

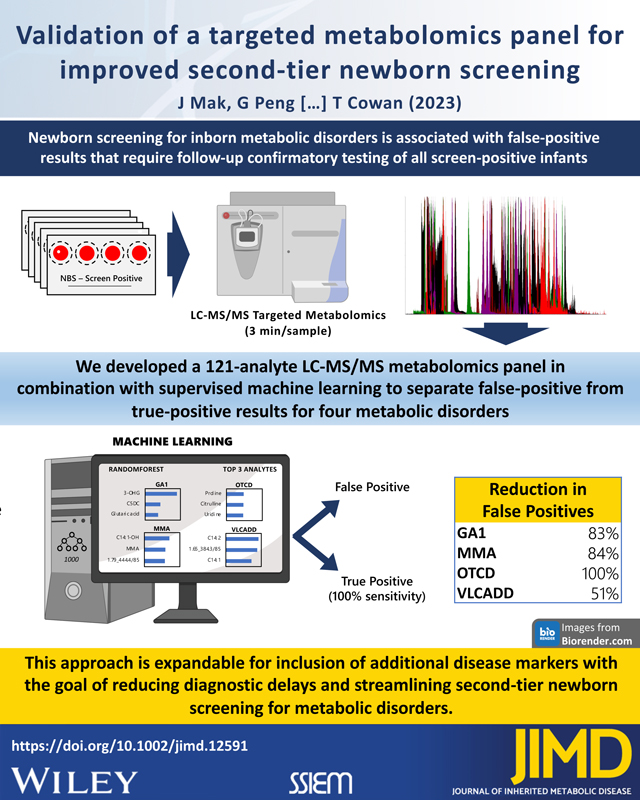
Abstract
Improved second-tier assays are needed to reduce the number of false positives in newborn screening (NBS) for inherited metabolic disorders including those on the Recommended Uniform Screening Panel (RUSP). We developed an expanded metabolite panel for second-tier testing of dried blood spot (DBS) samples from screen-positive cases reported by the California NBS program, consisting of true- and false-positives from four disorders: glutaric acidemia type 1 (GA1), methylmalonic acidemia (MMA), ornithine transcarbamylase deficiency (OTCD), and very long-chain acyl-CoA dehydrogenase deficiency (VLCADD). This panel was assembled from known disease markers and new features discovered by untargeted metabolomics and applied to second-tier analysis of single DBS punches using liquid chromatography-tandem mass spectrometry (LC-MS/MS) in a 3-min run. Additionally, we trained a Random Forest (RF) machine learning classifier to improve separation of true- and false positive cases. Targeted metabolomic analysis of 121 analytes from DBS extracts in combination with RF classification at a sensitivity of 100% reduced false positives for GA1 by 83%, MMA by 84%, OTCD by 100%, and VLCADD by 51%. This performance was driven by a combination of known disease markers (3-hydroxyglutaric acid, methylmalonic acid, citrulline, C14:1), other amino acids and acylcarnitines, and novel metabolites identified to be isobaric to several long-chain acylcarnitine and hydroxy-acylcarnitine species. These findings establish the effectiveness of this second-tier test to improve screening for these four conditions and demonstrate the utility of supervised machine learning in reducing false-positives for conditions lacking clearly discriminating markers, with future studies aimed at optimizing and expanding the panel to additional disease targets.
Keywords: Newborn Screening, Inborn Metabolic Disorders, Second-tier testing, Metabolomics, Tandem mass spectrometry, Machine learning, Supervised machine learning
Graphical Abstract
1. Introduction
Newborn screening (NBS) is an important public health activity for identifying newborns with inherited metabolic disorders, but with the corresponding detection of many false- positive cases. Confirmatory biochemical and/or genetic testing is required to exclude false-positive results, which occur in an estimated 0.02–1.5% of newborns1–4 and place excessive burden on patients, families, and the medical system5,6. Reducing the number and impact of false positives remain a central challenge to newborn screening and is the major purpose of second-tier testing of the original dried blood spot (DBS) from all newborns with a positive NBS result7,8. In contrast to first-tier screening using direct injection analysis, second-tier tests often use liquid chromatography-tandem mass spectrometry (LC-MS/MS) to increase specificity by distinguishing isomeric compounds9–12 and evaluating additional disease-related metabolites3,13.
False-positive cases also be reduced using post-analytic tools applied to first-tier screening data with the Collaborative Laboratory Integrated Reports (CLIR), 14–16 and more recently using machine learning-based methodologies17–20. However, the improvement in screening performance with these approaches varies widely among disorders on the Recommended Uniform Screening Panel (RUSP), suggesting that identification and clinical validation of additional markers could further improve screening performance. We therefore developed a rapid (3 min/sample) LC-MS/MS targeted metabolomics test that measures first-tier screening markers, known disease-related metabolites, and metabolites identified by untargeted metabolomics in a 121-analyte multiplex panel relevant to NBS disorders on the RUSP. To simulate second-tier testing, we used this method to analyze DBS from screen-positives for four inherited metabolic disorders, three on the RUSP - glutaric acidemia type 1 (GA1), methylmalonic acidemia (MMA), and very long-chain acyl-CoA dehydrogenase deficiency (VLCADD), and one additionally targeted by the California Newborn Screening Program - ornithine transcarbamylase deficiency (OTCD), and classified results as true- or false-positive using supervised machine learning. Results from this study have implications for second-tier screening with broader applicability to additional NBS disorders.
2. Materials and methods
2.1. Study population
Residual dried blood spot (DBS) specimens and their corresponding MS/MS screening results were obtained from the California Department of Public Health (CDPH) for 883 infants born between 2005–2015 comprising 178 true-positive cases of glutaric acidemia type I (GA1; n=43), methylmalonic acidemia (MMA; n=70 (22 mut−, 18 mut0, 30 cblCDF), ornithine transcarbamylase deficiency (OTCD; n=18), and very long-chain acyl-CoA dehydrogenase deficiency (VLCADD; n=47), as well as 613 false-positive cases (130 GA1, 334 MMA/PA, 70 OTCD, 79 VLCADD) and 92 true negative samples.
2.2. Standards, reagents, and quality control
Authentic standards were purchased Sigma-Aldrich (MO, USA), Cayman Chemicals (MI, USA), Santa Cruz Biotechnologies (SCBT; TX, USA), and IROA Technologies (MA, USA), and Labeled Carnitine Standards (NSK-B-1 and NSK-B-G1) and all isotopic internal standards were from Cambridge Isotopes (MA, USA). All solvents were LC-MS grade from Fisher Scientific. For both untargeted and targeted studies described below, we created pooled quality control samples as a mixture of sample extracts from several positive and negative samples and injected them at intervals of 12–24 injections. Sample injection order was randomized for all unknown samples.
2.3. Biomarker discovery using untargeted metabolomics
For each DBS, two 1/8-inch diameter circles were punched into a well of a 96-deepwell plate, mixed with 200μL of a DBS extraction solution of methanol/water (80:20 v/v). Plates were vortexed for 1 h and centrifuged at 1,800×g for 30 sec, and supernatants (180μL) transferred to a new 96-deepwell plate and evaporated under nitrogen to dryness. Samples were reconstituted with 180μL of a diluent solution containing internal standards (prepared as 400mL water, 100mL methanol, 200μL methylmalonic acid-D3 [1 mg/mL], 1mL of NSK-B-1 (Labeled Carnitine Standards containing free carnitine and C2, C3, C4, C5, C8, C14 and C16 acylcarnitines), and 0.5mL formic acid). All sample extracts were stored at −80°C. Untargeted LC-MS analyses were performed on an Acquity H-Class UPLC system (Waters, MA, USA) coupled to a Q Exactive Plus mass spectrometer (Thermo Fisher Scientific, CA, USA). Chromatographic separation was performed as previously described21 using a two-column method coupling a HSS T3 column (2.1 × 50 mm, Waters, MA, USA) in front of an AmazeHD column (2.1 × 50 mm, Helix Chromatography, IL, USA). The instrument collected positive and negative polarity electrospray ionization full-scan data (MS1) from m/z 70–900 and data-dependent fragmentation spectra (MS2). The runtime for each sample was 12 minutes.
2.4. Compound verification and blinded validation using targeted metabolomics
DBS from a subset of true-positive (TP) and false-positive (FP) samples (Table S1) were blinded and processed in one analytic batch. For each DBS, a single 1/8-inch diameter circle was punched into a 96-deepwell plate, mixed with 100 μL of a DBS extraction solution containing internal standards (prepared as 90mL methanol, 10mL water, 20μL formic acid, 40μL methylmalonic acid-D3 [1 mg/mL], 40μL each of NSK-B-1 and NSK-B-G1, 50μL of MSK-A2 Amino Acids mix, 8μL ornithine 13C5 [5 mg/mL], 2μL citrulline 13C5 [5 mg/mL], 2μL dimethylglycine D6 [1 mg/mL], 10μL uridine 13C5 [1 mg/mL], 40μL orotic acid D2 [6 mM]) and vortexed for 15 minutes. Following centrifugation at 1,800×g for 30 sec, supernatants (80μL) were transferred to a new 96-deepwell plate and evaporated under nitrogen to dryness, and then reconstituted with 100μL of a diluent solution (80% water, 20% methanol, 0.1% formic acid v/v/v). DBS extracts were injected onto an Acquity H-Class UPLC system and compounds were detected using a TQ-S Micro tandem quadrupole mass spectrometer (Waters, MA, USA) using multiple reaction monitoring (MRM). Chromatography was performed on a HSS T3 column (75mm) as previously described22 at flow rate 1.0 mL/min, with a 0.5 min isocratic hold followed by 1 min ramp from 0–100%B for total runtime of 3 min.
2.5. Characterization of acylcarnitine isobars
Acylcarnitine isobar characterization was performed using liquid chromatography ion-mobility high-resolution mass spectrometry. DBS extracts were injected onto an Acquity H-Class UPLC system and compounds were detected using a Vion ion mobility-qTOF mass spectrometer (Waters, MA, USA). The chromatographic setup was the same as in the targeted study, but the separation was 10 min. The mass spectrometer acquired MS1 from m/z 70–900 or 70–1200 and data dependent MS2 with ion mobility.
2.6. Statistical analysis
For untargeted studies, Progenesis (Waters, MA, USA) was used to open and align raw data, pick peaks, and export a dataset of ion features and their intensities in each sample. Internal standards were used for retention time alignment but not for data normalization. The data from positive- and negative-ionization acquisition modes were then merged into a master dataset, and ion features in the quality control samples with a coefficient of variation (CV) ≤ 30% were retained for data analysis. We used Random Forest (RF),23,24 a classification algorithm that combines the output of many random decision trees, to help identify those features that differed the most between true- and false-positive cases. To visualize the importance of each metabolite to classification, RF output was plotted as the Mean Decrease in Accuracy (MDA), a score chart that lists features in order of importance to the model. We manually reviewed this output and selected those features that clearly differed between true- and false-positives. Compounds were then identified using an in-house library consisting of MS1, MS2, and retention time data for a given chromatography method, and only those with tier 1 matches were carried forward for clinical test translation. For targeted studies, Targetlynx XS (Waters, MA, USA) was used to open the raw data, review peak integration, and export a dataset of compounds and their intensities in each sample. For compounds with a matching isotopic internal standard, peak area was normalized to internal standard peak area. All other compound responses were reported using raw peak area unless otherwise noted. To classify samples in the validation study, RF classification was applied as previously described19. Briefly, the performances of the RF models were estimated with leave-one-out cross-validation (LOOCV), which uses one sample as testing and all others as training to build the model for each disorder. This process was iterated for all samples and every sample was used as testing one time. Only RF assignments from testing cases (and not from training) were used for final outcome prediction. Positive predictive values (PPV=TP/TP+FP) with 100% classification sensitivity (i.e., correctly classify all true positives) and area under the receiver operating characteristic curves (AUC) were estimated from testing results. To show robustness, we repeated LOOCV 20 times and reported the median PPV and AUC results. RF classification analyses were done in R software 4.1.3 using these R packages: randomForest and pROC. Principal component analyses (PCA) and heatmaps with Ward’s hierarchical clustering using Euclidean distance were created using Metaboanalyst25. Boxplots were created using Microsoft Excel.
3. Results
3.1. Untargeted metabolomics identifies additional markers for second-tier analysis
To search for new screening markers in the four conditions, we performed untargeted metabolomics using NBS dried blood spots from 178 cases with a true-positive screen for GA1, MMA, OTCD, or VLCADD, as well as 613 false-positive screens for one of these conditions and 92 true-negative controls (see Materials and Methods). Following acquisition and processing of LC-MS raw data, we obtained a dataset of approximately 9,000 ion features, ranked the importance of each ion feature using RF, and manually reviewed the score matrix and feature abundance as described in Material and Methods. This identified 14 metabolites important to disease classification, including the primary NBS markers for the four conditions (C5DC, C3, citrulline, and C14:1), seven metabolites used in confirmatory testing (two saturated and unsaturated C14-acylcarnitines, glutaric acid, methylmalonic acid, methionine, glutamine, and orotic acid), proline and uridine associated with OTCD19,26, and one new metabolite, dimethylglycine, which was elevated in MMA subtype cblCDF. Patient data were normalized to true-negative group data to visualize metabolic differences (Figure S1). Results from the false-positive samples were not used for statistical comparisons, since these samples were processed separately and showed measurable batch effects.
3.2. Compound verification using targeted metabolomics
To verify discovery study results in a setting applicable to second-tier testing, we initially developed a high-throughput, targeted LC-MS/MS method capable of analyzing an expanded set of markers relevant to newborn screening in DBS, including 45 first-tier metabolic screening markers detected by California NBS and more than 50 additional analytes relevant to NBS disorders on the RUSP (Table 1). This method also separates many clinically important isomers including succinic/methylmalonic acids, isoleucine/leucine, and 2-hydroxyglutaric /3-hydroxyglutaric acids, at a runtime of 3 minutes per sample. Reanalyzing the same DBS extracts as above by targeted metabolomics resulted in chromatographic peaks for all amino acids and acylcarnitines analyzed in first tier screening and verified the relative intensities of the 14 metabolites from the discovery study.
Table 1. Analytes included in the 121-plex targeted metabolomics panel.
The multiplex panel includes first-tier screening markers, additional organic acids, acylcarnitines and acylglycines, and uncharacterized isobars. First-tier screening markers are defined as those included in our California Newborn Screening dataset.
| First-tier newborn screening markers and their isomers (n=45) | ||
|
| ||
| Alanine | C0 (free carnitine) | C10 |
| Arginine | C2 | C10:1 |
| Citrulline | C3 | C12 |
| Glycine | C3DC | C12:1 |
| Isoleucine/Alloisoleucine | C4 | C14 |
| Leucine | C4DC (Methylmalonylcarnitine) | C14:1 |
| Methionine | C4DC (Succinylcarnitine) | C14:2 |
| Ornithine | C5 (Isovaleryl/Valerylcarnitine) | C14-OH |
| Phenylalanine | C5 (Pivaloyl/2-methylbutyrylcarnitine) | C16 |
| Proline | C5:1 | C16:1 |
| Pyroglutamic acid | C5DC | C16-OH |
| Succinylacetone | C5-OH | C18 |
| Tyrosine | C6 | C18:1 |
| Valine | C8 | C18:1-OH |
| C8:1 | C18-OH | |
| C18:2 | ||
|
| ||
| Additional analytes (n=52) | ||
|
| ||
| 2-aminobutyric acid | C6-OH | Methylmalonic acid |
| 2-hydroxy-3-methylbutanoic acid | Creatine | Methylsuccinic/ethylmalonic acid |
| 2-hydroxyglutaric acid | Dimethylglycine | Octadecanedioic acid |
| 2-hydroxy-methylbutyric acid | Dodecanedioic acid | Orotic acid |
| 2-methylbutyrylglycine | Glutamic acid | Propionylglycine |
| 3-hydroxy-2-methylbutanoic acid | Glutamine | Sebacic acid |
| 3-hydroxyglutaric acid | Glutaric acid | Serine |
| 3-hydroxyisovaleric acid | Guanidinoacetic acid | Suberic acid |
| 3-hydroxypropionic acid | Hexadecanedioic acid | Suberylglycine |
| 3-methylcrotonylglycine | Hexanoylglycine | Succinic acid |
| 3-methylglutaconic acid | Histidine | Sulfocysteine |
| Acetoacetic acid | Homocitrulline | Tetradecanedioic acid |
| Adipic acid | Homocysteine | Threonine |
| Asparagine | Homocystine | Tryptophan |
| Aspartic acid | Hydroxyproline | Uridine |
| Butyryl/Isobutyrylglycine | Lactic acid | Valeryl/Isovaleryl/Methylbutyrylglycine |
| C14:1-OH | Lysine | |
| C16:1-OH | Methylcitric acid | |
|
| ||
| Uncharacterized ion features (isobaric to) (n=24) | ||
|
| ||
| 0.34_269/136 (homocystine) | 1.72_414.4/85 (C16:1-OH) | 1.91_384.3/85 (C14:2-OH) |
| 1.3_276.2/85 (C5DC) | 1.75_384.3/85 (C14:2-OH) | 2.0_386.3/85 (C14:1-OH) |
| 1.52_262.2/85 (C5-OH) | 1.75_442.3/85 (C18:1-OH) | 2.03_428.4/85 (C18) |
| 1.54_386.3/85 (C14:1-OH) | 1.78_416.4/85 (C16-OH) | 2.1_370.4/85 (C14:1) |
| 1.59_444.4/85 (C18-OH) | 1.79_444.4/85 (C18-OH) | 2.1_386.3/85 (C14:1-OH) |
| 1.65_384.3/85 (C14:2-OH) | 1.8_288.3/85 (C8) | 2.1_414.4/85 (C16:1-OH) |
| 1.66_386.3/85 (C14:1-OH) | 1.8_442.3/85 (C18:1-OH) | 2.18_414.4/85 (C16:1-OH) |
| 1.69_388.4/85 (C14-OH) | 1.86_444.4/85 (C18-OH) | 2.26_424.4/85 (C18:2) |
3.3. Targeted metabolomics reveals important acylcarnitine isobars
From this initial verification study, LC-MS/MS testing uncovered additional compounds isobaric to known targets, including two peaks eluting at 1.25 and 1.4 min at Multiple Reaction Monitoring (MRM) transition 276.2/85 (monitored to detect C5DC) among GA1 screen-positive samples. The first of these, identified as C5DC, was higher in true positive cases and the second, identified as C6-OH, was higher in false positives. New peaks were also seen at MRM transitions for several of the targeted long-chain acylcarnitine and hydroxy-acylcarnitine species. We characterized these compounds in VLCADD screen-positive samples using liquid chromatography ion-mobility high-resolution mass spectrometry to capture accurate mass and collisional cross section (CCS), a distinguishing property of ions. Extracted ion chromatograms (5-ppm window) revealed additional peaks for m/z representing C14-OH, C14:1-OH, C14:2-OH, C16-OH, C16:1-OH and C18:1-OH (Figure S2, S3). The peaks isobaric to C14-OH and C16-OH were distinct from their known 3S and 3R stereoisomers by both retention time and CCS measurements, despite having accurate masses within 5-ppm of their authentic standards. These compounds are therefore not the same as the authentic standards of C14-OH and C16-OH although they share major ion fragments in common with authentic standards including m/z 85, which is also shared by other acylcarnitine species (Figure S3).
3.4. Validation of targeted metabolomics for second-tier screening
To evaluate the performance of our targeted metabolomics assay as a second-tier test, we designed a blinded validation study using a subset of 285 screen-positive cases for each of the four target conditions, and 100 true-negative controls (Table S1). As configured for this study, the test targets 121 analytes comprising all metabolites from method verification including 24 features representing the uncharacterized isobars described above (Table 1). All DBS were processed and analyzed together to prevent batch effects. Principal component analysis of the resulting data showed tight clustering of quality control samples, indicating good technical reproducibility (Figure S4). On initial data review, two of the GA1 true-positive samples had C5DC peak areas indistinguishable from true negative controls (Patient 1: 0.32; Patient 2: 0.55; TN mean=0.27, SD=0.27) with normal levels of glutaric and 3-hydroxyglutaric acids and elevated C6, C8, C10 and C10:1-acylcarnitines, and likely were mislabeled cases of glutaric acidemia type II (multiple acyl-CoA dehydrogenase deficiency). These samples were therefore removed from further analysis. For the remaining dataset, heatmap analysis with Ward’s hierarchical clustering revealed clear separation along the horizontal axis between the true- and false-positive cases of GA1, VLCADD, and OTCD (Figure 1), suggesting that our expanded metabolite panel contained and detected the necessary markers to reduce false positive cases for multiple disorders.
Figure 1. Heatmap analysis of targeted validation results.
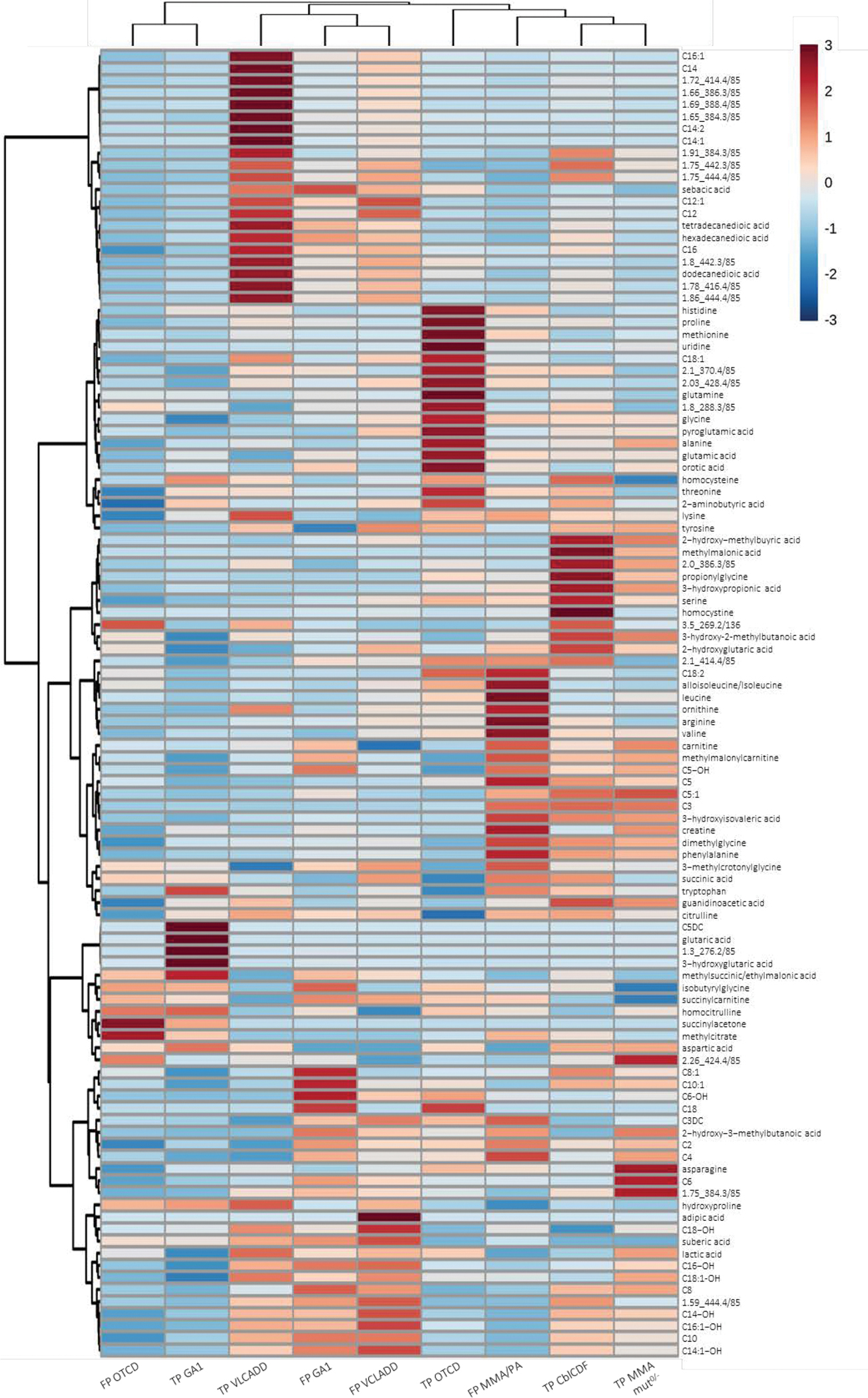
Profiles of the top 108 metabolites are shown for true-positive (TP) and false-positive (FP) cases from the four disorders. For each metabolite, data from each group were autoscaled to the data of the true-negative samples and the median response of each group was used.
3.5. Metabolic Pattern Analysis Using Random Forest
To demonstrate that machine learning using data from our expanded metabolite panel could improve discrimination between true- and false-positive cases without compromising sensitivity, we trained an RF classifier using our targeted metabolomics data (as either analyte peak areas or response ratios) for 285 screen-positive cases. With all true positives correctly identified (100% sensitivity), RF reduced the number of false positives by 83% for GA1, 84% for MMA, 100% for OTCD, and 51% for VLCADD (Table 2). Accordingly, AUC ranged from 0.93 (VLCADD) to 1.00 (OTCD) and PPV ranged from 36% (VLCADD) to 100% (OTCD). To evaluate precision of the classifier models we performed leave-one-out cross-validation 20 times to estimate the variation of AUC. The largest standard deviation of AUC results was 0.007 (GA1), demonstrating good stability and reproducibility (Figure S5).
Table 2. Targeted metabolomics reduces false-positive cases.
Number of true- and false-positives from first tier screening, and subsequent reduction of false positives, area under the receiver operating characteristic curve (AUC), and positive predictive value (PPV) following second-tier analysis using 121-analyte panel testing and RF classification at 100% sensitivity.
| First-Tier Analysis | Second-Tier Analysis (100% sensitivity) | |||||
|---|---|---|---|---|---|---|
|
| ||||||
| Disorder | True Positives | False Positives | False Positives Eliminated | % Reduction of False Positives | AUC | PPV |
|
| ||||||
| GA1 | 6 | 29 | 24 | 82.8% | 0.97 | 54.5% |
| MMA | 13 | 58 | 49 | 84.5% | 0.98 | 59.1% |
| OTCD | 3 | 16 | 16 | 100.0% | 1.00 | 100.0% |
| VLCADD | 13 | 47 | 24 | 51.1% | 0.93 | 36.1% |
3.6. Ranking of Metabolic Analytes
To identify the important analytes driving classification, we examined the Mean Decrease in Accuracy (MDA) rankings for the top-10 ranked analytes for each condition. This identified primary screening markers, additional known disease-associated metabolites (3-hydroxyglutaric acid [GA1], methylmalonic acid [MMA], uridine and proline [OTCD]), and several long-chain acylcarnitine and hydroxy-acylcarnitine isobars in VLCADD and MMA (Figure 2). In examining the top-3 analytes in each MDA chart, 3-hydroxyglutaric acid had the largest relative difference in importance, indicating that removal of this metabolite would substantially decrease GA1 classification accuracy. GA1 classification was also facilitated by chromatographic separation of C5DC from C6-OH, which was elevated in the false positives and could account for the overlap in C5DC between true- and false-positive cases in first-tier screening (Figure S6). For OTCD, proline, citrulline, and uridine showed clear separation between true- and false-positive cases supporting RF classification accuracy. For MMA, classification was driven by methylmalonic acid, long-chain acylcarnitine isobars, and free carnitine, although no single marker showed clear separation between true- and false-positive cases (Figure 2). Among MMA subtypes, homocystine was higher in MMA cblCDF than MMA mut− and mut0 by visual inspection of boxplots (Figure S7). In contrast, dimethylglycine, identified in the discovery study as a potential marker for cblCDF, was not among the top metabolites in the blinded validation study, possibly due to the small sample size. Finally, for VLCADD, the top features included saturated, and mono- and di-unsaturated C14-acylcarnitine along with several C14 and hydroxy-C14 isobars (Figure 2). None of the compounds showed complete group separation, suggesting that RF reduced false positives by recognizing complex patterns of multiple analytes. The top features for MMA and VLCAD included compounds isobaric to C18-OH and C14:2-OH respectively, although their identities could not be confirmed due to lack of available standards (fragmentation data is shown in Figure S8).
Figure 2. Analytes important for true- or false-positive classification.
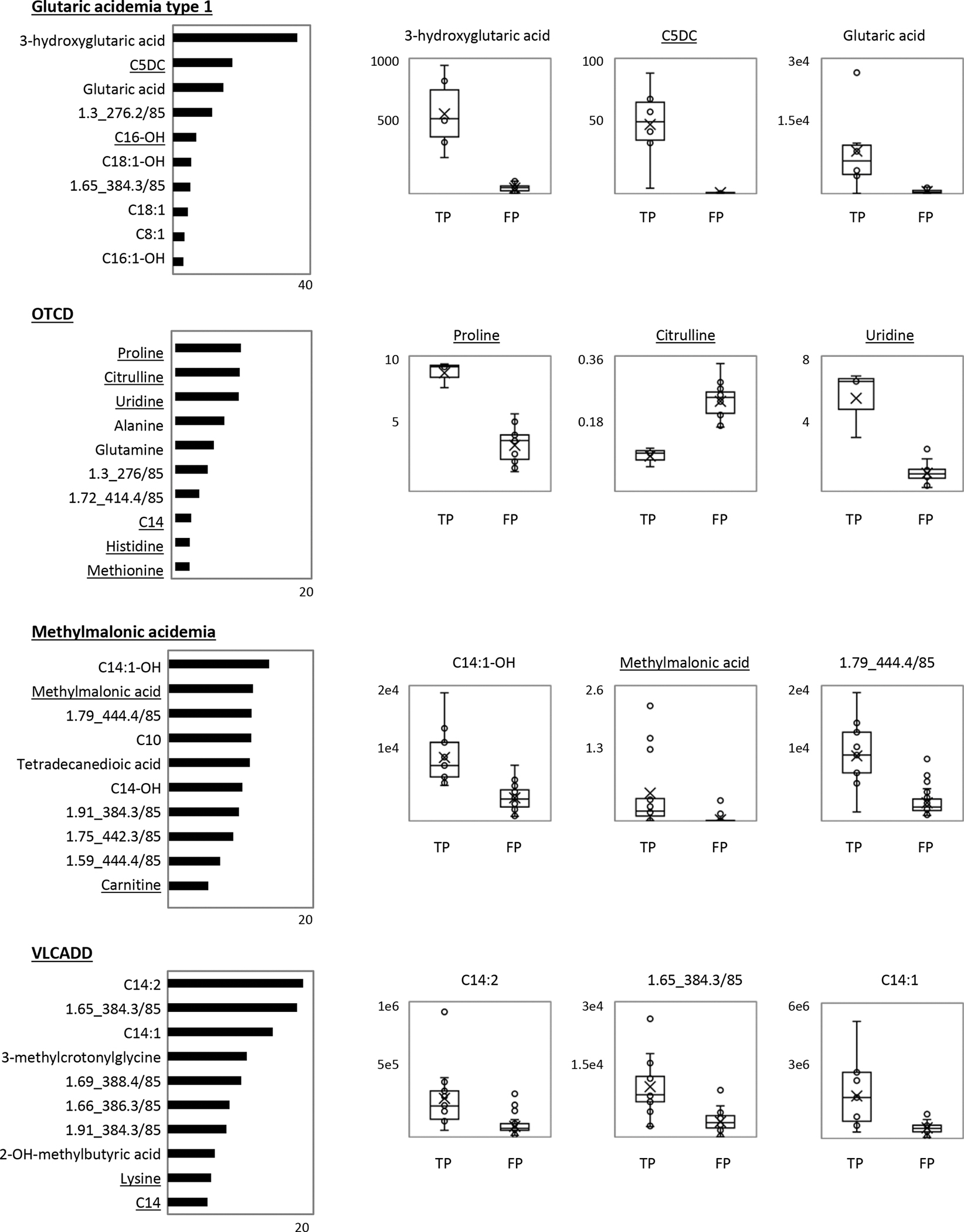
For each condition, a bar chart shows the Mean Decrease in Accuracy values (MDA; x-axis) for the 10 most important analytes for accurate random forest classification, with higher values representing higher importance for model accuracy. Corresponding boxplot analyses of the top-3 analytes for each condition show analyte peak area or response ratio (peak area divided by the peak area of the matching isotopic internal standard) for the true-positive (TP) and false-positive (FP) sample groups. The horizontal line indicates the group median, and the X indicates the group mean. Ion features are denoted by RT_MRM. Underlined analytes are those with a matching isotopic internal standard.
4. Discussion
Reducing the rate of false-positive results remains an important challenge in newborn screening for inborn metabolic disorders. Here we present an approach for second-tier testing using multiplex targeted metabolite profiling in combination with machine learning for the analysis of four metabolic disorders (GA1, MMA, OTCD, and VLCADD), each compromised by high false-positive rates and delays in case resolution following a positive newborn screen, with the goal of reducing parental anxiety and the costs of follow-up testing and management. At a screening sensitivity of 100%, our integrated approach of targeted metabolomics and RF classification reduced false positives for GA1 by 83%, MMA by 84%, OTCD by 100%, and VLCADD by 51% (Table 2). This performance was driven by (1) incorporation of additional disease markers, including those currently analyzed in reference metabolic testing; (2) chromatographic separation and measurement of clinically relevant isobars; and (3) analysis of complex metabolite profiles through machine learning. Here we show that chromatographic separation and incorporation of additional screening markers improves screening performance for the four target conditions and likely others. These results also support previous findings for improved prediction performance by utilizing the entire set of metabolites in machine learning-based disease classification19.
In this project, we had first set out to expand the number of markers available for second-tier screening of the four target conditions. Despite deep metabolic profiling, untargeted metabolomics contributed little beyond what was already known, such as the primary screening markers (C5DC, citrulline, C3 and C14:1), and additional known disease markers routinely used in confirmatory testing (e.g., orotic acid, MMA) and two recently described markers for OTC (proline, uridine)19,26. In turn, several known metabolites routinely used in confirmatory testing (e.g., 3-hydroxyglutaric acid, homocystine, methionine) escaped detection in the discovery study, which could be attributed to the age of some DBS specimens and potential degradation issues27,28. Consequently, we prioritized a list of 121 metabolites that included all known disease markers from primary screening, metabolites identified using untargeted metabolomics, and isobaric species revealed by chromatographic separation. To mimic a second-tier screening environment and rigorously evaluate our 121-plex metabolite panel, we then tested samples using a rapid and high-throughput LC-MS/MS method. Using this method, screen positives for the four target disorders can be followed up using a single instrument run (3-minute runtime/sample), highlighting the applicability for newborn screening settings. Additional studies will allow for refinement and focusing of this panel to include only those features needed for optimal screening performance for these and other disorders.
Targeted metabolomics analysis and machine learning revealed that long-chain acylcarnitine isobars were among the top features driving classification of true- and false-positives for all four conditions (Figure 2). Isomers (compounds with the same molecular formula) are well recognized for short- and medium-chain acylcarnitines including C5 and C5-OH 10, and long-chain acylcarnitine isomers or isobars (compounds differing in atomic number) have also been described for long-chain species in patients with various metabolic disorders 29–31. Most of our uncharacterized ion features (Table 1) likely represent isobars based on their CCS values compared with authentic standards for C14- and C16-acylcarnitines and their mono- and di-unsaturated forms, and C18:1-OH (Figures S2 and S3). In total, long-chain acylcarnitines and their isobars account for over half of the top-ten discriminating features for GA1, MMA and VLCADD (Figure 2) and represent a potential source of interference in first-tier screening. The identity and biologic significance of these isobars are currently unknown but could be investigated with additional studies of DBS metabolomic profiles and corresponding clinical and demographic data.
Second-tier analysis of MS/MS newborn screening data using machine learning has been shown to decrease false positives for the four disorders19, although the degree of improvement varied widely suggesting that measurement of additional markers could further improve screening performance. While the differences in sample size preclude direct comparison of our results with prior studies, our data indicate that the additional analytes detected in targeted metabolomics allowed machine learning to improve classification. For example, the combination of targeted metabolomics analysis with RF reduced false-positives by 51% for VLCADD and by 83% for MMA (Table 2), as compared to a 2% and 45% reduction, respectively, using RF analysis of first-tier screening data alone19. However, results also suggest that machine learning may not be necessary for every disorder and its use should be investigated for each screening marker. We used RF to classify all samples and corresponding conditions in this study, but closer examination of 3-hydroxylglutaric acid levels led us to question why RF did not eliminate all false-positive GA1 cases despite the clear differences in levels (Figure 2). We hypothesized that the RF model may have underperformed due to the combination of having only one fully discriminant marker (3-hydroxyglutaric acid) and small sample size, a known limitation to machine learning32. Since elevated 3-hydroxyglutaric acid has been measured in dried blood spots and dried urine spots, even in samples with lower levels of glutarylcarnitine, we predict that machine learning may not be needed to classify GA1 screen-positives33,34. Similarly, the presence of multiple clearly discriminant metabolites (proline, citrulline, and uridine) in OTCD suggests that machine learning has limited utility for improving classification of OTCD screen-positives. However, in contrast to cut-off-based methods, machine learning can be used to identify complex relationships among metabolites, such as in MMA and VLCADD. Furthermore, machine learning can be used to recognize metabolite level differences in relation to continuous and categorical covariates such as gestational age, birth weight, age at blood collection, sex, parent-reported ethnicity, nutritional therapy, or season of birth, which have been shown to reduce the accuracy of screening35–40.
Our study has several limitations. First, untargeted metabolomics analysis was performed using dried blood spots that were stored for a period of 7–17 years, and some extracts for up to 2 years. This may have led to degradation of some anticipated analytes including 3-hydroxyglutaric acid, homocystine, and methionine, and may have limited the discovery of novel metabolites. Future studies using fresh blood spots from screen positives could generate data that expand the number of candidate second-tier markers. Second, the processing of false-positive samples separately from true positives and true negatives in the discovery study led to measurable batch effects, which limited the statistical analysis and potential discovery of metabolite level differences between these groups. In contrast, samples for the validation study were processed in a single batch and incorporated numerous isotopic internal standards to prevent batch effects and reduce analytic variability. However, due to limitations in sample availability, the validation could not be performed on a completely independent set of samples as otherwise would be required. Full clinical implementation of this method will require independent validation as well as the use of as many isotopic standards as possible to enable long-term inter-laboratory comparisons. Third, false positives were reduced by 51–84 % for three of our four disorders with only OTCD achieving 100% PPV (Table 2), suggesting that there are additional factors influencing screening performance. Finally, our study cohort was relatively small and did not reflect the natural variability of the general population. Future studies should generate data from babies from different ancestries and consider other covariates that could lead to false-positive screens. 35–40
5. Conclusion
In performing effective second-tier testing for inborn metabolic disorders, NBS laboratories require a single, comprehensive panel of disease markers that can be assayed at high sensitivity and specificity from newborn dried blood spots. Here we developed a 121-metabolite panel and provide proof-of-principle that targeted metabolomics analysis can significantly improve screening performance for four metabolic disorders targeted by the California Newborn Screening Program. This metabolite panel is flexible, customizable, and expandable, allowing for insertion of additional markers, which will enhance the development of a larger panel, one that optimizes the multiplexing of additional markers for second-tier testing of other metabolic disorders.
Supplementary Material
Synopsis.
Robust, targeted metabolomics analysis with supervised machine learning reduces false-positive metabolic screening results.
Funding support:
This work was in parts funded by a grant from the National Institute of Child Health and Human Development (R01HD102537).
Footnotes
Conflicts of interest: Justin Mak, Gang Peng, Anthony Le, Neeru Gandotra, Gregory M. Enns, Curt Scharfe, and Tina M. Cowan declare that they have no conflict of interest.
Ethics approval: This study was overseen by the institutional review boards at Yale University (protocol 1505015917), Stanford University (protocol 30618), and the State of California Committee for the Protection of Human Subjects (protocol 13-05-1236).
Informed Consent Statement: Not applicable.
Data Sharing Statement:
The data used in this study were obtained from the California Biobank Program (CBP) under SIS request 886. The California Department of Public Health is not responsible for the results or conclusions drawn by the authors of this publication. Data can be obtained by others after submitting a new request to the CBP coordinator. Requests for data should be directed to CaliforniaBiobank@cdph.ca.gov.
References
- 1.Schulze A, Lindner M, Kohlmuller D, Olgemoller K, Mayatepek E, Hoffmann GF. Expanded newborn screening for inborn errors of metabolism by electrospray ionization-tandem mass spectrometry: results, outcome, and implications. Pediatrics. 2003;111(6 Pt 1):1399–1406. [DOI] [PubMed] [Google Scholar]
- 2.Tarini BA, Christakis DA, Welch HG. State newborn screening in the tandem mass spectrometry era: more tests, more false-positive results. Pediatrics. 2006;118(2):448–456. [DOI] [PubMed] [Google Scholar]
- 3.la Marca G, Malvagia S, Casetta B, Pasquini E, Donati MA, Zammarchi E. Progress in expanded newborn screening for metabolic conditions by LC-MS/MS in Tuscany: update on methods to reduce false tests. J Inherit Metab Dis. 2008;31 Suppl 2:S395–404. [DOI] [PubMed] [Google Scholar]
- 4.Lund A, Wibrand F, Skogstrand K, et al. Danish expanded newborn screening is a successful preventive public health programme. Dan Med J. 2020;67(1). [PubMed] [Google Scholar]
- 5.Schmidt JL, Castellanos-Brown K, Childress S, et al. The impact of false-positive newborn screening results on families: a qualitative study. Genet Med. 2012;14(1):76–80. [DOI] [PubMed] [Google Scholar]
- 6.Goldenberg AJ, Comeau AM, Grosse SD, et al. Evaluating Harms in the Assessment of Net Benefit: A Framework for Newborn Screening Condition Review. Matern Child Health J. 2016;20(3):693–700. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 7.Matern D, Tortorelli S, Oglesbee D, Gavrilov D, Rinaldo P. Reduction of the false-positive rate in newborn screening by implementation of MS/MS-based second-tier tests: the Mayo Clinic experience (2004–2007). J Inherit Metab Dis. 2007;30(4):585–592. [DOI] [PubMed] [Google Scholar]
- 8.Ombrone D, Giocaliere E, Forni G, Malvagia S, la Marca G. Expanded newborn screening by mass spectrometry: New tests, future perspectives. Mass Spectrom Rev. 2016;35(1):71–84. [DOI] [PubMed] [Google Scholar]
- 9.Oglesbee D, Sanders KA, Lacey JM, et al. Second-tier test for quantification of alloisoleucine and branched-chain amino acids in dried blood spots to improve newborn screening for maple syrup urine disease (MSUD). Clin Chem. 2008;54(3):542–549. [DOI] [PubMed] [Google Scholar]
- 10.Carling RS, Burden D, Hutton I, Randle R, John K, Bonham JR. Introduction of a Simple Second Tier Screening Test for C5 Isobars in Dried Blood Spots: Reducing the False Positive Rate for Isovaleric Acidaemia in Expanded Newborn Screening. JIMD Rep. 2018;38:75–80. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 11.Wojcik M, Morrissey M, Borden K, et al. Method modification to reduce false positives for newborn screening of guanidinoacetate methyltransferase deficiency. Mol Genet Metab. 2022;135(3):186–192. [DOI] [PubMed] [Google Scholar]
- 12.Monostori P, Godejohann M, Janda J, et al. Identification of potential interferents of methylmalonic acid: A previously unrecognized pitfall in clinical diagnostics and newborn screening. Clin Biochem. 2022. [DOI] [PubMed] [Google Scholar]
- 13.Hu Z, Yang J, Lin Y, et al. Determination of methylmalonic acid, 2-methylcitric acid, and total homocysteine in dried blood spots by liquid chromatography-tandem mass spectrometry: A reliable follow-up method for propionylcarnitine-related disorders in newborn screening. J Med Screen. 2021;28(2):93–99. [DOI] [PubMed] [Google Scholar]
- 14.Marquardt G, Currier R, McHugh DM, et al. Enhanced interpretation of newborn screening results without analyte cutoff values. Genet Med. 2012;14(7):648–655. [DOI] [PubMed] [Google Scholar]
- 15.Tortorelli S, Eckerman JS, Orsini JJ, et al. Moonlighting newborn screening markers: the incidental discovery of a second-tier test for Pompe disease. Genet Med. 2018;20(8):840–846. [DOI] [PubMed] [Google Scholar]
- 16.Minter Baerg MM, Stoway SD, Hart J, et al. Precision newborn screening for lysosomal disorders. Genet Med. 2018;20(8):847–854. [DOI] [PubMed] [Google Scholar]
- 17.Baumgartner C, Bohm C, Baumgartner D, et al. Supervised machine learning techniques for the classification of metabolic disorders in newborns. Bioinformatics. 2004;20(17):2985–2996. [DOI] [PubMed] [Google Scholar]
- 18.Chen WH, Hsieh SL, Hsu KP, et al. Web-based newborn screening system for metabolic diseases: machine learning versus clinicians. J Med Internet Res. 2013;15(5):e98. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 19.Peng G, Tang Y, Cowan TM, Enns GM, Zhao H, Scharfe C. Reducing False-Positive Results in Newborn Screening Using Machine Learning. Int J Neonatal Screen. 2020;6(1). [DOI] [PMC free article] [PubMed] [Google Scholar]
- 20.Zaunseder E, Haupt S, Mutze U, Garbade SF, Kolker S, Heuveline V. Opportunities and challenges in machine learning-based newborn screening-A systematic literature review. JIMD Rep. 2022;63(3):250–261. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 21.Le A, Mak J, Cowan TM. Metabolic profiling by reversed-phase/ion-exchange mass spectrometry. J Chromatogr B Analyt Technol Biomed Life Sci. 2020;1143:122072. [DOI] [PubMed] [Google Scholar]
- 22.Waters. Metabolomics & Lipidomics Application Notebook. https://wwwwaterscom/waters/libraryhtm?locale=en_US&lid=134841740. 2015.
- 23.Breiman L Random Forests. Machine Learning. 2001;45(1573–0565):5–32. [Google Scholar]
- 24.Liaw A, Wiener M. Classification and regression by randomForest. R news. 2002;2(3):18–22. [Google Scholar]
- 25.Pang Z, Chong J, Zhou G, et al. MetaboAnalyst 5.0: narrowing the gap between raw spectra and functional insights. Nucleic Acids Res. 2021;49(W1):W388–W396. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 26.Burrage LC, Thistlethwaite L, Stroup BM, et al. Untargeted metabolomic profiling reveals multiple pathway perturbations and new clinical biomarkers in urea cycle disorders. Genet Med. 2019;21(9):1977–1986. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 27.Golbahar J, Altayab DD, Carreon E. Short-term stability of amino acids and acylcarnitines in the dried blood spots used to screen newborns for metabolic disorders. J Med Screen. 2014;21(1):5–9. [DOI] [PubMed] [Google Scholar]
- 28.Drolet J, Tolstikov V, Williams BA, et al. Integrated Metabolomics Assessment of Human Dried Blood Spots and Urine Strips. Metabolites. 2017;7(3). [DOI] [PMC free article] [PubMed] [Google Scholar]
- 29.Peng M, Fang X, Huang Y, et al. Separation and identification of underivatized plasma acylcarnitine isomers using liquid chromatography-tandem mass spectrometry for the differential diagnosis of organic acidemias and fatty acid oxidation defects. J Chromatogr A. 2013;1319:97–106. [DOI] [PubMed] [Google Scholar]
- 30.Minkler PE, Stoll MS, Ingalls ST, Yang S, Kerner J, Hoppel CL. Quantification of carnitine and acylcarnitines in biological matrices by HPLC electrospray ionization-mass spectrometry. Clin Chem. 2008;54(9):1451–1462. [DOI] [PubMed] [Google Scholar]
- 31.Luna C, Griffin C, Miller MJ. A clinically validated method to separate and quantify underivatized acylcarnitines and carnitine metabolic intermediates using mixed-mode chromatography with tandem mass spectrometry. J Chromatogr A. 2022;1663:462749. [DOI] [PubMed] [Google Scholar]
- 32.Han S, Williamson BD, Fong Y. Improving random forest predictions in small datasets from two-phase sampling designs. BMC Med Inform Decis Mak. 2021;21(1):322. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 33.Al-Dirbashi OY, Kolker S, Ng D, et al. Diagnosis of glutaric aciduria type 1 by measuring 3-hydroxyglutaric acid in dried urine spots by liquid chromatography tandem mass spectrometry. J Inherit Metab Dis. 2011;34(1):173–180. [DOI] [PubMed] [Google Scholar]
- 34.Shigematsu Y, Yuasa M, Ishige N, Nakajima H, Tajima G. Development of Second-Tier Liquid Chromatography-Tandem Mass Spectrometry Analysis for Expanded Newborn Screening in Japan. Int J Neonatal Screen. 2021;7(3). [DOI] [PMC free article] [PubMed] [Google Scholar]
- 35.Ryckman KK, Berberich SL, Shchelochkov OA, Cook DE, Murray JC. Clinical and environmental influences on metabolic biomarkers collected for newborn screening. Clin Biochem. 2013;46(1–2):133–138. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 36.Hall PL, Marquardt G, McHugh DM, et al. Postanalytical tools improve performance of newborn screening by tandem mass spectrometry. Genet Med. 2014;16(12):889–895. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 37.Clark RH, Kelleher AS, Chace DH, Spitzer AR. Gestational age and age at sampling influence metabolic profiles in premature infants. Pediatrics. 2014;134(1):e37–46. [DOI] [PubMed] [Google Scholar]
- 38.Peng G, Tang Y, Gandotra N, et al. Ethnic variability in newborn metabolic screening markers associated with false-positive outcomes. J Inherit Metab Dis. 2020;43(5):934–943. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 39.Peng G, Tang Y, Cowan TM, Zhao H, Scharfe C. Timing of Newborn Blood Collection Alters Metabolic Disease Screening Performance. Front Pediatr. 2020;8:623184. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 40.Henderson MPA, McIntosh N, Chambers A, et al. Biotinidase activity is affected by both seasonal temperature and filter collection cards. Clin Biochem. 2022. [DOI] [PubMed] [Google Scholar]
Associated Data
This section collects any data citations, data availability statements, or supplementary materials included in this article.
Supplementary Materials
Data Availability Statement
The data used in this study were obtained from the California Biobank Program (CBP) under SIS request 886. The California Department of Public Health is not responsible for the results or conclusions drawn by the authors of this publication. Data can be obtained by others after submitting a new request to the CBP coordinator. Requests for data should be directed to CaliforniaBiobank@cdph.ca.gov.