

Abstract
Background & Aims:
Injury to biliary epithelial cells (BECs) lining the hepatic bile ducts leads to cholestatic liver diseases. Upon severe biliary damage, hepatocytes can convert to BECs, thereby contributing to liver recovery. Given a potential of augmenting this hepatocyte-to-BEC conversion as a therapeutic option for cholestatic liver diseases, it will be important to thoroughly understand the cellular and molecular mechanisms of the conversion process.
Approach & Results:
Towards this aim, we have established a zebrafish model for hepatocyte-to-BEC conversion by employing Tg(fabp10a:CFP-NTR) zebrafish with a temporal inhibition of Notch signaling during regeneration. Cre/loxP-mediated permanent and H2B-mCherry-mediated short-term lineage tracing revealed that in the model, all BECs originate from hepatocytes. During the conversion, BEC markers are sequentially induced in the order of Sox9b, Yap/Taz, Notch activity/epcam, and Alcama/krt18; the expression of the hepatocyte marker Bhmt disappears between the Sox9b and Yap/Taz induction. Importantly, live time-lapse imaging unambiguously revealed transdifferentiation of hepatocytes into BECs: hepatocytes convert to BECs without transitioning through a proliferative intermediate state. In addition, using compounds and transgenic and mutant lines that modulate Notch and Yap signaling, we found that both Notch and Yap signaling are required for the conversion even in Notch- and Yap-overactivating settings.
Conclusions:
Hepatocyte-to-BEC conversion occurs through transdifferentiation independently of proliferation, and Notch and Yap signaling control the process in parallel with a mutually positive interaction. The new zebrafish model will further contribute to a thorough understanding of the mechanisms of the conversion process.
Keywords: liver, regeneration, biliary epithelial cell, Notch, Yap
Introduction
Biliary epithelial cells (BECs; also called cholangiocytes) that line the hepatic bile ducts control bile composition and flow. Injury to the BECs leads to cholestasis, which can progress to fibrosis, cirrhosis, and liver failure (1). Cholestatic liver diseases are associated with high morbidity and mortality; however, few effective therapies are available. In fact, liver transplantation is the only life-extending treatment for end-stage cholestatic liver diseases, but the shortage of donated livers makes this therapy extremely limited (2). Upon severe biliary damage, hepatocytes can convert to BECs, thereby contributing to the restoration of bile flow. This conversion phenomenon was first reported more than 20 years ago in rats by Dr. Michalopoulos’ group (3) and later confirmed in mice by several other groups (4–8). Importantly, recent studies in mice have shown that hepatocyte-derived BECs contribute to the intrahepatic bile ducts, thereby restoring appropriate bile flow (9). Patients with biliary obstruction or cholangiopathies also exhibit biliary marker expression in hepatocytes, suggesting their conversion to BECs (10). Thus, augmenting innate hepatocyte-to-BEC conversion in cholestatic liver diseases is an attractive therapeutic alternative to ameliorate cholestasis and subsequent cirrhosis. To develop such therapy, it is crucial to better understand the cellular and molecular mechanisms underlying hepatocyte-to-BEC conversion.
Currently, (1) Notch, (2) Yap, (3) Wnt/β-catenin, and (4) TGFβ signaling are implicated in regulating the conversion process. (1) Hepatocyte-specific overexpression of Notch intracellular domain (NICD) induced hepatocyte-to-BEC conversion (4, 11, 12), while hepatocyte-specific deletion of Rbpj (the principal mediator of Notch signaling) (4) or Hes1 (a key effector of Notch signaling) (6) significantly reduced the number of hepatocyte-derived BECs in mice fed a 3,5-diethoxycarbonyl-1,4-dihydrocollidine (DDC) diet. (2) Similarly, hepatocyte-specific overexpression of constitutive-active YAP1 induced hepatocyte-to-BEC conversion (7, 13, 14), while hepatocyte-specific deletion of Yap1 greatly reduced ductular reactions and BEC marker induction in hepatocytes (15–17). Intriguingly, in the YAP1 overexpression setting, Notch signaling is a downstream target of YAP signaling (7), while in the NICD overexpression setting, Yap signaling is a downstream target of Notch signaling (12), suggesting reciprocal regulation between Notch and Yap signaling. (3) Hepatocyte-specific overexpression of a stabilized form of β-catenin in mice fed a DDC diet induced the expression of BEC markers in hepatocytes (18), suggesting the positive role of Wnt/β-catenin signaling in hepatocyte-to-BEC conversion. (4) Lastly, mice without Rbpj and Hnf6 in both hepatocytes and BECs initially lack peripheral bile ducts, but later, fully functional bile ducts recover through hepatocyte-to-BEC conversion (9). Additional deletion of Tgfbr2 blocked this bile duct recovery, while hepatocyte-specific overexpression of constitutive-active TGFBR1 promoted the recovery (9), indicating the essential role of TGFβ signaling in hepatocyte-to-BEC conversion in the absence of Notch signaling. However, other than these four signaling pathways, the molecular mechanisms of hepatocyte-to-BEC conversion remain largely unknown. Moreover, given the well-known effect of the four signaling pathways on cell proliferation, it is not clear whether the increased number of hepatocyte-derived BECs in the overexpression settings was due to enhanced conversion, proliferation, or both. The epistatic relationships among the signaling pathways in the conversion process (e.g., upstream, downstream, or in parallel) are also not well defined.
At the cellular level, two mechanisms have been suggested to illustrate how hepatocytes convert to BECs: (1) hepatocytes first dedifferentiate into liver progenitor cells (LPCs; also called oval cells or ductular reactions) and then LPCs differentiate into BECs; (2) hepatocytes directly convert to BECs without transitioning through an intermediate proliferative state, i.e., via transdifferentiation. The first mechanism has been suggested from the findings that hepatocytes gave rise to LPCs and functional BECs in mouse liver injury models inducing LPCs (4). However, this hepatocyte-lineage tracing cannot rule out the possibility that some hepatocytes directly converted to BECs, while other hepatocytes converted to LPCs. The second mechanism has been suggested from mouse studies with liver-specific Rbpj and Hnf6 double knockout mice (9). Clonal analyses of hepatocyte fates in these mice revealed a majority of hepatocyte-derived BECs as one-cell-size clones, suggesting transdifferentiation rather than proliferation-involved dedifferentiation and redifferentiation (9). Although this clonal analysis suggests transdifferentiation, live time-lapse imaging in vivo should unambiguously determine if hepatocytes convert to BECs with or without a proliferative intermediate state.
To better understand the cellular and molecular mechanisms of hepatocyte-to-BEC conversion, we have developed a novel zebrafish model in which the conversion occurs rapidly and robustly with all regenerated BECs originating from hepatocytes. In this model, hepatocyte ablation combined with temporal Notch inhibition during BEC-driven liver regeneration (HANI) generates zebrafish that completely lack BECs in the liver, and within two days after releasing Notch inhibition, BECs reappear through hepatocyte-to-BEC conversion. Using the HANI model, we here confirm the essential role of Notch and Yap signaling in hepatocyte-to-BEC conversion and clearly delineate the epistatic relationship between the two signaling pathways in the process. Furthermore, using live time-lapse imaging, we reveal that hepatocytes transdifferentiate into BECs without transitioning through an intermediate proliferative state.
Experimental procedures
Zebrafish lines
Experiments were performed with approval of the Institutional Animal Care and Use Committee at the University of Pittsburgh. Embryos and adult fish were raised and maintained under standard laboratory conditions (19). We used yap1bns19, fbxw7vu56, notch3fh332, and casper (mitfaw2;mpv17a9) mutant lines and following transgenic lines: Tg(krt18:Venus)pt622, Tg(Tp1:VenusPEST)s940, Tg(Tp1:H2B-mCherry)s939, Tg(fabp10a:H2B-mCherry)pt623, Tg(fabp10a:CFP-NTR)s931, TgBAC(sox9b:GAL4FF)pt621, Tg(UAS:Kaede)rk8, Tg(fabp10a:CreERT2)pt602, Tg(ubb:loxP-CFP-loxP-H2B-mCherry)jh63, Tg(hs:cayap1)zf622, Tg(hs:dnyap1)zf621, and Tg(hs:canotch3)co17. Their full names and references are listed in Table S1.
Hepatocyte ablation using Tg(fabp10a:CFP-NTR) larvae
To ablate hepatocytes, Tg(fabp10a:CFP-NTR) larvae were treated with 10 mM metronidazole (Mtz) in egg water supplemented with 0.2% dimethyl sulfoxide (DMSO) and 0.2 mM 1‐phenyl‐2‐thiourea from 3.5 to 5 days post-fertilization (dpf) for 36 hours, as previously described (20). The ablated larvae were further treated with 10 μM LY411575 from 4.7 to 6 dpf for 40 hours to generate livers without BECs.
Additional methods are available in Supporting Information.
Results
A zebrafish model for the robust conversion of hepatocytes to BECs
We have previously established the hepatocyte ablation model for BEC-driven liver regeneration: Tg(fabp10a:CFP-NTR)s931 fish that express bacterial nitroreductase (NTR) fused with a cyan fluorescent protein (CFP) under the hepatocyte-specific fabp10a promoter (20). NTR metabolizes nontoxic Mtz into a cytotoxic drug (21, 22); as such, Mtz treatment specifically ablates hepatocytes in the fish. In this model, preexisting hepatocytes all die; preexisting BECs all dedifferentiate into LPCs; and subsequently, the LPCs differentiate into either hepatocytes or BECs (20). Given the essential role of Notch signaling in BEC formation during development (23) and regeneration (4, 24), we anticipated that inhibiting Notch signaling during BEC-driven liver regeneration would generate zebrafish that lack BECs in the liver. Indeed, inhibiting Notch signaling with the γ-secretase inhibitor, LY411575 (25), during the regeneration generated livers with no BECs (Fig. 1A and B). Tg(fabp10a:CFP-NTR) larvae were treated with Mtz from 3.5 to 5 days dpf for 36 hours (ablation, A36h) and LY411575 from A20h to 6 dpf for 40 hours. Subsequent LY411575 washout was considered the beginning of biliary regeneration (BR0h). This LY411575 treatment completely blocked LPC-to-BEC differentiation but did not impair LPC-to-hepatocyte differentiation (Fig. S1A), thereby generating livers with no BECs. Following LY411575 washout (BR0h), BECs rapidly reappeared in the liver: 0-2 cells at BR24h, 15-20 cells at BR48h, and 35-50 cells at BR72h (Fig. 1A and B), as assessed by the expression of Tp1:VenusPEST and Tp1:H2B-mCherry, which reveal the cytoplasm and nuclei of BECs, respectively (20). The time-course analyses of the expression of multiple BEC markers (Sox9b, epcam, Alcama, Tp1:VenusPEST, and krt18:Venus) during biliary regeneration revealed the sequential induction of Sox9b, Notch activity (Tp1:VenusPEST)/epcam, and Alcama/krt18:Venus during hepatocyte-to-BEC conversion (Fig. 1B, S1B, S1D). Given the essential role of Yap signaling in BEC formation during development (26) and in hepatocyte-to-BEC reprogramming (7, 13), we also examined Yap/Taz expression using antibodies recognizing both Yap and Taz. Yap/Taz expression was detected specifically in BECs and sequentially after Sox9b but before Tp1:VenusPEST expression (Fig. 1B and S1C). Thus, hepatocyte-to-BEC conversion in the HANI model progresses through the sequential induction of Sox9b, Yap/Taz, Tp1:VenusPEST/epcam, and Alcama/krt18:Venus (Fig. 1C).
Figure 1. A zebrafish model for hepatocyte-to-BEC conversion.
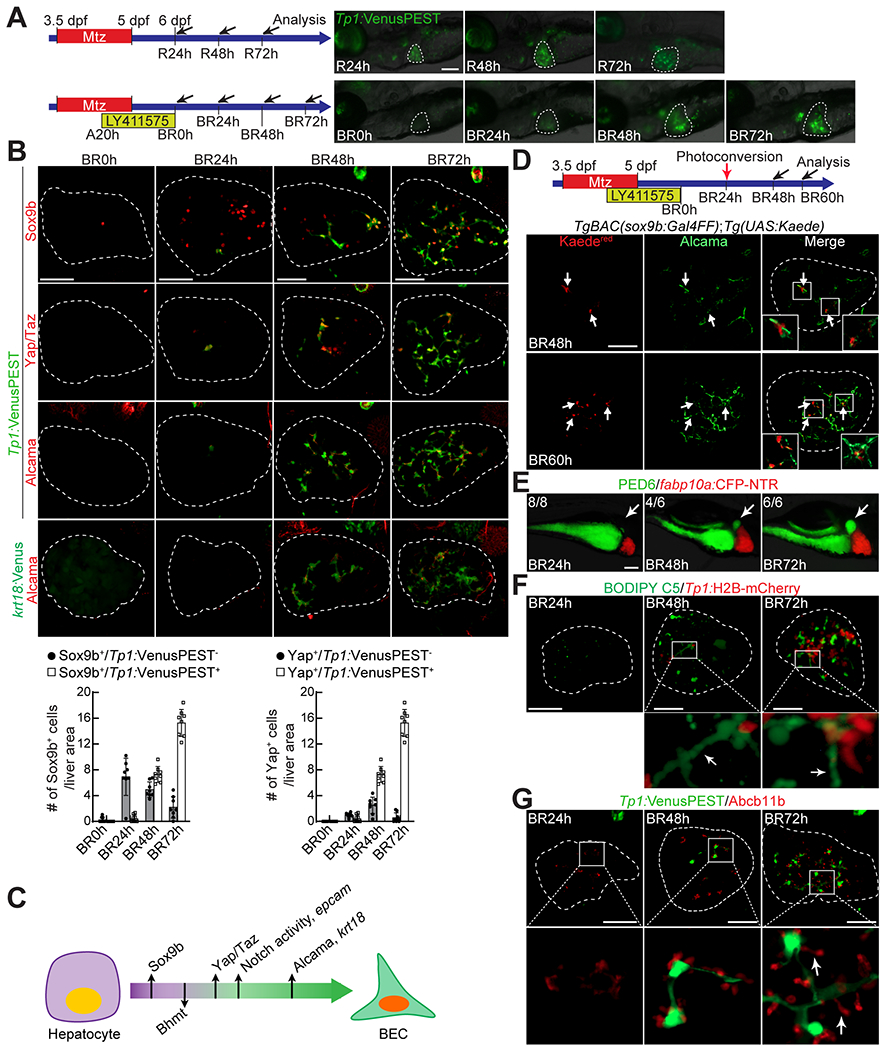
(A) Epifluorescence images showing Tp1:VenusPEST expression in regenerating larvae. Scheme illustrates the periods of Mtz and LY411575 treatments and the stages of liver and biliary regeneration (R and BR, respectively) and analysis (arrows). (B) Confocal projection images showing the expression of Sox9b, Yap/Taz, Alcama, Tp1:VenusPEST, and krt18:Venus in regenerating livers (dashed lines). The number of Sox9b+ or Yap/Taz+ cells with or without Tp1:VenusPEST expression is quantified and presented as mean ± SD. (C) Scheme illustrating the order of the induction of BEC markers and the disappearance of the hepatocyte marker during hepatocyte-to-BEC conversion. (D) Confocal projection images showing the expression of Alcama and Kaedered in regenerating livers. The TgBAC(sox9b:Gal4FF) and Tg(UAS:Kaede) lines were used to trace sox9b+ cells (arrows) by exposing the transgenic larvae to UV at BR24h. (E) Epifluorescence images showing PED6 accumulation in the gallbladder (arrows) and fabp10a:CFP-NTR expression in regenerating livers. Numbers in the upper left corner indicate the proportion of larvae exhibiting the same level of the gallbladder accumulation shown. (F) Confocal projection images showing BODIPY C5 staining and Tp1:H2B-mCherry expression in regenerating livers. Boxed regions are enlarged below; arrows point to well elongated bile ductules. (G) Confocal projection images showing Abcb11b and Tp1:VenusPEST expression in regenerating livers. Boxed regions are enlarged below; arrows point to well extended bile canaliculi. Dashed lines circle the livers. Scale bars: 50 (B,D,F,G), 100 (A,E) μm.
We also examined when the expression of the hepatocyte marker Bhmt disappears during the conversion. Bhmt was expressed in a subset of Sox9b+ cells at BR24h and BR48h (Fig. S1E). These Bhmt+/Sox9b+ cells are Tp1:VenusPEST− (Fig. S1E), indicating that Bhmt expression disappears following Sox9b induction but before Tp1:VenusPEST induction. Moreover, Bhmt expression was not detected in Yap/Taz+ cells at BR24h (Fig. S1F). Thus, Bhmt expression disappears following Sox9b but before Yap/Taz induction during hepatocyte-to-BEC conversion in the HANI model (Fig. 1C)
This sequential event suggests Sox9b as the earliest marker for hepatocyte-to-BEC conversion. However, it is possible that Sox9b+/Bhmt+/Tp1:VenusPEST− cells later lose their Sox9b expression and remain as hepatocytes, instead of turning on Tp1:VenusPEST expression and giving rise to BECs. To rule out this possibility, we traced the lineage of Sox9b+ cells present at BR24h using the photoconversion of Kaede proteins, which can be irreversibly converted from green (Kaedegreen) to red (Kaedered) fluorescence upon UV irradiation (27). The TgBAC(sox9b:Gal4FF) and Tg(UAS:Kaede) lines were used to express Kaede specifically in Sox9b-expressing cells; Kaede was photoconverted at BR24h (Fig. 1D). All Kaedered+ cells expressed Alcama at BR48h and BR60h (Fig. 1D), indicating that all Sox9b+ cells at BR24h gave rise to BECs. Using Sox9b as the earliest marker for hepatocyte-to-BEC conversion together with hematoxylin staining, we revealed that the nuclear morphology of hepatocytes converting to BECs changed from round to oval (Fig. S1G, arrowheads versus arrows). Moreover, phalloidin staining, which labels F-actin, revealed that the size of Sox9b+ cells decreased during hepatocyte-to-BEC conversion even before Notch signaling turns on (Fig. S1H).
Next, we examined when bile flow via the intrahepatic bile ducts recovers in the HANI model using a fluorescently labeled fatty acid reporter, PED6, which is metabolized into a component of bile in hepatocytes and accumulates in the gallbladder after biliary secretion (28). At BR24h, there was no PED6 accumulation in the gallbladder yet; however, at BR48h, most larvae exhibited some PED6 accumulation, and importantly at BR72h, all larvae exhibited strong PED6 accumulation (Fig. 1E, arrows), indicating a full recovery of bile flow at BR72h. Additionally, we used the fluorescently labeled lipid reporter BODIPY C5 to reveal intrahepatic bile conduits (29). At BR24h, such conduits were not revealed by BODIPY C5 fluorescence at all; however, at BR48h, some bile conduits were revealed, and at BR72h, more conduits were revealed (Fig. 1F, arrows). Given that bile is secreted from hepatocytes into bile ducts through bile canaliculi, we also examined bile canaliculi by assessing the expression of Abcb11b, a bile salt export pump present in the bile canaliculi of hepatocytes (30). At BR48h, Abcb11b expression was detected close to BECs, but bile canaliculi were not fully elongated yet. However, at BR72h, bile canaliculi were fully elongated (Fig. 1G, arrows), supporting a full recovery of bile flow at BR72h. Altogether, we have developed a zebrafish model for hepatocyte-to-BEC conversion, the HANI model, in which hepatocytes robustly convert to BECs in the liver that lacks BECs.
All BECs originate from hepatocytes in the HANI model
To unambiguously determine the origin of BECs in the HANI model, we first performed a short-term lineage tracing using the Tg(fabp10a:H2B-mCherry) line, which expresses H2B-mCherry fusion proteins under the hepatocyte-specific fabp10a promoter. The long half-life of histone H2B makes its fusion proteins very stable, allowing for a short-term lineage tracing. All BECs positive for krt18:Venus and Alcama at BR72h weakly expressed fabp10a:H2B-mCherry (Fig. 2A, arrows), indicating their hepatocyte origin. Second, we performed a permanent lineage tracing using the Cre/loxP system. The Tg(fabp10a:CreERT2)pt602 line (20) together with a Cre reporter line (31), Tg(ubb:loxP-CFP-loxP-H2B-mCherry)jh63, was used to permanently label hepatocytes with H2B-mCherry expression. Larvae were treated with 4-hydroxytamoxifen (4-OHT) from BR0h to BR6h for 6 hours to label hepatocytes before BEC formation. ~93% of hepatocytes expressing Bhmt were labeled with ubb:H2B-mCherry at BR24h; nearly all BECs expressing Alcama were positive for ubb:H2B-mCherry at BR72h (Fig. 2B, arrows). These two types of lineage tracing data demonstrate that in the HANI model, all BECs originate from hepatocytes.
Figure 2. All BECs originate from hepatocytes in the HANI model.
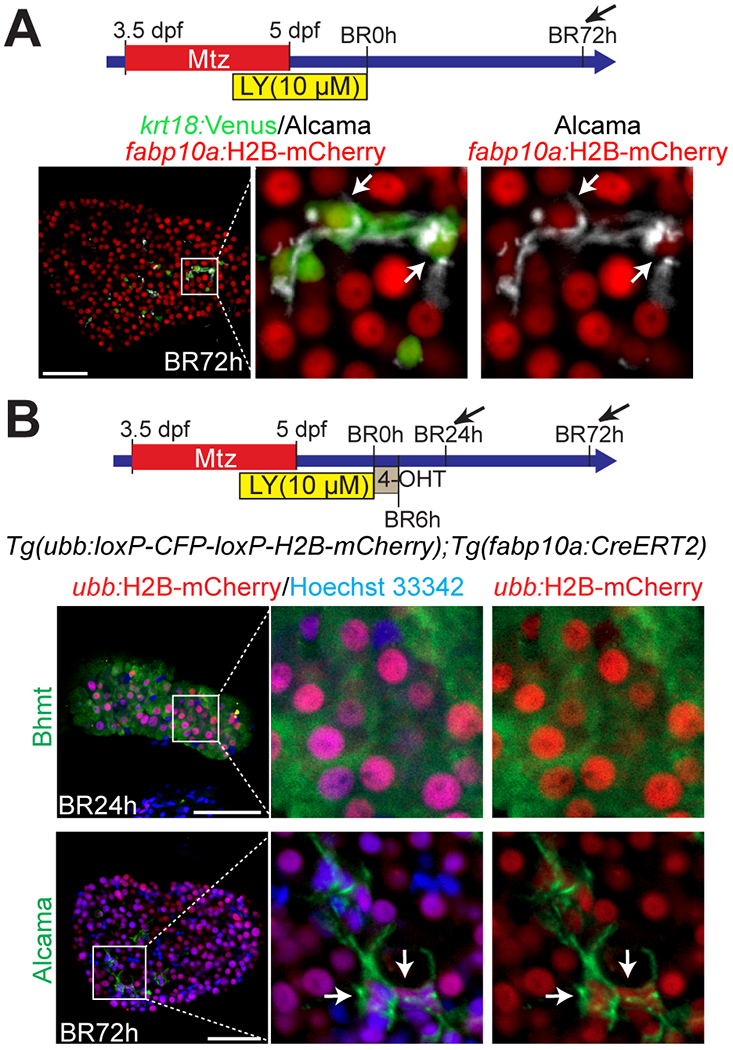
(A) Confocal single-optical section images showing the expression of fabp10a:H2B-mCherry, krt18:Venus, and Alcama in the regenerating liver at BR72h. All BECs weakly expressed fabp10a:H2B-mCherry (arrows). (B) Confocal single-optical section images showing Hoechst 33342 staining and the expression of H2B-mCherry and Bhmt or Alcama in regenerating livers. The Tg(ubb:loxP-CFP-loxP-H2B-mCherry) and Tg(fabp10a:CreERT2) lines were used to trace the lineage of hepatocytes. The transgenic larvae were treated with 4-OHT from BR0h to BR6h, which labeled ~93% of hepatocytes at BR24h. All Alcama+ BECs (arrows) expressed H2B-mCherry at BR72h, indicating their hepatocyte origin. Boxed regions are enlarged right. Scale bars: 50 μm.
Hepatocytes convert to BECs without transitioning through an intermediate proliferative state
It has been suggested that hepatocytes convert to BECs through an intermediate proliferative state, i.e., LPCs (4). On the contrary, it has also been suggested that hepatocytes transdifferentiate into BECs without proliferation (9). By performing live time-lapse imaging, we sought to determine if hepatocyte-to-BEC conversion in the HANI model involves an intermediate proliferative state. The Tg(fabp10a:H2B-mCherry) and Tg(Tp1:VenusPEST) lines were used to reveal hepatocytes and BECs, respectively. The casper line, in which mpv17 and mitfa double homozygous mutations make zebrafish transparent (32), was also used to improve the quality of live imaging. First, we imaged the regenerating larvae for 36 hours from BR24h, when BECs are barely present, to cover the earlier stages of BEC regeneration. A small subset of fabp10a:H2B-mCherry+ hepatocytes gradually turned on Tp1:VenusPEST expression and became BECs (Fig. 3A and Mov. S1–3). During this time period, Tp1:VenusPEST+ cells barely divided; thus, only 6.9% of BECs were generated by proliferation (Fig. 3A, arrows in #3 fish), revealing that hepatocyte-to-BEC conversion in the HANI model occurs regardless of proliferation. We then imaged the regenerating larvae for 33 hours from BR48h, when 10-15 BECs are already present in the liver, to cover the later stages of BEC regeneration. Intriguingly, a small subset of hepatocytes still converted to BECs (Fig. 3B, #1-2 fish; Mov. S4–5), contributing to 31.8% of the BECs. Among these newly converted BECs (34 BECs from total 6 fish), only one BEC divided, whereas 44% of BECs among preexisting BECs at BR48h (22 among 50 BECs from total 6 fish) divided (Fig. 3B, #3 fish; Mov. S6), contributing to 20.6% of the BECs. These live imaging data indicate that hepatocytes convert to BECs independently of proliferation, that the conversion occurs not simultaneously but rather over a longer period, and that a subset of hepatocyte-derived BECs proliferate later to contribute to BEC regeneration.
Figure 3. Hepatocytes convert to BECs independently of proliferation.
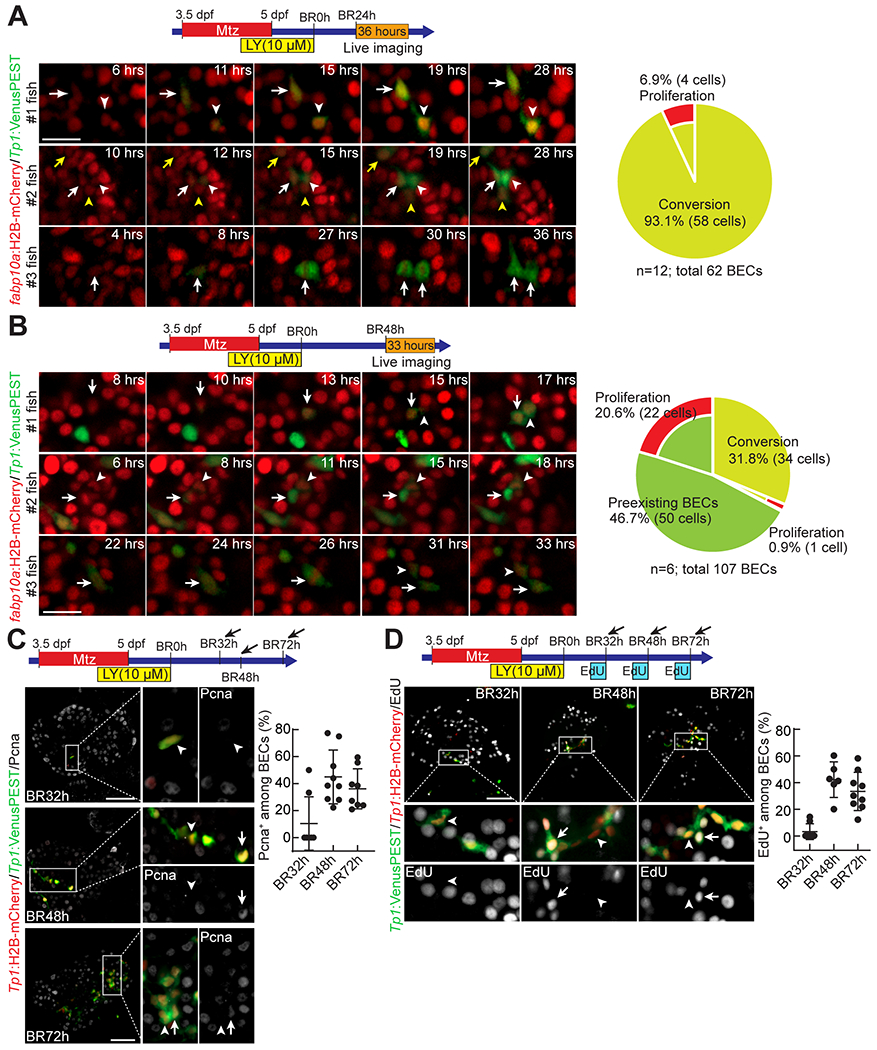
(A,B) Time-lapse confocal images showing fabp10a:H2B-mCherry and Tp1:VenusPEST expression in regenerating livers. Transgenic larvae were imaged from BR24h for 36 hours (A) or from BR48h for 33 hours (B). Cells exhibiting hepatocyte-to-BEC conversion are marked by arrows and arrowheads. Numbers in the upper right corner indicate the time imaged. The percentage of BECs generated by conversion or proliferation is quantified; n indicates the number of larvae analyzed for the quantification. (C) Confocal projection images showing the expression of Pcna, Tp1:H2B-mCherry, and Tp1:VenusPEST in regenerating livers. Boxed regions are enlarged right. Arrows point to Pcna+ BECs; arrowheads point to Pcna− BECs. The percentage of Pcna+ BECs is quantified and presented as mean ± SD. (D) Confocal projection images showing EdU labeling and the expression of Tp1:H2B-mCherry and Tp1:VenusPEST in regenerating livers. Boxed regions are enlarged below. Arrows point to EdU+ BECs; arrowheads point to EdU− BECs. The percentage of EdU+ BECs is quantified and presented as mean ± SD. Scale bars: 20 (A,B), 50 μm (C,D).
Given a possibility that proliferation may occur before Notch signaling turns on, we re-analyzed the above live imaging data, particularly 12-hour time-window before Tp1:VenusPEST induction. We also imaged regenerating larvae from BR12h for 18 hours. During this 12-hour time-window, ~11% of the hepatocytes that did not express Tp1:VenusPEST later (63 out of 565) divided, whereas none of the hepatocytes that later expressed Tp1:VenusPEST (0 out of 27) divided (Fig. S2 and Mov. S7). These data further support that hepatocyte-to-BEC conversion is independent of proliferation.
Given the involvement of BEC proliferation in the later stages of BEC regeneration, we further investigated BEC proliferation at multiple stages using anti-Pcna immunostaining (Fig. 3C) and EdU labeling (Fig. 3D). At BR32h, when 1-4 BECs are present in the liver, ~17% of BECs were positive for Pcna and ~7% of BECs were positive for EdU. However, later at BR48h, when 15-20 BECs are present, BEC proliferation was greatly increased with ~31% of BECs positive for Pcna and ~42% of BECs positive for EdU. At BR72h, the percentage of Pcna+ or EdU+ BECs rather decreased compared to at BR48h (Fig. 3C and D). These proliferation data further support that BEC proliferation contributes to BEC regeneration later compared to hepatocyte-to-BEC conversion. Although proliferation is not involved in hepatocyte-to-BEC conversion in the HANI model, it contributes to an increase in BEC number during the regeneration, albeit weakly.
Notch and Yap signaling positively control hepatocyte-to-BEC conversion
Notch signaling is a key determinant in BEC formation during development (23) and regeneration (4, 24); moreover, it induces hepatocyte-to-BEC conversion in mice (4, 11, 12). Given these known roles of Notch signaling in BEC formation, we sought to determine if Notch signaling also plays an essential role in the HANI model. First, we inhibited Notch signaling by treating the regenerating larvae with 1 μM LY411575 from BR0h onwards. This treatment completely blocked BEC formation in the regenerating livers at BR48h (Fig. 4A). Second, given the essential role of Notch3 in LPC-to-BEC differentiation during BEC-driven liver regeneration in zebrafish (33), we examined hepatocyte-to-BEC conversion in notch3 mutants. BEC number in the HANI model was greatly reduced in notch3 homozygous mutants; intriguingly, ~40% of notch3 heterozygous mutants also had fewer BECs than their wild-type siblings (Fig. 4B), indicating notch3 haploinsufficiency in hepatocyte-to-BEC conversion. Third, in parallel with the inhibition of Notch signaling, we enhanced Notch signaling in the regenerating larvae by inhibiting its negative regulator Cdk8 with senexin A (34). Senexin A treatment from BR0h onwards greatly increased BEC numbers in the regenerating livers at BR24h (Fig. 4C). Last, we also enhanced Notch signaling using the Tg(hs:canotch3) line, which expresses constitutive-active Notch3 under the heat-shock promoter (35). A heat-shock at BR0h greatly increased BEC numbers in the regenerating livers at BR24h (Fig. 4D). Collectively, these Notch signaling data demonstrate that Notch signaling is essential for hepatocyte-to-BEC conversion in the HANI model as in murine models.
Figure 4. Notch and Yap signaling positively control hepatocyte-to-BEC conversion.
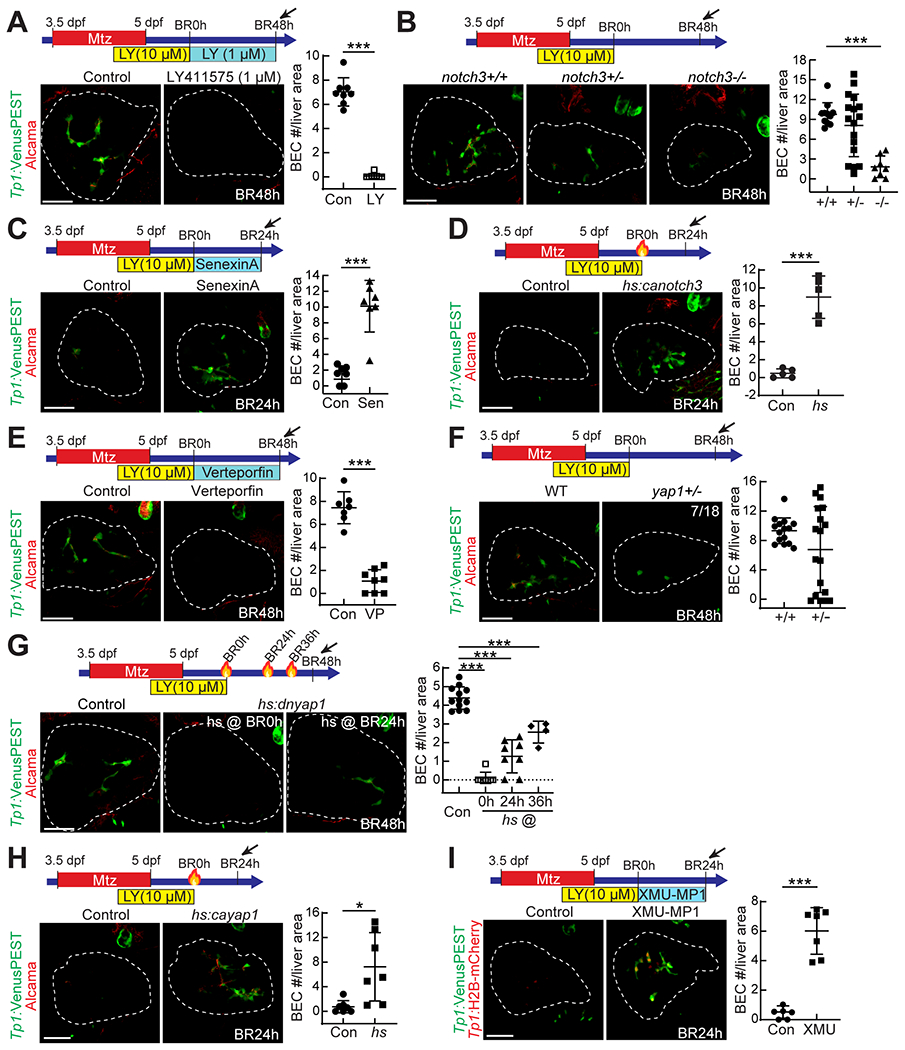
Confocal projection images showing the expression of Tp1:VenusPEST and Alcama (A-H) or Tp1:H2B-mCherry (I) in regenerating livers. The number of BECs per liver area is quantified and presented as mean ± SD. To suppress Notch signaling, LY411575 (A; 1 μM) and notch3 mutants (B) were used; to enhance Notch signaling, senexin A (C; 3 μM) and the Tg(hs:canotch3) line (D; heat-shock at BR0h) were used. To suppress Yap signaling, verteporfin (E; 1 μM), yap1 mutants (F), and the Tg(hs:dnyap1) line (G; heat-shock at BR0h, BR24h, or BR36h) were used; to enhance Yap signaling, the Tg(hs:cayap1) line (H; heat-shock at BR0h) and XMU-MP-1 (I; 15 μM) were used. *P<0.05, ***P<0.001; statistical significance was calculated using an unpaired two-tailed t-test (A,C-F,H,I) or one-way ANOVA (B,G). Scale bars: 50 μm.
In addition to Notch signaling, Yap signaling plays a crucial role in BEC formation during development (26) and in hepatocyte-to-BEC reprogramming (7, 12–14, 16). The overexpression of a stable form of YAP1 mutants in hepatocytes converts the hepatocytes to BECs in mice (7, 13, 14). We sought to determine if Yap signaling also plays a key role in the HANI model. First, we suppressed Yap signaling by treating the regenerating larvae with the Yap inhibitor verteporfin (36) from BR0h onwards. This treatment greatly reduced BEC numbers in the regenerating livers at BR48h (Fig. 4E). Second, we examined hepatocyte-to-BEC conversion in yap1 mutants. Although none of yap1 homozygous mutants survived in the HANI model, ~40% of yap1 heterozygous mutants exhibited reduced BEC number in the regenerating livers at BR48h (Fig. 4F). Third, we suppressed Yap signaling using the Tg(hs:dnyap1) line, which expresses dominant-negative Yap1 under the heat-shock promoter (37). A single heat-shock at BR0h completely blocked BEC formation in the regenerating livers at BR48h (Fig. 4G). A later heat-shock at BR24h or BR36h also reduced the BEC number albeit less effective than the early heat-shock (Fig. 4G). Fourth, in parallel with the suppression of Yap signaling, we enhanced Yap signaling using the Tg(hs:cayap1) line, which expresses constitutive-active Yap1 under the heat-shock promoter (37). A heat-shock at BR0h greatly increased BEC numbers in the regenerating livers at BR24h (Fig. 4H). Last, we enhanced Yap signaling by inhibiting its negative regulator Mst1/2 with XMU-MP-1 (38). XMU-MP-1 treatment from BR0h onwards greatly increased BEC numbers in the regenerating livers at BR24h (Fig. 4I). Collectively, these Yap signaling data demonstrate that Yap signaling is essential for hepatocyte-to-BEC conversion in the HANI model as in murine models.
Yap, but not Notch, overactivation also increases BEC proliferation
The increased BEC numbers at BR24h in the regenerating larvae overexpressing caNotch3 and caYap1 (Fig. 4D and H) indicates that both Notch and Yap overactivation promote hepatocyte-to-BEC conversion, thereby increasing BEC number. However, given the positive effect of Notch and Yap signaling on proliferation (39, 40), it is possible that the increased BEC number may be due to enhanced BEC proliferation as well as enhanced hepatocyte-to-BEC conversion. To test this possibility, we compared BEC proliferation between control and caNotch3- or caYap1-overexpressing larvae at BR48h, when a significant number of BECs are present in control regenerating livers, allowing for the comparison. A single heat-shock was given at BR36h; 12 hours later, larvae were analyzed for Pcna expression and BEC number. BEC numbers were increased in caNotch3-overexpressing larvae compared with controls, but the rate of BEC proliferation was not increased (Fig. 5A). In contrast, Yap overactivation significantly increased the rate of BEC proliferation with a trend of an increase in BEC number albeit not significant (Fig. 5B). Collectively, these data indicate in the HANI model that Yap overactivation increases BEC number by enhancing both hepatocyte-to-BEC conversion and BEC proliferation, while Notch overactivation increases BEC number by enhancing only hepatocyte-to-BEC conversion.
Figure 5. Yap, but not Notch, overactivation increases BEC proliferation.
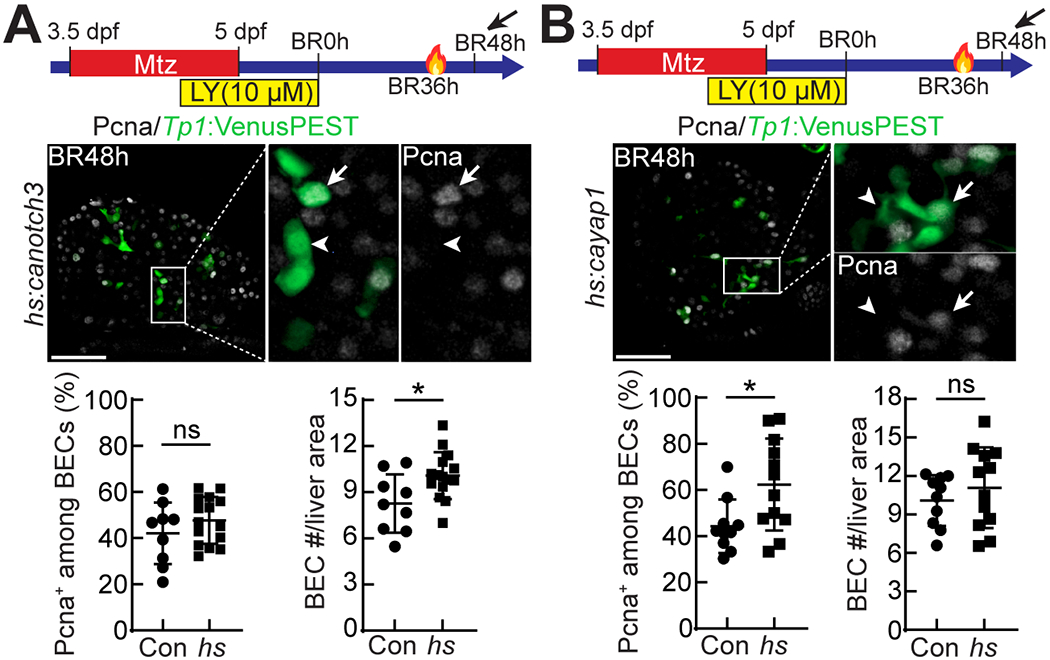
(A,B) Confocal single-optical section images showing Tp1:VenusPEST and Pcna expression in regenerating livers at BR48h. The Tg(hs:canotch3) and Tg(hs:cayap1) lines were used to enhance Notch (A) and Yap (B) signaling, respectively, with a single heat-shock at BR36h. Boxed regions are enlarged right. Arrows point to Pcna+ BECs; arrowheads point to Pcna− BECs. The percentage of Pcna+ BECs and the number of BECs per liver area are quantified and presented as mean ± SD. *P<0.05; statistical significance was calculated using an unpaired two-tailed t-test. Scale bars: 50 μm.
Notch and Yap signaling are equally required for hepatocyte-to-BEC conversion by functioning in parallel with a mutually positive interaction
The finding that the overexpression of a stable form of YAP1 mutants in hepatocytes induces Notch signaling through the upregulation of Notch2 expression (7) suggests that Notch signaling functions downstream of Yap signaling. However, it has also been reported that the overexpression of constitutive-active Notch in hepatocytes induces Yap signaling (12), suggesting that Yap signaling functions downstream of Notch signaling. Given the positive role of both Yap and Notch signaling in hepatocyte-to-BEC conversion in the HANI model (Fig. 4), we sought to clearly determine the epistatic relationship between these two signaling pathways in the conversion, i.e., one functions downstream of the other or they work in parallel. Given the increased BEC number in the regenerating livers with Notch (Fig. 4C, D) and Yap (Fig. 4H, I) overactivation, we first investigated if Yap signaling is necessary for the effect of Notch overactivation on hepatocyte-to-BEC conversion and vice versa. Notch overactivation with the Tg(hs:canotch3) line increased BEC number in the regenerating livers at BR48h; however, the suppression of Yap signaling with verteporfin or the Tg(hs:dnyap1) line blocked BEC formation in the caNotch3-overexpressing, regenerating livers (Fig. 6A, B). Similarly, Yap overactivation with the Tg(hs:cayap1) line increased BEC number in the regenerating livers at BR48h; however, the suppression of Notch signaling with LY411575 completely blocked BEC formation in the caYap1-overexpressing, regenerating livers (Fig. 6C). These epistasis data indicate that both Notch and Yap signaling are equally required for hepatocyte-to-BEC conversion regardless of driving forces of the conversion, e.g., Notch or Yap overactivation. We next investigated a genetic interaction between Notch and Yap signaling by treating the regenerating larvae from BR0h onwards with a suboptimal dose of LY411575 (50 nM) and verteporfin (500 nM). Each single treatment did not reduce BEC numbers in the regenerating livers at BR48h, whereas their cotreatment greatly reduced the BEC numbers (Fig. 6D), indicating a genetic interaction between Notch and Yap signaling in hepatocyte-to-BEC conversion.
Figure 6. Both Notch and Yap signaling are required for hepatocyte-to-BEC conversion.
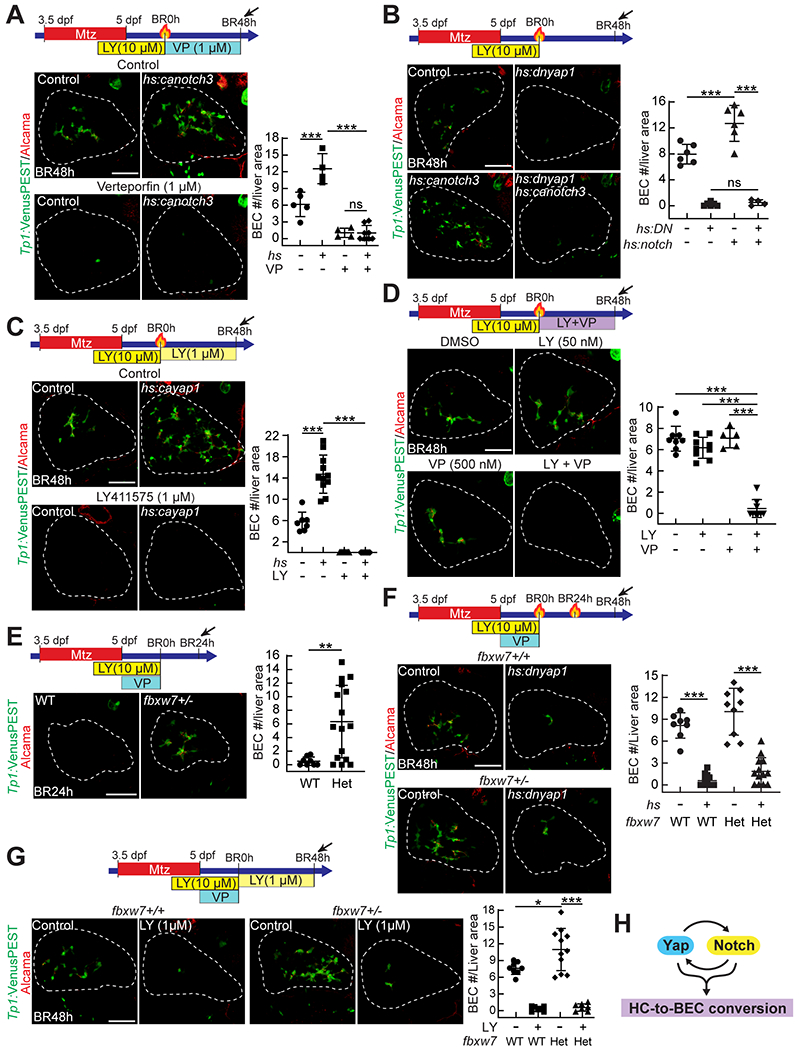
Confocal projection images showing the expression of Tp1:VenusPEST and Alcama in regenerating livers. (A, B) The Tg(hs:canotch3) line was used to enhance Notch signaling with a single heat-shock at BR0h; verteporfin (A) and the Tg(hs:dnyap1) line (B) were used to suppress Yap signaling. (C) The Tg(hs:cayap1) line was used to enhance Yap signaling with a single heat-shock at BR0h; LY411575 (1 μM) was treated from BR0h onwards to suppress Notch signaling. (D) Suboptimal doses of LY411575 (50 nM) and verteporfin (500 nM) were treated from BR0h onwards. (E-G) Hepatocyte-to-BEC conversion in fbxw7 heterozygous mutants. Given that Fbxw7 negatively regulates Yap signaling as well as Notch signaling, verteporfin (1 μM) was cotreated with LY411575 until BR0h, which permits one to determine the role of Fbxw7 in hepatocyte-to-BEC conversion. (H) A diagram depicting the epigenetic relationship between Notch and Yap signaling in hepatocyte-to-BEC conversion. The number of BECs per liver area is quantified and presented as mean ± SD. *P<0.05, **P<0.01, ***P<0.001; statistical significance was calculated using an unpaired two-tailed t-test (E) or one-way ANOVA (A-D,F,G). Scale bars: 50 μm.
As an E3 ubiquitin ligase substrate adaptor, Fbxw7 negatively regulates Notch and Yap signaling by promoting the degradation of activated Notch (41) and Yap1 (42), respectively. Moreover, zebrafish fbxw7 mutants exhibit enhanced Notch signaling in the liver (33). Using fbxw7 mutants, in which both Notch and Yap signaling are enhanced, we further investigated the requirement of Notch and Yap signaling in hepatocyte-to-BEC conversion. At BR0h, there were no BECs in the regenerating livers of fbxw7+/− mutants as in wild-type controls; however, unlike the controls, a subset of hepatocytes did not express Bhmt in the mutant regenerating livers, suggesting a delay in LPC-to-hepatocyte differentiation (Fig. S3A). Since this delay could be caused by enhanced Yap signaling in fbxw7+/− mutants, we additionally treated fbxw7+/− regenerating larvae with verteporfin from 5 dpf to BR0h for 24 hours. This treatment rescued the delay phenotype, making all hepatocytes in fbxw7+/− regenerating livers at BR0h express Bhmt (Fig. S3B). This setting allowed us to unambiguously determine the effect of fbxw7 haploinsufficiency on hepatocyte-to-BEC conversion. Indeed, BEC numbers greatly increased in ~85% of the fbxw7+/− regenerating larvae at BR24h compared with wild-type controls (Fig. 6E), probably due to enhanced Notch and Yap signaling from BR0h. Consistent with the above epistasis data (Fig. 6A–C), the suppression of Notch and Yap signaling with LY411575 and the Tg(hs:dnyap1) line, respectively, blocked BEC formation even in fbxw7+/− regenerating livers (Fig. 6F, G). Collectively, these epistasis data reveal that in hepatocyte-to-BEC conversion, Notch signaling does not function simply downstream or upstream of Yap signaling; instead, both signaling pathways are required for hepatocyte-to-BEC conversion and function in parallel with a mutually positive interaction (Fig. 6H).
Discussion
In this study, we have established a zebrafish model for hepatocyte-to-BEC conversion, the HANI model, in which a subset of hepatocytes rapidly convert to BECs. During the conversion, BEC markers are sequentially induced in the order of Sox9b, Yap/Taz, Notch activity/epcam, and Alcama/krt18; the expression of the hepatocyte marker Bhmt disappears between Sox9b and Yap/Taz induction. Using live time-lapse imaging, we provide the clear in vivo evidence of the transdifferentiation of hepatocytes into BECs: hepatocytes directly convert to BECs without transitioning through a proliferative intermediate state. Moreover, we delineate the epistatic relationship between Notch and Yap signaling in the conversion process: they function in parallel with a mutually positive interaction. These findings suggest the usefulness of the HANI model for elucidating the cellular and molecular mechanisms underlying hepatocyte-to-BEC conversion.
Consistent with the recent finding from mice that hepatocytes transdifferentiate into BECs (9), we found that hepatocytes transdifferentiate into BECs in the HANI model. In the mouse study, clonal analyses of the fate of sparsely labeled hepatocytes revealed no involvement of proliferation in hepatocyte-to-BEC conversion (9), suggesting transdifferentiation rather than proliferation-involved dedifferentiation and subsequent differentiation. Due to the transparency of casper zebrafish, the small size of regenerating livers, and the rapid conversion of hepatocytes to BECs in the HANI model, using a confocal microscope, we could image the conversion process in live animals. This live imaging revealed the direct conversion of hepatocytes to BECs without transitioning through a proliferative intermediate state, which is more convincing for transdifferentiation in hepatocyte-to-BEC conversion than the data from the mouse clonal analysis. In contrast to the direct conversion of hepatocytes to BECs, when BECs contribute to regenerating hepatocytes, BECs convert to hepatocytes through a proliferative intermediate state, i.e., LPCs (20). Namely, hepatocyte-to-BEC conversion does not involve proliferation, whereas BEC-to-hepatocyte conversion involves proliferation. Therefore, through this conversion, one hepatocyte generates one BEC, whereas one BEC generates multiple hepatocytes. Given that the number of hepatocytes is much higher than that of BECs in the liver, it appears to be efficient for proliferation to be involved in BEC-to-hepatocyte, but not hepatocyte-to-BEC, conversion.
It has been known from mouse studies that Notch (4, 6, 11, 12, 43) and Yap (7, 12–16) signaling positively regulate hepatocyte-to-BEC reprogramming. Their hepatocyte-specific overactivation induced hepatocyte-to-BEC conversion (4, 7, 11–14, 16), while their hepatocyte-specific inhibition suppressed the conversion (4, 6, 12, 14–16). However, the epistatic relationship between these two signaling pathways in the conversion process has remained unclear. In the HANI model, both Notch and Yap signaling are necessary and sufficient for hepatocyte-to-BEC conversion, permitting us to define the epistatic relationship between them. Indeed, we found that both signaling pathways are equally required for the conversion even in Notch or Yap overactivation settings and that they function in parallel with a mutually positive interaction. Moreover, given the positive role of Notch and Yap signaling in proliferation (39, 40), we determined whether the increased number of hepatocyte-derived BECs in the Notch or Yap overactivation settings was due to enhanced conversion alone or together with enhanced BEC proliferation. In fact, Notch overactivation did not increase BEC proliferation, whereas Yap overactivation increased its proliferation, indicating that Notch overactivation increases the number of hepatocyte-derived BECs solely by enhancing the conversion, while Yap overactivation increases the number by enhancing both the conversion and BEC proliferation.
In addition to Notch and Yap signaling, TGFβ signaling has been known to control hepatocyte-to-BEC conversion (9). In liver-specific Hnf6/Rbpj-double knockout mice, TGFβ signaling drives hepatocyte-to-BEC conversion (9). However, in the HANI model, inhibiting TGFβ signaling did not affect hepatocyte-to-BEC conversion (Fig. S4), suggesting no role of TGFβ signaling in the process. In the zebrafish model, Notch signaling is temporarily blocked and reactivated during biliary regeneration, whereas in the mouse model, Notch signaling is permanently blocked. It appears that Notch signaling plays a primary role in hepatocyte-to-BEC conversion and that TGFβ signaling plays a secondary role in the process. Thus, the positive effect of TGFβ signaling on hepatocyte-to-BEC conversion could be observed only in the setting with Notch signaling permanently off.
In the HANI model, a small subset of hepatocytes convert to BECs, raising an interesting question about how the hepatocytes are selected for the conversion. They might be selected because they are intrinsically different from other hepatocytes, because some extrinsic, inducing factors are available only to the hepatocytes, or because of random selection. Since Sox9b is the earliest marker for the conversion in the HANI model, investigating how Sox9b is induced will help answer to the question. Single-cell RNA-sequencing analysis will also help answer to the question by delineating the heterogeneity of hepatocytes in the HANI model.
In summary, we present a novel zebrafish model for hepatocyte-to-BEC conversion. Using the model, we provide the evidence of the transdifferentiation of hepatocytes into BECs and the epistatic relationship between Notch and Yap signaling in the process. The HANI model will not only contribute to a better understanding of the molecular mechanisms of hepatocyte-to-BEC conversion through the identification of novel genes that play key roles in the process, but also help identify conversion-promoting compounds, which have a potential for therapeutic drugs to treat patients with BEC paucity and cholestatic liver diseases.
Supplementary Material
Acknowledgments
We thank Michael Parsons for Tg(ubb:loxP-CFP-loxP-H2B-mCherry), David Kimelman and Ken Poss for Tg(hs:cayap1) and Tg(hs:dnyap1), and David Kimelman and Didier Stainier for yap1 mutant fish. We also thank Jinrong Peng for anti-Bhmt antibodies; Paul Monga, Sungjin Ko and Yoojeong Park for help on histology and immunohistochemistry; Neil Hukriede and Michael Tsang for discussions; Kari Nejak-Bowen and Mia Ikeda for critical reading of the manuscript. Graphical abstract was created using BioRender (biorender.com).
Financial Support:
The work was supported by NIH grants to D.S. (DK101426, DK132014).
List of abbreviations:
- BEC
biliary epithelial cell
- NICD
Notch intracellular domain
- DDC
3,5-diethoxycarbonyl-1,4-dihydrocollidine
- LPC
liver progenitor cell
- HANI
hepatocyte ablation combined with temporal Notch inhibition during BEC-driven liver regeneration
- Mtz
metronidazole
- DMSO
dimethyl sulfoxide
- dpf
days post-fertilization
- NTR
nitroreductase
- CFP
cyan fluorescent protein
- BR
biliary regeneration
- 4-OHT
4-hydroxytamoxifen
- caNotch3
constitutive-active Notch3
- dnYap1
dominant-negative Yap1
- caYap1
constitutive-active Yap1
References
Author names in bold designate shared co-first authorship.
- 1.Poupon R, Chazouilleres O, Poupon RE. Chronic cholestatic diseases. J Hepatol 2000;32:129–140. [DOI] [PubMed] [Google Scholar]
- 2.Lewis A, Koukoura A, Tsianos GI, Gargavanis AA, Nielsen AA, Vassiliadis E. Organ donation in the US and Europe: The supply vs demand imbalance. Transplant Rev (Orlando) 2021;35:100585. [DOI] [PubMed] [Google Scholar]
- 3.Michalopoulos GK, Bowen WC, Mule K, Lopez-Talavera JC, Mars W. Hepatocytes undergo phenotypic transformation to biliary epithelium in organoid cultures. Hepatology 2002;36:278–283. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 4.Yanger K, Zong Y, Maggs LR, Shapira SN, Maddipati R, Aiello NM, Thung SN, et al. Robust cellular reprogramming occurs spontaneously during liver regeneration. Genes Dev 2013;27:719–724. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 5.Nagahama Y, Sone M, Chen X, Okada Y, Yamamoto M, Xin B, Matsuo Y, et al. Contributions of hepatocytes and bile ductular cells in ductular reactions and remodeling of the biliary system after chronic liver injury. Am J Pathol 2014;184:3001–3012. [DOI] [PubMed] [Google Scholar]
- 6.Sekiya S, Suzuki A. Hepatocytes, rather than cholangiocytes, can be the major source of primitive ductules in the chronically injured mouse liver. Am J Pathol 2014;184:1468–1478. [DOI] [PubMed] [Google Scholar]
- 7.Yimlamai D, Christodoulou C, Galli GG, Yanger K, Pepe-Mooney B, Gurung B, Shrestha K, et al. Hippo pathway activity influences liver cell fate. Cell 2014;157:1324–1338. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 8.Font-Burgada J, Shalapour S, Ramaswamy S, Hsueh B, Rossell D, Umemura A, Taniguchi K, et al. Hybrid Periportal Hepatocytes Regenerate the Injured Liver without Giving Rise to Cancer. Cell 2015;162:766–779. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 9.Schaub JR, Huppert KA, Kurial SNT, Hsu BY, Cast AE, Donnelly B, Karns RA, et al. De novo formation of the biliary system by TGFbeta-mediated hepatocyte transdifferentiation. Nature 2018;557:247–251. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 10.Limaye PB, Alarcon G, Walls AL, Nalesnik MA, Michalopoulos GK, Demetris AJ, Ochoa ER. Expression of specific hepatocyte and cholangiocyte transcription factors in human liver disease and embryonic development. Lab Invest 2008;88:865–872. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 11.Fan B, Malato Y, Calvisi DF, Naqvi S, Razumilava N, Ribback S, Gores GJ, et al. Cholangiocarcinomas can originate from hepatocytes in mice. J Clin Invest 2012;122:2911–2915. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 12.Hu S, Molina L, Tao J, Liu S, Hassan M, Singh S, Poddar M, et al. NOTCH-YAP1/TEAD-DNMT1 Axis Drives Hepatocyte Reprogramming into Intrahepatic Cholangiocarcinoma. Gastroenterology 2022. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 13.Liu Y, Zhuo S, Zhou Y, Ma L, Sun Z, Wu X, Wang XW, et al. Yap-Sox9 signaling determines hepatocyte plasticity and lineage-specific hepatocarcinogenesis. J Hepatol 2022;76:652–664. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 14.Wang J, Dong M, Xu Z, Song X, Zhang S, Qiao Y, Che L, et al. Notch2 controls hepatocyte-derived cholangiocarcinoma formation in mice. Oncogene 2018;37:3229–3242. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 15.Planas-Paz L, Sun T, Pikiolek M, Cochran NR, Bergling S, Orsini V, Yang Z, et al. YAP, but Not RSPO-LGR4/5, Signaling in Biliary Epithelial Cells Promotes a Ductular Reaction in Response to Liver Injury. Cell Stem Cell 2019. [DOI] [PubMed] [Google Scholar]
- 16.Li W, Yang L, He Q, Hu C, Zhu L, Ma X, Ma X, et al. A Homeostatic Arid1a-Dependent Permissive Chromatin State Licenses Hepatocyte Responsiveness to Liver-Injury-Associated YAP Signaling. Cell Stem Cell 2019;25:54–68 e55. [DOI] [PubMed] [Google Scholar]
- 17.Pepe-Mooney BJ, Dill MT, Alemany A, Ordovas-Montanes J, Matsushita Y, Rao A, Sen A, et al. Single-Cell Analysis of the Liver Epithelium Reveals Dynamic Heterogeneity and an Essential Role for YAP in Homeostasis and Regeneration. Cell Stem Cell 2019;25:23–38 e28. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 18.Okabe H, Yang J, Sylakowski K, Yovchev M, Miyagawa Y, Nagarajan S, Chikina M, et al. Wnt signaling regulates hepatobiliary repair following cholestatic liver injury in mice. Hepatology 2016;64:1652–1666. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 19.Westerfield M The Zebrafish Book: A Guide for the Laboratory Use of Zebrafish (Danio rerio): University of Oregon Press, 2000. [Google Scholar]
- 20.Choi TY, Ninov N, Stainier DY, Shin D. Extensive conversion of hepatic biliary epithelial cells to hepatocytes after near total loss of hepatocytes in zebrafish. Gastroenterology 2014;146:776–788. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 21.Curado S, Stainier DYR, Anderson RM. Nitroreductase-mediated cell/tissue ablation in zebrafish: a spatially and temporally controlled ablation method with applications in developmental and regeneration studies. Nature Protocols 2008;3:948–954. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 22.Curado S, Anderson RM, Jungblut B, Mumm J, Schroeter E, Stainier DYR. Conditional targeted cell ablation in zebrafish: A new tool for regeneration studies. Developmental Dynamics 2007;236:1025–1035. [DOI] [PubMed] [Google Scholar]
- 23.Zong Y, Panikkar A, Xu J, Antoniou A, Raynaud P, Lemaigre F, Stanger BZ. Notch signaling controls liver development by regulating biliary differentiation. Development 2009;136:1727–1739. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 24.Geisler F, Strazzabosco M. Emerging roles of Notch signaling in liver disease. Hepatology 2015;61:382–392. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 25.Wong GT, Manfra D, Poulet FM, Zhang Q, Josien H, Bara T, Engstrom L, et al. Chronic treatment with the gamma-secretase inhibitor LY-411,575 inhibits beta-amyloid peptide production and alters lymphopoiesis and intestinal cell differentiation. J Biol Chem 2004;279:12876–12882. [DOI] [PubMed] [Google Scholar]
- 26.Molina LM, Zhu J, Li Q, Pradhan-Sundd T, Krutsenko Y, Sayed K, Jenkins N, et al. Compensatory hepatic adaptation accompanies permanent absence of intrahepatic biliary network due to YAP1 loss in liver progenitors. Cell Rep 2021;36:109310. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 27.Ando R, Hama H, Yamamoto-Hino M, Mizuno H, Miyawaki A. An optical marker based on the UV-induced green-to-red photoconversion of a fluorescent protein. Proceedings of the National Academy of Sciences of the United States of America 2002;99:12651–12656. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 28.Farber SA, Pack M, Ho SY, Johnson ID, Wagner DS, Dosch R, Mullins MC, et al. Genetic analysis of digestive physiology using fluorescent phospholipid reporters. Science 2001;292:1385–1388. [DOI] [PubMed] [Google Scholar]
- 29.Delous M, Yin C, Shin D, Ninov N, Debrito Carten J, Pan L, Ma TP, et al. Sox9b is a key regulator of pancreaticobiliary ductal system development. PLoS Genet 2012;8:e1002754. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 30.Gerloff T, Stieger B, Hagenbuch B, Madon J, Landmann L, Roth J, Hofmann AF, et al. The sister of P-glycoprotein represents the canalicular bile salt export pump of mammalian liver. J Biol Chem 1998;273:10046–10050. [DOI] [PubMed] [Google Scholar]
- 31.Wang YJ, Park JT, Parsons MJ, Leach SD. Fate mapping of ptf1a-expressing cells during pancreatic organogenesis and regeneration in zebrafish. Dev Dyn 2015;244:724–735. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 32.White RM, Sessa A, Burke C, Bowman T, LeBlanc J, Ceol C, Bourque C, et al. Transparent adult zebrafish as a tool for in vivo transplantation analysis. Cell Stem Cell 2008;2:183–189. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 33.Ko S, Russell JO, Tian J, Gao C, Kobayashi M, Feng R, Yuan X, et al. Hdac1 Regulates Differentiation of Bipotent Liver Progenitor Cells During Regeneration via Sox9b and Cdk8. Gastroenterology 2019;156:187–202 e114. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 34.Porter DC, Farmaki E, Altilia S, Schools GP, West DK, Chen M, Chang BD, et al. Cyclin-dependent kinase 8 mediates chemotherapy-induced tumor-promoting paracrine activities. Proc Natl Acad Sci U S A 2012;109:13799–13804. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 35.Wang Y, Pan L, Moens CB, Appel B. Notch3 establishes brain vascular integrity by regulating pericyte number. Development 2014;141:307–317. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 36.Liu-Chittenden Y, Huang B, Shim JS, Chen Q, Lee SJ, Anders RA, Liu JO, et al. Genetic and pharmacological disruption of the TEAD-YAP complex suppresses the oncogenic activity of YAP. Genes Dev 2012;26:1300–1305. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 37.Mateus R, Lourenco R, Fang Y, Brito G, Farinho A, Valerio F, Jacinto A. Control of tissue growth by Yap relies on cell density and F-actin in zebrafish fin regeneration. Development 2015;142:2752–2763. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 38.Fan F, He Z, Kong LL, Chen Q, Yuan Q, Zhang S, Ye J, et al. Pharmacological targeting of kinases MST1 and MST2 augments tissue repair and regeneration. Sci Transl Med 2016;8:352ra108. [DOI] [PubMed] [Google Scholar]
- 39.Patel SH, Camargo FD, Yimlamai D. Hippo Signaling in the Liver Regulates Organ Size, Cell Fate, and Carcinogenesis. Gastroenterology 2017;152:533–545. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 40.Ortica S, Tarantino N, Aulner N, Israel A, Gupta-Rossi N. The 4 Notch receptors play distinct and antagonistic roles in the proliferation and hepatocytic differentiation of liver progenitors. FASEB J 2014;28:603–614. [DOI] [PubMed] [Google Scholar]
- 41.Wu G, Lyapina S, Das I, Li J, Gurney M, Pauley A, Chui I, et al. SEL-10 is an inhibitor of notch signaling that targets notch for ubiquitin-mediated protein degradation. Mol Cell Biol 2001;21:7403–7415. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 42.Tu K, Yang W, Li C, Zheng X, Lu Z, Guo C, Yao Y, et al. Fbxw7 is an independent prognostic marker and induces apoptosis and growth arrest by regulating YAP abundance in hepatocellular carcinoma. Mol Cancer 2014;13:110. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 43.Terada M, Horisawa K, Miura S, Takashima Y, Ohkawa Y, Sekiya S, Matsuda-Ito K, et al. Kupffer cells induce Notch-mediated hepatocyte conversion in a common mouse model of intrahepatic cholangiocarcinoma. Sci Rep 2016;6:34691. [DOI] [PMC free article] [PubMed] [Google Scholar]
Associated Data
This section collects any data citations, data availability statements, or supplementary materials included in this article.