

Abstract
Treatment strategies with a strong scientific rationale based on specific biomarkers are needed to improve outcomes in patients with advanced sarcomas. Suppression of cell cycle progression through reactivation of the tumor suppressor retinoblastoma (Rb) using CDK4/6 inhibitors is a potential avenue for novel targeted therapies in sarcomas that harbor intact Rb signaling. Here, we evaluated combination treatment strategies (sequential and concomitant) with the CDK4/6 inhibitor abemacicib to identify optimal combination strategies. Expression of Rb was examined in 1043 sarcoma tumor specimens, and 50% were found to be Rb-positive. Using in vitro and in vivo models, an effective 2-step sequential combination strategy was developed. Abemaciclib was used first to prime Rb-positive sarcoma cells to reversibly arrest in G1-phase. Upon drug removal, cells synchronously traversed to S-phase, where a second treatment with S-phase targeted agents (gemcitabine or WEE1-kinase inhibitor) mediated a synergistic response by inducing DNA damage. The response to treatment could be non-invasively monitored using real-time positron emission tomography (PET) imaging and serum thymidine kinase activity. Collectively, these results show that a novel, sequential treatment strategy with a CDK4/6 inhibitor followed by a DNA damaging agent was effective, resulting in synergistic tumor cell killing. This approach can be readily translated into a clinical trial with non-invasive functional imaging and serum biomarkers as indicators of response and cell cycling.
Keywords: CDK4/6 inhibitor, soft tissue sarcomas, biomarkers, functional imaging, sequential combination treatment
Introduction
Soft tissue sarcomas (STS) are a rare and highly heterogeneous group of tumors that account for 1% of adult malignancies and 20% of pediatric cancers (1). There are more than 50 distinct subtypes of STS; leiomyosarcoma (LMS), liposarcoma (LPS), and rhabdomyosarcoma (RMS) represent the most common types of adult and pediatric cases (2,3). Surgical resection, with or without radiotherapy and chemotherapy, has been the cornerstone of therapeutic intervention for STS patients (4). In the advanced setting, first-line chemotherapy consists of doxorubicin (5), ifosfamide (6), or gemcitabine (7), given as single agents or in combination, with response rates in the range of 10%–25% (8,9). Prognosis is poor with a median overall survival (OS) of 12.8–14.3 months (10). Therefore, new treatment strategies that exploit specific targets/pathways are required to improve outcomes.
One such target is the retinoblastoma (Rb) protein and pathway, which are commonly altered in many adult and pediatric STSs. Rb is a tumor-suppressor protein; in its hypophosphorylated state, it sequesters the E2F transcription factor (11), inhibiting its ability to transcribe the genes required for G1 to S phase transition (12). Rb is sequentially phosphorylated by cyclin-dependent-kinase (CDK)4/cyclin D or CDK6/cyclin D and CDK2/cyclin E complexes at early and late G1 phases leading to transactivation of key cell cycle genes and progression of cells into S phase by releasing E2F from the Rb/E2F complex. Multiple STS subtypes, including some LPS, and RMS, exhibit amplification of chromosome 12q13–15, a region where the CDK4 gene is located (13,14). Overexpression or amplification of CDK4 results in inactivation of Rb as a tumor suppressor. Hence, the suppression of cell cycle progression through reactivation of the Rb pathway poses a potential avenue for novel targeted therapies in STS that harbor intact Rb signaling. Clinical efficacy data with single agent abemaciclib (CDK4/6 inhibitor) in adult dedifferentiated LPS patients (NCT02846987) showed median progression-free survival (PFS) was prolonged to 30.4 weeks but did not meet the criterion for partial response, underscoring the need for biomarkers and rational combination treatments for STS patients who receive CDK4/6 inhibitors (CDK4/6is) (15).
Concurrent combinations of CDK4/6is with chemotherapy have been examined in several pre-clinical models but antagonistic responses have been observed (16). One reason these concurrent combination therapies are not effective, is that CDK4/6is induce G1 arrest and prevent S phase specific chemotherapeutic agents from mediating their therapeutic effects (17). To address this challenge, a two-step sequential combination treatment strategy was designed in which CDK4/6is and chemotherapy dosing were separated (i.e. sequential) to induce a synergistic cell killing in STS models in vitro and in vivo. The impact of each agent on the cell cycle was evaluated real-time through functional imaging and non-invasive blood-based biomarkers, providing a potential treatment strategy for patients with STS harboring an intact Rb pathway.
Materials and methods
Tumor microarray
Formalin-fixed, paraffin-embedded (FFPE) sarcoma surgical specimens (n=1043) were retrieved from the University of Texas MD Anderson Cancer Center (MDACC, Houston, TX) pathology archives under an insititutional review board (IRB)-approved protocol and all patients provided written, informed consent. Clinical and pathological characteristics are summarized in Table S1. Whole slide H&E-stained sections were reviewed by a pathologist, and areas of viable tumor were selected and marked for sampling. Immunohistochemistry (IHC) was performed on FFPE tissue sections in the clinical IHC laboratory, which is certified under the provisions of the United States Clinical Laboratory Improvement Act and accredited by the College of American Pathologists. Purified mouse anti-human Rb protein (clone G3–245, BD Pharmagen™, RRID:AB_385259) antibodies were used for IHC staining (Supplementary Materials and Methods). All sections were scored independently by two pathologists to estimate the percentage of Rb-positive tumor cells. A cut-off point of 5% immunoreactive tumor cells was used to define a positive phenotype for Rb.
Cell lines and cell culture
Sarcoma cell lines were purchased from ATCC and maintained as described previously (18). Rhabdomyosarcoma cells were gifted by Dr. Peter J. Houghton (Greehey Children’s Cancer Research Institute, The University of Texas Health Science Center at San Antonio). Cell lines were regularly tested for Mycoplasma and authenticated regularly (every 6 months) by karyotype and short tandem repeat analysis at MDACC Characterized Cell Line Core Facility (Houston, TX). The detailed procedures for all in vitro assays (western blot analysis, high-through-put survival assay, cell cycle analysis, apoptosis assay, immunofluorescence staining, and immunohistochemical staining) were described previously (18–24) and are included in the Supplementary Materials and Methods.
Generation of CRISPR knock out clones and FUCCI cell lines
To generate RB1 knockout HT-1080 cell lines, sgRNA targeting exon 1 (GGTGGCGGCCGTTTTTCGGG) or exon 20 (GGATCTTCCTCATGCTGTTC) of human RB1 gene was cloned into pX330 plasmid (RRID:Addgene_101733). For human TP53 knockout, sgRNA targeting exon 9 (GGAGAGGAGCTGGTGTTGT) was used. FUCCI-expressing cells were generated by transducing HT-1080 cells with VSV-G pseudotyped lentiviral particles containing pBOB-EF1-FastFUCCI-Puro (Addgene; Plasmid #86849; RRID:Addgene_86849).
Combination index and synergy analysis
The combination index and synergy scores were analyzed using cell viability data obtained from high-throughput screening assay on a 96-well plate format following previously published protocols from our laboratory (20,21,25). To better predict synergism of drug combination treatment, cell viability data collected from combination treatment was processed by 3 different mathematical models (1) Bliss-independence (26,27), (2) Bayesian dose-response framework (28) and (3) Chou-Talalay (29) The combination indices or summary synergy scores for each drug combination is the average of individual synergy scores obtained from each of the dose combination measurements. The detailed procedures for combination treatments and combination index and synergy analysis are included in the Supplementary Materials and Methods.
In vitro radio-biotracer uptake study
For the in vitro radio-tracer uptake test, cells seeded in 12 well plates were treated with IC25 concentrations of abemaciclib for 6 days before the indicated recovery period in drug-free media (cell density and drug concentration are presented in table S5). In vitro radio-biotracer uptake was performed following the methods of previous publications (30,31) and are presented in Supplementary Materials and Methods.
In vivo [18]F-FLT PET imaging
Preclinical [18F]FLT–PET and CT imaging were performed using a Bruker Albira PET/SPECT/CT scanner. Tumor-bearing mice were anesthetized and injected with 100–150 μCi of [18F]FLT tracer intravenously. After 45 minutes, PET imaging was initiated, followed by a subsequent CT scan (10 minutes) for anatomical reference. The imaging process was completed within 2 hrs of the tracer injection. The PET analysis was performed with PMOD software (version 3.612, PMOD Technologies, Ltd., RRID:SCR_016547). [18F]FLT uptake in tumors was reported as a ratio of standardized uptake value (SUV) activity, obtained using the SUV of the most intense pixel in the tumor regions and normalized with the corresponding SUVmax obtained from the right/left leg muscle value from the same mouse. A series of PET/CT images were captured for each mouse over 4 different time points (baseline-before treatment initiated, following 6 days of abemaciclib treatment, 3 and 6 days following abemaciclib removal).
DiviTum® assay for serum TK1 activity measurement
Blood samples were collected from tumor bearing mice by retro-orbital bleeding or cardiac puncture at baseline, day 7 (6 days on), day 9 (3 days off), and day 12 (6 days off) of the treatment cycle. Serum was isolated from each blood draw collection, aliquoted, and stored at −80C for future studies. Serum enzymatic activity of TK1 was determined by a DiviTum® assay (Biovica International, Upsala, Sweden) using a refined ELISA based method, according to the manufacturer’s instructions (32).
Xenograft models
All mouse experiments were performed under Institutional Animal Care and Use Committee–approved protocols at the University of Texas MD Anderson Cancer Center. For cell line xenograft models, 1×106 HT-1080 (RRID:CVCL_0317) or HT-RB1-KO cells suspended in 150 µL of matrigel were injected subcutaneously into the right flank of 4- to 6-week-old female nude mice, which were randomized into treatment groups once the tumors reached 200 mm3. Leiomyosarcoma PDX lines were generated from surgical samples under an institutional review board–approved protocol (LAB07–0659). Rhabdomyosarcoma PDX lines (Rh30, RRID:CVCL_0041 and Rh36, RRID:CVCL_M599) were gifted by Dr. Peter J. Houghton (Greehey Children’s Cancer Research Institute, University of Texas Health Science Center at San Antonio). IHC conditions, including the source for all antibodies used, are included in Supplementary Table S6. The patient details can be found in Supplementary Table S3 and the in vivo therapeutic efficacy details can be found in Supplementary Materials and Methods.
Quantification and Statistical Analysis
Differences in the cohorts were evaluated using fisher’s exact tests for categorical variables and ANOVA for age. The endpoint was OS, calculated from the time of diagnosis to death or last follow-up date. Patients who did not experience the endpoint were censored at last follow-up. Median follow-up times were computed using the reverse Kaplan-Meier estimator. The Kaplan–Meier method was used to calculate the 5-year OS rate for each factor. The differences in survival curves were evaluated using log-rank test. Univariable regression analyses were performed using the Cox proportional hazards model. Stata statistical software (SE 16, StataCorp LP, College Station, TX; RRID:SCR_012763) was used for statistical analyses. All P values were two-tailed, and P ≤ 0.05 was considered statistically significant.
Linear regression and Pearson correlation analyses between protein expression and abemaciclib sensitivity were performed using GraphPad Prism 9 (RRID:SCR_000306). All P values were two-tailed with a 95% confidence interval.
All experiments were conducted with at least 3 biological replicates; each biological replicate had at least two technical replicates. The statistical analysis was carried out using Prism 9 (Graphpad Software Inc.). All statistical tests of comparative data were performed using a two-sided, unpaired Student’s t-test or one-way or two-way ANOVA for a differential comparison between two or more groups, respectively. Statistical significance was denoted as p-value > 0.05 (ns), ≤ 0.05 (*), ≤ 0.01 (**), ≤ 0.001 (***), and ≤0.0001 (****).
Data Availability Statement
The data generated in the study is available within the article and its supplemental data files.
Results
Rb as a stratifying biomarker for sarcoma subtypes
To determine whether Rb protein can be used as a stratifying biomarker for targeted therapy in STS patients, a Clinical Laboratory Improvement Amendments (CLIA) approved Rb immunohistochemistry (IHC) assay was used to examine the expression of Rb in 1043 sarcoma tumor specimens including rhabdomyosarcoma (RMS, n=46), angiosarcoma (AS, n=113), undifferentiated pleomorphic sarcoma (UPS, n=102), radiation associated sarcoma (RAS, n=62), liposarcoma (LPS, n=100), uterine leiomyosarcoma (ULMS, n=211), leiomyosarcoma (ST-LMS n=195), and osteosarcoma (OS, n=214). Approximately 50% (range: 37% for OS to 91.3% for RMS) of all tumor specimens stained positive for Rb (cutoff at >5% positivity) (Figure 1A-C). Overall survival (OS) was examined as a function of Rb expression, in three patient cohorts (AS, ST-LMS, and ULMS) that had complete annotated follow-up with clinical and pathological characteristics (Table S1). OS analysis revealed that Rb status was not significantly associated with outcome (Figure 1D). The median OS times for AS, ULMS, and ST-LMS patients were similar between the Rb positive [Rb(+ve)] and Rb negative [Rb(-ve)] cohorts within each subtype (Table S1). Assessment of Rb staining in matched primary and recurrent tumor specimens from 24 patients (5 AS, 4 UPS, and 15 ULMS) showed that 80% of tumors retained Rb positivity in matched samples (Figure 1E). Since Rb is the substrate for CDK4/6, we hypothesized Rb(+ve) STS would be good candidates for CDK4/6 inhibitor (CDK4/6i)-based therapies, which are currently FDA-approved in advanced hormone receptor-positive, HER2-negative (HR+, HER2-) breast cancer patients (33–35).
Figure 1: Retinoblastoma (Rb) as a stratifying biomarker for sarcoma patients across all subtypes.
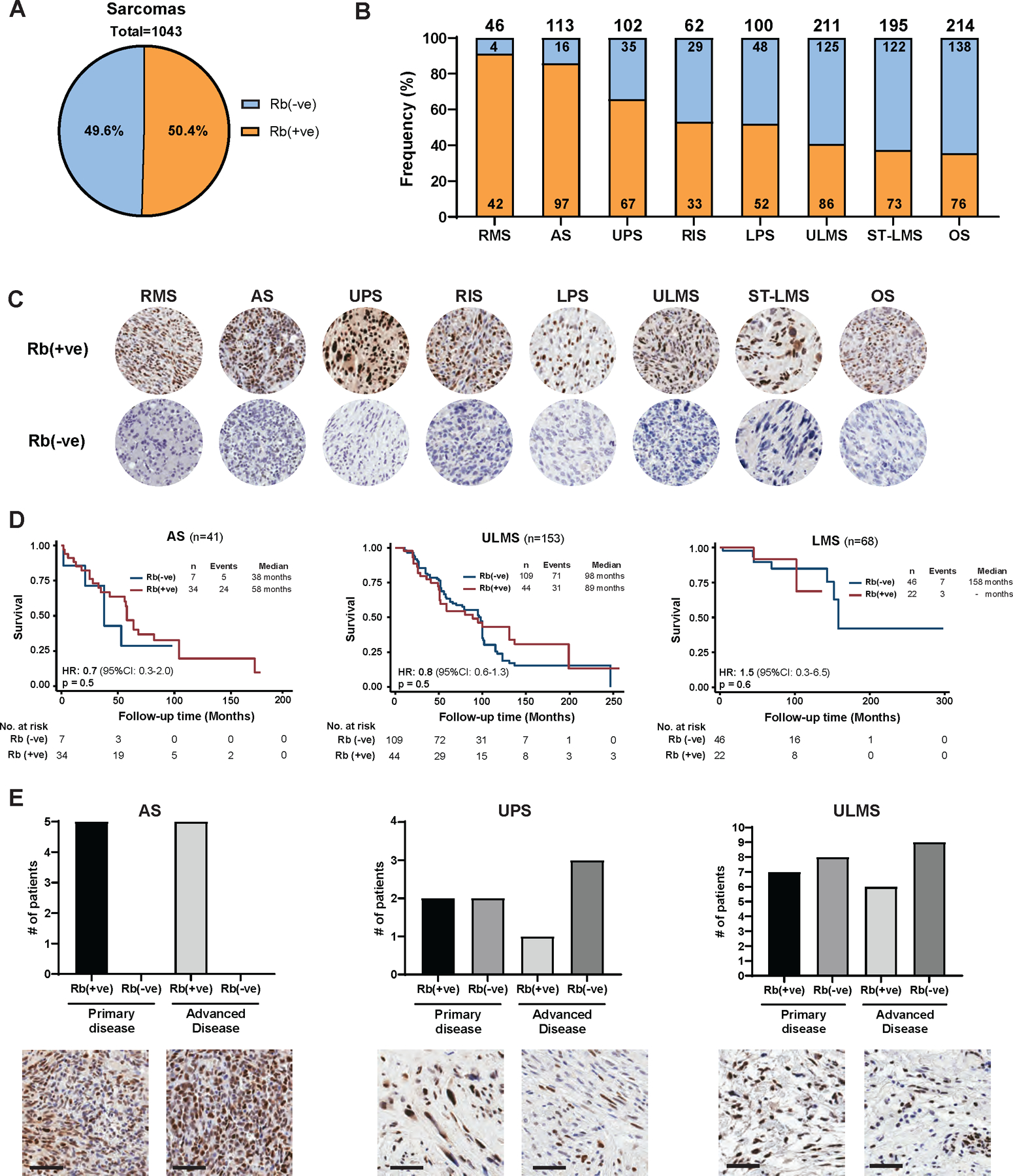
(A) Analysis of Rb status in tumor microarrays from a total of 1043 sarcoma tumor specimens (representing 7 different sarcoma subtypes: rhabdomyosarcoma (RMS), angiosarcoma (AS), undifferentiated pleomorphic sarcoma (UPS), radiation-associated sarcoma (RAS), liposarcoma (LPS), uterine leiomyosarcoma (ULMS), soft tissue leiomyosarcoma (ST-LMS), and osteosarcoma (OS) showing an average 50% of tumor samples stained positive for Rb (cut-off at >5% nuclear positivity). (B) Frequency of Rb-positive tumor samples in each sarcoma subtype. (C) Representative images of the Rb IHC from Rb-negative and Rb-postive samples are depicted for each sarcoma subtype (40X magnification). (D) Kaplan-Meier curves of overall survival as a function of Rb in 3 of the sarcoma subtypes. For this analysis, results from those patients who had already developed advanced disease were used. (E) Bar graphs showing the changes in Rb status detected in matched samples collected at primary and advanced stage (top). Representative images of the Rb IHC from a matched primary and advanced disease sample are depicted for each subtype (bottom, scale bar=60 µm).
Sarcoma cell line response to abemaciclib is dependent on Rb status
The CDK4/6is induce transient cell cycle inhibition, which can be reversed when the drug is removed. To sustain the therapeutic efficacy of CDK4/6is, a two-step sequential combination strategy was developed in which the CDK4/6i abemaciclib acts first to prime Rb-(+ve) sarcoma cells to reversibly arrest in G1 phase of the cell cycle (first-step) and upon drug removal induce synchronized traverse to S phase. The second drug, an S phase targeted agent (second-step) is then used to mediate a synergistic cell killing response (Figure S1A).
To identify Rb(+ve) and Rb(-ve) cell lines for analyzing drug sensitivity, four different sarcoma cell lines (OS, ST-LMS, UPS and RMS) (Table S2) were subjected to western blot analysis examining key cell cycle proteins governing the Rb pathway (Figure 2A). Total Rb and its phosphorylated forms (pRb) were expressed at high levels in HT-1080, SK-LMS1 and most RMS cell lines, whereas RD and SaOS2 exhibited low to undetectable protein levels (Figure 2A). Further analysis of the proteins that regulate the G1 to S transition (CDK4, CDK6, cyclin D1, p16 and, E2F1) revealed no detectable differences among the cell lines. The sensitivity of the cell lines to abemaciclib was examined and results revealed that while the doubling times (Td) of the cell lines at baseline were similar (47.3–54.8hrs) (Figure S1B) that there was a 10-fold difference in response to abemaciclib based on Rb status [IC50 in Rb(+ve) cell lines 0.1 µM-0.5 µM, and in Rb(-ve) cell lines 1 µM-5 µM] (Figure 2B). Pearson correlation analysis of the cell cycle protein levels (Figure 2A) and IC50 (Figure 2B) of all eight cell lines revealed that sensitivity to abemaciclib was significantly (p <0.05) associated with Rb and pRb status but not with any of the other cell cycle proteins examined (Figure 2C, Figure S1C). Knockout of Rb via CRIPSR/Cas9 in the Rb(+ve) HT-1080 cell line resulted in undetectable levels of Rb in HT-RB1-KO, resembling Rb(-ve) SaOS2 cells (Figure 2D, Figure S1D). The deletion of Rb did not modulate the cell cycle distribution or cell proliferation rates (Td = 39.3–48.5hrs) (Figure 2E-F, Figure S1D-E). However, with Rb deleted, HT-RB1-KO cells were resistant to abemaciclib with a 5-fold increase in IC50 (Figure 2G) (P<0.01). Compared to Rb knockout, p53 knockout had no effect on response to abemaciclib (Figure 2H-K). CRISPR/CAS9 knockout of p53 in the HT-1080 cell line (harboring wild-type p53) did not alter the Rb pathway protein expression (Figure 2H, Figure S1F), cell cycle profile (Figure 2I, Figure S1G), rate of proliferation (Figure 2J, Td = 45–48.5hrs) or response to abemaciclib (Figure 2K).
Figure 2. Rb-dependent response of sarcoma cell lines to abemaciclib.
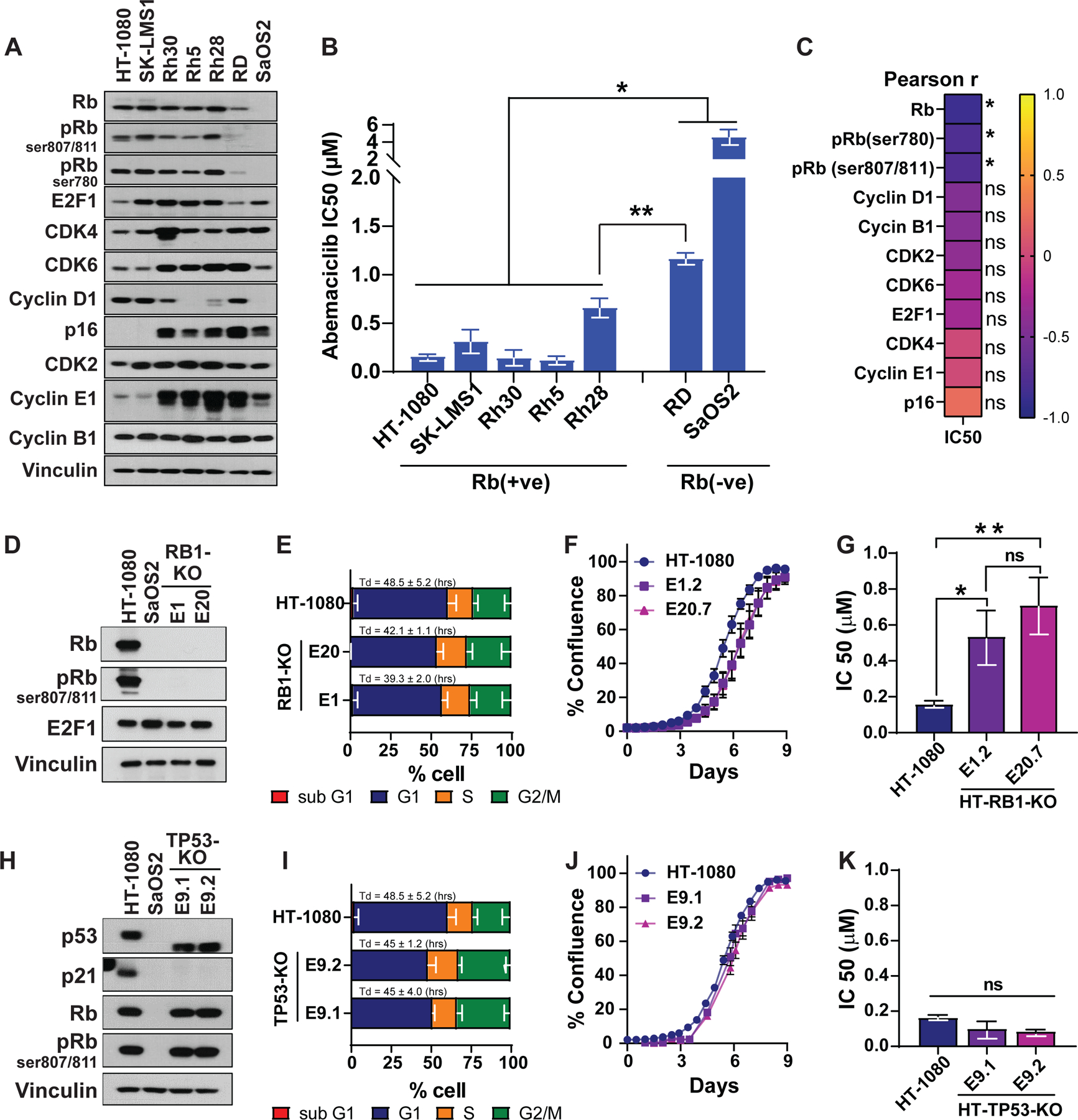
(A) Western blot analysis with the indicated antibodies to examine the status of the Rb pathway in a panel of sarcoma cell lines of differing histological subtypes (Table S2). (B) IC50 of abemaciclib following treatment of the 7 sarcoma cell lines for 6 days (n=3/cell line). (C) Pearson correlation (r) of abemaciclib IC50 in each cell line and the levels of indicated proteins, assessed through densitometry of the western blots from panel B. (D) Western blot analysis to confirm the successful deletion of RB1 (RB1-KO) in HT-1080 using CRISPR/Cas9 and sgRNA target exon 1 (E1) or exon 20 (E20) in an RB1-encoding gene. SaOS2 cells are used as a negative control for Rb expression. (E) Cell cycle analysis of parental HT-1080 and Rb-knockout clones show very similar doubling times among the three cell lines and similar changes in the cell cycle distribution at G1, S, and G2/M after Rb knockout (n=3). (F) Proliferation rates of HT-1080 parental and Rb-knockout cells showing very similar rates after RB1-KO. (G) IC50 of abemaciclib following 6 days of treatment of parental HT-1080 and the Rb-knockout clones (n=3). (H) Western blot analysis to confirm the successful deletion of TP53 in HT-1080 using CRISPR/Cas9 and sgRNA-targeting exon-9 in the TP53-encoding gene. (I) Cell cycle analysis of parental HT-1080 and TP53-knockout (TP53-KO) clones show very similar doubling times among the three cell lines with similar changes in cell cycle distribution at G1, S, and G2/M after TP53-KO (n=3). (J) Cell proliferation rates remained unchanged after p53 knockout. (K) IC50 of abemaciclib following 6 days of treatment of the parental HT-1080 and the TP53-KO clones (n=3). Experiments are representative of at least three independent biological replicates. The statistical analyses were performed using one- or two-way ANOVA. *p <0.05, **p ≤ 0.01.
Sequential treatment of Rb-positive sarcoma cells with abemaciclib, followed by DNA damaging agents results in synergistic cell killing
Rb status and transition of cells through each phase of the cell cycle were examined during and after treatment with abemaciclib. Treatment with increasing concentrations of abemaciclib for 6 days induced significant (p<0.05) G1-arrest in all Rb(+ve) cell lines (80%−90% G1 cells) but not in Rb-knockout (HT-RB1-KO E20 sgRNA) or Rb-deficient cells (SaOS2 and RD) (Figure 3A, B and Figure S2A).
Figure 3. Sequential treatment of Rb-positive sarcoma cells with abemaciclib, followed by DNA-damaging agents, is synergistic.
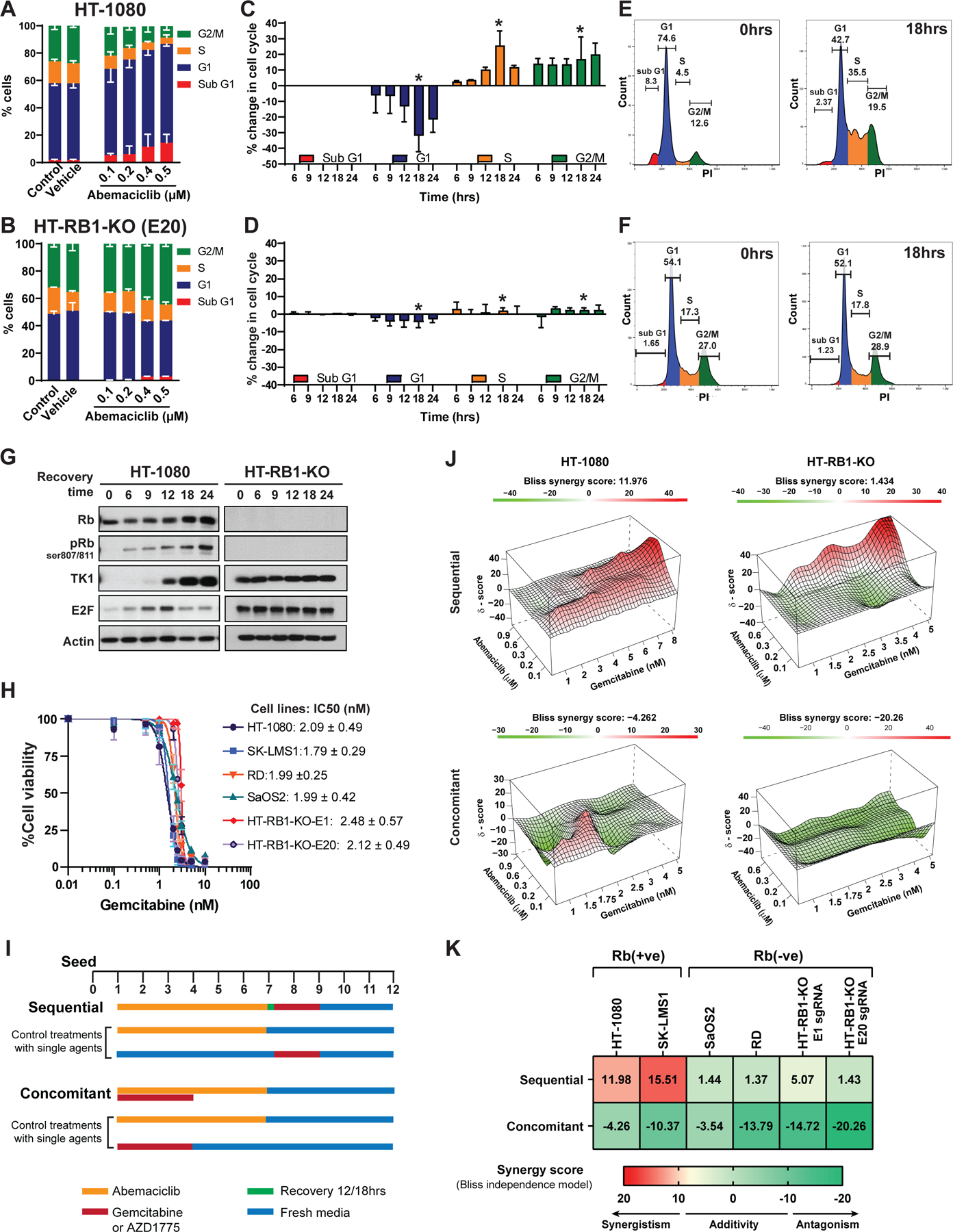
Cell cycle distribution of (A) HT-1080 [Rb(+ve)] and (B) HT-RB1-KO (E20) [Rb(-ve)] treated with the indicated concentrations of abemaciclib for 6 days prior to fixation and staining with PI (n = 3); the dose-dependent G1-arrest in the Rb(+ve) cell line is shown. (C-D) Kinetics of cell-cycle progression post-removal of abemaciclib treatment (n = 3) indicating that Rb(+ve) cells (C) synchronously progress through the S and G2/M phases of cell cycle after abemaciclib treatment. Cells were treated with the indicated concentrations of abemaciclib for 6 days, at which point the drug was removed and cells were allowed to recover in drug free media as indicated. Percentage change in the proportion of cells in each phase of the cell cycle was normalized to treated cells. (E-F) Representative cell cycle histograms at 0hrs and the maximum recovery time point, indicated by * in panels C and D. (G) Western blot analysis with the indicated proteins showing the expression of G1 checkpoint and E2F target proteins during abemaciclib recovery from cell lysates collected at the indicated time points from (C) and (D). (H) Dose response of sarcoma cell lines treated with gemcitabine. Cells were treated with different concentrations of gemcitabine for 72hrs followed by 9 days of recovery in drug free media. (I) Flow diagram depicting treatment schedules for the sequential and concomitant combination of abemaciclib and gemcitabine (or AZD1775). For sequential treatment, cells were treated with abemaciclib for 6 days, followed by recovery for 12–18hrs and gemcitabine for 3 days. For concomitant treatment, cells were treated with gemcitabine for 72hrs and abemaciclib for 6 days. Both treatments started at the same time on day 1. Control plates for cells treated with single agents were carried out in parallel with combination treatments under the same conditions. At the completion of drug treatment, cells were continued to be cultured in drug-free medium (also replaced every other day) until day 12, after which cell confluence was detected by Incucyte and crystal violet. All experiments were conducted with two biological replicates, each biological replicate contained 6 technical repeats. (J) 3D synergy maps highlighting synergistic (red) and antagonistic (green) dose regions of sequential and concomitant treatment in Rb(+ve) and Rb(-ve) cell lines analyzed by SynergyFinder using Bliss independence model. (K) Heatmap depicts the average synergy scores calculated using Bliss independence model for each pair of sequential or concomitant treatment using multiple drug concentrations for each agent. Synergism (synergy score >10), additivity (synergy score from −10 to 10) or antagonism (synergy score < −10) are indicated by red, yellow and green, respectively. The combination treatment is synergistic only in Rb(+ve) cell lines when they are treated with abemaciclib and gemcitabine sequentially. Experiments are representative of three independent biological replicates.
The next step was to determine whether the anti-proliferative effect of abemaciclib in Rb(+ve) cells is reversible and if this effect allows cells to synchronously re-enter the cell cycle upon drug removal. Results showed that Rb(+ve) cells (Rh30, SK-LMS1 and HT-1080) resumed cycling and synchronously entered S phase (Figure 3C-E, and Figure S2B-C). Within 9–12hrs after abemaciclib removal, the fraction of cells in S phase increased from 4% to 35% (i.e. 8.75-fold) between pre- and post-recovery time intervals in HT-1080 cells and increased 8.1- and 2.7-fold in SK-LMS1 and Rh30 cells, respectively. In contrast, the proportion of cells in S phase remained unchanged in all three Rb(-ve) cell lines (20%, 17.2% and 19.8 % in HT-RB1-KO, SaOS2, and RD cells, respectively) after 24hrs of drug removal (Figure 3D-F and Figure S2B-C).
The reversible G1 arrest following abemaciclib removal led to sequential changes in the expression of cell cycle regulators in Rb(+ve), but not Rb(-ve) cell lines, which can be rationally targeted using sequential combination treatment with S (gemcitabine) or G2/M (Wee-1 kinase inhibitor) targeting agents (Figure 3G, Figure S2D-F, Figure S3, S4, and S5). These changes included a reduction in Rb phosphorylation on serine 807/811 (CDK4/6 phosphorylated site), E2F1, and thymidine kinase 1 (TK1, an E2F1 target), followed by temporal inductions without any detectable changes in CDK4, CDK6, and cyclin D1 expression, only in Rb(+ve) cells (Figure S2E-F, Figure S3A-B). Examination of late S and G2/M regulatory proteins revealed an increase in cyclin B, CDK1, phospho-CDK1 (pCDK1), and Wee-1 kinase levels (Figure S5A-B). The expression of E2F1 and its downstream targets in Rb(-ve) cells remained unchanged during the recovery period (Figure 3G, Figure S2D-F, Figure S3A-B and Figure S5A-C).
Sequential combination treatment with abemaciclib, followed by S phase targeting agents was examined with gemcitabine, which is often utilized in STS patients, and a Wee-1 kinase inhibitor (AZD1775) as a late S and G2/M targeting agent for these studies. All sarcoma cell lines exhibited similar sensitivities to single agent gemcitabine (IC50 1.9–2.2 nM; (Figure 3H) or AZD1775 ( IC50 0.2–0.4 µM; Figure S5D), independent of Rb status.
Sequential and concomitant combination treatment with abemaciclib, followed by either gemcitabine or AZD1775, was examined in Rb(+ve) and Rb(-ve) cell lines as depicted by the treatment schedules shown in Figure 3I, using the high-throughput survival assay (HTSA) methodology. HTSA was used to increase the capacity of screening drug combination in many cell lines versus classical clonogenic assays, which is not conducive to high-throughput combination treatment experiments. However, in side-by-side comparisons, the HTSA and clonogenic assays are highly concordant (24,36–38) and HTSA has been routinely used to determine combination drug synergy in a variety of cell lines (23,39–43). The sensitivity of drug combination was initially accessed by comparing viability of cells treated with single agents versus combination treatment. The results showed that only Rb(+ve), but not Rb (-ve) cell lines, were significantly (p< 0.05) sensitive to sequential combination treatment of abemaciclib and gemcitabine (Figure S3C-D) or Wee-1 kinase inhibitor (Figure S3E-F).
To further infer the degree of drug interactions that contributes to the drug combination sensitivity, independent of the single drug effects, synergy scores of drug combinations were calculated using three different mathematical models (1) Bliss independence (by SynergyFinder) (44) and (2) Bayesian dose-response framework (using “d-chain” program) (28) for concomitant and sequential treatment strategies and (3) Chou-Talalay models for concomitant treatment strategies (using Calcusyn software) (45) (Figure S3J-K, Fgure S4A-B, and Figure S5E-H). The summary synergy scores for each drug combination is averaged over all the dose combination measurements for each method. Using Bliss independence model, our analysis showed that the sequential combination treatment with abemaciclib, followed by drug wash out, recovery and addition of gemcitabine or AZD1775, resulted in synergistic cell killing in Rb(+ve) with synergy scores greater than 10, indicating synergism of the sequential combination treatment in these cell lines (Figure 3J-K, Figure S4A, Figure S5E-F). In contrast, Rb(-ve) cell lines treated sequentially have synergy scores ranging from −10 to 7, underscoring the additive and antagonistic nature of the sequential combination treatments in these cell lines. Similarly, concomitant combination treatment failed to induce drug synergy in either Rb(+ve) or Rb(-ve) cell lines (Figure 3J-K, Figure S4A, Figure S5E-F).
Consistent to the synergy scores calculated by Bliss independence model, the synergy scores obtained from Bayesian dose-response framework for sequential drug treatment in Rb(+ve) cell line were at least 4-fold higher than those obtained in Rb(-ve) cell lines (Figure S4Cand S5H). Using the Chou-Talalay approach, the concomitant treatment with abemaciclib + gemcitabine or abemaciclib + AZD1775 showed antagonism in Rb(+ve) cells, with synergy score values greater than 1 for both HT-1080 and SK-LMS1 (Figure S4B and S5G). The sequential and concomitant combination treatment with abemaciclib and gemcitabine or AZD1775 were also antagonistic in Rb(-ve) cells (average synergy score=1.2 and 1.1 for sequential and concomitant treatment, respectively) and RB1-KO cells (average synergy score, 10–100) (Figure S4B and S5G). The most synergistic cell killing was observed in Rb(+ve) cell lines treated sequentially with abemaciclib and gemcitabine or AZD1775, with average synergy scores of 0.5 and 0.3 for SK-LMS1 and HT-1080 cells, respectively (Figure S4B and S5G). Collectively, all three mathematical models used in synergy score calculations confirmed that only when Rb (+ve) cells are treated sequentially with abemaciclib followed by gemcitabine or Wee1 kinase inhibitor, that the combination treatment is synergistic.
Sequential targeting of CDK4/6 and DNA synthesis pathways synergize by inducing DNA damage leading to increased lethality
The mode of cell death to the sequential combination of abemaciclib followed by gemcitabine was examined using the Annexin-V/PI assay (Figure S6A-B). Single agent abemaciclib or gemcitabine induced apoptotic cell death (12%−20%) in the Rb(+ve) cell lines HT-1080 and Rh30, however sequential treatment with abemaciclib followed by gemcitabine resulted in significantly higher rates of apoptosis (>80% for HT-1080 and >30% for Rh30, p < 0.0001). Concomitant treatment with abemaciclib+gemcitabine resulted in similar levels of apoptosis as treatment with single agents (i.e. 8%−22%; Figure S6A-B). Cell cycle analysis revealed an increase in the sub-G1 population of Rb(+ve) cell lines treated with sequential combination treatment but not in those cells treated with single agent or concomitant therapy (Figure S6C-D).
To monitor cells in each phase of the cell cycle in response to the different treatment strategies (monotherapy, sequential, or concomitant combination treatment), fluorescent ubiquitination-based cell cycle indicator (FUCCI)–expressing HT-1080 Rb(+ve) cell lines (Figure 4A) were generated, where cells emit red fluorescence during G1, yellow during early S, and green during late S/G2/M (Figure 4B). Treatment with abemaciclib induced G1 arrest in FUCCI-expressing cells, as demonstrated by 80% of cells emitting red fluorescence (Figure 4C-E). Eighteen hours after removal of abemaciclib, 60% of cells emitted yellow and green fluorescence, indicating enrichment of cells in S/G2/M phases of the cell cycle (Figure 4C). Treatment of cells with gemcitabine did not alter cell cycle distribution (Figure 4D-E and Table S3). However, treatment with abemaciclib followed by gemcitabine resulted in approximately 50% of cells enriched in early S (yellow) and G2/M (green) phases (Figure 4E). Concomitant combination treatment captured significantly (p<0.05) fewer cells in S phase (34%) with a smaller fraction of cells in G0/G1 (29%) (Figure 4E, Table S3).
Figure 4. Sequential targeting of CDK4/6 and DNA synthesis pathways synergize by inducing DNA damage leading to increased lethality.
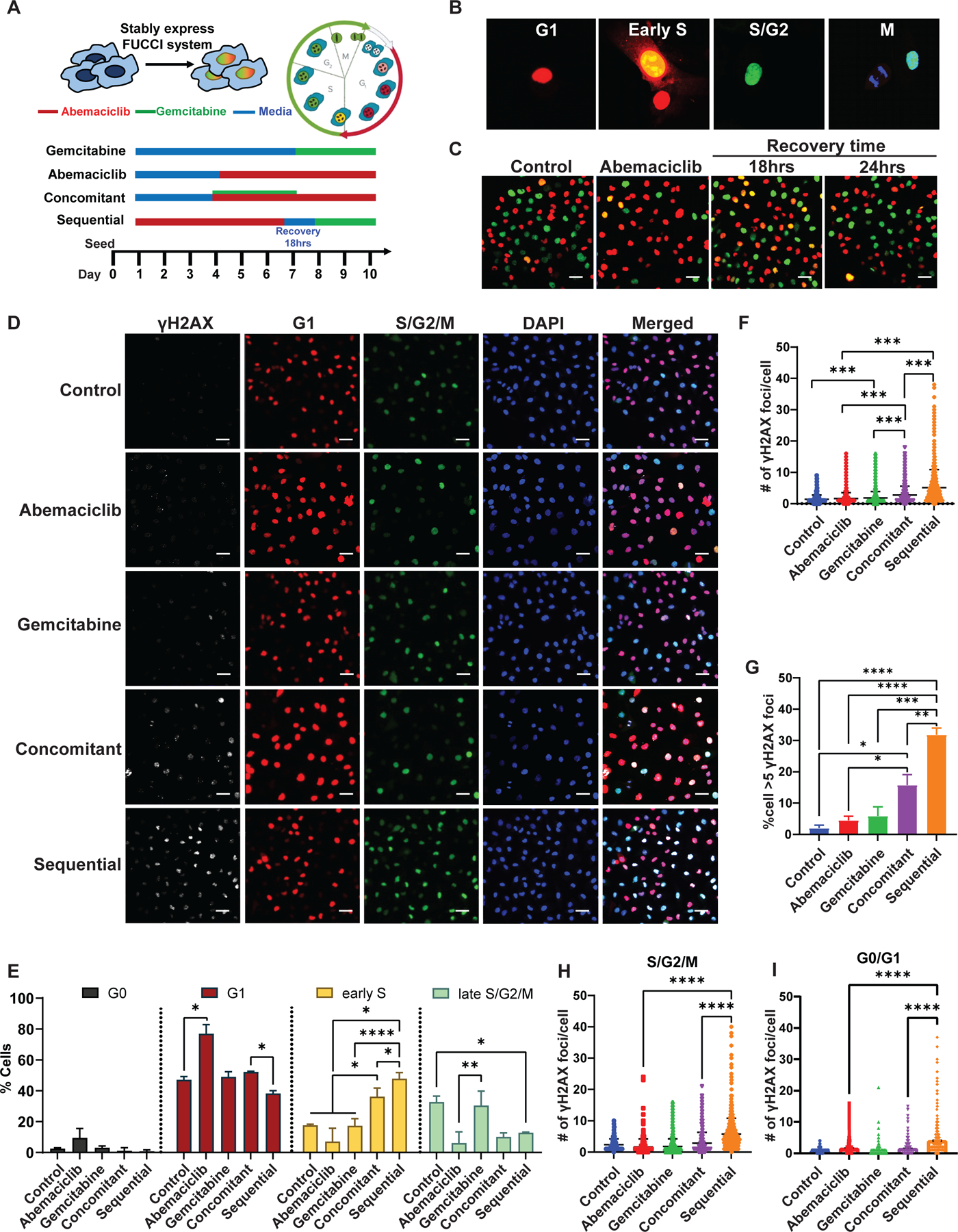
(A) Schema of the FUCCI system and the different treatment times of the HT-1080 cells with abemaciclib and gemcitabine as monotherapy or combination therapies. (B) FUCCI system stably expressed HT-1080 cells that emit different fluorescent signals at each cell cycle phase. G1-red, early S-yellow, and late S/G2/M-green (scale bar = 20 µm). (C) Cell cycle progression of the FUCCI system stably expressed HT-1080 cells before and after 6 days of treatment with 0.2 µM abemaciclib, followed by recovery in drug free medium for 18 and 24 hrs (scale bar = 50 µm). (D) Representative images of immunofluorescent (IF) analysis of γH2AX in FUCCI-labeled HT-1080 cells after the indicated treatments as depicted in panel (A) (scale bar = 50 µm). (E) Cell cycle distribution of FUCCI labeled HT-1080 cells after the indicated treatments (see Table S3). Assessment of DNA damage by quantification of γH2AX foci per cell (F) and % γH2AX-positive cells (foci>5, G), following each treatment condition (see table S4). Quantification of DNA damage marker γH2AX in proliferating cells (S/G2/M) (H) and non-proliferating cells (G0/G1) (I) following each treatment condition. Experiments are representative of three independent biological replicates. The statistical analyses were performed using one- or two-way ANOVA. *p <0.05, **p ≤ 0.01, ***p ≤ 0.001, ****p ≤ 0.0001.
DNA damage was assessed by quantitation of γH2AX (marker of DNA double stranded break) in FUCCI-expressing cells, after each treatment strategy (Figure 4D). In cells treated sequentially with abemaciclib followed by gemcitabine, the average number of γH2AX foci per cell was 5.1, significantly higher (p<0.001) than the concomitant (i.e. 2.7 foci/cell) or single-agent (i.e. < 1.6 and 1.8 foci/cell for abemaciclib and gemcitabine, respectively) treatment strategies (Figure 4F, Table S4). In the sequential treatment group, 40% of cells had more than five γH2AX foci, 2- and 3-fold higher than that of cells in the concomitant and single-agent treatment groups, respectively (Figure 4G). There was a significantly higher (p< 0.0001) number of γH2AX foci per cell induced by sequential treatment in both G0/G1 non-proliferative cells and S/G2/M proliferative cells (Figure 4H and 4I). These results imply that sequential targeting of the CDK4/6 and DNA synthesis pathways potentiates DNA damage in all phases of the cell cycle in which cells from the previous cycle may carry DNA damage upon re-entry into the next cell cycle. Consistently, the significant induction (p<0.001) of γH2AX foci was only observed in abemaciclib-treated cells but not in cells treated with other CDK4/6is such as palbociclib or ribociclib (Figure S7A-C).
Abemaciclib induces reversible G1 arrest in vivo in cell line and patient derived xenograft models
Cell line (CDX) and patient-derived xenograft (PDX) models were used to examine the treatment strategies in vivo. The time required for S phase re-entry in tumors cells in vivo was optimized using the schedule and dose of abemaciclib shown in Figure S8A. Treatment of mouse models with abemaciclib at 75 mg/kg for 6 days resulted in up to 25% regression of tumor growth compared to day 0) (Figure 5A).
Figure 5. Abemaciclib induces reversible G1 arrest in vivo in cell line xenografts (CDX) and patient derived xenograft (PDX) models.
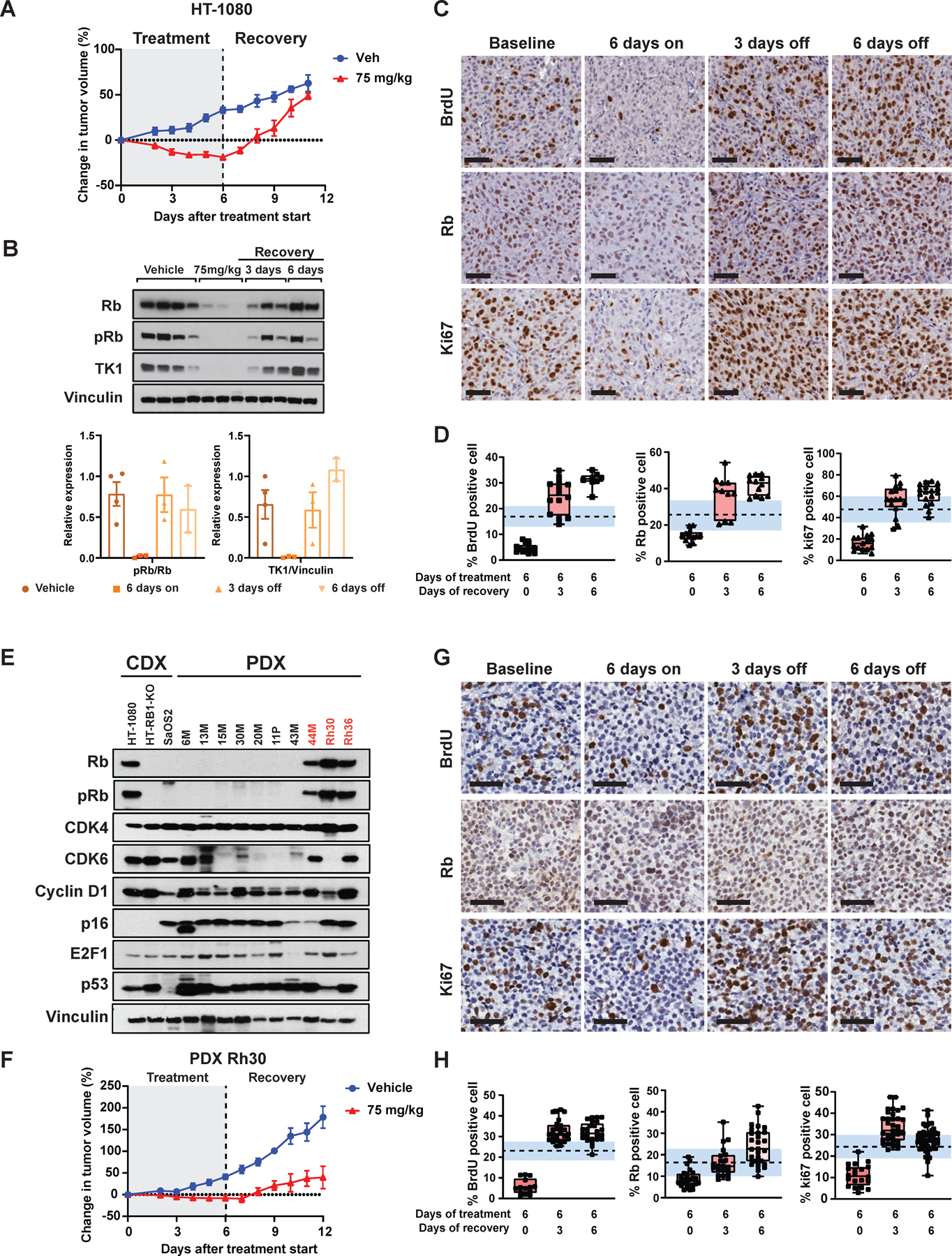
(A) Percent change in tumor volumes for mice (HT-1080 CDX model) treated with vehicle or 75 mg/kg of abemaciclib daily for 6 days (n=3 mice/arm) followed by 6 days of recovery. Tumors were measured daily with calipers. (B) Western blot analysis for the indicated protein in tumor samples collected after 6 days of treatment, 3 days of recovery and 6 days of recovery (n=3 each time point), top panel. The protein expression of the indicated protein was quantified by densitometry using Image J software, bottom panel. (C) Representative BrdU, Rb and Ki67 IHC images showing reversible G1 arrest of tumors when HT-1080 tumor bearing mice were treated with 75 mg/kg abemaciclib daily for 6 days (scale bar 50 µm). (D) Proportion of BrdU, Rb and Ki67-positive cells under each treatment condition. (E) Western blot analysis of 3 CDX and 10 LMS and RMS PDX tumors for key G1 regulatory proteins. (F) Percent change in tumor volumes for the Rh30 PDX model treated with vehicle, or the indicated doses of abemaciclib daily for 6 days (n=3 mice/arm) followed by 6 days of recovery. Tumors were measured daily with calipers. (G) Representative BrdU, Rb and Ki67 IHC images showing reversible G1 arrest of tumors when Rh30 tumor bearing mice were treated with 75 mg/kg abemaciclib daily for 6 days (scale bar 50 µm). (H) Proportion of BrdU, Rb and Ki67-positive cells under each treatment condition. Dashed line indicates the mean value of % positive cells in vehicle treated tumors at each time points, standard error is highlighted in shaded blue region. The percentage of positive cells that were quantified semi-manually by QuPath software using at least 5 images per slide, of which approximately 3000 cells per slide were counted as either positive or negative for staining for each mouse (n=3 mice per condition) for a total of 9000 data points for each bar graph presented. Experiments are representative of at least three independent biological replicates.
Rb, pRb, and TK1 protein levels were downregulated after 6 days of treatment with abemaciclib, with levels returning to baseline 3–6 days after drug removal (Figure 5B-D). BrdU labeling (as a measure of tumor cell proliferation) revealed that treatment with 75 mg/kg per day for 6 days was sufficient to inhibit S-phase fraction of the tumor cells (i.e. < 5% for the 75 mg/kg treatment group compared to 20% BrdU-positive cells in the vehicle arm). Levels returned to baseline at 3 (>25%) and 6 (30%) days of recovery, following abemaciclib removal (Figure 5C-D). Similar trends were detected when examining Ki67 and Rb status across all treatment groups (Figure 5C-D).
LMS and RMS PDX models comprising Rb(+ve) and Rb(-ve) subtypes were used to examine the experimental approach (Figure 5E and Figure S7B). Of 10 PDX models examined, 44M, Rh30, and Rh36 showed positive Rb and pRb expression accompanied by intact CDK4, CDK6, and cyclin D1, indicating that these models harbor a wildtype Rb pathway phenotype. Rh30 RMS tumor growth regressed and failed to increase in volume following 6-days of abemaciclib treatment (Figure 5F, Figure S8B). The expression of proliferative biomarkers such as TK1, BrdU, and Ki67 were consistent with tumor volume status, with enrichment in S phase detected after 3 days of recovery (Figure 5G-H, Figure S8B).
Sequential, but not concomitant treatment with abemaciclib and gemcitabine increases the survival in Rb(+ve) PDX mouse models
The sequential combination treatment strategy of abemaciclib followed by gemcitabine treatment was evaluated in vivo, using Rb(+ve) cell lines and PDX models (Figure 6 and Figure S9). Based on the ex vivo biomarker analysis (Figure 5) mice were treated with the sequential combination treatment for 12 days/cycle; each cycle comprised 6 consecutive days of treatment with abemaciclib (p.o., q.d., 75 mg/kg), recovery for 3 days and 1 day of gemcitabine treatment (i.p., q.d., 50mg/kg) (Figure S9A).
Figure 6. Sequential, but not concomitant combination treatment of abemaciclib + gemcitabine increases survival in Rb(+ve) CDX and PDX models.
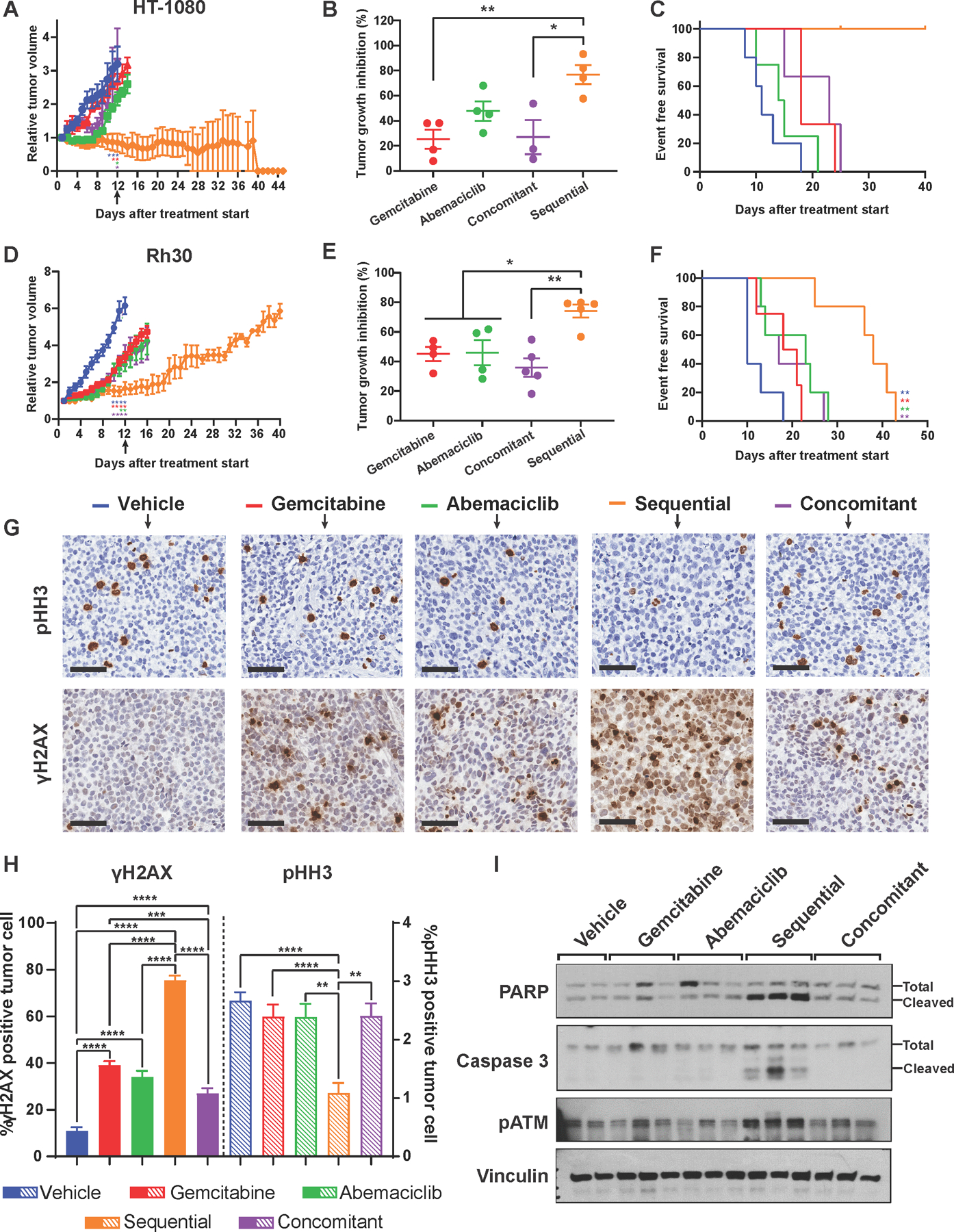
(A) Tumor growth curve showing relative tumor growth inhibition (over time) of the HT-1080 cell line xenograft (CDX) tumor model subjected to the indicated 5 treatment arms (12 days/cycle). Mice treated sequentially completed 3 cycles of treatment while other treatments arms stopped at cycle 1 or early cycle 2 due to tumor burden (n=4 or 5/treatment arm). (B) Percent of endpoint tumor growth inhibition of HT-1080 cell line xenograft tumor model subjected to the indicated 5 treatments for 1 cycle (day 12) as indicated by black arrow in (A) (n=4 or 5/treatment arm). (C) Survival curves of the HT-1080 tumor model showing that the sequential combination arm (orange) had the longest survival time compared to single agents or the concomitant combination. (D) Growth curve showing relative tumor growth inhibition over time of the Rh30 PDX tumor model subjected to the indicated 5 treatments (n=4 or 5/treatment arm, (12 days/cycle). Mice treated sequentially completed 3 cycles of treatment while other treatments arms were stopped at cycle 1 or early cycle 2 due to large tumor burden. (E) Percent of tumor growth inhibition, at the endpoint, of the Rh30 PDX tumor model subjected to the indicated 5 treatments for 1 cycle (day 12) as indicated by black arrow in (D) (n=4 or 5/treatment arm). (F) Survival curves of the Rh30 PDX tumor model showing that the sequential combination arm (orange) had the longest survival time compared to single agents or the concomitant combination. (G) IHC staining of pHH3 (proliferation marker) and γH2AX (DNA damage marker) in Rh30 tumors collected after 1 cycle as indicated by black arrow in (D) of the indicated treatments (scale bar 50 µm). (H) Quantification of the percent of γH2AX after 1 cycle of indicated treatments from the IHC analysis shown in (G) Solid bars refer to the γH2AX quantitation and dashed bars refer to the pHH3 quantitation. (I) Western blot analysis of Rh30 tumor samples showing upregulation of DNA damage repair markers (pATM and cleaved PARP) and apoptosis marker (cleaved caspase 3) in sequentially but not concomitantly treated tumors. Experiments are representative of 4 or 5 independent biological replicates. The statistical analyses were performed using one- or two-way ANOVA. *p <0.05, **p ≤ 0.01, ***p ≤ 0.001, ****p ≤ 0.0001.
In the HT-1080 model, there was regression of tumors in mice treated with sequential combination therapy for three cycles (Figure 6A-C). Concomitant treatment with abemaciclib and gemcitabine was similar to treatment with either single agent therapy (Figure 6A). There was 80% tumor growth inhibition (TGI) at the end of the first cycle with sequential combination treatment and only 25%−50% TGI with either a single-agent or concomitant treatment (Figures 6A and 6B). Similar results were observed with the treatment-naïve Rb(+ve) RMS Rh30 PDX model, where sequential combination treatment resulted in 80% TGI (first cycle) which was more effective than the single agent treatments (25% and 47% TGI for gemcitabine and abemaciclib, respectively) or concomitant treatment (26% TGI) (Figure 6D-F). Rh36, an RMS Rb(+ve) PDX model collected from a patient who experienced disease progression on chemotherapy (DITC/DOX), was resistant to gemcitabine (−32% TGI) but was sensitive to abemaciclib (22% TGI) (Figure S9B-D). This was enhanced when gemcitabine was administered sequentially following recovery from abemaciclib treatment (46% TGI).
Survival analysis performed after 3 cycles of treatment revealed that the sequential combination of abemaciclib and gemcitabine was more effective than individual drugs or concomitant treatment, resulting in longer survival times (Figure 6C, 6F and S9D). In the HT-1080 model, 100% of the mice in the sequential treatment group remained alive until day 40, while all of the mice in the single agent and concomitant treatment groups had to be euthanized by day 25, because of large tumor burden (Figure 6C). In the sequentially treated Rh30 PDX model there was an increase in survival duration (38 days) compared to the concomitant (27 days) or single-agent–treated mice (23 and 19.5 days for abemaciclib and gemcitabine, respectively) (Figure 6F). The Rh36 PDX model also benefitted from sequential combination treatment. The median survival time with sequential combination therapy was 27 days, compared to 17 days for concomitant, 13 days for gemcitabine, and 15 days for abemaciclib treatment alone (Figure S9D).
Levels of γH2AX, cleaved-PARP, cleaved caspase 3, and pATM all increased in mouse tumors treated sequentially with abemaciclib and gemcitabine compared with the single agent or concomitant treatment (Figure 6G-I and Figure S9E). There was a decrease in proliferation, as indicated by the percentage of phospho-Histone H3 (pHH3) positive cells, only evident in the sequential treatment cohort (Figure 6G-I).
Thymidine kinase 1, a cell-proliferative marker that facilitates differential 18F-FLT uptake to monitor the recovery of Rb(+ve) cells by positron emission tomography imaging
To translate this sequential treatment strategy clinically, in vivo real-time monitoring modalities were evaluated. TK1 is a key enzyme in the pyrimidine salvage pathway that takes part in the initial thymidine phosphorylation process required for DNA synthesis. TK1 expression is strongly associated with Rb(+ve) cell cycle progression both in vitro (Figure 3G, Figure S2D-F) and in vivo (Figure 7A-B, P<0.001). The changes in tumor TK1 can be captured by functional positron emission tomography (PET) imaging using [18]F-fluoro-3’-deoxy-3’-L-fluorothymidine ([18]F-FLT) as a TK1-regulated proliferative tracer and by circulating thymidine kinase activity (TKa) in the in vivo models (Figure 7C). While recovery from abemaciclib treatment did not alter [18]F-FLT uptake in Rb(-ve) and RB1-KO cells, a 10-fold increase in biotracer uptake after 12–18hrs of recovery was detected in Rb(+ve) cell lines that were pre-treated with abemaciclib (Figure 7D). Unlike [18]F-FLT, which directly measures thymidine uptake, neither [18]F-FDG, a glucose analogue, or its regulator hexokinases II (HKII) were able to capture the recovery of Rb(+ve) (Figure S10A-D).
Figure 7. Abemaciclib induces reversible G1 arrest in in vivo models as monitored by circulating TK1 and PET/CT imaging.
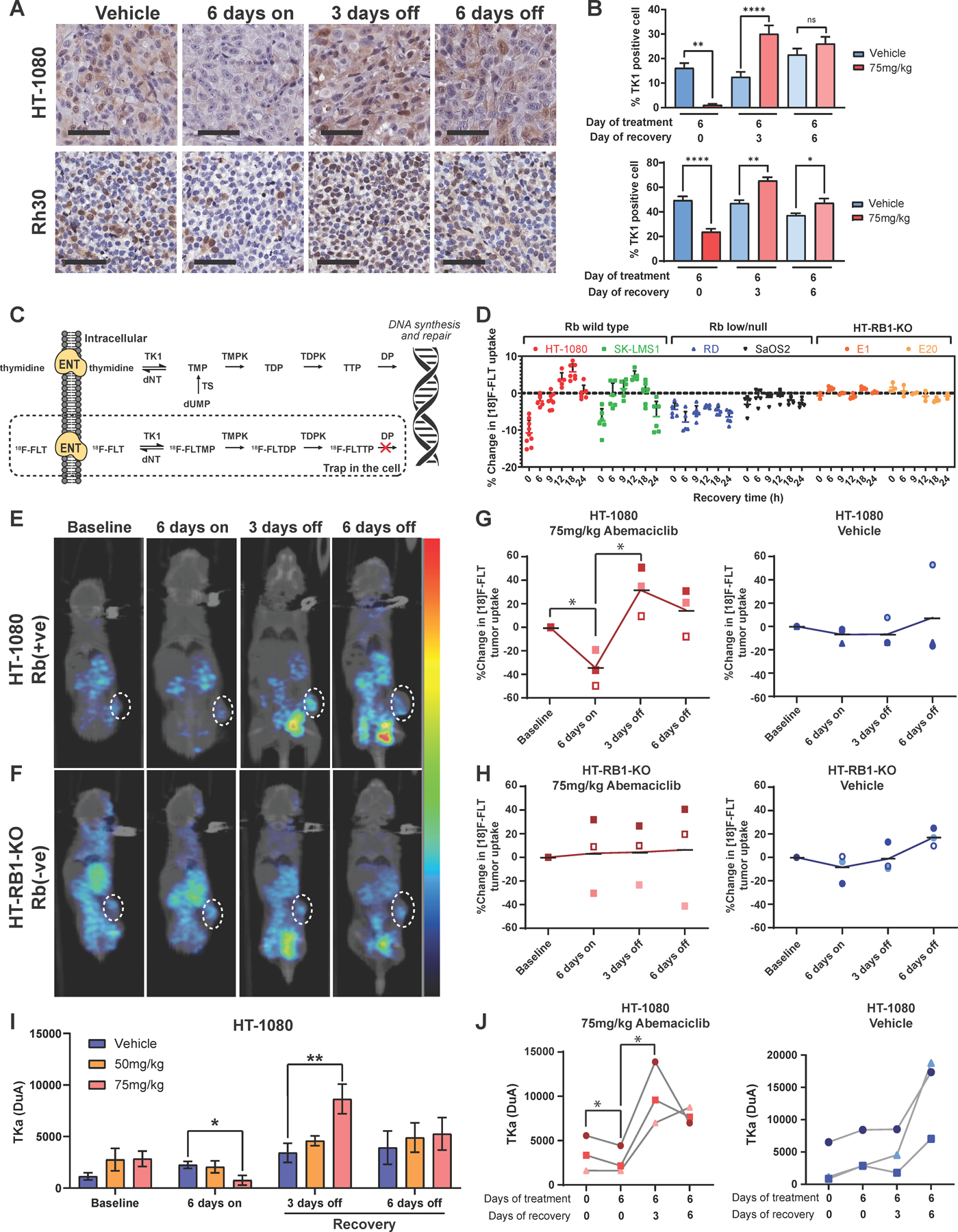
(A) Representative TK1 IHC images showing reversible G1 arrest of tumors when the HT-1080 CDX and Rh30 PDX Rb(+ve) model systems were treated with 75 mg/kg abemaciclib (scale bar = 60 µm). (B) Proportion of TK1-positive cells in tumor samples collected after 6 days of 75mg/kg abemaciclib treatment, 3 days of recovery, and 6 days of recovery (n=3 each time point). (C) Schematic showing the [18]F-FLT uptake mechanism through the thymidine salvage pathway. (D) Changes in [18]F-FLT uptake in Rb(+ve) and Rb(-ve) cells post-abemaciclib removal. For both experiments, cells were treated with IC50 of abemaciclib for 6 days before undergoing the indicated recovery period in drug free media. Cellular tracer uptake was corrected with [18]F decay time and normalized with total protein in cell lysates. The percent change in [18]F-FLT uptake was calculated by subtracting normalized [18]F-FLT uptake in drug-treated cells from normalized [18]F-FLT uptake in untreated cells at each indicated recovery time. (E) and (F) A series of representative PET/CT images of one Rb (+ve) (E) or Rb (-ve) (F) mouse demonstrated the kinetic changes in [18]F-FLT tumor uptake from individual abemaciclib-treated mice of a cohort of n=3. The results demonstrated a signficant decline of [18]F-FLT uptake on day 6 (6 days on) compared to baseline (p<0.05) followed by significant increase after 3 days of recovery (day 9; 3 days off) (p<0.05), and return to baseline uptake level following 6 days of recovery (day 12; 6 days off) in the Rb(+ve) model (E) but not the Rb(-ve) model (F), tumor regions are highlighted in white circles. (G) and (H) Percent changes in [18]F-FLT tumor uptake detected in the Rb(+ve) (G) and Rb(-ve) (H) model (n=3/model) after 6 days of treatment with 75mg/kg abemaciclib or vehicle, followed by 3 and 6 days of recovery. (I) Quantification of thymidine kinase activity (TKa) in plasma samples collected from mice (n=5/arm/timepoint) in the same experimental cohort as in (A). (J) Quantification of TKa in plasma samples collected longitudinally by retro-orbital bleeding (n=3). Each connected line (spider plot) depicts the kinetic changes in plasma TKa measured in individual abemaciclib-treated mice of a cohort of n=3. Experiments are representative of three independent biological replicates. The statistical analyses were performed using one- or two-way ANOVA. *p <0.05, **p ≤ 0.01, ***p ≤ 0.001, ****p ≤ 0.0001.
Finally, the utility of [18F]FLT–PET/CT imaging in monitoring the reversible antiproliferative activity of abemaciclib in vivo was examined. To demonstrate the kinetic changes in [18]F-FLT tumor uptake from individual abemaciclib-treated mice of a cohort of n=3/model, a series of images were taken for each mouse over 4 different time points (baseline, 6 days on, 3 days off, and 6 days off). Mice bearing Rb(+ve) tumors displayed a 40% reduction in tumor [18]F-FLT uptake compared with the vehicle-treated or Rb(-ve) cohorts (Figure 7E-H). Three days following drug removal, the intratumoral [18]F-FLT accumulations across all Rb(+ve) mice examined were elevated by 10-fold compared to mice on-treatment (6 days on) and 4-fold compared with no treatment (Figure 7E, G). Following six days of recovery, the [18]F-FLT signal returned to basal levels, indicating the return of tumor cells to an asynchronous proliferative state (Figure 7E, G). In HT-RB1-KO tumors, intratumoral [18]F-FLT accumulation was not responsive to abemaciclib treatment at any time interval during or after treatment (Figure 7F-H), consistent with the lack of change in TKI expression after abemaciclib treatment in vitro (Figure S2E-F).
TKa in the serum of treated mice was evaluated and correlated with[18]F-FLT uptake. TKa was measured using the DiviTum® assay (46) in sera collected from Rb(+ve) HT-1080 and Rb(-ve) HT-RB1-KO tumor-bearing mice (Figure 7I-J). This revealed that TKa mirrors [18]F-FLT uptake (Figure 7E, G) and is significantly associated with TK1 protein expression in tumors (Figure 7A, B; p<0.001). TKa changes were observed in Rb(+ve) models as a measure of response to abemaciclib, but no significant changes in TKa were observed in the Rb(-ve) tumor model (Figure S10E).
Discussion
The clinical management of sarcomas is challenging due to the rarity, heterogeneity, and aggressive nature of these tumors. Conventional chemotherapy is the mainstay of therapy, but survival benefit is limited and almost all patients progress on the few standard treatment options available. While molecularly targeted approaches to STS treatment have gained traction in recent years, the reported OS for targeted therapies, such as pazopanib, was only modestly prolonged compared to that of placebo (12.5 vs. 10.7 months) (47). There is a clear need for new rational biomarker-driven therapeutics to improve patient outcomes
CDK4/6 inhibitors (CDK4/6is) have shown significant clinical benefit in breast cancer patients with advanced or metastatic hormone receptor positive disease (33–35,48). These promising results have prompted the use of CDK4/6is in other cancers harboring deregulation of the cyclin D-CDK4/6-INK4-Rb pathway, including sarcomas (15,49–52). However, reports from phase II trials in advanced LPS sarcoma patients with CDK4 overexpression treated with palbociclib or abemaciclib showed only modest efficacy, with only 1 out of 60 palbociclib-treated patients showing response (15,52). Given the lack of effective therapeutic options for this LPS subtype, CDK4/6is are included as an option in the National Comprehensive Cancer Network (NCCN) guidelines even though not formally approved by any regulatory agencies. The use of CDK4 overexpression as a stratifying biomarker may have been the main limitation for patient selection in these studies, since upregulation of CDK4 or CDK6 contributes to the CDK4/6i resistance observed in various cancer types (53). Other biomarkers, such as CDKN2A loss/mutation or CCND1, 2, or 3 amplification, also failed to predict response to CDK4/6is (54–56). On the basis of these negative clinical findings, we hypothesized that a more functional marker for response to CDK4/6i in STS may be the Rb protein. Indeed, Rb was the only marker that persisted across all STS models we examined for response to abemaciclib. This finding provides a compelling rationale to evaluate abemaciclib activity in subtypes of sarcoma beyond those with CDK4 overexpression. None of the other G1/S regulators, including p16, cyclin D1, CDK4/6, and cyclin E1, showed correlation with response to abemaciclib across multiple sarcoma models (Figure 2).
There is precedent for using Rb as a biomarker for patient selection for CDK4/6is. Results from the PALOMA 1 and 2 trials in breast cancer patients revealed that PFS in palbociclib-treated patients whose tumors harbored RB1 loss was 3 times shorter than that in patients with intact RB1 (3.6 months vs 10.1 months) (54,56–59). These results suggest that RB1 loss is associated with resistance or lack of response to CDK4/6is in breast cancer patients. In sarcomas, only 20% of all STS cases examined exhibited RB1 loss/mutation at the genome level (60). The expression of Rb protein in over 1000 tumor specimens with different sarcoma histologies showed that over 50% retained Rb expression. There was no apparent survival benefit from standard of care management (surgery, chemotherapy, and radiotherapy) when patients were stratified based on Rb status, suggesting that other treatment strategies, such as CDK4/6is, which are likely to show response in the patients with Rb-positive tumors, could be offered to these patients. Notably, more than 90% of rhabdomyosarcoma and angiosarcoma tumors are Rb-positive, making these patient cohorts ideal candidates for clinical trials of CDK4/6is sequential combination therapy.
Since CDK4/6is are cytostatic agents that induce cell cycle arrest (i.e. G1) rather than cell death, combination strategies are needed to maximize therapeutic efficacy. However, the optimal treatment approach for combining CDK4/6is with chemotherapy, given sequentially or concurrently, has remained controversial. A recent study that examined 108,259 pairs of combination drug treatments across breast, colon, and pancreatic cancer cell lines revealed that the frequency of synergy observed in concurrent combination treatment is a rare event, with only 5.2% of all combinations examined showing synergistic response (61). Specifically, data from the concomitant combination of palbociclib with gemcitabine in 52 breast cancer cell lines was 18-fold less potent (median log2[∆IC50] = 4.2) compared to the expected combination response for synergy threshold (61). One reason for the lack of synergism of concurrent combination treatments with palbociclib may be that inhibition of CDK4/6 results in G1 arrest in cells, not allowing the chemotherapeutic agents that target S phase to mediate their cytotoxic activity (17).
A combination treatment strategy with CDK4/6is (followed by chemotherapeutic agents) that is biomarker-driven, mechanism-based and limits the overlapping toxicities of the two agents through sequential treatment is a novel approach, presented in this report. Specifically, a two-step sequential combination strategy in which abemaciclib acts to prime Rb-positive tumor cells to reversibly arrest at G1 phase, followed by a short recovery period to allow cells to synchronously enter S phase, at which point they are treated with S phase-targeting drugs, such as gemcitabine. The synergistic cell killing effects mediated by sequential combination treatment with abemaciclib and gemcitabine, are effective in several in vitro and in vivo models of sarcoma, including a chemotherapy resistant PDX model. The mechanism of this treatment strategy is through sustained induction of DNA damage in both proliferative and non-proliferative cells.
A challenge in the translation of the proposed sequential combination treatment is the identification of real-time pharmacodynamic biomarkers to ascertain when CDK4/6is induce G1 arrest and how long it takes for the tumor cells to enrich in S phase in patients undergoing such treatment strategies. One such biomarker is TK1, which can be measured in tumor tissues (Figure 7A-B). Its activity can be quantitated in serum (Figure 7I-J), and its effector uptake, as a biotracer, can be monitored through functional imaging (PET/CT, Figure 7E-H). The link between tumor cell proliferation and serum TK1 was reported with the neoadjuvant NeoPalAna phase II “To Reverse ENDocrine Resistance” (TREnd) trial (NCT02549430), in which changes in plasma TK1 were concordant to Ki67 and can be used to predict response to palbociclib (32,62). These clinical studies suggest that TK1 is a valid pharmaco-dynamic marker for tumor proliferation. TK1 levels and activity in Rb(+ve) models were highly concordant with tumor proliferation status pre- and post-abemaciclib treatment. As a result, real-time monitoring through functional PET imaging and a blood-based biomarker, in concert with the ex vivo biomarkers presented herein, can comprehensively monitor the early treatment response of abemaciclib-based sequential combination treatment, and translated into future clinical trials.
One limitation of our study is the reliance immunohistochemistry to identify Rb(+ve) tumors. It is important to also perform a genomic analysis of the RB1 gene and correlate its genetic loss or mutations to Rb protein expression. Our data shows that therapeutic benefits can be achieved in multiple PDX models as long as the Rb pathway is intact, although the mechanism of cell death induction will be different as each cancer type and CDK4/6i have distinct biological and pharmacodynamic profiles. In addition, the mechanism by which tumor cells secrete TK1 into the circulation and how CDK4/6is treatment affects such mechanisms remain unclear. These analyses will be instrumental in determining the utility of these biomarkers in patients with a functional Rb pathway who can be identified upfront as potential responders to CDK4/6i based sequential combination therapy.
In summary, a novel, sequential treatment strategy with abemaciclib followed by gemcitabine (or WEE1-kinase inhibitor) is effective in inhibiting tumor growth in vitro and in vivo. Rb is an important mediator of this synergistic activity and Rb expression can readily be assessed in tumors using immunohistochemistry. Finally, the cellular response to this sequential treatment can be monitored non-invasively by [18]F-FLT uptake or circulating TK1 activity in the serum, and through immunohistochemistry of TK1 in tumor tissues.
Supplementary Material
Statement of Sigfnificance:
An innovative sequential therapeutic strategy targeting Rb followed by treatment with agents that perturb DNA synthesis pathways results in synergistic killing of Rb-positive sarcomas that can be non-invasively monitored.
Acknowledgements:
We thank Dr. Natividata (Robert) Fuentes from MD Anderson Cancer Center, Department of Radiation Oncology for his advice on FUCCI system, cell confocal image analysis; we also thank Dr. Lucia Martini from MD Anderson Cancer Center, Department of Experimental Therapeutics for her advice on the PET/CT analysis on the mouse models. We are also grateful to the Biovica Inc (Dr. Amy Williams) who advised us on the use of and provided us with the DiviTum®TKa kits, free of charge, that we used for both in the in vitro and in vivo studies presented in this report.
Financial support:
Research reported in this article was supported by the NIH under award P30CA016672 to The University of Texas MD Anderson Cancer Center, the Department of Defense Breakthrough Post-Doctoral Fellowship BC170615 (to N. M. Kettner), NIH, National Cancer Institute R01 grants CA255960 and CA223772 to K. Keyomarsi, CPRIT multi-investigator grant RP180712 (to K. K. Hunt and K. Keyomarsi), CPRIT Research Training Program grant RP170067 and RP210028 (to N. M. Kettner and M. Li), the National Leiomyosarcoma Foundation (to K. Keyomarsi and K. K. Hunt) and the John Wayne Cancer Foundation (K. K. Hunt)
Footnotes
Conflict of interest: KKH – Medical Advisory Board (Armada Health, AstraZeneca, Merck & Co.); Research funding to MD Anderson Cancer Center (Cairn Surgical, Eli Lilly & Co., Lumicell). The other authors declare no potential conflicts of interest.
References:
- 1.de Pinieux G, Karanian M, Le Loarer F, Le Guellec S, Chabaud S, Terrier P, et al. Nationwide incidence of sarcomas and connective tissue tumors of intermediate malignancy over four years using an expert pathology review network. Plos One 2021;16 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 2.Hayat MJ, Howlader N, Reichman ME, Edwards BK. Cancer statistics, trends, and multiple primary cancer analyses from the surveillance, epidemiology, and end results (SEER) program. Oncologist 2007;12:20–37 [DOI] [PubMed] [Google Scholar]
- 3.Sangkhathat S Current management of pediatric soft tissue sarcomas. World J Clin Pediatr 2015;4:94–105 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 4.Ramu EM, Houdek MT, Isaac CE, Dickie CI, Ferguson PC, Wunder JS. Management of soft-tissue sarcomas; treatment strategies, staging, and outcomes. SICOT J 2017;3:20. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 5.Verschoor AJ, Litiere S, Marreaud S, Judson I, Toulmonde M, Wardelmann E, et al. Survival of soft tissue sarcoma patients after completing six cycles of first-line anthracycline containing treatment: an EORTC-STBSG database study. Clin Sarcoma Res 2020;10:18. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 6.Martin-Liberal J, Alam S, Constantinidou A, Fisher C, Khabra K, Messiou C, et al. Clinical activity and tolerability of a 14-day infusional Ifosfamide schedule in soft-tissue sarcoma. Sarcoma 2013;2013:868973. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 7.Choi Y, Yun MS, Lim SH, Lee J, Ahn JH, Kim YJ, et al. Gemcitabine and Docetaxel Combination for Advanced Soft Tissue Sarcoma: A Nationwide Retrospective Study. Cancer Res Treat 2018;50:175–82 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 8.Italiano A, Toulmonde M, Cioffi A, Penel N, Isambert N, Bompas E, et al. Advanced well-differentiated/dedifferentiated liposarcomas: role of chemotherapy and survival. Ann Oncol 2012;23:1601–7 [DOI] [PubMed] [Google Scholar]
- 9.Meyer M, Seetharam M. First-Line Therapy for Metastatic Soft Tissue Sarcoma. Curr Treat Options Oncol 2019;20:6. [DOI] [PubMed] [Google Scholar]
- 10.Italiano A, Mathoulin-Pelissier S, Cesne AL, Terrier P, Bonvalot S, Collin F, et al. Trends in survival for patients with metastatic soft-tissue sarcoma. Cancer 2011;117:1049–54 [DOI] [PubMed] [Google Scholar]
- 11.Herwig S, Strauss M. The retinoblastoma protein: a master regulator of cell cycle, differentiation and apoptosis. Eur J Biochem 1997;246:581–601 [DOI] [PubMed] [Google Scholar]
- 12.Dalton S Linking the Cell Cycle to Cell Fate Decisions. Trends Cell Biol 2015;25:592–600 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 13.Tseng WW, Somaiah N, Lazar AJ, Lev DC, Pollock RE. Novel systemic therapies in advanced liposarcoma: a review of recent clinical trial results. Cancers (Basel) 2013;5:529–49 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 14.Ragazzini P, Gamberi G, Pazzaglia L, Serra M, Magagnoli G, Ponticelli F, et al. Amplification of CDK4, MDM2, SAS and GLI genes in leiomyosarcoma, alveolar and embryonal rhabdomyosarcoma. Histol Histopathol 2004;19:401–11 [DOI] [PubMed] [Google Scholar]
- 15.Dickson MA, Koff A, D’Angelo SP, Gounder MM, Keohan ML, Kelly CM, et al. Phase 2 study of the CDK4 inhibitor abemaciclib in dedifferentiated liposarcoma. Journal of Clinical Oncology 2019;37
- 16.Roberts PJ, Kumarasamy V, Witkiewicz AK, Knudsen ES. Chemotherapy and CDK4/6 Inhibitors: Unexpected Bedfellows. Molecular Cancer Therapeutics 2020;19:1575–88 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 17.Jin D, Tran N, Thomas N, Tran DD. Combining CDK4/6 inhibitors ribociclib and palbociclib with cytotoxic agents does not enhance cytotoxicity. Plos One 2019;14:e0223555. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 18.Francis AM, Alexander A, Liu Y, Vijayaraghavan S, Low KH, Yang D, et al. CDK4/6 Inhibitors Sensitize Rb-positive Sarcoma Cells to Wee1 Kinase Inhibition through Reversible Cell-Cycle Arrest. Mol Cancer Ther 2017;16:1751–64 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 19.Luo L, Keyomarsi K. PARP inhibitors as single agents and in combination therapy: the most promising treatment strategies in clinical trials for BRCA-mutant ovarian and triple-negative breast cancers. Expert Opin Investig Drugs 2022:1–25 [DOI] [PMC free article] [PubMed]
- 20.Lulla AR, Akli S, Karakas C, Ha MJ, Fowlkes NW, Mitani Y, et al. LMW cyclin E and its novel catalytic partner CDK5 are therapeutic targets and prognostic biomarkers in salivary gland cancers. Oncogenesis 2021;10:40. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 21.Chen X, Yang D, Carey JPW, Karakas C, Albarracin C, Sahin AA, et al. Targeting Replicative Stress and DNA Repair by Combining PARP and Wee1 Kinase Inhibitors Is Synergistic in Triple Negative Breast Cancers with Cyclin E or BRCA1 Alteration. Cancers (Basel) 2021;13 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 22.Kettner NM, Vijayaraghavan S, Durak MG, Bui T, Kohansal M, Ha MJ, et al. Combined Inhibition of STAT3 and DNA Repair in Palbociclib-Resistant ER-Positive Breast Cancer. Clin Cancer Res 2019;25:3996–4013 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 23.Chen X, Low KH, Alexander A, Jiang Y, Karakas C, Hess KR, et al. Cyclin E Overexpression Sensitizes Triple-Negative Breast Cancer to Wee1 Kinase Inhibition. Clin Cancer Res 2018;24:6594–610 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 24.Vijayaraghavan S, Karakas C, Doostan I, Chen X, Bui T, Yi M, et al. CDK4/6 and autophagy inhibitors synergistically induce senescence in Rb positive cytoplasmic cyclin E negative cancers. Nat Commun 2017;8:15916. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 25.Mull BB, Livingston JA, Patel N, Bui T, Hunt KK, Keyomarsi K. Specific, reversible G1 arrest by UCN-01 in vivo provides cytostatic protection of normal cells against cytotoxic chemotherapy in breast cancer. Br J Cancer 2020;122:812–22 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 26.Ianevski A, He L, Aittokallio T, Tang J. SynergyFinder: a web application for analyzing drug combination dose-response matrix data. Bioinformatics 2017;33:2413–5 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 27.Ianevski A, Giri AK, Aittokallio T. SynergyFinder 3.0: an interactive analysis and consensus interpretation of multi-drug synergies across multiple samples. Nucleic Acids Res 2022 [DOI] [PMC free article] [PubMed]
- 28.Koplev S, Longden J, Ferkinghoff-Borg J, Blicher Bjerregard M, Cox TR, Erler JT, et al. Dynamic Rearrangement of Cell States Detected by Systematic Screening of Sequential Anticancer Treatments. Cell Rep 2017;20:2784–91 [DOI] [PubMed] [Google Scholar]
- 29.Chou TC. Drug combination studies and their synergy quantification using the Chou-Talalay method. Cancer Res 2010;70:440–6 [DOI] [PubMed] [Google Scholar]
- 30.Roppongi M, Izumisawa M, Terasaki K, Muraki Y, Shozushima M. (18)F-FDG and (11)C-choline uptake in proliferating tumor cells is dependent on the cell cycle in vitro. Ann Nucl Med 2019;33:237–43 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 31.Shangguan C, Gan G, Zhang J, Wu J, Miao Y, Zhang M, et al. Cancer-associated fibroblasts enhance tumor (18)F-FDG uptake and contribute to the intratumor heterogeneity of PET-CT. Theranostics 2018;8:1376–88 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 32.McCartney A, Bonechi M, De Luca F, Biagioni C, Curigliano G, Moretti E, et al. Plasma Thymidine Kinase Activity as a Biomarker in Patients with Luminal Metastatic Breast Cancer Treated with Palbociclib within the TREnd Trial. Clin Cancer Res 2020;26:2131–9 [DOI] [PubMed] [Google Scholar]
- 33.Johnston SRD, Harbeck N, Hegg R, Toi M, Martin M, Shao ZM, et al. Abemaciclib Combined With Endocrine Therapy for the Adjuvant Treatment of HR+, HER2-, Node-Positive, High-Risk, Early Breast Cancer (monarchE). J Clin Oncol 2020;38:3987–98 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 34.Turner NC, Slamon DJ, Ro J, Bondarenko I, Im SA, Masuda N, et al. Overall Survival with Palbociclib and Fulvestrant in Advanced Breast Cancer. N Engl J Med 2018;379:1926–36 [DOI] [PubMed] [Google Scholar]
- 35.Hindie E Ribociclib plus Fulvestrant in Advanced Breast Cancer. N Engl J Med 2020;382:e85. [DOI] [PubMed] [Google Scholar]
- 36.Nanos-Webb A, Jabbour NA, Multani AS, Wingate H, Oumata N, Galons H, et al. Targeting low molecular weight cyclin E (LMW-E) in breast cancer. Breast Cancer Res Treat 2012;132:575–88 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 37.Lambert LA, Qiao N, Hunt KK, Lambert DH, Mills GB, Meijer L, et al. Autophagy: a novel mechanism of synergistic cytotoxicity between doxorubicin and roscovitine in a sarcoma model. Cancer Res 2008;68:7966–74 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 38.Jabbour-Leung NA, Chen X, Bui T, Jiang Y, Yang D, Vijayaraghavan S, et al. Sequential Combination Therapy of CDK Inhibition and Doxorubicin Is Synthetically Lethal in p53-Mutant Triple-Negative Breast Cancer. Mol Cancer Ther 2016;15:593–607 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 39.Kettner NM, Vijayaraghavan S, Durak MG, Bui T, Kohansal M, Ha MJ, et al. Combined inhibition of STAT3 and DNA repair in palbociclib-resistant ER-positive breast cancer. Clin Cancer Res 2019 [DOI] [PMC free article] [PubMed]
- 40.Vijayaraghavan S, Moulder S, Keyomarsi K, Layman RM. Inhibiting CDK in Cancer Therapy: Current Evidence and Future Directions. Target Oncol 2018;13:21–38 [DOI] [PubMed] [Google Scholar]
- 41.Carey JPW, Karakas C, Bui T, Chen X, Vijayaraghavan S, Zhao Y, et al. Synthetic Lethality of PARP Inhibitors in Combination with MYC Blockade Is Independent of BRCA Status in Triple-Negative Breast Cancer. Cancer Res 2018;78:742–57 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 42.Alexander A, Karakas C, Chen X, Carey JP, Yi M, Bondy M, et al. Cyclin E overexpression as a biomarker for combination treatment strategies in inflammatory breast cancer. Oncotarget 2017;8:14897–911 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 43.Lucenay KS, Doostan I, Karakas C, Bui T, Ding Z, Mills GB, et al. Cyclin E Associates with the Lipogenic Enzyme ATP-Citrate Lyase to Enable Malignant Growth of Breast Cancer Cells. Cancer Res 2016;76:2406–18 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 44.Duarte D, Vale N. Evaluation of synergism in drug combinations and reference models for future orientations in oncology. Curr Res Pharmacol Drug Discov 2022;3:100110. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 45.Chou TC, Talalay P. Quantitative analysis of dose-effect relationships: the combined effects of multiple drugs or enzyme inhibitors. Adv Enzyme Regul 1984;22:27–55 [DOI] [PubMed] [Google Scholar]
- 46.Bacovsky J, Myslivecek M, Minarik J, Scudla V, Pika T, Zapletalova J, et al. Analysis of thymidine kinase serum levels by novel method DiviTum in multiple myeloma and monoclonal gammopathy of undetermined significance - comparison with imaging methods 99mTc-MIBI scintigraphy and 18F-FDG PET/CT. Biomed Pap Med Fac Univ Palacky Olomouc Czech Repub 2015;159:135–8 [DOI] [PubMed] [Google Scholar]
- 47.van der Graaf WT, Blay JY, Chawla SP, Kim DW, Bui-Nguyen B, Casali PG, et al. Pazopanib for metastatic soft-tissue sarcoma (PALETTE): a randomised, double-blind, placebo-controlled phase 3 trial. Lancet 2012;379:1879–86 [DOI] [PubMed] [Google Scholar]
- 48.Neven P, Rugo HS, Tolaney SM, Iwata H, Toi M, Goetz MP, et al. Abemaciclib plus fulvestrant in hormone receptor-positive, human epidermal growth factor receptor 2-negative advanced breast cancer in premenopausal women: subgroup analysis from the MONARCH 2 trial. Breast Cancer Res 2021;23:87. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 49.Patnaik A, Rosen LS, Tolaney SM, Tolcher AW, Goldman JW, Gandhi L, et al. Efficacy and Safety of Abemaciclib, an Inhibitor of CDK4 and CDK6, for Patients with Breast Cancer, Non-Small Cell Lung Cancer, and Other Solid Tumors. Cancer Discov 2016;6:740–53 [DOI] [PubMed] [Google Scholar]
- 50.Al Baghdadi T, Halabi S, Garrett-Mayer E, Mangat PK, Ahn ER, Sahai V, et al. Palbociclib in Patients With Pancreatic and Biliary Cancer With CDKN2A Alterations: Results From the Targeted Agent and Profiling Utilization Registry Study. JCO Precis Oncol 2019;3:1–8 [DOI] [PubMed] [Google Scholar]
- 51.Gopalan PK, Villegas AG, Cao C, Pinder-Schenck M, Chiappori A, Hou W, et al. CDK4/6 inhibition stabilizes disease in patients with p16-null non-small cell lung cancer and is synergistic with mTOR inhibition. Oncotarget 2018;9:37352–66 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 52.Dickson MA, Schwartz GK, Keohan ML, D’Angelo SP, Gounder MM, Chi P, et al. Progression-Free Survival Among Patients With Well-Differentiated or Dedifferentiated Liposarcoma Treated With CDK4 Inhibitor Palbociclib: A Phase 2 Clinical Trial. JAMA Oncol 2016;2:937–40 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 53.Asghar US, Kanani R, Roylance R, Mittnacht S. Systematic Review of Molecular Biomarkers Predictive of Resistance to CDK4/6 Inhibition in Metastatic Breast Cancer. JCO Precis Oncol 2022;6:e2100002. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 54.Schoninger SF, Blain SW. The Ongoing Search for Biomarkers of CDK4/6 Inhibitor Responsiveness in Breast Cancer. Mol Cancer Ther 2020;19:3–12 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 55.DeMichele A, Clark AS, Tan KS, Heitjan DF, Gramlich K, Gallagher M, et al. CDK 4/6 inhibitor palbociclib (PD0332991) in Rb+ advanced breast cancer: phase II activity, safety, and predictive biomarker assessment. Clin Cancer Res 2015;21:995–1001 [DOI] [PubMed] [Google Scholar]
- 56.Finn RS, Crown JP, Lang I, Boer K, Bondarenko IM, Kulyk SO, et al. The cyclin-dependent kinase 4/6 inhibitor palbociclib in combination with letrozole versus letrozole alone as first-line treatment of oestrogen receptor-positive, HER2-negative, advanced breast cancer (PALOMA-1/TRIO-18): a randomised phase 2 study. Lancet Oncol 2015;16:25–35 [DOI] [PubMed] [Google Scholar]
- 57.Finn RS, Cristofanilli M, Ettl J, Gelmon KA, Colleoni M, Giorgetti C, et al. Treatment effect of palbociclib plus endocrine therapy by prognostic and intrinsic subtype and biomarker analysis in patients with bone-only disease: a joint analysis of PALOMA-2 and PALOMA-3 clinical trials. Breast Cancer Res Treat 2020;184:23–35 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 58.Finn RS, Crown JP, Ettl J, Schmidt M, Bondarenko IM, Lang I, et al. Efficacy and safety of palbociclib in combination with letrozole as first-line treatment of ER-positive, HER2-negative, advanced breast cancer: expanded analyses of subgroups from the randomized pivotal trial PALOMA-1/TRIO-18. Breast Cancer Res 2016;18:67. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 59.Finn RS, Liu Y, Zhu Z, Martin M, Rugo HS, Dieras V, et al. Biomarker Analyses of Response to Cyclin-Dependent Kinase 4/6 Inhibition and Endocrine Therapy in Women with Treatment-Naive Metastatic Breast Cancer. Clin Cancer Res 2020;26:110–21 [DOI] [PubMed] [Google Scholar]
- 60.Hsu JY, Seligson ND, Hays JL, Miles WO, Chen JL. Clinical Utility of CDK4/6 Inhibitors in Sarcoma: Successes and Future Challenges. JCO Precis Oncol 2022;6:e2100211. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 61.Jaaks P, Coker EA, Vis DJ, Edwards O, Carpenter EF, Leto SM, et al. Effective drug combinations in breast, colon and pancreatic cancer cells. Nature 2022;603:166–73 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 62.Bagegni N, Thomas S, Liu N, Luo J, Hoog J, Northfelt DW, et al. Serum thymidine kinase 1 activity as a pharmacodynamic marker of cyclin-dependent kinase 4/6 inhibition in patients with early-stage breast cancer receiving neoadjuvant palbociclib. Breast Cancer Res 2017;19:123. [DOI] [PMC free article] [PubMed] [Google Scholar]
Associated Data
This section collects any data citations, data availability statements, or supplementary materials included in this article.
Supplementary Materials
Data Availability Statement
The data generated in the study is available within the article and its supplemental data files.