

Abstract
We present a ligand platform featuring appended ditopic Lewis acids to facilitate capture/activation of diatomic substrates. We show that incorporation of two 9-borabicyclo[3.3.1]nonane (9-BBN) units on a single carbon tethered to a pyridine pyrazole scaffold maintains a set of unquenched nitrogen donors available to coordinate Fe(II), Zn(II), and Ni(II). Using hydride ion affinity and competition experiments, we establish an additive effect for ditopic secondary sphere boranes, compared to the monotopic analogue. These effects are exploited to achieve high selectivity for binding NO2- in the presence of competitive anions such as F- and NO3-. Finally, we demonstrate hydrazine capture within the second-sphere of metal complexes, followed by unique activation pathways to generate hydrazido and diazene ligands on Zn and Fe, respectively.
Keywords: Lewis acids, ditopic boranes, chemoselective anion binding, secondary coordination sphere, hydrazine functionalization
Graphical Abstract
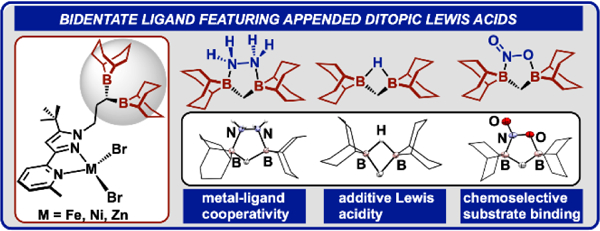
We report the synthesis of a bidentate ligand featuring secondary sphere ditopic Lewis acids. We verify a Lewis acid additivity effect for the ditopic boranes compared to a monotopic analogue using hydride ion affinity and competition studies. We show chemoselective nitrite capture in the presence of other anions. Pre-organized hydrazine adducts in the second sphere of Zn and Fe are functionalized to hydrazido and diazene ligands, respectively.
To capture and reduce biologically relevant substrates with high specificity, the active site(s) of metalloenzymes often feature surrounding secondary sphere groups that accommodate multiple points of binding (multitopic hydrogen bond donors/acceptors).[1] These units facilitate selective substrate binding with high affinity at sites that might otherwise bind with low affinity. Substrate binding/recognition serves as the key first step to dictate structure, function, and product distribution.[2] One strategy to emulate these design principles within a synthetic system is to couple a Lewis acidic group with a metal site via a covalently linked tether.[3] While H-bonds are routinely used within enzyme active sites, Lewis acids provide a complementary approach because of their steric/electronic versatility and their stability to highly reducing environments.[4] This approach has facilitated C-C bond coupling of CO ligands,[5] ligand exchange,[6] oxidative addition,[7] and hydride transfer.[8] However few systems contain secondary sphere groups that mimic di/poly-topic units.[3] To distinguish this work from reported ligand scaffolds containing multiple second-sphere boranes, we define ditopic boranes as a pre-organized unit with two boranes predisposed for multi-point binding of substrate. Our group has previously investigated cooperative binding modes and subsequent reactivity enabled by Lewis acidic borane-appended ligands.[4, 9] To complement these efforts and parallel bifurcated H-bonds, we targeted a ditopic borane Lewis acid analogue amenable to incorporation within a ligand platform to ultimately facilitate cooperative substrate binding/activation.
1,1-diborylalkane units (Figure 1) have been previously used for a range of electrophilic couplings, nucleophilic additions, and catalytic borylation of arenes.[10] Preorganized ditopic boranes enable CO2 activation in the presence of tBu3P,[11] and 1e- and 2e- trapping.[12] Ditopic boranes of linkages B(Cn)B (n>1) have been exploited for anion recognition,[13] hydrazine capture,[14] and olefin polymerization cocatalysts.[15] Despite the versatility of ditopic boranes, their extension to cooperative substrate binding/functionalization is under-explored, and they have never been incorporated into a ligand scaffold for this purpose. Herein, we report the synthesis of a ligand platform containing bifurcated second-sphere Lewis acids, evaluate the additive Lewis acidity and selective substrate binding, and describe their associated metal complexes as well as subsequent reactivity toward small molecules.
Figure 1.
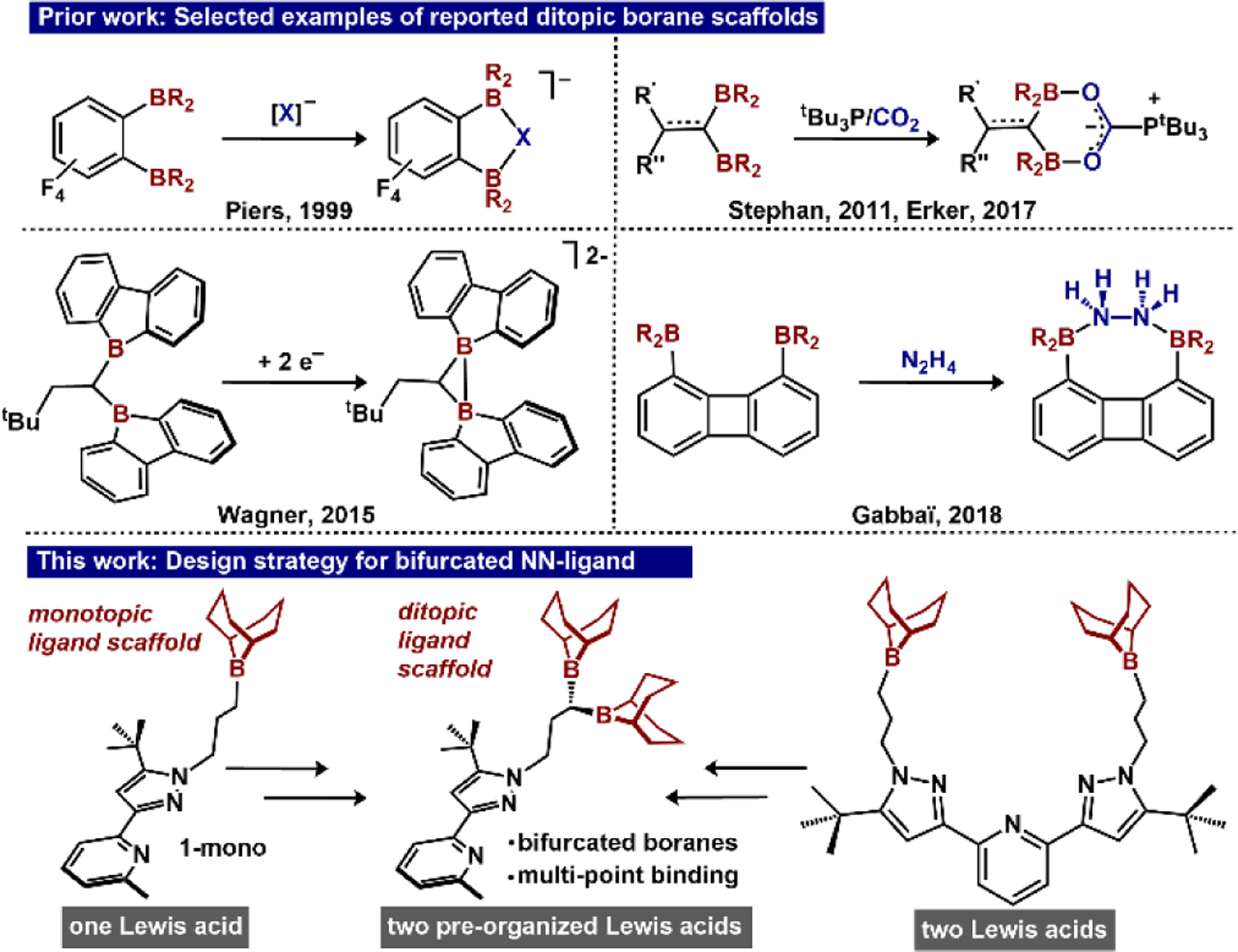
Selected examples of known ditopic boranes,[11, 12b, 14, 15e] and design strategy outlining the ditopic ligand used in this work from prior work using a bidentate ligand with single second-sphere Lewis acid,[9a-c] and a tridentate ligand with two Lewis acids,[4, 9d, 9e]
We targeted the diborane-appended pyridine pyrazole ligand through alkyne double hydroboration. The key synthon, a progaryl-appended ligand (propargylNNtBu) was prepared in one step using a modification of a route previously used by our group for a related single borane Lewis acid.[9b] Addition of two equivalents of 9-borabicyclo[3.3.1]nonane (9-BBN) to propargylNNtBu at 55 °C in C6H6 solvent resulted in the double anti-Markovnikov hydroboration product to afford ((BBN)2NNtBu) (1). Despite the presence of Lewis basic pyridine and pyrazole groups, both boranes were unquenched, as assessed by a broad resonance at 89.3 ppm in the 11B NMR spectrum. This result contrasts with the monoborylated variant (1-mono) which features a quenched (intramolecular Lewis acid/base interaction) borane (11B = 39.2 ppm): a difference we attribute to an increased steric profile of the Lewis acids. We found that the outcome of the hydroboration reaction was influenced by whether the ligand was first metalated. When (propargylNNtBu) was combined with MBr2 (M = Fe or Zn), forming ((propargyl)NNtBu)MBr2 (Figure 2a), and then treated with 2 equivalents 9-BBN, the alkyne underwent a single hydroboration as established by single crystal X-ray diffraction (XRD) (M = Fe) and 1H NMR spectroscopy (M = Zn, Fe) to generate the hydroborated alkene complexes ((ene)NNtBu)MBr2 (Figure 2).[16]
Figure 2.
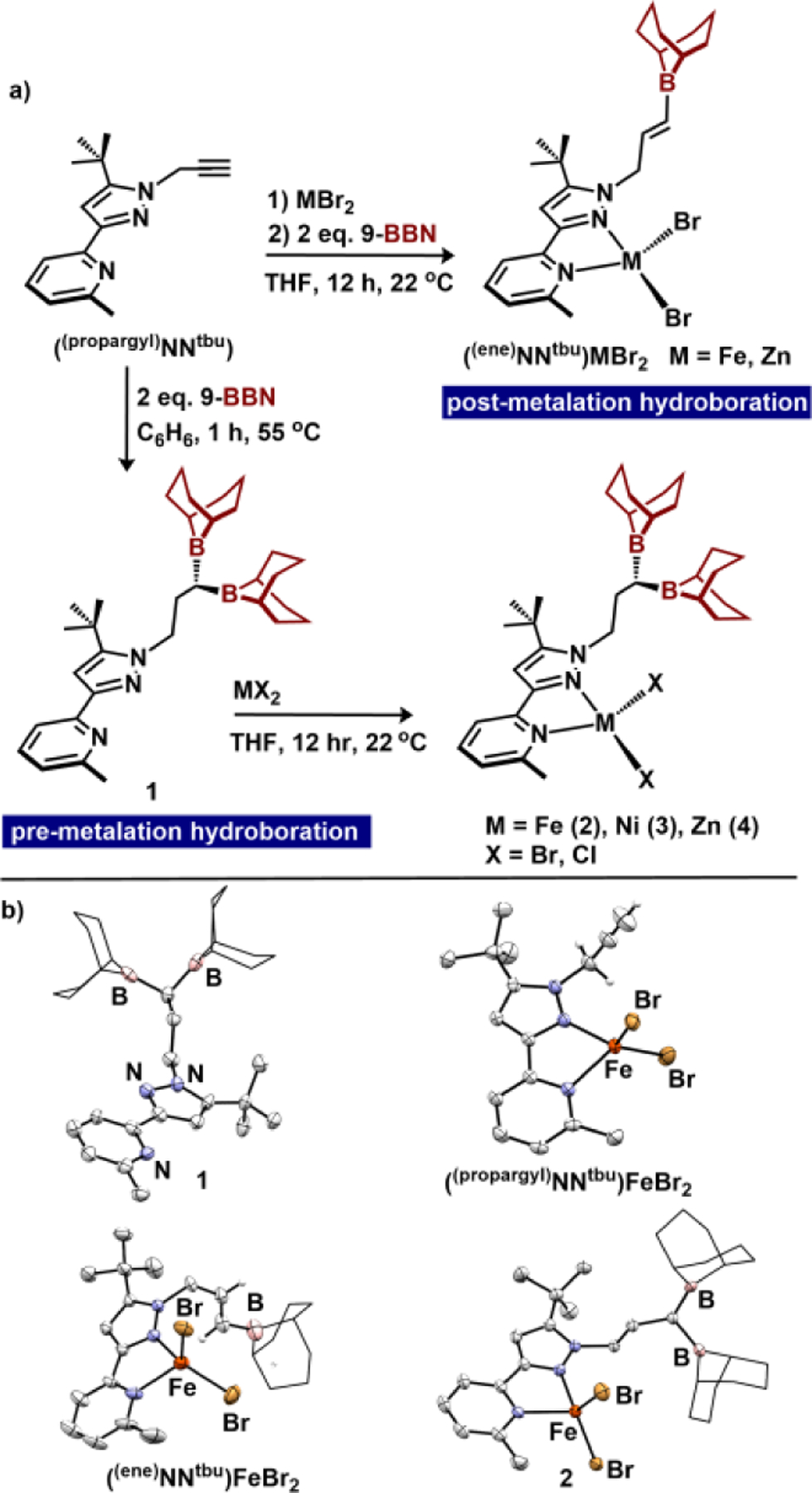
a) Synthesis of (ene)BBNtBuFeBr2 via post-metalation hydroboration and synthesis of 1 and 2–4 via pre-metalation hydroboration b) molecular structures (50% probability ellipsoids) of 1, (propargyl)BBNtBuFeBr2, (ene)BBNtBuFeBr2, and 2. H-atoms excluding (ene) and (propargyl) groups are omitted, and the 9-BBN substituents are displayed in wireframe for improved clarity.
To assess the competency of 1 to engage in bidentate binding to metal centers, we targeted metalation of Fe, Ni and Zn. Metalation of 1 with FeBr2, (DME)NiBr2, or ZnX2 (X = Cl, Br) in THF solvent afforded the respective divalent metal complexes in high yields (Figure 2a). Complex ((BBN)2NNtBu)FeBr2 (2) is high-spin (μB = 5.4, THF, 22 °C) and displays solution Cs symmetry with 1H NMR resonances between −27 and +53 ppm. 2 exhibits a reversible reduction wave in the cyclic voltammogram at −2.23 V (vs. Fc/Fc+; 0.1 M [Bu4N][PF6] in THF). This potential is cathodically shifted by 110 mV compared to singly borylated ((BBN)NNtBu)FeBr2, consistent with modest electronic differences between the two ligands. Complex ((BBN)2NNtBu)NiBr2 (3) is high-spin (μB = 2.9, THF, 22 °C), and replacement of Fe with Ni results in an 880 mV anodic shift of the M(II)/M(I) redox couple. Finally, we prepared the Zn complexes ((BBN)2NNtBu)ZnX2 (4-X; X=Br, Cl) to examine the system in the absence of a redox-active metal. The molecular structures of complexes 2–4 were established using single crystal XRD. Each isostructural complex displays tetrahedral geometry about the metal center (τ4 = 0.84–0.89), and the secondary sphere diboranes are geometrically unperturbed after metalation. The bifurcated Lewis acids exhibit average B-B bond distances of 2.486 Å, with an average ΣB = 359.41°, indicative of unquenched, trigonal planar boranes.
To assess the ability of 1 to serve as a ditopic acid and exert a Lewis acid additive effect compared to 1-mono (Figure 1), we evaluated its Lewis acidity and substrate binding properties. Although Brønsted acidity is routinely assessed by pKa values, Lewis acidity is substrate dependent, and defined against a reference Lewis base (e.g. Fˉ, O=PEt3, Hˉ).[17] One of most commonly used methods to evaluate Lewis acidity is measurement of 31P NMR chemical shifts of O=PEt3/Lewis acid adducts that are transformed into an acceptor number (AN) (Gutmann-Beckett).[17a] We obtained opposing values when we used this method for a mono- and double-BBN containing ligand. Although 1-mono provided an acceptor number (AN) of 23.8 (consistent with similar 9-BBN boranes),[9a] the AN of 1 was significantly lower and on par with that of solvent (5.9). Because a requirement of obtaining high ANs is formation of a strong acid/base adduct we attribute the low AN of 1 to the large steric profile of both –BBN groups, which we propose impedes OPEt3 adduct formation. To overcome these steric constraints, we assessed Lewis acidity using another commonly used method: fluoride ion affinity (FIA). The low steric profile of Fˉ allows binding in the bifurcated pocket of 1; however, 1-mono exhibited a larger FIA than 1 by 7.00 kcal/mol. We attribute the lower FIA of 1 to a geometric mismatch between the 1,1-diborylalkane empty p-orbitals and Fˉ lone pairs.
To assess Lewis acidity using a base with no orbital directionality, we calculated the hydride ion affinities (HIA) of 1 and 1-mono. The size and spherical electron distribution of Hˉ mitigates both issues encountered using either the Guttman-Beckett method or FIA as metrical parameters for evaluating Lewis acidity. We found that the HIA of 1 is 15.7 kcal/mol greater than that of 1-mono, implicating an additive effect exerted by the two second-sphere boranes (Figure 3c). For context, the 15.7 kcal/mol difference is similar to the hydride affinity difference between BH3 and BEt3.[18] Overall, these results underpin the complexity of Lewis acidity determination for gem-ditopic boranes, and suggest HIA is one of the few methods that allows direct Lewis acidity quantification across mono and ditopic acids.
Figure 3.
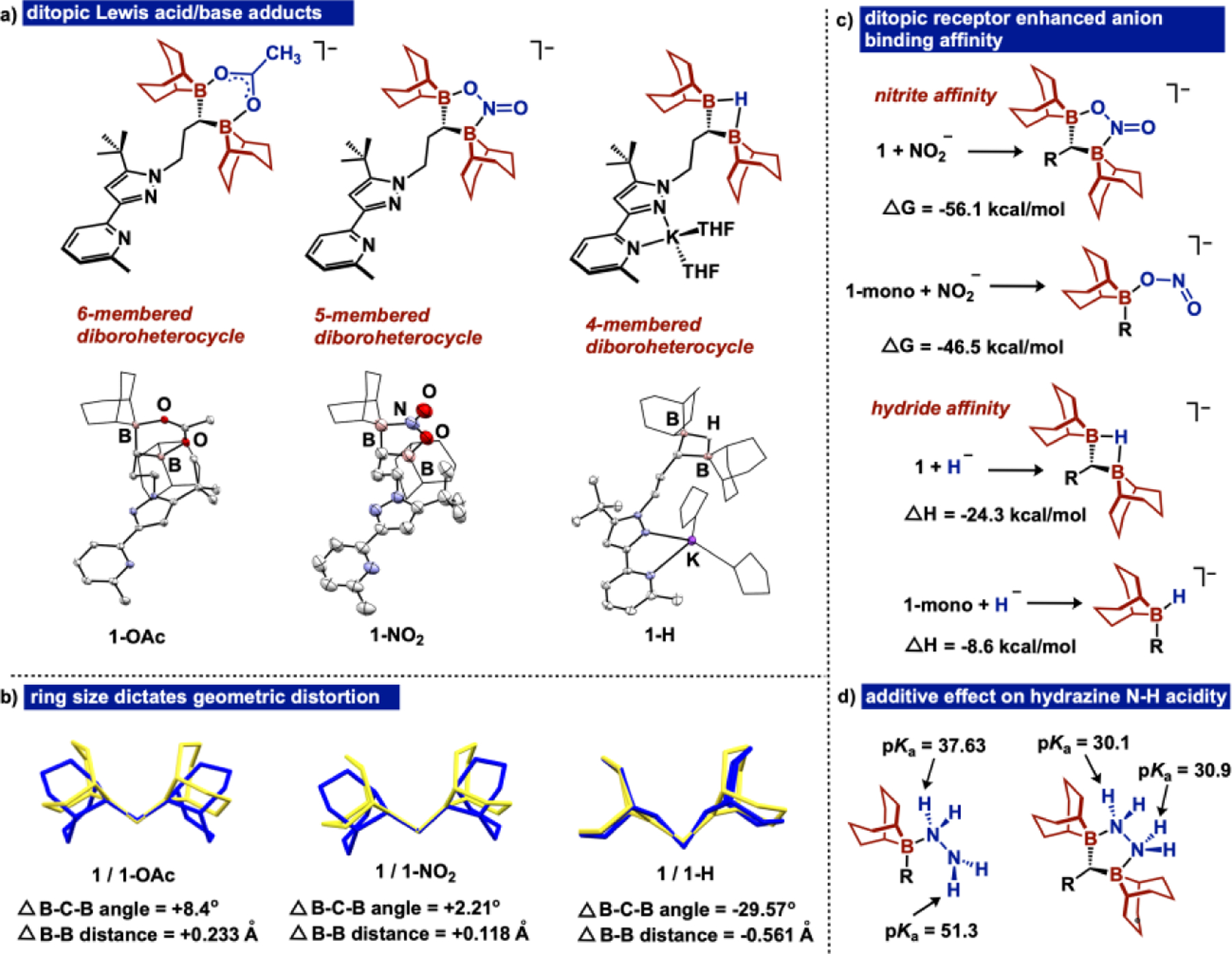
a) Anion capture mediated by 1 yielding ditopic boron containing heterocyles 1-OAc, 1-NO2, and 1-H and their respective molecular structures b) Comparison vs. free ligand (yellow) of distortion for ditopic borane unit with each heterocycle (blue) c) Comparative thermodynamics for nitrite and hydride capture for 1 and 1-mono and d) Comparative pKa values for hydrazine N-H protons. R represents the remainder of free ligand. Thermal ellipsoids displayed at 50% probability. H-atoms and counterions (in 1-OAc and 1-NO2) are omitted, and the 9-BBN substituents and THF ligands in 1-H are displayed in wireframe for improved clarity.
Due to the high HIA of 1, we targeted a 4-membered ring using hydride as a single atom bridge. Addition of 1 equiv. KEt3BH to 1 in THF generates a new species consistent with hydride adduct formation by 1H and 11B NMR spectroscopy.[19] The molecular structure of 1-H (Fig 3a) confirms ditopic hydride capture. In the absence of 18-crown-6, the bidentate NN binding pocket sequesters the K+ ion.
After assessing the Lewis acidity and HIA of 1, we targeted adduct formation to evaluate the geometric preference of the ditopic diborane. We chose acetate as the ditopic Lewis base to first address the ability of 1 to form a 6-membered diboroheterocyle. Addition of KOAc to 1 in the presence of 1 equiv 18-crown-6 in THF solvent cleanly formed a new species (1-OAc), as indicated by 1H and 11B NMR spectroscopy. The molecular structure (Figure 3a) of 1-OAc establishes κ2-acetate capture, forming a 6-membered ring. We subsequently assessed binding with nitrite, which can coordinate either κ2-N,O to form a 5-membered ring or κ2-O,O to form a 6-membered ring. Upon addition of NaNO2, nitrite binding to 1 occurs within 1 hour in THF at room temperature, and despite the oxophilicity of boron, the molecular structure of 1-NO2 (Figure 3a) indicates a κ2-N,O coordination mode of nitrite. The structurally characterized isomer was confirmed as the global minimum by density functional theory (DFT) (B3LYP-D3BJ/6–311g(d,p)) with κ2-N,O coordination favored over κ2-O,O by 4.7 kcal/mol.
Evaluation of the structural metrics of the 4-, 5-, and 6-membered diboroheterocycles (Figure 3b) indicates that the 2-atom bridge (nitrite) minimizes structural distortion of the ditopic unit when compared to 1, exhibiting a slightly elongated B-B distance (+0.118 Å) and widening of the B-C-B angle (2.21°). The 6-membered ring in 1-OAc affects B-B lengthening (+0.233 Å) and B-C-B angle widening (+8.4°) to a greater extent than 1-NO2. To accommodate a single atom bridge in 1-H, the ditopic borane unit is significantly distorted compared to 1, exemplifying B-B distance shortening (−0.561 Å) and considerable B-C-B angle contraction (−29.57°). To establish the extent to which the chelate effect of ditopic borane Lewis acid promoted adduct formation with ditopic Lewis bases, we used DFT to evaluate the thermodynamics for formation of both the acetate and nitrite adducts with 1 vs. 2 second-sphere boranes (Figure 3c). Notably, the formation of adducts with ditopic acids are more exergonic (compared to monotopic counterparts) by 9.6 kcal/mol for nitrite and 12.1 kcal/mol for acetate.
To experimentally corroborate the 15.7 kcal/mol difference in HIA between 1 and 1-mono, we executed both hydride competition and hydride transfer experiments. Addition of 1 equiv. of KEt3BH to a −78 °C THF solution containing 1 equiv. of 1 and 1-mono selectively generates 1-H with unreacted 1-mono by 1H NMR spectroscopy. Further, addition of an isolated sample of monoborylated hydride 1-mono-H to 1 in THF at 22 °C generates 1-H and 1-mono, substantiating the calculated HIA and implicating a low kinetic barrier for hydride transfer. To experimentally validate our hypothesis that pre-organized ditopic boranes can selectively coordinate ditopic Lewis bases through multi-point binding, we performed competition experiments with nitrite. Addition of 1 equiv. NaNO2 to a THF solution containing 18-crown-6 with 1 equiv. of 1 and 1-mono resulted in a color change to yellow, characteristic of 1-NO2. The 1H NMR spectrum collected after 1 hour confirmed selective capture of NO2ˉ by 1, rather than 1-mono. To assess the ability of the ditopic borane to selectively coordinate a dibasic substrate of higher Lewis basicity, we executed an analogous competition experiment with hydrazine (N2H4). Titration of N2H4 (two 0.5 equiv. intervals) to a THF solution containing 1 equiv. of both 1 and 1-mono selectively generates 1-N2H4 (Figure 4) with 1-mono unchanged as judged by 1H NMR spectroscopy when integrated against a (SiMe3)2O internal standard. To ensure that the self-quenching of 1-mono was not affecting the competition experiment, we executed the same experiment with 4 and the monoborylated analogue 4-mono. Similar to the free ligand, N2H4 selectively coordinates the ditopic receptor in 4. The calculated DFT thermodynamics and competition experiments confirm a selective preference of ditopic Lewis bases to form chelates with the ditopic Lewis acids, compared to monotopic analogues.
Figure 4.
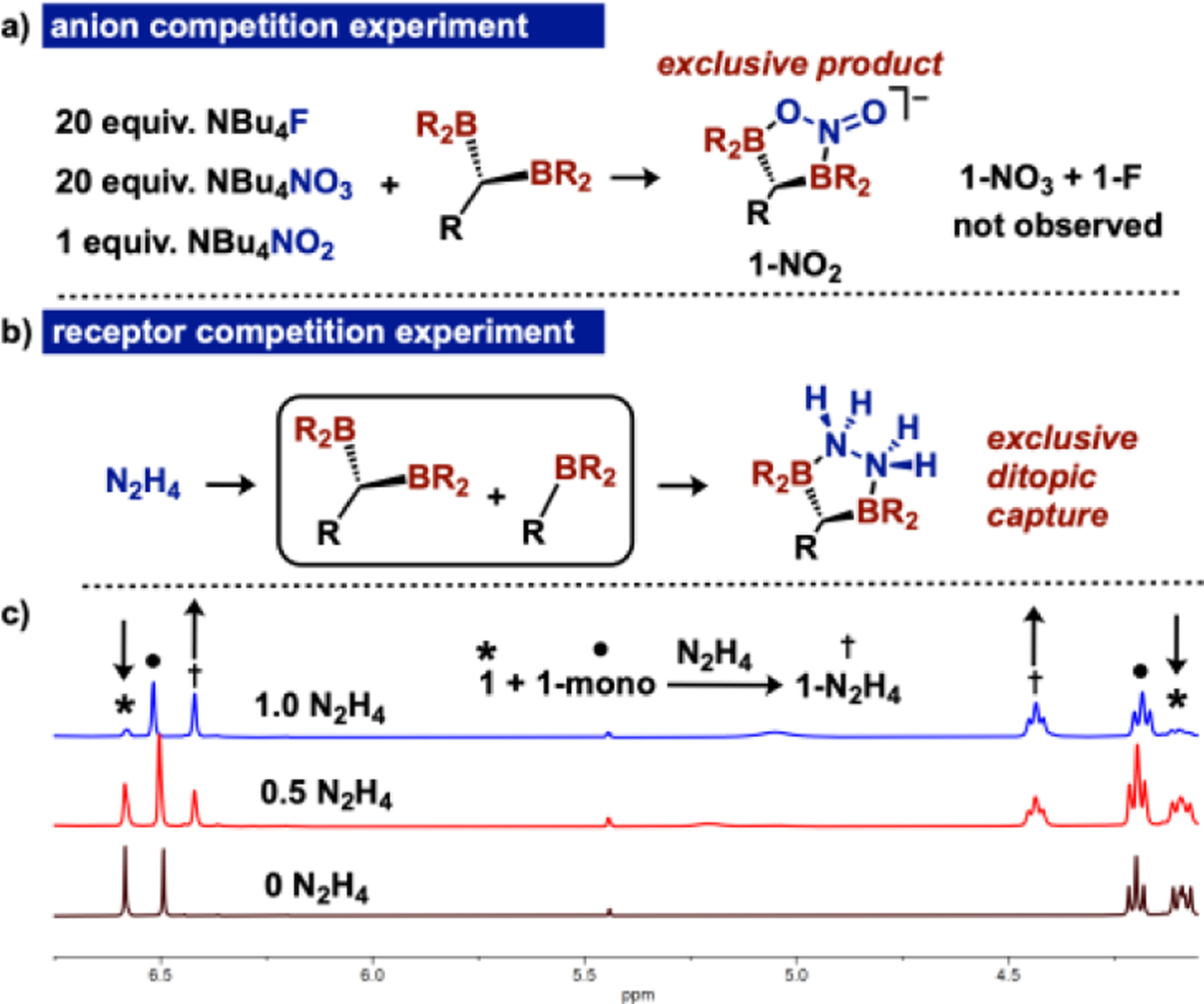
Anion competition reaction showing selective nitrite capture by 1 in the presence of fluoride and nitrate (top), selectivity of hydrazine binding to 1 in the presence of 1-mono (middle) and 1H NMR of hydrazine competition experiment (bottom) (N-CH2 and pyz-CH shown). BR2 = 9-BBN and R represents the remainder of the free ligand.
To evaluate the extent to which 1 could be used for selective anion recognition, we performed an anion competition experiment. In contrast to ortho-phenylene and naphthalene based ditopic receptors, which have been reported as Fˉ sensors,[13] 1 exhibits a distinct selectivity preference for nitrite. Addition of a clear THF solution containing a mixture of 20 equiv. Fˉ, 20 equiv. NO3ˉ, and 1 equiv. NO2ˉ (as [NBu4]+ salts) to a THF solution of 1 resulted in a color change to yellow. 1H NMR spectroscopy confirmed a single product: 1-NO2 (Figure 4).[20] This experiment indicates high NO2ˉ affinity of 1, even in the presence of anions that typically bind tightly to boranes, such as Fˉ. Overall, these results indicate that 1,1-diborylalkanes can be used as chemoselective molecular sensors for nitrite, which contrasts with the high Fˉ affinity of known ditopic borane receptors.
Upon establishing the viability of selective substrate capture using 1, we examined the extent to which ditopic boranes could enable binding of reactive substrates adjacent to a metal site that would otherwise be unstable. Our group previously reported N2H4 Lewis acid/base adducts using Fe and Zn,[4a] and related work by the Gabbaï group showed μ1,2-hydrazine complexation using diboranes with large bite angles (B-B distances > 4.5 Å).[14] Treating 2–4 with anhydrous N2H4 in THF proceeded smoothly to furnish the hydrazine adducts, 2-N2H4, 3-N2H4, and 4-N2H4, respectively (Figure 5). The IR spectra contain two νNH bands between 3206 and 3296 cm−1, and for 4-N2H4, the 1H NMR spectrum features a broad hydrazine N-H resonance at 6.44 ppm, while the 11B NMR spectrum indicates equivalent tetrahedral boron atoms (1.3 ppm). The μ1,2 binding mode of hydrazine was unequivocally confirmed through SCXRD, with the molecular structures of the hydrazine adducts exhibiting isostructural second-sphere hydrazine coordination.
Figure 5.
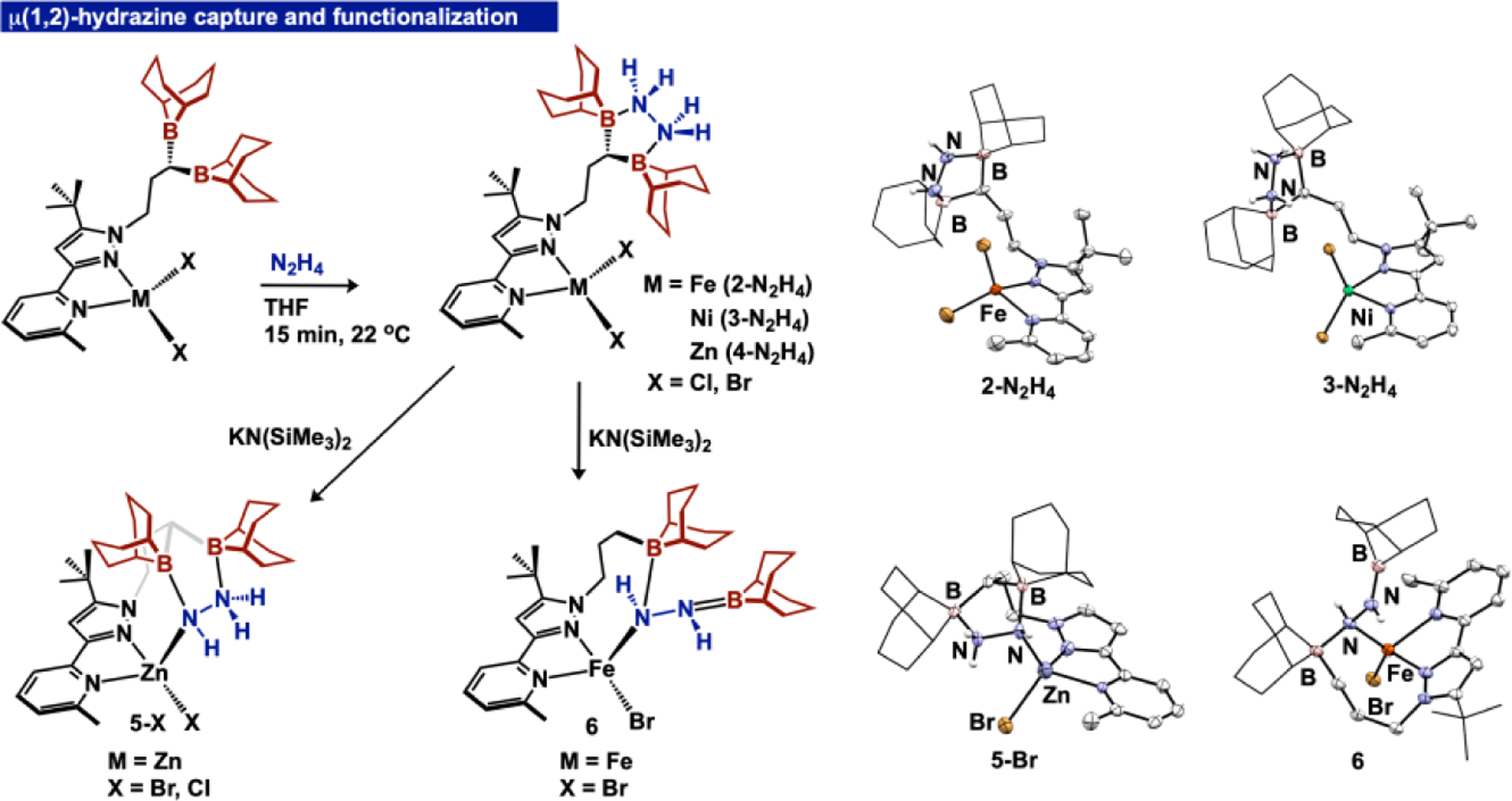
μ(1,2)-hydrazine capture using 2–4 to yield 2–4-N2H4 and hydrazine reactivity of 2-N2H4 and 4-N2H4 to generate hydrazido and diazene complexes 5-X and 6 (X = Br, Cl). Molecular structures of 2-N2H4, 3-N2H4, 5-Br and 6 (right). Thermal ellipsoids displayed at 50% probability. Non-diazene or hydrazine H-atoms are omitted, and the 9-BBN substituents are displayed in wireframe for improved clarity.
To further evaluate the additive effect of the ditopic boranes, we computed the pKa values of the N-H protons of the hydrazine adduct 1-N2H4 (Figure 3d). The N-H protons of the hydrazine unit are more greatly acidified in 1-N2H4 (pKa units = 30.9 and 30.1) compared to the monoborylated hydrazine adduct (pKa units = 37.63 and 51.3). These data support an additive Lewis acidity effect for coordinated substrates to the ditopic borane unit, which complements prior reports for an additive effect through boron/zinc cooperativity.[9b]
After verifying the additive acidification of the N-H protons of chelated N2H4 adducts, we attempted selective deprotonation of ditopic borane hydrazine adducts. Addition of KN(SiMe3)2 to 1-N2H4 generates a mixture of products; however, deprotonation of metalated hydrazine adducts 2-N2H4 and 4-N2H4 afforded a distinct outcome. Dropwise addition of a freshly thawed KN(SiMe3)2 THF solution to a cold solution of 4-N2H4 afforded a white precipitate. The 1H NMR and 11B NMR spectra revealed reduced symmetry of the diborane tether, and the IR spectrum (KBr) exhibited νNH bands at 3292 and 3215 cm−1, slightly shifted from 4-N2H4 (3288 and 3228 cm−1). Two independent XRD experiments established connectivity and assignment of the hydrazido complexes ((BBN)2NNtBu)ZnX(N2H3) (5-X, X = Br, Cl) (Figure 5), with the molecular structure of 5-Br (Figure 5) showing retention of bifurcated acid/base binding with the N2H3ˉ ligand. We propose the difference in reaction outcomes between 1-N2H4 and 4-N2H4 is dictated by the placement of the generated highly reactive hydrazido unit, which in the latter case is directed toward the metal center, enabling cooperative stabilization by the metal and the ditopic second-sphere boranes. Complexes 5-Br and 5-Cl represent the first ditopic Lewis acid stabilized hydrazido ligands, an extension of prior work which stabilizes the Zn-N2H3 moiety through monotopic borane binding.[9a]
Replacement of Zn with Fe resulted in a distinct outcome. Addition of KN(SiMe3)2 to 2-N2H4 afforded a yellow compound whose solution structure (NMR) indicated reduced symmetry. The νNH bands in the IR spectrum appear at 3224, 3290 cm−1, and are shifted from 2-N2H4 (3297, 3224 cm−1). The molecular structure established through SCXRD revealed that the ligand underwent 9-BBN migration to cap the Fe-N2H2 diazene unit in 6 (Figure 5), with proton migration to the tether arm. The tethered 9-BBN interacts with the Fe coordinated α-N (B-N = 1.635(3) Å), while the β-N is π-bound to the capped 9-BBN (B-N = 1.384(3) Å). The N-N distance (1.449(2) Å) is elongated compared to a similar π-delocalized Fe-N2H2 unit.[21] For electronic structure elucidation, we modelled 6 using DFT (See SI), and located the B-N π-bond in the HOMO-1. A comparison of the relative energies of isomeric 6 and the unobserved Fe-hydrazido complex indicate that 6 is more stable by 28.96 kcal/mol. The divergent product distribution for reactivity of 2-N2H4 and 4-N2H4 suggests that the incorporation of a redox-active metal unlocks mechanistic pathways for ligand rearrangement that are not viable with zinc.
In conclusion, we report the synthesis of the first ligand with ditopic Lewis acids in the outer sphere. Metalation with iron, nickel, and zinc yields stable, tetrahedral complexes. The second-sphere mediates selective substrate capture including hydrazine, and functionalization of hydrazine leads to a hydrazido ligand on zinc and a diazene ligand on iron. An additive effect of the ditopic boranes was verified experimentally and computationally for substrate capture/activation. Given the appropriate substrate and synthetic conditions, 1 represents an attractive new class of ligands featuring bifurcated Lewis acids that can be exploited for small molecule activation.
Supplementary Material
Acknowledgements
This work was supported by the NIH (1R35GM13660–01). N. K. S. is a Camille Dreyfus Teacher-Scholar. X-ray diffraction equipment at Purdue University was supported by the National Science Foundation through the Major Research Instrumentation Program under Grant No. CHE 1625543. The authors thank Fengrui Qu for SCXRD data collection of 1, 1-OAc, 1-NO2, 1-H, 2-Cl, 3, 3-N2H4, 4, 4-N2H4, 5-Cl, 5-Br, and 6.
Footnotes
Supporting information for this article is given via a link at the end of the document.
Notes and references
- [1].a) Borovik AS, Acc. Chem. Res 2005, 38, 54–61; [DOI] [PubMed] [Google Scholar]; b) Spatzal T, Perez KA, Einsle O, Howard JB, Rees DC, Science 2014, 345, 1620–1623; [DOI] [PMC free article] [PubMed] [Google Scholar]; c) Sippel D, Rohde M, Netzer J, Trncik C, Gies J, Grunau K, Djurdjevic I, Decamps L, Andrade SLA, Einsle O, Science 2018, 359, 1484–1489; [DOI] [PubMed] [Google Scholar]; d) Van Stappen C, Deng Y, Liu Y, Heidari H, Wang J-X, Zhou Y, Ledray AP, Lu Y, Chem. Rev 2022, 122, 11974–12045; [DOI] [PMC free article] [PubMed] [Google Scholar]; e) Stripp ST, Duffus BR, Fourmond V, Léger C, Leimkühler S, Hirota S, Hu Y, Jasniewski A, Ogata H, Ribbe MW, Chem. Rev 2022, 122, 11900–11973. [DOI] [PMC free article] [PubMed] [Google Scholar]
- [2].a) Dos Santos PC, Igarashi RY, Lee H-I, Hoffman BM, Seefeldt LC, Dean DR, Acc. Chem. Res 2005, 38, 208–214; [DOI] [PubMed] [Google Scholar]; b) Zhang X, Houk KN, Acc. Chem. Res 2005, 38, 379–385; [DOI] [PubMed] [Google Scholar]; c) Prejanò M, Medina FE, Ramos MJ, Russo N, Fernandes PA, Marino T, ACS Catal 2020, 10, 2872–2881; [DOI] [PMC free article] [PubMed] [Google Scholar]; d) Warshel A, Sharma PK, Kato M, Xiang Y, Liu H, Olsson MHM, Chem. Rev 2006, 106, 3210–3235. [DOI] [PubMed] [Google Scholar]
- [3].Drover MW, Chem. Soc. Rev 2022, 51, 1861–1880. [DOI] [PubMed] [Google Scholar]
- [4].a) Kiernicki JJ, Zeller M, Szymczak NK, J. Am. Chem. Soc 2017, 139, 18194–18197; [DOI] [PMC free article] [PubMed] [Google Scholar]; b) Kiernicki JJ, Shanahan JP, Zeller M, Szymczak NK, Chem. Sci 2019, 10, 5539–5545. [DOI] [PMC free article] [PubMed] [Google Scholar]
- [5].Miller AJM, Labinger JA, Bercaw JE, J. Am. Chem. Soc 2008, 130, 11874–11875. [DOI] [PubMed] [Google Scholar]
- [6].a) Cowie BE, Emslie DJH, Organometallics 2015, 34, 2737–2746; [Google Scholar]; b) Cowie BE, Emslie DJH, Can. J. Chem 2018, 96, 484–491. [Google Scholar]
- [7].Zurakowski JA, Austen BJH, Dufour MC, Spasyuk DM, Nelson DJ, Drover MW, Chem. Eur. J 2021, 27, 16021–16027. [DOI] [PubMed] [Google Scholar]
- [8].Zurakowski JA, Bhattacharyya M, Spasyuk DM, Drover MW, Inorg. Chem 2021, 60, 37–41. [DOI] [PubMed] [Google Scholar]
- [9].a) Kiernicki JJ, Norwine EE, Lovasz MA, Zeller M, Szymczak NK, Chem. Commun 2020, 56, 13105–13108; [DOI] [PMC free article] [PubMed] [Google Scholar]; b) Kiernicki JJ, Norwine EE, Zeller M, Szymczak NK, Chem. Commun 2019, 55, 11896–11899; [DOI] [PMC free article] [PubMed] [Google Scholar]; c) Kiernicki JJ, Norwine EE, Zeller M, Szymczak NK, Inorg. Chem 2021, 60, 13806–13810; [DOI] [PubMed] [Google Scholar]; d) Kiernicki JJ, Zeller M, Szymczak NK, Inorg. Chem 2019, 58, 1147–1154; [DOI] [PMC free article] [PubMed] [Google Scholar]; e) Kiernicki JJ, Zeller M, Szymczak NK, Inorg. Chem 2020, 59, 9279–9286; [DOI] [PMC free article] [PubMed] [Google Scholar]; f) Norwine EE, Kiernicki JJ, Zeller M, Szymczak NK, J. Am. Chem. Soc 2022, 144, 15038–15046; [DOI] [PMC free article] [PubMed] [Google Scholar]; g) Wang B, Seo CSG, Zhang C, Chu J, Szymczak NK, J. Am. Chem. Soc 2022, 144, 15793–15802. [DOI] [PMC free article] [PubMed] [Google Scholar]
- [10].a) Miralles N, Maza RJ, Fernández E, Adv. Synth. Catal 2018,360, 1306–1327; [Google Scholar]; b) Nallagonda R, Padala K, Masarwa A, Org. Bioml. Chem 2018, 16, 1050–1064; [DOI] [PubMed] [Google Scholar]; c) Liu Y-L, Kehr G, Daniliuc CG, Erker G, Chem. Eur. J 2017, 23, 12141–12144. [DOI] [PubMed] [Google Scholar]
- [11].a) Zhao X, Stephan DW, Chem Commun 2011, 47, 1833–1835; [DOI] [PubMed] [Google Scholar]; b) Liu Y-L, Kehr G, Daniliuc CG, Erker G, Chem. Sci 2017, 8, 1097–1104. [DOI] [PMC free article] [PubMed] [Google Scholar]
- [12].a) Hoefelmeyer JD, Gabbaï FP, J. Am. Chem. Soc 2000, 122, 9054–9055; [Google Scholar]; b) Hübner A, Kaese T, Diefenbach M, Endeward B, Bolte M, Lerner H-W, Holthausen MC, Wagner M, J. Am. Chem. Soc 2015, 137, 3705–3714. [DOI] [PubMed] [Google Scholar]
- [13].a) Chen C-H, Gabbaï FP, Angew. Chem. Int. Ed 2018, 57, 521–525; [DOI] [PubMed] [Google Scholar]; b) Melaïmi M, Solé S, Chiu C-W, Wang H, Gabbaï FP, Inorg. Chem 2006, 45, 8136–8143; [DOI] [PubMed] [Google Scholar]; c) Solé S, Gabbaï FP, Chem. Commun 2004, 1284–1285. [DOI] [PubMed] [Google Scholar]
- [14].Chen C-H, Gabbaï FP, Chem. Sci 2018, 9, 6210–6218. [DOI] [PMC free article] [PubMed] [Google Scholar]
- [15].a) Chai J, Lewis SP, Collins S, Sciarone TJJ, Henderson LD, Chase PA, Irvine GJ, Piers WE, Elsegood MRJ, Clegg W, Organometallics 2007, 26, 5667–5679; [Google Scholar]; b) Köhler K, Piers WE, Jarvis AP, Xin S, Feng Y, Bravakis AM, Collins S, Clegg W, Yap GPA, Marder TB, Organometallics 1998, 17, 3557–3566; [Google Scholar]; c) Lewis SP, Henderson LD, Chandler BD, Parvez M, Piers WE, Collins S, J. Am. Chem. Soc 2005, 127, 46–47; [DOI] [PubMed] [Google Scholar]; d) Lewis SP, Taylor NJ, Piers WE, Collins S, J. Am. Chem. Soc 2003, 125, 14686–14687; [DOI] [PubMed] [Google Scholar]; e) Williams VC, Piers WE, Clegg W, Elsegood MRJ, Collins S, Marder TB, J. Am. Chem. Soc 1999, 121, 3244–3245. [Google Scholar]
- [16]. See supporting information Pg S14 for more details of single vs. double hydroboration.
- [17].a) Beckett MA, Strickland GC, Holland JR, Sukumar Varma K, Polymer 1996, 37, 4629–4631; [Google Scholar]; b) Lathem AP, Heiden ZM, Dalton Trans 2017, 46, 5976–5985; [DOI] [PubMed] [Google Scholar]; c) Müller LO, Himmel D, Stauffer J, Steinfeld G, Slattery J, Santiso-Quiñones G, Brecht V, Krossing I, Angew. Chem. Int. Ed 2008, 47, 7659–7663; [DOI] [PubMed] [Google Scholar]; d) Mallouk TE, Rosenthal GL, Mueller G, Brusasco R, Bartlett N, Inorg. Chem 1984, 23, 3167–3173. [Google Scholar]
- [18].Mock MT, Potter RG, Camaioni DM, Li J, Dougherty WG, Kassel WS, Twamley B, DuBois DL, J. Am. Chem. Soc 2009, 131, 14454–14465. [DOI] [PubMed] [Google Scholar]
- [19]. The 11B NMR and 1H NMR (hydride) signals observed are both broad singlets and show no B-H coupling, which is consistent with similar 9-BBN hydrides reported from our group (Refs. 9f and 4b).
- [20]. Free fluoride observed by 19F spectroscopy (see SI for more details)
- [21].Saouma CT, Kinney RA, Hoffman BM, Peters JC, Angew. Chem. Int. Ed 2011, 50, 3446–3449. [DOI] [PMC free article] [PubMed] [Google Scholar]
- [22]. Deposition numbers 2214449, 2214450, 2214451, 2214452, 2214453, 2214454, 2214455, 2214456, 2214457, 2214458, 2214459, 2214460, 2214461, 2214462, 2214463 contain the supplementary crystallographic data for this paper. These data are provided free of charge by the joint Cambridge Crystallographic Data Centre and Fachinformationszentrum Karlsruhe Access Structures service.
Associated Data
This section collects any data citations, data availability statements, or supplementary materials included in this article.