologies, Woburn, MA, USA) and 200 μM ascorbic acid (Sigma-Aldrich, Natick, MA, USA)] supplemented with 0.1 μM SAG and 50 ng/ml FGF8 (ProSpect, Rocky Hill, CT, USA). From day 22 to day 28, cells were cultured in N2AA media supplemented with 5 ng/ml glial cell–derived neurotrophic factor (GDNF, ProSpect, Rocky Hill, CT, USA) and 5 ng/ml brain derived neurotrophic factor (BDNF, ProSpect, Rocky Hill, CT, USA). From day 29, cells were maintained in B27GB media (DMEM/F12 supplemented with B27 supplement (1:100, Thermo Fisher, Waltham, MA, USA), 5 ng/ml GDNF and 5 ng/ml BDNF). To chemically mature cINs (Ni et al., 2019), a combination of chemicals (1% CultureOne (Thermo Fisher, Waltham, MA, USA), 10μM DAPT (Sigma-Aldrich, Natick, MA, USA) and 2 μM PD0332991(Sigma-Aldrich, Natick, MA, US)) was added to the media for one week starting on day 22 in order to induce synchronous cell cycle exit of these progenitor cells, as the MGE progenitor phenotype is fully established by day 2130.
Immunocytochemistry of cultured neurons
Cultured cells grown on PLO/FN (Poly-L-Ornithine, 15 ug/ml, Sigma-Aldrich, Natick; Fibronectin, 10 μg/ml, Thermo Fisher, Waltham, MA, USA)-coated coverslips were fixed in 4% paraformaldehyde (PFA, Electron Microscopy Sciences, Hatfield, PA) for 10 min, blocked in PBS containing 10% normal serum and 0.1% Triton X-100 for 10 min at room temperature and incubated with primary antibodies diluted in PBS with 2% normal serum overnight at 4°C. The information of primary antibodies is listed in the Key Resource Table. After rinsing with PBS three times, cells were incubated with fluorescent secondary antibodies and DAPI (Invitrogen, Waltham, MA, USA) in PBS with 2% normal serum for 1h at room temperature. After washing in PBS, samples were mounted with Fluoromount-G (SouthernBiotech, Birmingham, AL, USA). Fluorescent images were taken by EVOS FL Auto microscope (Life Technologies, Carlsbad, CA) and Leica SPE Confocal Laser Scanning Microscopes (Leica microsystems, Wetzlar, Germany). For cell counting, the multi-point tool in Image J software (Version 1.51p, NIH, Bethesda, MD, USA) was used. Percentages of positive cells for each marker were quantified in relation to DAPI-stained nuclei from at least three independent pictures, with a total of at least 300 cells counted for each group.
KEY RESOURCES TABLE.
| REAGENT or RESOURCE | SOURCE | IDENTIFIER |
|---|---|---|
| Antibodies | ||
| Rabbit polyclonal anti- FOXG1 | Abcam | Cat# ab18259; RRID:AB_732415 |
| Mouse polyclonal anti-NKX2.1(TTF-1) | SCBT | Cat# sc-13040, RRID:AB_793532 |
| Rabbit polyclonal anti-Sox6 | Millipore | Cat# AB5805, RRID:AB_2302618 |
| Rabbit polyclonal anti-Pan-DLX | A kind gift from Dr. Morozov at Yale University | |
| Mouse monoclonal anti-PAX6 | DSHB | Cat# pax6, RRID:AB_528427 |
| Mouse monoclonal anti-Nestin | BD Biosciences | Cat# 611658, RRID:AB_399176 |
| Mouse monoclonal anti-NeuN | Millipore | Cat# MAB377, RRID:AB_2298772 |
| Recombinant Anti-Ki67 antibody | Abcam | Cat# ab16667, RRID:AB_302459 |
| Rabbit polyclonal anti-GABA | Immunostar | Cat# 20094, RRID:AB_572234 |
| Human-specific NCAM-Alexa594 | SCBT | Cat# sc-106, RRID:AB_627128 |
| Human-specific Nucleus | Millipore | Cat# MAB1281, RRID: AB_94090 |
| Rabbit polyclonal anti-VGAT | Synaptic system | Cat# 131 003, RRID:AB_887869 |
| Mouse monoclonal anti-Gephyrin | SCBT | Cat# sc-25311, RRID:AB_627670 |
| Rabbit polyclonal anti-Synaptophysin | Thermo Fisher Scientific | Cat# PA1-1043, RRID:AB_2199026 |
| Mouse monoclonal anti-PSD95 | UC Davis/NIH NeuroMab | Cat# 75-028, RRID:AB_2292909 |
| Rabbit polyclonal anti- Tubulin β-3 | Biolegend | Cat# 802001, RRID:AB_2564645 |
| Rabbit polyclonal anti- Calbindin D | Swant | Cat# CB38, RRID:AB_10000340 |
| Goat polyclonal anti- Somatostatin | SCBT | Cat# sc-7819, RRID:AB_2302603) |
| Rabbit Polyclonal anti-parvalbumin antibody | Swant | Cat# PV 25, RRID:AB_10000344 |
| Rabbit Polyclonal anti-VIP | Immunostar | Cat# 20077, RRID:AB_572270 |
| Rabbit anti-LHX6 | A kind gift from Dr. Pachnis at the Crick Institute | |
| Mouse monoclonal anti-GFAP | UC Davis/NIH NeuroMab Facility | Cat# 73-240, RRID:AB_10672298 |
| Rabbit Polyclonal anti-TH | Pel-Freez | Cat# P40101-150, RRID:AB_2617184 |
| Rabbit Polyclonal anti-DARPP32 | SCBT | Cat# sc-11365, RRID:AB_639000 |
| Goat Polyclonal anti-CHAT | Chemicon | Cat# AB144, RRID:AB_90650 |
| Rabbit monoclonal anti-TBR2 | Abcam | Cat# ab31940, RRID:AB_2200219 |
| Rabbit Polyclonal anti-5HT | Immunostar | Cat# 20080, RRID:AB_572263 |
| Rabbit Polyclonal anti-ZNT3 | Alomone Lab | Cat# AZT-013, RRID:AB_2756813 |
| Rabbit Polyclonal anti-IBA1 | FUJIFILM Wako | Cat# 019-19741, RRID:AB_839504 |
| Rabbit Monoclonal anti-cleaved-caspase | Cell Signaling | Cat# 9661, RRID:AB_2341188 |
| Mouse Monoclonal anti-CAMKII | Millipore | Cat# 05-532, RRID:AB_309787 |
| Rabbit Polyclonal anti- OCT4 | Cell Signaling | Cat# 2750, RRID:AB_823583 |
| Rabbit Polyclonal anti-KCC2 | Abcam | Cat# ab97502 RRID: AB_10866258 |
| Bacterial and virus strains | ||
| EnvA G-Deleted Rabies-mCherry | Salk Institute for Biological Studies | Addgene 32636 |
| Biological samples | ||
| Chemicals, peptides, and recombinant proteins | ||
| Geltrex | Thermo Fischer Scientific | Cat#A1413201 |
| Y27632 | ApexBio | Cat# A3008 |
| β-Mercaptoethanol | Thermo Fischer Scientific | Cat# 21985023 |
| LDN-193189 | Selleck Chem | Cat# S2618 |
| SB431542 | Tocris Bioscience | Cat# 1614 |
| SAG | Selleck Chem | Cat# S7779 |
| IWP-2 | Selleck Chem | Cat# S7085 |
| N-2 Supplement | Life Technologies | Cat#17502048 |
| L-Ascorbic acid | Sigma-Aldrich | Cat# A8960-5G |
| FGF8 | Prospecbio | Cat# cyt-839 |
| GDNF | Prospecbio | Cat# cyt-305 |
| BDNF | Prospecbio | Cssat# cyt-207 |
| B27 Supplement | Thermo Fischer Scientific | Cat# 17504001 |
| CultureOne | Thermo Fischer Scientific | Cat# A3320201 |
| DAPT | Sigma-Aldrich | Cat# D5942 |
| PD033299 | Sigma-Aldrich | Cat# PZ0199 |
| Poly-L-Ornithine | Sigma-Aldrich | Cat# P4957 |
| Fibronectin | Thermo Fischer Scientific | Cat# J66903 |
| Paraformaldehyde | Electron Microscopy science | Cat# NC0179595 |
| DAPI | Invitrogen | Cat# 10236276001 |
| Fluoromount-G | SouthernBiotech | Cat# 0100-01 |
| Pilocarpine hydrochloride | Sigma-Aldrich | Cat# P6503 |
| Diazepam | Henry Schein | Cat# 1278188 |
| Kainic acid | Hello Bio | Cat# HB0355 |
| Sucrose | Boston Bioproduct | Cat# P-908 |
| Trehalose | Sigma-Aldrich | Cat# A9165 |
| Boc-Asp (OMe)-fluoromethyl ketone | Sigma-Aldrich | Cat# B2682 |
| Critical commercial assays | ||
| Deposited data | ||
| Experimental models: Cell lines | ||
| human iPSC 317 | Shao et al.,2019 | |
| Human H9 ESCs | Wicell | Wae009-A |
| Experimental models: Organisms/strains | ||
| NOD.Cg-Prkdc<scid> Il2rg<tm1Wjl>/SzJ | The Jackson Laboratory | Strain #:005557 RRID:IMSR_JAX:005557 |
| Oligonucleotides | ||
| Recombinant DNA | ||
| pBOB-synP-HTB | Addgene | Cat# 30195 |
| pLenti-hSyn-eNpHR3.0-EYFP | Addgene | Cat# 26775 |
| pLenti-Synapsin-hChR2(H134R)-EYFP-WPRE | Addgene | Cat# 20945 |
| Software and algorithms | ||
| AcqKnowledge 4.0 EEG Acquisition and Reader Software | BIOPAC Systems Inc | Cat# UPGACQ |
| Anymaze video-tracking system | Stoelting, Wood Dale, IL | Version 5 |
| Sirenia® Software | Pinnacle technology | Sirenia version 2.2.5. |
| Ethovision software | Noldus Technology | EthoVision XT 15.0.1418 |
| Image J | NIH | ImageJ version 1.53 |
| IMARIS software | Bitplane | Cat# BPI-IM90-SF-N |
| GraphPad Prism | Graph Pad | GraphPad Prism version 8.0.0 |
| Clampex software | Molecular Devices | Version 10 and 11 |
| Clampfit software | Molecular Devices | Version 10 |
| StereoInvestigator | MBF Bioscience | SS-15 |
| PASS software | NCSS, LLC | Version 12.0.5 |
| Other | ||
Generation of TLE models in NSG mice
For induction of PILO-TLE mice, five-week-old male and female NSG mice (The Jackson Laboratory) were administered with methyl scopolamine (Sigma-Aldrich; 1.7 mg/kg; i.p.), and 15 min later, injected with pilocarpine hydrochloride (Sigma-Aldrich; 315 mg/kg; i.p.). The seizures were scored behaviorally using a modified Racine scale76. If a mouse did not exhibit any stage 4 or stage 5 seizures within 20 minutes of pilocarpine injection, a supplemental booster of 1/10 of regular dose was given with a maximum of three boosters. Seizures were terminated by injecting diazepam (Henry Schein Animal Health; 10 mg/kg; i.p.) 60 mins after SE initiation. Seven days after pilocarpine injection, mice were subject to continuous video screening of development of spontaneous recurrent seizures (SRS) using Lorex 4K Ultra HD Security Camera System (Lorex Technology) for a week. Mice that showed SRS with stage 4 or stage 5 seizures during the 7-day recording period were designated as “PILO-TLE mice” and randomly assigned for subsequent experiments. For the generation of KA-TLE mice, four-week-old NSG mice were used for intrahippocampal KA injection. NSG mice were anesthetized in an induction chamber supplied with 2% isoflurane mixed with 2.0–2.5 L/min oxygen using a calibrated vaporizer, and then administered with a maintenance dose of continuous isoflurane (1%) mixed with oxygen (2.0–2.5 L/min) via snout mask. Designated doses of KA for initial optimization and then 0.8 nmol KA for the rest of the studies were injected into the hippocampus of NSG mice using a Hamilton syringe mounted on a Kopf stereotaxic instrument (Kopf, Tujunga, CA) with a mouse adapter (Stoelting, Wood Dale, IL) at the following coordinate: AP −3.0 mm, L 2.7 mm, V −3.7 mm. Mice were used for further experiments at least three weeks post KA injection when SRS is fully established. The development of SRS was confirmed within a week after grafting and EEG electrode implantation as described below. Only those mice with confirmed SRS through EEG recordings, with the EEG seizure criteria as described below, were designated as “KA-TLE mice” and used for further analysis. Both pilocarpine-injected mice and KA-injected mice developed SRS in >90% of injected cases.
Transplantation of human cINs and implantation of EEG electrodes
NSG mice were anesthetized in an induction chamber supplied with 2% isoflurane mixed with 2.0–2.5 L/min oxygen using a calibrated vaporizer, and then administered with a maintenance dose of continuous isoflurane (1%) mixed with oxygen (2.0–2.5 L/min) via a snout mask. 0.5 μl of human cINs (1×105/μl) per site in transplantation media (Hank’s balanced salt solution (Thermo Fisher, Waltham, MA, USA) supplemented with 4.5mg/ml sucrose,100mM trehalose (Sigma-Aldrich, Natick, MA, USA), 10 ng/μl GDNF, 10 ng/μl BDNF, 20 μM Boc-Asp(OMe)-fluoromethyl ketone (BAF, Sigma-Aldrich, Natick, MA, US) and 10 μM Y27632), or the same volume of transplantation media as controls, were injected into the hippocampus of NSG mice using a Hamilton syringe mounted on a Kopf stereotaxic instrument (Kopf, Tujunga, CA) with a mouse adapter (Stoelting, Wood Dale, IL) at the multiple sites as following coordinates: for PILO-TLE mice bilateral injection, AP −2.2 mm, L ±2.4 mm, V −2.2 mm for rostral location; AP −3.30 mm, L ±3.3 mm, V −3.2 mm, −3.7 mm and −4.2 mm for the three caudal locations and for KA-TLE mice unilateral injection, AP −2.2 mm, L 2.4 mm, V −2.2 mm for rostral location; AP −3.30 mm, L 3.3 mm, V −3.2 mm, −3.7 mm and −4.2 mm for the three caudal locations.
For EEG recording of PILO-TLE mice, sterilized stainless steel bone screw recording electrodes (diameter 0.5mm, length 1.1mm; Plastics One) soldered with lead wire were placed epidurally through rostral burr holes in the skull, grounding electrodes were mounted rostral to bregma, and reference electrodes were implanted caudal to lambda. Electrodes were cemented in place with a rapid-curing dental cement (DenMat Holdings, Lompoc, CA). For EEG recording of KA-TLE mice, 0.005” stainless steel wire depth recording electrodes (P1 Technologies) were used instead of screw surface recording electrodes, and inserted into the following coordinates: AP −3.0 mm, L 2.7 mm, V −3.7 mm. Grounding electrodes were mounted rostral to bregma, and reference electrodes were implanted caudal to lambda. Electrodes were cemented in place with a rapid-curing dental cement (DenMat Holdings, Lompoc, CA).
For closed-loop optogenetic analysis, human cINs infected with lentiviruses that express ChR2 or HR were injected into the same grafting coordinates as described above. Wire EEG electrode attached to the cannula for optic fibers were implanted ipsilateral to the injection site (AP −3.3 mm, L 3.3 mm, V−3.2 mm). Grounding electrodes were mounted rostral to bregma, and reference electrodes were implanted caudal to lambda. Electrodes and cannula were cemented in place with a rapid-curing dental cement (DenMat Holdings, Lompoc, CA).
For Rabies retrograde monosynaptic tracing, five- to six-week-old male and female NSG mice were anesthetized by isoflurane gas anesthesia. 1.0 μl of 105/μl human cINs infected with lentivirus that express TVA-P2A-eGFP-2A-oG were injected into the hippocampus of NSG mice using a Hamilton syringe mounted on a Kopf stereotaxic instrument at the following coordinates: AP −3.3 mm, L 3.3 mm, V −3.7. Three months after the transplantation, 0.5μl of EnvA-pseudotyped, G-protein deleted mCherry-expressing rabies viral vector (1.0×10e5 unit/μl from the Salk Virus Core) were stereotactically injected into the same coordinates (AP −3.3 mm, L 3.3 mm, V −3.7 mm), followed by perfusion one week after rabies injection.
Behavioral analysis
For continuous Video-EEG analysis, seizure activity in mice was analyzed using a MP150 Biopac data acquisition System, EEG100C EEG amplifier module, and AcqKnowledge 4.0 EEG Acquisition and Reader Software (BIOPAC Systems Inc.) along with Eco Black Box security camera system (Lorex Technology) by investigators who were blind to treatment conditions. We used the criteria of high-frequency, high-voltage synchronized polyspike profiles with amplitudes greater than 2-fold of background activity lasting more than 15 seconds. Seizure activity detected during EEG recording was confirmed by time-locked video analysis for PILO-TLE mice. Each animal was subject to more than 12 days of continuous video-EEG recording.
For closed-loop optogenetic analysis, first the LED optic fiber that was connected to the headstage was inserted into guide cannula (the tip of optic fiber was located 0.5 mm below the tip of cannula). The headstage, which amplifies and filters signals and is connected to the head-mount on the mouse’s head, was connected to a data conditioning/acquisition module and an optogenetic interface module via a commutator. Optogenetics Interface Module allows for the precise control of the duration and intensity of optical stimulation. The data conditioning/acquisition module and optogenetic interface module were connected to the computer and controlled by Sirenia Seizure feedback software for closed loop optogenetics. This software monitored EEG seizures and, upon seizure detection, triggered LED light illumination. The software randomly triggers light for 50% of seizure events and does not trigger light for the other 50% of seizure events, which we used as internal negative controls (no light controls). Upon seizure detection, the LED optic fiber delivered 100mW/mm2 of blue light (465 nm) for ChR2-expressing cIN grafts and 100mW/mm2 of yellow light (590 nm) for HR-expressing cIN grafts at 30 Hz for 10 seconds for random 50% of detected seizure events.
To assess anxiety-like behavior, we used Elevated Plus Maze (EPM), equipped with Anymaze video-tracking system (v.5; Stoelting, Wood Dale, IL). The maze was located away from walls in a brightly lit room to ensure accurate contrast for the tracking system and provide an aversive environment in the open arms. The mouse was placed at the end of one of the open arms and allowed to explore the maze freely for three minutes. Each animal was subjected to a single trial only. The maze was cleaned with 70% alcohol after each animal. We evaluated total time on open arms for each animal as a measure of anxiety. Hyperlocomotion and thigmotaxis often observed in PILO-TLE mice did not affect their overall EPM performance, as demonstrated by similar total distance traveled in all groups (Fig. 2C). Thigmotaxis seems not to have affected their EPM performance, since two arms of EPM have no walls and the other two arms have narrow walls. No PILO-TLE mice showed behavioral seizures during EPM sessions, reflecting an overall low seizure frequency in this model.
We performed the Novel Object Recognition test to analyze memory and cognition of grafted mice. First, mice were placed in an open field arena for five minutes for acclimation to the open field. The next day, mice were placed in the open field and allowed to explore two identical objects placed in distant areas of the open field. After 24 hours, one of the objects was replaced with a novel object (NO) while the other object remained in its location (familiar object, FO). Trials were video recorded and analyzed by Ethovision software (Noldus IT) to quantify the time spent with NO vs. FO. Based on the quantification, the discrimination index was calculated by using the following formula: (time spent with the NO / total object exploration time) × 100. After each test, before placing the next animal, the apparatus was wiped with 70% alcohol to remove the odor of the previous animal. Some KA-LTE mice sometimes displayed hyperlocomotion, especially in a novel environment, in which case we increased the number of acclimation sessions as needed. The majority of seizures in KA-TLE mice are focal seizures without motor components, so there may have been some focal seizures during the behavioral assays; however, we did not observe any generalized motor seizures during the assays. For each behavioral assay, we monitored total distance traveled to check for any obvious abnormalities in overall mice behavior/movement, and we did not see any significant changes (Fig. S9).
The Y-maze test was used to measure spatial working memory. The apparatus consisted of three identical arms (45 × 12 × 35 cm) diverging at 120° angles one to the other and an equilateral triangular central area. Each animal was placed in the center of the Y-maze and allowed to explore the maze arena for five minutes. Each trial was recorded using a video camera, and percentages of alteration were analyzed by Ethovision as a measure of short-term memory. After each test, before placing next animal, the apparatus was wiped with 70% alcohol to remove the odor of previous animal.
For the Social Interaction test, an experimental mouse was placed at the center of the open field to explore whole apparatus for five minutes for acclimatization. After this five-minute period, the experimental mouse was placed back in the middle of the open field, which now contained a stranger mouse confined in a 9.7cm diameter round cage, and an object. The stranger mouse and the object were placed far from each other in the open field. The experimental mouse was allowed to freely explore for a total period of 10 min. Each trial was video-recorded and analyzed by Ethovision software to determine the amount of time spent in a close proximity with a mouse compared to an object. The social interaction index was calculated by the following formula: time spent with mouse / (time spent with mouse + time spent with object) × 100. After each test, before placing the next animal, the apparatus was wiped with 70% alcohol to remove the odor of the previous animal.
A Sucrose Preference test was performed to determine anhedonic behaviors in grafted mice. All mice received two bottles, one with 2% sucrose solution (Sigma-Aldrich) and another with water. During the 24 hour training phase, the position of each bottle was switched after 12 hours to avoid the possible effects of location preference. After training, a 24-hour testing period started with two pre-weighed identical bottles that contained either 2% sucrose solution or water. Water and sucrose solution consumption was calculated by measuring the change in the weight of fluid consumed. Sucrose preference rate was calculated using the following formula: sucrose consumption / (water consumption + sucrose consumption) × 100.
Histological analysis
Transplanted mice were sacrificed at designated time points post-transplant and perfused transcardially with 0.1 M PBS followed by 4% PFA. After isolation, brains were postfixed in 4% PFA overnight, and then transferred to 30% sucrose. 40 μm coronal brain sections were cut on a Leica CM1850 cryostat (Leica Biosystem, Buffalo Grove, IL, USA). For immunohistochemistry, brain sections were blocked in PBS containing 10% normal serum and 0.1% Triton X-100 for 10 minutes at room temperature and incubated with primary antibodies diluted in PBS with 2% normal serum overnight at 4°C. The information on primary antibodies is listed in the Key Resource Table. After rinsing with PBS, sections were incubated with fluorescent secondary antibodies and DAPI (Invitrogen, Waltham, MA, USA) in PBS with 2% normal serum for 1h at room temperature. After washing in PBS, sections were mounted on slide glass and a coverslip was placed with Fluoromount-G (SouthernBiotech, Birmingham, AL, USA). Fluorescent images were captured by EVOS FL Auto microscope (Life Technologies, Carlsbad, CA) and Leica SPE Confocal Laser Scanning Microscopes (Leica microsystems, Wetzlar, Germany). For phenotype analysis of grafted human cINs, the multi-point tool in Image J software (Version 1.51p, NIH, Bethesda, MD, USA) was used for cell counting. Percentages of positive cells for each marker were quantified in relation to hNCAM+ or human Nucleus+ human graft cell numbers.
For stereological analysis of grafted human cINs, StereoInvestigator image-capture equipment and software (Microbright Field, Williston, VT) were used to count and estimate the cell numbers within the graft core (area of high grafted cell density defined by >13 grafted cells in 75mm × 75mm counting frame) vs. migrated cells outside the graft cores. Stereological estimation of the total survived graft cell numbers was done using the optical fractionator workflow of StereoInvestigator software from every 12th 40 μm thick coronal brain section. A 300 μm × 300 μm grid was used along with a 75 μm × 75 μm counting frame. Stereological estimation of the total grafted volume was done using Cavalieri estimator option in StereoInvestigator software from every 12th 40μm thick coronal brain section.
For synapse analysis, images were taken using Leica SPE Confocal Laser Scanning Microscopes (Leica microsystems, Wetzlar, Germany) with 100x objective and processed using IMARIS software (Bitplane, Switzerland), which allows objective counting of synaptic puncta based upon absolute fluorescent intensity. In the dentate gyrus, synaptic puncta positive for VGAT (inhibitory presynaptic), gephyrin (inhibitory postsynaptic), synaptophysin (excitatory presynaptic) or PSD95 (excitatory postsynaptic) were identified with spot diameters of 0.5 μm. For synapses, a juxtaposition of VGAT+ puncta and gephyrin+ puncta were identified as inhibitory synapses, and a juxtaposition of synaptophysin+ puncta and PSD95+ puncta was identified as excitatory synapses. The result was expressed as synapse numbers per brain area (100 μm2).
For 3D grafted cell density analysis of post-electrophysiology brain slices, z-stack images of hNCAM+ human grafted neurons surrounding biocytin+ recorded host neurons through the entire depth of 300 μm brain slices were taken using Leica SPE Confocal Laser Scanning Microscopes (Leica microsystems, Wetzlar, Germany) with a 20x objective. Cell bodies of human NCAM+ graft neurons were determined with spot diameters of 10 μm using IMARIS software (Bitplane, Switzerland). The density was calculated as grafted cell numbers per imaged brain area (mm3). Calculated human cIN density in each brain slice was used to determine the relationship between grafted cell density and inhibition onto host neurons using simple linear regression.
For quantitative intensity analysis of GFAP and IBA-1, Image-J software was used on 1200 μm × 800 μm images taken using 10x objective. Mean fluorescence intensity (MFI) was obtained from each picture using measure option and used for comparison among groups.
Whole-cell patch-clamp recordings in brain slices
For electrophysiological studies, human cINs were infected with lentivirus, so that they expressed GFP protein under the control of the synapsin promoter (Addgene #20945). Ten months after transplantation into the hippocampus of NSG mice, acute brain slices containing the hippocampus were prepared using a vibrating microtome. After recovery, brain slices were placed in the recording chamber and continuously perfused at the rate of 1 mL per minute with artificial cerebrospinal fluid containing 130 mM NaCl, 2.5 mM KCl, 2.5 mM CaCl2, 1 mM MgSO4, 1.25 mM NaH2PO4, 26 mM NaHCO3, and 10 mM glucose with 95 % O2 and 5 % CO2. Whole-cell patch-clamp recordings were performed at 23–24°C using a Multiclamp 700B amplifier, a Digidata 1550A digitizer, and Clampex 10 software (Molecular Devices). The patch electrodes (2–4 MΩ resistance) were filled with solution containing 150 mM K-gluconate, 5 mM NaCl, 1 mM MgCl2, 10 mM HEPES, 0.2 EGTA, 2 mM MgATP, and 0.5 mM NaGTP (290 mOsm, adjusted to pH 7.3 with KOH). Liquid junction potential of 15.5 mV was corrected. For experiments described in Fig. 3B–C, whole-cell membrane capacitance (Cm) was compensated with the amplifier. Series (access) resistance was not compensated. Offline analysis of electrophysiological data was performed using Clampfit 10 program (Molecular Devices).
To calculate membrane resistance (Rm) and membrane capacitance (Cm) of grafted human interneurons in Fig, 3A, we applied hyperpolarizing voltage pulses (−5 mV, 50 ms duration) from the holding voltage of −85 mV and recorded currents in voltage-clamp mode with a sampling rate of 100 kHz in grafted cells. The peak amplitude of the transient capacitive current (Ipeak) was used to estimate apparent access resistance (Ra) using the equation, Ra = −5 mV / Ipeak. Steady-state current (Iss) is the average current from the baseline for the last 5 ms of voltage pulse and was used to calculate membrane resistance (Rm) using the equation, Rm = (−5 mV / Iss) − Ra. Transient capacitive current was fitted with a double exponential function, I(t) = If exp(−t/τf) + Is exp(−t/τs), where If/τf and Is/τs are fast and slow components of peak amplitudes and decay time constants of transient capacitive current. The weighted mean decay time constant (τw) was calculated from the equation, τw = τf [If/(If + Is)] + τs [Is/(If + Is)] and then was used to calculate membrane capacitance (Cm) from the equation, Cm = t w × (Ra + Rm) / (Ra × Rm).
In Fig. 3D–E, the membrane potential was held at approximately −85 mV in current-clamp mode, and square pulses of depolarizing currents (10–90 pA with increments of 10 pA, 0.5 s duration) were applied. The first APs induced by depolarizing current injection near threshold was used to calculate afterhyperpolarization. Spontaneous AP firings were recorded in grafted cells in current-clamp mode without current injection or withdrawal (Fig. 3F). The resting membrane potential was estimated as the average membrane potential when the recorded graft cells did not fire APs spontaneously. Spontaneous EPSCs (sEPSC) were recorded in grafted cells at −85 mV in voltage-clamp mode (Fig. 3G).
IPSCs of host granule cells were recorded with whole cell patch clamp with voltage clamp mode. The patched neurons were voltage-clamped at −60 mV. The patch electrodes were filled with solution containing 135 mM CsMeSO2, 10 mM HEPES, 2 mM Mg-ATP, 0.3 mM Na-GTP, 0.5 mM EGTA, and 1 mM QX-314. Osmolarity is adjusted to 275 mOsm and pH to 7.25. Pipette resistance are 4 – 5 MΩ when filled with the solution. Since the pipette solution has a low concentration of chloride, the IPSCs are manifested as outward currents.
Optogenetic whole-cell patch-clamp analysis
Human cINs infected with lentiviruses that express ChR2 or HR on PLO/FN-coated glass coverslips were transferred to a chamber mounted on an upright fixed-stage microscope (Olympus BX50WI). The chamber was perfused at 1–2 ml/min with oxygenated artificial cerebrospinal fluid (ACSF) containing (in mM) 124 NaCl, 3 KCl, 1.2 NaH2PO4, 2.0 CaCl2, 1.2 MgSO4, 26 NaHCO3 and 10 dextrose (295–305 mOsm). Cells were visualized with a 40X water immersion objective (0.8 NA) using near infrared, differential interference contrast optics and an Electro CCD camera (Teledyne Qimaging). Borosilicate micropipettes (3–6 MΩ; AM systems) were pulled with a Sutter Instruments P97 horizontal puller and filled with a solution containing (in mM): 144 K-gluconate, 3 MgCl2, 10 HEPES, 0.3 NaGTP, and 4 Na2ATP (310 mOsm) with Alex 594-biocytin (100 μM). Pipettes were positioned with an MP-285 micromanipulator (Sutter Instruments) and gigaseal whole-cell recordings were made using an Axopatch 200A amplifier (Molecular Devices) operated in voltage clamp or “I-clamp fast” mode, low-pass filtered at 2 or 5 KHz and sampled at 50 KHz with a Digidata 1550B (Molecular Devices) controlled by Clampex 11 software (Molecular Devices) running on a PC.
ChR2 and HR were stimulated by computer-triggered light flashes delivered by a UHP-T-SR white LED source (Prizmatix, Israel) directly mounted to the microscope. Wavelength was selected with a filter cube in the light path (ChR2: 480 ± 20 nm, Chroma 41001; HR 560 ± 22 nm Chroma 41004). Flash intensity was controlled using the 10-turn potentiometer on the LED controller and the intensity was selected to produce a maximal photocurrent for each recording. Photocurrents and most other experiments were conducted at room temperature (23 ± 2° C) except for some experiments examining the firing properties which were done with the ACSF maintained at 32 ± 1° C. Values are reported as Mean ± SEM and membrane potentials are corrected for an estimated junction potential of −10 mV.
QUANTIFICATION AND STATISTICAL ANALYSIS
Statistics
GraphPad Prism8 (GraphPad Software, La Jolla, CA) was used for statistical analysis. To compare mean differences between two groups with normal distributions, an unpaired t-test was used with a significance level preset to 0.05; for those with non-normal distributions, the Mann-Whitney test was used with a significance level of 0.05. For a comparison of multiple groups with normal distributions, one-way ANOVA was used with a significance level of 0.05; for those with non-normal distributions, a non-parametric Kruskal-Wallis test was used with a significance level of 0.05. If significant differences were observed from one-way ANOVA or the Kruskal-Willis test, post hoc analysis was done using Dunnett’s multiple comparisons test with a significance level of 0.05. Time course analysis of seizure frequencies in each group was done by repeated measures ANOVA with a significance level of 0.05, followed by post hoc analysis using Bonferroni’s multiple comparisons test with a significance level of 0.05. Post hoc Power analyses were performed using PASS software (version 12.0.5, NCSS, LLC. Kaysville, Utah) and were based on two-sample t-test or ANOVA with a significance level of 0.05, with the given means and standard deviations. No statistical method was used to pre-determine the sample sizes. However, the sample sizes we used in this study were similar to the previous publication18. Detailed descriptions of statistical analysis used are in the Table S1.
Supplementary Material
Highlights.
Lasting efficacy and safety of chemically matured human cINs
Host inhibition did not increase with increasing grafted cIN densities
Closed-loop optogenetic activation of grafted human cINs aborted seizure activity
Extensive and specific synaptic connections with host neurons
Acknowledgements
This study was supported by MH107884 (S.C.), NS121541 (S.C.), NYSTEM C32607GG (S.C.), NS092786 (J.V.), NS118337 (L.V.), the Institute for Organic Chemistry and Biochemistry, Czech Academy of Sciences (L.V.), AMZELL (L.V.), MH118339 (J.H.C), MH113894 (J.H.C) and NS027881 (C.S.L).
Footnotes
Declaration of interests
The authors have no conflicts of interest to declare.
Publisher's Disclaimer: This is a PDF file of an unedited manuscript that has been accepted for publication. As a service to our customers we are providing this early version of the manuscript. The manuscript will undergo copyediting, typesetting, and review of the resulting proof before it is published in its final form. Please note that during the production process errors may be discovered which could affect the content, and all legal disclaimers that apply to the journal pertain.
References
- 1.Fisher RS, van Emde Boas W, Blume W, Elger C, Genton P, Lee P, and Engel J Jr. (2005). Epileptic seizures and epilepsy: definitions proposed by the International League Against Epilepsy (ILAE) and the International Bureau for Epilepsy (IBE). Epilepsia 46, 470–472. 10.1111/j.0013-9580.2005.66104.x [DOI] [PubMed] [Google Scholar]
- 2.Goldberg EM, and Coulter DA (2013). Mechanisms of epileptogenesis: a convergence on neural circuit dysfunction. Nat Rev Neurosci 14, 337–349. 10.1038/nrn3482 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 3.Ngugi AK, Bottomley C, Kleinschmidt I, Sander JW, and Newton CR (2010). Estimation of the burden of active and life-time epilepsy: a meta-analytic approach. Epilepsia 51, 883–890. 10.1111/j.1528-1167.2009.02481.x [DOI] [PMC free article] [PubMed] [Google Scholar]
- 4.Rho JM, and Boison D (2022). The metabolic basis of epilepsy. Nat Rev Neurol 18, 333–347. 10.1038/s41582-022-00651-8 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 5.Singh A, and Trevick S (2016). The Epidemiology of Global Epilepsy. Neurol Clin 34, 837–847. 10.1016/j.ncl.2016.06.015 [DOI] [PubMed] [Google Scholar]
- 6.Organizzazione mondiale della s., and Levi-Montalcini R (2006). Neurological disorders : public health challenges (WHO; ). [Google Scholar]
- 7.Chisholm D, and Who C (2005). Cost-effectiveness of first-line antiepileptic drug treatments in the developing world: a population-level analysis. Epilepsia 46, 751–759. 10.1111/j.1528-1167.2005.52704.x [DOI] [PubMed] [Google Scholar]
- 8.Berg AT (2009). Identification of pharmacoresistant epilepsy. Neurol Clin 27, 1003–1013. 10.1016/j.ncl.2009.06.001 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 9.Löscher W, Potschka H, Sisodiya SM, and Vezzani A (2020). Drug Resistance in Epilepsy: Clinical Impact, Potential Mechanisms, and New Innovative Treatment Options. Pharmacol Rev 72, 606–638. 10.1124/pr.120.019539 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 10.Jette N, Reid AY, and Wiebe S (2014). Surgical management of epilepsy. CMAJ 186, 997–1004. 10.1503/cmaj.121291 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 11.DeGiorgio CM, and Krahl SE (2013). Neurostimulation for drug-resistant epilepsy. Continuum (Minneap Minn) 19, 743–755. 10.1212/01.CON.0000431397.61970.2b [DOI] [PMC free article] [PubMed] [Google Scholar]
- 12.Buzsaki G, Kaila K, and Raichle M (2007). Inhibition and brain work. Neuron 56, 771–783. 10.1016/j.neuron.2007.11.008 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 13.Pelkey KA, Chittajallu R, Craig MT, Tricoire L, Wester JC, and McBain CJ (2017). Hippocampal GABAergic Inhibitory Interneurons. Physiol Rev 97, 1619–1747. 10.1152/physrev.00007.2017 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 14.de Lanerolle NC, Kim JH, Robbins RJ, and Spencer DD (1989). Hippocampal interneuron loss and plasticity in human temporal lobe epilepsy. Brain Res 495, 387–395. 10.1016/0006-8993(89)90234-5 [DOI] [PubMed] [Google Scholar]
- 15.Ribak CE, Bradburne RM, and Harris AB (1982). A preferential loss of GABAergic, symmetric synapses in epileptic foci: a quantitative ultrastructural analysis of monkey neocortex. J Neurosci 2, 1725–1735. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 16.Sun C, Mtchedlishvili Z, Bertram EH, Erisir A, and Kapur J (2007). Selective loss of dentate hilar interneurons contributes to reduced synaptic inhibition of granule cells in an electrical stimulation-based animal model of temporal lobe epilepsy. J Comp Neurol 500, 876–893. 10.1002/cne.21207 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 17.Wiebe S (2000). Epidemiology of temporal lobe epilepsy. Can J Neurol Sci 27 Suppl 1, S6–10; discussion S20–11. 10.1017/s0317167100000561 [DOI] [PubMed] [Google Scholar]
- 18.Cunningham M, Cho JH, Leung A, Savvidis G, Ahn S, Moon M, Lee PK, Han JJ, Azimi N, Kim KS, et al. (2014). hPSC-derived maturing GABAergic interneurons ameliorate seizures and abnormal behavior in epileptic mice. Cell Stem Cell 15, 559–573. 10.1016/j.stem.2014.10.006 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 19.Upadhya D, Hattiangady B, Castro OW, Shuai B, Kodali M, Attaluri S, Bates A, Dong Y, Zhang SC, Prockop DJ, and Shetty AK (2019). Human induced pluripotent stem cell-derived MGE cell grafting after status epilepticus attenuates chronic epilepsy and comorbidities via synaptic integration. Proc Natl Acad Sci U S A 116, 287–296. 10.1073/pnas.1814185115 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 20.Volarevic V, Markovic BS, Gazdic M, Volarevic A, Jovicic N, Arsenijevic N, Armstrong L, Djonov V, Lako M, and Stojkovic M (2018). Ethical and Safety Issues of Stem Cell-Based Therapy. Int J Med Sci 15, 36–45. 10.7150/ijms.21666 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 21.Ni P, Noh H, Shao Z, Zhu Q, Guan Y, Park JJ, Arif F, Park JM, Abani C, Beaudreault C, et al. (2019). Large-Scale Generation and Characterization of Homogeneous Populations of Migratory Cortical Interneurons from Human Pluripotent Stem Cells. Mol Ther Methods Clin Dev 13, 414–430. 10.1016/j.omtm.2019.04.002 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 22.Friedman A, and Dingledine R (2011). Molecular cascades that mediate the influence of inflammation on epilepsy. Epilepsia 52 Suppl 3, 33–39. 10.1111/j.1528-1167.2011.03034.x [DOI] [PMC free article] [PubMed] [Google Scholar]
- 23.Henderson KW, Gupta J, Tagliatela S, Litvina E, Zheng X, Van Zandt MA, Woods N, Grund E, Lin D, Royston S, et al. (2014). Long-term seizure suppression and optogenetic analyses of synaptic connectivity in epileptic mice with hippocampal grafts of GABAergic interneurons. J Neurosci 34, 13492–13504. 10.1523/JNEUROSCI.0005-14.2014 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 24.Olsen R, and DeLorey T (1999). GABA Receptor Physiology and Pharmacology. In Basic Neurochemistry: Molecular, Cellular and Medical Aspects, GJ A.B.a.A.R. S., ed. (Lippincott-Raven; ). [Google Scholar]
- 25.Hainmueller T, and Bartos M (2020). Dentate gyrus circuits for encoding, retrieval and discrimination of episodic memories. Nat Rev Neurosci 21, 153–168. 10.1038/s41583-019-0260-z [DOI] [PMC free article] [PubMed] [Google Scholar]
- 26.Sun Y, Nguyen AQ, Nguyen JP, Le L, Saur D, Choi J, Callaway EM, and Xu X (2014). Cell-type-specific circuit connectivity of hippocampal CA1 revealed through Cre-dependent rabies tracing. Cell Rep 7, 269–280. 10.1016/j.celrep.2014.02.030 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 27.Leung A, Ahn S, Savvidis G, Kim Y, Iskandar D, Luna MJ, Kim KS, Cunningham M, and Chung S (2015). Optimization of pilocarpine-mediated seizure induction in immunodeficient NodScid mice. Epilepsy Res 109, 114–118. 10.1016/j.eplepsyres.2014.10.016 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 28.Flames N, Pla R, Gelman DM, Rubenstein JL, Puelles L, and Marín O (2007). Delineation of multiple subpallial progenitor domains by the combinatorial expression of transcriptional codes. J Neurosci 27, 9682–9695. 10.1523/jneurosci.2750-07.2007 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 29.Wonders CP, Taylor L, Welagen J, Mbata IC, Xiang JZ, and Anderson SA (2008). A spatial bias for the origins of interneuron subgroups within the medial ganglionic eminence. Dev Biol 314, 127–136. 10.1016/j.ydbio.2007.11.018 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 30.Kim TG, Yao R, Monnell T, Cho JH, Vasudevan A, Koh A, Peeyush KT, Moon M, Datta D, Bolshakov VY, et al. (2014). Efficient specification of interneurons from human pluripotent stem cells by dorsoventral and rostrocaudal modulation. Stem Cells 32, 1789–1804. 10.1002/stem.1704 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 31.Shao Z, Noh H, Bin Kim W, Ni P, Nguyen C, Cote SE, Noyes E, Zhao J, Parsons T, Park JM, et al. (2019). Dysregulated protocadherin-pathway activity as an intrinsic defect in induced pluripotent stem cell-derived cortical interneurons from subjects with schizophrenia. Nat Neurosci 22, 229–242. 10.1038/s41593-018-0313-z [DOI] [PMC free article] [PubMed] [Google Scholar]
- 32.Nicholas CR, Chen J, Tang Y, Southwell DG, Chalmers N, Vogt D, Arnold CM, Chen YJ, Stanley EG, Elefanty AG, et al. (2013). Functional maturation of hPSC-derived forebrain interneurons requires an extended timeline and mimics human neural development. Cell Stem Cell 12, 573–586. 10.1016/j.stem.2013.04.005 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 33.Gonchar Y, Wang Q, and Burkhalter A (2007). Multiple distinct subtypes of GABAergic neurons in mouse visual cortex identified by triple immunostaining. Front Neuroanat 1, 3. 10.3389/neuro.05.003.2007 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 34.Kanner AM (2011). Anxiety disorders in epilepsy: the forgotten psychiatric comorbidity. Epilepsy Curr 11, 90–91. 10.5698/1535-7511-11.3.90 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 35.Kobayashi M, and Buckmaster PS (2003). Reduced inhibition of dentate granule cells in a model of temporal lobe epilepsy. J Neurosci 23, 2440–2452. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 36.Zhang W, Huguenard JR, and Buckmaster PS (2012). Increased excitatory synaptic input to granule cells from hilar and CA3 regions in a rat model of temporal lobe epilepsy. J Neurosci 32, 1183–1196. 10.1523/JNEUROSCI.5342-11.2012 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 37.Twele F, Schidlitzki A, Töllner K, and Löscher W (2017). The intrahippocampal kainate mouse model of mesial temporal lobe epilepsy: Lack of electrographic seizure-like events in sham controls. Epilepsia Open 2, 180–187. 10.1002/epi4.12044 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 38.Godale CM, and Danzer SC (2018). Signaling Pathways and Cellular Mechanisms Regulating Mossy Fiber Sprouting in the Development of Epilepsy. Front Neurol 9, 298. 10.3389/fneur.2018.00298 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 39.Krook-Magnuson E, Armstrong C, Oijala M, and Soltesz I (2013). On-demand optogenetic control of spontaneous seizures in temporal lobe epilepsy. Nat Commun 4, 1376. 10.1038/ncomms2376 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 40.Paz JT, Davidson TJ, Frechette ES, Delord B, Parada I, Peng K, Deisseroth K, and Huguenard JR (2013). Closed-loop optogenetic control of thalamus as a tool for interrupting seizures after cortical injury. Nat Neurosci 16, 64–70. 10.1038/nn.3269 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 41.Gröticke I, Hoffmann K, and Löscher W (2008). Behavioral alterations in a mouse model of temporal lobe epilepsy induced by intrahippocampal injection of kainate. Exp Neurol 213, 71–83. 10.1016/j.expneurol.2008.04.036 [DOI] [PubMed] [Google Scholar]
- 42.Alhashimi R, Thoota S, Ashok T, Palyam V, Azam AT, Odeyinka O, and Sange I (2022). Comorbidity of Epilepsy and Depression: Associated Pathophysiology and Management. Cureus 14, e21527. 10.7759/cureus.21527 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 43.Manouze H, Ghestem A, Poillerat V, Bennis M, Ba-M’hamed S, Benoliel JJ, Becker C, and Bernard C (2019). Effects of Single Cage Housing on Stress, Cognitive, and Seizure Parameters in the Rat and Mouse Pilocarpine Models of Epilepsy. eNeuro 6. 10.1523/eneuro.0179-18.2019 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 44.Zeidler Z, Brandt-Fontaine M, Leintz C, Krook-Magnuson C, Netoff T, and Krook-Magnuson E (2018). Targeting the Mouse Ventral Hippocampus in the Intrahippocampal Kainic Acid Model of Temporal Lobe Epilepsy. eNeuro 5. 10.1523/eneuro.0158-18.2018 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 45.Ives-Deliperi VL, and Jokeit H (2019). Impaired Social Cognition in Epilepsy: A Review of What We Have Learnt From Neuroimaging Studies. Front Neurol 10, 940. 10.3389/fneur.2019.00940 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 46.Rini JF, and Ochoa J (2020). Behavioral implications of temporal lobe epilepsy on social contingency. Epilepsy Behav 110, 107101. 10.1016/j.yebeh.2020.107101 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 47.Stewart E, Catroppa C, and Lah S (2016). Theory of Mind in Patients with Epilepsy: a Systematic Review and Meta-analysis. Neuropsychol Rev 26, 3–24. 10.1007/s11065-015-9313-x [DOI] [PubMed] [Google Scholar]
- 48.Inostroza M, Cid E, Menendez de la Prida L, and Sandi C (2012). Different emotional disturbances in two experimental models of temporal lobe epilepsy in rats. PLoS One 7, e38959. 10.1371/journal.pone.0038959 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 49.Seo J, Jung S, Lee SY, Yang H, Kim BS, Choi J, Bang M, Shin HS, and Jeon D (2013). Early deficits in social behavior and cortical rhythms in pilocarpine-induced mouse model of temporal lobe epilepsy. Exp Neurol 241, 38–44. 10.1016/j.expneurol.2012.11.024 [DOI] [PubMed] [Google Scholar]
- 50.Ives-Deliperi V, and Butler JT (2021). Mechanisms of cognitive impairment in temporal lobe epilepsy: A systematic review of resting-state functional connectivity studies. Epilepsy Behav 115, 107686. 10.1016/j.yebeh.2020.107686 [DOI] [PubMed] [Google Scholar]
- 51.Broekaart DW, Bertran A, Jia S, Korotkov A, Senkov O, Bongaarts A, Mills JD, Anink JJ, Seco J, Baayen JC, et al. (2021). The matrix metalloproteinase inhibitor IPR-179 has antiseizure and antiepileptogenic effects. J Clin Invest 131. 10.1172/jci138332 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 52.Carron S, Dezsi G, Ozturk E, Nithianantharajah J, and Jones NC (2019). Cognitive deficits in a rat model of temporal lobe epilepsy using touchscreen-based translational tools. Epilepsia 60, 1650–1660. 10.1111/epi.16291 [DOI] [PubMed] [Google Scholar]
- 53.Ikegaya Y, Nishiyama N, and Matsuki N (2000). L-type Ca(2+) channel blocker inhibits mossy fiber sprouting and cognitive deficits following pilocarpine seizures in immature mice. Neuroscience 98, 647–659. 10.1016/s0306-4522(00)00188-3 [DOI] [PubMed] [Google Scholar]
- 54.Li F, and Liu L (2019). Comparison of kainate-induced seizures, cognitive impairment and hippocampal damage in male and female mice. Life Sci 232, 116621. 10.1016/j.lfs.2019.116621 [DOI] [PubMed] [Google Scholar]
- 55.Pearson JN, Schulz KM, and Patel M (2014). Specific alterations in the performance of learning and memory tasks in models of chemoconvulsant-induced status epilepticus. Epilepsy Res 108, 1032–1040. 10.1016/j.eplepsyres.2014.04.003 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 56.Wickersham IR, Lyon DC, Barnard RJ, Mori T, Finke S, Conzelmann KK, Young JA, and Callaway EM (2007). Monosynaptic restriction of transsynaptic tracing from single, genetically targeted neurons. Neuron 53, 639–647. 10.1016/j.neuron.2007.01.033 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 57.Gong X, Mendoza-Halliday D, Ting JT, Kaiser T, Sun X, Bastos AM, Wimmer RD, Guo B, Chen Q, Zhou Y, et al. (2020). An Ultra-Sensitive Step-Function Opsin for Minimally Invasive Optogenetic Stimulation in Mice and Macaques. Neuron 107, 38–51.e38. 10.1016/j.neuron.2020.03.032 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 58.Vrinda M, Sasidharan A, Aparna S, Srikumar BN, Kutty BM, and Shankaranarayana Rao BS (2017). Enriched environment attenuates behavioral seizures and depression in chronic temporal lobe epilepsy. Epilepsia 58, 1148–1158. 10.1111/epi.13767 [DOI] [PubMed] [Google Scholar]
- 59.Parfitt GM, Nguyen R, Bang JY, Aqrabawi AJ, Tran MM, Seo DK, Richards BA, and Kim JC (2017). Bidirectional Control of Anxiety-Related Behaviors in Mice: Role of Inputs Arising from the Ventral Hippocampus to the Lateral Septum and Medial Prefrontal Cortex. Neuropsychopharmacology 42, 1715–1728. 10.1038/npp.2017.56 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 60.Shultz LD, Lyons BL, Burzenski LM, Gott B, Chen X, Chaleff S, Kotb M, Gillies SD, King M, Mangada J, et al. (2005). Human lymphoid and myeloid cell development in NOD/LtSz-scid IL2R gamma null mice engrafted with mobilized human hemopoietic stem cells. J Immunol 174, 6477–6489. 10.4049/jimmunol.174.10.6477 [DOI] [PubMed] [Google Scholar]
- 61.Shabab T, Khanabdali R, Moghadamtousi SZ, Kadir HA, and Mohan G (2017). Neuroinflammation pathways: a general review. Int J Neurosci 127, 624–633. 10.1080/00207454.2016.1212854 [DOI] [PubMed] [Google Scholar]
- 62.Lindvall O, and Björklund A (2004). Cell therapy in Parkinson’s disease. NeuroRx 1, 382–393. 10.1602/neurorx.1.4.382 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 63.Björklund A, and Lindvall O (2017). Replacing Dopamine Neurons in Parkinson’s Disease: How did it happen? J Parkinsons Dis 7, S21–s31. 10.3233/jpd-179002 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 64.Blum B, and Benvenisty N (2008). The tumorigenicity of human embryonic stem cells. Adv Cancer Res 100, 133–158. 10.1016/s0065-230x(08)00005-5 [DOI] [PubMed] [Google Scholar]
- 65.Miura K, Okada Y, Aoi T, Okada A, Takahashi K, Okita K, Nakagawa M, Koyanagi M, Tanabe K, Ohnuki M, et al. (2009). Variation in the safety of induced pluripotent stem cell lines. Nat Biotechnol 27, 743–745. 10.1038/nbt.1554 [DOI] [PubMed] [Google Scholar]
- 66.Roy NS, Cleren C, Singh SK, Yang L, Beal MF, and Goldman SA (2006). Functional engraftment of human ES cell-derived dopaminergic neurons enriched by coculture with telomerase-immortalized midbrain astrocytes. Nat Med 12, 1259–1268. 10.1038/nm1495 [DOI] [PubMed] [Google Scholar]
- 67.Hallett PJ, Deleidi M, Astradsson A, Smith GA, Cooper O, Osborn TM, Sundberg M, Moore MA, Perez-Torres E, Brownell AL, et al. (2015). Successful function of autologous iPSC-derived dopamine neurons following transplantation in a non-human primate model of Parkinson’s disease. Cell Stem Cell 16, 269–274. 10.1016/j.stem.2015.01.018 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 68.Kim TW, Piao J, Koo SY, Kriks S, Chung SY, Betel D, Socci ND, Choi SJ, Zabierowski S, Dubose BN, et al. (2021). Biphasic Activation of WNT Signaling Facilitates the Derivation of Midbrain Dopamine Neurons from hESCs for Translational Use. Cell Stem Cell 28, 343–355.e345. 10.1016/j.stem.2021.01.005 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 69.Piao J, Zabierowski S, Dubose BN, Hill EJ, Navare M, Claros N, Rosen S, Ramnarine K, Horn C, Fredrickson C, et al. (2021). Preclinical Efficacy and Safety of a Human Embryonic Stem Cell-Derived Midbrain Dopamine Progenitor Product, MSK-DA01. Cell Stem Cell 28, 217–229.e217. 10.1016/j.stem.2021.01.004 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 70.Schweitzer JS, Song B, Herrington TM, Park TY, Lee N, Ko S, Jeon J, Cha Y, Kim K, Li Q, et al. (2020). Personalized iPSC-Derived Dopamine Progenitor Cells for Parkinson’s Disease. N Engl J Med 382, 1926–1932. 10.1056/NEJMoa1915872 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 71.Xu H, Wang B, Ono M, Kagita A, Fujii K, Sasakawa N, Ueda T, Gee P, Nishikawa M, Nomura M, et al. (2019). Targeted Disruption of HLA Genes via CRISPR-Cas9 Generates iPSCs with Enhanced Immune Compatibility. Cell Stem Cell 24, 566–578.e567. 10.1016/j.stem.2019.02.005 [DOI] [PubMed] [Google Scholar]
- 72.Hallett PJ, Cooper O, Sadi D, Robertson H, Mendez I, and Isacson O (2014). Long-term health of dopaminergic neuron transplants in Parkinson’s disease patients. Cell Rep 7, 1755–1761. 10.1016/j.celrep.2014.05.027 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 73.Li W, Englund E, Widner H, Mattsson B, van Westen D, Lätt J, Rehncrona S, Brundin P, Björklund A, Lindvall O, and Li JY (2016). Extensive graft-derived dopaminergic innervation is maintained 24 years after transplantation in the degenerating parkinsonian brain. Proc Natl Acad Sci U S A 113, 6544–6549. 10.1073/pnas.1605245113 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 74.Piccini P, Brooks DJ, Björklund A, Gunn RN, Grasby PM, Rimoldi O, Brundin P, Hagell P, Rehncrona S, Widner H, and Lindvall O (1999). Dopamine release from nigral transplants visualized in vivo in a Parkinson’s patient. Nat Neurosci 2, 1137–1140. 10.1038/16060 [DOI] [PubMed] [Google Scholar]
- 75.Southwell DG, Nicholas CR, Basbaum AI, Stryker MP, Kriegstein AR, Rubenstein JL, and Alvarez-Buylla A (2014). Interneurons from embryonic development to cell-based therapy. Science 344, 1240622. 10.1126/science.1240622 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 76.Shibley H, and Smith BN (2002). Pilocarpine-induced status epilepticus results in mossy fiber sprouting and spontaneous seizures in C57BL/6 and CD-1 mice. Epilepsy Res 49, 109–120. 10.1016/s0920-1211(02)00012-8 [DOI] [PubMed] [Google Scholar]
Associated Data
This section collects any data citations, data availability statements, or supplementary materials included in this article.
Supplementary Materials
Data Availability Statement
Any additional information required to reanalyze the data reported in this paper is available from the lead contact upon request. This paper does not report original code.