

Abstract
Background:
A recent study suggests that systemic hypoxemia in adult male mice can induce cardiac myocytes to proliferate. The goal of the present experiments was to confirm these results, provide new insights on the mechanisms that induce adult cardiomyocyte cell cycle re-entry, and to determine if hypoxemia also induces cardiomyocyte proliferation in female mice.
Methods:
EdU-containing mini pumps were implanted in 3-month-old, male and female C57BL/6 mice. Mice were placed in a hypoxia chamber and the oxygen was lowered by 1% every day for 14 days to reach 7% oxygen. The animals remained in 7% oxygen for 2 weeks before terminal studies. Myocyte proliferation was also studied with a mosaic analysis with double markers (MADM) mouse model.
Results:
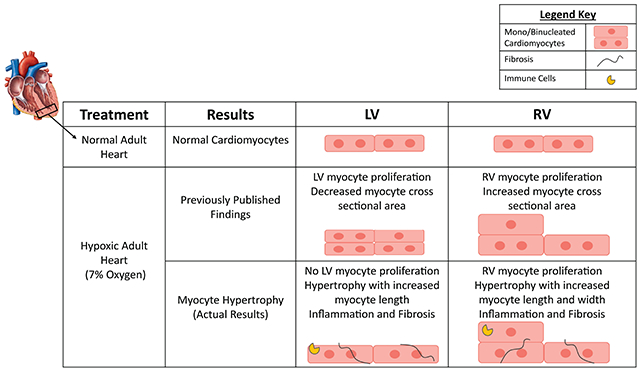
Hypoxia induced cardiac hypertrophy in both left ventricular (LV) and right ventricular (RV) myocytes, with LV myocytes lengthening and RV myocytes widening and lengthening. Hypoxia induced an increase (0.01±0.01% in normoxia to 0.11±0.09% in hypoxia) in the number of EdU+ RV cardiomyocytes, with no effect on LV myocytes in male C57BL/6 mice. Similar results were observed in female mice. Furthermore, in MADM mice, hypoxia induced a significant increase in RV myocyte proliferation (0.03±0.03% in normoxia to 0.32±0.15% in hypoxia of RFP+ myocytes), with no significant change in LV myocyte proliferation. RNA-sequencing showed upregulation of mitotic cell cycle genes and a downregulation of Cullin genes, that promote the G1 to S phase transition in hypoxic mice. There was significant proliferation of non-myocytes and mild cardiac fibrosis in hypoxic mice that did not disrupt cardiac function. Male and female mice exhibited similar gene expression following hypoxia.
Conclusion:
Systemic hypoxia induces a global hypertrophic stress response that was associated with increased RV proliferation, and while LV myocytes did not show increased proliferation, our results minimally confirm previous reports that hypoxia can induce cardiomyocyte cell cycle activity in vivo.
Keywords: Hypoxia, Cardiomyocyte Proliferation, Cardiomyocyte Cytokinesis, Cardiac Remodeling, Angiogenesis
Graphical Abstract
Introduction
Cardiovascular disease (CVD) remains as a prominent cause of death in the United States despite many advances1. Myocardial infarction (MI) can result from CVD. Cardiac injury caused by MI leads to the death of heart tissue, including cardiomyocytes2. The tissue death during and after MI leads to adverse structural and functional remodeling that culminates in reduced cardiac pump function3–5. Over time, these detrimental changes progress and lead to heart failure (HF) and premature mortality6, 7. Current treatments for MI include reperfusion therapy that can salvage some cardiomyocytes in the damaged area, but reperfusion injury can also cause some myocyte damage8. Other HF therapies largely treat neurohormonal imbalances secondary to HF but do not address the primary problem that cardiac myocytes have died after MI. True repair of the infarcted heart requires replacement of dead tissue, including their myocytes. Many recent studies have explored new approaches to repopulate the heart with new myocytes and supportive tissues9–11. These approaches have had marginal success12, 13.
Research on zebrafish and newts has shown that after injury pre-existing cardiomyocytes can re-enter the cell cycle and divide to repopulate the damaged region with new cardiac myocytes14–16. In mammals, myocyte proliferation is abundant during embryonic development and for up to 7 days after birth but declines thereafter to a very low rate17–20. It is clear that in adult mammals, cardiac myocyte regeneration is not a major contributor to post MI cardiac repair. Novel strategies to induce adult cardiac myocytes to reenter the cell cycle and form new cardiac muscle cells are being tested. These approaches include genetic manipulation of cell cycle genes and signaling pathways, alteration of the cardiomyocyte metabolism, and changes in environmental factors such as hypoxia21, 22.
A recent study showed that chronic hypoxemia in young adult male mice induces their cardiomyocytes to re-enter the cell cycle and proliferate23. The goals of our study are to first confirm previous results in male mice, to determine if similar effects are present in female mice, and then to gain more insight into the molecular bases for hypoxemia-induced adult cardiomyocyte proliferation. A novel aspect of this study was the utilization of Mosaic analysis with double markers (MADM) mice24 to confirm myocyte proliferation.
The effects of chronic hypoxia on male and female mice were determined. Structural changes and hypertrophy of the left ventricle (LV) and the right ventricle (RV) were observed. Chronic hypoxemia also caused cardiomyocyte hypertrophy which took a different form in the RV and LV, with LV myocytes having an increase in their length and RV myocytes exhibiting an increase in width and length. Myocyte proliferation induced by reducing inspired oxygen levels to 7% was measured with three independent techniques: direct myocyte counting, myocyte EdU incorporation, or with MADM mice. Comparison of EdU labeled myocytes undergoing DNA synthesis and single color labeled myocytes in MADM mice revealed that hypoxia did not induce significant increases in LV myocyte proliferation but did cause small but significant increases in RV myocyte proliferation. Similar results were observed in male and female mice. Hypoxemia also induced angiogenesis, fibrosis, apoptosis, and immune cell infiltration in the heart with proliferation of endothelial and immune cells, and fibroblast activation.
Collectively, our studies show that systemic hypoxemia induces a small increase in RV myocyte proliferation with no significant increase in LV myocyte proliferation. The hypoxia protocol used also caused modest cardiac fibrosis, myocyte apoptosis and immune cell infiltration consistent with dysfunctional remodeling.
Methods
Data Availability.
The data, materials, and methods used in this study to perform research will be available to any researcher for purposes of reproducing the results.
Data Summarry.
For detailed Materials and Methods, refer to the Online Data Supplement, and please see the Major Resources Table in the Supplemental Materials.
Briefly, all animal protocols were approved by the Institutional Animal Care and Use Committee at Temple University Lewis Katz School of Medicine. Male and female mice of C57BL/6J and MADM strains were purchased from Jackson Laboratory at 9-12 weeks of age. EdU-containing osmotic minipumps were implanted in all mice and tamoxifen was also administered to MADM mice as described. Mice were subjected to chronic hypoxia at 7% oxygen for 2 weeks, where oxygen was lowered by 1% daily from 20.9% to 7% oxygen for 2 weeks and then maintained at 7% oxygen for an additional 2 weeks. Echocardiography was used to measure systolic and diastolic structural and functional parameters before and after hypoxia treatment. Following tissue harvest, organ weight measurements, tissue processing, cardiomyocyte isolation, histology, immunostaining, and RNA sequencing were performed to determine cardiac hypertrophy, cardiomyocyte and non-myocyte cell proliferation, angiogenesis, fibrosis, immune cell infiltration, apoptosis, and differential gene expression in male and female hearts. Investigators were blinded to the treatment group of the animals during the study. Data are represented as mean±SD. For unpaired t-test and one-way ANOVA, Shapiro-Wilk test was used to test for Normality. Statistical analysis was performed using one-or two-way ANOVA followed by Turkey’s or Sidak’s multiple comparisons test, and t-test or Mann-Whitney test depending on the distribution of the data. All data were analyzed using GraphPad Prism software (Version 9.3.1, GraphPad Software Inc., San Diego, CA). All representative images chosen were the best quality images that represent the mean of the data.
Results
Systemic hypoxemia induces different types of cardiac myocyte hypertrophy in the left ventricle and right ventricle
Effects of Hypoxemia on adult male mice:
Adult male mice were subjected to chronic hypoxia of 7% oxygen for 2 weeks using an approach described previously23. Mice were placed in a hypoxia chamber and the oxygen partial pressure was lowered by 1% every day for 2 weeks until it reached 7%; Inspired oxygen then remained at 7% for an additional 2 weeks for chronic exposure (Figure 1A). Hypoxemia caused a 17% increase in heart weight (HW), an increase in heart weight to body weight (HW/BW), and a 17% increase in heart weight to tibia length (HW/TL) ratios (Figure 1B) documenting organ hypertrophy.
Figure 1. Chronic hypoxia induces different forms of LV and RV hypertrophy in male mice.
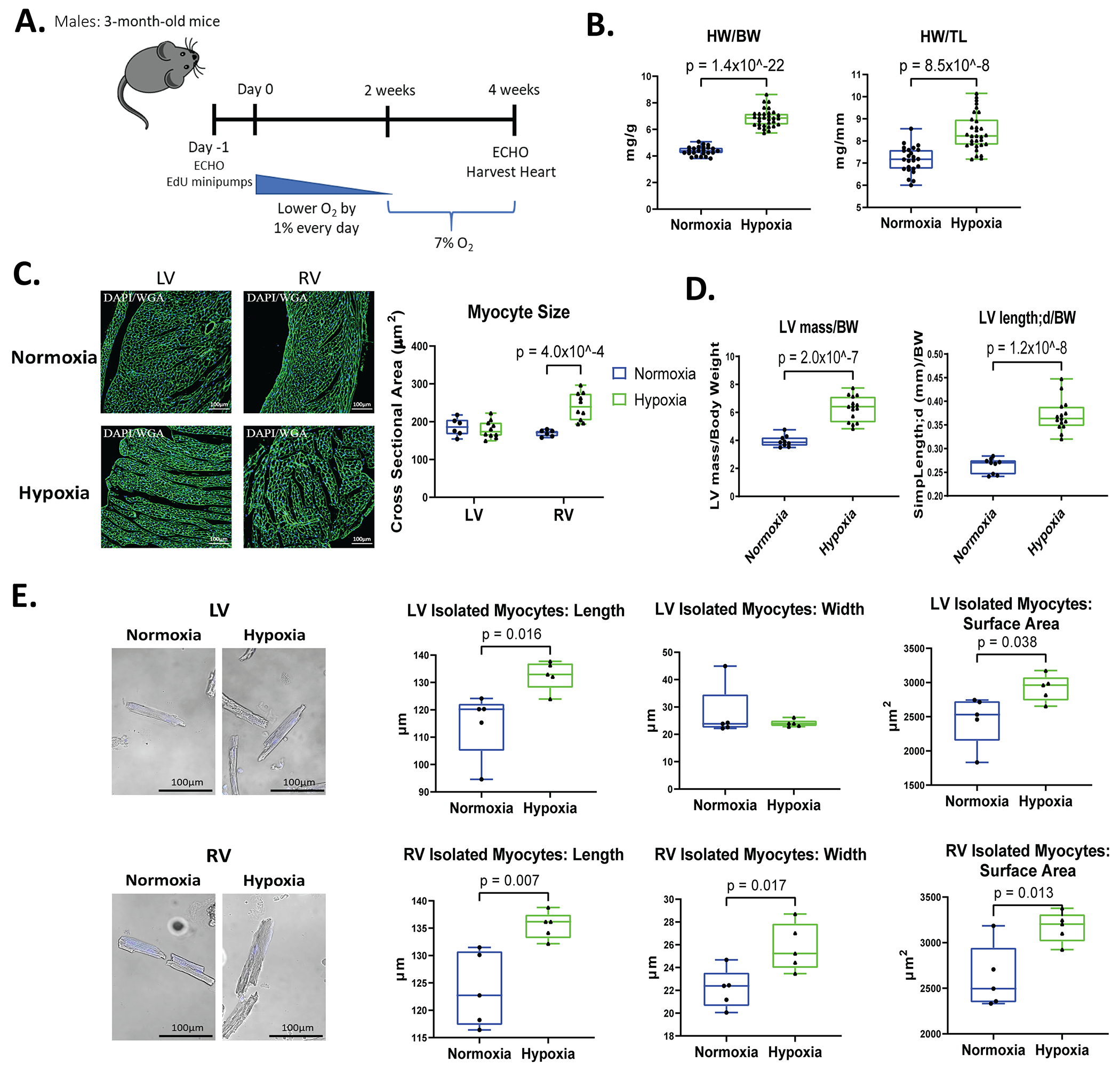
A: Schematic of hypoxia protocol in male mice with gradual reduction in oxygen levels to reach 7% for 2 weeks. B: Heart weight to body weight (HW/BW) and heart weight to tibia length (HW/TL) were measured at the time of harvest (Normoxia n=24, Hypoxia n=31). C: Myocyte size was determined from wheat germ agglutinin (WGA) staining of hearts and measurement of cross-sectional area of left ventricular (LV) and right ventricular (RV) myocytes with visible nuclei (normoxia n=6, hypoxia n=10). D: Echocardiography was performed during terminal studies. LV mass and the length of the LV measured by parasternal long axis (PSLAX) view relative to body weight was determined. E: Cardiac myocytes were isolated from the LV and RV of the heart and stained with DAPI to label nuclei. Myocyte length, width, and surface area (SA) were measured in at least 100 ventricular cardiomyocytes of the LV and RV from each mouse (n=5). Data represented as mean±SD.
Mice subjected to hypoxemia consumed less food and lost body weight during their 4-week exposure to low oxygen levels when compared to normoxia controls (Figure S1A). In order to eliminate the effects of weight loss in hypoxia mice on heart size (HW/BW), we also tested the effects of hypoxemia against a food restricted control group which were fed the same amount of food that the hypoxia mice consumed, as has been reported in previous studies23. The food restricted controls consumed the same average amount of food as the hypoxia mice during the 4-week study and exhibited a decrease in body weight (Figure S1B). Hypoxia mice still had a significant increase in HW/BW and HW/TL ratios when compared to food restricted controls (Figure S1C).
Cardiomyocyte cross sectional area (CSA) was measured in heart tissue from all groups. CSA was significantly increased in right ventricular (RV) myocytes of hypoxia mice compared to normoxia hearts, but the left ventricular (LV) myocyte CSA of the hypoxic hearts was not changed significantly (Figure 1C). LV mass measured by echocardiography was significantly increased in hypoxic mice, and the length of the LV measured from parasternal long axis (PSLAX) view was larger in hypoxia mice when normalized to body weight and compared to normoxia mice (Figure 1D).
Myocyte length, width, and surface area were measured in cardiomyocytes isolated from hypoxia and normoxia hearts. LV myocytes from hypoxemia hearts were significantly longer and had a greater surface area than LV myocytes from normoxia hearts. RV myocytes from hypoxia hearts had significant increases in cell width, length, and surface area when compared to normoxia hearts (Figure 1E). Overall, hypoxia induced an increase in myocyte size, as assessed by myocyte surface area, with an 18.84% increase in LV myocyte size and 21.23% increase in RV myocyte size.
Collectively, these data show that hypoxemia induces cardiac hypertrophy, but the nature of the hypertrophy is different in LV and RV cardiac myocytes. These differences likely result from the effects of hypoxia to increase cardiac work as evidenced by the increase in cardiac output (Figure 2A) that affects both chambers and from the pressure overload on the RV secondary to the increased pulmonary vascular resistance that is known to be caused by pulmonary hypoxemia25–27.
Figure 2. Male mice have normal cardiac function, no changes in the number of LV and RV myocytes, and only RV cardiomyocytes have increased EdU labeling after hypoxia.
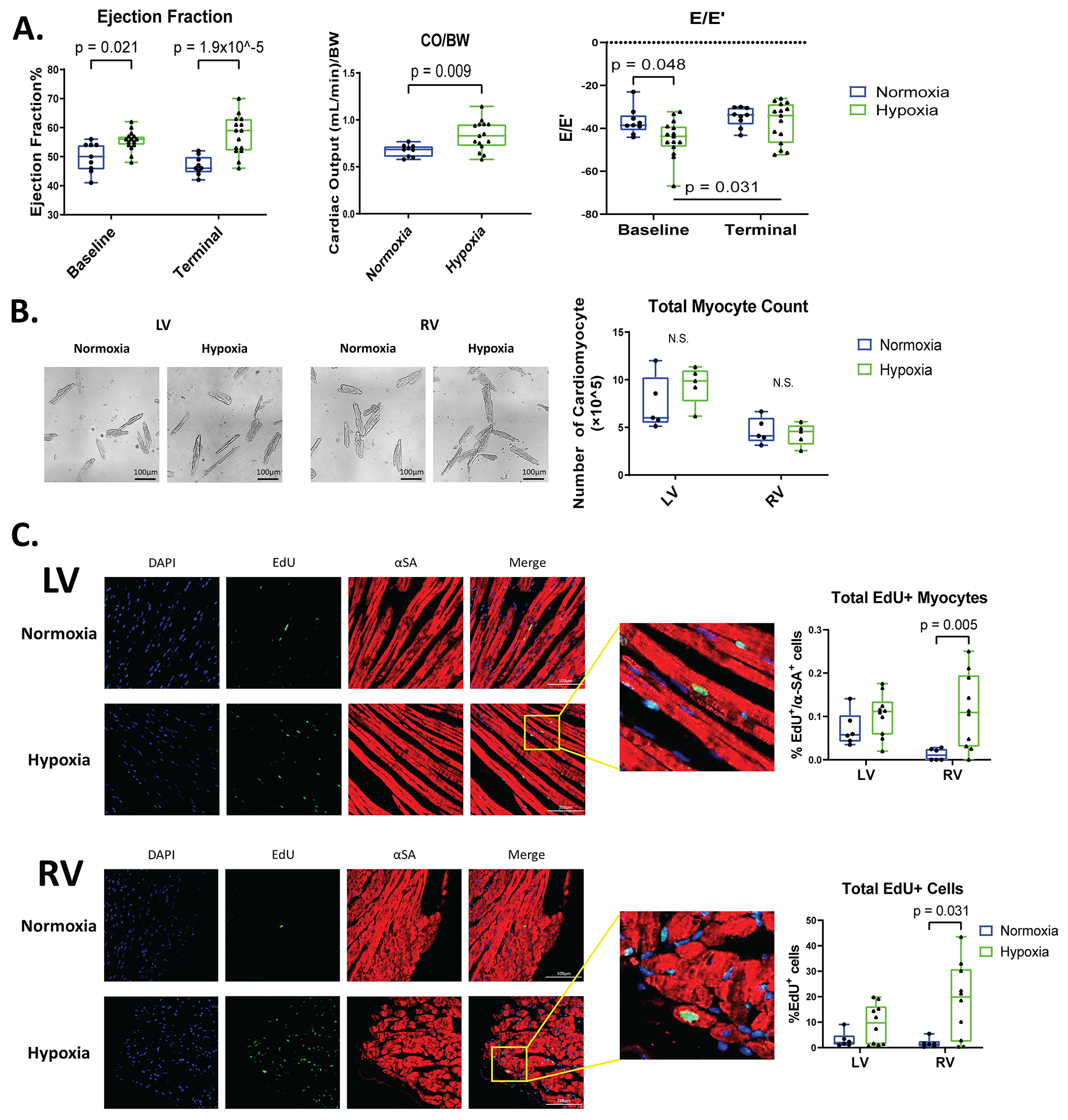
A: Ejection fraction (EF), cardiac output relative to body weight (CO/BW), and E/E’ were measured with Echocardiography at baseline and during terminal studies (n=9-15). B: Cardiomyocytes were isolated from the LV and RV of these hearts and the total number of cardiomyocytes were quantified. C: Mouse hearts were stained for alpha-sarcomeric actin (αSA, red, label cardiomyocytes), EdU (green, DNA synthesis), and DAPI (blue, nuclei) to identify myocytes and nonmyocytes undergoing DNA synthesis. Total EdU+ cardiomyocytes and total EdU+ cells were quantified in LV and RV tissue sections (n=6-10). Data are shown as mean±SD.
2 weeks at 7% oxygen does not disrupt cardiac function
Functional assessment of the heart by echocardiography revealed that 7% inspired oxygen levels induced significant increases in ejection fraction (EF) and cardiac output/body weight ratio (Figure 2A). There was no change in the E/E’ ratio following hypoxia. These results show that 2 weeks of chronic hypoxemia did not result in abnormal systolic or diastolic function. The food restricted controls versus hypoxia mice also showed similar results (Figure S1D), indicating that weight loss in hypoxia mice had no effects on cardiac function. These findings show that systolic and diastolic cardiac function were maintained in mice with two weeks of hypoxia.
Chronic hypoxia induces a small increase in the number of adult RV cardiomyocytes that make new DNA (EdU+)
Following the 4-week hypoxia study, cardiomyocytes were isolated from the LV and RV of normoxia and hypoxia mice to determine whether hypoxia increased the number of isolated LV or RV myocytes and the percentage of EdU+ myocytes. There was no significant difference in the total number of cardiomyocytes isolated from both ventricles of normoxia and hypoxia hearts (Figure 2B). These results suggest that the number of cardiac myocytes in the ventricles does not change after hypoxia.
EdU minipumps were placed in hypoxia and normoxia mice to label cells undergoing DNA synthesis during the duration of the 4-week study (Figure 1A). The ability of the 28-day EdU minipump regimen to label proliferative cells was validated by measuring EdU+ labeling of proliferative intestinal epithelium after 4-weeks of minipump implantation (Figure S2A). To identify EdU+ cardiomyocytes, histological heart sections were stained with wheat germ agglutin (WGA) to clearly define the membranes of myocytes with EdU+ nuclei (Figure S2B). In the LV of hypoxia hearts, there was no significant increase in EdU+ cardiomyocytes when compared to normoxia hearts. There was a small but significant increase in EdU+ RV myocytes following hypoxia (Figure 2C). Total EdU+ cells (myocytes and non-myocytes) were greater in the LV after hypoxia, and these changes were not significantly greater than in controls. The total EdU+ cells in the RV were significantly increased in hypoxia versus normoxia hearts (Figure 2C). Similar results were observed in the Atria following hypoxia (Figure S2C).
Ki67+ and pH3+ cells were also measured in the LV and RV of normoxic and hypoxic hearts. No significant change in Ki67+ myocytes in the LV (Figure S2D) was observed, and a small but significant increase in Ki67+ myocytes were seen in the RV. These results are consistent with the EdU labeling in these same hearts.
EdU+ nuclei were also measured in isolated cardiac myocytes (Figure S2E). Small numbers of EdU+ myocyte nuclei were observed and there was no significant difference between normoxic and hypoxic myocytes (Figure S2E). There was also no significant change in myocyte nucleation (mono versus binucleated), but most of the EdU+ cardiomyocytes were mononucleated in the hypoxemia group (0.24±0.34% in normoxia to 1.47±2.27% in hypoxia of EdU+ mononucleated myocytes) (Figure S2E) which is consistent with previous studies28–31. These results show that hypoxemia in adult male mice had an effect on inducing DNA synthesis in RV cardiac myocytes but no significant effect in LV myocytes.
The Effects of Hypoxemia in Female Mice:
Adult female mice were subjected to the identical hypoxia protocols (Figure 3A). Hypoxia induced significant amounts of cardiac hypertrophy in female hearts, as evidenced by increased HW/BW and HW/TL ratios (Figure 3B). Echocardiography measurements revealed female mice at 7% oxygen had increased LV mass and RV wall thickness (RVWT) when compared to female mice under normoxia (20.9% oxygen) (Figure 3C). There was a significant increase in EF between hypoxia and normoxia female mice, and there was no diastolic dysfunction (Figure 3D). These results show that hypoxia induced cardiac hypertrophy in female hearts without reducing pump function. There were no differences in these parameters in male and female mice subjected to hypoxia.
Figure 3. Female mice have normal cardiac function and only RV cardiomyocytes have increased EdU labeling after hypoxia.
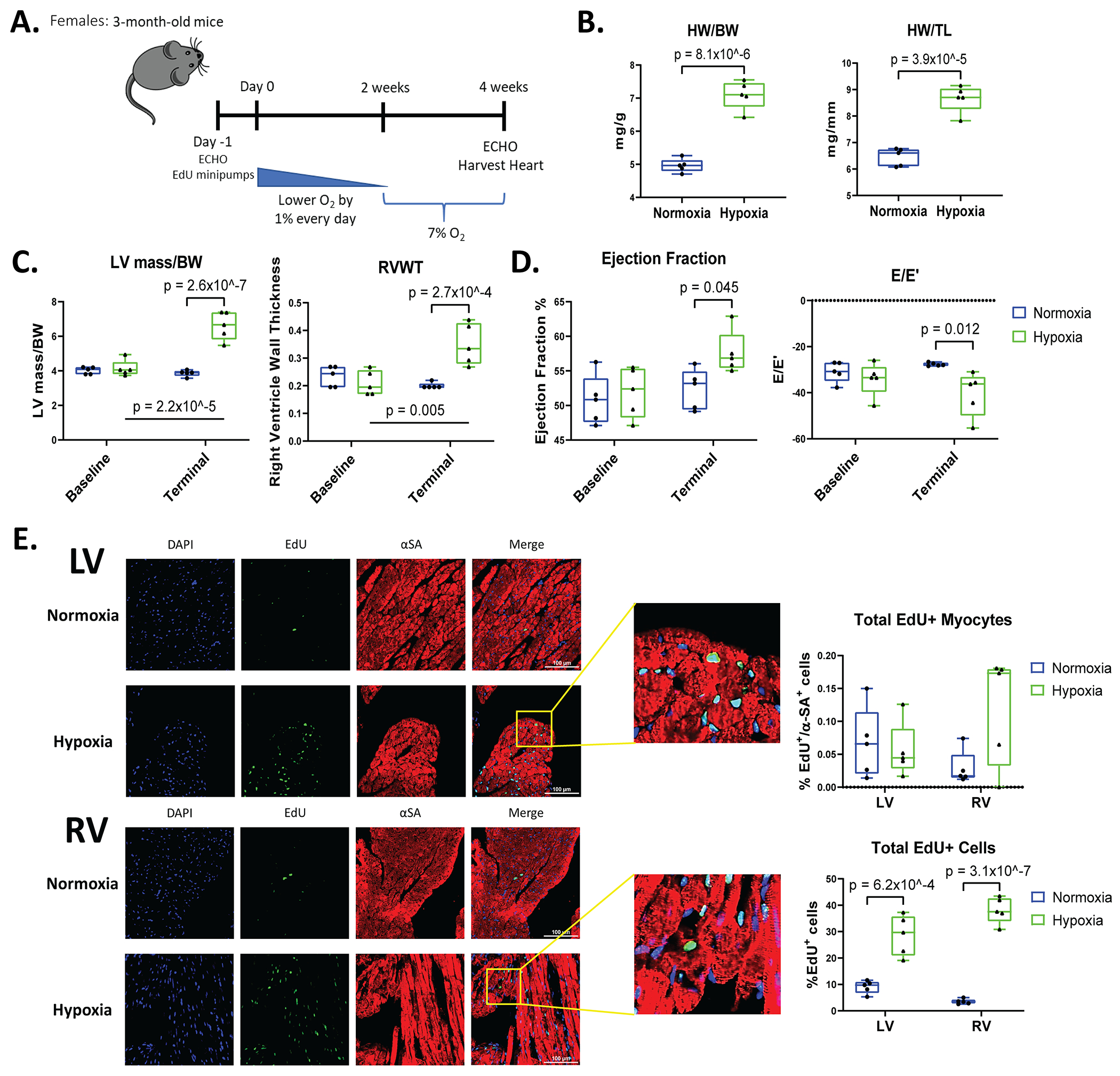
A: Schematic of hypoxia (7% oxygen) treatment in female mice. B: HW/BW and HW/TL were recorded during terminal studies. C, D: LV mass relative to body weight (BW), right ventricular wall thickness (RVWT), EF, and E/E’ obtained from echocardiography at baseline and terminal. E: Heart sections of the LV and RV were stained for αSA (red), EdU (green), and DAPI (blue) to determine myocyte and nonmyocyte EdU labeling. Total EdU+ cardiomyocytes and total EdU+ cells were quantified in the ventricles (n=5). Data represented as mean±SD.
Myocyte proliferation was also studied in female mice using EdU labeling. There was no significant increase in EdU+ cardiomyocytes in the LV in hypoxia versus normoxia hearts. There was a small increase in EdU+ myocytes in the RV of the hypoxia hearts (Figure 3E), which did not reach significance but had a similar trend as was seen in male mice. Female mice subjected to hypoxemia had increased EdU labeling of total cells (mainly nonmyocytes) in both the LV and RV of the heart (Figure 3E). The total number of EdU+ cells in female hypoxic hearts was almost 2-fold higher when compared to male mice under hypoxic conditions. Overall, systemic hypoxemia in female mice induced hypertrophy of the LV and RV with normal cardiac function, and an increase in the number of proliferative nonmyocytes. Collectively, our EdU results in both females and males show that there was a small increase in RV cardiomyocytes that synthesized new DNA after hypoxia, with no significant effects observed in LV myocytes.
Hypoxia induces very few myocytes to complete cell division (cytokinesis) in MADM mice
Systemic hypoxemia induced a small increase in RV cardiomyocyte EdU labeling in male and female hearts with no significant increase in EdU labeling of LV cardiomyocytes (Figure 2C and 3E). EdU is incorporated into nascent DNA during DNA synthesis and its presence suggests that the labeled myocytes are in the S-phase of the cell cycle. Cardiomyocytes could also have increased EdU labeling if hypoxemia induced DNA damage32–34. EdU can also cause DNA damage, inhibition of cell cycle progression, and apoptosis35, 36. Our results are not well explained by DNA damage/repair. Therefore, we used a different approach to assess myocyte proliferation in hypoxic mice.
To further study the idea that hypoxemia can induce cardiomyocyte proliferation we employed the Mosaic analysis with double markers (MADM) mouse model24. Myh6-MerCreMer mice were crossed into MADM mice to generate Myh6-MerCreMer::MADM (referred as MADMMyh6-MerCreMer) mice. Mice were administrated tamoxifen to induce cardiac myocyte specific recombination between the LoxP sites, which restored GFP and RFP expression in the affected myocytes. Recombination induced in G1 or post-mitotic G0 will induce GFP and RFP double positive cells (yellow color). Recombination induced in the G2 phase of the cell cycle generates two daughter cells by (1) X-segregation of recombinant chromatid resulting in one cell labeled with only GFP and the other with only RFP, or (2) Z-segregation of recombinant chromatids yielding one unlabeled cell and a double positive (yellow) cell37. It has been shown that X-segregation and Z-segregation events occur at the same rate during cell division38. Thus, the number of double positive (yellow) cells generated from the true cell division is expected to be the same as the number of single RFP or GFP cells. It is important to note that although high doses of tamoxifen can cause DNA damage39, we used a low dose of tamoxifen, 50ug/g, 5-6.25 mg/mouse in total, which is a lower dose than other reports (10.5-13 mg/mouse in total)40.
Male and female MADMMyh6-MerCreMer mice were placed under chronic hypoxia and were administered 5 injections of tamoxifen (one injection every 3 days) during the last two weeks at 7% oxygen (Figure 4A). Heart weight was increased in the hypoxic mice, documenting hypertrophy, and LV mass was significantly increased in MADMMyh6-MerCreMer mice under low oxygen when compared to normoxia mice (Figure 4B). Evaluation of single-labeled GFP and RFP cardiomyocytes in MADMMyh6-MerCreMer mice showed significantly increased RFP+ myocytes in the RV of hypoxic mice. There was no significant increase in the RFP+ LV myocytes in hypoxic hearts (Figure 4C). There was no change in GFP myocytes within either the LV or RV of the hypoxic heart versus normoxic hearts (Figure 4C). The lower levels of GFP+ versus RFP+ myocytes may be due to measurement challenges secondary to the shorter half-life of GFP41. These results document a small amount of cardiomyocyte proliferation in hypoxic MADMMyh6-MerCreMer mice.
Figure 4. Hypoxia increases the number of labeled cardiac myocytes after tamoxifen injection in MADM mice and identifies an increased number of newly formed myocytes in the RV.
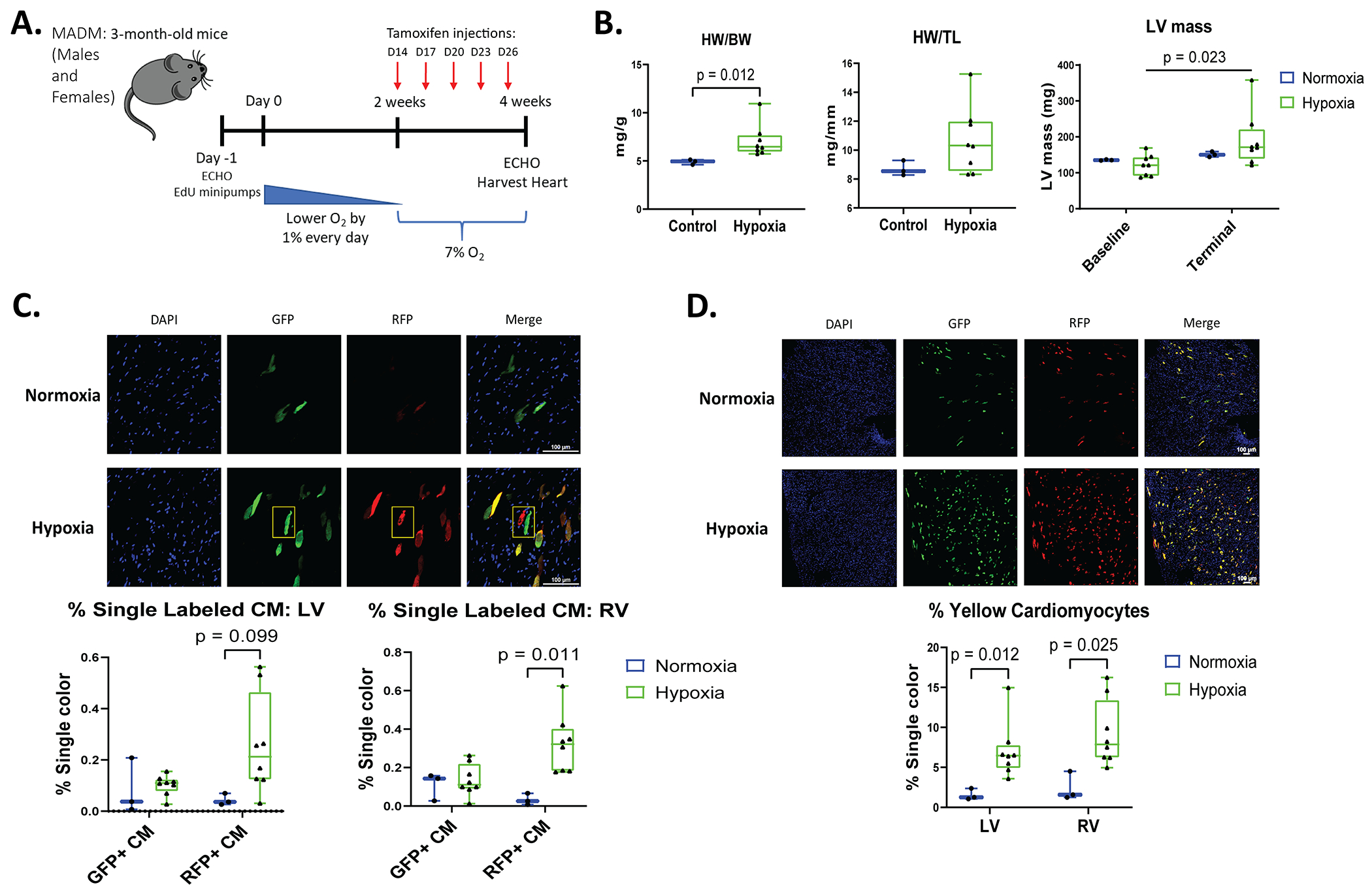
A: Schematic of experimental design in Mosaic analysis with double markers (MADMMyh6-MerCreMer) mice. Five injections of tamoxifen were administered to MADMMyh6-MerCreMer mice every 3 days (D) on day 14, 17, 20, 23, and 26. B: HW/BW and HW/TL was determined during terminal studies. Echocardiography was performed at baseline and during terminal studies to measure LV mass. C and D: Following tamoxifen injections, myocytes that completed cell division were single-labeled GFP+ or RFP+ only (“twin spots” highlighted by the yellow box in D). Hypoxia increased the number of affected cardiac myocytes that were labeled yellow (GFP and RFP double positive) in the LV and RV. Quantification of green (GFP+), red (RFP+), and yellow myocytes in male and female mice (normoxia n=3, hypoxia n=8). Data represented as mean±SD.
EdU labeling of MADMMyh6-MerCreMer hearts showed EdU+ myocyte numbers that were consistent with Red only/Green only myocytes. There were no changes in EdU+ cardiomyocytes within the LV in hypoxia, but a small and insignificant increase in EdU+ cardiomyocytes in the RV was observed in MADMMyh6-MerCreMer hearts after hypoxemia; Total EdU+ cells (nonmyocytes) were significantly increased in both ventricles of mice with hypoxia (Figure S3A). Our results suggest that those few myocytes that enter S phase and make new DNA (EdU+) go on to complete the cell cycle and divide, because EdU+ myocyte number is generally consistent with the number of Red only MADMMyh6-MerCreMer myocytes. These experiments reveal that both EdU and MADM analysis can be used to document myocyte proliferation. When used to study the effects of hypoxia our results show no significant increase in LV myocyte proliferation and a small but significant increase in the number of proliferative RV myocytes.
Analysis of MADMMyh6-MerCreMer mice also revealed that hypoxia significantly increased the % of yellow-labeled cardiomyocytes (myocytes containing both GFP and RFP) in both the LV and RV (1.57±0.71% in normoxia to 7.06±3.48% in hypoxia in the LV and 2.44±1.80% in normoxia to 9.24±4.12% in hypoxia in the RV) (Figure 4D). When considering that X- and Z-segregation rates are similar38, the number of yellow cells generated after cell division should be identical to the number of single RFP and GFP cells. As a result, the total number of proliferative myocytes, which include single-colored cells and a subset of yellow cells is still a small amount of myocyte proliferation under hypoxia. Consequently, when we subtract the subset of yellow cardiomyocytes that completed cytokinesis, the majority of yellow myocytes are a result of myocyte recombination in G0/G1 phase of the cell cycle. No nonmyocytes were found to be GFP/RFP+, documenting the cardiac specificity of the recombination.
The number of tamoxifen-induced recombined myocytes was increased by hypoxia. Specifically, the 7-9% yellow cardiomyocytes measured in hypoxic MADMMyh6-MerCreMer mice is significantly greater than observed in normoxia mice (Figure 4D). Given the greater % of MADM myocytes that became recombined (Yellow) in G0/G1 mitotic phase under hypoxia, a greater number of Red or Green only myocytes would be expected, even if the % of proliferative myocytes were unchanged.
The reason(s) why hypoxia increased tamoxifen induced myocyte recombination is unclear. Since hypoxia decreases global metabolism in mice23, we measured levels of Tamoxifen (TAM) and its metabolite, 4-hydroxytamoxifen (4OHTAM), to determine if hypoxemia caused tamoxifen to be more slowly metabolized (tamoxifen half-life was extended) in vivo and thus increased myocyte recombination. Liquid chromatography-Mass spectrometry (LC-MS) was performed on normoxia and hypoxia plasma samples collected at terminal study (2-3 days after the last tamoxifen injection). While there was a trend for increased TAM and 4OHTAM levels in mice exposed to hypoxia, there was no significant difference in levels, likely because of the fact that these measurements were made 2-3 days after the injection (Figure S3B). This issue deserves further study, with the temporal decay of TAM and 4OHTAM levels determined.
Nonetheless, results with MADM mice provide additional support that hypoxia did not induce increased proliferation of LV myocytes. Instead, there was a small increase in RV myocyte proliferation which is consistent with our EdU-labeled myocytes data.
Hypoxia induces angiogenesis, fibrotic pathological remodeling, and proliferation of other cell types in adult mice
Adult mice breathing 7% oxygen had an increased capillary cross sectional area in the LV and in the RV, but these changes did not reach significance (Figure 5A). Although there was no difference in capillary density in hypoxia mice versus normoxia mice, there was a small and insignificant increase in the RV of hypoxic mice (Figure 5A).
Figure 5. Systemic Hypoxemia stimulates cardiac remodeling with fibrosis and non-myocyte cell proliferation.
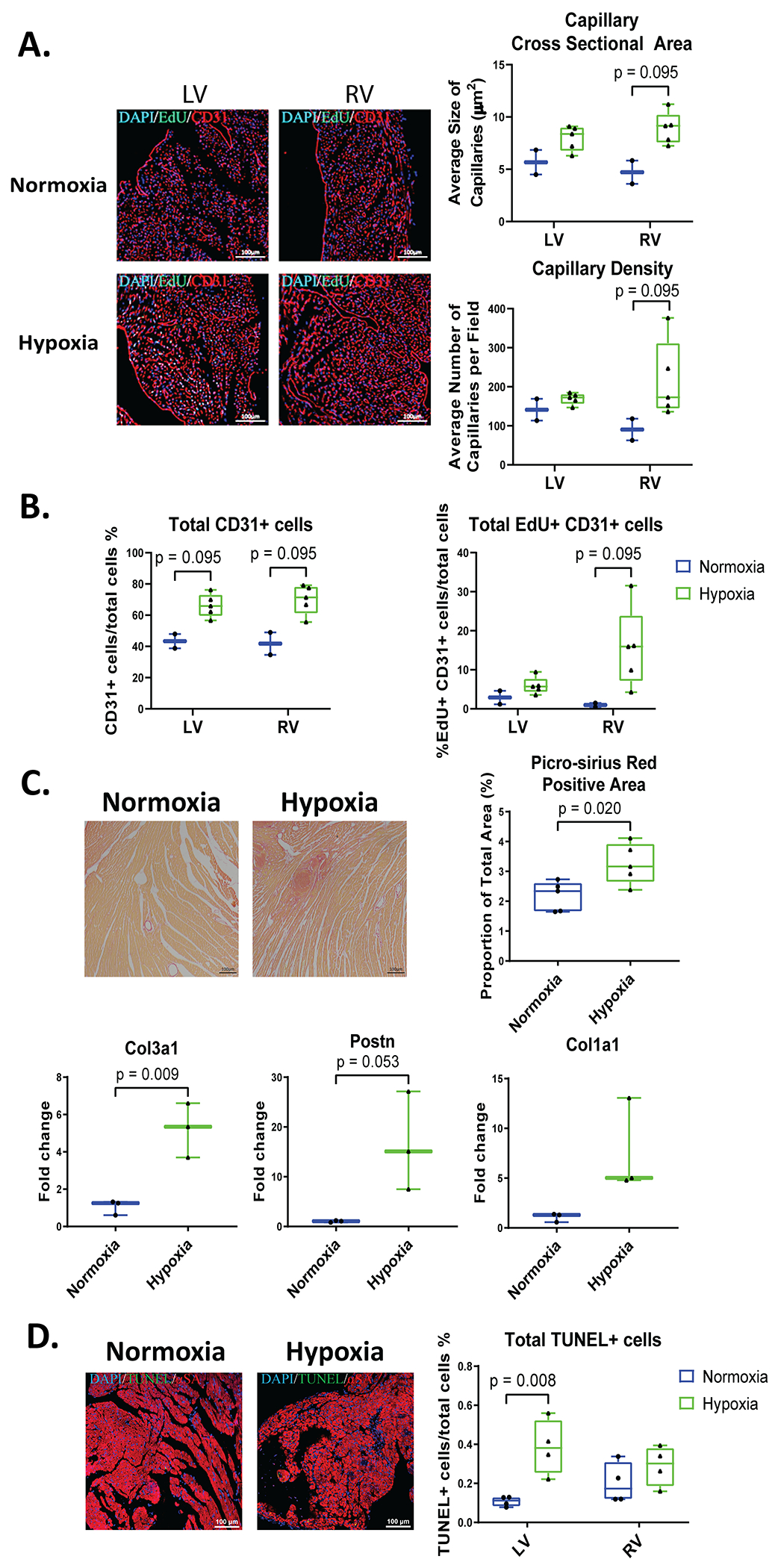
Adult hearts were stained for CD31 (red, labels endothelial cells), EdU (green), and DAPI (blue). A: Capillary density and size was measured in LV and RV sections of the heart. B: Total CD31+ cells and total EdU+ CD31+ cells were quantified. C: Fibrosis was analyzed and quantified by Picro Sirius Red staining and gene expression level of collagen 3a1 (col3a1), collagen 1a1 (col1a1), and Periostin (Postn) by RT-PCR. D: Heart sections were stained for αSA (red, labels cardiomyocytes), TUNEL (green, apoptotic cells), and DAPI (blue, nuclei) to label total apoptotic (TUNEL+) cells (n=4-5 mice). Data represented as mean±SD.
There was a trend for more endothelial cells in hypoxic hearts with a small increase in EdU+ endothelial cells, which may account for the majority of total EdU+ cells seen in hypoxia hearts (Figure 5B).
Hypoxia caused a significant increase in cardiac picro-sirius red staining, suggesting that fibrosis was present. Consistent with this idea was the finding of increased expression of Collagen types 3a1 and 1a1, and Periostin genes (Figure 5C) in hypoxemia heart tissue. There was also a small, but insignificant increase in smooth muscle actin+ fibroblasts in hypoxia mice (Figure S4A). Finally, there were significantly more TUNEL-positive cells in the LV but no significant change in the RV (Figure 5D) consistent with an early cell death phenotype.
To explore the idea that hypoxemia induces a proinflammatory environment, CD45+ cells were measured in heart tissues. There was an increase in CD45+ cells in both ventricles of hypoxia mice and an increase in EdU+ CD45+ cells (Figure S4B).
Overall, hypoxia stimulated moderate angiogenesis in the RV, mild apoptosis in the LV, and moderate fibrosis and inflammation in both ventricles. In addition, hypoxia induced proliferation of various cell types including endothelial cells and immune cells, which contribute to the total EdU+ cell population seen in hypoxic mice.
Hypoxia induces the upregulation of mitotic cell cycle genes
RNA-sequencing was performed on normoxic and hypoxic heart tissue to define differential gene expression in male (M) and female (F) heart tissues under normoxia control (C) or hypoxia (H) conditions. The following comparisons were made: MH vs. MC, FH vs. FC, and FH vs. MH. Hypoxia in male mice induced an upregulation of 673 genes and a downregulation of 285 genes when compared to normoxic male mice with a cutoff of FDR=0.1, FC=2 (MH vs. MC) (Figure 6A). Hypoxia in females (FH vs. FC) induced a similar pattern of gene expression but with a reduced number of differentially expressed genes (Figure 6A). When comparing female to male mice (FH vs. MH), there were minor differences with <30 upregulated genes and <70 downregulated genes under hypoxic conditions (Figure 6A), supporting the idea that hypoxia induced similar cardiac gene expression changes in both male and female hearts. Overall, these findings suggest that chronic hypoxemia induces a similar cardiac gene expression profile in male and female mice.
Figure 6. Hypoxia induces upregulation of mitotic cell cycle genes with similar gene expression profile in both sexes.
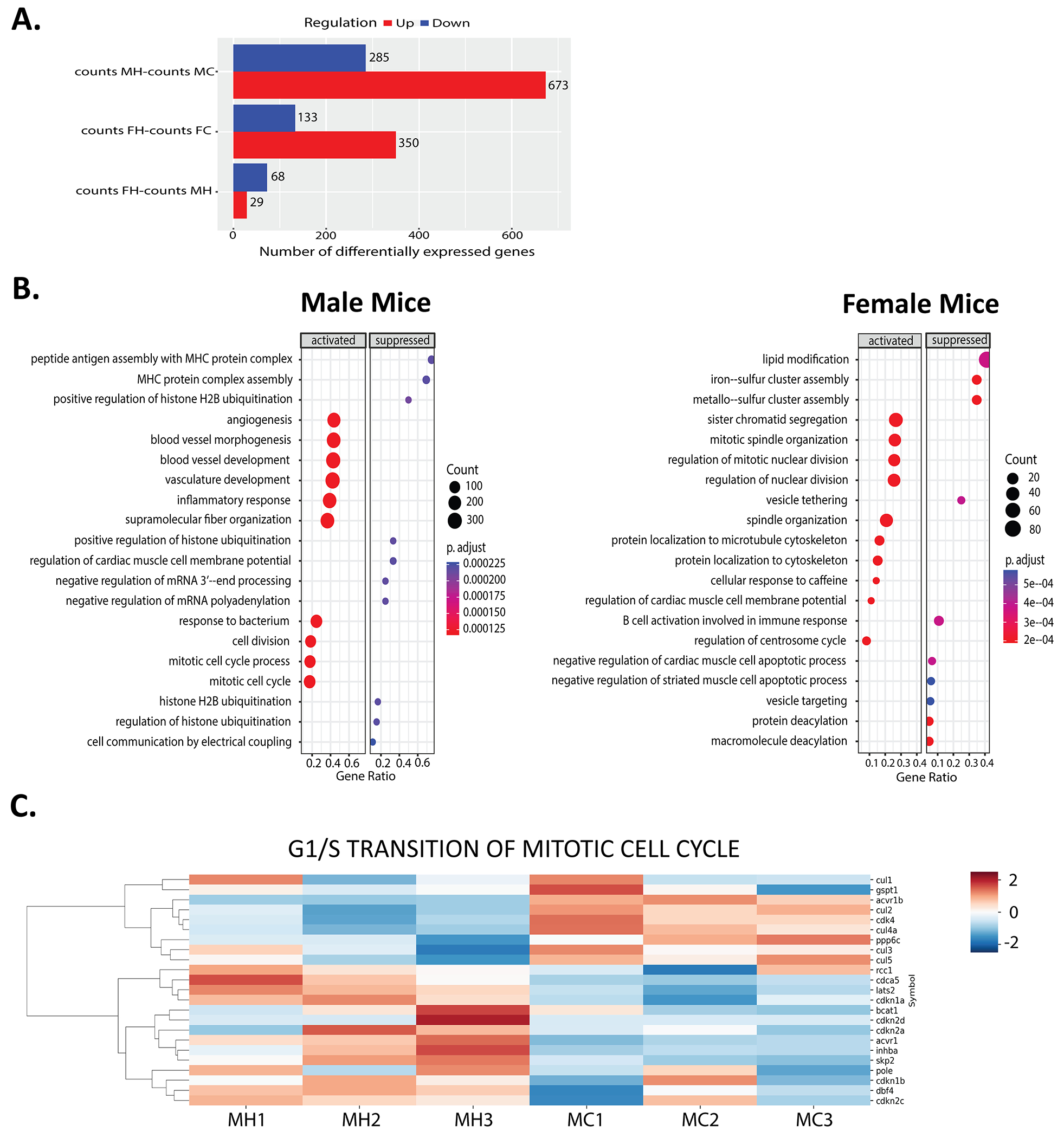
RNA sequencing analysis on male (M) and female (F) hearts in normoxia control (C) or hypoxia (H) group. A: Number of differentially expressed genes in MH vs. MC, FH vs. FC, and FH vs MH mice as presented in bar graphs. B: GO analysis depicted dot plots of male and female mice with most significantly activated or suppressed biological processes including mitosis, angiogenesis, apoptosis, and immune cell response. C: Heat maps of relative expression level of genes involved in G1 to S phase transition (GO:0000082) in MH vs. MC mice. Upregulated and downregulated genes shown as red and blue, respectively (n=3 each group).
Gene set enrichment analysis (GSEA) based on gene ontology gene sets revealed that among the most significantly upregulated biological processes, mitotic cell cycle, cell/nuclear division, and blood vessel development have higher expression and gene counts in hypoxic male and female mice versus controls (Figure 6B). These data support our findings of increased non myocyte cell proliferation under low oxygen conditions (Figure 2C and 3E). Furthermore, the activation of angiogenesis and inflammatory responses, as well as the suppression of the negative regulation of cardiac muscle cell apoptotic process, is consistent with other current results (Figure 6B, 5A, 5B, 5D, and Figure S4B). Heatmaps of data from male mice were generated in previous reports23 and showed similar trends, with an upregulation of genes in the mitotic cell cycle, positive regulation of proliferation, angiogenesis, and vascular morphogenesis; There was a downregulation of genes involved in oxidation reduction, mitochondrion organization, electron transport chain, and cardiomyocyte hypertrophy (Figure S5, Data Set S1).
The most significant finding in the current study is that hypoxemia did not induce significant increases in LV myocyte proliferation. Examination of the cell cycle genes of male heart tissues showed a downregulation of genes that promote the G1 to S phase transition in hypoxic mice (e.g. Cullin genes42–44; cul), and an upregulation of genes that inhibit the G1 to S phase transition (e.g. CDK inhibitors45–47; cdkn) as depicted in a heatmap (Figure 6C). One hypoxia mouse, MH1, has two-fifths of its cullin genes and CDK inhibitor genes with similar expression patterns as normoxia (MC) mice. However, the majority of the cullin genes are downregulated and most of its CDK inhibitor genes are upregulated, and thus mimic the other hypoxia mice. Therefore, low oxygen levels caused changes in gene expression of those molecules involved in G1 to S phase transition.
There were also differential expression patterns of S phase genes and upregulation of expression of G2/M phase genes in hypoxia mice versus normoxia mice (Figure S6A and S6B). These results demonstrate that many genes involved with regulation of the mitotic cell cycle are altered in hypoxia hearts (Figure S6C), but our results suggest that these changes do not induce LV myocyte proliferation.
Severe Hypoxemia decreases mice survival and impairs cell proliferation
We were concerned that the hypoxemia levels we employed (7% oxygen), while identical to the levels used in a previous study23, might not have been sufficient to induce the myocyte cell cycle activity that causes cells to complete the cell cycle and form new myocytes. Therefore, a group of mice were exposed to 5% oxygen (Figure S7A). Briefly, mice were placed in the hypoxia chamber at 19% oxygen which was lowered by 1% per day for 2 weeks. Once the chamber reached 5% oxygen, the animals began to die (Figure S7B). Consequently, the remaining survivors were sacrificed after 6 days at 5% oxygen (Figure S7A). EdU labeling was used to evaluate myocytes undergoing DNA synthesis. Mice breathing 5% oxygen had a significant decrease in EdU+ LV myocytes when compared to normoxia controls (Figure S7C). There was no change in EdU+ myocytes in the RV and the total EdU+ cells did not increase following low oxygen levels (Figure S7C). These studies showed that reducing oxygen below 7% caused significant mortality and did not induce non-myocyte and myocyte cell cycle re-entry, but rather reduced the already low level of myocyte proliferation in the heart.
Discussion
Cardiovascular Disease (CVD) remains the leading cause of death in Western society1. MI, secondary to CVD, results in the death of cardiomyocytes and supportive tissue which causes deterioration of cardiac pump function and detrimental remodeling of the cardiac chambers2, 48. The consensus of many previous studies is that adult mammalian cardiomyocytes are largely terminally differentiated and myocyte proliferation does not result in repopulation of the heart with new myocytes after injury21. Fetal and neonatal mammalian cardiomyocytes proliferate during development49–51 and they also proliferate following cardiac injury to regenerate normal myocardium14, 17. Adult mammalian myocytes appear to undergo a very low rate of self-renewal, even following MI19, 20.
Novel approaches to repopulate the injured adult heart with new myocytes would have a significant positive impact on patients with post MI heart failure. Many studies have explored novel therapeutics to promote cardiac regeneration to replenish cardiomyocyte numbers21, 52, 53. One study tested the novel concept that recapitulating the embryonic hypoxic environment, where low oxygen levels in the fetus supports the embryo development and cardiac growth, could reawaken adult myocyte proliferative properties23. In that study adult male mice were subjected to hypoxia (7% oxygen) and the results suggested an induction of large amounts of cardiomyocyte proliferation 23. In this study23, chronic hypoxia resulted in increased numbers of adult myocytes (870,000 normoxic isolated myocytes to 1,900,000 hypoxic isolated myocytes) with a decrease in their CSA. Evidence of cardiomyocytes in active mitosis was revealed by labeling with proliferative markers such as BrdU, pH3, and Aurora B. Additionally, there was reduced oxidative DNA damage in cardiomyocytes due to the hypoxia-induced decrease of mitochondrial-derived reactive oxygen species (ROS). The results of this study23 suggest that hypoxia induces new myocyte formation in the adult male mouse heart. In addition, when male mice were subjected to hypoxia after MI, the resulting cardiomyocyte proliferation was associated with improved cardiac function, myocardial repair, and reduced DNA damage23. These results suggest that hypoxia could be used clinically to induce myocyte regeneration after MI.
The present study reexamined the effects of hypoxia on cardiomyocyte proliferation in both male and female adult mice. We sought to provide more mechanistic insight on myocyte proliferation with hypoxemia. The novelty of this study is that we utilized both male and female mice and we also employed MADM mice, to have an independent technique to complement EdU/BrdU nuclear labeling to determine if adult cardiomyocytes proliferate. Effects of hypoxia on cardiomyocytes in the LV vs. RV were also studied. Comparison of results from the present study to previously published data23 on the effects of hypoxemia in uninjured mice are listed in Table 1. Many similarities are noted with the major difference being that our study did not observe an increase in LV myocyte proliferation or myocyte number with hypoxia challenge.
Table 1.
Comparison of literature on uninjured mice subjected to Hypoxia
| Category | Nakada et al. Research Paper | Johnson et al. Research Paper | |
|---|---|---|---|
| Methods | Sex | Males only | Males and Females |
| Hypoxia | Experimental: 7% O2, Tested: 10% O2 | Experimental: 7% O2, Tested: 5% O2 | |
| Analysis | LV and RV analyzed together | LV and RV analyzed separately | |
| Cardiac Morphology | Heart size (HW/BW) | Increased (5 mg/g Normoxia vs. 8 mg/g Hypoxia*) | Increased (4.3 mg/g Normoxia vs. 6.8 mg/g Hypoxia*) |
| CM Cross Sectional Area | Decreased in LV*; Increased in RV* | No change in LV; Increased in RV* | |
| Isolated Cardiomyocyte Size | Not measured | Increased L and SA in LV*; Increased L, W, and SA in RV* | |
| Isolated Myocyte Number | Increased in Ventricles* | No change in LV; No change in RV | |
| CM Nucleation | Increased mononucleated CM* and Decreased binucleated CM* | No change in Nucleation but Increased mononucleated EdU+ isolated CM | |
| Cardiac Myocyte Proliferation | DNA Synthesis-drug given continuously | Increased (0.1% BrdU+ CMs*) | Increased (0.1% EdU+ CMs in LV; 0.1% EdU+ CMs in RV*) |
| DNA Synthesis at Terminal | Not measured | Increased (0.01% Ki67+ CM in LV; 0.02% Ki67+ CM in RV*) | |
| Mitosis at Terminal | Increased (0.06% pH3+ CMs*) | No change (0.002% pH3+ CM in LV; 0% pH3+ CM in RV) | |
| Cytokinesis | Increased (0.0028% Aurora B+ CM*) at Terminal | Increased (MADM mice showed 0.1-0.3% GFP/RFP+ CM in LV and RV*); tamoxifen given during 2 weeks at 7% Oxygen | |
| Cardiac Remodeling | Angiogenesis | No change in Capillary Density; Increased Capillary size* | Increased Capillary density in RV; Increased Capillary Size in LV and RV |
| Fibrosis | Not measured in uninjured mice | Increased in Ventricles* | |
| Apoptosis | Not measured in uninjured mice | Increased in LV*; No change in RV | |
| Cell Proliferation | Global Proliferation of cardiac cells | Not measured | Increased in LV* and RV* (shown as EdU+, Ki67+, and pH3+ cells) |
| Endothelial Cells | Images shown but no Quantification | Increased EdU+ endothelial cells in LV and RV | |
| RNA Sequencing Analysis | Mitochondrial Metabolism | Decreased in LV | Decreased in both ventricles |
| Cell Cycle Regulation | Increased in LV | Increased in both ventricles; Dysregulated G1 to S phase transition | |
| Angiogenesis | Increased in LV | Increased in both ventricles |
Numbers above reflect the estimated averages seen from figures in each paper.
represents significantly different between normoxia and hypoxia mice. CM, cardiomyocytes; L, length; W, width; SA, surface area.
Hypoxia induced cardiac hypertrophy and hypertrophy of LV and RV myocytes
Adult male mice exposed to chronic hypoxemia (Figure 1A) had increased HW/BW and HW/TL ratios (Figure 1B), documenting cardiac hypertrophy. The HW was increased by 17% in hypoxia vs control hearts. Female mice also had significant cardiac hypertrophy following hypoxia (Figure 3A and 3B). In addition to gross heart weights measured in terminal studies, echocardiography measurements showed significant increases in LV mass in male and female mice (Figure 1D and 3C). Myocyte CSA measured in fixed tissues revealed an increase in RV CSA, with no change in the LV myocyte CSA (Figure 1C).
The nature of the myocyte hypertrophy in LV and RV myocytes was further studied by measuring myocyte length, width, and surface area in isolated ventricular myocytes. These studies showed a significant increase in LV myocyte length and surface area, with significant increases in RV myocyte width, length, and surface area in hypoxic mice (Figure 1E). The increase in myocyte SA was 21.23% in RV and 18.84% in LV myocytes (Figure 1E). These results show that the increase in LV and RV myocyte size can fully account for the 17% increase in heart weight that we observed.
These results also show that hypoxia caused LV myocytes to lengthen in a fashion reminiscent of the eccentric hypertrophy seen in volume overload25, 27. RV myocytes were wider and longer which resembles a mixed concentric/eccentric hypertrophy response. The changes in LV and RV myocytes length/width likely result from hypoxia-induced effects on cardiac work, including increased cardiac output to support tissue oxygenation. It is likely that the RV is also subjected to pressure overload due to increases in pulmonary vascular resistance induced by chronic hypoxia25–27. Collectively, our results show that hypoxia-induced cardiac hypertrophy can be accounted for by the increased size of LV and RV myocytes.
Hypoxemia in adult mice did not cause cardiac pump dysfunction
Echocardiography measurements (Figure 1A) showed a small but significant increase in the EF of male and female hypoxic mice when compared to normoxic mice (Figure 2A and 3D). The cardiac output was significantly increased in hypoxia, documenting the need for an increased blood flow to maintain tissue oxygenation (Figure 2A). The diastolic function of male and female mice following hypoxia was unchanged (Figure 2A and 3D). These results show that chronic hypoxemia, for the time frame used, did not negatively disrupt cardiac function in either male or female mice.
Chronic hypoxia does not induce LV myocyte proliferation
Results of a previous study suggested that hypoxemia induced a large increase in ventricular myocyte number (870,000 normoxic isolated myocytes to 1,900,000 hypoxic isolated myocytes)23. We were unable to confirm these results in either males or females. In the present study there was no significant change in total cardiomyocyte numbers when we isolated myocytes from the LV and RV of normal and hypoxia hearts (Figure 2B). Furthermore, when EdU minipumps were implanted in male and female mice to label cells undergoing DNA synthesis, no significant increase in EdU+ myocytes in the LV of male and female hearts subjected to hypoxia was observed (Figure 2C and 3E). In male mice, a small but significant increase in EdU+ RV myocytes was found after hypoxemia (Figure 2C). In female mice, there was a similar trend of increased EdU+ myocytes in the RV of hypoxic hearts, but these changes do not reach statistical significance due to the smaller sample size and variability in female mice (Figure 3E).
The finding of a small amount of myocyte proliferation in the RV but not the LV in hypoxia may be due to the fact that only the RV was subjected to pressure overload54. It is important to point out that the 0.1% of EdU+ myocytes in the hypoxic RV seen in our study is similar to the 0.1% of BrdU+ ventricular myocytes in the Nakada et al. study23 (Table 1). In the current study no change in myocyte nucleation was observed, but most of the EdU+ myocytes were mononucleated as seen in previous studies (Figure S2E)28–31. Collectively, our results show small and similar numbers of EdU+ LV myocytes in normal and hypoxic hearts, consistent with numbers reported by others23. We did not find that hypoxia induced increased numbers of LV myocytes.
We performed an additional study to assess myocyte proliferation using male and female MADMMyh6-MerCreMer mice. In these experiments, the tamoxifen treatment protocol employed induced significantly more recombination in hypoxemia hearts than in normoxic controls (of yellow myocytes: 1.57±0.71% in normoxia to 7.06±3.48% in hypoxia in the LV and 2.44±1.80% in normoxia to 9.24±4.12% in hypoxia in the RV) (Figure 4D). The presence (7-9%) of a yellow color in the MADM model is consistent with myocytes in the G0/G1 phase of the cell cycle. The reason why tamoxifen caused more myocyte recombination in hypoxic hearts is not entirely clear and deserves attention. Given the slow metabolism in hypoxia animals23 we suggest that tamoxifen metabolism may have been slowed in animals in hypoxic environments. Our preliminary studies provide some support for this notion.
Myocytes exhibiting a single color (red or green only) and a subset of yellow myocytes had recombination induced while in the G2/M phase of the cell cycle37. Single labeled GFP and RFP cardiomyocytes and a yellow cell subset, signify completion of the cell cycle and cell division. We found no significant increases in RFP-only myocytes in the LV and small but significant increases in RFP myocytes in the RV of hypoxic mice (Figure 4C). Myocytes that were only GFP+ were observed but at a somewhat lower number than RFP+ only myocytes in both ventricles. The reasons for this are unclear but could be due to a more rapid decay of the GFP versus RFP fluorescence signals under our conditions41. When we evaluate the number of yellow myocytes that also completed cytokinesis, this number is identical to the % of single-labeled GFP and RFP myocytes, due to the same segregation rate in the MADMMyh6-MerCreMer mouse model38. Nevertheless, the total number of myocytes that completed cell division following hypoxemia was small (< 1%) in both the LV and RV.
Our studies with MADM mice indicate that hypoxemia did not induce significant increases in LV myocyte proliferation but did induce small amounts of RV myocyte proliferation (Figure 4C). EdU labeling in MADM mice provided complementary results supporting the conclusion that hypoxia in adult mice does not induce the proliferation of LV myocytes.
RNA sequencing results suggest that the failure of most myocytes to complete the cell cycle and form new myocytes in hypoxic conditions may be due to a downregulation in genes that promote the G1 to S phase transition, such as Cullin genes42–44, and an upregulation of the cell cycle inhibitor genes such as CDK inhibitors45–47 (Figure 6C). Thus, hypoxia induces changes in myocyte gene expression that suppress rather than enhance the transition from G0/G1 to S phase and then progression to complete cell division and form new myocytes.
Based on our data and recently published results55, it appears that hypoxemia reduces mitochondrial ROS production, which is essential for progression of cells through G0/G1 phase of mitosis. Higher levels of ROS production are required for cells to progress through S phase and G2/M phase. Therefore, low oxygen levels may also contribute to inhibition of myocyte progression from G1 to S and G2/M phases of the cell cycle.
Our results also showed that cardiomyocyte DNA synthesis (EdU labeling) occurred globally throughout the heart with evidence of EdU+ myocytes in the LV, RV, and Atria (Figure 2C, 3E, and Figure S2C). Evidence of myocyte cell cycle activity was observed throughout the heart, in the endocardium, midmyocardium, and epicardium (Figure 4C and 4D). These results are different than those reporting a majority of proliferating cardiomyocytes to be located in the subendocardial muscle of the LV56.
Systemic hypoxia induced cardiac remodeling with increased proliferation of multiple types of nonmyocytes
Hypoxia induced angiogenesis in the heart with increased capillary size in the LV and increases in RV capillary cross sectional area (Figure 5A). There was a trend for increased capillary density in the RV with no change in the LV (Figure 5A). This is consistent with studies that shows hypoxia induces angiogenesis57–59, and an increase in ventricular capillary size23. There was an increase in EdU+ endothelial cells, which accounts for much of the total EdU+ non-myocytes observed (Figure 5B). These results show that hypoxia causes moderate angiogenesis in the heart and the resulting increase in myocardial blood flow may have contributed to the maintenance of cardiac pump function in female and male mice (Figure 2A and 3D).
Mild fibrosis was observed in hypoxic hearts with increased deposition of collagen and heightened expression of collagen genes and periostin (Figure 5C). Activated fibroblasts were also present in hypoxic heart tissues (Figure S4A). This mild fibrosis in hypoxic hearts was not sufficient to disrupt cardiac function (Figure 2A and 3D). However, longer hypoxic challenges could result in significant cardiac fibrosis with associated abnormalities of cardiac diastolic function.
A small but significant increase in myocyte apoptosis was observed in the LV of hypoxic hearts with no change in the RV (Figure 5D). In addition, there were more inflammatory cells in the LV and RV of mice subjected to hypoxia (Figure S4B). These results suggest that longer periods of hypoxemia could have damaging effects on the heart.
There was a small increase in EdU+ nonmyocytes in the LV, with a larger, significant increase in EdU+ nonmyocytes in the RV of hypoxic male mice versus normoxic controls (Figure 2C). Similarly, female hypoxic hearts had significant increases in EdU+ nonmyocytes in the LV and RV, which was nearly 2-fold higher than in male hypoxic hearts (Figure 3E). The reasons for this are not clear and require further study. These results show that systemic hypoxia induces proliferative changes in almost all cardiac cell types.
Hypoxia induced similar changes in gene expression in male and female hearts
RNA sequencing in tissues from male and female hearts revealed that hypoxia induced similar gene expression patterns in males and females (Figure 6A). Of the top upregulated biological processes, genes involved in mitotic cell cycle and cell/nuclear division were significantly upregulated in hypoxia versus normoxic mice (Figure 6B). Likewise, there were similar trends of upregulated genes involved in positive regulation of proliferation processes and angiogenesis in hypoxic mice (Figure S5), consistent with previous studies23. These results likely explain our findings that chronic hypoxemia caused an increase in proliferation of non-myocyte cardiac cells and could contribute to the small yet significant increase in RV myocyte proliferation (Figure 2C, 3E, 4C, and 4D). Interestingly, there was a downregulation in genes that promote the G1 to S phase transition (Figure 6C). These changes in expression of genes that regulate the G1 to S phase transition of the cell cycle, together with the reduced levels of mitochondrial ROS production55 may help explain why so few of the hypoxic RV and LV myocytes demonstrated DNA synthesis and cell division (Figure 2C, 3E, 4C, and 4D). Cardiomyocyte hypertrophy in hypoxia mice (Figure 1E) may also be related to an increase in genes involved in DNA synthesis60. Collectively our results showed that systemic hypoxemia upregulated mitotic gene expression and increased cell cycle activity of multiple cardiac cells in both male and female mice, but few RV cardiac myocytes entered S phase and went on to form 2 daughter cells. Most importantly, we did not find a significant increase in LV myocyte proliferation with either EdU labeling or in MADM mice subjected to hypoxic stress.
Severe Hypoxia reduces cell proliferation and increases mortality in mice
A recent study demonstrated that moderate hypoxemia (10% oxygen) induced a trend for increased myocyte DNA synthesis but did not improve cardiac function following MI like treatment with 7% oxygen23. These studies suggest that the level of inspired oxygen is likely to be critical in determining the cardiac myocyte hypoxic response. We were concerned that 7% oxygen might not be sufficient to induce adult LV myocyte proliferation. Therefore, we tested more severe hypoxia (5% oxygen) in adult mice. Our results showed that severe hypoxemia caused mice to die during the two-week test period. Since animals were dying over time with this oxygen level, we studied a small cohort of animals after 6 days at 5% oxygen (Figure S7B). There was a significant reduction in LV myocyte DNA synthesis in these mice, and no change in proliferation of RV myocytes and non-myocyte cells in hypoxic mice versus normoxic mice (Figure S7C). These results showed that 5% oxygen increased mortality and impaired hypoxia-induced cell proliferation in the heart with a more pronounced reduction in LV myocyte proliferation.
Additional studies are needed to determine if and how modulation of cell cycle checkpoint genes in hypoxic conditions modulates new myocyte formation.
Summary and Conclusions
Our study did not confirm that chronic hypoxemia (7% oxygen) induces LV myocytes of adult male and female mice to proliferate. We used three independent techniques (direct counting of isolated myocytes, EdU incorporation, and MADM mice) to show that hypoxia does not induce LV myocyte proliferation. A small but significant increase in RV myocyte proliferation was observed. Since RV myocytes were subjected to hypoxia and RV pressure overload, the small amount of proliferation likely resulted from the effects of pressure overload on the myocytes in this chamber. The low production of ROS in hypoxia55 could prevent an increase in the activation of G1 to S phase transition genes, but this will require additional study.
Hypoxia increased non-myocyte cardiac cell proliferation. Similar results were seen in male and female mice where there was a significant upregulation of mitotic cell cycle genes, and angiogenesis. Hypoxic hearts also exhibited different types of cardiomyocyte hypertrophy in the LV and RV, with small increases in fibrosis, apoptosis, and immune cell infiltration. Cardiac function was not significantly reduced in hypoxic mice.
In summary, chronic hypoxemia did not induce LV cardiomyocytes to re-enter the cell cycle and proliferate. The increase in heart size following hypoxia can be fully accounted for by cardiomyocyte hypertrophy. Given the modest fibrotic effects of short-term hypoxia and the lack of effect on LV myocyte proliferation, the idea of using hypoxia as a therapy to regenerate the heart after MI should be more carefully tested.
Supplementary Material
Novelty and Significance.
What is Known?
Myocardial Infarction (MI) and other cardiovascular diseases, cause the death of cardiac muscle cells and this can lead to abnormal heart function, heart failure and premature death.
The adult heart is unable to replenish cardiac muscle cells that die with disease stress, in large part because adult cardiac myocytes rarely proliferate.
A recent study23 showed that chronic systemic hypoxia (7% oxygen) in adult male mice induces ventricular myocyte proliferation, suggesting that hypoxia could be used as a therapy to replace ventricular myocytes lost with disease.
What new information does this article contribute?
Hypoxemia in both adult male and female mice caused a small increase in the proliferation of right ventricular (RV) cardiomyocytes but had no effect on left ventricular (LV) myocyte proliferation.
Systemic hypoxia induced cardiac and cardiomyocyte hypertrophy in both ventricles with increased width and length of RV myocytes and increased length of LV myocytes
The increase in cardiac myocyte size (myocyte hypertrophy) was sufficient to account for the increase in heart size (cardiac hypertrophy).
Diseases that induce cardiomyocyte death can lead to cardiac dysfunction and heart failure. Since adult cardiomyocytes rarely proliferate, novel therapies that induce adult cardiomyocyte proliferation after injury could replenish myocyte numbers. A previous study23 suggested that systemic hypoxemia in adult male mice caused a large increase in ventricular cardiomyocyte proliferation. The present study examined the effects of systemic hypoxia on LV and RV myocyte size and proliferation in adult male and female mice. We found that hypoxia caused cardiac hypertrophy in both sexes. However, the numbers of LV and RV myocytes in hypoxia versus normoxia hearts were not different. Both LV and RV myocytes from hypoxia hearts were increased in size and this increase was sufficient to account for the organ hypertrophy. A small increase in RV myocyte proliferation was observed but there was no increase in LV myocyte proliferation. Hypoxemia caused proliferation of other cardiac cells, moderate fibrosis, and mild inflammation. These results suggest that hypoxia does not induce LV myocyte proliferation but causes some pathological remodeling. The hypothesis that hypoxia causes adult ventricular myocytes to proliferate requires further study before it should be considered as a therapy to treat patients with cardiac diseases.
Sources of Funding:
Jaslyn Johnson was supported by the National Institutes of Health (5F31HL143865-03). S.R.H. is funded by a grant from the National Institutes of Health (HL132391). S.M. is supported by the National Institutes of Health (HL137850). Y.T. is funded by the National Institutes of Health (RO1-HL150587).
Non-standard Abbreviations and Acronyms:
- MADM
Mosaic analysis with double markers
- LV
Left ventricle
- RV
Right ventricle
- HW
Heart weight
- BW
Body weight
- TL
Tibia length
- CSA
Cross-sectional area
- WGA
Wheat germ agglutinin
- RVWT
Right ventricle wall thickness
- PSLAX
Parasternal long axis
- EF
Ejection fraction
- Myh6
Myosin heavy chain 6
- GFP
Green fluorescent protein
- RFP
Red fluorescent protein
- LC-MS
Liquid chromatography-Mass spectrometry
- MC
Male control
- MH
Male hypoxia
- FC
Female control
- FH
Female hypoxia
- GO
Gene ontology
- CDK
Cyclin-dependent kinases
- ROS
Reactive oxygen species
- SA
Surface area
- αSA
Alpha-sarcomeric actin
- αSMA
Alpha-smooth muscle actin
- PSR
Picro-sirius red
Footnotes
Disclosures: The authors have declared that they have no conflict of interest.
References
- 1.Tsao CW, Aday AW, Almarzooq ZI, Alonso A, Beaton AZ, Bittencourt MS, Boehme AK, Buxton AE, Carson AP, Commodore-Mensah Y, et al. Heart disease and stroke statistics-2022 update: A report from the american heart association. Circulation. 2022;145:e153–e639 [DOI] [PubMed] [Google Scholar]
- 2.Frangogiannis NG. Pathophysiology of myocardial infarction. Compr Physiol 2015;5:1841–1875 [DOI] [PubMed] [Google Scholar]
- 3.Richards DA, Denniss AR. Assessment, significance and mechanism of ventricular electrical instability after myocardial infarction. Heart Lung Circ. 2007;16:149–155 [DOI] [PubMed] [Google Scholar]
- 4.Swan HJ. Left ventricular systolic and diastolic dysfunction in the acute phases of myocardial ischaemia and infarction, and in the later phases of recovery. Function follows morphology. Eur Heart J. 1993;14 Suppl A:48–56 [DOI] [PubMed] [Google Scholar]
- 5.Thune JJ, Solomon SD. Left ventricular diastolic function following myocardial infarction. Curr Heart Fail Rep. 2006;3:170–174 [DOI] [PubMed] [Google Scholar]
- 6.Bahit MC, Kochar A, Granger CB. Post-myocardial infarction heart failure. JACC Heart Fail. 2018;6:179–186 [DOI] [PubMed] [Google Scholar]
- 7.Jenča D, Melenovský V, Stehlik J, Staněk V, Kettner J, Kautzner J, Adámková V, Wohlfahrt P. Heart failure after myocardial infarction: Incidence and predictors. ESC Heart Fail. 2021;8:222–237 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 8.Wu MY, Yiang GT, Liao WT, Tsai AP, Cheng YL, Cheng PW, Li CY, Li CJ. Current mechanistic concepts in ischemia and reperfusion injury. Cell Physiol Biochem. 2018;46:1650–1667 [DOI] [PubMed] [Google Scholar]
- 9.Luo L, Li TS. Mini review: Recent advances in the cell-based therapies for cardiac regeneration. Curr Stem Cell Res Ther. 2020;15:649–660 [DOI] [PubMed] [Google Scholar]
- 10.Tzahor E, Poss KD. Cardiac regeneration strategies: Staying young at heart. Science. 2017;356:1035–1039 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 11.Zhang Y, Mignone J, MacLellan WR. Cardiac regeneration and stem cells. Physiol Rev. 2015;95:1189–1204 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 12.Giacca M Cardiac regeneration after myocardial infarction: An approachable goal. Curr Cardiol Rep. 2020;22:122. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 13.Hashimoto H, Olson EN, Bassel-Duby R. Therapeutic approaches for cardiac regeneration and repair. Nat Rev Cardiol. 2018;15:585–600 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 14.de Wit L, Fang J, Neef K, Xiao J, P AD, Schiffelers RM, Lei Z, Sluijter JPG. Cellular and molecular mechanism of cardiac regeneration: A comparison of newts, zebrafish, and mammals. Biomolecules. 2020;10. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 15.Jopling C, Sleep E, Raya M, Martí M, Raya A, Izpisúa Belmonte JC. Zebrafish heart regeneration occurs by cardiomyocyte dedifferentiation and proliferation. Nature. 2010;464:606–609 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 16.Sanz-Morejón A, Mercader N. Recent insights into zebrafish cardiac regeneration. Curr Opin Genet Dev. 2020;64:37–43 [DOI] [PubMed] [Google Scholar]
- 17.Porrello ER, Mahmoud AI, Simpson E, Hill JA, Richardson JA, Olson EN, Sadek HA. Transient regenerative potential of the neonatal mouse heart. Science. 2011;331:1078–1080 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 18.Uygur A, Lee RT. Mechanisms of cardiac regeneration. Dev Cell. 2016;36:362–374 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 19.Bergmann O, Bhardwaj RD, Bernard S, Zdunek S, Barnabé-Heider F, Walsh S, Zupicich J, Alkass K, Buchholz BA, Druid H, et al. Evidence for cardiomyocyte renewal in humans. Science. 2009;324:98–102 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 20.Bergmann O, Zdunek S, Felker A, Salehpour M, Alkass K, Bernard S, Sjostrom SL, Szewczykowska M, Jackowska T, Dos Remedios C, et al. Dynamics of cell generation and turnover in the human heart. Cell. 2015;161:1566–1575 [DOI] [PubMed] [Google Scholar]
- 21.Johnson J, Mohsin S, Houser SR. Cardiomyocyte proliferation as a source of new myocyte development in the adult heart. Int J Mol Sci. 2021;22. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 22.Zhu F, Meng Q, Yu Y, Shao L, Shen Z. Adult cardiomyocyte proliferation: A new insight for myocardial infarction therapy. J Cardiovasc Transl Res. 2021;14:457–466 [DOI] [PubMed] [Google Scholar]
- 23.Nakada Y, Canseco DC, Thet S, Abdisalaam S, Asaithamby A, Santos CX, Shah AM, Zhang H, Faber JE, Kinter MT, et al. Hypoxia induces heart regeneration in adult mice. Nature. 2017;541:222–227 [DOI] [PubMed] [Google Scholar]
- 24.Henner A, Ventura PB, Jiang Y, Zong H. Madm-ml, a mouse genetic mosaic system with increased clonal efficiency. PLoS One. 2013;8:e77672. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 25.Cantor EJ, Babick AP, Vasanji Z, Dhalla NS, Netticadan T. A comparative serial echocardiographic analysis of cardiac structure and function in rats subjected to pressure or volume overload. J Mol Cell Cardiol. 2005;38:777–786 [DOI] [PubMed] [Google Scholar]
- 26.Shimoda LA. Cellular pathways promoting pulmonary vascular remodeling by hypoxia. Physiology (Bethesda). 2020;35:222–233 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 27.Julian RJ. The response of the heart and pulmonary arteries to hypoxia, pressure, and volume. A short review. Poult Sci. 2007;86:1006–1011 [DOI] [PubMed] [Google Scholar]
- 28.Becker C, Hesse M. Role of mononuclear cardiomyocytes in cardiac turnover and regeneration. Curr Cardiol Rep. 2020;22:39. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 29.Bersell K, Arab S, Haring B, Kühn B. Neuregulin1/erbb4 signaling induces cardiomyocyte proliferation and repair of heart injury. Cell. 2009;138:257–270 [DOI] [PubMed] [Google Scholar]
- 30.Kimura W, Xiao F, Canseco DC, Muralidhar S, Thet S, Zhang HM, Abderrahman Y, Chen R, Garcia JA, Shelton JM, et al. Hypoxia fate mapping identifies cycling cardiomyocytes in the adult heart. Nature. 2015;523:226–230 [DOI] [PubMed] [Google Scholar]
- 31.Liao HS, Kang PM, Nagashima H, Yamasaki N, Usheva A, Ding B, Lorell BH, Izumo S. Cardiac-specific overexpression of cyclin-dependent kinase 2 increases smaller mononuclear cardiomyocytes. Circ Res. 2001;88:443–450 [DOI] [PubMed] [Google Scholar]
- 32.Barr AR, Cooper S, Heldt FS, Butera F, Stoy H, Mansfeld J, Novák B, Bakal C. DNA damage during s-phase mediates the proliferation-quiescence decision in the subsequent g1 via p21 expression. Nat Commun. 2017;8:14728. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 33.Buck SB, Bradford J, Gee KR, Agnew BJ, Clarke ST, Salic A. Detection of s-phase cell cycle progression using 5-ethynyl-2’-deoxyuridine incorporation with click chemistry, an alternative to using 5-bromo-2’-deoxyuridine antibodies. Biotechniques. 2008;44:927–929 [DOI] [PubMed] [Google Scholar]
- 34.Salic A, Mitchison TJ. A chemical method for fast and sensitive detection of DNA synthesis in vivo. Proc Natl Acad Sci U S A. 2008;105:2415–2420 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 35.Kohlmeier F, Maya-Mendoza A, Jackson DA. Edu induces DNA damage response and cell death in mesc in culture. Chromosome Res. 2013;21:87–100 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 36.Zhao H, Halicka HD, Li J, Biela E, Berniak K, Dobrucki J, Darzynkiewicz Z. DNA damage signaling, impairment of cell cycle progression, and apoptosis triggered by 5-ethynyl-2’-deoxyuridine incorporated into DNA. Cytometry A. 2013;83:979–988 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 37.Tasic B, Miyamichi K, Hippenmeyer S, Dani VS, Zeng H, Joo W, Zong H, Chen-Tsai Y, Luo L. Extensions of madm (mosaic analysis with double markers) in mice. PLoS One. 2012;7:e33332. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 38.Chua P, Jinks-Robertson S. Segregation of recombinant chromatids following mitotic crossing over in yeast. Genetics. 1991;129:359–369 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 39.Bersell K, Choudhury S, Mollova M, Polizzotti BD, Ganapathy B, Walsh S, Wadugu B, Arab S, Kühn B. Moderate and high amounts of tamoxifen in αmhc-mercremer mice induce a DNA damage response, leading to heart failure and death. Dis Model Mech. 2013;6:1459–1469 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 40.Cardoso AC, Lam NT, Savla JJ, Nakada Y, Pereira AHM, Elnwasany A, Menendez-Montes I, Ensley EL, Petric UB, Sharma G, et al. Mitochondrial substrate utilization regulates cardiomyocyte cell cycle progression. Nat Metab. 2020;2:167–178 [PMC free article] [PubMed] [Google Scholar]
- 41.He L, Binari R, Huang J, Falo-Sanjuan J, Perrimon N. In vivo study of gene expression with an enhanced dual-color fluorescent transcriptional timer. Elife. 2019;8. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 42.Jang SM, Redon CE, Thakur BL, Bahta MK, Aladjem MI. Regulation of cell cycle drivers by cullin-ring ubiquitin ligases. Exp Mol Med. 2020;52:1637–1651 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 43.Li T, Wu S, Jia L, Cao W, Yao Y, Zhao G, Li H. Cul4 e3 ligase regulates the proliferation and apoptosis of lung squamous cell carcinoma and small cell lung carcinoma. Cancer Biol Med. 2020;17:357–370 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 44.Meyer SJ, Böser A, Korn MA, Koller C, Bertocci B, Reimann L, Warscheid B, Nitschke L. Cullin 3 is crucial for pro-b cell proliferation, interacts with cd22, and controls cd22 internalization on b cells. J Immunol. 2020;204:3360–3374 [DOI] [PubMed] [Google Scholar]
- 45.Hatzistergos KE, Williams AR, Dykxhoorn D, Bellio MA, Yu W, Hare JM. Tumor suppressors rb1 and cdkn2a cooperatively regulate cell-cycle progression and differentiation during cardiomyocyte development and repair. Circ Res. 2019;124:1184–1197 [DOI] [PubMed] [Google Scholar]
- 46.Lee J, Kim K, Ryu TY, Jung CR, Lee MS, Lim JH, Park K, Kim DS, Son MY, Hamamoto R, et al. Ehmt1 knockdown induces apoptosis and cell cycle arrest in lung cancer cells by increasing cdkn1a expression. Mol Oncol. 2021;15:2989–3002 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 47.Yang Z, Liao B, Xiang X, Ke S. Mir-21-5p promotes cell proliferation and g1/s transition in melanoma by targeting cdkn2c. FEBS Open Bio. 2020;10:752–760 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 48.Prabhu SD, Frangogiannis NG. The biological basis for cardiac repair after myocardial infarction: From inflammation to fibrosis. Circ Res. 2016;119:91–112 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 49.Barak Y, Hemberger M, Sucov HM. Phases and mechanisms of embryonic cardiomyocyte proliferation and ventricular wall morphogenesis. Pediatr Cardiol. 2019;40:1359–1366 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 50.Barak Y, Hemberger M, Sucov HM. Correction to: Phases and mechanisms of embryonic cardiomyocyte proliferation and ventricular wall morphogenesis. Pediatr Cardiol. 2020;41:220. [DOI] [PubMed] [Google Scholar]
- 51.Samsa LA, Yang B, Liu J. Embryonic cardiac chamber maturation: Trabeculation, conduction, and cardiomyocyte proliferation. Am J Med Genet C Semin Med Genet. 2013;163c:157–168 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 52.Salama ABM, Gebreil A, Mohamed TMA, Abouleisa RRE. Induced cardiomyocyte proliferation: A promising approach to cure heart failure. Int J Mol Sci. 2021;22. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 53.Vagnozzi RJ, Molkentin JD, Houser SR. New myocyte formation in the adult heart: Endogenous sources and therapeutic implications. Circ Res. 2018;123:159–176 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 54.Gu J, Chen X, Jin Y, Liu M, Xu Q, Liu X, Luo Z, Ling S, Liu N, Liu S. A neonatal mouse model for pressure overload: Myocardial response corresponds to severity. Front Cardiovasc Med. 2021;8:660246. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 55.Kirova DG, Judasova K, Vorhauser J, Zerjatke T, Leung JK, Glauche I, Mansfeld J. A ros-dependent mechanism promotes cdk2 phosphorylation to drive progression through s phase. Dev Cell. 2022;57:1712–1727.e1719 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 56.Liu X, Pu W, He L, Li Y, Zhao H, Li Y, Liu K, Huang X, Weng W, Wang QD, et al. Cell proliferation fate mapping reveals regional cardiomyocyte cell-cycle activity in subendocardial muscle of left ventricle. Nat Commun. 2021;12:5784. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 57.Fajersztajn L, Veras MM. Hypoxia: From placental development to fetal programming. Birth Defects Res. 2017;109:1377–1385 [DOI] [PubMed] [Google Scholar]
- 58.Fong GH. Mechanisms of adaptive angiogenesis to tissue hypoxia. Angiogenesis. 2008;11:121–140 [DOI] [PubMed] [Google Scholar]
- 59.Forsythe JA, Jiang BH, Iyer NV, Agani F, Leung SW, Koos RD, Semenza GL. Activation of vascular endothelial growth factor gene transcription by hypoxia-inducible factor 1. Mol Cell Biol. 1996;16:4604–4613 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 60.Ahuja P, Perriard E, Pedrazzini T, Satoh S, Perriard JC, Ehler E. Re-expression of proteins involved in cytokinesis during cardiac hypertrophy. Exp Cell Res. 2007;313:1270–1283 [DOI] [PubMed] [Google Scholar]
- 61.Ali SR, Hippenmeyer S, Saadat LV, Luo L, Weissman IL, Ardehali R. Existing cardiomyocytes generate cardiomyocytes at a low rate after birth in mice. Proc Natl Acad Sci U S A. 2014;111:8850–8855 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 62.Sohal DS, Nghiem M, Crackower MA, Witt SA, Kimball TR, Tymitz KM, Penninger JM, Molkentin JD. Temporally regulated and tissue-specific gene manipulations in the adult and embryonic heart using a tamoxifen-inducible cre protein. Circ Res. 2001;89:20–25 [DOI] [PubMed] [Google Scholar]
- 63.Boehm M, Lawrie A, Wilhelm J, Ghofrani HA, Grimminger F, Weissmann N, Seeger W, Schermuly RT, Kojonazarov B. Maintained right ventricular pressure overload induces ventricular-arterial decoupling in mice. Exp Physiol. 2017;102:180–189 [DOI] [PubMed] [Google Scholar]
- 64.Cheng HW, Fisch S, Cheng S, Bauer M, Ngoy S, Qiu Y, Guan J, Mishra S, Mbah C, Liao R. Assessment of right ventricular structure and function in mouse model of pulmonary artery constriction by transthoracic echocardiography. J Vis Exp. 2014:e51041. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 65.Makarewich CA, Zhang H, Davis J, Correll RN, Trappanese DM, Hoffman NE, Troupes CD, Berretta RM, Kubo H, Madesh M, et al. Transient receptor potential channels contribute to pathological structural and functional remodeling after myocardial infarction. Circ Res. 2014;115:567–580 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 66.Wallner M, Duran JM, Mohsin S, Troupes CD, Vanhoutte D, Borghetti G, Vagnozzi RJ, Gross P, Yu D, Trappanese DM, et al. Acute catecholamine exposure causes reversible myocyte injury without cardiac regeneration. Circ Res. 2016;119:865–879 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 67.Zhang H, Makarewich CA, Kubo H, Wang W, Duran JM, Li Y, Berretta RM, Koch WJ, Chen X, Gao E, et al. Hyperphosphorylation of the cardiac ryanodine receptor at serine 2808 is not involved in cardiac dysfunction after myocardial infarction. Circ Res. 2012;110:831–840 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 68.Ge SX, Son EW, Yao R. iDEP: an integrated web application for differential expression and pathway analysis of RNA-Seq data. BMC Bioinformatics. 2018;19:534. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 69.Edgar R, Domrachev M, Lash AE. Gene Expression Omnibus: NCBI gene expression and hybridization array data repository. Nucleic Acids Res. 2002;30:207–210 [DOI] [PMC free article] [PubMed] [Google Scholar]
Associated Data
This section collects any data citations, data availability statements, or supplementary materials included in this article.
Supplementary Materials
Data Availability Statement
The data, materials, and methods used in this study to perform research will be available to any researcher for purposes of reproducing the results.