

Abstract
Head and neck squamous cell carcinoma (HNSCC), a highly heterogeneous disease that involves multiple anatomic sites, is a leading cause of cancer-related mortality worldwide. Although the utility of noninvasive biomarkers based on circulating cell-free DNA (cfDNA) methylation profiling has been widely recognized, limited studies have been reported so far regarding the dynamics of cfDNA methylome in oral cavity squamous cell carcinoma (OCSCC). It is hypothesized in this study that comparison of methylation profiles in pre- and post-surgery plasma samples will reveal OCSCC-specific prognostic and diagnostic biomarkers. As a strategy to further prioritize tumor-specific targets, top differential methylated regions (DMRs) were called by reanalyzing methylation data from paired tumor and normal tissue collected in the TCGA head and neck cancer cohort. Matched plasma samples from eight patients with OCSCC were collected at Moffitt Cancer Center before and after surgical resection. Plasma-derived cfDNA was analyzed by cfMBD-seq, which is a high-sensitive methylation profiling assay. Differential methylation analysis was then performed based on the matched samples profiled. In the top 200 HNSCC-specific DMRs detected based on the TCGA dataset, a total of 23 regions reached significance in the plasma-based DMR test. The top five validated DMR regions (ranked by the significance in the plasma study) are located in the promoter regions of genes PENK, NXPH1, ZIK1, TBXT and CDO1, respectively. The genome-wide cfDNA DMR analysis further highlighted candidate biomarkers located in genes SFRP4, SOX1, IRF4 and PCDH17. The prognostic relevance of candidate genes was confirmed by survival analysis using the TCGA data. This study supports the utility of cfDNA-based methylome profiling as a promising noninvasive biomarker source for OCSCC and HNSCC.
Keywords: cell-free DNA, head and neck cancer, methylation biomarkers, oral cavity squamous cell carcinoma, plasma
INTRODUCTION
Head and neck squamous cell carcinoma (HNSCC) is a highly heterogeneous disease that involves multiple anatomic sites including oral cavity, larynx, and pharynx. Outcomes of patients with HNSCC have not improved significantly over the past decade, with an overall five-year survival rate around 50%. There is a pressing need in the area to develop reliable prognostic and diagnostic biomarkers to enable better patient management, from early detection of disease to efficient monitoring of cancer recurrence following treatment. Despite their established role in cancer development1, epigenetic biomarkers and DNA methylation (DNAme) remain understudied in HNSCC research. Currently, TCGA-HNSC is the only publicly available resource for mining DNAme patterns in head and neck cancer. Cell free DNA (cfDNA) includes both genetic and epigenetic information2 and offers several advantages including monitoring tumor burden3, and novel discovery of biomarkers for diagnosis and prognosis.4 cfDNA is thought to potentially incorporate metastatic sites thus addressing tumor heterogeneity.5,6 Aberrant DNA methylation changes are thought to occur early during tumorigenesis and enables tumor progression7 and thus may be a more specific and sensitive approach to identify minimal residual disease and prognosis.8,9 While genetic analysis of cfDNA can be challenging due to its low yield and being highly fragmented,10 plasma cfDNA next generation assays are starting to be utilized in routine clinical use for solid malignancies such as lung11 and colon cancers12 to make treatment decisions.
cfDNA has been reported to decrease to background level following surgery.13 Therefore, we hypothesized that comparing methylation profiles in pre- and post-surgery plasma samples will help validate HNSCC-specific prognostic and diagnostic biomarkers, and provides an opportunity for novel biomarker discovery. Here, we focus on single anatomic site and assess the feasibility of detecting cfDNA methylome in patients with locoregional oral cavity squamous cell carcinomas (OCSCC) and the methylome dynamics in post-operative setting. A high-sensitive cfDNA methylome profiling technique called cfMBD-seq was applied on collected plasma samples. Different from bisulfite conversion-based sequencing methods, cfMBD-seq capture and quantify methylated DNA by methyl-CpG binding protein (MBD). cfMBD-seq is able to generate high-quality sequencing read with ultra-low amount of input DNA (2–10 ng per ml), and has demonstrated better performance in terms of enrichment of CpG islands compared to similar protocols such as cfMeDIP-seq.14 To facilitate cfDNA methylation biomarker prioritization with limited sample size, we first conducted a bioinformatics analysis to detect differentially methylated regions (DMRs) based on the matched tumor-normal tissue data collected from the TCGA-HNSC project. We hypothesized that the top cancer-specific DMRs detected in the TCGA-HNSC cohort will also exhibit differential methylation patterns between before- and after-surgery plasma samples. As an alternative strategy, we performed a genome-wide search of top DMRs based on the matched cfDNA methylation profiles. DMR analyses were conducted on different patient subgroups as a sensitivity analysis considering the presence of patient and sample heterogeneity. Once we identified top DMRs, we also examined their prognostic relevance in the TCGA patient data, as well as their performance in discriminating pre- and post-treatment plasma samples.
MATERIALS AND METHODS
Patient sample collection
To detect HNSC-specific cfDNA biomarkers, we studied the plasma samples collected from a cohort of head and neck cancer patients treated at Moffitt Cancer Center (Tampa, USA). In this pilot study, a total of 16 matched plasma samples were collected from 8 patients before and at least 4 weeks after surgery. The basic clinical characteristics of these patients are displayed in Table 1. The study was approved by Institutional Review Board at Moffitt. All patients were consented to the protocol and all samples are de-identified during the methylation profiling process and in the downstream analysis.
Table 1. Baseline clinical characteristics and treatment of patients included in the plasma cfMBD-seq analysis.
| Previous treatment† | |||||||||||||
|---|---|---|---|---|---|---|---|---|---|---|---|---|---|
| P1 | Male | 56–60 | Mandible | 4a | 3b | 0 | 5.3 | 30 | Y | N | 2.0 | Y | Y |
| P2 | Male | 51–55 | Mandible | 4a | 3b | 0 | 4 | 28 | N | N | 2.5 | Y | N |
| P3 | Male | 56–60 | Tongue | 4a | 3b | 0 | 6.5 | 35 | Y | Y | 1.9 | Y | Y |
| P4 | Female | 65–70 | Maxilla | 1 | X | 0 | 0.3 | 1 | N | N | N/A | N/A | N |
| P5 | Male | 76–80 | Tongue | 3 | 1 | 0 | 3.6 | 14 | Y | N | 0.9 | N | N |
| P6 | Female | 41–45 | Tongue | 2 | 0 | 0 | 1.2 | 7 | Y | Y | N/A | N/A | N |
| P7 | Male | 76–80 | Tongue | 4a | 2a | 0 | 5.2 | 41 | Y | Y | 1.7 | Y | Y |
| P8 | Female | 76–80 | Buccal Mucosa | 1 | 1 | 0 | 2 | 2 | N | N | 0.6 | N | N |
P1 and P3 had undergone previous surgery and P7 had previously received cisplatin and radiation therapy.
pN – pathologic nodal classification; pT – pathologic tumor classification; M – Metastasis; PNI – Perineural invasion; LVI – Lymphovascular invasion; LN – Lymph Node; ENE – Extranodal extension
TCGA-HNSC analysis
To facilitate cfDNA methylation biomarker discovery, we first conducted a bioinformatics analysis to detect differentially methylated regions (DMRs) based on the tumor tissue methylation data collected from the TCGA-HNSC project. A total of 580 samples were profiled by the Illumina Infinium HumanMethylation450 BeadChip (450K array) in this cohort. Because the goal is to identify cancer-specific regions, our DMR analysis focuses on the 100 paired tumor and normal tissue methylation data collected from 50 patients. We downloaded level 3 DNA methylation data (beta value) from Broad Firehose web portal (gdac.broadinstitute.org) and all clinical data from GDC data portal (portal.gdc.cancer.gov). DMR analysis was performed using the bumphunter function implemented in the R package “minfi”, with the effect size cutoff set at 0.3 and the resampling number at 1000. A significant DMR is called if a region with family wise error rate (FWER) is less than 0.05. We restricted downstream analyses on regions > 5 bp in length, in which all regions contain at least two probes (L≥2). The detected regions were annotated against genome build UCSC hg19 by using the annotateDMRInfo function implemented in the “methyAnalysis” package.
Plasma cfDNA methylome profiling by cfMBD-seq
Cell-free DNA in plasma were profiled by cfMBD-seq, which is an enrichment-based ultra-low input cfDNA methylation profiling method recently developed by our team at Moffitt14. Briefly, Maxwell RSC ccf DNA Plasma kit was used to extract the cfDNA from 1 ml of plasma. If one sample contains less than 5ng DNA, we extract DNA from another 1 ml plasma sample. We then combined the cfDNA from the first and second extraction (if needed for a patient sample) for the methylation enrichment and sequencing library preparation. Methylated DNA fragments were enriched and captured by using a MethylCap Kit. cfMBD libraries were prepared and quantified following the steps as previously described in the cfMBD-seq protocol, and sequenced by Illumina NextSeq 500/550 High Output Kit (75 cycles).
After fastq file merging and adapter trimming steps, sequence reads were aligned to hg19 assembly using BWA MEM (v0.7.10). Mapped reads were further sorted and filtered to remove low-quality and duplicated reads using samtools (v1.9) and picard tools (v1.82). R package “MEDIPS” was used to conduct coverage saturation analysis and downstream QC analysis. The “qsea” R package was used to calculate normalized methylation (beta) levels both at a genome-wide level and in targeted ROI regions (such as promoter regions). We applied fitNBglm function in “qsea” to perform the differential coverage analysis. The function fits a negative-binomial model for each genomic widow similar to the differential expression analysis function implemented in the package “edgeR”. To maximize the statistical power, the design matrix in the DMR analysis was formed in a paired DMR setting, in which an additive model formula is formed to include both treatment effect and subject effect. For statistical testing, a reduced model was fit by the function without the treatment term and the p-value is generated by comparing the likelihood ratio of the models against a Chi-square distribution.15,16 To remove potential noise regions with low coverage, we only consider regions (1kb window) with a minimum of 50 reads in all the DMR tests.
Prioritizing candidate cfDNA methylation biomarkers
Because the current study has limited sample size and HNSC samples exhibit high heterogeneity in nature, we propose two schemes to efficiently prioritize the most robust cfDNA biomarker panels while minimizing the false discovery markers or ones with weak clinical significance. As illustrated in the Fig 1A, the first biomarker discovery scheme only investigates DMRs that have been detected based on the analysis using the TCGA data. We hypothesize that the top cancer-specific DMRs detected based on the matched tumor-normal tissues will also exhibit differential methylation patterns between before- and after-surgery plasma samples. The stringent genome-wide multiple-testing correction is not required in this setting because it becomes a targeted biomarker validation analysis. In the second scheme, we perform genome-wide differential coverage analysis on cfDNA methylation data by only considering regions that are located in or nearby the promoter of known genes (defined as 5kb upstream and 2kb downstream of the TSS regions). Furthermore, as will be explained more in the next section, we considered different patient subgroups for the pre- and post-treatment DMR analysis. In each test, regions with an adjusted p-value less than 0.1 or unadjusted p-values less than 1×10–6 will be reported. Similar to the TCGA analysis, the detected regions were annotated using the “methyAnalysis” package. In summary, we reason that both schemes are useful in identifying promising targets that could be further tested as diagnostic and prognostic markers in managing HNSC patients.
Figure 1. Overview of the proposed DMR analysis on OCSCC plasma samples.
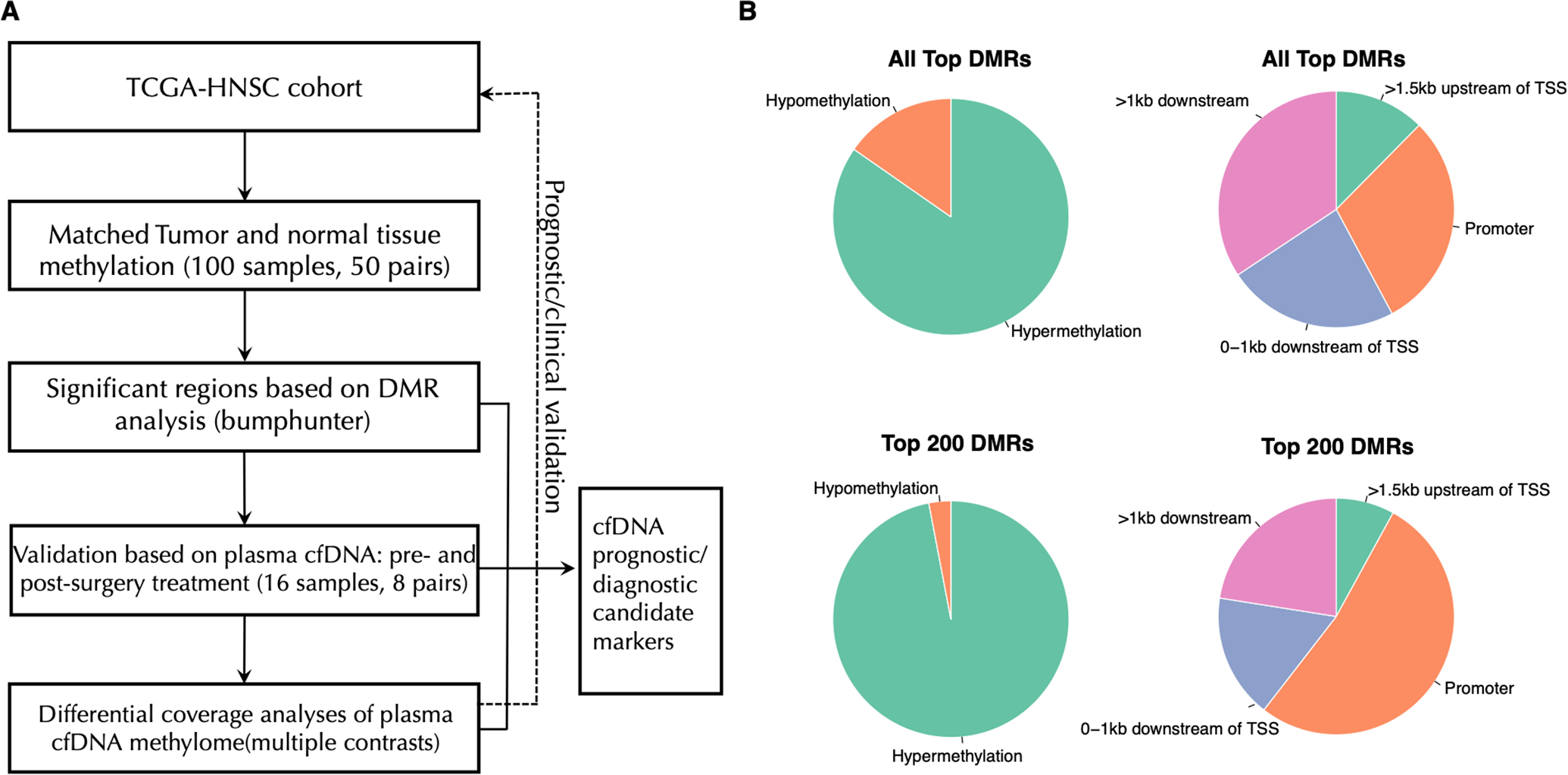
A. The experimental design and overall analytical workflow for cfDNA methylation profiling on pre- and post-treatment OCSCC patient samples.
B. Pie charts showing the distribution of methylation status and genomic locations in the top detected DMRs.
RESULTS
Candidate regions based on TCGA-HSNC analysis
The DMR analysis by comparing the TCGA-HNSC matched tumor and normal methylation profiles identified a total of 1468 significant regions (effect size cutoff of 0.3 and FWER adjusted p value<0.01) (Supplementary Table 1). As shown in Figure 1B, the majority (84.7%) of these top regions are hypermethylated DMRs; and more than half of regions are located in promoter (29.8%) or nearly (0–1kb) downstream regions of TSS (23.5%). When we narrow the list to the top 200 DMRs only, the proportion of hypermethylated DMRs and DMRs in promoter region further increased to 97% and 52.5%, respectively. The average length of top 200 DMRs is 461 bp. The top five DMRs in the promoter region are located in genes MARCHF11, ZNF154, ELMO1, ADCYAP1 and PIEZO2. A summary of top DMRs and their associated genes is provided in Table 2. Interestingly, we observed that many zinc-finger genes were enriched in the top DMR list, to only list those in the top 100 list: ZNF154, ZNF582, ZNF135, ZNF136, ZNF577, ZNF781, ZNF529, ZNF132, ZNF85, ZNF583, ZNF471, and ZNF665.
Table 2. Table of top genes containing significant DMRs based on TCGA-HNSC matched tumor and normal tissue methylation profiles.
| Rank methods | Top genes* |
|---|---|
| Genes contain top 100 DMRs (promoter regions) | MARCHF11, ZNF154, ELMO1, ADCYAP1, PIEZO2, CCDC181, KCNA3, OTX2-AS1, PCDHGC4, DPP10-AS1, MIR129–2, ZNF582, WT1, DRD5, CBLN4, DPP6, SORCS3, ZNF135, GALR1, BOLL, ZSCAN18, BARHL2, ZNF577, CLVS2, ABCC9, INSC, GRM6, ZNF781, ZNF529, TBXT, ITGA8, GCSAML, CDO1, ZNF132, IRF8, NKX2–6, ZC4H2, ZSCAN1, NKAPL, NPY, PENK, ZNF85, ADAMTS16, EVX2, NETO1, ZNF583, VAX1, HOXD9, KLHL34, CFTR, PCSK1, ZNF471, ZIK1, SHISA3, SIX6, ZNF665 |
| Genes contain top 100 DMRs (1kb downstream of TSS) | HOXD9, TRH, GRIA4, ZNF542P, PAX6, GSC, NTM, PPFIA3, PABPC5, SFTA3, NID2, HOXA11-AS, ASCL1, MAGI2-AS3, LOC100289656 |
| Genes contain top 100 DMRs (other regions) | EN1, SOX17, LHX1, PANTR1, NXPH1, ZNF136, LINC00461, PAX2, SEPTIN9, VAX1, ZIC4, HOTAIR, UBD, GBX2, ZNF559-ZNF177, NKX2–2, SOX1, SOX11, GSC, LINC01304, LINC01623, PITX2, PCDHA13, RMCX5-GPRASP2 |
| Top genes ranked by number of DMRs (>3 DMRs; No. of DMRs indicated in parentheses; Genes containing top 100 DMRs are highlighted in bold) | ZIC4 (11), ZIC1 (9), EN1(8), SOX1(8), TBX5-AS1(8), SOX17(7), BARHL2(6), ZBED9(6), EVX2(5), HOXB3(5), HOXD10(5), LINC00461(5), OTX2(5), PAX2(5), SOX3(5), VAX1(5), HOTAIR(4), HOXA3(4), HOXC4(4), HOXC6(4), LHX8(4), MIR124–2HG(4), MKI67(4), NXPH1(4), ONECUT2(4), PAX6(6), PAX6-AS1(4), PDX1(4), PITX2(4), PTF1A(4), PTPRN2(4), TBX15(4), TLX1(4), TLX3(4), UBD(4) |
Full list of DMRs and their locations are listed in Supplementary Table 1.
Targeted DMR validation on plasma cfDNA methylomes
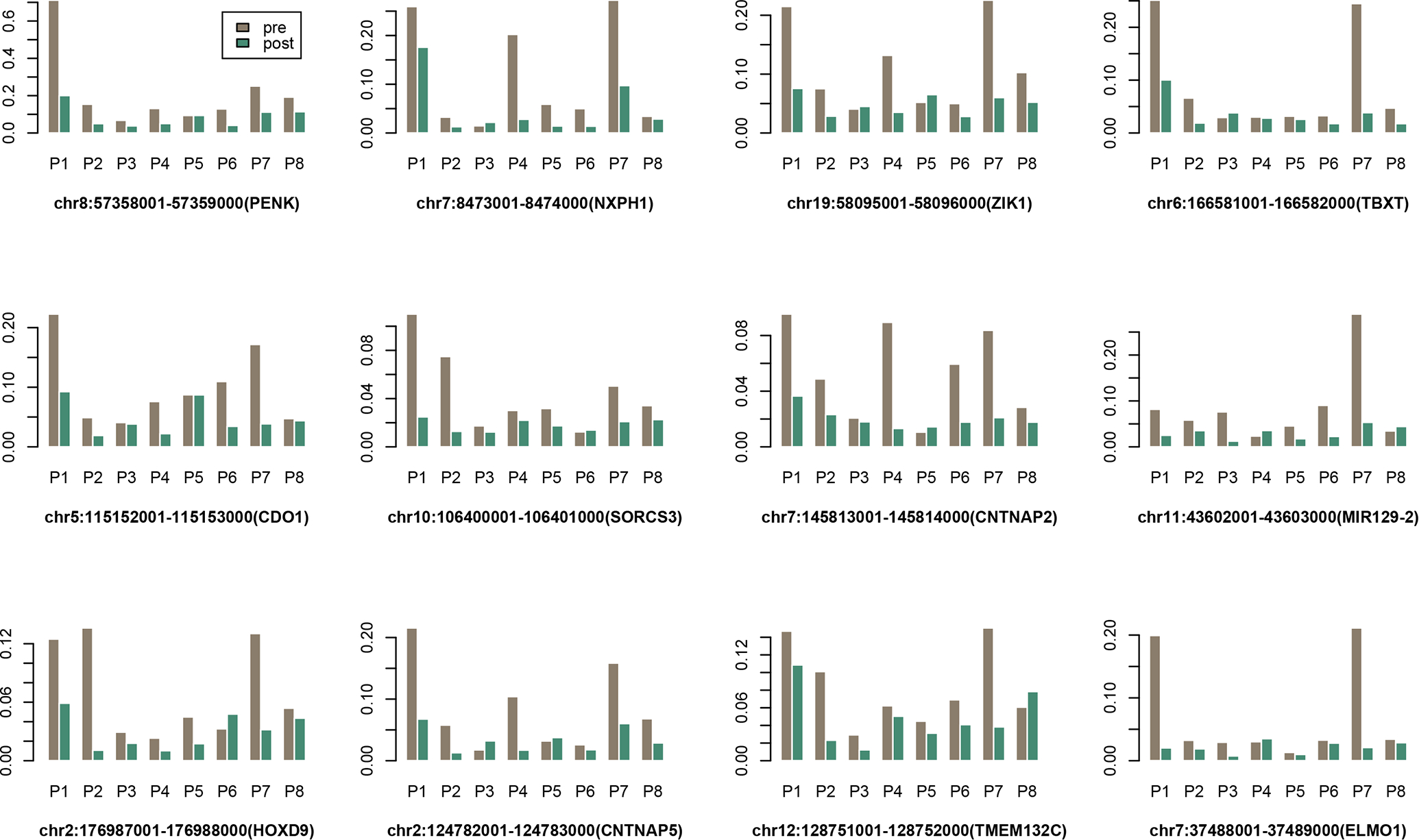
In this section, we focus on validating the significant DMRs detected between tumor and normal tissues. We first performed a paired DMR screening test by comparing the matched pre- and post-treatment cfMBD-seq data, using a fixed window size at 1kb. In the top 200 DMRs detected in the TCGA dataset, we found a total of 23 overlapping gene regions reached significance (p<0.05) in the plasma DMR test (Supplementary Table 2), including the two regions in ZNF154 and ELMO1 (top five candidate DMRs from the TCGA analysis). Another two regions ADCYAP1 and PIEZO2 from the top five candidate DMRs also reached marginal significance (p-value ~0.06). The normalized methylation levels at these top regions across the 16 plasma samples are depicted in Figure 2 (ranked by plasma DMR p-values). The top five validated regions are located in the promoter regions of genes PENK, NXPH1, ZIK1, TBXT and CDO1. A clear pattern revealed by Figure 2 is that the TCGA-based DMRs showed the best discriminating power between pre- and post-treatment samples for patients P1 and P7, followed by P4 and P8. Overall, patients P1 and P7 showed the most drastic methylation changes between the pre- and post-treatment samples across most targeted DMRs, potentially due to the fact that both patients (both are male) and had T4 tumors. The pattern of methylation is heterogenous between different patients as evidenced by results in Figure 2. For example, changes in methylation in patient P4 are more pronounced in DMRs in NXPH1, ZIK1 and CNTNAP2, while changes in P2 (also with a T4 tumor) are more pronounced in HOXD9, TMEM132C and SORCS3.
Figure 2. The normalized methylation levels of top DMRs across the matched plasma samples from 8 patients.
Genome-wide cfDNA DMR analysis on patient subgroups
Given the heterogeneity of head and neck cancer at the molecular level and the limited sample size, we reason that it is more powerful to perform DMR analyses based on subsets of patients based on their clinical characteristics. The patient subset information and the resulted top significant or suggestive DMRs (defined as a p-value < 0.1 level for adjusted p-value or at 10−7 level for unadjusted p-value) are listed in Table 3. It is interesting to note that the DMR test based on the four patients (P1, P4, P7 and P8) that had the most concordant patterns with TCGA data resulted in the highest number of significant DMRs, followed by the two patient subsets containing T4 tumors (P1, P2, P3 and P7). We excluded patient P5 in most tests (except for the tongue-site subgroup) because the genome-wide PCA analysis (Supplementary Figure S1) indicated that the pre-treatment cfDNA methylation profiles could be a potential outlier, which may explain why there was no significant DMR when all patients are included in the test. The multiple-contrast DMR tests can also be considered as sensitivity analyses that provide further confidence for overlapped findings. It was observed that the genomic regions in gene PENK, SFRP4 and SOX17 were selected in at least three tests, suggesting they might be further prioritized for biomarker validation. Figure 3A shows the detailed methylation levels in top regions that were identified based on the TCGA-concordant subgroup, including two top-ranked validated DMRs listed in Figure 2 (PENK and ZIK1). Similar to the pattern observed in Figure 2, patients P1 and P7 showed the most drastic changes between the pre- and post-treatment samples. Through assessing the gene expression and methylation levels of these top genes in TCGA-HNSC data, we observed that four genes (ZIK1, IRF4, PCDH17 and PENK) demonstrated significant or suggestive association with patient overall survival at both gene expression level (Figure 3B–E) and CpG level (Supplementary Figure S2). Collectively, these findings suggest that these four genes may have tumor suppressor functions and are often hypermethylated in HNSC tumor samples or pre-treatment plasma samples.
Table 3. Table of top cfDNA DMRs identified based on comparing pre- and post-treatment cfDNA methylation profiles by considering different patient subgroups.
| Patient group/subgroup | Subset rationale | Top cfDNA DMRs (hg19)† |
|---|---|---|
| Paired samples from P1, P4, P7, and P8 | Patterns most consistent with TCGA results | chr7:37956001-37957000 (SFRP4) chr8:67873001-67874000 (TCF24) chr2:45171001-45172000 (SIX3*) chr11:32458001-32459000 (WT1-AS) chr8:53478001-53479000 (ALKAL1) chr8:57358001-57359000 (PENK) chr13:93879001-93880000 (GPC6) chr8:55370001-55371000 (SOX17) chr4:183062001-183063000 (TENM3-AS1*) chr1:119535001-119536000 (TBX15*) chr19:58095001-58096000 (ZIK1) |
| Paired samples from P1, P2, P3, and P7 | Patients with T4 tumors (male patients) | chr7:37956001-37957000 (SFRP4) chr1:47696001-47697000 (TAL1) chr5:1594001-1595000(SDHAP3) chr7:87257001-87258000 (ABCB1/RUNDC3B) chr8:57358001-57359000(PENK) |
| Paired samples from P3, P5, P6, and P7 | Tongue site only | chr11:43602001-43603000 (MIR129-2) |
| P4, P6, and P8 | Female patients only | No region significant at 10−7 level |
| Paired samples from P1, P2, P3, P4 and P7 | All T4 patients+ P4 (signature patient) | chr7:37956001-37957000 (SFRP4) chr8:57358001-57359000 (PENK) chr8:55370001-55371000 (SOX17) chr3:129693001-129694000 (TRH) chr14:48143001-48144000 (MDGA2*) chr14:48145001-48146000 (MDGA2) |
| Paired samples from P1, P2, P3, P4, P6, P7, and P8 | All patients except P5 (potential outlier) | chr8:57358001-57359000 (PENK) chr7:37956001-37957000 (SFRP4) chr8:55370001-55371000 (SOX17) |
Unless otherwise specific, DMRs are significant at 0.1 level for adjusted p-value or at 10−7 level for unadjusted p-value. DMRs not in the promoter region of the gene are indicated by “*”.
Figure 3. cfDNA DMR subgroup prioritization and validation.
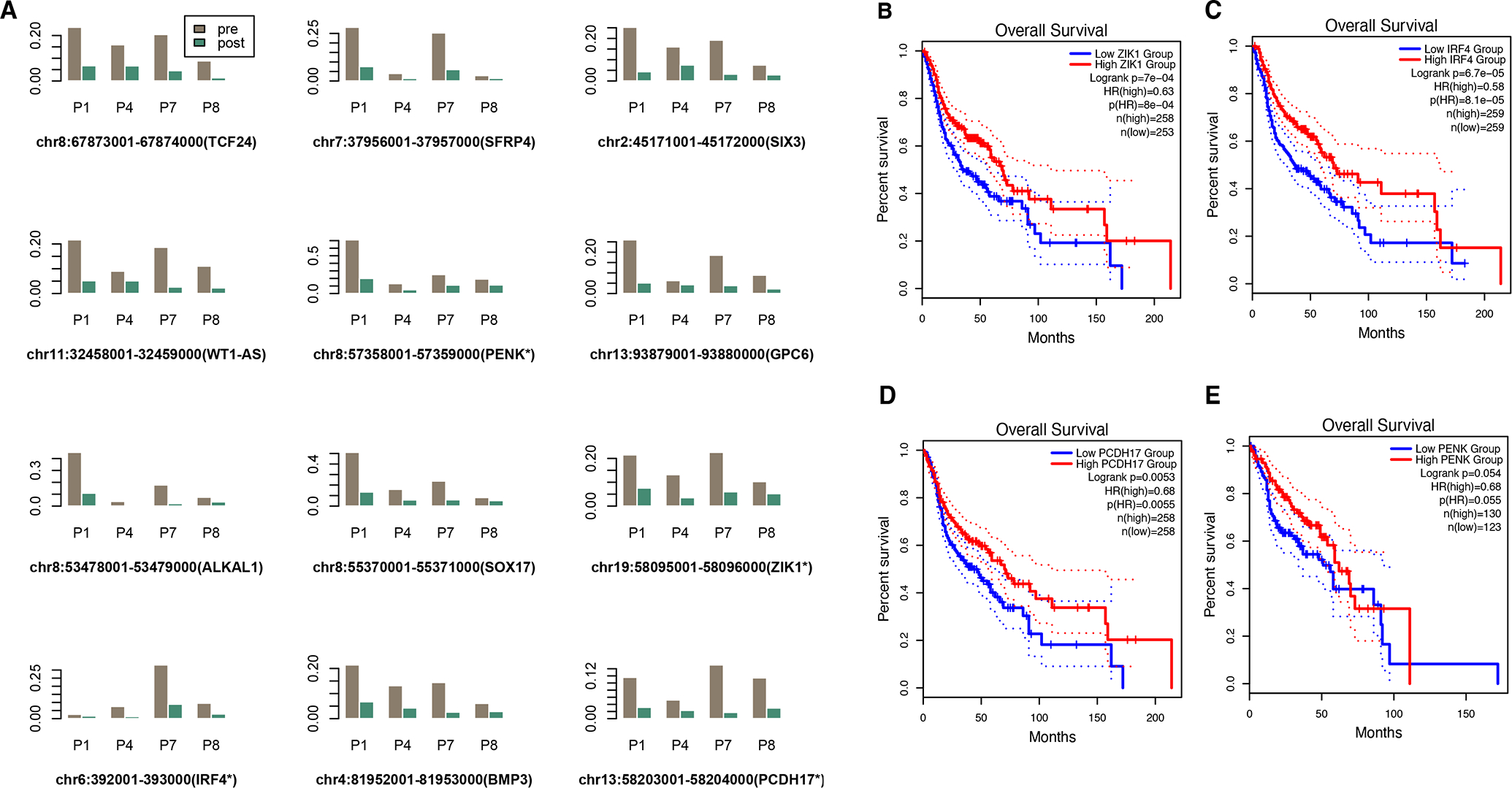
A. Methylation levels in top regions that were identified based on the TCGA-concordant patient subgroup (P1, P4, P7 and P8)
B. (C,D,E) Kaplan-Meier plots validating the prognostic significance of four genes that include detected DMRs (based on the gene expression and survival data from the TCGA-HNSC data).
Clustering analysis of targeted plasma cfDNA methylation regions
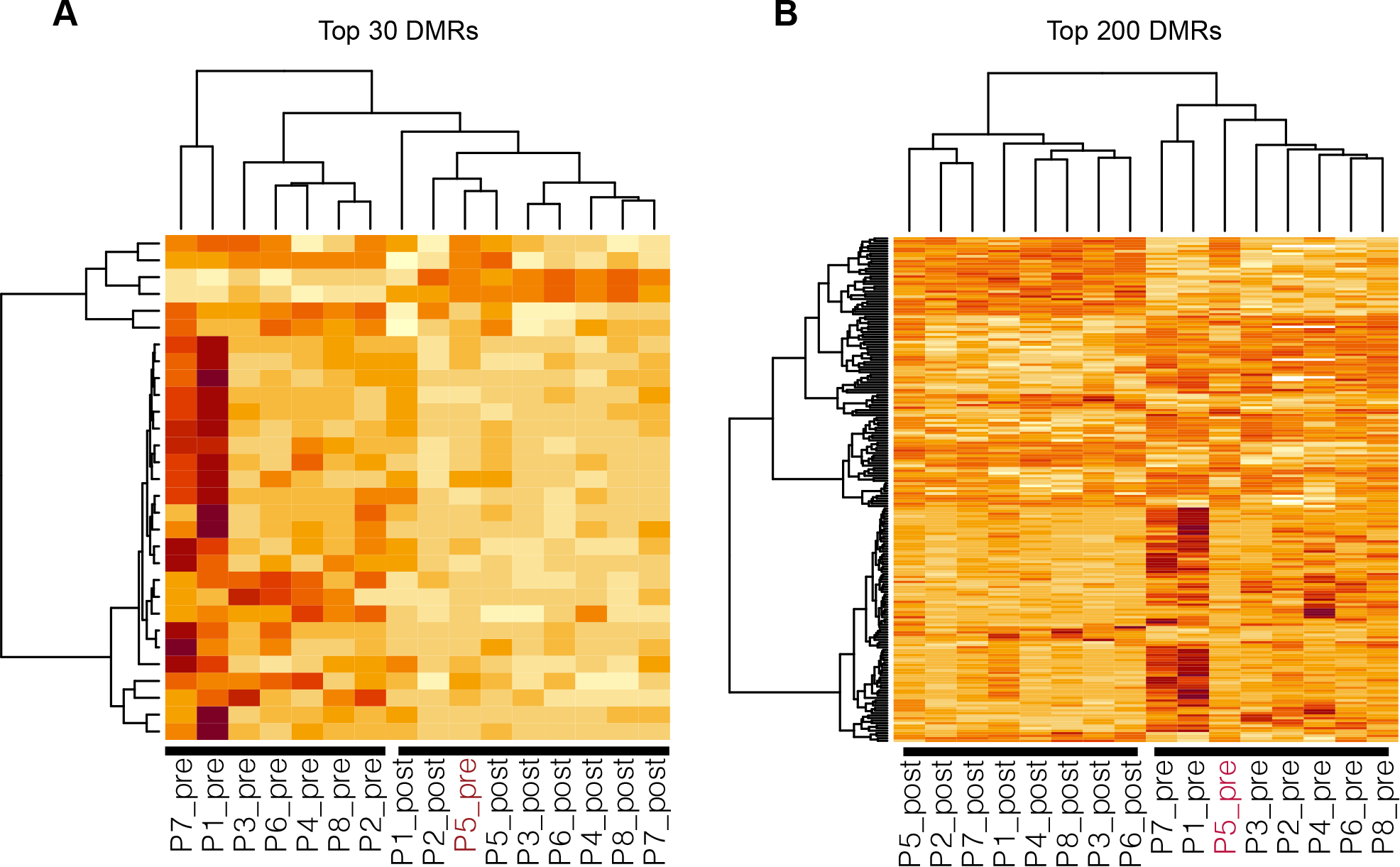
Finally, we tested the performance of top DMRs (as well as the model saturation in terms of the number of biomarkers included) in discriminating pre- and post-treatment plasma samples. The two heatmaps in Figure 4 illustrate the unsupervised clustering results generated based on top 30 DMRs and top 200 DMRs (from the DMR test using all samples but P5), respectively. It shows that the top 30 regions (most of them are hypermethylated in pre-treatment samples) are already sufficient to separate pre- and post-treatment plasma samples except for the P5 pre-treatment sample. This was expected, because the global PCA analysis also indicated that this sample could be a potential outlier. But when top 200 regions were included, this sample, together with all other samples, can be correctly separated. Finally, we performed the clustering analysis to the same set of CpGs (in the top DMRs discovered from the cfDNA data) with the TCGA matched tumor and normal samples. These CpGs (from either top 30 or 200 DMRs) can provide a clear separation of tumor samples from the matched normal samples, as seen in Supplementary Figure S3.
Figure 4. The unsupervised clustering results generated based on top 30 DMRs (A) and top 200 DMRs (B) showing the separation of pre- and post-treatment samples.
DISCUSSION
In this study, we successfully demonstrate the feasibility of isolating cfDNA from plasma and a two-pronged approach in identifying top candidate biomarkers by first identifying DMRs from the TCGA dataset and then validating them in our cfDNA samples. A biomarker for minimal residual disease is advantageous, especially in the setting of oral cavity squamous cell carcinoma patients, a population where 5-year survival rates are estimated between 40–60% for patients with advanced-stage disease, and the majority of the recurrence occurs in the first 2 years.17–20 Moreover, if the biomarkers can predict tumor immune response it would be even more preferable.21 HNSCC is a heterogenous disease and thus focusing on cfDNA DNAme may be advantageous to increase specificity. Previous studies have focused on targeted panels of methylation in either serum/plasma of HNSCC patients.22–28 Recently, a study by Burgener et al. did demonstrate tumor-naïve detection of ctDNA by simultaneously profiling mutations and methylation.29
Our study also suggests that top 30 DMRs are sufficient to differentiate between pre-treatment and post-treatment samples suggesting that a signature based on these DMRs maybe sufficient to determine minimal residual disease. Many genes in the top DMR list have also been suggested as liquid biopsy methylation biomarkers in other cancer types, such as ZNF154 for multiple cancers,30 ELMO1 for gastric cancer31, suggesting that they are reliable cancer-relevant epigenetic biomarkers. IRF4 and PCDH17 have been previously reported as liquid biopsy biomarkers for colorectal32,33 and bladder cancers.34
Even with a limited sample size, the results from comparing the DMRs obtained from the matched samples in TCGA and our data successfully demonstrated that methylation-based head and neck cancer biomarkers can be robustly and reliably detected across studies and methylation profiling platforms. The top five validated regions were in the promoter regions of genes PENK, NXPH1, ZIK1, TBXT and CDO1. Four candidate genes were identified that may have prognostic value in addition to their role in determining minimal residual disease. ZIK1 (ZNF762) is part of the Zinc Finger protein group with a KRAB-A domain.35 KRAB box-A is a transcription repressor module35 and it is plausible that ZIK1 is epigenetically regulated tumor suppressor gene.36–38 Interferon regulatory factor 4 (IRF4) is a member of the Interferon family and is specifically expressed in lymphocytes39 regulating immune responses, immune cell proliferation and differentiation.40 While its role in hematologic malignancies has been described previously,41 IRF4 expression in lung adenocarcinoma has been associated with favorable prognosis.42 Protocadherin 17 (PCDH17) is part of the cadherin superfamily responsible for cell adhesion and possible tumor growth, migration and invasion.43,44 PCDH17 methylation has been noted in multiple cancers including esophageal,45 gastric,46 colon,46 and bladder cancers.47 Proenkephalin (PENK) is expressed in nervous and neuroendocrine systems as part of the opioid pathway48, but is also involved in cell cycle regulation and implicated in head and neck,49 gastric,50 colon,51 breast,52 pancreatic,53 osteosarcoma,54 and bladder55 cancers. NXPH1 is primarily expressed in nervous system and is a secreted glycoprotein that forms complexes with alpha neurexins – a group of protein that promote adhesion between dendrites and axons56. In breast cancer, NXPH1 methylation levels were lower compared to normal tissues and was more likely to be methylated in low-grade dysplasia than in high-grade dysplasia.57 In prostate cancers with Gleason score ≥ 7, NXPH1 expression was upregulated and was incorporated in a 10-gene signature that predicted biochemical recurrence.58 However, a negative correlation was noted in patients with pancreatic cancer with regards to lymph node metastasis.59 NXPH1 methylation has also been implicated in neuroblastoma60 and was incorporated in a 5 gene prognostic signature where it was down regulated suggestive of playing a tumor suppressive role.61 TBXT expression has been reported in a number of solid malignancies including head and neck62, lung,63 breast,64 colon,64 prostate65 and chordoma66 – with hypothesis that it promotes epithelial-mesenchymal transition and targeting it may help in cancer control.67 Promoter methylation of CDO1 has also been previously identified as diagnostic biomarkers in lung cancer.9
Limitations of this study include limited samples size to draw definitive prognostic conclusion. cfDNA has been correlated with overall stage and subsite. Our study included primarily advanced stage disease and mainly oral cavity squamous cell carcinomas. It is well established that advanced stage cancers9 and different subsites will have different methylation pattern37 and lymphatic drainage, thus it is plausible that similar results may not be evident in lower stage disease. Additionally, patients’ peripheral methylation profiles could be altered by previous treatment such as platinum-based chemotherapy. Future studies with expanded patient samples will be necessary to evaluate this effect, because only one patient (P7) in our study had previously received cisplatin therapy.
In summary, in this study we identified multiple candidate DMRs that allowed distinction between pre-treatment and post-treatment plasma samples suggesting its utility for minimal residual disease and potential as prognostic biomarker.
Supplementary Material
Funding support:
This work has been supported in part by Moffitt Clinical Science Fund (to. K.B.P.); a National Institute of Health grant R01DE030493 (to X.W.); an NCI grant R01CA212097 (to L.W.); and Moffitt’s Biostatistics and Bioinformatics Shared Resources, Tissue Core, and Genomics Core Facilities at the H. Lee Moffitt Cancer Center & Research Institute, an NCI-designated Comprehensive Cancer Center (P30-CA076292).
Footnotes
Conflicts of interest disclosure: None declared
REFERENCES
- 1.Klutstein M, Nejman D, Greenfield R, et al. : DNA Methylation in Cancer and Aging. Cancer Res 76:3446–50, 2016 [DOI] [PubMed] [Google Scholar]
- 2.Thierry AR, El Messaoudi S, Gahan PB, et al. : Origins, structures, and functions of circulating DNA in oncology. Cancer Metastasis Rev 35:347–76, 2016 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 3.Diehl F, Schmidt K, Choti MA, et al. : Circulating mutant DNA to assess tumor dynamics. Nat Med 14:985–90, 2008 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 4.Schwarzenbach H, Hoon DS, Pantel K: Cell-free nucleic acids as biomarkers in cancer patients. Nat Rev Cancer 11:426–37, 2011 [DOI] [PubMed] [Google Scholar]
- 5.Jamal-Hanjani M, Wilson GA, Horswell S, et al. : Detection of ubiquitous and heterogeneous mutations in cell-free DNA from patients with early-stage non-small-cell lung cancer. Ann Oncol 27:862–7, 2016 [DOI] [PubMed] [Google Scholar]
- 6.De Mattos-Arruda L, Weigelt B, Cortes J, et al. : Capturing intra-tumor genetic heterogeneity by de novo mutation profiling of circulating cell-free tumor DNA: a proof-of-principle. Ann Oncol 25:1729–1735, 2014 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 7.Baylin SB, Esteller M, Rountree MR, et al. : Aberrant patterns of DNA methylation, chromatin formation and gene expression in cancer. Hum Mol Genet 10:687–92, 2001 [DOI] [PubMed] [Google Scholar]
- 8.Vrba L, Futscher BW: A suite of DNA methylation markers that can detect most common human cancers. Epigenetics 13:61–72, 2018 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 9.Constancio V, Nunes SP, Henrique R, et al. : DNA Methylation-Based Testing in Liquid Biopsies as Detection and Prognostic Biomarkers for the Four Major Cancer Types. Cells 9, 2020 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 10.El Messaoudi S, Rolet F, Mouliere F, et al. : Circulating cell free DNA: Preanalytical considerations. Clin Chim Acta 424:222–30, 2013 [DOI] [PubMed] [Google Scholar]
- 11.Powrozek T, Krawczyk P, Kucharczyk T, et al. : Septin 9 promoter region methylation in free circulating DNA-potential role in noninvasive diagnosis of lung cancer: preliminary report. Med Oncol 31:917, 2014 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 12.Song L, Yu H, Jia J, et al. : A systematic review of the performance of the SEPT9 gene methylation assay in colorectal cancer screening, monitoring, diagnosis and prognosis. Cancer Biomark 18:425–432, 2017 [DOI] [PubMed] [Google Scholar]
- 13.Catarino R, Ferreira MM, Rodrigues H, et al. : Quantification of free circulating tumor DNA as a diagnostic marker for breast cancer. DNA Cell Biol 27:415–21, 2008 [DOI] [PubMed] [Google Scholar]
- 14.Huang J, Soupir AC, Wang L: Cell-free DNA methylome profiling by MBD-seq with ultra-low input. Epigenetics 17:239–252, 2022 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 15.Lienhard M, Grimm C, Morkel M, et al. : MEDIPS: genome-wide differential coverage analysis of sequencing data derived from DNA enrichment experiments. Bioinformatics 30:284–286, 2014 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 16.Lienhard M, Grasse S, Rolff J, et al. : QSEA—modelling of genome-wide DNA methylation from sequencing enrichment experiments. Nucleic acids research 45:e44–e44, 2017 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 17.Leon X, Quer M, Orus C, et al. : Distant metastases in head and neck cancer patients who achieved loco-regional control. Head Neck 22:680–6, 2000 [DOI] [PubMed] [Google Scholar]
- 18.Patel KB, Martin D, Zhao S, et al. : Impact of age and comorbidity on survival among patients with oral cavity squamous cell carcinoma. Head Neck, 2020 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 19.Wang B, Zhang S, Yue K, et al. : The recurrence and survival of oral squamous cell carcinoma: a report of 275 cases. Chin J Cancer 32:614–8, 2013 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 20.Brands MT, Smeekens EAJ, Takes RP, et al. : Time patterns of recurrence and second primary tumors in a large cohort of patients treated for oral cavity cancer. Cancer Med 8:5810–5819, 2019 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 21.Jeschke J, Bizet M, Desmedt C, et al. : DNA methylation-based immune response signature improves patient diagnosis in multiple cancers. J Clin Invest 127:3090–3102, 2017 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 22.Carvalho AL, Jeronimo C, Kim MM, et al. : Evaluation of promoter hypermethylation detection in body fluids as a screening/diagnosis tool for head and neck squamous cell carcinoma. Clin Cancer Res 14:97–107, 2008 [DOI] [PubMed] [Google Scholar]
- 23.Mydlarz WK, Hennessey PT, Wang H, et al. : Serum biomarkers for detection of head and neck squamous cell carcinoma. Head Neck 38:9–14, 2016 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 24.Schrock A, Leisse A, de Vos L, et al. : Free-Circulating Methylated DNA in Blood for Diagnosis, Staging, Prognosis, and Monitoring of Head and Neck Squamous Cell Carcinoma Patients: An Observational Prospective Cohort Study. Clin Chem 63:1288–1296, 2017 [DOI] [PubMed] [Google Scholar]
- 25.Nakahara Y, Shintani S, Mihara M, et al. : Detection of p16 promoter methylation in the serum of oral cancer patients. Int J Oral Maxillofac Surg 35:362–5, 2006 [DOI] [PubMed] [Google Scholar]
- 26.Sanchez-Cespedes M, Esteller M, Wu L, et al. : Gene promoter hypermethylation in tumors and serum of head and neck cancer patients. Cancer Res 60:892–5, 2000 [PubMed] [Google Scholar]
- 27.Danstrup CS, Marcussen M, Pedersen IS, et al. : DNA methylation biomarkers in peripheral blood of patients with head and neck squamous cell carcinomas. A systematic review. PLoS One 15:e0244101, 2020 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 28.Ha PK, Califano JA: Promoter methylation and inactivation of tumour-suppressor genes in oral squamous-cell carcinoma. Lancet Oncol 7:77–82, 2006 [DOI] [PubMed] [Google Scholar]
- 29.Burgener JM, Zou J, Zhao Z, et al. : Tumor-Naive Multimodal Profiling of Circulating Tumor DNA in Head and Neck Squamous Cell Carcinoma. Clin Cancer Res 27:4230–4244, 2021 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 30.Miller BF, Petrykowska HM, Elnitski L: Assessing ZNF154 methylation in patient plasma as a multicancer marker in liquid biopsies from colon, liver, ovarian and pancreatic cancer patients. Sci Rep 11:221, 2021 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 31.Anderson BW, Suh YS, Choi B, et al. : Detection of Gastric Cancer with Novel Methylated DNA Markers: Discovery, Tissue Validation, and Pilot Testing in Plasma. Clin Cancer Res 24:5724–5734, 2018 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 32.Bach S, Sluiter NR, Beagan JJ, et al. : Circulating Tumor DNA Analysis: Clinical Implications for Colorectal Cancer Patients. A Systematic Review. JNCI Cancer Spectr 3:pkz042, 2019 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 33.Young GP, Symonds EL, Nielsen HJ, et al. : Evaluation of a panel of tumor-specific differentially-methylated DNA regions in IRF4, IKZF1 and BCAT1 for blood-based detection of colorectal cancer. Clin Epigenetics 13:14, 2021 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 34.Wang Y, Yu Y, Ye R, et al. : An epigenetic biomarker combination of PCDH17 and POU4F2 detects bladder cancer accurately by methylation analyses of urine sediment DNA in Han Chinese. Oncotarget 7:2754–64, 2016 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 35.Grimwood J, Gordon LA, Olsen A, et al. : The DNA sequence and biology of human chromosome 19. Nature 428:529–35, 2004 [DOI] [PubMed] [Google Scholar]
- 36.Lleras RA, Adrien LR, Smith RV, et al. : Hypermethylation of a cluster of Kruppel-type zinc finger protein genes on chromosome 19q13 in oropharyngeal squamous cell carcinoma. Am J Pathol 178:1965–74, 2011 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 37.Lleras RA, Smith RV, Adrien LR, et al. : Unique DNA methylation loci distinguish anatomic site and HPV status in head and neck squamous cell carcinoma. Clin Cancer Res 19:5444–55, 2013 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 38.Gaykalova DA, Vatapalli R, Wei Y, et al. : Outlier Analysis Defines Zinc Finger Gene Family DNA Methylation in Tumors and Saliva of Head and Neck Cancer Patients. PLoS One 10:e0142148, 2015 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 39.Eisenbeis CF, Singh H, Storb U: Pip, a novel IRF family member, is a lymphoid-specific, PU.1-dependent transcriptional activator. Genes Dev 9:1377–87, 1995 [DOI] [PubMed] [Google Scholar]
- 40.Nam S, Lim JS: Essential role of interferon regulatory factor 4 (IRF4) in immune cell development. Arch Pharm Res 39:1548–1555, 2016 [DOI] [PubMed] [Google Scholar]
- 41.Wong RWJ, Tan TK, Amanda S, et al. : Feed-forward regulatory loop driven by IRF4 and NF-kappaB in adult T-cell leukemia/lymphoma. Blood 135:934–947, 2020 [DOI] [PubMed] [Google Scholar]
- 42.Li X, Zhai S, Zhang J, et al. : Interferon Regulatory Factor 4 Correlated With Immune Cells Infiltration Could Predict Prognosis for Patients With Lung Adenocarcinoma. Front Oncol 11:698465, 2021 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 43.Theveneau E, Mayor R: Cadherins in collective cell migration of mesenchymal cells. Curr Opin Cell Biol 24:677–84, 2012 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 44.Lin YL, Li ZG, He ZK, et al. : Clinical and prognostic significance of protocadherin-10 (PCDH10) promoter methylation in bladder cancer. J Int Med Res 40:2117–23, 2012 [DOI] [PubMed] [Google Scholar]
- 45.Haruki S, Imoto I, Kozaki K, et al. : Frequent silencing of protocadherin 17, a candidate tumour suppressor for esophageal squamous cell carcinoma. Carcinogenesis 31:1027–36, 2010 [DOI] [PubMed] [Google Scholar]
- 46.Hu X, Sui X, Li L, et al. : Protocadherin 17 acts as a tumour suppressor inducing tumour cell apoptosis and autophagy, and is frequently methylated in gastric and colorectal cancers. J Pathol 229:62–73, 2013 [DOI] [PubMed] [Google Scholar]
- 47.Wang XB, Lin YL, Li ZG, et al. : Protocadherin 17 promoter methylation in tumour tissue from patients with bladder transitional cell carcinoma. J Int Med Res 42:292–9, 2014 [DOI] [PubMed] [Google Scholar]
- 48.Tegeder I, Geisslinger G: Opioids as modulators of cell death and survival--unraveling mechanisms and revealing new indications. Pharmacol Rev 56:351–69, 2004 [DOI] [PubMed] [Google Scholar]
- 49.McLaughlin PJ, Stucki JK, Zagon IS: Modulation of the opioid growth factor ([Met(5)]-enkephalin)-opioid growth factor receptor axis: novel therapies for squamous cell carcinoma of the head and neck. Head Neck 34:513–9, 2012 [DOI] [PubMed] [Google Scholar]
- 50.Suzuki M, Chiwaki F, Sawada Y, et al. : Peripheral opioid antagonist enhances the effect of anti-tumor drug by blocking a cell growth-suppressive pathway in vivo. PLoS One 10:e0123407, 2015 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 51.Roperch JP, Incitti R, Forbin S, et al. : Aberrant methylation of NPY, PENK, and WIF1 as a promising marker for blood-based diagnosis of colorectal cancer. BMC Cancer 13:566, 2013 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 52.Salhia B, Kiefer J, Ross JT, et al. : Integrated genomic and epigenomic analysis of breast cancer brain metastasis. PLoS One 9:e85448, 2014 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 53.Zagon IS, Smith JP, McLaughlin PJ: Human pancreatic cancer cell proliferation in tissue culture is tonically inhibited by opioid growth factor. Int J Oncol 14:577–84, 1999 [DOI] [PubMed] [Google Scholar]
- 54.Zhang HP, Yu ZL, Wu BB, et al. : PENK inhibits osteosarcoma cell migration by activating the PI3K/Akt signaling pathway. J Orthop Surg Res 15:162, 2020 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 55.Zhang Y, Fang L, Zang Y, et al. : Identification of Core Genes and Key Pathways via Integrated Analysis of Gene Expression and DNA Methylation Profiles in Bladder Cancer. Med Sci Monit 24:3024–3033, 2018 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 56.Missler M, Sudhof TC: Neurexophilins form a conserved family of neuropeptide-like glycoproteins. J Neurosci 18:3630–8, 1998 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 57.Faryna M, Konermann C, Aulmann S, et al. : Genome-wide methylation screen in low-grade breast cancer identifies novel epigenetically altered genes as potential biomarkers for tumor diagnosis. FASEB J 26:4937–50, 2012 [DOI] [PubMed] [Google Scholar]
- 58.Wu X, Lv D, Lei M, et al. : A 10-gene signature as a predictor of biochemical recurrence after radical prostatectomy in patients with prostate cancer and a Gleason score >/=7. Oncol Lett 20:2906–2918, 2020 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 59.Jin JS, Tsai WC: The detection of tumor location and lymph node metastasis by aberrant NXPH1 and NXPH2 expressions in pancreatic ductal adenocarcinomas. Chin J Physiol 59:348–354, 2016 [DOI] [PubMed] [Google Scholar]
- 60.Decock A, Ongenaert M, Cannoodt R, et al. : Methyl-CpG-binding domain sequencing reveals a prognostic methylation signature in neuroblastoma. Oncotarget 7:1960–72, 2016 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 61.Xia Y, Li X, Tian X, et al. : Identification of a Five-Gene Signature Derived From MYCN Amplification and Establishment of a Nomogram for Predicting the Prognosis of Neuroblastoma. Front Mol Biosci 8:769661, 2021 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 62.Imajyo I, Sugiura T, Kobayashi Y, et al. : T-box transcription factor Brachyury expression is correlated with epithelial-mesenchymal transition and lymph node metastasis in oral squamous cell carcinoma. Int J Oncol 41:1985–95, 2012 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 63.Roselli M, Fernando RI, Guadagni F, et al. : Brachyury, a driver of the epithelial-mesenchymal transition, is overexpressed in human lung tumors: an opportunity for novel interventions against lung cancer. Clin Cancer Res 18:3868–79, 2012 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 64.Palena C, Polev DE, Tsang KY, et al. : The human T-box mesodermal transcription factor Brachyury is a candidate target for T-cell-mediated cancer immunotherapy. Clin Cancer Res 13:2471–8, 2007 [DOI] [PubMed] [Google Scholar]
- 65.Pinto F, Pertega-Gomes N, Pereira MS, et al. : T-box transcription factor brachyury is associated with prostate cancer progression and aggressiveness. Clin Cancer Res 20:4949–61, 2014 [DOI] [PubMed] [Google Scholar]
- 66.Cottone L, Cribbs AP, Khandelwal G, et al. : Inhibition of Histone H3K27 Demethylases Inactivates Brachyury (TBXT) and Promotes Chordoma Cell Death. Cancer Res 80:4540–4551, 2020 [DOI] [PubMed] [Google Scholar]
- 67.Hamilton DH, David JM, Dominguez C, et al. : Development of Cancer Vaccines Targeting Brachyury, a Transcription Factor Associated with Tumor Epithelial-Mesenchymal Transition. Cells Tissues Organs 203:128–138, 2017 [DOI] [PMC free article] [PubMed] [Google Scholar]
Associated Data
This section collects any data citations, data availability statements, or supplementary materials included in this article.