

Summary
Increased glucose metabolism and uptake are characteristic of many tumors and used clinically to diagnose and monitor cancer progression. In addition to cancer cells, the tumor microenvironment (TME) encompasses a wide range of stromal, innate, and adaptive immune cells. Cooperation and competition between these cell populations supports tumor proliferation, progression, metastasis, and immune evasion. Cellular heterogeneity leads to metabolic heterogeneity, as metabolic programs within the tumor are dependent not only on the TME cellular composition, but also on cell states, location, and nutrient availability. In addition to driving metabolic plasticity of cancer cells, altered nutrients and signals in the TME can lead to metabolic immune suppression of effector cells and promote regulatory immune cells. Here we discuss how metabolic programming of cells within the TME promotes tumor proliferation, progression, and metastasis. We also discuss how targeting metabolic heterogeneity may offer therapeutic opportunities to overcome immune suppression and augment immunotherapies.
Keywords: Metabolism, Tumor Microenvironment, Metastasis, Immune, Plasticity
eTOC blurb/In Brief summary
Arner and Rathmell discuss how metabolic programs within tumors depend not only on the tumor microenvironment (TME) cellular composition, but also on cell states, location, and nutrient availability. Altered nutrient availability and signals in the TME lead to metabolic immune suppression which drives immune evasion, tumor progression, and metastasis. Understanding and targeting metabolic heterogeneity within the TME may offer therapeutic opportunities to overcome immune suppression and augment immunotherapies.
1. Introduction
Tumors require an abundance of energy for malignant growth, proliferation, and metastasis. To sustain these processes, oncogenic signals drive diverse anabolic metabolic pathways including glycolysis, one-carbon metabolism, the tricarboxylic acid (TCA) cycle, and fatty acid synthesis that generate essential energetic, biosynthetic precursors, and signaling molecules. It is now apparent, however, that these same metabolic pathways are important not only for cancer cell growth but for proliferative cells in general, including both pro-tumorigenic and anti-tumorigenic immune cells1–3. This shared demand for similar nutrients results in a potentially competitive tumor microenvironment and immune suppression.
The tumor microenvironment (TME) is composed of many different cells that support or restrain tumorigenesis. Just as the TME includes a diversity of cells, it is metabolically and spatially heterogeneous (Figure 1). Cancer cells and resulting tumor progression are greatly affected by metabolic stress within various regions of the TME, in part through spatial heterogeneity across tumors as cell populations compete for limiting oxygen and nutrient supply in poorly vascularized or highly metabolically active areas. The TME is also often characterized by the accumulation of metabolic waste and an unfavorable pH and harsh environment, where nutrients are limited4,5. Because nutrients are also dependent on systemic and whole tissue factors, tumor type, location of the tumor within the primary tissue, and host diet and nutritional state will affect nutrient availability within the TME6. Additionally metabolic plasticity may occur, as cells within the TME adapt to utilize different nutrients depending on availability. Metabolic heterogeneity within the TME not only supports the proliferation of transformed cells, but also aids in metastatic disease, as multiple metabolic processes influence each step of the metastatic cascade, both within cancer cells and the local TME as cancer cells traverse and escape tumors.
Figure 1. Metabolic Heterogeneity in the Tumor Microenvironment is Location Dependent.
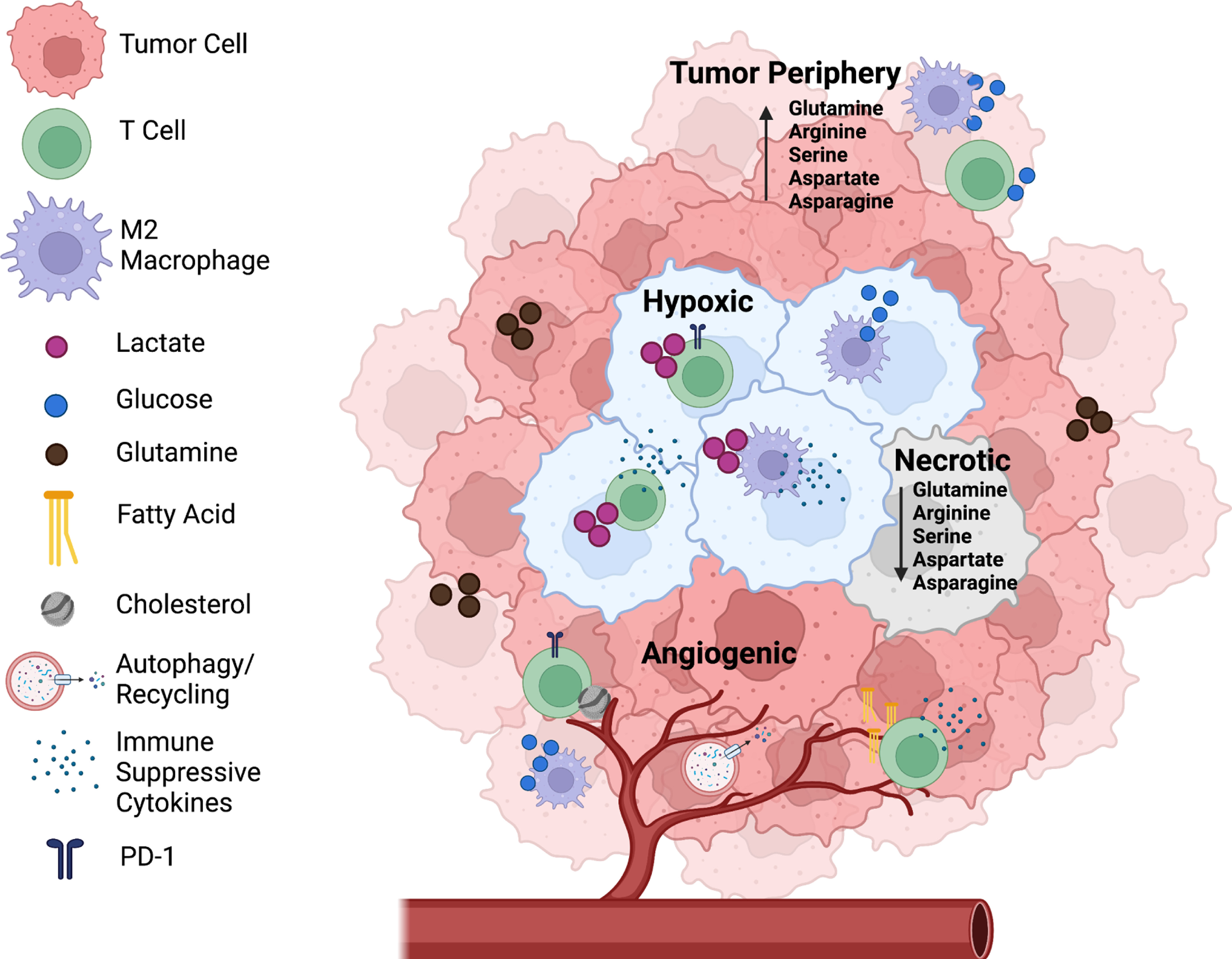
The tumor microenvironment (TME) is composed of many different cells that support or restrain tumorigenesis, including cancer cells and pro- and anti-tumor immune cells. Just as the TME has a diversity of cells, the TME is also metabolically heterogeneous. In hypoxic regions of the tumor (blue cancer cells), cancer cells often secrete lactate, which has been shown to inhibit effector T cell activation while promoting suppressive Tregs to drive immune suppression. Additionally, lactate promotes differentiation and polarization of TAMs towards a more pro-tumorigenic M2-like phenotype, which secrete immunosuppressive cytokines. In angiogenic tumors, blood vessels branch irregularly which leads to regions with increased autophagy to make up for the lack of delivered nutrients. Less angiogenic regions typically use glucose as their main source of energy. Necrotic tumor regions arise in part because of severe lack of nutrients and have been shown to have decreased levels of amino acids such as glutamine, arginine, asparagine, serine, and aspartate compared to tumor peripheries.
This complex landscape can be detrimental for treatment, as inhibitors that suppress the growth or metastasis of cancer cells can also inhibit or change the effectiveness of anti-tumor immune cells. Conversely, this opens new opportunities to specifically activate the immune cells in immunotherapy by interfering with their metabolic programs. Indeed, while current immunotherapies blocking PD-1 or CTLA-4 can activate anti-tumor T cells in many patients across multiple cancer types, treatments frequently fail, and some tumors become resistant after initial response. Mechanisms of resistance are not fully understood, but metabolic pathways are fundamentally involved in cell fate and cell program determinations7 and immunotherapies have been long appreciated to influence T cell metabolism8–11. Metabolic adaptations to the TME, however, may impede the effectiveness of immune checkpoint blockade in a “metabolic immune suppression” that impairs metabolic reprogramming necessary for effector function12–14. Improving immunotherapies and reducing resistance are of high priority, but it is essential to first understand how immune cells function and interact with cancer and other cells in the TME, in particular with regard to shared nutrient and resource utilization to overcome metabolic immune suppression in the TME.
The diverse metabolic programs of cancer and tumor associated immune cells may explain some failed immunotherapies and provide new avenues to prevent metastasis and limit immune suppression. Increased glucose uptake of tumors is the basis of 18F-2-deoxyglucose (18FDG) Positron Emission Tomography (PET) imaging that is widely used to diagnose and monitor progression of a wide range of tumor types. While these tests demonstrate high accumulation of glucose in tumors as bulk tissues, they do not resolve the metabolic activities and differences of the various individual cell types in the TME. Heterogeneity of cell metabolism across tumors and how nutrients and metabolic pathways influence immunotherapy remains poorly understood. Nevertheless, metabolic immune suppression of T cells leads to dysregulated and fragmented mitochondria12, increased production of reactive oxygen species (ROS)15, and reduced glycolysis14,16. Importantly, these metabolic changes directly impair T cells, as metabolic rescue can improve T cell effector activity and ability to control tumors12,17,18. Here we discuss how metabolic diversity in the TME may support metastasis and re-program the metabolism of immune cells within the TME to prevent their anti-tumor functions and how these changes open new therapeutic doors for cancer patients.
2. Tumor Metabolic heterogeneity
Proliferating cells require abundant and diverse nutrients to support bioenergetic demands, growth, and replication while maintaining necessary cell functions or migratory properties. The fuel for these processes comes from increased glycolysis, glutaminolysis, and lipolysis. Increased glycolysis that occurs in tumors despite the presence of oxygen is referred to as the “Warburg effect”, where glucose is converted to lactate via aerobic glycolysis19. Indeed, glucose uptake and lactate production are often high in tumor tissues, which can leave extracellular levels of glucose reduced and lactate elevated6,20. Many factors contribute to the rewiring of tumors to undergo aerobic glycolysis, including oncogenic signaling or loss of tumor suppressors such as VHL or p53, mitochondrial alterations, up-regulation of glycolytic enzymes, or hypoxia signaling21. These factors are coupled, as oncogenic drivers including PI3Kα or MYC directly drive cell-intrinsic growth factor signals that promote aerobic glycolysis. In addition, when hypoxia occurs during tumorigenesis or loss of VHL, the transcription factor HIF1α is stabilized, activating the transcription of several glycolytic transporters and enzymes, including, GLUT1, HK1, HK2, PKM2, PDK1, and LDHA22,23. HIF1α also reduces mitochondrial activity and ROS, which is typically generated from oxidative phosphorylation24.
Beyond aerobic glycolysis, anabolic metabolism requires a wide range of metabolic changes. The pentose phosphate pathway (PPP) is essential for generation of NADPH and ribose sugars within cancer cells, which are needed for nucleotide synthesis, ATP production, lipogenesis, and eliminating oxidative stress. Lipid and cholesterol are also utilized by cancer cells to support tumorigenesis, which can either be taken up in the cells from free fatty acids, or produced de novo by fatty acid synthesis and the mevalonate pathway25. Additionally, cancer cells often increase both the uptake and synthesis of glutamine and glutamate as alternative carbon sources for amino acid and nucleotide biosynthesis, and to feed the TCA cycle to produce ATP through anaplerosis26. The metabolism of other amino acids, such as arginine, tryptophan, glycine, and serine also play a key role in tumor metabolism and growth within the TME27–29, highlighting the range of metabolic pathways that play essential cancer cell intrinsic roles in tumorigenesis.
Pathways such as one-carbon metabolism, glycolysis, and the TCA cycle are important not only for cancer cell growth, but also for the function of both pro- and anti-tumorigenic immune cells1–3. The TME is made up of many different types of cells, including immune cells, such as B cells, T cells, neutrophils, natural killer (NK) cells, and macrophages, and stromal cells, such as fibroblasts, adipocytes, endothelial cells, and pericytes30. Simplistically, endothelial and stromal cells have been thought to aid tumor growth, whereas immune cells are largely considered to be inflammatory and anti-tumorigenic. Although generally true for cytotoxic T cells, different immune cell populations can have both anti-tumorigenic and pro-tumorigenic effects, which can change depending on temporal and context-dependent metabolic programs.
Cancer cells can shape the microenvironment to help support tumorigenesis and evade the immune system via suppression. One way in which this can occur is through shuttling of metabolic intermediates and nutrients between cancer cells and other cell types within the TME, often leading to metabolic heterogeneity, which can lead to immunosuppression as nutrients are not uniformly available31,32. There is also substantial crosstalk between other types of immune cells. For example, myeloid-derived suppressive cells (MDSCs) can inhibit cytotoxic T cells and NK cells while inducing regulatory T cells (Tregs) and regulatory B cells to drive an immunosuppressive TME33–35. In addition to immunosuppressive effects, tumor-associated macrophages (TAMs) may promote cancer proliferation, tumor expansion, invasion, and immune escape to drive tumorigenesis36,37. This crosstalk and heterogeneity provides a technical barrier for determining which metabolites are from the cancer cells or other cells within the TME and has limited our understanding of tumor metabolic heterogeneity. A key challenge in tumor metabolism and immunotherapy, therefore, is how to deal with the diversity of cell types and metabolic programs in the TME. types and metabolic programs in the TME.
3. Nutrient Availability within the TME
Location Dependency
Tumor type, location within the organ, and diet of the host will all affect nutrient availability for cells within the tumor microenvironment6. In hypoxic tumor regions, cancer cells secrete lactate that can block monocyte and dendritic cell differentiation and inhibit effector T cell activation while promoting suppressive Tregs to thus impede immunosurveillance38–41 (Figure 1). Additionally, lactate promotes differentiation and polarization of TAMs towards a more pro-tumorigenic, or M2-like, phenotype with increased levels of arginase-1 (ARG1) and mannose receptor C type 1 (CD206). These M2-like TAMs secrete immunosuppressive IL-10 and metabolites, such as polyamines, which promote cell division of cancer cells42. In hypoxic niches, TAMs acquire pro-angiogenic and pro-invasive phenotypes by altering their metabolism to adapt to the low oxygen environment43. The key nutrient and energy sensor, mTOR, regulates metabolic pathways such as glycolysis, de novo lipogenesis, protein synthesis, and transcription. In hypoxic states, mTOR complex 1 (mTORC1) function is inhibited by REDD144, thus leading to metabolically rewiring cancer cells to use other pathways, such as glutaminolysis and reductive carboxylation45–47.
Tumor angiogenesis often leads to blood vessels in solid tumors that branch irregularly and can be inefficient at delivering nutrients or removing waste. This exacerbates metabolic heterogeneity within the tumor and leads to regions or microenvironments with limited vascular exchange of nutrients and waste. While increased autophagy and lysosome activity via AMPK can partially compensate for the lack of extracellular nutrients48–51, these regions can experience extreme stress. Well-vascularized tumor areas in human non-small cell lung cancer (NSCLC) patients have been shown to use multiple metabolites whereas less angiogenic regions may be restricted in the diversity of nutrients available for metabolism52. Necrotic tumor regions may arise in part because of reduced blood supply and thus severe or prolonged lack of nutrients. In fact, the centers of tumors often have decreased levels of amino acids such as glutamine, arginine, asparagine, serine, and aspartate compared to tumor peripheries27. Interestingly, a recent study by Fu et al.53, observed that viable renal cell carcinoma (RCC) cells in necrotic regions had higher clonal diversity than the periphery of the tumor, suggesting that a harsh environment creates selective pressure for cancer cell survival53,54. It is possible that deprivation of nutrients within these necrotic regions and subsequent metabolic stress may provide surviving cancer cells with selective pressure to render them more fit for metastatic activity.
Different nutrients for different cells
Classically, cancer metabolism studies have used bulk tumor tissues to define basic pathways and potential vulnerabilities55. Based on the activity of oncogenic drivers that dysregulate cell metabolism, cancer cells were considered the dominant consumers of glucose and producers of lactate in the TME. This approach has been highly valuable but is complicated by increased recognition that tumors are comprised of many cell types beyond the cancer cells themselves. With limited vascular exchange in tumors, nutrients may become limiting and force cells to compete for access. T cells may thus be prevented from acquiring sufficient glucose if cancer cells capture and consume this nutrient16,56. However, a direct study of glucose uptake in the TME using 18F-2DG and PET-imaging based techniques revealed that across a variety of mouse cancer models, it was not cancer cells, but rather myeloid cells that demonstrated the biggest consumption of glucose, followed by T cells, then cancer cells. Instead, cancer cells showed the highest uptake of glutamine20. Blocking glutamine uptake increased glucose uptake in all cell types, demonstrating that glucose is not widely limited in the TME and can be accessible when cell demands shift. These data also suggest that glutamine rather than glucose uptake may be limiting, and cells adapt to lower glutamine with increased glucose metabolism. As such, targeting glutamine metabolism increases anti-tumor immunity in mouse models by increasing the mitochondrial metabolism of cytotoxic T cells57,58.
Because of interplay between available nutrients and metabolic activities, the metabolism of any given cell type in the TME may affect the metabolic features of other cells in close proximity. Consistent with this model, studies have shown that selectively targeting glutaminolysis in cancer cells can enhance T cell metabolism and anti-tumor immunity59. Similarly, increased uptake of methionine by cancer cells due to elevated levels of the methionine transporter SLC43A2 leads to decreased methionine availability for cytotoxic T cells, which can then result in reduced cytotoxic T cell function and suppression of effector T cells60. Tregs play a direct role in promoting immune evasion and tumorigenesis61,62. As Tregs are abundant even in the metabolically unfavorable TME, they have been shown to be dependent on mitochondrial oxidative metabolism of lipids to retain their immunosuppressive function63–66. It is unclear how Tregs metabolically differ to utilize mitochondrial oxidation for proliferation compared to cytotoxic T cells, however Tregs within the TME indirectly promote immunosuppressive TAMs by increasing SREBP1-dependent lipid metabolism and decreasing the CD8+ T-produced IFNγ67.
In macrophages, not only can oxygen availability influence metabolism, but activation by different cytokines in the TME can drive differing metabolic programs. For example, in vitro, pro-inflammatory macrophages (M1) switch their metabolism towards aerobic glycolysis, pentose phosphate pathway activation, and protein and fatty acid synthesis68. However, when macrophages are activated by cytokines that induce a pro-tumorigenic or M2-like phenotype, such as IL-4, IL-10, or IL-13, macrophages adapt to use oxidative phosphorylation to meet their metabolic demands69. Either phenotype may provide benefits in distinct regions of a given tumor. Comprehensively understanding these inter-relationships presents a new and unique opportunity to manipulate cellular metabolism, influencing numerous cell types simultaneously.
“Waste” Products
Cancer cells also suppress T cells through secreted “waste” products. Metabolic end products are secreted to allow continued flux, but rather than simple waste, they can signal or be used by neighboring cells. For example, the glycolytic end-product lactate can serve as a fuel for the TCA cycle in some cancers, such as NSCLC70. Additionally, while a certain level of lactate may enhance CD8+ T cell “stemness” and memory71, high lactate can directly inhibit effector T cells39. Production of IFNγ may be particularly sensitive to reduced glucose and increased lactate72,73. Increased lactate also promotes Tregs that utilize and promote mitochondrial oxidative pathways67,74,75 and suppress anti-tumor immunity40,41,76. Additionally, lactate promotes differentiation and polarization of TAMs towards an M2-like phenotype, which can secrete immunosuppressive or pro-tumor cytokines42.
It is important to point out that lactate may not be solely pro-tumorigenic, as lactate is associated with reduced pH within the TME which leads to oxidative stress77 and therefore may be anti-tumorigenic in some cancers. Interestingly, in contrast to cancer where lactate is reported to be immune suppressive in multiple cancer types, during acute exercise lactate is associated with an increase in CD8+ T cells78. This phenomenon suggests that the influence of lactate on T cell function may be contextually plastic. Perhaps in the hypoxic state of the TME, where constant stimulation leads to mitochondrial stress and T cell exhaustion15, lactate functions in an immunosuppressive manner. However, in a normoxic state with plentiful oxygen and normal T cell function, lactate can fuel the TCA cycle and support effector function. More studies are needed to fully understand this dynamic effect of lactate on immunometabolism.
Another example of waste products within the TME that affect the metabolism of other cells is nucleoside adenosine. Adenosine is generated by both cancer cells and Tregs through CD39 and CD73, which convert ATP to adenosine, which binds to adenosine 2A receptors (A2AR) on cytotoxic T cells and NK cells and inhibits antitumor immunity through the suppression of NFκB signaling79,80. As such, A2AR blockade is being tested as an immunotherapy for treatment resistant RCC81. Succinate is also secreted by cancer cells and can act as an epigenetic modifier in immune cells82. Succinate can be taken up or activate the succinate receptor, GPR91. Once activated, GPR91 can signal through the PI3K-HIF1α axis to polarize TAMs to M2-like leading to pro-tumorigenic functions such as immune suppression and increased metastasis83. The oncometabolite (R)-2-hydroxyglutaraate (R-2-HG), which is produced by isocitrate dehydrogenase mutations in some tumors such as glioblastoma, has been shown to be taken up by T cells where it reduces T cell transcriptional activity and polyamine biosynthesis, thus suppressing cytotoxic T cell function84. Lastly, cholesterol can accumulate in tumors to cause endoplasmic reticulum (ER) stress in T cells, ultimately preventing T cells from secreting anti-tumor effector cytokines. This ER stress response in T cells also upregulates XBP-1, which promotes the expression of the immunosuppressive molecules PD-1, TIM-3, and LAG-3 to promote T cell exhaustion85.
Deficient Nutrients within the TME
Within the TME, amino acids such as arginine, tryptophan, serine, cysteine, and alanine can be limited and may lead to competition between cancer cells and cytotoxic T cells, which both rely on these nutrients for proliferation6. Some cancer cells are dependent on extracellular arginine29 due to the lack of sufficient urea cycle enzyme Arginosuccinate Synthetase 1 that prevents cancer cells from synthesizing endogenous arginine86,87. This causes increased consumption of extracellular arginine by the cancer cells that ultimately leads to a reduction of available arginine for T cells29,79. Deficient arginine availability reduces T cell mTORC1 activity88 and results in an increase of memory-like T cells while T cell effector functions and immunosurveillance are reduced89. Pro-tumorigenic TAMs also consume L-arginine via Arginase-1, which further reduces available arginine for T cell consumption90.
Tryptophan is an essential amino acid only available from diet but that can also be depleted in the tumor due to competition within the TME. Cancer cells take up and catabolize the limited tryptophan within the TME, which produces kynurenine, a ligand of aryl hydrocarbon receptor, AHR91. The activation of AHR in CD4+ T cells results in differentiation into immunosuppressive Tregs92–94. Kynurenine can also induce the expression of PD-1 on CD8+ T cells, ultimately leading the suppression of cytotoxic function95 (Figure 2). Inhibition of tryptophan catabolism combined with checkpoint blockade is currently being tested in clinical trials96. Like tryptophan and arginine, some cancer cells consume copious amounts of serine. Serine is a non-essential amino acid that is converted to glycine and one-carbon units to build nucleotides and maintain mitochondria redox homeostasis28. When cancer cells take up the majority of extracellular serine, limited amounts are left for the T cells which have been shown to depend on extracellular serine for T cell expansion and effector functions97. Some tumors have been shown to import extracellular cysteine, which acts as an antioxidant and reduces ferroptosis, leading to tumor progression98. Cancer cell uptake of cysteine leads to limiting amounts of extracellular cysteine, which is required for T cell activation and function, and as such, cysteinase treatment in mice has been shown to synergize with cytotoxic T cell anti-tumor function in mice by enhancing ferroptosis99.
Figure 2. Dysregulated Cytotoxic T Cell Metabolism.
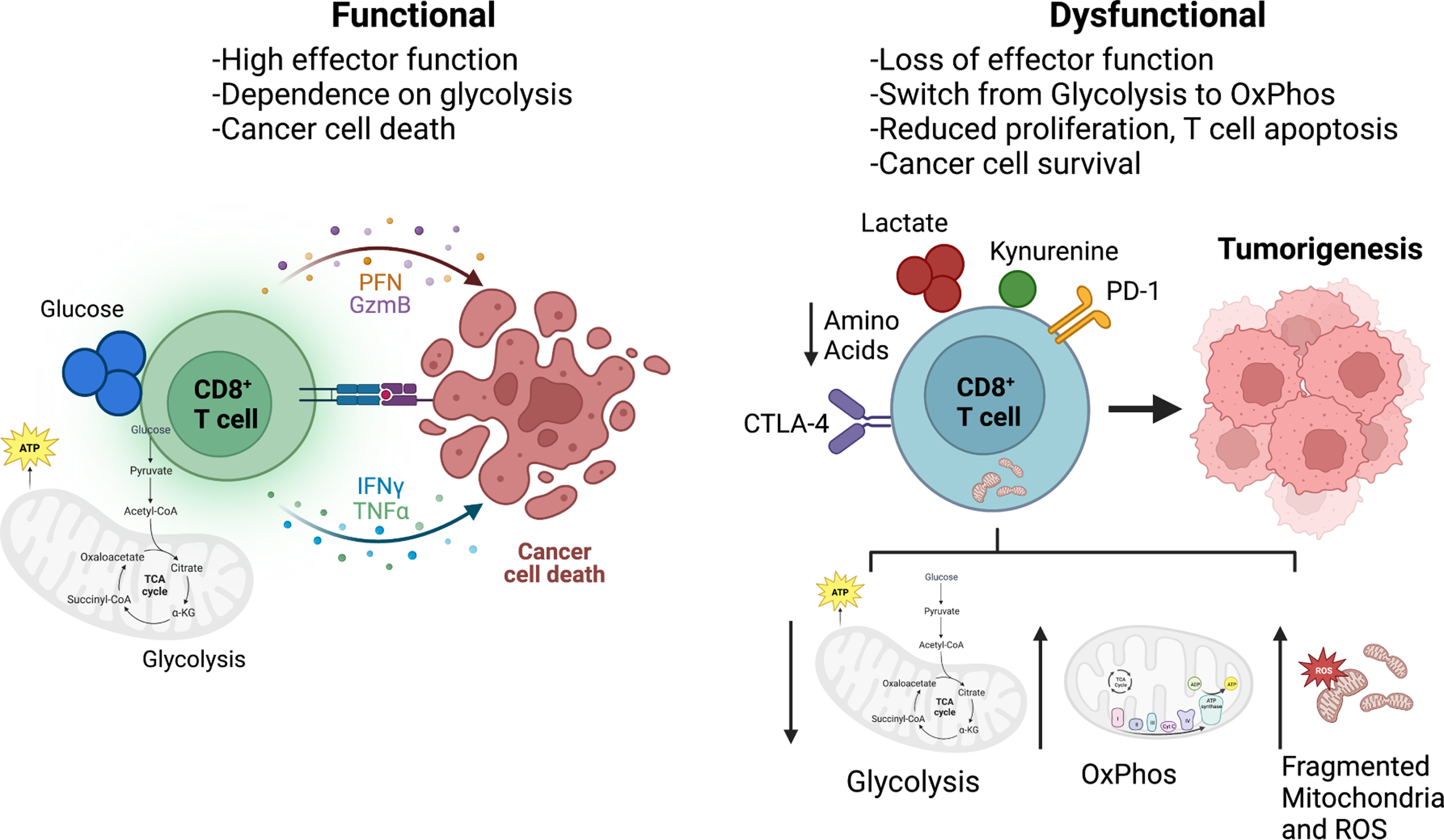
Cytotoxic T cells act directly to kill cancer cells by secreting inflammatory cytokines as well as cell lytic molecules such as granzyme. In a highly functioning state, CD8+ effector T cells are dependent on glycolysis, which promotes inflammation. Metabolic immune suppression occurs when T cells from tumors develop a wide range of metabolic adaptations or dysfunctions that prevent anti-tumor activity. These include dysregulated and fragmented mitochondria, increased ROS production, and reduced glycolysis. In the TME, cancer cells secrete metabolites that effect the function of immune cells within the TME and promote T cell exhaustion such as lactate, cholesterol, and kynurenine, a by-product of tryptophan catabolism. Once exhausted, T cells are unable survive and function to secrete cytokines and express inhibitory receptors such as PD-1 and CTLA-4, which reduce the function of effector T cells by inhibiting glycolysis and upregulating oxidative phosphorylation of the cells. Additionally, amino acids such as arginine, tryptophan, and serine may be limited in the TME, thus inhibiting cytotoxic T cells as they rely on these nutrients for proliferation and function.
This body of data begs the question, can amino acids be supplemented in patients to promote anti-tumor T cells? Results from Geiger et al.89, revealed that the supplementation of L-arginine both in vitro and in vivo did indeed drive T cell anti-tumor immunity by enhancing memory function and mitochondrial respiration in a murine melanoma model89. Additional studies are needed to determine if cancer cells would also benefit from amino acid supplementation and grow faster that could counter-balance any benefit to immune cells.
4. Immunotherapy and Metabolic Immune Suppression
T cell activation to gain effector function requires signals through the antigen receptor (signal 1), co-stimulatory receptors (signal 2), and cytokine growth factors (signal 3) that promote proliferation, survival, and differentiation. Additionally, activation of metabolic pathways within T cells is also essential as a “signal 4”. Each of these signals can be modified in the TME and be a target for immune checkpoint or adoptive T cell immunotherapy. While many T cells in the TME are exhausted and unable to readily recover function caused by chronic stimulation and TME factors such as hypoxia15,100, these immune therapies seek to reinvigorate or induce new tumor specific T cells. Metabolic immune suppression contributes to exhaustion and provides an additional barrier to T cell reactivation12,14,16,101. Most approaches to overcome T cell dysfunction and metabolic barriers in the TME have focused on modifying antigen receptor signaling or co-stimulation.
The most common immunotherapies in current use are checkpoint blockade of PD-1 or CTLA-4 signaling, both of which are inhibitory co-stimulatory CD28 family members that restrain T cell effector functions102. PD-1 can be induced by lactate76 and inhibits PI3K and mTORC1 signaling to reduce anabolic pathways including glycolysis while enhancing lipid oxidation that can promote longer T cell survival103,104. Likewise, CTLA-4 suppresses effector T cell glycolysis and can promote Treg stability in the TME10,105. Thus, checkpoint blockade of these two prominent immunotherapies has profound immunometabolic implications for tumor specific T cells. Given the need of proliferative T cells for glucose uptake that supports biosynthesis and energy generation, the metabolic actions of immune checkpoints are likely fundamental to patient therapeutic responses. Consistent with a connection between metabolism in the TME and immunotherapies, inhibiting glycolysis of T cells in RCC strongly suppressed proliferation and effector function106 and tumor hypoxia can influence or impair tumor immunotherapy13. An unknown, however, is how PD-1 or CTLA-4 signals effect the metabolism and function of other cell types in the TME. Indeed, macrophages can express PD-1, and this checkpoint molecule can inhibit phagocytosis and inflammation107,108.
Adoptive T cell therapies target T cells to tumor-specific antigens by identifying and expanding neoantigen-specific T cells or engineering T cells to express chimeric antigen receptors (CAR) specific to tumor antigens (Figure 3)109,110. In each case, T cells must induce an anabolic metabolism that may be suppressed in the TME. To allow T cells to best survive and function in the TME, adoptive T cell therapies have shown that increased mitochondrial capacity and quality is critical101. This can be developed by adapting T cells to use mitochondrial pathways by inhibition of glycolysis or glutamine metabolism in vitro prior to adoptive transfer to patients or through genetic engineering14,58. Indeed, T cells with low mitochondrial potential, and thus tightly coupled or efficient mitochondria, can provide superior responses in adoptive T cell therapies111. Consistent with mitochondria quality control as a key factor for long-term T cell memory, CD8+ T cells with the greatest mitochondrial turn-over were shown to have the highest levels of memory, while CD8+ T cells with low mitochondrial turn-over had short lifespans112.
Figure 3. Metabolic Rewiring to Reduce Metabolic Immune Suppression.
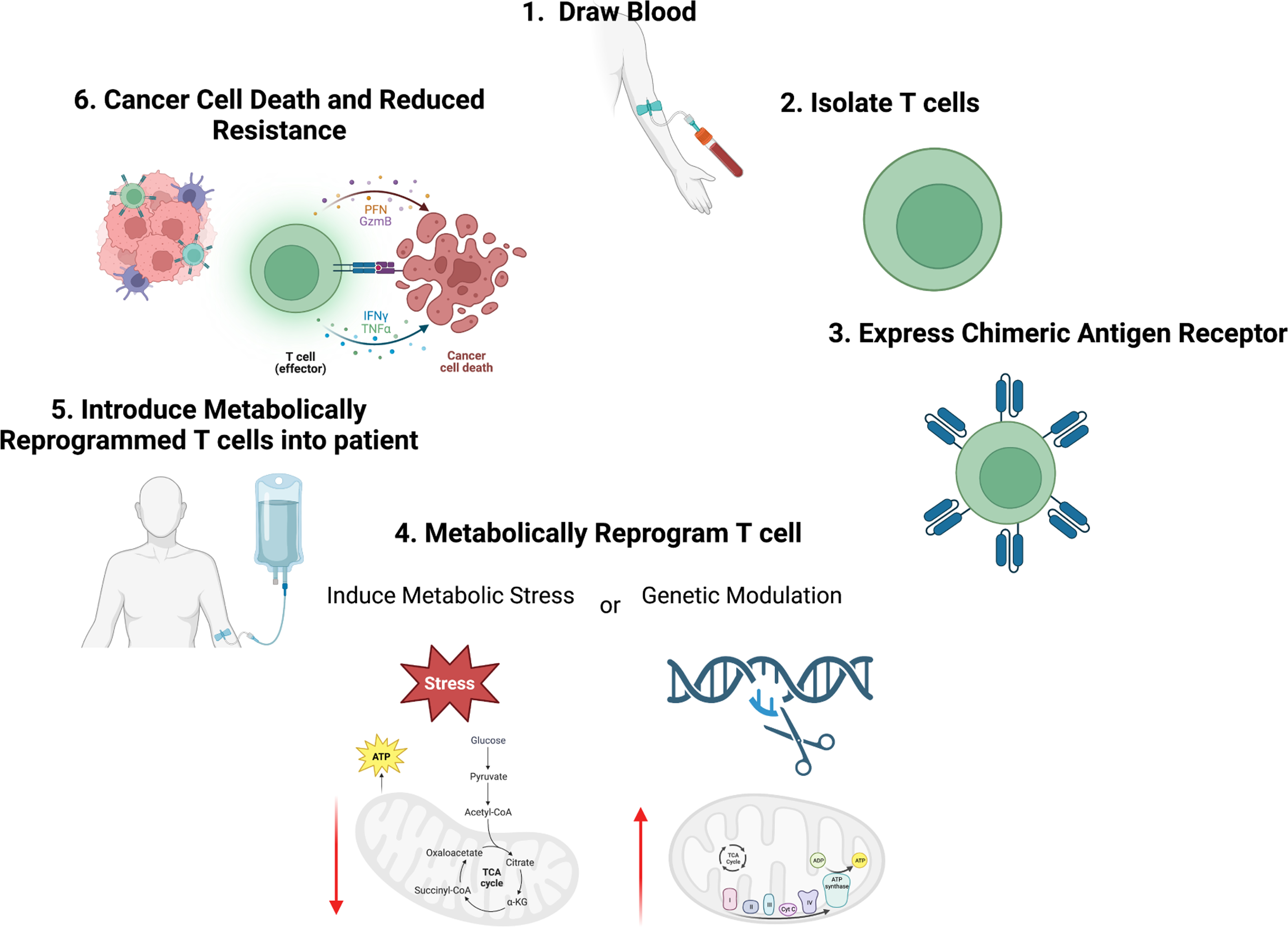
Adoptive T cell therapies target cytotoxic T cells by engineering T cells from patients to express chimeric antigen receptors (CAR) specific to tumor antigens. To reduce metabolic immune suppression of T cells in the TME and increase their metabolic capacity, it may be possible to metabolically rewire T cells either by inducing metabolic stress or through genetic modification. These strategies may be able to increase T cell metabolic plasticity and fitness to turn tumors that are typically immune “cold” due to poor nutrient conditions to immune “hot”, thus increasing anti-tumor immunity and reducing resistance to therapies such as immune checkpoint.
In CAR T cells, the CAR signaling domain consists of both antigen receptor and co-stimulatory components. It is now clear that CD28 signaling domains promote high rates of glycolysis and rapid expansion at the expense of a short lifespan while 4–1BB domains promote mitochondrial metabolism and CAR T longevity at the expense of lower effector function113. The poor responses of CAR T cells in solid tumors, however, demonstrates that the TME can overcome even this programming. It remains unclear to what extent metabolic immune suppression contributes to solid tumor resistance to CAR T cells. A likely contributing factor, however, is the metabolic heterogeneity of solid tumors and the need for adoptively transferred T cells to function in a variety of nutrient and metabolic environments. Metabolic plasticity and efficiency, therefore, may be the ultimate goal rather than forced metabolic programs based on fixed CAR signaling characteristics. Additionally, recent studies using CRISPR-based screening methods, have identified gain-of-function targets for CAR T engineering, such as PRODH2114, a proline dehydrogenase that has been shown to improve mitochondrial function in CD8+ T cells and anti-tumor activity in mice. These methods illustrate that genetically engineering CAR T cells ex vivo for adoptive transfer may also be a valuable therapy option in the future.
5. Metabolic Plasticity and Metastasis
Despite research efforts, metastasis and relapse remain the primary cause of cancer related deaths. The metastasis of solid tumors requires cancer cells to pass heterogeneous immune and metabolic microenvironments as they escape the primary tumor, invade surrounding stroma, enter the bloodstream or lymphatic vessels (referred to as intravasation), survive circulation, and extravasate to secondary sites to colonize and expand as metastases115 (Figure 4). This escape requires that cancer cells survive in multiple metabolic microenvironments. Metastasis is facilitated by tumor cell epithelial plasticity, resulting in epithelial cells adopting mesenchymal-like features that enable cell migration and invasion, referred to as epithelial-to-mesenchymal transition or EMT. Epithelial plasticity includes EMT and the reverse, mesenchymal-to-epithelial transition (MET). MET is reported to be required for successful metastatic colonization as distant metastases in carcinoma patients often present with epithelial features having a similar pathology as the tissue of origin116. Only a small percentage of cancer cells successfully complete this process, as cancer cells must adapt to different nutrient environments and resist anti-tumor immunity to survive and thrive. Cancer cells undergoing EMT may secrete cytokines that lead to suppressive immune cell recruitment, such as macrophages, MDSCs, and Tregs117 to enhance cancer cell survival and metastasis. There is also high production of transforming growth factor (TGF)-β during EMT, which has been shown to exclude cytotoxic T cells and induce Tregs to support immune suppression118. It can be imagined that interactions within tumors and multiple metabolic processes of immune cells in the TME may dictate this plasticity at each step of the metastatic cascade, although there are limited studies thus far that evaluate the metabolic plasticity of EMT and MET or the role of immunometabolism in metastasis.
Figure 4. Metabolic Plasticity in Metastasis.
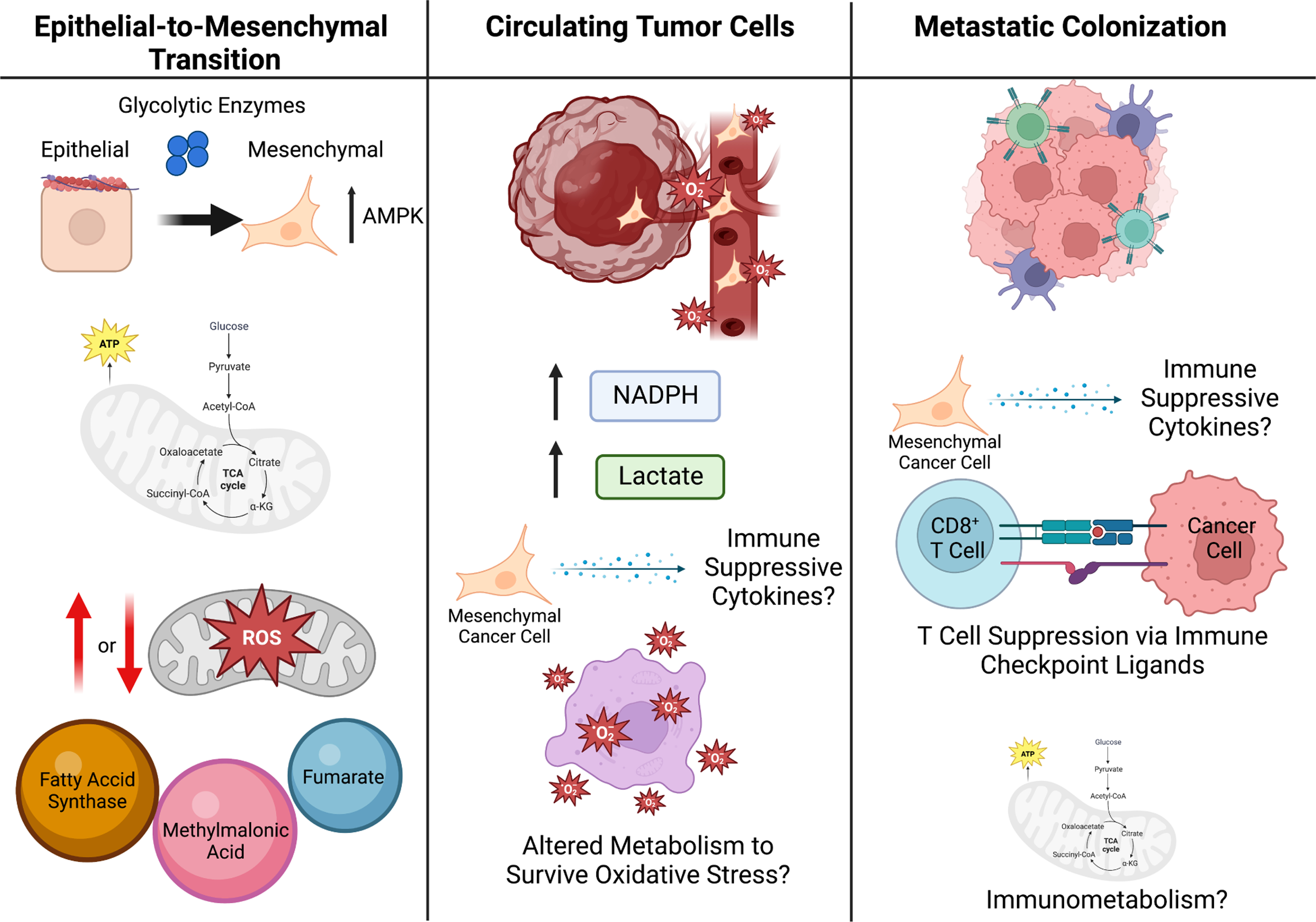
To escape the primary tumor and form metastases, cells undergo EMT (left panel). Glycolytic enzymes are attached to the cell cytoskeleton, as such it is possible that during cytoskeleton rearrangement in the EMT process, these glycolytic enzymes are released to promote glycolysis during EMT. The production of mitochondrial ROS within the cancer cells has been suggested to both promote and reduce EMT and metastatic potential of cancer cells, highlighting the likelihood that this process is highly dependent on the genetic context and local TME. Additionally, some metabolites act as pro-EMT signaling molecules, such as fumarate, methylmalonic acid, and fatty-acid synthase (FASN). Once in circulation (middle panel), cancer cells undergo oxidative stress and thus cell-death, therefore it is likely the cancer cells that are able to survive oxidative stress have a metabolic advantage resulting in their survival. Additionally, CTCs have been shown to upregulate NADPH production and lactate uptake, which diverts glucose carbon into the oxidate PPP and increases their antioxidant capacity. It is likely that cytokines and metabolites within the blood stream may influence both immune surveillance of CTCs as well as cancer cell immune evasion. Once cancer cells enter the metastatic site (right panel), they must act on the tumor microenvironment within the metastatic site to suppress immune surveillance, which can be done metabolically, such as by presenting immune checkpoint ligands. Additionally, immunometabolism likely plays a significant role in this process and may provide a therapeutic vulnerability for the treatment of metastatic cancer.
Metabolism of EMT
The metastatic potential of cancer cells has been linked to mitochondrial function, as ROS homeostasis is essential for cancer cell survival. While moderate increases in ROS stimulates cell proliferation, excess ROS can cause oxidative stress and lead to cell death119. The production of mitochondrial ROS within the cancer cells has been suggested to both promote120,121 or reduce122,123 EMT and metastatic potential of cancer cells, highlighting the likelihood that this process is dependent on the type of cancer, genetic context, and local TME. For example, metastatic fibrosarcoma cells prevent ROS-mediated cell death by stabilizing BACH1 downstream of nuclear factor-erythroid 2-related factor 2 (NRF2) activated heme oxygenase 1 (HO-1)123. Ultimately accumulation of BACH1 may promote glycolysis-dependent metastasis of lung cancer cells124,125.
Some metabolites have also been shown to promote metastasis by acting as pro-EMT signaling molecules. In a study by Sciacovelli et al.126, fumarate was found to drive EMT via inhibition of Tet-mediated demethylation of the miRNA cluster mir-200baa429 and subsequent transcription of EMT Transcription factors in kidney cancer126. Additionally, the induction of glycolysis via the ECM component hyaluronidase in cancer cells has been shown to drive EMT and cell migration127. In another study, Gomes et al.128, show methylmalonic acid 128, a by-product of propionate metabolism, was elevated in the serum of older people and associated with increased tumor progression. Mechanistic studies revealed that MMA induced SOX4 and thus increased the transcription of EMT transcription factors to drive EMT and invasion128. In addition, fatty-acid synthase (FASN), an enzyme that catalyzes the final step of fatty acid synthesis, has been shown to be strongly associated with tumorigenesis, EMT, and metastasis in HCC129, ovarian cancer130, glioblastoma131, and prostate cancer132. Genetic inhibition of FASN suppressed invasion and migration in HCC metastasis129, while increased expression of FASN may induce EMT and peritoneal metastasis in ovarian cancer130.
Metabolic Plasticity in the Metastatic Cascade
Metastasis requires cancer cells traverse multiple nutrient microenvironments and interact with a variety of cells. Glycolytic enzymes are attached to the cytoskeleton and during cell growth these enzymes function to increase glycolysis. During cytoskeleton rearrangement in the EMT process these enzymes may be released, thus promoting glycolysis in cells actively undergoing EMT98. Additionally, AMPK is often activated in metastatic cancer cells that have detached from the extracellular matrix, resulting in decreased NADPH consumption in fatty acid synthesis and increased NADPH generation via fatty acid oxidation (FAO)133. Low PHGDH and the hexosamine pathway also can promote metastasis through aberrant integrin glycosylation134. Although there is limited evidence, one could speculate that the high energy demands and metabolic plasticity of cells undergoing EMT may limit available nutrients for cytotoxic T cells, thus leading to immune suppression during the escape of cancer cells from the primary tumor. Additionally, TAMs within the hypoxic TME have been shown to promote the formation of blood vessels, thus providing an escape route for cancer cells43.
Although little is known about the metabolism of circulating tumor cells (CTCs), cancer cells are likely to enter a catabolic state once they enter the blood stream to survive due to the different metabolites that are available135. While in circulation, cancer cells can undergo oxidative stress and cell death136, and cancer cells best able to survive oxidative stress due to enhanced redox capacity may have a metabolic advantage. For example, metastatic melanoma cells upregulate NADPH production and lactate uptake, which diverts glucose carbon into the oxidate PPP and increases their antioxidant capacity137. Consistent with this model, dietary antioxidants have been shown to reduce oxidative stress and increase metastasis in mouse models of lung cancer and melanoma138,139. CTCs increase PGC1α-dependent mitochondrial biogenesis to maintain redox and energetic homeostasis140. It is possible that cytokines and metabolites within the blood stream may influence both immune surveillance of circulating cancer cells as well as cancer cell immune evasion, although studies are needed to determine if this is the case.
Once cells enter the metastatic site, metabolic plasticity is required to both survive and colonize within the new metastatic niche, as different nutrients will likely be available compared to both the primary tumor and circulation. For example, the brain is a common metastatic site in breast cancer patients and has a vastly different tumor microenvironment than breast cancer. In the brain, both serine and fatty acids are decreased, thus breast cancer cells that can adapt and colonize in the brain upregulate PHGDH to increase glucose-dependent serine and glycine production141. Additionally, cancer cells must act on the tumor microenvironment within the metastatic site to suppress immune surveillance, which may occur through metabolic suppression as discussed previously.
Given the potential for limiting nutrients and metabolic signaling in the TME, it is likely that metabolic plasticity contributes to EMT and supports the energetic demands of cell migration. How these changes affect cancer cell interactions with immune cells in the TME or at metastatic niches remain uncertain. Immunometabolism likely plays a significant role in this process and may provide a therapeutic vulnerability for the treatment of metastatic cancer. It Is possible that metabolic vulnerabilities can be utilized to target both the primary and metastatic lesions. Future studies will determine how the immune system can best be activated to target metastases both before they arise and after they form a new metastatic niche. Perhaps rewiring the metabolism of cancer cells or immune cells within the TME before the cancer spreads will prevent metastasis or recurrence of high-risk cancers.
6. Conclusion
We present here recent findings of differential nutrient uptake due to cellular and metabolic heterogeneity within tumors. Tumor type, location of the tumor, and diet of the host will all affect nutrient availability within the tumor microenvironment6. This is not only critical for the proliferation of the cancer cells but also tumor infiltrating immune cells and likely influences metastasis. As the metabolism of immune cells likely plays a significant role in metastasis, further research is needed to determine if metabolic vulnerabilities within the primary tumor and metastatic lesions can be utilized to target metastasis before they arise as well as after they form and colonize a new metastatic niche by utilizing metabolic dependencies and immunotherapy.
The diversity of cell types and metabolic landscapes in the TME can be detrimental for treatment and allow tumor recurrence. One challenge is that similar metabolic pathways are used to support cell growth and inhibitors that suppress the growth of cancer cells can also inhibit anti-tumor immunity. Conversely, the different metabolic programs and regions within a given TME challenge approaches that target single pathways. This metabolic heterogeneity, however, opens new opportunities for selective targeting of specific cell types or conditions. Immunotherapies that target T cells to reduce resistance are currently being investigated, such as adoptive T cell therapies, and metabolic immune suppression should be considered. To further improve anti-tumor immunity, T cells can be expanded in the presence of metabolic stress to promote metabolic plasticity as immune cells are forced to adapt and utilize multiple nutrients depending on availability, to support better effector function across a wide range of nutrient microenvironmental conditions. Perhaps these strategies will increase T cell metabolic fitness to turn tumors that are typically immune “cold” tumors due to poor nutrient conditions, into immune “hot” tumors, thereby increasing the effectiveness of immune checkpoint blockade, such as anti-PD-1. Although anti-cancer therapies targeting metabolic heterogeneity are still in early stages of development, additional studies that further our understanding of metabolism both contextually and spatially are sure to lead to new efficacious therapies for cancer patients.
Acknowledgements
We thank members of the Rathmell lab and W.K. Rathmell for valuable input. We apologize to the colleagues whose work could not be included due to space limitations. Figures were created with Biorender.com. This work was supported by K00 CA253718 (E.N.A.), R01 CA217987 (J.C.R.), R01 DK105550 (J.C.R.), and the Vanderbilt-Incyte Alliance (J.C.R.).
Footnotes
Declaration of Interests
J.C.R. holds stock equity in Sitryx and Caribou, and within the past two years has received unrelated research support, travel or honoraria from Sitryx, Caribou, Nirogy, Kadmon, Calithera, Tempest, Merck, Mitobridge and Pfizer. E.N.A declares no competing interests.
Publisher's Disclaimer: This is a PDF file of an unedited manuscript that has been accepted for publication. As a service to our customers we are providing this early version of the manuscript. The manuscript will undergo copyediting, typesetting, and review of the resulting proof before it is published in its final form. Please note that during the production process errors may be discovered which could affect the content, and all legal disclaimers that apply to the journal pertain.
References
- 1.Li X, Sun X, and Carmeliet P. (2019). Hallmarks of Endothelial Cell Metabolism in Health and Disease. Cell Metab 30, 414–433. 10.1016/j.cmet.2019.08.011. [DOI] [PubMed] [Google Scholar]
- 2.Ryan DG, and O’Neill LAJ (2020). Krebs Cycle Reborn in Macrophage Immunometabolism. Annu Rev Immunol 38, 289–313. 10.1146/annurev-immunol-081619-104850. [DOI] [PubMed] [Google Scholar]
- 3.Makowski L, Chaib M, and Rathmell JC (2020). Immunometabolism: From basic mechanisms to translation. Immunol Rev 295, 5–14. 10.1111/imr.12858. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 4.DeBerardinis RJ, and Chandel NS (2016). Fundamentals of cancer metabolism. Sci Adv 2, e1600200. 10.1126/sciadv.1600200. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 5.Gouirand V, Guillaumond F, and Vasseur S. (2018). Influence of the Tumor Microenvironment on Cancer Cells Metabolic Reprogramming. Front Oncol 8, 117. 10.3389/fonc.2018.00117. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 6.Sullivan MR., Danai LV., Lewis CA., Chan SH., Gui DY., Kunchok T., Dennstedt EA., Vander Heiden MG., and Muir A. (2019). Quantification of microenvironmental metabolites in murine cancers reveals determinants of tumor nutrient availability. Elife 8. 10.7554/eLife.44235. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 7.Bader JE, Voss K, and Rathmell JC (2020). Targeting Metabolism to Improve the Tumor Microenvironment for Cancer Immunotherapy. Mol Cell 78, 1019–1033. 10.1016/j.molcel.2020.05.034. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 8.Klein Geltink RI, O’Sullivan D, Corrado M, Bremser A, Buck MD, Buescher JM, Firat E, Zhu X, Niedermann G, Caputa G, et al. (2017). Mitochondrial Priming by CD28. Cell 171, 385–397 e311. 10.1016/j.cell.2017.08.018. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 9.Parry RV, Chemnitz JM, Frauwirth KA, Lanfranco AR, Braunstein I, Kobayashi SV, Linsley PS, Thompson CB, and Riley JL (2005). CTLA-4 and PD-1 receptors inhibit T-cell activation by distinct mechanisms. Mol Cell Biol 25, 9543–9553. 10.1128/MCB.25.21.9543-9553.2005. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 10.Frauwirth KA, Riley JL, Harris MH, Parry RV, Rathmell JC, Plas DR, Elstrom RL, June CH, and Thompson CB (2002). The CD28 signaling pathway regulates glucose metabolism. Immunity 16, 769–777. 10.1016/s1074-7613(02)00323-0. [DOI] [PubMed] [Google Scholar]
- 11.Jacobs SR, Herman CE, Maciver NJ, Wofford JA, Wieman HL, Hammen JJ, and Rathmell JC (2008). Glucose uptake is limiting in T cell activation and requires CD28-mediated Akt-dependent and independent pathways. J Immunol 180, 4476–4486. 10.4049/jimmunol.180.7.4476. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 12.Siska PJ, Beckermann KE, Mason FM, Andrejeva G, Greenplate AR, Sendor AB, Chiang YJ, Corona AL, Gemta LF, Vincent BG, et al. (2017). Mitochondrial dysregulation and glycolytic insufficiency functionally impair CD8 T cells infiltrating human renal cell carcinoma. JCI Insight 2. 10.1172/jci.insight.93411. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 13.Zandberg DP, Menk AV, Velez M, Normolle D, DePeaux K, Liu A, Ferris RL, and Delgoffe GM (2021). Tumor hypoxia is associated with resistance to PD-1 blockade in squamous cell carcinoma of the head and neck. J Immunother Cancer 9. 10.1136/jitc-2020-002088. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 14.DePeaux K, and Delgoffe GM (2021). Metabolic barriers to cancer immunotherapy. Nat Rev Immunol. 10.1038/s41577-021-00541-y. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 15.Scharping NE, Rivadeneira DB, Menk AV, Vignali PDA, Ford BR, Rittenhouse NL, Peralta R, Wang Y, Wang Y, DePeaux K, et al. (2021). Mitochondrial stress induced by continuous stimulation under hypoxia rapidly drives T cell exhaustion. Nat Immunol. 10.1038/s41590-020-00834-9. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 16.Chang CH, Qiu J, O’Sullivan D, Buck MD, Noguchi T, Curtis JD, Chen Q, Gindin M, Gubin MM, van der Windt GJ, et al. (2015). Metabolic Competition in the Tumor Microenvironment Is a Driver of Cancer Progression. Cell 162, 1229–1241. 10.1016/j.cell.2015.08.016. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 17.Xu S., Chaudhary O., Rodriguez-Morales P., Sun X., Chen D., Zappasodi R., Xu Z., Pinto AFM., Williams A., Schulze I., et al. (2021). Uptake of oxidized lipids by the scavenger receptor CD36 promotes lipid peroxidation and dysfunction in CD8(+) T cells in tumors. Immunity 54, 1561–1577 e1567. 10.1016/j.immuni.2021.05.003. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 18.Bengsch B, Johnson AL, Kurachi M, Odorizzi PM, Pauken KE, Attanasio J, Stelekati E, McLane LM, Paley MA, Delgoffe GM, and Wherry EJ (2016). Bioenergetic Insufficiencies Due to Metabolic Alterations Regulated by the Inhibitory Receptor PD-1 Are an Early Driver of CD8(+) T Cell Exhaustion. Immunity 45, 358–373. 10.1016/j.immuni.2016.07.008. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 19.Warburg OH, Dickens F, and Dickens F. (1930). The metabolism of tumours (Constable). [Google Scholar]
- 20.Reinfeld BI, Madden MZ, Wolf MM, Chytil A, Bader JE, Patterson AR, Sugiura A, Cohen AS, Ali A, Do BT, et al. (2021). Cell-programmed nutrient partitioning in the tumour microenvironment. Nature 593, 282–288. 10.1038/s41586-021-03442-1. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 21.Gupta S, Roy A, and Dwarakanath BS (2017). Metabolic Cooperation and Competition in the Tumor Microenvironment: Implications for Therapy. Front Oncol 7, 68. 10.3389/fonc.2017.00068. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 22.Papandreou I, Cairns RA, Fontana L, Lim AL, and Denko NC (2006). HIF-1 mediates adaptation to hypoxia by actively downregulating mitochondrial oxygen consumption. Cell Metab 3, 187–197. 10.1016/j.cmet.2006.01.012. [DOI] [PubMed] [Google Scholar]
- 23.Kim JW, Tchernyshyov I, Semenza GL, and Dang CV (2006). HIF-1-mediated expression of pyruvate dehydrogenase kinase: a metabolic switch required for cellular adaptation to hypoxia. Cell Metab 3, 177–185. 10.1016/j.cmet.2006.02.002. [DOI] [PubMed] [Google Scholar]
- 24.Fukuda R, Zhang H, Kim JW, Shimoda L, Dang CV, and Semenza GL (2007). HIF-1 regulates cytochrome oxidase subunits to optimize efficiency of respiration in hypoxic cells. Cell 129, 111–122. 10.1016/j.cell.2007.01.047. [DOI] [PubMed] [Google Scholar]
- 25.Huang B, Song BL, and Xu C. (2020). Cholesterol metabolism in cancer: mechanisms and therapeutic opportunities. Nat Metab 2, 132–141. 10.1038/s42255-020-0174-0. [DOI] [PubMed] [Google Scholar]
- 26.Hensley CT, Wasti AT, and DeBerardinis RJ (2013). Glutamine and cancer: cell biology, physiology, and clinical opportunities. J Clin Invest 123, 3678–3684. 10.1172/JCI69600. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 27.Pan M., Reid MA., Lowman XH., Kulkarni RP., Tran TQ., Liu X., Yang Y., Hernandez-Davies JE., Rosales KK., Li H., et al. (2016). Regional glutamine deficiency in tumours promotes dedifferentiation through inhibition of histone demethylation. Nat Cell Biol 18, 1090–1101. 10.1038/ncb3410. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 28.Yang L, Garcia Canaveras JC, Chen Z, Wang L, Liang L, Jang C, Mayr JA, Zhang Z, Ghergurovich JM, Zhan L, et al. (2020). Serine Catabolism Feeds NADH when Respiration Is Impaired. Cell Metab 31, 809–821 e806. 10.1016/j.cmet.2020.02.017. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 29.Patil MD, Bhaumik J, Babykutty S, Banerjee UC, and Fukumura D. (2016). Arginine dependence of tumor cells: targeting a chink in cancer’s armor. Oncogene 35, 4957–4972. 10.1038/onc.2016.37. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 30.Amatangelo MD, Bassi DE, Klein-Szanto AJ, and Cukierman E. (2005). Stroma-derived three-dimensional matrices are necessary and sufficient to promote desmoplastic differentiation of normal fibroblasts. Am J Pathol 167, 475–488. 10.1016/S0002-9440(10)62991-4. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 31.Buck MD, O’Sullivan D, and Pearce EL (2015). T cell metabolism drives immunity. J Exp Med 212, 1345–1360. 10.1084/jem.20151159. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 32.Eckert AW, Wickenhauser C, Salins PC, Kappler M, Bukur J, and Seliger B. (2016). Clinical relevance of the tumor microenvironment and immune escape of oral squamous cell carcinoma. J Transl Med 14, 85. 10.1186/s12967-016-0828-6. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 33.Grover A, Sanseviero E, Timosenko E, and Gabrilovich DI (2021). Myeloid-Derived Suppressor Cells: A Propitious Road to Clinic. Cancer Discov 11, 2693–2706. 10.1158/2159-8290.CD-21-0764. [DOI] [PubMed] [Google Scholar]
- 34.Trovato R, Cane S, Petrova V, Sartoris S, Ugel S, and De Sanctis F. (2020). The Engagement Between MDSCs and Metastases: Partners in Crime. Front Oncol 10, 165. 10.3389/fonc.2020.00165. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 35.Pastaki Khoshbin A, Eskian M, Keshavarz-Fathi M, and Rezaei N. (2019). Roles of Myeloid-Derived Suppressor Cells in Cancer Metastasis: Immunosuppression and Beyond. Arch Immunol Ther Exp (Warsz) 67, 89–102. 10.1007/s00005-018-0531-9. [DOI] [PubMed] [Google Scholar]
- 36.Pergamo M, and Miller G. (2017). Myeloid-derived suppressor cells and their role in pancreatic cancer. Cancer Gene Ther 24, 100–105. 10.1038/cgt.2016.65. [DOI] [PubMed] [Google Scholar]
- 37.Malek E, de Lima M, Letterio JJ, Kim BG, Finke JH, Driscoll JJ, and Giralt SA (2016). Myeloid-derived suppressor cells: The green light for myeloma immune escape. Blood Rev 30, 341–348. 10.1016/j.blre.2016.04.002. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 38.Fischer K, Hoffmann P, Voelkl S, Meidenbauer N, Ammer J, Edinger M, Gottfried E, Schwarz S, Rothe G, Hoves S, et al. (2007). Inhibitory effect of tumor cell-derived lactic acid on human T cells. Blood 109, 3812–3819. 10.1182/blood-2006-07-035972. [DOI] [PubMed] [Google Scholar]
- 39.Brand A, Singer K, Koehl GE, Kolitzus M, Schoenhammer G, Thiel A, Matos C, Bruss C, Klobuch S, Peter K, et al. (2016). LDHA-Associated Lactic Acid Production Blunts Tumor Immunosurveillance by T and NK Cells. Cell Metab 24, 657–671. 10.1016/j.cmet.2016.08.011. [DOI] [PubMed] [Google Scholar]
- 40.Watson MJ., Vignali PDA., Mullett SJ., Overacre-Delgoffe AE., Peralta RM., Grebinoski S., Menk AV., Rittenhouse NL., DePeaux K., Whetstone RD., et al. (2021). Metabolic support of tumour-infiltrating regulatory T cells by lactic acid. Nature 591, 645–651. 10.1038/s41586-020-03045-2. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 41.Angelin A, Gil-de-Gomez L, Dahiya S, Jiao J, Guo L, Levine MH, Wang Z, Quinn WJ 3rd, Kopinski PK, Wang L, et al. (2017). Foxp3 Reprograms T Cell Metabolism to Function in Low-Glucose, High-Lactate Environments. Cell Metab 25, 1282–1293 e1287. 10.1016/j.cmet.2016.12.018. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 42.Colegio OR, Chu NQ, Szabo AL, Chu T, Rhebergen AM, Jairam V, Cyrus N, Brokowski CE, Eisenbarth SC, Phillips GM, et al. (2014). Functional polarization of tumour-associated macrophages by tumour-derived lactic acid. Nature 513, 559–563. 10.1038/nature13490. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 43.Flerin NC, Pinioti S, Menga A, Castegna A, and Mazzone M. (2020). Impact of Immunometabolism on Cancer Metastasis: A Focus on T Cells and Macrophages. Cold Spring Harb Perspect Med 10. 10.1101/cshperspect.a037044. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 44.Brugarolas J, Lei K, Hurley RL, Manning BD, Reiling JH, Hafen E, Witters LA, Ellisen LW, and Kaelin WG Jr. (2004). Regulation of mTOR function in response to hypoxia by REDD1 and the TSC1/TSC2 tumor suppressor complex. Genes Dev 18, 2893–2904. 10.1101/gad.1256804. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 45.Wang Y, Bai C, Ruan Y, Liu M, Chu Q, Qiu L, Yang C, and Li B. (2019). Coordinative metabolism of glutamine carbon and nitrogen in proliferating cancer cells under hypoxia. Nat Commun 10, 201. 10.1038/s41467-018-08033-9. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 46.Mullen AR, Wheaton WW, Jin ES, Chen PH, Sullivan LB, Cheng T, Yang Y, Linehan WM, Chandel NS, and DeBerardinis RJ (2011). Reductive carboxylation supports growth in tumour cells with defective mitochondria. Nature 481, 385–388. 10.1038/nature10642. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 47.Metallo CM, Gameiro PA, Bell EL, Mattaini KR, Yang J, Hiller K, Jewell CM, Johnson ZR, Irvine DJ, Guarente L, et al. (2011). Reductive glutamine metabolism by IDH1 mediates lipogenesis under hypoxia. Nature 481, 380–384. 10.1038/nature10602. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 48.Poillet-Perez L, and White E. (2019). Role of tumor and host autophagy in cancer metabolism. Genes Dev 33, 610–619. 10.1101/gad.325514.119. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 49.Eichner LJ, Brun SN, Herzig S, Young NP, Curtis SD, Shackelford DB, Shokhirev MN, Leblanc M, Vera LI, Hutchins A, et al. (2019). Genetic Analysis Reveals AMPK Is Required to Support Tumor Growth in Murine Kras-Dependent Lung Cancer Models. Cell Metab 29, 285–302 e287. 10.1016/j.cmet.2018.10.005. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 50.Amaravadi RK, Kimmelman AC, and Debnath J. (2019). Targeting Autophagy in Cancer: Recent Advances and Future Directions. Cancer Discov 9, 1167–1181. 10.1158/2159-8290.CD-19-0292. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 51.Bosc C., Broin N., Fanjul M., Saland E., Farge T., Courdy C., Batut A., Masoud R., Larrue C., Skuli S., et al. (2020). Autophagy regulates fatty acid availability for oxidative phosphorylation through mitochondria-endoplasmic reticulum contact sites. Nat Commun 11, 4056. 10.1038/s41467-020-17882-2. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 52.Hensley CT, Faubert B, Yuan Q, Lev-Cohain N, Jin E, Kim J, Jiang L, Ko B, Skelton R, Loudat L, et al. (2016). Metabolic Heterogeneity in Human Lung Tumors. Cell 164, 681–694. 10.1016/j.cell.2015.12.034. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 53.Fu X, Zhao Y, Lopez JI, Rowan A, Au L, Fendler A, Hazell S, Xu H, Horswell S, Shepherd STC, et al. (2022). Spatial patterns of tumour growth impact clonal diversification in a computational model and the TRACERx Renal study. Nat Ecol Evol 6, 88–102. 10.1038/s41559-021-01586-x. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 54.Gaines DK, and Rathmell WK (2022). Spatial models of tumour evolution. Nat Ecol Evol 6, 26–27. 10.1038/s41559-021-01584-z. [DOI] [PubMed] [Google Scholar]
- 55.Vander Heiden MG, and DeBerardinis RJ (2017). Understanding the Intersections between Metabolism and Cancer Biology. Cell 168, 657–669. 10.1016/j.cell.2016.12.039. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 56.Ho PC, Bihuniak JD, Macintyre AN, Staron M, Liu X, Amezquita R, Tsui YC, Cui G, Micevic G, Perales JC, et al. (2015). Phosphoenolpyruvate Is a Metabolic Checkpoint of Anti-tumor T Cell Responses. Cell 162, 1217–1228. 10.1016/j.cell.2015.08.012. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 57.Leone RD, and Powell JD (2020). Metabolism of immune cells in cancer. Nat Rev Cancer 20, 516–531. 10.1038/s41568-020-0273-y. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 58.Leone RD, Zhao L, Englert JM, Sun IM, Oh MH, Sun IH, Arwood ML, Bettencourt IA, Patel CH, Wen J, et al. (2019). Glutamine blockade induces divergent metabolic programs to overcome tumor immune evasion. Science 366, 1013–1021. 10.1126/science.aav2588. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 59.Edwards DN, Ngwa VM, Raybuck AL, Wang S, Hwang Y, Kim LC, Cho SH, Paik Y, Wang Q, Zhang S, et al. (2021). Selective glutamine metabolism inhibition in tumor cells improves antitumor T lymphocyte activity in triple-negative breast cancer. J Clin Invest 131. 10.1172/JCI140100. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 60.Bian Y, Li W, Kremer DM, Sajjakulnukit P, Li S, Crespo J, Nwosu ZC, Zhang L, Czerwonka A, Pawlowska A, et al. (2020). Cancer SLC43A2 alters T cell methionine metabolism and histone methylation. Nature 585, 277–282. 10.1038/s41586-020-2682-1. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 61.Bos PD, Plitas G, Rudra D, Lee SY, and Rudensky AY (2013). Transient regulatory T cell ablation deters oncogene-driven breast cancer and enhances radiotherapy. J Exp Med 210, 2435–2466. 10.1084/jem.20130762. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 62.Delgoffe GM., Woo SR., Turnis ME., Gravano DM., Guy C., Overacre AE., Bettini ML., Vogel P., Finkelstein D., Bonnevier J., et al. (2013). Stability and function of regulatory T cells is maintained by a neuropilin-1-semaphorin-4a axis. Nature 501, 252–256. 10.1038/nature12428. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 63.Weinberg SE, Singer BD, Steinert EM, Martinez CA, Mehta MM, Martinez-Reyes I, Gao P, Helmin KA, Abdala-Valencia H, Sena LA, et al. (2019). Mitochondrial complex III is essential for suppressive function of regulatory T cells. Nature 565, 495–499. 10.1038/s41586-018-0846-z. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 64.Beier UH, Angelin A, Akimova T, Wang L, Liu Y, Xiao H, Koike MA, Hancock SA, Bhatti TR, Han R, et al. (2015). Essential role of mitochondrial energy metabolism in Foxp3(+) T-regulatory cell function and allograft survival. FASEB J 29, 2315–2326. 10.1096/fj.14-268409. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 65.Michalek RD, Gerriets VA, Jacobs SR, Macintyre AN, MacIver NJ, Mason EF, Sullivan SA, Nichols AG, and Rathmell JC (2011). Cutting edge: distinct glycolytic and lipid oxidative metabolic programs are essential for effector and regulatory CD4+ T cell subsets. J Immunol 186, 3299–3303. 10.4049/jimmunol.1003613. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 66.Shi LZ, Wang R, Huang G, Vogel P, Neale G, Green DR, and Chi H. (2011). HIF1alpha-dependent glycolytic pathway orchestrates a metabolic checkpoint for the differentiation of TH17 and Treg cells. J Exp Med 208, 1367–1376. 10.1084/jem.20110278. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 67.Liu C, Chikina M, Deshpande R, Menk AV, Wang T, Tabib T, Brunazzi EA, Vignali KM, Sun M, Stolz DB, et al. (2019). Treg Cells Promote the SREBP1-Dependent Metabolic Fitness of Tumor-Promoting Macrophages via Repression of CD8(+) T Cell-Derived Interferon-gamma. Immunity 51, 381–397 e386. 10.1016/j.immuni.2019.06.017. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 68.Tannahill GM, Curtis AM, Adamik J, Palsson-McDermott EM, McGettrick AF, Goel G, Frezza C, Bernard NJ, Kelly B, Foley NH, et al. (2013). Succinate is an inflammatory signal that induces IL-1beta through HIF-1alpha. Nature 496, 238–242. 10.1038/nature11986. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 69.Vats D, Mukundan L, Odegaard JI, Zhang L, Smith KL, Morel CR, Wagner RA, Greaves DR, Murray PJ, and Chawla A. (2006). Oxidative metabolism and PGC-1beta attenuate macrophage-mediated inflammation. Cell Metab 4, 13–24. 10.1016/j.cmet.2006.05.011. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 70.Faubert B, Li KY, Cai L, Hensley CT, Kim J, Zacharias LG, Yang C, Do QN, Doucette S, Burguete D, et al. (2017). Lactate Metabolism in Human Lung Tumors. Cell 171, 358–371 e359. 10.1016/j.cell.2017.09.019. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 71.Feng Q., Liu Z., Yu X., Huang T., Chen J., Wang J., Wilhelm J., Li S., Song J., Li W., et al. (2022). Lactate increases stemness of CD8 + T cells to augment anti-tumor immunity. Nat Commun 13, 4981. 10.1038/s41467-022-32521-8. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 72.Chang CH, Curtis JD, Maggi LB Jr., Faubert B, Villarino AV, O’Sullivan D, Huang SC, van der Windt GJ, Blagih J, Qiu J, et al. (2013). Posttranscriptional control of T cell effector function by aerobic glycolysis. Cell 153, 1239–1251. 10.1016/j.cell.2013.05.016. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 73.Peng M, Yin N, Chhangawala S, Xu K, Leslie CS, and Li MO (2016). Aerobic glycolysis promotes T helper 1 cell differentiation through an epigenetic mechanism. Science 354, 481–484. 10.1126/science.aaf6284. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 74.Xu C, Sun S, Johnson T, Qi R, Zhang S, Zhang J, and Yang K. (2021). The glutathione peroxidase Gpx4 prevents lipid peroxidation and ferroptosis to sustain Treg cell activation and suppression of antitumor immunity. Cell Rep 35, 109235. 10.1016/j.celrep.2021.109235. [DOI] [PubMed] [Google Scholar]
- 75.Gerriets VA, Kishton RJ, Johnson MO, Cohen S, Siska PJ, Nichols AG, Warmoes MO, de Cubas AA, MacIver NJ, Locasale JW, et al. (2016). Foxp3 and Toll-like receptor signaling balance Treg cell anabolic metabolism for suppression. Nat Immunol 17, 1459–1466. 10.1038/ni.3577. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 76.Kumagai S, Koyama S, Itahashi K, Tanegashima T, Lin YT, Togashi Y, Kamada T, Irie T, Okumura G, Kono H, et al. (2022). Lactic acid promotes PD-1 expression in regulatory T cells in highly glycolytic tumor microenvironments. Cancer Cell 40, 201–218 e209. 10.1016/j.ccell.2022.01.001. [DOI] [PubMed] [Google Scholar]
- 77.de la Cruz-Lopez KG, Castro-Munoz LJ, Reyes-Hernandez DO, Garcia-Carranca A, and Manzo-Merino J. (2019). Lactate in the Regulation of Tumor Microenvironment and Therapeutic Approaches. Front Oncol 9, 1143. 10.3389/fonc.2019.01143. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 78.Baker FL, Smith KA, Zuniga TM, Batatinha H, Niemiro GM, Pedlar CR, Burgess SC, Katsanis E, and Simpson RJ (2021). Acute exercise increases immune responses to SARS CoV-2 in a previously infected man. Brain Behav Immun Health 18, 100343. 10.1016/j.bbih.2021.100343. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 79.Li F, and Simon MC (2020). Cancer Cells Don’t Live Alone: Metabolic Communication within Tumor Microenvironments. Dev Cell 54, 183–195. 10.1016/j.devcel.2020.06.018. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 80.Ohta A, Madasu M, Subramanian M, Kini R, Jones G, Chouker A, Ohta A, and Sitkovsky M. (2014). Hypoxia-induced and A2A adenosine receptor-independent T-cell suppression is short lived and easily reversible. Int Immunol 26, 83–91. 10.1093/intimm/dxt045. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 81.Fong L, Hotson A, Powderly JD, Sznol M, Heist RS, Choueiri TK, George S, Hughes BGM, Hellmann MD, Shepard DR, et al. (2020). Adenosine 2A Receptor Blockade as an Immunotherapy for Treatment-Refractory Renal Cell Cancer. Cancer Discov 10, 40–53. 10.1158/2159-8290.CD-19-0980. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 82.Liu JY, and Wellen KE (2020). Advances into understanding metabolites as signaling molecules in cancer progression. Curr Opin Cell Biol 63, 144–153. 10.1016/j.ceb.2020.01.013. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 83.Wu JY., Huang TW., Hsieh YT., Wang YF., Yen CC., Lee GL., Yeh CC., Peng YJ., Kuo YY., Wen HT., et al. (2020). Cancer-Derived Succinate Promotes Macrophage Polarization and Cancer Metastasis via Succinate Receptor. Mol Cell 77, 213–227 e215. 10.1016/j.molcel.2019.10.023. [DOI] [PubMed] [Google Scholar]
- 84.Bunse L, Pusch S, Bunse T, Sahm F, Sanghvi K, Friedrich M, Alansary D, Sonner JK, Green E, Deumelandt K, et al. (2018). Suppression of antitumor T cell immunity by the oncometabolite (R)-2-hydroxyglutarate. Nat Med 24, 1192–1203. 10.1038/s41591-018-0095-6. [DOI] [PubMed] [Google Scholar]
- 85.Ma X, Bi E, Lu Y, Su P, Huang C, Liu L, Wang Q, Yang M, Kalady MF, Qian J, et al. (2019). Cholesterol Induces CD8(+) T Cell Exhaustion in the Tumor Microenvironment. Cell Metab 30, 143–156 e145. 10.1016/j.cmet.2019.04.002. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 86.Keshet R, Szlosarek P, Carracedo A, and Erez A. (2018). Rewiring urea cycle metabolism in cancer to support anabolism. Nat Rev Cancer 18, 634–645. 10.1038/s41568-018-0054-z. [DOI] [PubMed] [Google Scholar]
- 87.Ochocki JD, Khare S, Hess M, Ackerman D, Qiu B, Daisak JI, Worth AJ, Lin N, Lee P, Xie H, et al. (2018). Arginase 2 Suppresses Renal Carcinoma Progression via Biosynthetic Cofactor Pyridoxal Phosphate Depletion and Increased Polyamine Toxicity. Cell Metab 27, 1263–1280 e1266. 10.1016/j.cmet.2018.04.009. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 88.Chantranupong L, Scaria SM, Saxton RA, Gygi MP, Shen K, Wyant GA, Wang T, Harper JW, Gygi SP, and Sabatini DM (2016). The CASTOR Proteins Are Arginine Sensors for the mTORC1 Pathway. Cell 165, 153–164. 10.1016/j.cell.2016.02.035. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 89.Geiger R, Rieckmann JC, Wolf T, Basso C, Feng Y, Fuhrer T, Kogadeeva M, Picotti P, Meissner F, Mann M, et al. (2016). L-Arginine Modulates T Cell Metabolism and Enhances Survival and Anti-tumor Activity. Cell 167, 829–842 e813. 10.1016/j.cell.2016.09.031. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 90.Mazzone M, Menga A, and Castegna A. (2018). Metabolism and TAM functions-it takes two to tango. FEBS J 285, 700–716. 10.1111/febs.14295. [DOI] [PubMed] [Google Scholar]
- 91.Opitz CA, Litzenburger UM, Sahm F, Ott M, Tritschler I, Trump S, Schumacher T, Jestaedt L, Schrenk D, Weller M, et al. (2011). An endogenous tumour-promoting ligand of the human aryl hydrocarbon receptor. Nature 478, 197–203. 10.1038/nature10491. [DOI] [PubMed] [Google Scholar]
- 92.Rothhammer V, and Quintana FJ (2019). The aryl hydrocarbon receptor: an environmental sensor integrating immune responses in health and disease. Nat Rev Immunol 19, 184–197. 10.1038/s41577-019-0125-8. [DOI] [PubMed] [Google Scholar]
- 93.Mezrich JD, Fechner JH, Zhang X, Johnson BP, Burlingham WJ, and Bradfield CA (2010). An interaction between kynurenine and the aryl hydrocarbon receptor can generate regulatory T cells. J Immunol 185, 3190–3198. 10.4049/jimmunol.0903670. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 94.Mondanelli G., Ugel S., Grohmann U., and Bronte V. (2017). The immune regulation in cancer by the amino acid metabolizing enzymes ARG and IDO. Curr Opin Pharmacol 35, 30–39. 10.1016/j.coph.2017.05.002. [DOI] [PubMed] [Google Scholar]
- 95.Liu Y, Liang X, Dong W, Fang Y, Lv J, Zhang T, Fiskesund R, Xie J, Liu J, Yin X, et al. (2018). Tumor-Repopulating Cells Induce PD-1 Expression in CD8(+) T Cells by Transferring Kynurenine and AhR Activation. Cancer Cell 33, 480–494 e487. 10.1016/j.ccell.2018.02.005. [DOI] [PubMed] [Google Scholar]
- 96.Li X, Wenes M, Romero P, Huang SC, Fendt SM, and Ho PC (2019). Navigating metabolic pathways to enhance antitumour immunity and immunotherapy. Nat Rev Clin Oncol 16, 425–441. 10.1038/s41571-019-0203-7. [DOI] [PubMed] [Google Scholar]
- 97.Ma EH, Bantug G, Griss T, Condotta S, Johnson RM, Samborska B, Mainolfi N, Suri V, Guak H, Balmer ML, et al. (2017). Serine Is an Essential Metabolite for Effector T Cell Expansion. Cell Metab 25, 482. 10.1016/j.cmet.2017.01.014. [DOI] [PubMed] [Google Scholar]
- 98.Martinez-Reyes I, and Chandel NS (2021). Cancer metabolism: looking forward. Nat Rev Cancer 21, 669–680. 10.1038/s41568-021-00378-6. [DOI] [PubMed] [Google Scholar]
- 99.Wang W, Green M, Choi JE, Gijon M, Kennedy PD, Johnson JK, Liao P, Lang X, Kryczek I, Sell A, et al. (2019). CD8(+) T cells regulate tumour ferroptosis during cancer immunotherapy. Nature 569, 270–274. 10.1038/s41586-019-1170-y. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 100.Philip M, and Schietinger A. (2022). CD8(+) T cell differentiation and dysfunction in cancer. Nat Rev Immunol 22, 209–223. 10.1038/s41577-021-00574-3. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 101.Scharping NE, Menk AV, Moreci RS, Whetstone RD, Dadey RE, Watkins SC, Ferris RL, and Delgoffe GM (2016). The Tumor Microenvironment Represses T Cell Mitochondrial Biogenesis to Drive Intratumoral T Cell Metabolic Insufficiency and Dysfunction. Immunity 45, 701–703. 10.1016/j.immuni.2016.08.009. [DOI] [PubMed] [Google Scholar]
- 102.Morad G, Helmink BA, Sharma P, and Wargo JA (2021). Hallmarks of response, resistance, and toxicity to immune checkpoint blockade. Cell 184, 5309–5337. 10.1016/j.cell.2021.09.020. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 103.Patsoukis N, Bardhan K, Chatterjee P, Sari D, Liu B, Bell LN, Karoly ED, Freeman GJ, Petkova V, Seth P, et al. (2015). PD-1 alters T-cell metabolic reprogramming by inhibiting glycolysis and promoting lipolysis and fatty acid oxidation. Nat Commun 6, 6692. 10.1038/ncomms7692. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 104.Kalia V, Yuzefpolskiy Y, Vegaraju A, Xiao H, Baumann F, Jatav S, Church C, Prlic M, Jha A, Nghiem P, et al. (2021). Metabolic regulation by PD-1 signaling promotes long-lived quiescent CD8 T cell memory in mice. Sci Transl Med 13, eaba6006. 10.1126/scitranslmed.aba6006. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 105.Zappasodi R, Serganova I, Cohen IJ, Maeda M, Shindo M, Senbabaoglu Y, Watson MJ, Leftin A, Maniyar R, Verma S, et al. (2021). CTLA-4 blockade drives loss of Treg stability in glycolysis-low tumours. Nature. 10.1038/s41586-021-03326-4. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 106.Beckermann KE, Hongo R, Ye X, Young K, Carbonell K, Healey DCC, Siska PJ, Barone S, Roe CE, Smith CC, et al. (2020). CD28 costimulation drives tumor-infiltrating T cell glycolysis to promote inflammation. JCI Insight 5. 10.1172/jci.insight.138729. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 107.Gordon SR., Maute RL., Dulken BW., Hutter G., George BM., McCracken MN., Gupta R., Tsai JM., Sinha R., Corey D., et al. (2017). PD-1 expression by tumour-associated macrophages inhibits phagocytosis and tumour immunity. Nature 545, 495–499. 10.1038/nature22396. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 108.Strauss L, Mahmoud MAA, Weaver JD, Tijaro-Ovalle NM, Christofides A, Wang Q, Pal R, Yuan M, Asara J, Patsoukis N, and Boussiotis VA (2020). Targeted deletion of PD-1 in myeloid cells induces antitumor immunity. Sci Immunol 5. 10.1126/sciimmunol.aay1863. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 109.Finck AV, Blanchard T, Roselle CP, Golinelli G, and June CH (2022). Engineered cellular immunotherapies in cancer and beyond. Nat Med 28, 678–689. 10.1038/s41591-022-01765-8. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 110.Rosenberg SA, and Restifo NP (2015). Adoptive cell transfer as personalized immunotherapy for human cancer. Science 348, 62–68. 10.1126/science.aaa4967. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 111.Sukumar M, Liu J, Mehta GU, Patel SJ, Roychoudhuri R, Crompton JG, Klebanoff CA, Ji Y, Li P, Yu Z, et al. (2016). Mitochondrial Membrane Potential Identifies Cells with Enhanced Stemness for Cellular Therapy. Cell Metab 23, 63–76. 10.1016/j.cmet.2015.11.002. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 112.Adams WC, Chen YH, Kratchmarov R, Yen B, Nish SA, Lin WW, Rothman NJ, Luchsinger LL, Klein U, Busslinger M, et al. (2016). Anabolism-Associated Mitochondrial Stasis Driving Lymphocyte Differentiation over Self-Renewal. Cell Rep 17, 3142–3152. 10.1016/j.celrep.2016.11.065. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 113.Kawalekar OU, O’Connor RS, Fraietta JA, Guo L, McGettigan SE, Posey AD Jr., Patel PR, Guedan S, Scholler J, Keith B, et al. (2016). Distinct Signaling of Coreceptors Regulates Specific Metabolism Pathways and Impacts Memory Development in CAR T Cells. Immunity 44, 380–390. 10.1016/j.immuni.2016.01.021. [DOI] [PubMed] [Google Scholar]
- 114.Ye L, Park JJ, Peng L, Yang Q, Chow RD, Dong MB, Lam SZ, Guo J, Tang E, Zhang Y, et al. (2022). A genome-scale gain-of-function CRISPR screen in CD8 T cells identifies proline metabolism as a means to enhance CAR-T therapy. Cell Metab 34, 595–614 e514. 10.1016/j.cmet.2022.02.009. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 115.Arner EN, Du W, and Brekken RA (2019). Behind the Wheel of Epithelial Plasticity in KRAS-Driven Cancers. Front Oncol 9, 1049. 10.3389/fonc.2019.01049. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 116.Pei D, Shu X, Gassama-Diagne A, and Thiery JP (2019). Mesenchymal-epithelial transition in development and reprogramming. Nat Cell Biol 21, 44–53. 10.1038/s41556-018-0195-z. [DOI] [PubMed] [Google Scholar]
- 117.Dongre A, and Weinberg RA (2019). New insights into the mechanisms of epithelial-mesenchymal transition and implications for cancer. Nat Rev Mol Cell Biol 20, 69–84. 10.1038/s41580-018-0080-4. [DOI] [PubMed] [Google Scholar]
- 118.El-Kenawi A, Hanggi K, and Ruffell B. (2020). The Immune Microenvironment and Cancer Metastasis. Cold Spring Harb Perspect Med 10. 10.1101/cshperspect.a037424. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 119.Marengo B., Nitti M., Furfaro AL., Colla R., Ciucis CD., Marinari UM., Pronzato MA., Traverso N., and Domenicotti C. (2016). Redox Homeostasis and Cellular Antioxidant Systems: Crucial Players in Cancer Growth and Therapy. Oxid Med Cell Longev 2016, 6235641. 10.1155/2016/6235641. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 120.Schworer S, Berisa M, Violante S, Qin W, Zhu J, Hendrickson RC, Cross JR, and Thompson CB (2020). Proline biosynthesis is a vent for TGFbeta-induced mitochondrial redox stress. EMBO J 39, e103334. 10.15252/embj.2019103334. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 121.Cheung EC, DeNicola GM, Nixon C, Blyth K, Labuschagne CF, Tuveson DA, and Vousden KH (2020). Dynamic ROS Control by TIGAR Regulates the Initiation and Progression of Pancreatic Cancer. Cancer Cell 37, 168–182 e164. 10.1016/j.ccell.2019.12.012. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 122.Xu WN, Yang RZ, Zheng HL, Jiang LS, and Jiang SD (2020). NDUFA4L2 Regulated by HIF-1alpha Promotes Metastasis and Epithelial-Mesenchymal Transition of Osteosarcoma Cells Through Inhibiting ROS Production. Front Cell Dev Biol 8, 515051. 10.3389/fcell.2020.515051. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 123.Dey S, Sayers CM, Verginadis II, Lehman SL, Cheng Y, Cerniglia GJ, Tuttle SW, Feldman MD, Zhang PJ, Fuchs SY, et al. (2015). ATF4-dependent induction of heme oxygenase 1 prevents anoikis and promotes metastasis. J Clin Invest 125, 2592–2608. 10.1172/JCI78031. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 124.Lignitto L, LeBoeuf SE, Homer H, Jiang S, Askenazi M, Karakousi TR, Pass HI, Bhutkar AJ, Tsirigos A, Ueberheide B, et al. (2019). Nrf2 Activation Promotes Lung Cancer Metastasis by Inhibiting the Degradation of Bach1. Cell 178, 316–329 e318. 10.1016/j.cell.2019.06.003. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 125.Wiel C, Le Gal K, Ibrahim MX, Jahangir CA, Kashif M, Yao H, Ziegler DV, Xu X, Ghosh T, Mondal T, et al. (2019). BACH1 Stabilization by Antioxidants Stimulates Lung Cancer Metastasis. Cell 178, 330–345 e322. 10.1016/j.cell.2019.06.005. [DOI] [PubMed] [Google Scholar]
- 126.Sciacovelli M, Goncalves E, Johnson TI, Zecchini VR, da Costa AS, Gaude E, Drubbel AV, Theobald SJ, Abbo SR, Tran MG, et al. (2016). Fumarate is an epigenetic modifier that elicits epithelial-to-mesenchymal transition. Nature 537, 544–547. 10.1038/nature19353. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 127.Sullivan WJ., Mullen PJ., Schmid EW., Flores A., Momcilovic M., Sharpley MS., Jelinek D., Whiteley AE., Maxwell MB., Wilde BR., et al. (2018). Extracellular Matrix Remodeling Regulates Glucose Metabolism through TXNIP Destabilization. Cell 175, 117–132 e121. 10.1016/j.cell.2018.08.017. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 128.Gomes AP, Ilter D, Low V, Endress JE, Fernandez-Garcia J, Rosenzweig A, Schild T, Broekaert D, Ahmed A, Planque M, et al. (2020). Age-induced accumulation of methylmalonic acid promotes tumour progression. Nature 585, 283–287. 10.1038/s41586-020-2630-0. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 129.Hao Q, Li T, Zhang X, Gao P, Qiao P, Li S, and Geng Z. (2014). Expression and roles of fatty acid synthase in hepatocellular carcinoma. Oncol Rep 32, 2471–2476. 10.3892/or.2014.3484. [DOI] [PubMed] [Google Scholar]
- 130.Jiang L, Wang H, Li J, Fang X, Pan H, Yuan X, and Zhang P. (2014). Upregulated FASN expression promotes transcoelomic metastasis of ovarian cancer cell through epithelial-mesenchymal transition. Int J Mol Sci 15, 11539–11554. 10.3390/ijms150711539. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 131.Yasumoto Y, Miyazaki H, Vaidyan LK, Kagawa Y, Ebrahimi M, Yamamoto Y, Ogata M, Katsuyama Y, Sadahiro H, Suzuki M, and Owada Y. (2016). Inhibition of Fatty Acid Synthase Decreases Expression of Stemness Markers in Glioma Stem Cells. PLoS One 11, e0147717. 10.1371/journal.pone.0147717. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 132.Ahmad I, Mui E, Galbraith L, Patel R, Tan EH, Salji M, Rust AG, Repiscak P, Hedley A, Markert E, et al. (2016). Sleeping Beauty screen reveals Pparg activation in metastatic prostate cancer. Proc Natl Acad Sci U S A 113, 8290–8295. 10.1073/pnas.1601571113. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 133.Jeon SM, Chandel NS, and Hay N. (2012). AMPK regulates NADPH homeostasis to promote tumour cell survival during energy stress. Nature 485, 661–665. 10.1038/nature11066. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 134.Rossi M, Altea-Manzano P, Demicco M, Doglioni G, Bornes L, Fukano M, Vandekeere A, Cuadros AM, Fernandez-Garcia J, Riera-Domingo C, et al. (2022). PHGDH heterogeneity potentiates cancer cell dissemination and metastasis. Nature 605, 747–753. 10.1038/s41586-022-04758-2. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 135.Ubellacker JM, and Morrison SJ (2019). Metabolic Adaptation Fuels Lymph Node Metastasis. Cell Metab 29, 785–786. 10.1016/j.cmet.2019.03.006. [DOI] [PubMed] [Google Scholar]
- 136.Labuschagne CF, Cheung EC, Blagih J, Domart MC, and Vousden KH (2019). Cell Clustering Promotes a Metabolic Switch that Supports Metastatic Colonization. Cell Metab 30, 720–734 e725. 10.1016/j.cmet.2019.07.014. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 137.Tasdogan A, Faubert B, Ramesh V, Ubellacker JM, Shen B, Solmonson A, Murphy MM, Gu Z, Gu W, Martin M, et al. (2020). Metabolic heterogeneity confers differences in melanoma metastatic potential. Nature 577, 115–120. 10.1038/s41586-019-1847-2. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 138.Le Gal K, Ibrahim MX, Wiel C, Sayin VI, Akula MK, Karlsson C, Dalin MG, Akyurek LM, Lindahl P, Nilsson J, and Bergo MO (2015). Antioxidants can increase melanoma metastasis in mice. Sci Transl Med 7, 308re308. 10.1126/scitranslmed.aad3740. [DOI] [PubMed] [Google Scholar]
- 139.Sayin VI, Ibrahim MX, Larsson E, Nilsson JA, Lindahl P, and Bergo MO (2014). Antioxidants accelerate lung cancer progression in mice. Sci Transl Med 6, 221ra215. 10.1126/scitranslmed.3007653. [DOI] [PubMed] [Google Scholar]
- 140.LeBleu VS, O’Connell JT, Gonzalez Herrera KN, Wikman H, Pantel K, Haigis MC, de Carvalho FM, Damascena A, Domingos Chinen LT, Rocha RM, et al. (2014). PGC-1alpha mediates mitochondrial biogenesis and oxidative phosphorylation in cancer cells to promote metastasis. Nat Cell Biol 16, 992–1003, 1001–1015. 10.1038/ncb3039. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 141.Ngo B., Kim E., Osorio-Vasquez V., Doll S., Bustraan S., Liang RJ., Luengo A., Davidson SM., Ali A., Ferraro GB., et al. (2020). Limited Environmental Serine and Glycine Confer Brain Metastasis Sensitivity to PHGDH Inhibition. Cancer Discov 10, 1352–1373. 10.1158/2159-8290.CD-19-1228. [DOI] [PMC free article] [PubMed] [Google Scholar]