

Abstract
Although photopolymerization reactions are commonly used to form hydrogels, these strategies rely on light and may not be suitable for delivering therapeutics in a minimally invasive manner. Here, hyaluronic acid (HA) macromers are modified with Nor or Tet and upon mixing click into covalently crosslinked Nor-Tet hydrogels via a Diels-Alder reaction. By incorporating a high degree of Nor and Tet substitution, Nor-Tet hydrogels with a broad range in elastic moduli (5 to 30 kPa) and fast gelation times (1 to 5 minutes) are achieved. By pre-coupling methacrylated HANor macromers with thiolated peptides via a Michael addition reaction, Nor-Tet hydrogels are peptide-functionalized without affecting their physical properties. Mesenchymal stem cells (MSCs) on RGD-functionalized Nor-Tet hydrogels adhere and exhibit stiffness-dependent differences in matrix mechanosensing. Fluid properties of Nor-Tet hydrogel solutions allow for injections through narrow syringe needles and can locally deliver viable cells and peptides. Substituting HA with enzymatically degradable gelatin also results in cell-responsive Nor-Tet hydrogels, and MSCs encapsulated in Nor-Tet hydrogels preferentially differentiate into adipocytes or osteoblasts, based on 3D cellular spreading regulated by stable (HA) and degradable (gelatin) macromers.
Keywords: injectable hydrogels, hyaluronic acid, gelatin, biochemical modifications, peptides, matrix mechanosensing
Graphical Abstract
Here, injectable hydrogels are developed with independently tunable biophysical and biochemical properties. Hydrogel stiffness and gelation time are controlled by varying concentration and ratios between norbornene and tetrazine macromers, and hydrogel functionalization with bioactive peptides has no effect on hydrogel mechanics. Cells adhere on 2D hydrogels, and 3D differentiation is regulated via cell-mediated degradation of enzymatically sensitive macromers.
1. Introduction
Hydrogels are three-dimensional and highly hydrated crosslinked polymer networks that are used in various biomedical applications including tissue engineering, drug delivery, and regenerative medicine.[1–5] To synthesize hydrogels, free-radical photopolymerization reactions using visible or ultraviolet light are commonly used due to fast gelation under physiological conditions.[6] Depending on the moities present, hydrogels can be formed via chain-growth or step-growth photopolymerization.[7,8] Free-radical chain-growth photopolymerization of acrylated macromers form hydrogels with polydisperse kinetic chains, resulting in local differences in crosslink density,[7,9] which introduces heterogeneity that could unpredictably influence cell-hydrogel interactions of encapsulated cells. In contrast, free-radical step-growth photopolymerization between molecules containing thiol and vinyl (ene) groups result in one-to-one click reactions that form hydrogels with a homogenous network structure.[7,8] Photopolymerized hydrogels have many advantages, including high biocompatibility and fast gelation times. Step-growth hydrogels also benefit from the potential for photopatterning which introduces heterogeneity to an otherwise homogeneous material.[10] Despite these advantages, free-radical photopolymerization reactions require light and thus are limited to applications where light is readily available or to applications where precise control of the gelation time is not needed.
Alternatives to hydrogel photopolymerization rely on other catalysts including pH, electrostatic interactions, and temperature.[11] For example, Michael-addition reactions between thiols and acrylates form hydrogels with tunable mechanical properties, and gelation rates can be decreased with increasing pH.[12,13] Michael-addition reactions are robust, and have been used to form hydrogels and have been used for static and dynamic 2D cell culture studies.[14] However, due to slow gelation times and the need for high pH buffers, these hydrogels are limited to either acellular or 2D cell culture. Alginate is an anionic biopolymer that forms ionically crosslinked hydrogels when mixed with divalent cations (e.g., Ca2+ or Zn2+).[15] Ionically crosslinked hydrogels form rapidly, and alginate hydrogels specifically have been extensively used for tissue engineering and cellular delivery applications.[16] Despite their high biocompatibility and ease of use, ionically crosslinked hydrogels generally feature low mechanical properties and are unstable due to the diffusion of divalent cations over time. Thermoresponsive hydrogels transition from liquid to hydrogel above a lower critical solution temperature (LCST) or below an upper critical solution temperature (UCST).[17,18] Sala et. al developed thermosensitive poly(N-vinylcaprolactam) (PNVCL) hydrogels that are liquid at room temperature and gel at a physiologic LCST.[19] Chondrocytes and mesenchymal stem cells encapsulated in these hydrogels exhibit high viability and cartilage extracellular matrix formation in vitro and in vivo. Gelatin is a thermoresponsive polymer that has a concentration-dependent UCST of ~25 to 30 °C,[20] which is not suitable for in vivo applications but could be leveraged for additive manufacturing processes. Although not many synthetic polymers with physiologic UCSTs exist,[21] Boustta and colleagues developed poly(N-acryloyl glycinamide) (PNAGA) hydrogels that gel when cooled to 37 °C as a potential injectable drug-releasing material.[22]
While catalyst-based hydrogel polymerization reactions can create hydrogels with myriad properties, these hydrogel techniques rely on an external input (e.g., light, pH, cations, temperature), which limits their use to conditions where a catalyst is present. Diels-Alder reactions are highly specific cycloadditions between dienes and dienophiles that do not require light or other stimuli in aqueous environments.[23,24] For example, hyaluronic acid (HA) macromers modified with furan (diene) were reacted with di-maleimide (dienophile) poly(ethylene glycol) (PEG) crosslinkers to create self-forming hydrogels.[25] Since furan-maleimide hydrogels take several hours to form at a neutral pH, these hydrogels are typically synthesized in acidic conditions not suitable for 3D cell culture. By adding an electron-donating methyl group to furan dienes, methylfuran-dialdehyde hydrogels can be synthesized in under 15 minutes at a physiologic pH.[26] However, a major limitation of Diels-Alder hydrogels formed between furans and maleimides is that these reactions are reversible and prone to hydrolysis, resulting in uncontrollable changes to hydrogel properties including swelling, stiffness, and porosity.[26,27] By using electron-rich cyclic fulvenes in lieu of furan dienes, Madl & Heilshorn formed more stable fulvene-maleimide hydrogels that support 3D cell culture.[28]
Although these Diels-Alder reactions can create biocompatible, self-forming hydrogels, hydrogel mechanics are low (storage modulus, G′ < 1,000 Pa) and gelation times are high (upwards of 3 hours). Diels-Alder reactions between tetrazine (Tet, diene) and norbornene (Nor, dienophile) are irreversible and offer an alternative chemistry for creating self-forming hydrogels with tunable properties.[29–31] Hydrogels that use this chemistry were first reported by Alge et. al, by reacting 4-arm PEG-Tet with di-Nor peptides.[29] By including Nor-modified RGD, biocompatible self-forming hydrogels with adhesive domains were created, with storage moduli ranging from 225 to 2,345 Pa by varying PEG-Tet concentration. In another study, by modifying alginate biopolymers with Nor or Tet moieties (5% Nor and Tet substitution), hydrogels with increased mechanical properties (elastic modulus ~15 kPa for 4% w/v hydrogels) were formed.[30] Although these hydrogels are structurally stable and support 3D cell culture, hydrogel polymerization is slow (> 1 hour), which limits their clinical use and could cause an inhomogeneous cell distribution during gelation. By using gelatin macromers with higher Nor (GelNor) and Tet (GelTet) substitution (~20%), Koshy et al. synthesized rapid self-forming hydrogels that gel as little as ~5 minutes with a G′ of ~4,000 Pa.[31]
We hypothesize that by increasing the degree of substitution of Nor and Tet moieties, self-forming hydrogels with fast gelation times and superior mechanics can be achieved. To our knowledge, peptide coupling to Nor-Tet hydrogels either lowers overall hydrogel mechanics or necessitates a secondary photocoupling step, thus independent control over physical and biochemical hydrogel properties is not possible. Towards addressing this current limitation, we also hypothesize that macromers pre-coupled with peptides can be used to incorporate bioactive motifs to Nor-Tet hydrogels without impacting mechanical properties or gelation rates. In this study, carboxyl groups of hyaluronic acid (HA) macromers were modified with Nor (HANor, 50% substitution) or Tet (HATet, 40% substitution) to create self-forming Nor-Tet hydrogels. Mechanical properties and gelation kinetics were controlled by tuning the total macromer concentration and the stoichiometric ratio between HANor and HATet macromers. By modifying HANor hydroxyl groups with methacrylates (HANorMe), thiol-containing peptides were pre-coupled to methacrylates in HANorMe macromers and used to form peptide-functionalized Nor-Tet hydrogels. Using this platform, we investigated matrix mechanosensing of mesenchymal stem cells (MSCs) on 2D RGD-functionalized Nor-Tet hydrogels and the protective effects of Nor-Tet hydrogel solutions on MSCs injected from needles of clinically relevant dimensions. Further, by substituting HA in HANor with enzymatically degradable gelatin (GelNor), we controlled MSC spreading and MSC differentiation into adipogenic (round, HANor) or osteogenic (spread, GelNor) lineages of encapsulated MSCs cultured in bipotential adipogenic/osteogenic (AD/OS) induction medium.
2. Results and Discussion
2.1. Macromer Synthesis and Nor-Tet Hydrogel Polymerization
HANor macromers were synthesized by converting sodium hyaluronate (HA) to its tetrabutylammonium salt (HATBA), followed by an anhydrous benzotriazole-1-yl-oxy-tris(dimethylamino)-phosphonium hexafluorophosphate (BOP) reaction in dimethyl sulfoxide (DMSO) between carboxyl groups in HATBA and amines in norbornene amine (Nor-NH2) (Figure 1a). HATet macromers were synthesized by reacting carboxyl residues in HATBA with amines in tetrazine amine (Tet-NH2) in the presence of 1-(3-dimethylaminopropyl)-3-ethylcarbodiimide (EDC) and N-hydroxysuccinimide (NHS) (Figure 1b). The anhydrous BOP reaction between carboxyl groups in HATBA and amines in Nor-NH2 to form HANor and the aqueous EDC/NHS reaction between carboxyl groups in the macromer backbone and amines in Tet-NH2 to form HATet were adapted from previous studies that have demonstrated successful tethering of Nor and Tet moieties to macromer backbones with high reproducibility.[10,30]
Figure 1.
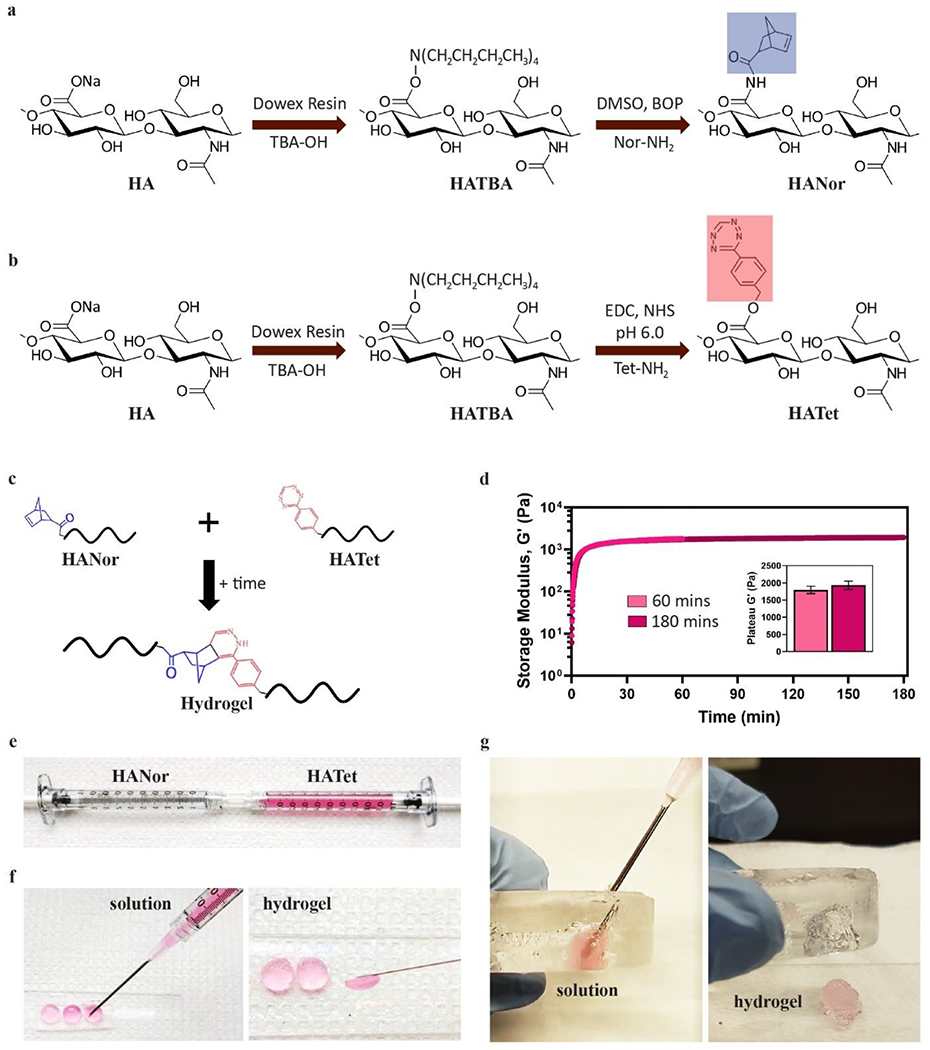
Macromer synthesis and Nor-Tet hydrogel polymerization. (a) HA macromers modified with Nor (HANor) or (b) Tet (HATet) (c) spontaneously from stable crosslinked hydrogels when combined. (d) Representative time sweep rheology plot of 2% w/v Nor-Tet hydrogels polymerizing at 37 °C for 60 minutes and 180 minutes. (e) HANor and HATet macromers can be dissolved rapidly at room temperature and loaded into syringes for mixing through a Luer-Lock coupler. Mixed macromers remain in solution long enough for extrusion into (f) molds or (g) cavities, resulting in self-forming hydrogels that conform to the shape of space they occupy.
Upon mixing, HANor and HATet macromers self-form into stable Nor-Tet hydrogels (Figure 1c). At 37 °C, the storage modulus (G´) of 2% w/v hydrogels after 60 (1,800 ± 100 Pa) and 180 (1,930 ± 120 Pa) minutes is not statistically significant (Figure 1d), demonstrating that Nor-Tet hydrogels self-form in under an hour. To evaluate the specificity of Diels-Alder reactions between HANor and HATet, two tests were performed. HANor or HATet were first mixed with unmodified HA, resulting in no hydrogel formation (Figure S1, Supporting Information). HANor and HATet were also mixed in the presence of either Nor-NH2 or Tet-NH2 molecules in excess (1,000x). Here, the unbound Nor and Tet molecules bind to their respective Diels-Alder partners, preventing hydrogels from forming (Figure S2, Supporting Information). These findings demonstrate that the Diels-Alder reactions between Nor and Tet are highly specific and necessary for Nor-Tet hydrogels to self-form.
We next sought to determine how the inclusion of Nor and Tet moieties could impact fluid viscosity of HANor and HATet macromers in solution since this is an important parameter for developing injectable materials and bioinks for 3D printing. Alge et al. showed that solutions containing gelatin functionalized with 20% Nor and Tet pendant groups were less viscous than solutions of unmodified gelatin,[31] and this may be due to the presence of Nor and Tet interrupting with physical interactions required for the thermogelation of gelatin.[32] HANor and HATet macromers used in this study have approximately 50% and 40% of their repeat units functionalized with Nor and Tet, respectively, as confirmed by 1H NMR (400 MHz, D2O) (Figure S3, Supporting Information), which is significantly higher than Nor and Tet substitutions used by other Diels-Alder Nor-Tet hydrogels.[30,31] At 37 °C, the viscosity of unmodified HA ranges from 3.3 to 76.2 mPa s for 2% to 6% w/v solutions (Table 1). Nor and Tet substitutions result in a modest decrease in viscosity for 2% and 4% w/v HANor and HATet solutions, and a much larger decrease in viscosity for 6% w/v HANor (37.8 mPa s) and HATet (36.3 mPa s) solutions (Table 1). The presence of Nor and Tet results in a decrease in viscosity, and the viscosities for 2%, 4%, and 6% w/v HANor and HATet solutions are well below the upper limit of viscous bioinks and fluids (> 300 mPa s)[33,34] that can result in needle clogging and heterogeneous mixing post-hydrogel extrusion.[35]
Table 1.
Viscosity of polymers used in hydrogel formation at 37 °C. Viscosities of HA, HANor, and HATet at 2, 4, and 6% w/v.
| Polymer | 2% w/v [mPa s] | 4% w/v [mPa s] | 6% w/v [mPa s] |
|---|---|---|---|
| HA | 3.3 ± 0.7 | 17.3 ± 2.7 | 76.2 ± 10.2 |
| HANor | 3.1 ± 0.5 | 16.4 ± 2.4 | 37.8 ± 5.6 |
| HATet | 2.4 ± 0.4 | 15.1 ± 1.7 | 36.3 ± 4.7 |
HANor and HATet macromers can be dissolved at room temperature in aqueous media, loaded into a syringe, and mixed with a syringe coupler (Figure 1e). Upon mixing, the hydrogel solution will begin to polymerize while allowing for sufficient time to extrude through a needle (about 5 minutes for a 2 wt% Nor-Tet hydrogel) prior to self-forming into a hydrogel that conforms into any shape ranging from cylindrical molds (Figure 1f) to amorphous cavities (Figure 1g). Gelation time is an important parameter of self-forming hydrogels and ideally, injectable hydrogels should polymerize within a few minutes post-injection under physiological conditions.[36] For in vivo applications, hydrogels that polymerize too slowly could diffuse into surrounding tissues, leading to the unwanted presence of hydrogel material away from the target site, whereas hydrogels that form too quickly can polymerize prematurely in the needle prior to reaching the targeted site. Nor-Tet hydrogels with 2% to 6% w/v macromer concentration can be readily dissolved and mixed with sufficient time to locally deliver hydrogel that is contained within a region of interest (e.g., molds, cavities).
2.2. Physical Properties of Self-Forming Nor-Tet Hydrogels are Highly Tunable
To evaluate mechanics and gelation properties of self-forming hydrogels, HANor and HATet macromers were mixed at 2%, 4%, or 6% total w/v macromer concentration. Frequency sweep rheology (0.1 to 10 Hz) at 37 °C shows that the storage modulus (G´) is above the loss modulus (G″) and that G´ is constant across a range of oscillatory frequencies (Figure 2a), demonstrating the formation of stable hydrogels post-mixing. Nor-Tet hydrogel mechanics also increase with increasing macromer concentration (Figure 2b). The range in hydrogel mechanics achieved is significant, with G´ values of 1,800 ± 100 Pa (2% w/v), 6,300 ± 1,000 Pa (4% w/v), and 12,500 ± 1,100 Pa (6% w/v) (Figure 2c). To determine the elastic modulus (E), cylindrical Nor-Tet hydrogels (8 mm diameter, 2 mm height) were subject to compression testing, with E values ranging from ~5 kPa (2% w/v) to ~30 kPa (6% w/v) (Figure S4, Supporting Information). Gelation rates also decreased with increasing macromer content. For 2% w/v Nor-Tet hydrogels, it took 5.80 ± 0.24 minutes to reach 50% of their final G´, and gelation time decreased for 4% w/v (2.04 ± 0.34 min) and 6% w/v (0.83 ± 0.44 min) hydrogels (Figure 2d).
Figure 2.
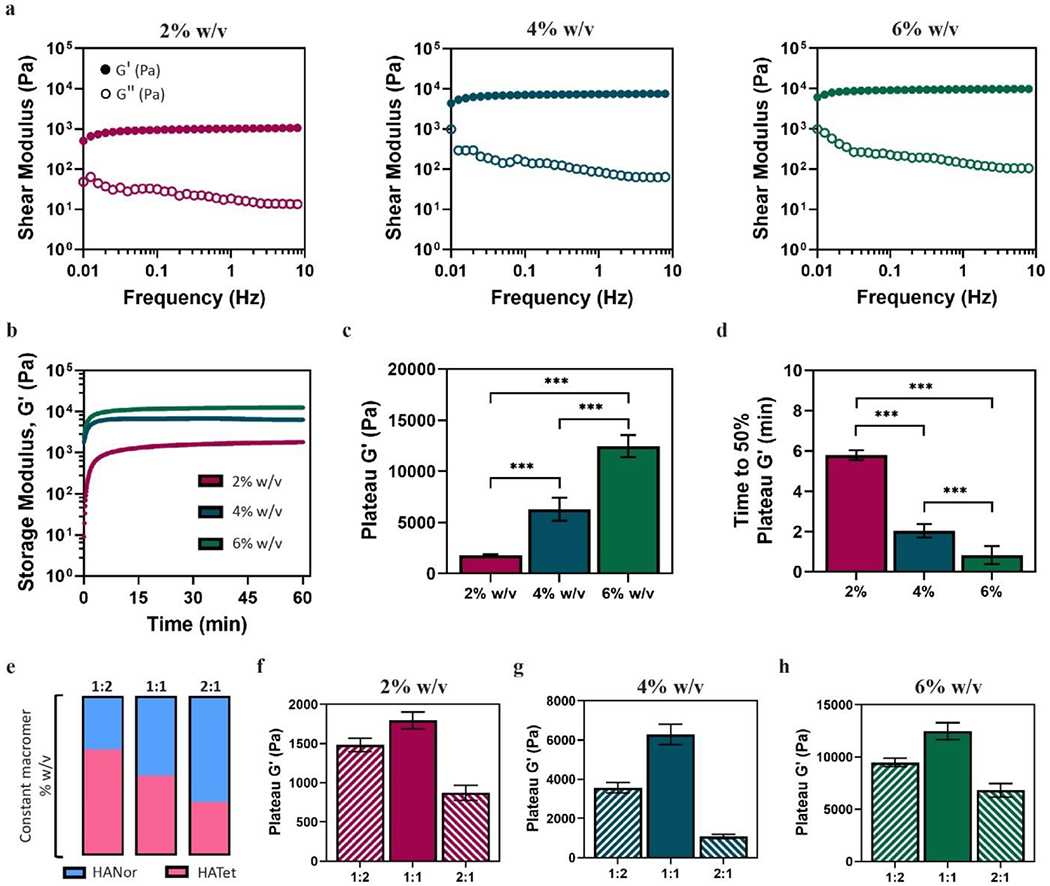
Physical characterization of self-forming hydrogels at 37 °C. (a) Representative frequency sweep rheology plot shows storage (G′) and loss moduli (G″) of 2%, 4%, and 6% w/v Nor-Tet hydrogels at a 1:1 stoichiometric ratio. (b) Representative time sweep rheology plot was used to determine (c) plateau G′ and (d) time to 50% plateau G′ of 2%, 4%, and 6% w/v Nor-Tet hydrogels at a 1:1 stoichiometric ratio. (e) HANor and HATet were mixed at a 1:2, 1:1, or 2:1 stoichiometric ratio and the plateau G′ was determined for (f) 2%, (g) 4%, and (h) 6% w/v Nor-Tet hydrogels. Bar graphs shown as mean ± SD (n ≥ 3 samples per condition) with significant differences determined with ANOVA followed by Tukey’s post hoc test where ***p < 0.001.
The range in Nor-Tet hydrogel mechanics achieved by simply increasing the total macromer concentration is significantly larger than previous systems that use a Diels-Alder chemistry (Table S1, Supporting Information). This could be due to irreversible bonds formed between Nor and Tet and the high degree of Nor and Tet substitution in the HANor and HATet macromers. For instance, Diels-Alder reactions between furan or methylfuran and maleimides are reversible, resulting in hydrogels that are unstable over time with low mechanical properties. While replacing furans with fulvenes increases the stability of self-forming hydrogels, the mechanical properties are still low (< 1,000 Pa), possibly due to a low amount of maleimides and fulvenes present.[28] Additionally, hydrogel formation is inherently slow, which could result in low retention at a target site post-injection and in cell sedimentation leading to an uneven distribution of encapsulated cells throughout the hydrogel.[28] By using Tet as the diene and Nor as the dienophile, irreversible bonds form during gelation, resulting in mechanically stable, biocompatible self-forming hydrogels. Further, by using HANor and HATet macromers with a high degree of Nor (50%) and Tet (40%) substitution, rapidly forming hydrogels with a large range of mechanics were formed, and it is expected that an even broader range of mechanical properties is possible by increasing the number of Nor and Tet moieties, either by further increasing the degree of substitution, or by using higher molecular weight HA macromers.
To further explore the physical properties of self-forming Nor-Tet hydrogels, HANor and HATet were mixed at different stoichiometric ratios (HANor:HATet) at a constant total macromer concentration (Figure 2e). For 2% w/v Nor-Tet hydrogels, the plateau G´ varied for different HANor:HATet ratios, with the highest mechanics observed at a 1:1 ratio (1,800 ± 110 Pa), followed by 1:2 (1,500 ± 90 Pa) and 2:1 (900 ± 100 Pa) ratios (Figure 2f). Similarly, for 4% and 6% w/v Nor-Tet hydrogels, highest mechanics were seen at a 1:1 ratio, with second and third highest G´ observed in 1:2 and 2:1 HANor:HATet ratios, respectively (Figure 2g and 2h). The range in mechanics by simply varying the HANor:HATet ratio was significant at a constant macromer concentration. In 4% w/t Nor-Tet hydrogels there was an over 5-fold change (1,100 ± 110 to 6,300 ± 500 Pa) and in 6% w/v Nor-Tet hydrogels an almost 2-fold change (6,800 ± 700 to 12,500 ± 800 Pa) in G´ between 2:1 and 1:1 HANor:HATet ratios. Nor-Tet hydrogels formed by reacting HANor and HATet at a 1:2 stoichiometric ratio resulted in higher mechanics than those formed at a 2:1 ratio regardless of total macromer concentration, and this could be due to the difference in percent modifications of HANor (50% mod) and HATet (40% mod). These findings demonstrate that mechanical properties can be independently controlled while maintaining biopolymer concentration constant. This is useful in tuning hydrogel mechanics without changing fluid properties (e.g., viscosity) that could impact extrusion parameters.
Interestingly, the rate of hydrogel formation also changed with varying HANor:HATet ratios at a constant biopolymer concentration. For 2% w/v Nor-Tet hydrogels, gelation time increased from 5.80 minutes at a 1:1 Nor:Tet ratio to 12.30 minutes for a 2:1 Nor:Tet ratio (Figure S5a, Supporting Information). The same trends were observed for 4% and 6% w/v Nor-Tet hydrogels, with the shortest gelation time seen at a 1:1 Nor:Tet ratio, and second and third longest gelation times at 1:2 and 2:1 Nor:Tet ratios, respectively (Figure S5b and Figure S5c, Supporting Information). An increase in total macromer concentration corresponds to higher mechanics and a decrease in gelation kinetics,[30,31] and this observation shows that changing the amount of Nor and Tet moieties at a fixed macromer concentration also regulates gelation time.
At a 1:1 Nor:Tet ratio, there is approximately 25% more Nor than Tet moieties, since the degree of substitution for the HANor and HATet macromers used is 50% and 40%, respectively. Thus, it is expected that the theoretical maximum stiffness (and minimum gelation time) would occur at a ratio where Nor and Tet moieties are equal. Based on 50% HANor and 40% HATet modifications, this corresponds to a 1:1.25 HANor:HATet ratio. At this optimal ratio, mechanics for 2%, 4%, and 6% w/v Nor-Tet hydrogels were higher than Nor-Tet hydrogels formed at a 1:1 HANor:HATet stoichiometric ratio (Figure S6, Supplemental Information). The storage modulus G′ increased from 1,800 to 2,100 Pa for 2% w/v, 6,300 to 8,000 Pa for 4% v/w, and 12,500 to 14,000 Pa for 6% w/v Nor-Tet hydrogels. These findings show that Diels-Alder Nor-Tet reactions are highly specific, and that maximal mechanics are achieved when the amount of Nor and Tet moieties present is equal.
2.3. Self-Forming Hydrogels can be Peptide Functionalized without Changing Mechanics
Hydrogels formed with a Diels-Alder Nor-Tet chemistry cannot be peptide-functionalized without affecting crosslinking density or requiring a secondary photoreaction post-hydrogel polymerization.[29,30] To functionalize Nor-Tet hydrogels with adhesive RGD peptides, Alge and coworkers bound Nor-RGD to multi-arm PEG-Tet, resulting in competition between Nor-RGD and di-Nor crosslinker peptides for available Tet sites.[29] In another study, Desai et al. used a thiol-ene photopolymerization reaction to couple thiolated RGD peptides to pendant Nor moieties in pre-formed alginate Nor-Tet hydrogels.[30]. As an alternate strategy, in this study Nor-Tet hydrogels with independent control over peptide functionalization were created by tethering thiolated RGD peptides to HANor via a two-step process (Figure 3).
Figure 3.
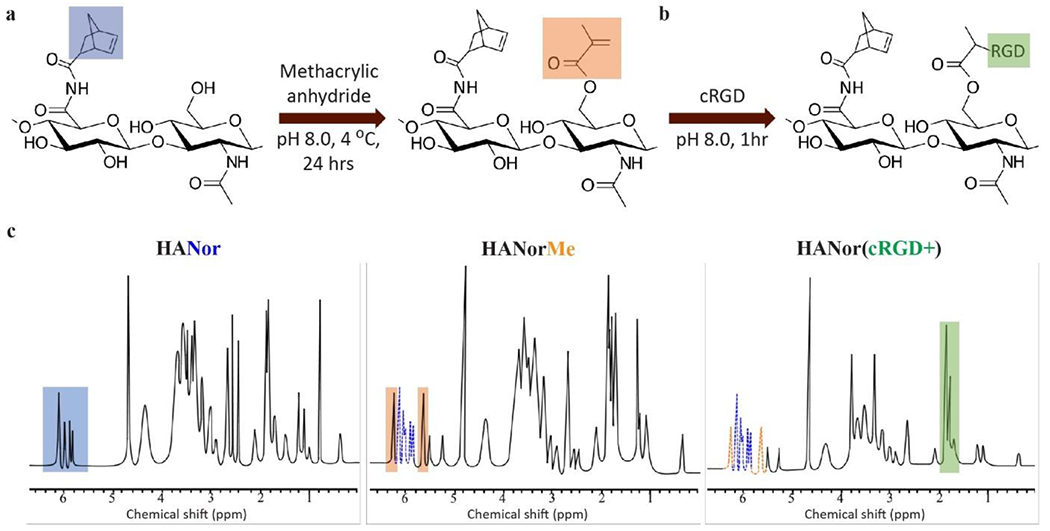
HANor macromers functionalized with thiolated peptides. (a) Hydroxyl groups in HANor macromers are coupled with methacrylic anhydride to form HANorMe macromers. (b) HANorMe macromers are then mixed with thiolated RGD adhesive peptide (cRGD) to form peptide functionalized HANor(cRGD+) macromers. (c) 1H NMR spectra shows peaks corresponding to norbornene (blue, left), methacrylate (orange, middle), and cRGD (green, right) modifications to the HA backbone.
First, hydroxyl groups in HANor were modified with methacrylates (Me) via esterification with methacrylic anhydride,[37] resulting in an HANorMe macromer (Figure 3a). Next, Me groups in HANorMe and thiols in thiolated RGD (cRGD) peptides were covalently bound via a Michael-addition reaction,[38] resulting in HANor(cRGD+) (Figure 3b), an HANor macromer that is peptide-functionalized without altering Nor groups dedicated for Nor-Tet hydrogel formation. By varying the amount of cRGD reacted with a constant amount of HANorMe, the effective concentration of RGD was kept at 2 mM for 2%, 4%, and 6% w/v Nor-Tet hydrogels (Table S2 Supporting Information). MSCs adhere to HA hydrogels functionalized with RGD at a 2 mM concentration, as shown by our group and others.[39–41] 1H NMR was used to confirm Nor (50%), Me (78%), and cRGD modification (Figure 3c). To maintain a constant effective concentration, the amount of cRGD peptide added to HANorMe during synthesis was adjusted accordingly. Less peptide coupling to HANorMe should occur for increasing total macromer concentration, which was confirmed by 1H NMR to be 17%, 9%, and 6% cRGD modification for 2%, 4%, and 6% w/v Nor-Tet hydrogels, respectively (Figure S7, Supporting Information). At least 60% of Me sites are available for coupling after functionalization with cRGD, and these unreacted Me moieties can be used to either increase the effective concentration of cRGD or modify biochemical properties by adding other thiolated peptides.
To confirm that peptide modifications to the hydroxyl group do not impact Nor moieties used for Nor-Tet binding, HATet was mixed with either HANor, HANorMe, or HANor(cRGD+) at 2%, 4%, or 6% total macromer w/v at a 1:1 macromer ratio. Time sweep rheology shows that G′ increases with increasing w/v, and that no discernable changes in gelation from Me substitution or RGD peptide functionalization are observed (Figure 4a). Final mechanical properties (plateau G´) did not change across the macromer groups for 2% (1,800 ± 110 Pa), 4% (6,300 ± 1,000 Pa), and 6% (12,500 ± 1,100 Pa) w/v Nor-Tet hydrogels (Figure 4b). Additionally, peptide functionalization did not have a significant effect on gelation times, with times to 50% plateau G´ of 5.80 ± 0.24, 2.04 ± 0.34, and 0.83 ± 0.44 minutes for 2%, 4%, and 6% w/v Nor-Tet hydrogels, respectively (Figure 4c). Elastic moduli (E) of fully formed cylindrical hydrogels yielded consistent results across the different macromer groups for 2% (4.6 ± 1.2 kPa), 4% (26.1 ± 2.7 kPa), and 6% (45.2 ± 4.1 kPa) w/v Nor-Tet hydrogels (Figure 4d). These results show that Nor-Tet hydrogels can be biofunctionalized with thiolated peptides without affecting mechanical properties and gelation kinetics. Although cRGD was used, this procedure can be applied to any thiol-containing molecule. The effective peptide concentration can also be easily tuned by varying the amount of peptide mixed with HANorMe during thiol-methacrylate peptide coupling.
Figure 4.
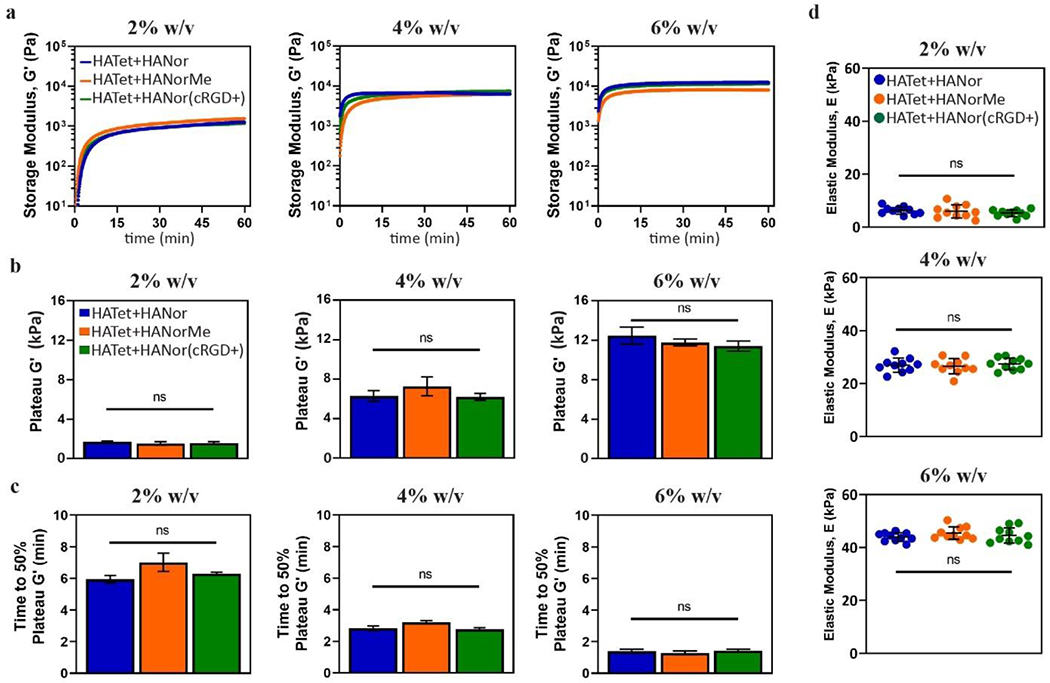
Peptide coupling does not affect mechanical properties of Nor-Tet hydrogels. (a) Representative time sweep rheology of G′ kinetics for 2%, 4%, and 6% w/v Nor-Tet hydrogels was used to determine (b) plateau G′ and (c) time to 50% plateau G′. (d) Compression testing of 2%, 4%, and 6% w/v Nor-Tet hydrogels formed in cylindrical molds was used to determine elastic modulus (E). Bar graphs shown as mean ± SD (n ≥ 3 samples per condition) with significant differences determined with ANOVA followed by Tukey’s post hoc test where ns is not significant.
2.4. Cells Adhere and Proliferate on RGD-Functionalized Nor-Tet Hydrogels
After demonstrating that Nor-Tet hydrogels can be independently functionalized with peptides without altering mechanics, their use as a biocompatible cell culture platform was investigated. Since cellular adhesion to unmodified HA hydrogels is poor,[42,43] adhesive molecules need to either be passively adsorbed or chemically bound to HA hydrogels to support cell adhesion. To confirm the bioactivity of adhesive RGD peptides, HANor with or without coupled cRGD was mixed with HATet and extruded into silicone molds (8 mm diameter, 0.5 mm height), to form adhesive (cRGD+) or non-adhesive (cRGD-) Nor-Tet hydrogels (Figure 5a). Human mesenchymal stem cells (MSCs, 3,000 cells cm−2) were then cultured on top of Soft (2% w/v, 4.6 kPa E) or Stiff (6% w/t, 45.2 kPa E) self-forming Nor-Tet hydrogels (Figure 5b) and cellular adhesion and proliferation were evaluated.
Figure 5.
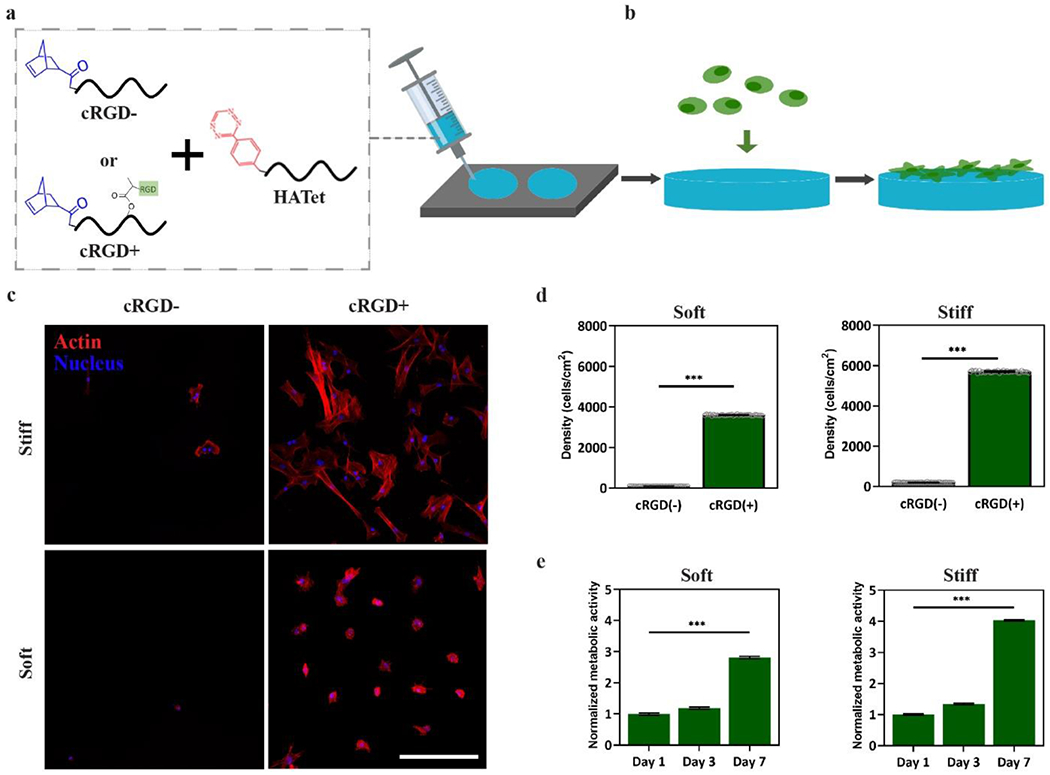
2D cell attachment and proliferation on cRGD-functionalized Nor-Tet hydrogels. (a) HANor macromers with or without cRGD functionalization were mixed with HATet, injected into cylindrical molds, and (b) MSCs were cultured on cRGD+ and cRGD- Nor-Tet hydrogels. (c) Representative maximum projection images (actin, red; nuclei, blue) of MSCs on Soft or Stiff Nor-Tet hydrogels with or without covalently bound cRGD. Confocal images were used to determine MSC Density (number of nuclei cm−2) on (d) Soft and Stiff Nor-Tet hydrogels with or without cRGD. AlamarBlue assay was performed on day 1, 3, and 7 to quantify normalized metabolic activity of MSCs on RGD-functionalized (e) Soft and Stiff Nor-Tet hydrogels. Bar graphs shown as mean ± SD (n ≥ 3 samples per condition) with significant differences determined with ANOVA followed by Tukey’s post hoc test where ***p < 0.001. Scale bar: 500 μm.
After three days in culture, representative fluorescent images of MSCs show minimal cell attachment on Soft and Stiff cRGD- hydrogels, while significant cell atttachment is observed on Soft and Stiff Nor-Tet hydrogels functionalized with RGD (Figure 5c). Quantification of cell density (number of cells cm−2) of MSCs on Soft Nor-Tet hydrogels confirms poor cell attachment on cRGD- hydrogels (100 ± 5 cells cm−2), whereas significant attachment was observed on cRGD+ Nor-Tet hydrogels (3,600 ± 20 cells cm−2) (Figure 5d). Cell counts on Stiff Nor-Tet hydrogels were also higher for MSCs on cRGD+ (5,700 ± 40 cells cm−2) hydrogels in comparison to cRGD- (201 ± 11 cells cm−2) Nor-Tet hydrogels. The cell density on Soft and Stiff cRGD+ hydrogels was higher than the initial seeding density (3,000 cells cm−2), suggesting that MSCs adhere and proliferate on RGD-functionalized 2D Nor-Tet hydrogels. To evaluate cell proliferation over time, MSCs were cultured on Soft and Stiff cRGD+ Nor-Tet hydrogels and metabolic activity was measured after 1, 3, and 7 days in culture using an alamarBlue assay. On both Soft and Stiff Nor-Tet hydrogels, there was a subtle increase in metabolic activity between one and three days, followed by a much larger increase between 3 and 7 days (Figure 5e). While Soft and Stiff hydrogels support MSC adhesion and proliferation, Stiff Nor-Tet hydrogels displayed higher MSC densities and metabolic activity, which is due to stiffness-dependent differences in matrix mechanosensing.[44]
2.5. Matrix Stiffness Regulates MSC Morphology and Matrix Mechanosensing on RGD-Functionalized Nor-Tet Hydrogels
An important characteristic of stem cells is their ability to sense and respond to mechanical cues.[45] For example, MSCs on soft hydrogels that mimic the stiffness of brain tissue express neural biomarkers, whereas MSCs cultured on more rigid hydrogels preferentially differentiate into cell types present in stiffer tissues, like muscle or bone.[46] At the cellular level, changes in cell shape and spreading occur in response to mechanical cues shortly after making initial contact with their new environment.[46,47] On a molecular level, matrix mechanosensing is led by several mechano-transducer proteins that collectively induce changes in focal adhesion maturation,[48] cytoskeletal contractility,[49] and nuclear Yes-associated protein (YAP) localization.[50] While these differences have been observed in photopolymerized hydrogels, stiffness-dependent differences in cellular matrix mechanosensing on Nor-Tet hydrogels has not been demonstrated. To highlight the use of the self-forming Nor-Tet hydrogels presented as a facile platform for matrix mechanosensing studies, MSCs were cultured on Soft and Stiff RGD-functionalized hydrogels for three days and stiffness-dependent changes in morphology (area, roundness, aspect ratio) and mechanosensing (nuclear YAP localization, focal adhesion maturation, actin anisotropy) were evaluated.
Representative silhouettes of MSCs on Soft and Stiff hydrogels show large differences in morphology induced by Nor-Tet hydrogel stiffness (Figure 6a). MSCs on Soft Nor-Tet hydrogels were significantly smaller (1,500 ± 600 μm2) than MSCs on Stiff Nor-Tet hydrogels (3,500 ± 300 μm2) (Figure 6b). MSCs were also signficantly rounder on Soft Nor-Tet hydrogels (Figure 6c) and had a larger aspect ratio on Stiff Nor-Tet hydrogels (Figure 6d). These results are consistent with morphology of MSCs on photopolymerized HA hydrogels of comparable stiffness.[40,51] For example, MSCs on softer (1 kPa) HANor hydrogels had an approximate average area of 650 μm2, while on stiff (20 kPa) hydrogels MSC area increased to ~3,750 μm2.[40] Similarly, on soft (~3 kPa) o-Nitrobenzyl-Methacrylate-HA hydrogels MSCs had an average area of about 1,500 μm2 while on stiff (~15 kPa) hydrogels the area doubled to ~3,300 μm2.[51] Although the Nor-Tet hydrogels were formed with a Diels-Alder click chemistry and not using free-radical polymerization between acrylamide and bis-acrylamide or photopolymerization reactions, stiffness-dependent changes in morphology were conserved.
Figure 6.
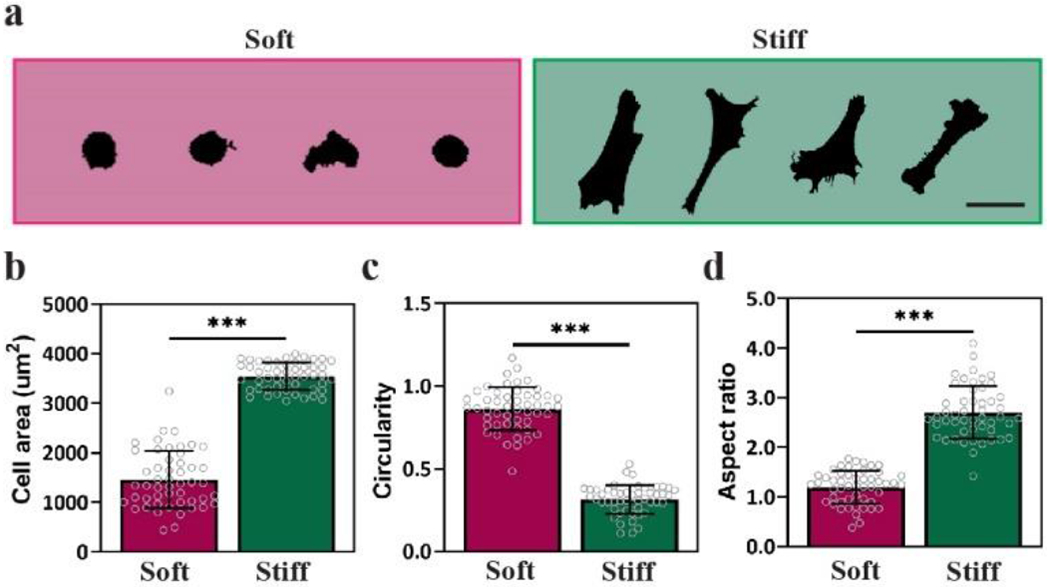
Cells on 2D Nor-Tet hydrogels display stiffness-dependent changes in cell morphology. (a) Representative silhouettes of MSCs on Soft and Stiff Nor-Tet hydrogels. Bar graphs show (b) Area, (c) Circularity, and (d) Aspect ratio of MSCs on Soft and Stiff Nor-Tet hydrogels. Bar graphs shown as mean ± SD (n ≥ 3 samples per condition) with significant differences determined with ANOVA followed by Tukey’s post hoc test where ***p < 0.001. Scale bar: 100 μm.
After establishing that MSC morphology changes in a stiffness-dependent manner on 2D Nor-Tet hydrogels, matrix mechanosensing on self-forming hydrogels was evaluated. Yes-associated protein (YAP) is a key transducer of outside-in mechanical signals,[50] and cytosolic YAP translocates to MSC nuclei with increasing stiffness on polyacrylamide and photopolymerized 2D hydrogels.[40,50] The nuclear YAP of MSCs on Stiff Nor-Tet hydrogels was significantly higher than MSCs on Soft Nor-Tet hydrogels (Figure 7a). At the cell-hydrogel interface, focal adhesions act as intermediaries that connect the local extracellular environment to the actin cytoskeleton, consequently activating mechanotransductive pathways.[52] Specifically, focal adhesion kinase (FAK) activation through the phosphorylation of tyrosine residues (pFAK) plays a role in matrix mechanosensing by responding to substrate stiffness and cytoskeletal contractility.[53] As such, pFAK maturation (length, count) and cytoskeletal alignment (actin anisotropy) were used as additional proxies for stiffness-dependent mechanosensing.
Figure 7.
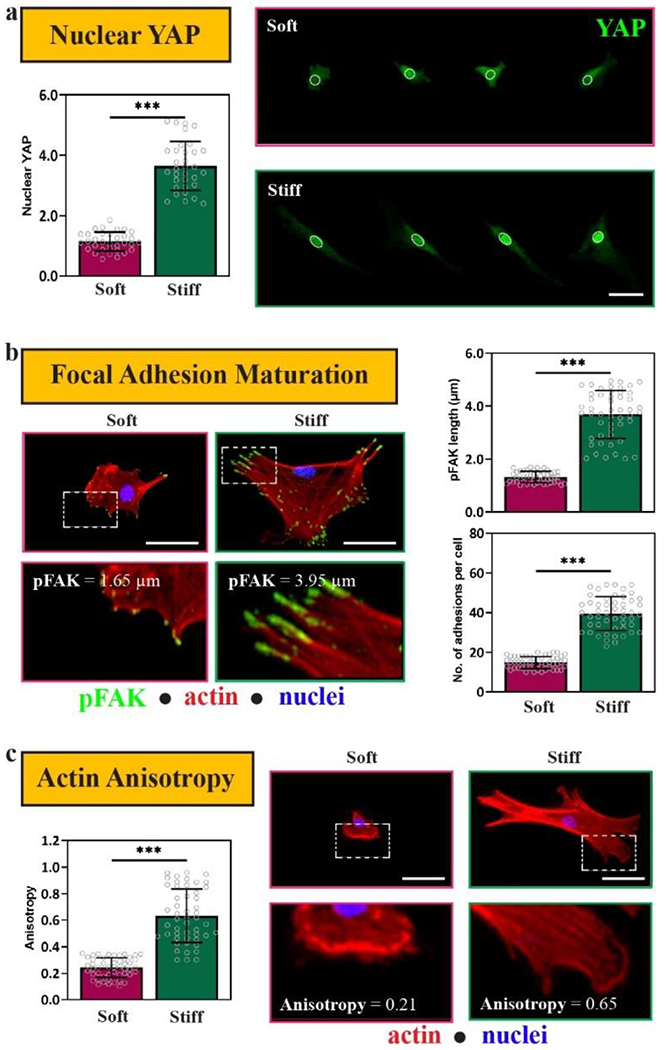
MSCs are mechanosensitive on 2D Soft and Stiff Nor-Tet hydrogels. (a) Representative images and Nuclear YAP (green) quantification of MSCs on Soft and Stiff Nor-Tet hydrogels. (b) Representative images of MSCs stained for pFAK (green), phalloidin (red), and nuclei (blue) and quantification of pFAK maturation (length, average number of adhesions per cell) of MSCs on Soft and Stiff Nor-Tet hydrogels. Bar graphs shown as mean ± SD (n ≥ 3 samples per condition) with significant differences determined with ANOVA followed by Tukey’s post hoc test where ***p < 0.001. Scale bars: a, b, c 50 μm.
The length of pFAK increased with increasing Nor-Tet hydrogel stiffness, and so did the average number of focal adhesions per cell (Figure 7b). Actin anisotropy (actin fiber alignment) is a way to evaluate the organization of actin stress fibers, and there was a significant increase in actin anisotropy (0.24 ± 0.07 vs. 0.63 ± 0.20) between MSCs on Soft and Stiff Nor-Tet hydrogels (Figure 7c). These results show that MSCs respond to mechanical cues on 2D Nor-Tet hydrogels which highlights the versatility of these catalyst-free hydrogels as a platform to study and control matrix mechanosensing.
2.6. Nor-Tet Hydrogels as a Delivery Vehicle of Highly Viable Cells
While cell injections are a widely used method to transplant cells, low cell viability and poor cell retention at the site of injection limit the efficacy of this minimally invasive technique.[54,55] Hydrogels address the limitations of liquid cell injections by shielding cells from shear stresses during extrusion and by keeping cells at the target site as the hydrogel polymerizes. To evaluate the use of Nor-Tet hydrogels as a biocompatible and injectable cell carrier, MSCs were suspended in culture medium or an RGD-functionalized Nor-Tet hydrogel solution that was loaded into a syringe and extruded through a needle tip onto cylindrical molds (Figure 8a). Syringe needles with varying inner diameters were used ranging from 160 μm (30 G) to 1,190 μm (16 G) (Figure S8a, Supporting Information). One (D1), three (D3), and seven (D7) days post-injection, the percentage of live cells extruded through different size needles was determined for Soft (2% w/v) and Stiff (6% w/v) Nor-Tet hydrogels by analyzing confocal images stained with a Live/Dead assay (live, green; dead, red) (Figure 8b, 8c; Figure S8b, S8c, Supporting Information). One day post-injection, cells extruded through different size needles are highly viable (> 85% viability: 16G, 20G, 26G and > 70% viability: 30G) for Soft and Stiff groups (Figure 8d, 8e). Meanwhile, MSCs injected using culture medium (C) had 75% (16G), 66% (20G), 64% (26G), and 42% (30G) viable cells one day post-injection. MSCs in Soft and Stiff Nor-Tet hydrogels also remain more viable than MSCs injected using culture medium only after at least seven days in culture. The Soft and Stiff Nor-Tet hydrogels also retained their shape seven days post-injection, which demonstrates that viable MSCs can be extruded from a range of syringe needle sizes through a solution that self-forms into mechanically stable hydrogels.
Figure 8.
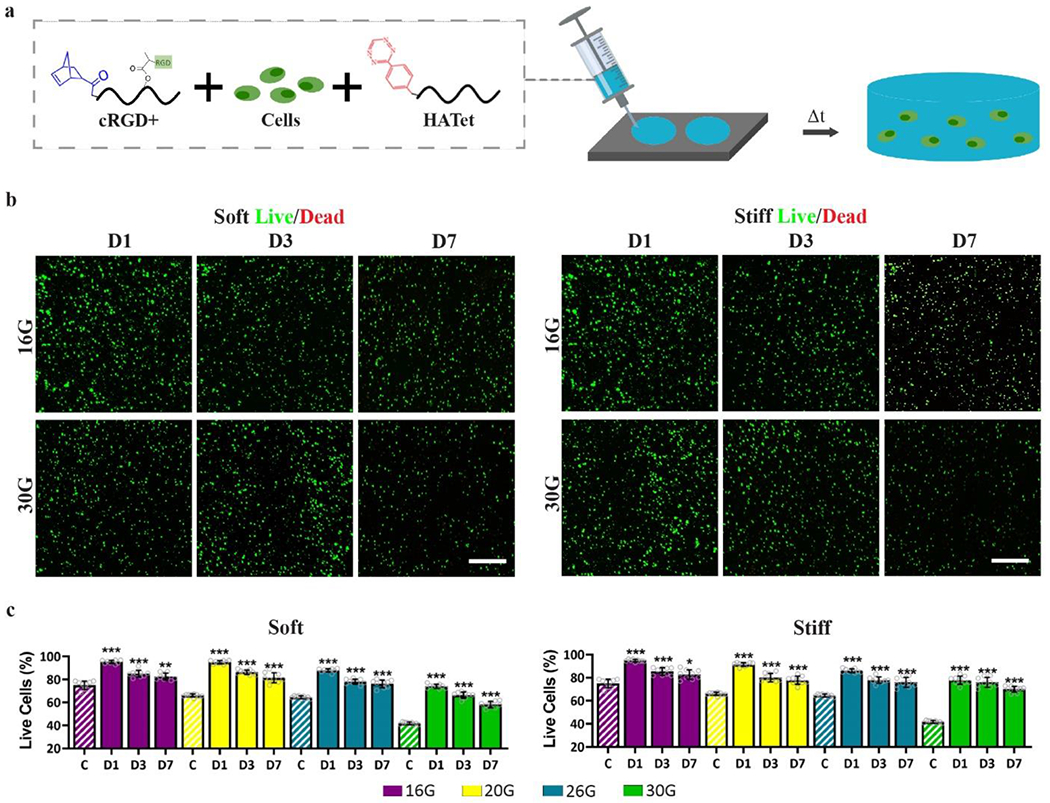
MSCs in injectable 3D Nor-Tet hydrogels are highly viable. (a) Nor-Tet hydrogel solutions consisting of MSCs mixed with RGD-functionalized HANor and HATet were injected into cylindrical molds via extrusion of various clinically relevant syringe needle sizes. Representative live (green) and dead (red) staining of MSCs cultured in (b) Soft and (c) Stiff Nor-Tet hydrogels. Bar graphs show percentage of live cells in (d) Soft and (e) Stiff Nor-Tet hydrogels from confocal image analysis. Bar graphs shown as mean ± SD (n ≥ 3 samples per condition) with significant differences determined with ANOVA followed by Tukey’s post hoc test where *p < 0.05, **p < 0.01, ***p < 0.001 compared to the C group (MSCs injected through cell culture medium). Scale bars: 500 μm.
MSCs encapsulated in Soft and Stiff 3D Nor-Tet hydrogels remain spherical after 7 days in culture (Figure S9a, Supporting Information). Further, no difference in cellular volume (Figure S9b, Supporting Information) or sphericity (Figure S9c, Supporting Information) were observed between MSCs in 3D Soft and Stiff Nor-Tet hydrogels. These findings are consistent with previous studies showing that cells encapsulated in covalently crosslinked hydrogels remain spherical, independent of stiffness.[56] As expected for spherical cells, nuclear YAP ratios were close to unity and did not change between Soft and Stiff Nor-Tet hydrogels (Figure S10, Supporting Information).
2.7. 3D Cell-Mediated Nor-Tet Hydrogel Degradation Directs Stem Cell Differentiation
Stem cells can sense biophysical signals, leading to numerous cell functions including differentiation. On 2D substrates, MSC lineage commitment is dictated in part by stiffness, and this is due to stiffness-dependent changes in matrix mechanosensing.[46,57] MSCs on 2D soft materials are round and exhibit low traction forces whereas MSCs on 2D stiff substrates are spread and more contractile. While MSCs spread with increasing stiffness on 2D, this is not the case in 3D. For 3D morphology-mediated MSC differentiation to occur, encapsulated MSCs need to locally remodel their surrounding hydrogel matrix, which can be enabled by either by tuning hydrogel viscoelasticity,[58,59] or hydrogel degradation.[40,56] To demonstrate the effects of cell-mediated hydrogel degradation on MSC differentiation, Khetan et al. encapsulated MSCs in hydrogels that restrict or permit 3D cellular spreading.[56] In bipotential AD/OS (adipogenic/osteogenic) Medium, MSCs spread and preferentially differentiate into osteoblasts in 3D enzymatically degradable hydrogels, whereas MSCs in 3D non-degradable hydrogels are round and commit to an adipogenic phenotype.[56]
MSCs encapsulated in covalently crosslinked hydrogels can spread if the hydrogel contains protease-degradable components. Gelatin is enzymatically degradable and cells encapsulated in gelatin hydrogels spread,[60] while MSCs encapsulated in HA hydrogels do not spread in vitro cultures in the absence of hyaluronidase. Thus, the balance between stable HA and enzymatically degradable gelatin can be leveraged to modulate the extent of 3D spreading-mediated differentiation within injectable Nor Tet hydrogels. Stable Nor-Tet hydrogels were formed by mixing HANor, HATet, and MSCs. Stiffness-matched (4.6 kPa E) Degradable Nor-Tet hydrogels were formed by mixing enzymatically degradable GelNor with HATet and MSCs. One- and seven-days post-encapsulation, the morphology of MSCs (Volume, Sphericity) were determined for Stable and Degradable Nor-Tet groups. As expected, MSCs in Stable Nor-Tet hydrogels remained small (Volume 5,198 ± 700 μm3 on Day 1 and 5,862 ± 686 μm3 on Day 7) and round (Sphericity 0.65 ± 0.09 on Day 1 and 0.65 ± 0.08 on Day 7) (Figure 9a). MSCs in Degradable Nor-Tet hydrogels had comparable volume (5,947 ± 849 μm3) and sphericity (0.65 ± 0.09) after 1 day in culture, however, after 7 days there was an almost two-fold increase in volume (10,275 ± 758 μm3) and a significant drop in sphericity (0.27 ± 0.05) (Figure 9a). Representative images of MSCs in Stable hydrogels show that they remain round for at least 7 days (Figure 9b), whereas MSCs in Degradable hydrogels are larger and display branching protrusions on Day 7 (Figure 9c). These findings show that 3D cellular spreading in Nor-Tet hydrogels can be induced simply by substituting HANor with enzymatically degradable GelNor macromer.
Figure 9.
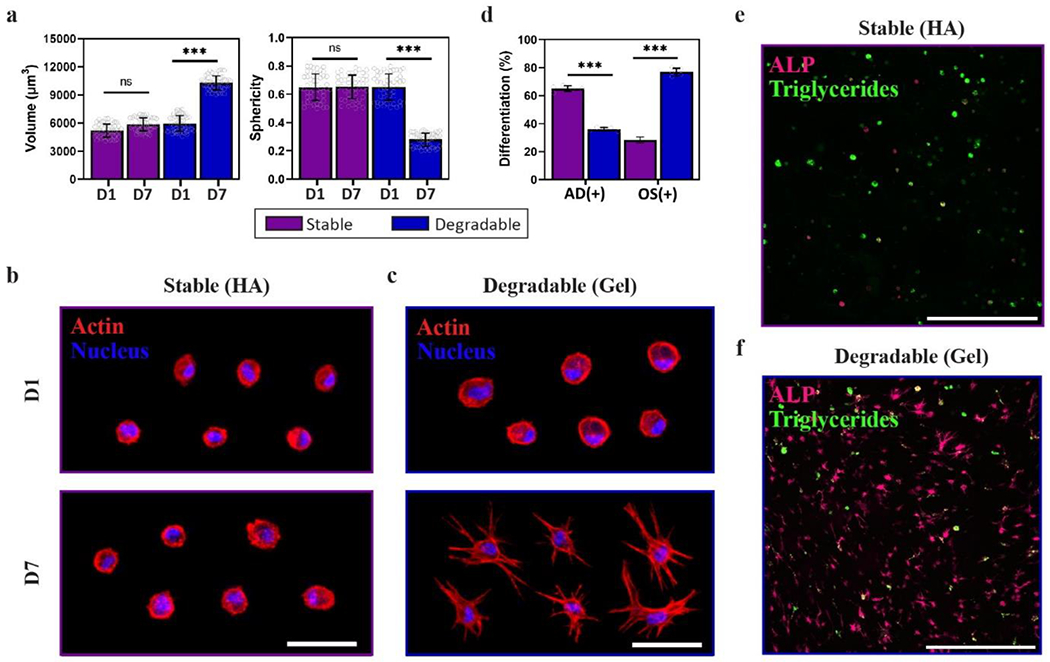
MSC spreading and differentiation can be controlled in 3D Nor-Tet hydrogels. (a) Quantification of MSC Volume (left) and Sphericity (right) of MSCs in Stable or Degradable Nor-Tet hydrogels after 1 and 7 days in culture. Representative images of MSCs stained for actin (red) and nuclei (blue) after 1 and 7 days in culture inside (b) Stable or (c) Degradable Nor-Tet hydrogels. (d) Quantification of percentage of AD(+) and OS(+) cells in Stable HA and Degradable Gel Nor-Tet hydrogels after 7 days in culture. Representative images of cells stained for ALP (magenta) and lipid droplets (green) in (e) Stable or (f) Degradable Nor-Tet hydrogels after 7 days in culture. Bar graphs shown as mean ± SD (n ≥ 3 samples per condition) with significant differences determined with ANOVA followed by Tukey’s post hoc test where ***p < 0.001 and ns is not significant. Scale bars: d 50 μm; e 500 μm.
After demonstrating that encapsulated MSCs in Nor-Tet hydrogels can take on spread or round morphologies depending on the polymer backbone used, the effects of 3D cellular spreading on MSC lineage commitment were investigated. MSCs were encapsulated in Stable or Degradable Nor-Tet hydrogels, cultured in AD/OS Medium for seven days, and co-stained with alkaline phosphatase (ALP, OS biomarker) and intracellular triglycerides (AD biomarker) to identify OS(+) and AD(+) cells, respectively. After seven days in culture, 65 ± 2% of cells in Stable HA hydrogels are AD(+) which is over a 1.8-fold increase compared to cells in Degradable gelatin hydrogels (36 ± 1%) (Figure 9d). In contrast, only 28 ± 2% of cells in Stable HA hydrogels are OS(+) while 77 ± 3% of cells in Degradable gelatin hydrogels are OS(+) (Figure 9d). Representative image of cells in Stable HA Nor-Tet hydrogels co-stained with ALP and lipid droplets show that cells retain a spherical morphology and most cells are AD(+) (Figure 9e). In contrast, representative image of cells in Degradable Gel Nor-Tet hydrogels are highly spread and most cells display intracellular ALP (Figure 9f). These findings show that the Nor-Tet hydrogels presented are biocompatible and can regulate 3D cellular spreading and downstream differentiation by incorporating enzymatically degradable GelNor in lieu of HANor.
3. Conclusion
In this study, biocompatible Nor-Tet hydrogels are developed with tunable physical and biochemical properties. Mechanical parameters of these self-forming hydrogels are controlled by varying macromer concentration and stoichiometric ratios between Nor and Tet moieties. By modifying Nor containing macromers with thiolated peptides, bioactivity is incorporated independent of stiffness or gelation time. MSCs on 2D RGD-functionalized Nor-Tet hydrogels adhere, proliferate, and are mechanically sensitive to changes in hydrogel stiffness. MSCs in 3D Nor-Tet hydrogel solutions are more viable than MSCs injected in liquid, and 3D cellular spreading-mediated differentiation is controlled by simply substituting stable HANor with enzymatically degradable GelNor. These self-forming Nor-Tet hydrogels feature a wide range of physicochemical properties and have broad applicability in fundamental and translational research.
4. Experimental Section
HANor and HATet macromer synthesis:
To synthesize HANor, carboxyl groups in HA were modified with norbornene (Nor) as previously described.[61] Briefly, sodium hyaluronate (NaHA, Lifecore, 60 kDa) was converted to its tetrabutylammonium salt (HATBA) by dissolving in distilled water (2% w/v) and mixing with Dowex resin for two hours at room temperature. The resin was then vacuum filtered, and the pH was adjusted to 7.02 using tetrabutylammonium hydroxide (TBA-OH, ~2.4 mmol per g of HA) diluted in water (1:1 v/v). The resulting HATBA solution was then frozen and lyophilized. Carboxyl groups in HATBA were then modified with Nor via amidation with 5-norbornene-2-methylamine (Nor-NH2, 0.4 mmol per gram of dry HATBA) in anhydrous dimethyl sulfoxide (DMSO, 2% w/v) and benzotriazole-1-yl-oxy-tris-(dimethylamino)-phosphonium hexafluorophosphate (BOP) under nitrogen for two hours at room temperature. The reaction was quenched with cold distilled water, dialyzed (SpectraPor, 6-8 kDa molecular weight cutoff), frozen, and lyophilized. The synthesized HANor macromer had ~50% of its repeat units functionalized with Nor, as analyzed with 1H NMR spectroscopy (Figure S3a, Supporting Information). The percentage of modification was calculated by comparing the integral of the methyl HA peaks between δ 1.8-2.0 ppm to the vinyl proton peaks of norbornene between δ 6.2-6.3 ppm.[62]
To synthesize HATet macromers, carboxyl groups in HA were modified with tetrazine (Tet) using a modified procedure described by Desai and coworkers.[63] Briefly, HATBA was dissolved (1% w/v) in 100 mM β-(N-morpholino)ethanesulfonic acid (MES) buffer (pH 6) and 1-(3-dimethylaminopropyl)-3-ethylcarbodiimide (EDC), N-hydroxysuccinimide (NHS), and tetrazine-amine (Tet-NH2) were added at a 1:4:1 molar ratio at 0.5 mmol Tet per gram of HATBA and reacted overnight at room temperature. The HATet solution was then dialyzed (SpectraPor, 6-8 kDa molecular weight cutoff), frozen, and lyophilized. 1H NMR spectroscopy analysis confirmed that ~40% of HATet repeat units were functionalized with Tet (Figure S3b, Supporting Information). The percentage of modification was calculated by comparing the integral of the methyl HA peaks between δ 1.8-2.0 ppm to the aromatic proton peaks of tetrazine between δ10.0-10.5 ppm and between δ 7.0-8.5 ppm.[31]
GelNor macromer synthesis:
To synthesize GelNor macromers, carboxyl groups in gelatin were modified with Nor using a procedure previously described by Koshy and coworkers.[31] Briefly, gelatin (Type A, 300 bloom) was dissolved (1% w/v) in 100 mM β-(N-morpholino)ethanesulfonic acid (MES) buffer (pH 6) and 1-(3-dimethylaminopropyl)-3-ethylcarbodiimide (EDC), N-hydroxysuccinimide (NHS), and Nor-NH2 was added at a 2:1:1 molar ratio at 2 mmol Nor-NH2 per gram of gelatin and reacted for 4 hours at 37 °C. The GelNor solution was then dialyzed (SpectraPor, 6-8 kDa molecular weight cutoff), frozen, and lyophilized. 1H NMR spectroscopy analysis confirmed that ~15% of GelNor repeat units were functionalized with Nor (Figure S11, Supporting Information). The percentage of modification was calculated by comparing the integral of the aromatic amino acid peaks between δ 7.0-7.5 ppm to the vinyl proton peaks of norbornene between δ 6.0-6.5 ppm.[31]
HANor(cRGD+) synthesis:
To synthesize HANor(cRGD+) macromers, hydroxyl groups in HANor were first modified with methacrylates (Me) to form HANorMe via esterification with methacrylic anhydride (MA) by adapting a previously described protocol.[37] HANor was dissolved (1% w/v) in distilled water at 4 °C. A 15-fold molar excess of MA was added dropwise while maintaining pH between 8.5-9.0. After all MA was added, the solution was left stirring overnight at room temperature. The HANorMe solution was then dialyzed (SpectraPor, 6-8 kDa molecular weight cutoff), frozen, and lyophilized. 1H NMR spectroscopy analysis confirmed that ~78% of HANor repeat units were functionalized with Me. The percentage of Me modification (amount of hydroxyl groups in HA coupled with Me) was calculated by comparing the integral of the HA peaks between δ 1.8-2.0 ppm to the alkene proton peaks of methacrylates between δ 5.5-6.5 ppm.[64]
To peptide-functionalize HANorMe, thiols in cysteine-containing peptides were coupled to Me groups in HANorMe via an aqueous Michael addition reaction.[65] Briefly, HANorMe was dissolved (1% w/v) in 200 mM triethanolamine (TEOA) buffer (pH 8) at room temperature. Thiolated RGD peptide (sequence: GCGYGRGDSPG, cRGD) in solution (50 mM in PBS) was added dropwise to reach a 2 mM effective cRGD concentration in 2%, 4%, and 6% w/v Nor-Tet hydrogels (parameters in Table S2, Supporting Information). The HANor(cRGD+) solution was then dialyzed (SpectraPor, 6-8 kDa molecular weight cutoff), frozen, and lyophilized. 1H NMR spectroscopy analysis confirmed that 17%, 9%, and 6% of Me groups were coupled with cRGD for 2%, 4%, and 6% w/v HANor(cRGD+) macromers (Figure S7, Supporting Information). The percentage of Me-cRGD coupling was calculated by comparing integral of the alkene proton peaks of methacrylate between δ 5.5-6.5 ppm[64] to the β and γ ethyl protons of the arginine moiety between δ 1.50-2.00 ppm.[66]
Preparation and characterization of Nor-Tet hydrogels:
Rheological properties of Nor-Tet hydrogels were measured using a Discovery Hybrid Rheometer (DHR-3, TA Instruments) with a 20 mm diameter 1° cone upper plate geometry and the lower plate was heated to a physiological temperature of 37 °C. Samples were prepared by dissolving HANor and HATet separately in PBS, followed by mixing, resulting in 2%, 4%, or 6% final w/v. Immediately after mixing, 40 μl of the solution was pipetted to the center of the rheometer stage and the shear storage (G´) and loss (G″) moduli were monitored. To confirm that click crosslinking between Nor and Tet moieties creates stable hydrogels, frequency sweeps were performed at 1% oscillatory strain while varying the frequency from 0.01 to 10 Hz. To measure plateau G′ and gelation kinetics, time sweeps were performed for 1 hour at 1 Hz and 1% oscillatory strain. Bulk mechanical properties of self-forming hydrogels were measured using a Shimadzu EZ-SX Mechanical Tester equipped with a 50 N compression load. Briefly, hydrated HANor and HATet were mixed and pipetted into cylindrical silicone molds (2 mm height, 8 mm diameter). The hydrogels were allowed to form for 30 minutes before soaking in PBS overnight at 37 °C. Formed cylindrical Nor-Tet hydrogels were compressed until 30% strain, and the elastic modulus was determined using the slope of the stress-strain curve between 10% and 20% strain.
2D cell attachment and proliferation on Nor-Tet hydrogels:
Thin hydrogels were formed by dissolving HANor (with or without cRGD functionalization) and HATet separately in Growth Medium (α-Minimum Essential Medium (αMEM) supplemented with 10% fetal bovine serum (FBS, Lonza)), followed by mixing, resulting in Soft (2% w/v) and Stiff (6% w/v) hydrogel solutions. Immediately after mixing, 70 μl of the solution was pipetted into cylindrical polydimethylsiloxane (PDMS) molds (11 mm diameter, 1 mm height). The thin hydrogels were allowed to crosslink for 2 minutes followed by gently adding Growth Medium to prevent hydrogels from drying out as they continue to polymerize. To evaluate cell adhesion and proliferation, human primary MSCs (from bone marrow, Lonza) were expanded in 100 mm petri dishes in Growth Medium. MSCs were seeded on top of 2D Nor-Tet hydrogels at a density of 3,000 cells per cm2. To evaluate cell attachment, cells were fixed after 3 days in culture using 10% neutral buffered formalin for 10 minutes at room temperature. Samples were washed twice with PBS after fixation and kept at 4 °C until immunostaining was performed. To evaluate cell proliferation, culture media was removed and replaced with alamarBlue working solution (Growth Medium supplemented with 10% alamarBlue Reagent, Invitrogen) after 1, 3, and 7 days in culture, and kept in the cell incubator for 4 h. Samples of the media were collected (50 μl in triplicate) and fluorescence was measured with a plate reader (560 nm excitation, 590 nm emission).
3D cell encapsulation in Nor-Tet hydrogels:
For cell encapsulation, cells were resuspended in RGD-functionalized HANor dissolved in Growth Medium at a density of 106 cells per mL. HATet dissolved in Growth Medium was mixed, resulting in a 2% w/v hydrogel solution, which was injected into a PDMS mold (8 mm diameter, 2 mm height). The hydrogels were allowed to crosslink for 2 minutes followed by gently adding Growth Medium to prevent hydrogels from drying out as they continue to polymerize. To evaluate cell viability, culture media was removed and replaced with Live/Dead Viability working solution (Growth Medium supplemented with 1:1,000 calcein AM and 1:1,000 ethidium homodimer, Invitrogen) and kept in a cell incubator for 30 minutes prior to confocal imaging and imaging-based analysis.
Immunostaining and imaging-based analysis:
After 3 days in culture, MSC-laden hydrogels were fixed in 10% formalin for 10 minutes at room temperature. Samples were then permeabilized with 0.1% Triton X-100 for 2 minutes and blocked with 3% bovine serum albumin in PBS for 30 minutes. Primary YAP or pFAK antibody (Santa Cruz Biotechnologies, 1:200) was added for 1 hour, followed by Alexa Fluor 488 secondary antibody (Life Technologies, 1:200) for 2 hours at room temperature. To visualize individual cells and nuclei, samples were stained for actin (Alexa Flour 568 phalloidin, 20 minutes, 1:100) and double stranded DNA (Hoechst, 5 minutes, 1:1,000), respectively. Confocal imaging was performed with a Nikon A1 confocal microscope.
Imaging-based cell analysis was performed using ImageJ software (National Institutes of Health). For 2D morphology analysis, the wand tracing tool was used to select cell outlines from the actin channel and the measure function was used to calculate cell area, circularity, and aspect ratio. For 2D YAP analysis, nuclear YAP localization was determined by first measuring the integrated density of YAP of five regions of interest (ROI) on the cytoplasm and nucleus of each cell, respectively. The ratio between the nuclear and cytosolic integrated densities was defined as the nuclear YAP value. To evaluate focal adhesion maturation, the number of pFAK adhesions per cell were counted using the Find Maxima feature, and at least 10 focal adhesion lengths were measured per cell. Actin anisotropy was quantified by determining the common directionality of actin fibers within manually-defined ROIs (at least three ROIs surrounding the cell nucleus) using the FibrilTool plugin.[67]
For 3D morphology analysis, z-stacks of the actin channel were binarized using the Otsu thresholding method. Cell volume and surface area were determined using the 3D Objects Counter feature, and these values were used to calculate sphericity as previously reported.[40] For 3D YAP analysis, actin cytoskeleton and nucleus z-stacks were binarized using the Otsu thresholding method. These cellular and nuclear ROIs were then superimposed with the YAP channel to obtain 3D YAP stacks of the cytoplasmic and nuclear space. The integrated density of YAP was then quantified using the 3D Objects Counter feature and the ratio between the nuclear and cytoplasmic YAP intensity was reported as the nuclear YAP ratio.
MSC syringe needle flow and encapsulation viability study:
MSCs were resuspended at a cell density of 106 cells per mL in either Growth Medium or an RGD-functionalized Nor-Tet hydrogel solution. The suspensions were loaded into a 1 mL syringe with an appropriate gauge needle and ejected onto a sterile glass coverslip (for cells suspended in Growth Medium) or into PDMS molds (for cells suspended in hydrogel solution) at a volumetric flow rate of 3,000 μL per min. The hydrogel groups were allowed to crosslink for 2 minutes followed by gently adding Growth Medium to prevent hydrogels from drying out as they continue to polymerize. To evaluate cell viability, culture media was removed and replaced with Live/Dead Viability working solution (Growth Medium supplemented with 1:1,000 Calcein AM and 1:1,000 ethidium homodimer, Invitrogen) and kept in a cell incubator for 30 minutes prior to confocal imaging. For cells ejected onto glass coverslips, three regions on every glass coverslip were imaged. For cells suspended in hydrogel solution, three 100-μm z-stacks were imaged across each hydrogel and a maximum z-projection was attained using ImageJ software to create 2-dimensional images. To quantify viability, the number of cells stained green (live) or red (dead) were determined for every image. The percentage of live cells on every image was then determined by calculating the ratio of live cells to the total count of live and dead cells.
MSC differentiation study:
MSCs were suspended in RGD-functionalized HANor (Stable) or GelNor (Degradable) dissolved in Growth Medium at a density of 106 cells per mL. To maintain GelNor in liquid phase, GelNor solutions were maintained at 37 °C prior to and during mixing with HATet. HATet dissolved in Growth Medium was then mixed with HANor or GelNor solution (2% w/v total macromer concentration), which was injected into a PDMS mold (8 mm diameter, 2 mm height). The hydrogels were allowed to crosslink for 2 minutes followed by washes in Growth Medium. MSC-laden hydrogels were cultured in either Growth Medium, OS Medium (hMSC Osteogenic Differentiation BulletKit™ Medium, Lonza), AD Medium (hMSC Adipogenic Differentiation BulletKit™ Medium, Lonza) or bipotential AD/OS Medium (1:1 OS Medium and AD Medium) for 7 days. To evaluate cell differentiation, cell-laden hydrogels were fixed with 10% neutral buffered formalin for 30 minutes. Fixed samples were co-stained with BODIPY (1:2500 dilution) to visualize triglycerides and Vector® Blue Alkaline Phosphatase Substrate prepared according to manufacturer specifications to visualize ALP. Samples were counterstained with Hoescht to visualize nuclei, and the percentage of OS(+) and AD(+) cells was determined by counting the number of cells that stained positive for ALP and BODIPY, respectively.
Statistics:
The statistical analysis was performed using GraphPad Prism 9.3.1 software. All experiments were carried out in triplicates and single cell analysis was done with at least 50 cells per group. All graphs represent mean ± standard deviation (SD). For comparisons of three or more groups: normally distributed populations were analyzed via analysis of variance (ANOVA) with a Tukey’s post hoc test to correct for multiple comparisons. Differences among groups are stated as p < 0.05 (*), p < 0.01 (**), p < 0.001 (***), and stated as (ns) when differences between groups are not statistically significant.
Supplementary Material
Acknowledgements
This work was supported by grants from the National Institutes of Health (R21DC018818) and the Camden Health Research Initiative.
Footnotes
Supporting Information
Supporting Information is available from the Wiley Online Library or from the author.
Contributor Information
Kirstene A. Gultian, Department of Biomedical Engineering, Rowan University, Glassboro NJ 08028, USA
Roshni Gandhi, Department of Biomedical Engineering, Rowan University, Glassboro NJ 08028, USA.
Tae Won B. Kim, Department of Orthopaedic Surgery, Cooper Medical School of Rowan University, Camden NJ 08103
Sebastián L. Vega, Department of Biomedical Engineering, Rowan University, Glassboro NJ 08028, USA
References
- [1].Hoffman AS, Adv. Drug Deliv. Rev 2012, 64, 18. [Google Scholar]
- [2].Lee KY, Mooney DJ, Chem. Rev 2001, 101, 1869. [DOI] [PubMed] [Google Scholar]
- [3].Drury JL, Mooney DJ, Biomaterials 2003, 24, 4337. [DOI] [PubMed] [Google Scholar]
- [4].Hoare TR, Kohane DS, Polymer (Guildf). 2008, 49, 1993. [Google Scholar]
- [5].Annabi N, Tamayol A, Uquillas JA, Akbari M, Bertassoni LE, Cha C, Camci-Unal G, Dokmeci MR, Peppas NA, Khademhosseini A, Adv. Mater 2014, 26, 85. [DOI] [PMC free article] [PubMed] [Google Scholar]
- [6].Nguyen KT, West JL, Biomaterials 2002, 23, 4307. [DOI] [PubMed] [Google Scholar]
- [7].April Kloxin BM, Kloxin CJ, Bowman CN, Anseth KS, Kloxin AM, Kloxin CJ, Bowman CN, Anseth KS, Adv. Mater 2010, 22, 3484. [DOI] [PMC free article] [PubMed] [Google Scholar]
- [8].Fairbanks BD, Schwartz MP, Halevi AE, Nuttelman CR, Bowman CN, Anseth KS, Adv. Mater 2009, 21, 5005. [DOI] [PMC free article] [PubMed] [Google Scholar]
- [9].Lin-Gibson S, Jones RL, Washburn NR, Horkay F, Macromolecules 2005, 38, 2897. [Google Scholar]
- [10].Gramlich WM, Kim IL, Burdick JA, Biomaterials 2013, 34, 9803. [DOI] [PMC free article] [PubMed] [Google Scholar]
- [11].Yang J-A, Yeom J, Woo Hwang B, Hoffman AS, Hahn SK, Prog. Polym. Sci 2014, 39, 1973. [Google Scholar]
- [12].Liu ZQ, Wei Z, Zhu XL, Huang GY, Xu F, Yang JH, Osada Y, Zrínyi M, Li JH, Chen YM, Colloids Surfaces B Biointerfaces 2015, 128, 140. [DOI] [PubMed] [Google Scholar]
- [13].Hiemstra C, Van Der Aa LJ, Zhong Z, Dijkstra PJ, Feijen J, Macromolecules 2007, 40, 1165. [Google Scholar]
- [14].Guvendiren M, Burdick JA, Nat. Commun 2012, 3, 792. [DOI] [PubMed] [Google Scholar]
- [15].Drury JL, Dennis RG, Mooney DJ, Biomaterials 2004, 25, 3187. [DOI] [PubMed] [Google Scholar]
- [16].Bidarra SJ, Barrias CC, Granja PL, Acta Biomater. 2014, 10, 1646. [DOI] [PubMed] [Google Scholar]
- [17].Klouda L, Mikos AG, Eur. J. Pharm. Biopharm 2008, 68, 34. [DOI] [PMC free article] [PubMed] [Google Scholar]
- [18].Peppas NA, Bures P, Leobandung W, Ichikawa H, Eur. J. Pharm. Biopharm 2000, 50, 27. [DOI] [PubMed] [Google Scholar]
- [19].Sala RL, Kwon MY, Kim M, Gullbrand SE, Henning EA, Mauck RL, Camargo ER, Burdick JA, Tissue Eng. Part A 2017, 23, 935. [DOI] [PMC free article] [PubMed] [Google Scholar]
- [20].Bohidar HB, Jena SS, J. Chem. Phys 1998, 98, 8970. [Google Scholar]
- [21].Seuring J, Agarwal S, ACS Macro Lett. 2013, 2, 597. [DOI] [PubMed] [Google Scholar]
- [22].Boustta M, Colombo PE, Lenglet S, Poujol S, Vert M, J. Control. Release 2014, 174, 1. [DOI] [PubMed] [Google Scholar]
- [23].Tietze LF, Kettschau G, Springer, Berlin, Heidelberg, 1997, pp. 1–120. [Google Scholar]
- [24].Rideout DC, Breslow R, J. Am. Chem. Soc 2002, 102, 7816. [Google Scholar]
- [25].Nimmo CM, Owen SC, Shoichet MS, Biomacromolecules 2011, 12, 824. [DOI] [PubMed] [Google Scholar]
- [26].Smith LJ, Taimoory SM, Tam RY, Baker AEG, Binth Mohammad N, Trant JF, Shoichet MS, Biomacromolecules 2018, 19, 926. [DOI] [PubMed] [Google Scholar]
- [27].Kirchhof S, Brandl FP, Hammer N, Goepferich AM, J. Mater. Chem. B 2013, 1, 4855. [DOI] [PubMed] [Google Scholar]
- [28].Madl CM, Heilshorn SC, Chem. Mater 2019, 31, 8035. [DOI] [PMC free article] [PubMed] [Google Scholar]
- [29].Alge DL, Azagarsamy MA, Donohue DF, Anseth KS, Biomacromolecules 2013, 14, 949. [DOI] [PMC free article] [PubMed] [Google Scholar]
- [30].Desai RM, Koshy ST, Hilderbrand SA, Mooney DJ, Joshi NS, Biomaterials 2015, 50, 30. [DOI] [PubMed] [Google Scholar]
- [31].Koshy ST, Desai RM, Joly P, Li J, Bagrodia RK, Lewin SA, Joshi NS, Mooney DJ, Adv. Healthc. Mater 2016, 5, 541. [DOI] [PMC free article] [PubMed] [Google Scholar]
- [32].Guo L, Colby RH, Lusignan CP, Howe AM, Macromolecules 2003, 36, 10009. [Google Scholar]
- [33].Miri AK, Khalilpour A, Cecen B, Maharjan S, Shin SR, Khademhosseini A, Biomaterials 2019, 198, 204. [DOI] [PMC free article] [PubMed] [Google Scholar]
- [34].Hölzl K, Lin S, Tytgat L, Van Vlierberghe S, Gu L, Ovsianikov A, Biofabrication 2016, 8, 1. [DOI] [PubMed] [Google Scholar]
- [35].Wüst S, Godla ME, Müller R, Hofmann S, Acta Biomater. 2014, 10, 630. [DOI] [PubMed] [Google Scholar]
- [36].Patenaude M, Smeets NMB, Hoare T, Macromol. Rapid Commun 2014, 35, 598. [DOI] [PubMed] [Google Scholar]
- [37].Smeds KA, Grinstaff MW, J. Biomed. Mater. Res 2001, 54, 115. [DOI] [PubMed] [Google Scholar]
- [38].Nair DP, Podgórski MP, Chatani S, Gong T, Xi W, Fenoli CR, Bowman CN, Chem. Mater 2013, DOI 10.1021/cm402180t. [DOI] [Google Scholar]
- [39].Gultian KA, Gandhi R, Sarin K, Sladkova-Faure M, Zimmer M, de Peppo GM, Vega SL, Sci. Reports 2022 121 2022, 12, 1. [DOI] [PMC free article] [PubMed] [Google Scholar]
- [40].Caliari SR, Vega SL, Kwon M, Soulas EM, Burdick JA, Biomaterials 2016, 103, 314. [DOI] [PMC free article] [PubMed] [Google Scholar]
- [41].Holloway JL, Ma H, Rai R, Hankenson KD, Burdick JA, Macromol. Biosci 2015, 15, 1218. [DOI] [PMC free article] [PubMed] [Google Scholar]
- [42].Burdick JA, Prestwich GD, Adv. Mater 2011, 23, 41. [DOI] [PMC free article] [PubMed] [Google Scholar]
- [43].Glass JR, Dickerson KT, Stecker K, Polarek JW, Biomaterials 1996, 17, 1101. [DOI] [PubMed] [Google Scholar]
- [44].Wells RG, Hepatology 2008, 47, 1394. [DOI] [PubMed] [Google Scholar]
- [45].Guilak F, Cohen DM, Estes BT, Gimble JM, Liedtke W, Chen CS, Cell Stem Cell 2009, 5, 17. [DOI] [PMC free article] [PubMed] [Google Scholar]
- [46].Engler AJ, Sen S, Sweeney HL, Discher DE, Cell 2006, 126, 677. [DOI] [PubMed] [Google Scholar]
- [47].Provenzano PP, Keely PJ, J. Cell Sci 2011, 124, 1195. [DOI] [PMC free article] [PubMed] [Google Scholar]
- [48].Pasapera AM, Plotnikov SV, Fischer RS, Case LB, Egelhoff TT, Waterman CM, Curr. Biol 2015, 25, 175. [DOI] [PMC free article] [PubMed] [Google Scholar]
- [49].McBeath R, Pirone DM, Nelson CM, Bhadriraju K, Chen CS, Dev. Cell 2004, 6, 483. [DOI] [PubMed] [Google Scholar]
- [50].Dupont S, Morsut L, Aragona M, Enzo E, Giulitti S, Cordenonsi M, Zanconato F, Le Digabel J, Forcato M, Bicciato S, et al. , Nature 2011, 474, 179. [DOI] [PubMed] [Google Scholar]
- [51].Rosales AM, Vega SLSL, DelRio FW, Burdick JA, Anseth KS, 2017, 129, 12300. [Google Scholar]
- [52].Geiger B, Spatz JP, Bershadsky AD, Nat. Rev. Mol. Cell Biol 2009, 10, 21. [DOI] [PubMed] [Google Scholar]
- [53].Chen CS, Tan J, Tien J, Annu. Rev. Biomed. Eng 2004, 6, 275. [DOI] [PubMed] [Google Scholar]
- [54].Aguado BA, Mulyasasmita W, Su J, Lampe KJ, Heilshorn SC, Tissue Eng. - Part A 2012, 18, 806. [DOI] [PMC free article] [PubMed] [Google Scholar]
- [55].Cai L, Dewi RE, Heilshorn SC, Adv. Funct. Mater 2015, 25, 1344. [DOI] [PMC free article] [PubMed] [Google Scholar]
- [56].Khetan S, Guvendiren M, Legant WR, Cohen DM, Chen CS, Burdick JA, Nat. Mater 2013, 12, 458. [DOI] [PMC free article] [PubMed] [Google Scholar]
- [57].Discher DE, Janmey P, Wang Y-L, Science 2005, 310, 1139. [DOI] [PubMed] [Google Scholar]
- [58].Chaudhuri O, Gu L, Klumpers D, Darnell M, Bencherif SA, Weaver JC, Huebsch N, Lee H, Lippens E, Duda GN, et al. , Nat. Mater 2016, 15, 326. [DOI] [PMC free article] [PubMed] [Google Scholar]
- [59].Das RK, Gocheva V, Hammink R, Zouani OF, Rowan AE, Nat. Mater 2016, 15, 318. [DOI] [PubMed] [Google Scholar]
- [60].Yue K, Trujillo-de Santiago G, Alvarez MM, Tamayol A, Annabi N, Khademhosseini A, Biomaterials 2015, 73, 254. [DOI] [PMC free article] [PubMed] [Google Scholar]
- [61].Vega SL, Kwon MY, Song KH, Wang C, Mauck RL, Han L, Burdick JA, Nat. Commun 2018, 9, 614. [DOI] [PMC free article] [PubMed] [Google Scholar]
- [62].Miao W-K, Yi A, Yan Y-K, Ren L-J, Chen D, Wang C-H, Wang W, Polym. Chem 2015, 6, 7418. [Google Scholar]
- [63].Desai RM, Koshy ST, Hilderbrand SA, Mooney DJ, Joshi NS, Biomaterials 2015, 50, 30. [DOI] [PubMed] [Google Scholar]
- [64].Yousefi F, Kandel S, Pleshko N, Appl. Spectrosc 2018, 72, 1455. [DOI] [PubMed] [Google Scholar]
- [65].Li G-Z, Randev RK, Soeriyadi AH, Rees G, Boyer C, Tong Z, Davis TP, Remzi Becer C, Haddleton DM, Polym. Chem 2010, 1, 1196. [Google Scholar]
- [66].Vida Y, Collado D, Najera F, Claros S, Becerra J, Andrades JA, Perez-Inestrosa E, RSC Adv 2016, 6, 49839. [Google Scholar]
- [67].Boudaoud A, Burian A, Borowska-Wykręt D, Uyttewaal M, Wrzalik R, Kwiatkowska D, Hamant O, Nat. Protoc 2014, 9, 457. [DOI] [PubMed] [Google Scholar]
Associated Data
This section collects any data citations, data availability statements, or supplementary materials included in this article.