

Abstract
As the COVID-19 pathogen, SARS-CoV-2 relies on its main protease (MPro) for pathogenesis and replication. During the crystallographic analyses of MPro crystals that were exposed to the air, a uniquely Y-shaped, S-O-N-O-S-bridged posttranslational crosslink that connects three residues C22, C44, and K61 at their side chains was frequently observed. As a novel covalent modification, this crosslink serves potentially as a redox switch to regulate the catalytic activity of MPro, a demonstrated drug target of COVID-19. The formation of this linkage leads to a much more opened active site that can be potentially targeted for the development of novel SARS-CoV-2 antivirals. The structural rearrangement of MPro by this crosslink indicates that small molecules that lock MPro in the crosslinked form can be potentially used with other active site-targeting molecules such as paxlovid for synergistic effects in inhibiting the SARS-CoV-2 viral replication.
Graphical Abstract
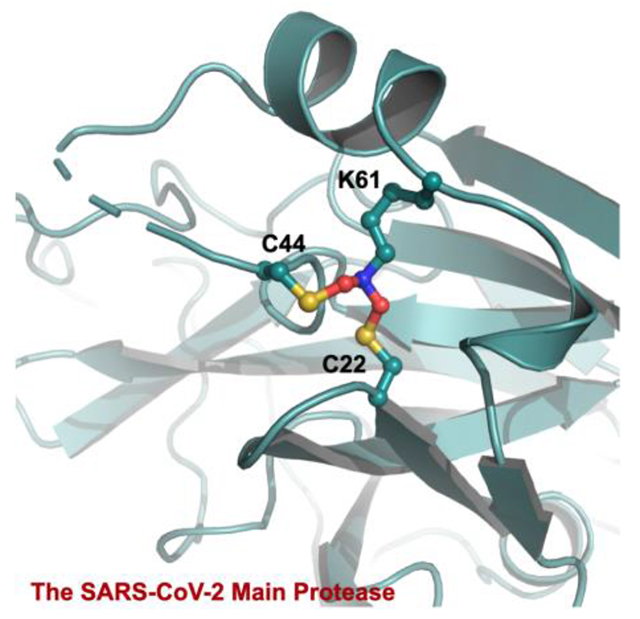
SARS-CoV-2 is the viral pathogen of COVID-19 that has ravaged the whole world for more than two years. Effective vaccines that target the membrane Spike protein of SARS-CoV-2 have been developed and widely adopted for human immunization.1 However, the continuous emergence of new SARS-CoV-2 variants with mutations at Spike has led to viral evasion of vaccines and consequently infection surges.2 The situation has called for the search for SARS-CoV-2 antivirals and led to the recent success of the development of Pfizer’s paxlovid.3–4 SARS-CoV-2 is an RNA virus with a positive-sense RNA genome. It contains a big 20-kb open reading frame ORF1ab that is translated alternatively to form two large polypeptides pp1a and pp1ab in infected human cells.5–6 These two polypeptides need to be hydrolyzed to form 16 nonstructural proteins (nsps) that are key to viral biology including the formation of the RNA-dependent RNA polymerase complex for the replication of the viral genome and subgenomic RNAs. The maturation of pp1a and pp1ab is catalyzed by two internal nsp fragments. One of them is main protease (MPro) that processes 13 out of the total 16 nsps.7 Paxlovid is a combination therapy with two chemical components. One component is nirmatrelvir that potently inhibits MPro to prevent viral replication in infected human cells.4
As an essential enzyme for SARS-CoV-2, MPro is an established target for the development of SARS-CoV-2 antivirals.8–12 Many academic research groups and a number of pharmaceutical companies have been working on the development of MPro inhibitors.13–24 A large international COVID Moonshot project for the development of MPro inhibitors has also been organized.25 As a key tool for the structure-based drug discovery, X-ray protein crystallography has been frequently employed to determine structures of MPro-inhibitor complexes to assist further inhibitor optimization. This is also the approach that we have adopted for the development of MPro inhibitors.26 To facilitate a quick determination of MPro-inhibitor complexes, we have been crystalizing the apo form of MPro and then soaking the crystals with different inhibitors for the ensuing X-ray protein crystallographic analysis.26–31 Our obtained apo-MPro crystals have a C121 or I121 space group that contains an MPro dimer or monomer, respectively, in an asymmetric unit. These crystals are different from other reported MPro crystal forms. In both crystal forms, MPro has a relatively loose and open packaging pattern that makes the active site more solvent accessible (Figure S1). In the I121 space group, two monomers from two asymmetric units form a tightly bound dimer. In both C121 and I121 space groups, a tightly bound dimer interacts with other MPro dimers within the crystals at two regions, aa53-60 and aa216-222 (Figure S2). The aa216-222 region is in the noncatalytic C-terminal domain (Figure S3). Although the aa53-60 region is within the catalytic N-terminal domain, it is distant to the active site (Figure 1A).26 The MPro dimer in these crystals has an open active site whose structure rearrangements are not expected to be limited by the protein packaging in crystals (Figure S1, PDB ID: 7RVN). Therefore, these crystals are ideal for a soaking-based structural determination of MPro-inhibitor complexes. This relatively open form of MPro is considered more desired than a closed form in representing MPro in solutions as well since MPro dimers in solutions are not expected to interact with each other. Therefore, we have been using these apo-MPro crystals and soaking them with different inhibitors for the determination of crystal structures of more than 30 MPro-inhibitor complexes.26,30–31 One unique observation that we have made for almost all our determined MPro-inhibitor structures was a poorly defined conformation for the aa46-50 region. This region is part of the MPro active site, serving as a cap of the protein S2 site to bind the P2 residue in a peptide substrate (Figure 1A).26,30–31 The electron density of this region was very weak in all determined MPro-inhibitor structures indicating a highly flexible conformation (Figure 1B and Figure S4). There have been many other MPro-inhibitor complexes whose crystal structures have been determined and deposited into the Protein Data Bank.8–24 A lot of them have a clearly defined conformation for aa46-50. However, crystals for most of these structures either had a closed form around the active site due to the protein packaging in crystals or were obtained by co-crystallization with ligands. We reason that in the closed form of the MPro crystals the protein packaging limits the structure rearrangement of aa46-50 and prevents it from adopting a flexible conformation. However, in solutions this limitation does not apply. This structural rearrangement is apparently related to the soaking process since the apo-MPro crystals that did not undergo soaking show a well-defined confirmation for aa46-50 and MPro-inhibitor complexes in co-crystalized crystals did not show this rearrangement either.8–10, 26
Figure 1.
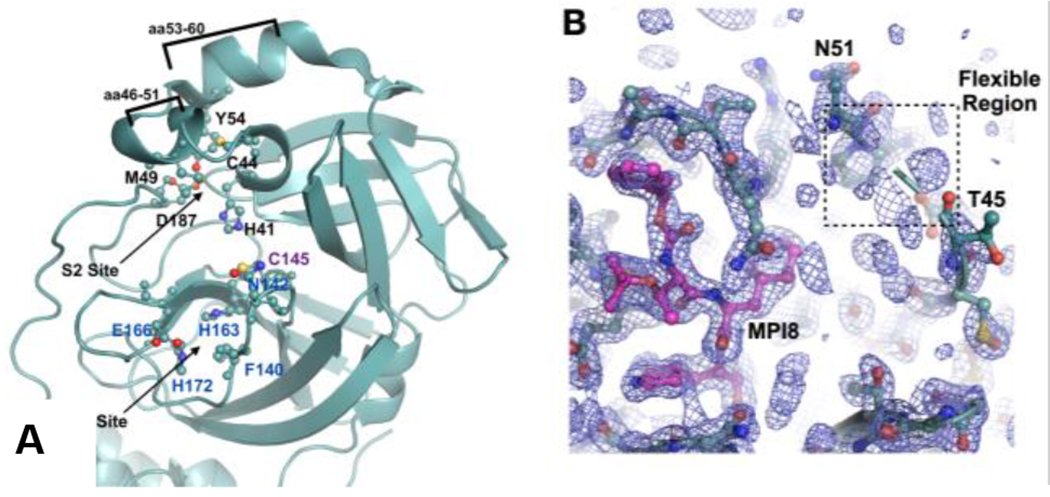
(A) The MPro active site based on the pdb entry 7JPY. C145 is the catalytic cysteine. Residues that form the binding sites for P1 and P2 residues in a substrate are labeled. The aa46-51 and aa53-60 are highlighted. (B) The low electron density map around aa46-50 in a representative MPro-inhibitor complex. The ligand MPI8 that is covalently bound to C145 is shown in a color code in which carbon atoms are shown in red. For the protein, carbon atoms are shown in light teal. The aa46-50 flexible region is indicated in a dashed square. The electron density was contoured at 1σ.
MPro-MPI8 was an early determined MPro-inhibitor complex structure. In this crystal structure, besides a structurally undefined aa46-50 region, we noticed the uniquely connected electron density at residues C22, C44 and K61. As shown in Figure 2A, 2Fo-Fc electron density contoured at 1σ around the ends of side chains from all three residues clearly merged indicating that the three residues were covalently connected to form a Y-shaped tri-residue crosslink. Directly connecting three residues at their terminal heteroatoms to form a Y-shaped S-N-S crosslink did not fit the calculated electron density. Although there was no literature report about a Y-shaped crosslink between one lysine and two cysteine residues in a protein, a recent Nature article by Wensien et al. described a novel posttranslational N-O-S-bridged lysine-cysteine crosslink as a redox switch for the transaldolase enzyme from Neisseria gonorrhoeae.32 N. gonorrhoeae transaldolase undergoes oxidation to form an N-O-S crosslink in which an oxygen atom covalently bridges the K8 side chain amine and C38 side chain thiol. The consequential oxidized enzyme has a substantially lower activity than the reduced form. As a newly identified posttranslational modification, the N-O-S crosslink via oxidation serves as a regulatory mechanism for N. gonorrhoeae transaldolase. Since in MPro-MPI8, the K61 side chain amine connected with two side chain thiol groups from C22 and C44, respectively, to form a Y-shaped crosslink instead of a linear S-O-N crosslink as observe between K8 and C38 in N. gonorrhoeae transaldolase, we suspected that a S-O-N-O-S-bridged, Y-shaped tri-residue crosslink in which two oxygen atoms bridged two cysteine thiol groups separately with the K61 side chain amine existed in our MPro-MPI8 structure. Adding this crosslink into the structure for undergoing the structure refinement fit the calculated electron density perfectly. The same S-O-N-O-S crosslink was found for a deposited MPro-inhibitor structure 7JR4.33 It was assigned as two methylene-crosslinked lysine-cysteine bonds by the depositors and later analyzed as a S-O-N-O-S bond by Tittmann et al.33
Figure 2.
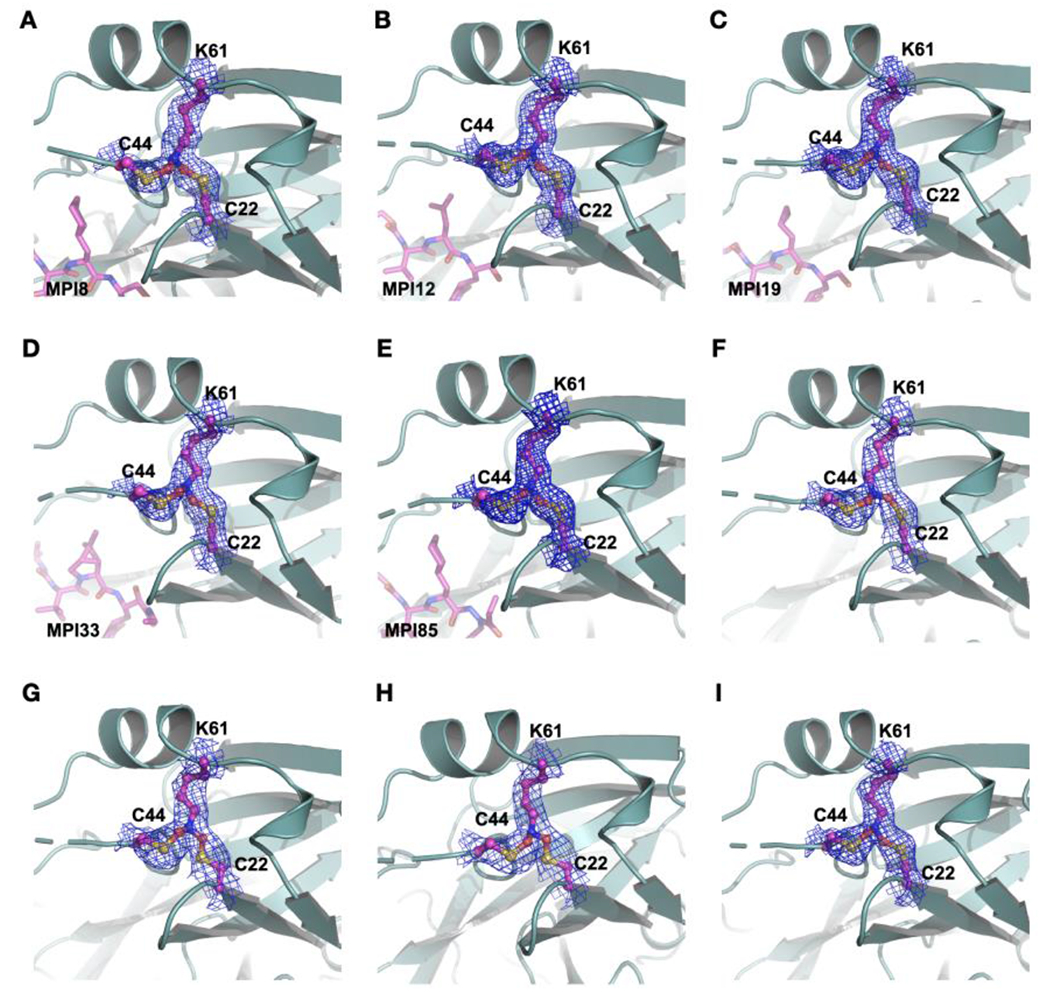
Five MPro-ligand complexes and four apo enzyme structures show a SONOS-bridged crosslink between three residues including C22, C44 and K61. The electron density was contoured at 1 σ.
Among about 10 early crystal structures of MPro-inhibitor complexes, MPro-MPI8 was the only one that contained the Y-shaped crosslink. However, this crosslink was subsequently also observed in several MPro-inhibitor complexes determined later. Figures 2B–E show crystal structures for four other MPro complexes with MPI12, MPI19, MPI33, and MPI85, respectively. They all show a well-defined Y-shaped, S-O-N-O-S-bridged crosslink between K61 and two cysteine residues C22 and C44. We suspected that the inhibitor binding process might have triggered the formation of the crosslink, however, a careful search of our other determined structures revealed four apo-MPro structures that contained this crosslink as well. As shown in Figures 2F–H, all four structures showed clearly connected electron density at the ends of three residues C22, C44 and K61. These four apo-MPro structures were determined from apo-MPro crystals that were soaked with chemical fragments, but no bound ligands were observed. So far, all structures that showed this crosslink were determined from crystals that were exposed to the air due to the soaking process.
We also determined X-ray crystal structures for several apo-MPro crystals that were not exposed to the air. They all had a structurally defined aa46-50 region and exhibited no crosslink between C22, C44 and K61 (PDB entry: 7UU6, 7UU7, 7UU8, 7UU9). One of these structures was previously deposited into the Protein Data Bank (PDB entry: 7JPY).26 As shown in Figure 3A, the superposition of 7JPY over an apo-MPro structure with the Y-shaped crosslink reveals a large structural rearrangement at the region aa43-52 between two structures. In 7JPY, the side chain of C44 points toward the active site. The small aa46-51 helix tucks C44 toward the active site and makes it a key component to form the S2 binding pocket for a substrate P2 residue. The thiol group of C44 is 9.1 Å away from the K61 side chain amine (Figure 3A in limon). This distance is too far to form any possible direct interaction between the two residues. For C44 to physically meet K61 for a covalent interaction, the C44 backbone α-carbon moved 4.1 Å closer to K61 and rotated its side chain almost 180° to adopt a conformation to form a covalent adduct with the K61 side chain amine as shown in the crosslinked apo-MPro (Figure 3A in light teal). This structural rearrangement also pushed the whole aa43-52 region away from the MPro active site, making the active site much more open. For C44 to translocate and rotate, a large structural rearrangement for the whole aa43-52 region is required. This explains why the Y-shaped S-O-N-O-S crosslink was observed in our crystal structures but not in most other determined MPro structures since the protein packaging in our MPro crystals allowed the large structure rearrangement at aa43-52. In 7JPY, C22 and K61 are in a close distance to each other. This close distance will likely make them form an N-O-S-bridged crosslink first and then a structurally flipped C44 will be engaged to generate the second N-O-S bridge. Since all our MPro structures that contained the Y-shaped posttranslational crosslink were determined from crystals that were exposed to the air, the molecular oxygen was most likely the reagent that generated the Y-shaped S-O-N-O-S bridge. Although how exactly the molecular oxygen oxidizes the three residues to form the crosslink needs to be further explored and confirmed, we propose a likely mechanism as shown in Figure 3B. In this mechanism, the C22 thiolate reacts with oxygen to form a peroxysulfane that then reacts with lysine to generate the first S-O-N crosslink. After C44 flips toward K61, its thiolate can react with oxygen to form the second peroxysulfane that then reacts with the first S-O-N crosslink to form the Y-shaped, S-O-N-O-S-bridged tri-residue crosslink. One puzzling fact was that all crystals were preserved in 1 mM DTT even in the soaking environments to maintain a highly reductive environment. How this crosslink formed in this highly reductive condition is intriguing.
Figure 3.
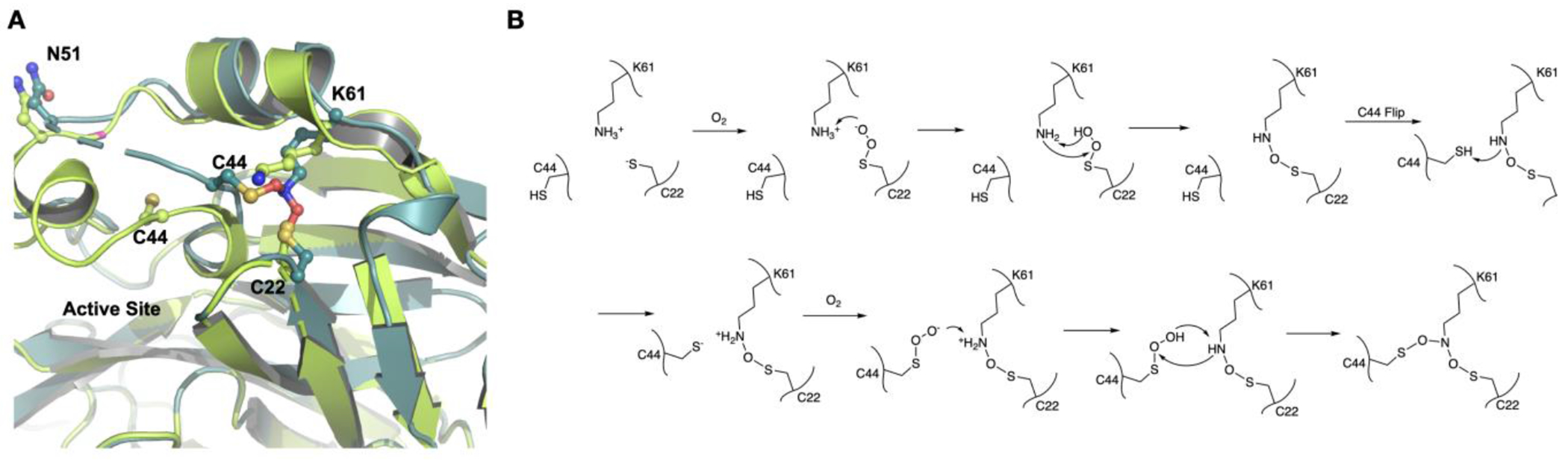
(A) The superposition of two different apo-MPro structures. One doesn’t have the S-O-N-O-S-bridged triresidue crosslink (limon) and the other one does (light teal). (B) A proposed scheme for the formation of the S-O-N-O-S-bridged triresidue crosslink in MPro.
Since there is no information such as chemical stability about the S-O-N-O-S crosslink in the literature, we conducted a native mass spectrometry analysis of the freshly purified MPro to keep the potential S-O-N-O-S crosslink intact. Our collected spectrum of MPro at 25°C clearly showed the MPro dimer, the MPro dimer with two additional oxygen atoms, and the MPro dimer with four additional oxygen atoms (Figures 4 and S5). The MPro dimer with four additional oxygen atoms is equivalent to MPro with the Y-shaped crosslink. The MPro dimer with two additional oxygen atoms is likely an intermediate state that has either just one MPro monomer with the S-O-N-O-S crosslink or both MPro monomers with just the S-O-N crosslink. An interesting observation was also made during the native mass spectrometry analysis of MPro when we did the in-instrument heating of MPro by sequentially increasing the temperature by 5°C and waiting for 30 min before the native mass spectrometry analysis was conducted. When the temperature was increased to 40°C, the majority of the MPro dimer was converted to the one containing four additional oxygen atoms. When 5 mM DTT was used during the analysis, the oxidation was not observed indicating that the oxidation is reversible (Figure S6). To demonstrate this oxidation is related to the crosslink formation, we generated the MPro K61A mutant and used it for the same mass spectrometry analysis without adding 5 mM DTT. Since K61 bridges the S-O-N-O-S crosslink with C22 and C44, its mutation to alanine was expected to destroy the crosslink formation. As expected, the MPro K61A mutant did not show any oxidation during the temperature treatment associated with the native mass spectrometry analysis (Figure S7), supporting the direct involvement of K61 in the crosslink formation. One likely explanation of all mass spectrometry results is that the protein at 40°C has enhanced structural dynamics that assists the formation of the Y-shaped S-O-N-O-S crosslink. One needs to caution that the mass spectrometry results provide indirect support of the S-O-N-O-S formation. The definitive formation of the S-O-N-O-S bridge will still need to be further confirmed. Since the bond is weak, we have not been able to use tandem mass spectrometry analysis to independently validate the S-O-N-O-S bridge formation between the three residues.
Figure 4.
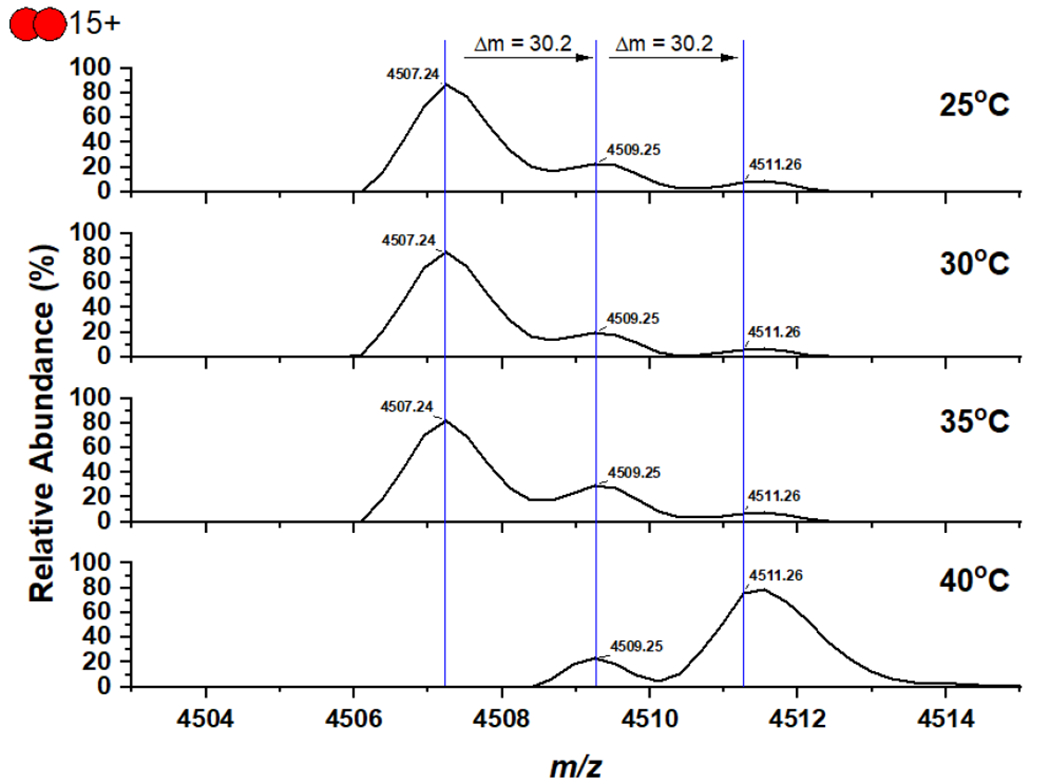
Native mass spectra of MPro that was freshly purified without a reducing reagent in lysis and purification buffers and sequentially treated in the instrument by increasing the temperature by 5°C and waiting for 30 min.
In summary, we have observed a uniquely Y-shaped, S-O-N-O-S-bridged crosslink between three residues C22, C44 and K61 in the SARS-CoV-2 MPro. As a novel posttranslational modification, this crosslink significantly changes the SARS-CoV-2 MPro structure. Our finding is important in assisting the development of MPro inhibitors as SARS-CoV-2 antivirals. The generation of the crosslink leaves a more open, larger active site allowing for the design and testing of many new inhibitors. One significant benefit of inhibitors that target this more open active site is their potential synergy in driving and stabilizing the crosslink formation together with small molecules that could permanently lock C22, C44 and K61 in their crosslinked form. Exploring this direction will likely lead to the development of a totally new group of SARS-CoV-2 antivirals.
Methods
Recombinant MPro protein expression and purification.
The pET28a-His-SUMO-MPro expression and purification are according to our previous report.26 The pET28a-His-SUMO-MPro construct was transformed into E. coli BL21(DE3) cells. Transformed cells were cultured at 37°C in 2xYT medium with kanamycin (50 g/mL) until OD600 reaching 0.6, then induced with 1 mM isopropyl-D-1-thiogalactoside (IPTG) at 37 °C. After 3 hours, cells were harvested and lysed in buffer A (20 mM Tris, 100 mM NaCl, 10 mM imidazole, pH 8.0). The supernatant was loaded onto a nickel-chelating column (GenScript) washed with buffer A, followed by elution with buffer B (20 mM Tris, 100 mM NaCl, 250 mM imidazole, pH 8.0). The eluted protein solution was desalted to buffer C (20 mM Tris, 10 mM NaCl, pH 8.0) by HiPrep 26/10 desalting column (GE Healthcare). The His-SUMO-MPro proteins were digested with SUMO protease overnight at 4°C. The digested protein was applied to nickel-chelating column again to remove the His-tagged SUMO protease, the His-SUMO tag, and the expressed protein with uncleaved His-SUMO tag. The tag-free MPro protein was loaded to the size exclusion column HiPrep 16/60 Sephacryl S-100 HR (GE Healthcare) pre-equilibrated with buffer D (20 mM Tris, 100 mM NaCl, 1 mM EDTA, pH 7.8). The eluted MPro protein was stored with buffer D in −80 °C for further use. Site directed mutagenesis was used to introduce the K61A mutation into the plasmid pET28a-His-SUMO-MPro. The afforded plasmid was used to express MPro K61A by following the same procedures as for the wildtype MPro.
X-Ray Crystallography Analysis.
The production of crystals of apo MPro and MPro-inhibitor complexes was following the previous protocols with the crystal growing condition of 0.1 M Bis-tris, pH6.5, 16% w/v PEG10k.26 The data of MPro with MPI12 was collected on a Rigaku R-AXIS IV++ image plate detector, the data of MPro with MPI19 was collected at the Advanced Light Source (ALS) beamline 5.0.2 using a Pilatus3 6M detector, all the other data were collected on a Bruker Photon II detector. The diffraction data were indexed, integrated and scaled with iMosflm or PROTEUM3.34 The structure was determined by molecular replacement using the structure model of the free enzyme of the SARS-CoV-2 Mpro [Protein Data Bank (PDB) ID code 7JPY] as the search model using Phaser in the Phenix package.26, 35 JLigand and Sketcher from the CCP4 suite were employed for the generation of PDB and geometric restraints for the inhibitors. The inhibitors were built into the Fo-Fc density by using Coot.36 Refinement of all the structures was performed with Real-space Refinement in Phenix.35 Details of data quality and structure refinement are summarized in Table S1. All structural figures were generated with PyMOL (https://www.pymol.org).
Native Mass Spectrometry Materials.
Ammonium acetate (99.999% trace metals basis) was purchased from Sigma Aldrich (St. Louis, MO). MPro was expressed as described. Sample aliquots were stored at −80°C before freshly thawed for immediate analysis. MPro was buffer exchanged to 200 mM ammonium acetate (pH = 6.8) by using Micro Biospin P-6 gel column (BioRad) with final concentration 4 μM. The exchanged MPro solution was equilibrated for 15 min at room temperature prior to mass spectrometry analysis.
Native Mass Spectrometry.
Native mass spectrometry (nMS) analysis was performed on a Q Exactive UHMR Hybrid Quadruple-Orbitrap Mass Spectrometer (ThermoFisher) with m/z range was set from 1,000 to 10,000 and with resolution set to 12,500 (at m/z 400). 10 μL of the sample was loaded to a borosilicate glass capillary tip (Sutter, CA) with 1100 to 1500 V spray voltage supplied by an inserted platinum wire. Important parameters to reduce non-specific adducts include capillary temperature 100°C, in-source trapping and activation −10 V, ion transfer high m/z, collision-induced dissociation (CID) 10 eV, and higher energy dissociation (HCD) 30 V. In variable-temperature electrospray ionization (vT-ESI) experiment, the temperature of solution was controlled as previously described.37 The temperature was varied from 25°C to 40°C with 5°C increments and the equilibrium time at each temperature was 5 min. Additional characterizations were performed on an Exactive Plus EMR orbitrap mass spectrometer (ThermoFisher) with set from 1,000 to 10,000 and with resolution set to 17, 500 (at m/z 400). 10 μL of the sample was loaded to a borosilicate glass capillary tip (Sutter, CA) with 1.0 to 1.2 kV spray voltage supplied by an inserted platinum wire. Important parameters to reduce non-specific adducts include: capillary temperature 50°C, collision-induced dissociation (CID) 10 eV, higher energy dissociation (HCD) 50 V, and trapping gas set to 5 (arbitrary unit). In variable-temperature electrospray ionization (vT-ESI) experiment, the temperature of solution was controlled as previously described.37 The temperature was varied from 25°C to 40°C with 5°C increments and the equilibrium time at each temperature was 5 min.
Supplementary Material
ACKNOWLEDGMENT
The work was supported by Welch Foundation (Grants A-1715 and A-2089), National Institutes of Health (Grants R21AI164088, R21EB032983, R35GM143047, R35GM145351, P41GM128577, and R01GM138863), and the Texas A&M X Grants Mechanism.
Footnotes
Supporting Information
The Supporting Information is available free of charge on the ACS Publications website.
Supplementary Figures and Supplementary Table 1 that contains statistics of crystallographic analysis.
Conflict of Interests
The authors declare no competing financial interest.
REFERENCES
- 1.Martinez-Flores D; Zepeda-Cervantes J; Cruz-Resendiz A; Aguirre-Sampieri S; Sampieri A; Vaca L, SARS-CoV-2 Vaccines Based on the Spike Glycoprotein and Implications of New Viral Variants. Front. Immunol 2021, 12, 701501. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 2.Mengist HM; Kombe Kombe AJ; Mekonnen D; Abebaw A; Getachew M; Jin T, Mutations of SARS-CoV-2 spike protein: Implications on immune evasion and vaccine-induced immunity. Semin. Immunol 2021, 55, 101533. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 3.Mahase E, Covid-19: Pfizer’s paxlovid is 89% effective in patients at risk of serious illness, company reports. BMJ 2021, 375, n2713. [DOI] [PubMed] [Google Scholar]
- 4.Owen DR; Allerton CMN; Anderson AS; Aschenbrenner L; Avery M; Berritt S; Boras B; Cardin RD; Carlo A; Coffman KJ; Dantonio A; Di L; Eng H; Ferre R; Gajiwala KS; Gibson SA; Greasley SE; Hurst BL; Kadar EP; Kalgutkar AS; Lee JC; Lee J; Liu W; Mason SW; Noell S; Novak JJ; Obach RS; Ogilvie K; Patel NC; Pettersson M; Rai DK; Reese MR; Sammons MF; Sathish JG; Singh RSP; Steppan CM; Stewart AE; Tuttle JB; Updyke L; Verhoest PR; Wei L; Yang Q; Zhu Y, An oral SARS-CoV-2 M(pro) inhibitor clinical candidate for the treatment of COVID-19. Science 2021, 374 (6575), 1586–1593. [DOI] [PubMed] [Google Scholar]
- 5.Kim D; Lee JY; Yang JS; Kim JW; Kim VN; Chang H, The Architecture of SARS-CoV-2 Transcriptome. Cell 2020, 181 (4), 914–921 e10. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 6.Zhou P; Yang XL; Wang XG; Hu B; Zhang L; Zhang W; Si HR; Zhu Y; Li B; Huang CL; Chen HD; Chen J; Luo Y; Guo H; Jiang RD; Liu MQ; Chen Y; Shen XR; Wang X; Zheng XS; Zhao K; Chen QJ; Deng F; Liu LL; Yan B; Zhan FX; Wang YY; Xiao GF; Shi ZL, A pneumonia outbreak associated with a new coronavirus of probable bat origin. Nature 2020, 579 (7798), 270–273. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 7.Morse JS; Lalonde T; Xu S; Liu WR, Learning from the Past: Possible Urgent Prevention and Treatment Options for Severe Acute Respiratory Infections Caused by 2019-nCoV. ChemBioChem 2020, 21 (5), 730–738. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 8.Dai W; Zhang B; Jiang XM; Su H; Li J; Zhao Y; Xie X; Jin Z; Peng J; Liu F; Li C; Li Y; Bai F; Wang H; Cheng X; Cen X; Hu S; Yang X; Wang J; Liu X; Xiao G; Jiang H; Rao Z; Zhang LK; Xu Y; Yang H; Liu H, Structure-based design of antiviral drug candidates targeting the SARS-CoV-2 main protease. Science 2020, 368 (6497), 1331–1335. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 9.Jin Z; Du X; Xu Y; Deng Y; Liu M; Zhao Y; Zhang B; Li X; Zhang L; Peng C; Duan Y; Yu J; Wang L; Yang K; Liu F; Jiang R; Yang X; You T; Liu X; Yang X; Bai F; Liu H; Liu X; Guddat LW; Xu W; Xiao G; Qin C; Shi Z; Jiang H; Rao Z; Yang H, Structure of M(pro) from SARS-CoV-2 and discovery of its inhibitors. Nature 2020, 582 (7811), 289–293. [DOI] [PubMed] [Google Scholar]
- 10.Zhang L; Lin D; Sun X; Curth U; Drosten C; Sauerhering L; Becker S; Rox K; Hilgenfeld R, Crystal structure of SARS-CoV-2 main protease provides a basis for design of improved alpha-ketoamide inhibitors. Science 2020, 368 (6489), 409–412. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 11.Gunther S; Reinke PYA; Fernandez-Garcia Y; Lieske J; Lane TJ; Ginn HM; Koua FHM; Ehrt C; Ewert W; Oberthuer D; Yefanov O; Meier S; Lorenzen K; Krichel B; Kopicki JD; Gelisio L; Brehm W; Dunkel I; Seychell B; Gieseler H; Norton-Baker B; Escudero-Perez B; Domaracky M; Saouane S; Tolstikova A; White TA; Hanle A; Groessler M; Fleckenstein H; Trost F; Galchenkova M; Gevorkov Y; Li C; Awel S; Peck A; Barthelmess M; Schlunzen F; Lourdu Xavier P; Werner N; Andaleeb H; Ullah N; Falke S; Srinivasan V; Franca BA; Schwinzer M; Brognaro H; Rogers C; Melo D; Zaitseva-Doyle JJ; Knoska J; Pena-Murillo GE; Mashhour AR; Hennicke V; Fischer P; Hakanpaa J; Meyer J; Gribbon P; Ellinger B; Kuzikov M; Wolf M; Beccari AR; Bourenkov G; von Stetten D; Pompidor G; Bento I; Panneerselvam S; Karpics I; Schneider TR; Garcia-Alai MM; Niebling S; Gunther C; Schmidt C; Schubert R; Han H; Boger J; Monteiro DCF; Zhang L; Sun X; Pletzer-Zelgert J; Wollenhaupt J; Feiler CG; Weiss MS; Schulz EC; Mehrabi P; Karnicar K; Usenik A; Loboda J; Tidow H; Chari A; Hilgenfeld R; Uetrecht C; Cox R; Zaliani A; Beck T; Rarey M; Gunther S; Turk D; Hinrichs W; Chapman HN; Pearson AR; Betzel C; Meents A, X-ray screening identifies active site and allosteric inhibitors of SARS-CoV-2 main protease. Science 2021, 372 (6542), 642–646. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 12.Qiao J; Li YS; Zeng R; Liu FL; Luo RH; Huang C; Wang YF; Zhang J; Quan B; Shen C; Mao X; Liu X; Sun W; Yang W; Ni X; Wang K; Xu L; Duan ZL; Zou QC; Zhang HL; Qu W; Long YH; Li MH; Yang RC; Liu X; You J; Zhou Y; Yao R; Li WP; Liu JM; Chen P; Liu Y; Lin GF; Yang X; Zou J; Li L; Hu Y; Lu GW; Li WM; Wei YQ; Zheng YT; Lei J; Yang S, SARS-CoV-2 M(pro) inhibitors with antiviral activity in a transgenic mouse model. Science 2021, 371 (6536), 1374–1378. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 13.Dai W; Jochmans D; Xie H; Yang H; Li J; Su H; Chang D; Wang J; Peng J; Zhu L; Nian Y; Hilgenfeld R; Jiang H; Chen K; Zhang L; Xu Y; Neyts J; Liu H, Design, Synthesis, and Biological Evaluation of Peptidomimetic Aldehydes as Broad-Spectrum Inhibitors against Enterovirus and SARS-CoV-2. J. Med. Chem 2022, 65 (4), 2794–2808. [DOI] [PubMed] [Google Scholar]
- 14.Ma C; Sacco MD; Hurst B; Townsend JA; Hu Y; Szeto T; Zhang X; Tarbet B; Marty MT; Chen Y; Wang J, Boceprevir GC -376, and calpain inhibitors II, XII inhibit SARS-CoV-2 viral replication by targeting the viral main protease. Cell Res. 2020, 30 (8), 678–692. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 15.Ghahremanpour MM; Tirado-Rives J; Deshmukh M; Ippolito JA; Zhang CH; Cabeza de Vaca I; Liosi ME; Anderson KS; Jorgensen WL , Identification of 14 Known Drugs as Inhibitors of the Main Protease of SARS-CoV-2. ACS Med. Chem. Lett 2020, 11 (12), 2526–2533. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 16.Zhu W; Xu M; Chen CZ; Guo H; Shen M; Hu X; Shinn P; Klumpp-Thomas C; Michael SG; Zheng W, Identification of SARS-CoV-2 3CL Protease Inhibitors by a Quantitative High-Throughput Screening. ACS Pharmacol. Transl. Sci 2020, 3 (5), 1008–1016. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 17.Hoffman RL; Kania RS; Brothers MA; Davies JF; Ferre RA; Gajiwala KS; He M; Hogan RJ; Kozminski K; Li LY; Lockner JW; Lou J; Marra MT; Mitchell LJ Jr.; Murray BW; Nieman JA; Noell S; Planken SP; Rowe T; Ryan K; Smith GJ 3rd; Solowiej JE; Steppan CM; Taggart B, Discovery of Ketone-Based Covalent Inhibitors of Coronavirus 3CL Proteases for the Potential Therapeutic Treatment of COVID-19. J. Med. Chem 2020, 63 (21), 12725–12747. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 18.Bai B; Arutyunova E; Khan MB; Lu J; Joyce MA; Saffran HA; Shields JA; Kandadai AS; Belovodskiy A; Hena M; Vuong W; Lamer T; Young HS; Vederas JC; Tyrrell DL; Lemieux MJ; Nieman JA, Peptidomimetic nitrile warheads as SARS-CoV-2 3CL protease inhibitors. RSC Med. Chem 2021, 12 (10), 1722–1730. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 19.Bai B; Belovodskiy A; Hena M; Kandadai AS; Joyce MA; Saffran HA; Shields JA; Khan MB; Arutyunova E; Lu J; Bajwa SK; Hockman D; Fischer C; Lamer T; Vuong W; van Belkum MJ; Gu Z; Lin F; Du Y; Xu J; Rahim M; Young HS; Vederas JC; Tyrrell DL; Lemieux MJ; Nieman JA, Peptidomimetic alpha-Acyloxymethylketone Warheads with Six-Membered Lactam P1 Glutamine Mimic: SARS-CoV-2 3CL Protease Inhibition, Coronavirus Antiviral Activity, and in Vitro Biological Stability. J. Med. Chem 2022, 65 (4), 2905–2925. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 20.Breidenbach J; Lemke C; Pillaiyar T; Schakel L; Al Hamwi G; Diett M; Gedschold R; Geiger N; Lopez V; Mirza S; Namasivayam V; Schiedel AC; Sylvester K; Thimm D; Vielmuth C; Phuong Vu L; Zyulina M; Bodem J; Gutschow M; Muller CE, Targeting the Main Protease of SARS-CoV-2: From the Establishment of High Throughput Screening to the Design of Tailored Inhibitors. Angew. Chem. Int. Ed 2021, 60 (18), 10423–10429. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 21.Dampalla CS; Zheng J; Perera KD; Wong LR; Meyerholz DK; Nguyen HN; Kashipathy MM; Battaile KP; Lovell S; Kim Y; Perlman S; Groutas WC; Chang KO, Postinfection treatment with a protease inhibitor increases survival of mice with a fatal SARS-CoV-2 infection. Proc. Natl. Acad. Sci. U. S. A 2021, 118 (29), e2101555118. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 22.Ghosh AK; Raghavaiah J; Shahabi D; Yadav M; Anson BJ; Lendy EK; Hattori SI; Higashi-Kuwata N; Mitsuya H; Mesecar AD, Indole Chloropyridinyl Ester-Derived SARS-CoV-2 3CLpro Inhibitors: Enzyme Inhibition, Antiviral Efficacy, Structure-Activity Relationship, and X-ray Structural Studies. J. Med. Chem 2021, 64 (19), 14702–14714. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 23.Ma C; Xia Z; Sacco MD; Hu Y; Townsend JA; Meng X; Choza J; Tan H; Jang J; Gongora MV; Zhang X; Zhang F; Xiang Y; Marty MT; Chen Y; Wang J, Discovery of Di- and Trihaloacetamides as Covalent SARS-CoV-2 Main Protease Inhibitors with High Target Specificity. J. Am. Chem. Soc 2021, 143 (49), 20697–20709. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 24.Unoh Y; Uehara S; Nakahara K; Nobori H; Yamatsu Y; Yamamoto S; Maruyama Y; Taoda Y; Kasamatsu K; Suto T; Kouki K; Nakahashi A; Kawashima S; Sanaki T; Toba S; Uemura K; Mizutare T; Ando S; Sasaki M; Orba Y; Sawa H; Sato A; Sato T; Kato T; Tachibana Y, Discovery of S-217622, a Noncovalent Oral SARS-CoV-2 3CL Protease Inhibitor Clinical Candidate for Treating COVID-19. J. Med. Chem 2022, e2024358118. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 25.Kantsadi AL; Cattermole E; Matsoukas MT; Spyroulias GA; Vakonakis I, A COVID moonshot: assessment of ligand binding to the SARS-CoV-2 main protease by saturation transfer difference NMR spectroscopy. J. Biomol. NMR 2021, 75 (4–5), 167–178. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 26.Yang KS; Ma XR; Ma Y; Alugubelli YR; Scott DA; Vatansever EC; Drelich AK; Sankaran B; Geng ZZ; Blankenship LR; Ward HE; Sheng YJ; Hsu JC; Kratch KC; Zhao B; Hayatshahi HS; Liu J; Li P; Fierke CA; Tseng CK; Xu S; Liu WR, A Quick Route to Multiple Highly Potent SARS-CoV-2 Main Protease Inhibitors. ChemMedChem 2021, 16 (6), 942–948. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 27.Li L; Chenna BC; Yang KS; Cole TR; Goodall ZT; Giardini M; Moghadamchargari Z; Hernandez EA; Gomez J; Calvet CM; Bernatchez JA; Mellott DM; Zhu J; Rademacher A; Thomas D; Blankenship LR; Drelich A; Laganowsky A; Tseng CK; Liu WR; Wand AJ; Cruz-Reyes J; Siqueira-Neto JL; Meek TD, Self-Masked Aldehyde Inhibitors: A Novel Strategy for Inhibiting Cysteine Proteases. J. Med. Chem 2021, 64 (15), 11267–11287. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 28.Li SG; Yang KS; Blankenship LR; Cho CD; Xu S; Wang H; Liu WR, An Enhanced Hybrid Screening Approach to Identify Potent Inhibitors for the SARS-CoV-2 Main Protease From the NCI Compound Library. Front Chem 2022, 10, 816576. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 29.Cao W; Cho CD; Geng ZZ; Shaabani N; Ma XR; Vatansever EC; Alugubelli YR; Ma Y; Chaki SP; Ellenburg WH; Yang KS; Qiao Y; Allen R; Neuman BW; Ji H; Xu S; Liu WR, Evaluation of SARS-CoV-2 Main Protease Inhibitors Using a Novel Cell-Based Assay. ACS Cent. Sci 2022, 8 (2), 192–204. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 30.Ma Y; Yang KS; Geng ZZ; Alugubelli YR; Shaabani N; Vatansever EC; Ma XR; Cho CC; Khatua K; Xiao J; Blankenship LR; Yu G; Sankaran B; Li P; Allen R; Ji H; Xu S; Liu WR, A multi-pronged evaluation of aldehyde-based tripeptidyl main protease inhibitors as SARS-CoV-2 antivirals. Eur. J. Med. Chem 2022, 240, 114570. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 31.Alugubelli YR; Geng ZZ; Yang KS; Shaabani N; Khatua K; Ma XR; Vatansever EC; Cho CC; Ma Y; Xiao J; Blankenship LR; Yu G; Sankaran B; Li P; Allen R; Ji H; Xu S; Liu WR, A systematic exploration of boceprevir-based main protease inhibitors as SARS-CoV-2 antivirals. Eur. J. Med. Chem 2022, 240, 114596. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 32.Wensien M; von Pappenheim FR; Funk LM; Kloskowski P; Curth U; Diederichsen U; Uranga J; Ye J; Fang P; Pan KT; Urlaub H; Mata RA; Sautner V; Tittmann K, A lysine-cysteine redox switch with an NOS bridge regulates enzyme function. Nature 2021, 593 (7859), 460–464. [DOI] [PubMed] [Google Scholar]
- 33.Rabe von Pappenheim F; Wensien M; Ye J; Uranga J; Irisarri I; de Vries J; Funk LM; Mata RA; Tittmann K, Widespread occurrence of covalent lysine-cysteine redox switches in proteins. Nat. Chem. Biol 2022, 18 (4), 368–375. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 34.Winn MD; Ballard CC; Cowtan KD; Dodson EJ; Emsley P; Evans PR; Keegan RM; Krissinel EB; Leslie AG; McCoy A; McNicholas SJ; Murshudov GN; Pannu NS; Potterton EA; Powell HR; Read RJ; Vagin A; Wilson KS, Overview of the CCP4 suite and current developments. Acta Crystallogr. D Biol. Crystallogr 2011, 67 (Pt 4), 235–42. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 35.Adams PD; Afonine PV; Bunkoczi G; Chen VB; Davis IW; Echols N; Headd JJ; Hung LW; Kapral GJ; Grosse-Kunstleve RW; McCoy AJ; Moriarty NW; Oeffner R; Read RJ; Richardson DC; Richardson JS; Terwilliger TC; Zwart PH, PHENIX: a comprehensive Python-based system for macromolecular structure solution. Acta Crystallogr. D Biol. Crystallogr 2010, 66 (Pt 2), 213–21. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 36.Emsley P; Lohkamp B; Scott WG; Cowtan K, Features and development of Coot. Acta Crystallogr. D Biol. Crystallogr 2010, 66 (Pt 4), 486–501. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 37.McCabe JW; Shirzadeh M; Walker TE; Lin CW; Jones BJ; Wysocki VH; Barondeau DP; Clemmer DE; Laganowsky A; Russell DH, Variable-Temperature Electrospray Ionization for Temperature-Dependent Folding/Refolding Reactions of Proteins and Ligand Binding. Anal. Chem 2021, 93 (18), 6924–6931. [DOI] [PMC free article] [PubMed] [Google Scholar]
Associated Data
This section collects any data citations, data availability statements, or supplementary materials included in this article.