

Key Points
-
•
RUNX1 isoform disequilibrium toward RUNX1A and its interaction with MYC:MAX are key in the pathogenesis of trisomy 21–associated ML.
-
•
Restoration of RUNX1A:RUNX1C equilibrium and pharmacological interference with MYC:MAX dimerization reverses the oncogenic phenotype.
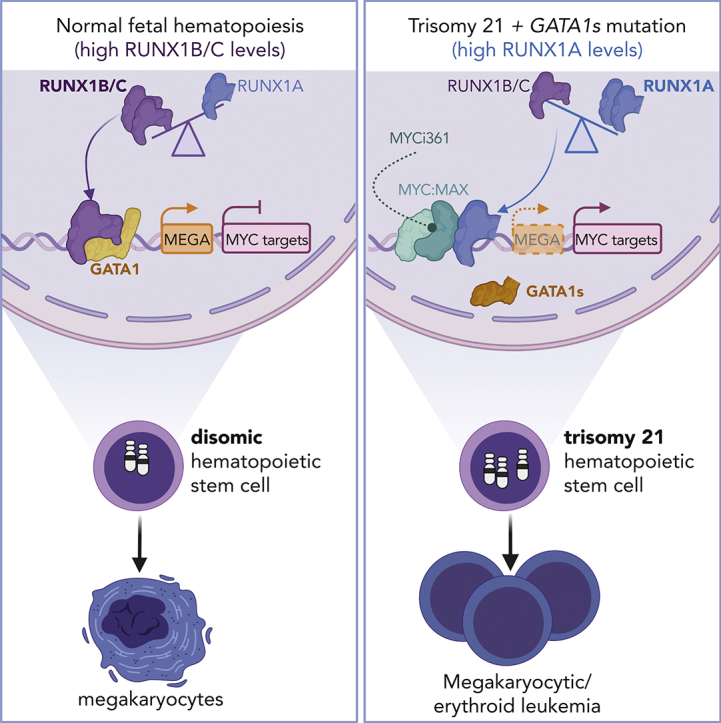
Visual Abstract
Abstract
Gain of chromosome 21 (Hsa21) is among the most frequent aneuploidies in leukemia. However, it remains unclear how partial or complete amplifications of Hsa21 promote leukemogenesis and why children with Down syndrome (DS) (ie, trisomy 21) are particularly at risk of leukemia development. Here, we propose that RUNX1 isoform disequilibrium with RUNX1A bias is key to DS-associated myeloid leukemia (ML-DS). Starting with Hsa21-focused CRISPR–CRISPR-associated protein 9 screens, we uncovered a strong and specific RUNX1 dependency in ML-DS cells. Expression of the RUNX1A isoform is elevated in patients with ML-DS, and mechanistic studies using murine ML-DS models and patient-derived xenografts revealed that excess RUNX1A synergizes with the pathognomonic Gata1s mutation during leukemogenesis by displacing RUNX1C from its endogenous binding sites and inducing oncogenic programs in complex with the MYC cofactor MAX. These effects were reversed by restoring the RUNX1A:RUNX1C equilibrium in patient-derived xenografts in vitro and in vivo. Moreover, pharmacological interference with MYC:MAX dimerization using MYCi361 exerted strong antileukemic effects. Thus, our study highlights the importance of alternative splicing in leukemogenesis, even on a background of aneuploidy, and paves the way for the development of specific and targeted therapies for ML-DS, as well as for other leukemias with Hsa21 aneuploidy or RUNX1 isoform disequilibrium.
Trisomy 21 (+21) is a frequent alteration in acute myeloid leukemias (AML) and constitutive in Down syndrome myeloid leukemia (ML-DS). In this Plenary Paper, Gialesaki et al show that ML-DS blasts are dependent on RUNX1 and reveal that disequilibrium among RUNX1 transcripts with disproportionately increased level of RUNX1A is pivotal for DS leukemogenesis and prominent in sporadic AML. This validates a new model for the early stages of ML-DS development and suggests new therapeutic targets for +21 AMLs in general.
Introduction
Major multiomic efforts have mapped the cytogenetic, mutational, and epigenetic landscape of many cancers, including acute myeloid leukemia (AML).1,2 Subsequent functional studies involving disease modeling in mice or in human cells have pointed toward complex cooperation between common fusion oncogenes and recurrently mutated genes during disease initiation and progression. However, the contribution of aneuploidy to oncogenesis remains poorly understood3 because of context-dependent effects, technical challenges, and a lack of appropriate models. Gain of chromosome 21 (Hsa21), one of the most frequent numerical alterations in leukemia,4,5 is no exception.
Myeloid leukemia associated with Down syndrome (ML-DS) and its preleukemic antecedent, transient abnormal myelopoiesis (TAM), are excellent paradigms for studying leukemic progression associated with trisomy 21. TAM is caused by a single genetic mutation in the transcription factor GATA1 in trisomic fetal stem and/or progenitor cells, which causes the exclusive expression of a shorter isoform known as GATA1s.6 We, and others, have shed light on the additional mutational events that are required for progression to overt ML-DS,7,8 however, far less is known about the role of trisomy 21 in the development of the TAM/ML-DS disease phenotype. A critical region has been identified on Hsa21 that mediates the expansion of early hematopoietic progenitors observed in patients with DS9, 10, 11 and several Hsa21 genes, such as ERG,12,13 DYRK1A,14 CHAF1B,15 and microRNA 125b (miR-125b),16,17 have been postulated to play a role in leukemogenesis. In contrast, Ts65dn mice that are trisomic for 104 orthologs of Hsa21 genes do not fully recapitulate the human phenotype in association with GATA1s,18 and the postulated factors are either located outside of the critical region, not overexpressed in trisomic fetal progenitor cells,11 or lack full leukemic potential in humans when combined with mutated GATA1s.14,19,20
The transcription factor RUNX1, which is essential for the establishment of definitive hematopoiesis,21, 22, 23 has attracted considerable attention as a candidate Hsa21 oncogene. Although RUNX1 has been extensively studied in leukemia development,24,25 its expression is reduced in trisomic fetal progenitor cells11 and its trisomy is dispensable for myeloproliferative disease in elderly Ts65dn mice.24 Despite initial hints pointing toward differential roles for alternatively spliced RUNX1 isoforms in ML-DS,26 studies, to date, have not accounted for the fact that RUNX1 is transcribed from 2 distinct promoters and undergoes alternative splicing, giving rise to 3 main isoforms with diverse effects on hematopoiesis27: RUNX1C, transcribed from the P1 promoter, is the most abundant isoform in definitive hematopoiesis. RUNX1A and RUNX1B, both transcribed from the P2 promoter, are differentially expressed throughout hematopoietic differentiation.28, 29, 30 Interestingly, because of the lack of a splice acceptor site in an isoform-specific exon, mice do not express RUNX1A, the short RUNX1 isoform that lacks the transactivation domain, underlining important species-specific differences31 and contributing, in part, to the incomplete understanding of isoform-specific roles for RUNX1.
In this study, we systematically investigated protein-coding genes on Hsa21 for dependency in ML-DS and found that disequilibrium of the RUNX1 isoforms, specifically excess RUNX1A, is key to trisomy 21–associated leukemogenesis.
Methods
Reagents and resources
Supplemental Table 1, available on the Blood website, contains a list of all relevant reagents.
Patient samples
Samples from pediatric patients with AML were collected from patients enrolled in the AML Berlin-Frankfurt-Münster treatment protocols for children and adolescents. Written, informed consent was obtained from all patients and custodians in accordance with the Declaration of Helsinki and local laws and regulations, and the study was approved by the institutional review boards of all participating centers. For details, refer to supplemental Table 2.
Animal studies
All animal experiments were performed according to protocols approved by the local authorities (Niedersächsisches Landesamt für Verbraucherschutz und Lebensmittelsicherheit, Landesverwaltungsamt Sachsen-Anhalt, and Regierungspräsidium Darmstadt). The animals were maintained under pathogen-free conditions.
Statistical analysis
Statistical evaluation was performed using the Student t test, the Mann-Whitney U test, and 1-way or 2-way analysis of variance. The Kaplan-Meier method and log-rank tests were used to estimate overall survival and to compare differences between survival curves, respectively. All data are presented as mean ± standard deviation. Calculations were performed using GraphPad Prism version 8/9 (STATCON). All statistical tests and sample numbers are disclosed in the respective figure legends/supplemental tables.
Additional detailed methods can be found in the supplemental Methods.
Results
CRISPR-Cas9 screen reveals RUNX1 dependency in ML-DS
To identify potential oncogenes on Hsa21 that contribute to the pathogenesis of TAM and ML-DS, we generated a lentiviral CRISPR–CRISPR-associated protein 9 (CRISPR-Cas9) library (1090 single-guide RNA [sgRNA]) targeting the 218 annotated coding genes on Hsa21 and performed a dropout screen in the ML-DS cell line CMK, as well as in the erythroleukemia cell line K562 (Figure 1A). We identified 19 genes specifically required for the survival of CMK cells (Figure 1B; supplemental Table 3). Interestingly, we observed a strong depletion of RUNX1-targeting sgRNAs in CMK but not in K562 cells (Figure 1B-C). Subsequent flow cytometry–based depletion assays using individual sgRNAs found RUNX1 to be the most specific ML-DS dependency in our screen, with the sgRNAs’ impact on cell survival corresponding to RUNX1 knockdown levels (Figure 1C; supplemental Figure 1A-D). These results were recapitulated in vitro in stable Cas9-expressing blasts derived from patients with ML-DS and in vivo by fluorescence-based competitive transplantation assays (Figure 1C-E; supplemental Figure 1E-G; supplemental Table 2).
Figure 1.

CRISPR-Cas9 screen reveals RUNX1 dependency in ML-DS. (A) Schema of the high-throughput Hsa21 CRISPR-Cas9 in vitro screen. (B) Dot plots showing the log10 fold change and −log10 (P 2-sided) of the Hsa21 sgRNAs in CMK (left), K562 (middle), and CMK + K562 (right; maximum likelihood estimation [MLE] β score) cell lines. Dark green dots represent highly depleting sgRNAs in CMK cells that also deplete in K562s. Light green dots represent positive controls (MYB, MYC, ACTB, SF3B3, RPL9, and POLR2A). Blue dots represent sgRNAs targeting RUNX1. (C) Dot plot showing the number of sgRNA-transduced (control sgRNA [sgCtrl] and sgRUNX1.1-1.5 indicated by shape) CMK and K562 cells (with stable Cas9 expression) after 14 days of culture, normalized to day 0 (n = 2 per sgRNA, 2-tailed unpaired t test). (D) Experimental setup for evaluating RUNX1 knockout in vivo. ML-DS blasts (with stable Cas9 expression) were transduced with sgRUNX1 (E2Crimson+ and sgRUNX1.1) or a sgCtrl (E2Crimson+) and mixed 1:1 with sgCtrl-transduced blasts (GFP+), before transplantation into sublethally irradiated recipient mice. (E) Dot plot showing the ratio of E2Crimson+ to green fluorescent protein–positive (GFP+) cells after 12 days of culture, normalized to day 0 (left; n = 4, 2-tailed unpaired t test) and in the bone marrow of mice euthanized 6 to 8 weeks after transplantation (right; n = 5, Mann Whitney U test). (F) Schematic representation of the human RUNX1 genomic locus (top) and the 3 main RUNX1 transcript isoforms (bottom; not to scale). Functional exons encoding the DNA-binding domain (Runt; red) and transactivation domain (TAD; blue) are indicated. Bar graph showing RUNX1 isoform distribution (Oxford Nanopore sequencing) in polyA+-enriched RNA samples from TAM and ML-DS samples. (G) Expression of RUNX1A and RUNX1C isoforms normalized to the expression of β2-microglobulin (B2M) (left graph; 2-way analysis of variance [ANOVA]) and RUNX1A to RUNX1C ratios (right graph; 1-way ANOVA) in CD34+ HSPCs, erythrocytes, megakaryocytes, granulocytes, and monocytes isolated from healthy donors, as well as in leukemic blasts from patients with TAM/ML-DS. gDNA, genomic DNA; NGS, next-generation sequencing; PCR, polymerase chain reaction.
Considering that former studies have disputed the role of RUNX1 in TAM/ML-DS pathogenesis, and given that Bourquin et al hypothesized that alternatively spliced RUNX1 isoforms may lead the reduced expression of RUNX1 targets in ML-DS,11,24,26 we wondered whether deregulation of the RUNX1 isoforms, rather than altered overall expression, might underlie its dependency phenotype in ML-DS cells. Nanopore full-length RNA sequencing (RNA-seq) revealed that the main isoforms, RUNX1A, RUNX1B, and RUNX1C, are predominant in TAM and ML-DS (Figure 1F). Thus, we further quantified the expression of these 3 isoforms in healthy hematopoietic cells (hematopoietic stem and progenitor cells [HSPCs], erythrocytes, megakaryocytes, granulocytes, and monocytes) and TAM/ML-DS blasts. RUNX1C is the predominant isoform in all cell types, but ML-DS and trisomy 21 samples presented with elevated expression of the RUNX1A isoform, resulting in a significantly higher RUNX1A:RUNX1B/C ratio compared with HSPCs or terminally differentiated cells (Figure 1G; supplemental Figure 1H-K).
Notably, trisomy 21 is associated with an elevated RUNX1A:RUNX1B/C ratio, as determined by comparing fetal trisomy 21 and nontrisomic CD34+ HSPCs (supplemental Figure 1H-I). Interestingly, this imbalance can be induced by elevated levels of the splicing factor subunit U2AF1, which is encoded by chromosome 21 (supplemental Figure 2A). Moreover, GATA1s’ ability to regulate RUNX1 isoforms appears to be impaired compared with full-length GATA1; it shows reduced occupancy at the RUNX1 P1 und P2 promoter regions in Gata1s–fetal liver cells (FLCs) (supplemental Figure 2B-D) and causes higher RUNX1A levels in CRISPR-Cas9–edited human megakaryocytic/erythroid precursors cultured in vitro compared with wild-type controls (supplemental Figure 2E). Induced expression of GATA1 and GATA1s in K562 cells activated transcription from the P2 promoter, leading to increased RUNX1A and RUNX1B expression, whereas RUNX1C transcription from the P1 promoter was enhanced by GATA1 but repressed by GATA1s (supplemental Figure 2F). Altogether, these data suggest a mechanism in which trisomy 21 and GATA1s synergize to amplify RUNX1 isoform imbalance.
Thus, our CRISPR-Cas9 screen of Hsa21 genes and subsequent RUNX1 isoform expression analyses in patient samples suggest that RUNX1 isoform disequilibrium and RUNX1A bias contribute to the pathogenesis of TAM and ML-DS.
Increased RUNX1A:RUNX1C ratio induces malignant ML-DS phenotype
To probe the functional relevance of unbalanced RUNX1 isoform expression in TAM and ML-DS development, we modulated the RUNX1A:RUNX1C ratio in fetal and neonatal CD34+ HSPCs of human origin and in patient-derived ML-DS blasts (supplemental Figure 3A). Methylcellulose-based colony-forming unit assays showed the sustained growth of RUNX1A-expressing CD34+ HSPCs, as evidenced by increased replating capacity (Figure 2A). In culture conditions that promote megakaryocytic differentiation, RUNX1A expression induced the expansion of CD34+ immature cells and blocked their differentiation to CD41+/CD61+CD42b+ megakaryocytes (Figure 2B-C; supplemental Figure 3B), effects that were more pronounced in fetal cells and that we confirmed in collagen-based megakaryocytic colony-forming unit assays (supplemental Figure 3C). To better understand the effects of RUNX1A and RUNX1C on lineage fate decision, we also cultured CD34+ HSPCs in conditions allowing for differentiation along the erythroid as well as megakaryocytic lineages. RUNX1C and RUNX1A conferred a lineage bias toward megakaryopoiesis (CD41+/CD235a−) over erythropoiesis (CD41−/CD235a+) in neonatal CD34+ HSPCs. RUNX1A halted cells in an undifferentiated CD117+ stage (supplemental Figure 3D-E).
Figure 2.

Increased RUNX1A:RUNX1C ratio induces malignant ML-DS phenotype. (A-C) Neonatal (top) and fetal (bottom) CD34+ HSPCs were lentivirally transduced with RUNX1A, RUNX1C, or EV control. (A) Colonies after plating CD34+ HSPCs from 2 independent donors in methylcellulose-based colony-forming unit (CFU) assays (mean ± standard deviation [SD], n = 2, 1-way ANOVA). Percentage of immature (CD41+CD61+/CD42−) and mature megakaryocytes (CD41+CD61+/CD42+) (B) and immature CD34+ cells after 7 days in media promoting megakaryocytic differentiation (mean ± SD, n = 5, 2-way ANOVA) (C). (D-F) Cells derived from patients with ML-DS were lentivirally transduced with RUNX1A, RUNX1C, or EV control. (D) Percentage of GFP+ transduced cells normalized to day 0 after transduction (mean ± SD, n = 3, 1-way ANOVA, ∗∗P < .01). Bar graphs show the percentage of CD41+CD117+ and CD41+CD117− megakaryocytic cells (E) and CD33+CD117+ and CD33+CD117− myeloid cells (F) 5 days after transduction (mean ± SD, n = 3, 2-way ANOVA). (G) Experimental setup for evaluating RUNX1A:RUNX1C restoration in vivo. ML-DS blasts from 2 patients were transduced with RUNX1A (GFP+), RUNX1C (GFP+), or EV control (GFP+) and mixed 1:1 with EV control–transduced blasts (dTomato+), before transplantation into sublethally irradiated recipient mice. (H) Ratio of GFP+ to dTomato+ cells in the bone marrow of mice euthanized 4 to 8 weeks after transplantation (n = 5, Kruskal-Wallis test). dT, dTomato; ns, not significant.
Importantly, restoring the RUNX1A:RUNX1C equilibrium through RUNX1C expression in patient-derived ML-DS blasts (supplemental Figure 3A) halted proliferation and accelerated differentiation, as indicated by loss of CD117 and gain of CD33 and CD41 expression (Figure 2D-F). Inversely, further increasing the expression of RUNX1A conferred a mild growth advantage in ML-DS blasts and an increase in the fraction of cells with an immature phenotype (CD33+CD117+ or CD41+CD42b−CD117+ myeloid/megakaryocytic blasts; Figure 2D-F). Lastly, we evaluated restoration of the RUNX1A:RUNX1C ratio in vivo through fluorescence-based competitive transplantation assays using 2 patient-derived xenografts from patients with ML-DS (Figure 2G). In both cases, RUNX1C-expressing leukemic blasts were significantly diminished in the bone marrow of recipient mice at the experimental end point, whereas RUNX1A-expressing leukemic blasts were unchanged compared with control-transduced blasts (Figure 2H).
In summary, these experiments in primary human cells depict a landscape of perturbed differentiation and accelerated proliferation guided by RUNX1A. Our data further demonstrate that restoring RUNX1A:RUNX1C equilibrium can overcome differentiation arrest in ML-DS blasts, leading to their depletion.
RUNX1A synergizes with Gata1s in the leukemic transformation of FLCs
Because TAM occurs in utero, implying a fetal origin for this disease, we employed a murine FLC-based in vitro model to study the oncogenic potential of RUNX1 isoform imbalance in concert with mutated Gata1s (supplemental Figure 3A).7 Notably, mice do not express the RUNX1A isoform,31 underlining important species-specific differences that could explain the inability of previous DS mouse models to recapitulate the human TAM/ML-DS phenotype.18,32 Combined lentiviral transduction of Cas9-knockin FLCs with RUNX1A and a sgRNA targeting exon 2 of Gata1, thereby introducing Gata1s mutations, indeed resulted in a robust hyperproliferative phenotype (Figure 3A; 4140-fold) compared with cells transduced with a combination of a nontargeting control sgRNA and an empty vector (EV). Individually, Gata1s (sgGata1 + EV) and RUNX1A expression (control sgRNA + RUNX1A) also enhanced growth (Figure 3A; 366-fold and 140-fold, respectively), but the effect was less pronounced than in combination, suggesting synergy between Gata1s and RUNX1A. Inversely, ectopic expression of RUNX1C or RUNX1B alone or in combination with Gata1s mutations resulted in a growth disadvantage (Figure 3A; supplemental Figure 4A). We note that none of the other Hsa21 candidate genes selected from our sgRNA dropout screen produced a synergistic hyperproliferative phenotype with Gata1s (supplemental Figure 4B). Immunophenotypically, the hyperproliferative RUNX1A Gata1s-FLCs were mainly CD117+CD41+ and partially CD71+Ter119+ (Figure 3B), corresponding to megakaryocytic progenitors with erythroid features, a hallmark of TAM/ML-DS. The expression of erythroid markers was increased upon RUNX1A expression compared with control Gata1s-FLCs (Figure 3B). The synergistic effect of RUNX1A and Gata1s on the proliferation of FLCs was also observed in culture conditions promoting the expansion and differentiation of erythroid progenitor cells but not in conditions promoting myeloid expansion and differentiation (supplemental Figure 4C).
Figure 3.

RUNX1A synergizes with Gata1s in leukemic transformation of murine FLCs. Cas9-knockin Ter119− FLCs were lentivirally transduced with either Gata1 (sgGata1s) or sgCtrls, as well as with RUNX1A, RUNX1B, RUNX1C, or EV control. (A) Absolute cell number of murine FLCs after combined transduction with lentiviral vectors and maintenance in liquid cultures. Data from 1 representative experiment performed in replicates are shown as mean ± SD (2-way ANOVA); ∗P < .05, ∗∗P < .001, ∗∗∗∗P < .0001. (B) Flow cytometry plots of murine sgGata1s + RUNX1A, sgGata1s + EV, and sgCtrl + RUNX1A transduced FLCs in liquid culture. The percentage of cells belonging to each immunophenotype is indicated in the corresponding gates. Data from 3 replicates ± SD are shown. (C-D) Bar graphs showing NESs of significantly upregulated or downregulated gene sets associated with differentiation and oncogenic cellular programs, in Gata1s- (sgGata1s + EV, orange), RUNX1A- (sgCtrl + RUNX1A, blue), and Gata1s- and RUNX1A- (sgGATA1s + RUNX1A, red) murine FLCs compared with sgCtrl + EV control FLCs. This analysis highlights the unique and synergistic functional features of Gata1s and RUNX1A. ∗False discovery rate (FDR) q < .25; ∗∗FDR q < .05. (E) Bar graph showing NESs of significantly upregulated or downregulated gene sets in (red) Gata1s + RUNX1A– and (green) Gata1s + RUNX1C–expressing FLCs compared with control sgCtrl + EV FLCs. This analysis highlights the contrasting functional features of RUNX1A and RUNX1C on a Gata1s background. ∗FDR q < .25; ∗∗FDR q < .05. (F-H) Analysis of mice transplanted with RUNX1A- or EV-transduced Gata1s-FLCs (n = 10 per group), including comparisons of Kaplan-Meier survival curves (log-rank test) (F), spleen weights (unpaired Student t test) (G), and flow cytometry on bone marrow cells from the diseased mice (2-way ANOVA) (H). (I) Heat map representation of RNA-seq data, showing unsupervised hierarchical clustering on the 2824 most variable genes (SD > 1) across 5 bone marrow samples from leukemic mice and fluorescence-activated cell sorter–sorted murine fetal liver HSCs [LSK (Lin−Sca1+cKit+), common myeloid progenitors (CMPs) (Lin−Sca1−cKit+CD34+ CD16/32med), granulocyte-monocyte progenitors (GMPs) (Lin−Sca1−cKit+CD34+ CD16/32+), and megakaryocyte-erythroid progenitors (MEPs) (Lin−Sca1−cKit+CD34− CD16/32low)]. The sample types are color coded on the bottom of the heat map and z scores are indicated by the legend at the top of the panel. (J) Bar graph showing NESs of significantly upregulated or downregulated gene sets in the murine leukemia samples compared with all progenitor populations. ∗FDR q < .25; ∗∗FDR q < .05. LT-HSCs, long-term hematopoietic stem cells.
Comparative transcriptomic analysis of RNA-seq data substantiated the synergy between Gata1s mutations and RUNX1A expression. As previously described, gene set enrichment analysis revealed a reduced expression of erythroid genes and concurrent activation of proproliferative genes, including MYC and E2F targets, in Gata1s mutated FLCs (Figure 3C-D; supplemental Table 4).16,18,33 Although RUNX1A expression mitigated the reduced expression of erythroid genes caused by Gata1s, it triggered a similar upregulation of MYC and E2F target genes, suggesting convergence on these oncogenic pathways. In addition, RUNX1A expression in the background of Gata1s induced target genes of EVI1, one of the most invasive proto-oncogenes in human leukemia34 and a long-term hematopoietic stem cell signature, whereas the opposite was true upon RUNX1C expression (Figure 3D-E; supplemental Table 4). Hence, the synergistic oncogenic expression program of Gata1s and RUNX1A is characterized by the induction of EVI1, MYC, and E2F target genes, together with a long-term hematopoietic stem cell signature and the concomitant repression of erythroid and megakaryocytic differentiation signatures.
To further explore the leukemogenic potential of RUNX1 isoform disequilibrium in the pathogenesis of DS-associated leukemia, we performed in vivo experiments using Gata1s-FLCs. Upon transplantation into sublethally irradiated syngeneic recipients (C57Bl/6J), Gata1s-FLCs typically become transiently abundant in the peripheral blood.7 In contrast, RUNX1A-expressing Gata1s-FLCs caused high-penetrance leukemia (100%) with a short latency (median survival, 39 days) and organ infiltration (Figure 3F-G; supplemental Figure 4D). Detailed flow cytometry analysis revealed cells of a megakaryocytic progenitor–like phenotype (CD41+CD117+CD34−CD16/32low) resembling TAM and ML-DS (Figure 3H; supplemental Figure 4E), which reinitiated disease when transplanted into secondary recipients (supplemental Figure 4F). Notably, RUNX1A expression did not cause leukemia in wild-type Gata1-FLCs (supplemental Figure 4G). Transcriptomic analysis of RNA-seq data from the murine leukemias compared with stringently sorted murine fetal liver stem and/or progenitor populations confirmed the megakaryocytic progenitor–like phenotype and the ML-DS–like gene expression profile26,35 (Figure 3I-J; supplemental Table 5). Importantly, Cre recombinase–mediated excision of the LoxP-flanked RUNX1A complementary DNA induced rapid depletion of the leukemic blasts in vitro, underlining their dependency on RUNX1A and its importance in ML-DS pathogenesis (supplemental Figure 4H). Of note, miR-125 further accelerated the leukemic transformation of Gata1s-FLCs by RUNX1A, suggesting synergy between RUNX1A and another well-characterized oncogene on chromosome 21 (supplemental Figure 4I).16,36
These data demonstrate that the interplay between Gata1s and RUNX1 isoform disequilibrium results in the proliferation and accumulation of immature megakaryocytic progenitors in vitro, and in ML-DS–like leukemia or an aggressive form of TAM in vivo.
Distinct RUNX1A and RUNX1C protein interaction networks
To better understand the oncogenic mechanisms mediated by RUNX1A in TAM/ML-DS, we compared RUNX1A and RUNX1C protein interaction networks via coimmunoprecipitation of doxycycline-inducible hemagglutinin (HA)-tagged RUNX1A and RUNX1C in CMK cells, followed by mass spectrometry of the bound cofactors (Figure 4A; supplemental Figure 5A). In total, 98 and 57 proteins were significantly bound by RUNX1C and RUNX1A, respectively (Figure 4B; supplemental Figure 5B-C). Of these, 45 proteins were commonly bound by both RUNX1 isoforms, including CBFβ and other bona fide RUNX1 interaction partners, whereas 53 protein interactions were unique to RUNX1C. RUNX1C-specific cofactors were enriched for proteins involved in the regulation of cell cycle, gene expression, protein and messenger RNA metabolism, as well as chromosome organization (Figure 4B). Interestingly, members of the spliceosome A/C and NSL complexes (eg, SF3B1, WDR5, and KANSL2) were among the RUNX1C-specific cofactors (supplemental Figure 5D), suggesting an active role in splicing and chromosomal organization that is lost in RUNX1A, as demonstrated with the RUNX1::RUNX1T1 fusion oncoprotein.37 In contrast, the 12 RUNX1A-specific cofactors were involved in active transcription/replication and G1-S transition, including the megakaryocytic transcription factor NFE2, which is mutated in a subset of patients with myeloid neoplasms and is functionally involved in the megakaryocyte differentiation blockage of GATA1s pluripotent stem cells (Figure 4B).38, 39, 40 Importantly, MAX, a crucial cofactor of MYC, was among the RUNX1A-specific interacting proteins, as verified via western blot (Figure 4C). GATA1s coimmunoprecipitated with RUNX1A and RUNX1C, albeit not at a significant enrichment level (data not shown). As GATA1 is a well-described RUNX1 interaction partner41 and as GATA1s mutations characterize TAM/ML-DS,42,43 we investigated their putative differential binding of RUNX1A and RUNX1C in more detail. To this end, we pulled down doxycycline-induced HA-tagged GATA1 or GATA1s in stably transduced CMK cells, which harbor endogenous GATA1s mutations and, hence, exclusively express GATA1s; this is an important point, because GATA1 forms homodimers44,45 precluding the enrichment of GATA1s-specific protein complexes in the presence of GATA1. We found that both GATA1 and GATA1s interact with RUNX1C, as previously described.46 However, neither GATA1 nor GATA1s appear to interact with RUNX1A (Figure 4D). Thus, our proteomic data suggest an altered protein interaction network, which may contribute to the TAM/ML-DS–specific phenotype of RUNX1A in GATA1-mutated cells.
Figure 4.

Distinct RUNX1A and RUNX1C protein interaction networks. (A) Experimental setup for isolating HA-RUNX1A– or HA-RUNX1C–containing protein complexes from CMK cells. (B) RUNX1A and RUNX1C complex composition in CMK cells with functional grouping. The Venn diagram shows significantly enriched interactors (log2 fold change > 1; P < .05). (C) Western blot confirming coimmunoprecipitation of MAX and RUNX1A using anti-MAX and anti-RUNX1 antibodies. Representative picture of 2 independent experiments using K562 cells are shown. A 10% input was used as the loading control. (D) Western blot showing RUNX1A/C isoforms coimmunoprecipitated with doxycycline-inducible HA-tagged GATA1 and GATA1s or the EV vector. Representative pictures of 3 independent experiments using CMK cells are shown. A 5% input was used as the loading control. IP, immunoprecipitation; mRNA, messenger RNA.
RUNX1A affects gene regulation by displacing endogenous RUNX1C
To further interrogate the RUNX1A-centered protein interaction network and determine the consequences of its inability to form a complex with GATA1 or GATA1s, we performed CUT&RUN on endogenous GATA1s and RUNX1C (using a C-terminal antibody that does not recognize exogenous RUNX1A) in Gata1s-FLC with or without exogenous expression of doxycycline-inducible HA-tagged RUNX1A or RUNX1C (Figure 5A; supplemental Figure 6A)47. We found that 33% of the promoter or enhancer regions occupied by endogenous RUNX1C were also occupied by HA-RUNX1A, and that half (52%) of the RUNX1C/RUNX1A-occupied regulatory regions were cobound by GATA1s (Figure 5B; supplemental Table 6), corroborating the known GATA1-RUNX1 interplay in transcriptional regulation.48
Figure 5.

RUNX1A affects gene regulation by displacing endogenous RUNX1C. (A) Experimental setup of CUT&RUN on murine Gata1s-FLCs after doxycycline-induced expression of HA-RUNX1A/RUNX1C. CUT&RUN was performed using anti-HA, anti-RUNX1(C-term), and anti-GATA1 antibodies. Peaks were called by SEACR47 using EV control cells as background. Data represent 1 experiment. (B) Venn diagram showing the number of genomic regions bound by endogenous RUNX1, endogenous GATA1s, and/or HA-RUNX1A after HA-RUNX1A overexpression. (C) Heat maps depicting the colocalization of endogenous GATA1s (orange, left), endogenous RUNX1 (green, middle), and HA-RUNX1A/-RUNX1C (red, right) signals after doxycycline-induced HA-RUNX1A or HA-RUNX1C expression in Gata1s-FLCs. EV-transduced cells were used as a control. Regions ±2.5 kilobases (kb) of the peak center are shown. Binding intensities are represented as normalized reads per kilobase of transcript per million reads mapped values (bars below the blots). (D) Binding intensities (normalized read density) of RUNX1 (left) and GATA1s (right), in EV control and doxycycline-induced HA-RUNX1A and HA-RUNX1C expressing cells. (E) Transcription factor motif enrichment analysis under RUNX1 peaks, at the promoter regions of genes that are differentially expressed upon doxycycline-induced RUNX1A expression (log2 fold change = 1). Human promoter regions were used as background. z score intensities are shown. (F) Integrative Genomics Viewer snapshots of Mxd1 and Lyl1 gene promoters showing occupancy of endogenous GATA1s, RUNX1, and HA-RUNX1A/-RUNX1C in Gata1s-FLCs after doxycycline-induced HA-RUNX1A/HA-RUNX1C expression or in EV control–expressing cells. The tracks display coverage (reads per kilobase of transcript per million reads mapped) (left). Scale and chromosome location are shown (top).
Unsupervised clustering of peaks at regulatory regions bound by endogenous RUNX1C, GATA1, and HA-RUNX1A revealed 3 clusters (Figure 5C). De novo motif discovery uncovered an enrichment of RUNX family motifs in clusters 1 and 3, whereas GATA family motifs were most abundant in cluster 2 (supplemental Figure 6B-C). Importantly, we observed a global reduction of endogenous RUNX1 occupancy upon HA-RUNX1A expression across all clusters (Figure 5D; supplemental Figure 6D; supplemental Table 6). Consistent with our protein-protein interaction studies, GATA1s binding increased globally upon expression of HA-RUNX1C, compared with HA-RUNX1A and the EV control (Figure 5D; supplemental Figure 6E).
Interestingly, most of the genes in the 3 CUT&RUN clusters were upregulated upon HA-RUNX1A expression (67.6% to 81.5%) (supplemental Figure 6C). Transcription factor motif analysis of peaks in the promoter regions of differentially expressed genes revealed an overrepresentation of the MYC-coactivation factor E2F, as well as of single MYC, MAX, and MYC:MAX dimeric motifs (Figure 5E; supplemental Table 7). The median distance between the RUNX1 and MYC:MAX binding motifs (under HA-RUNX1A peaks) was 298 base pairs. Among the RUNX1A-induced genes with MAX or MYC:MAX binding motifs in their promoter regions are Max dimerization protein 1 (Mxd1 [Mad1]), MYC target 1 (Myct1), known oncogenic drivers such as Lyl1 and Malat1, as well as the megakaryocyte gene Itga2b (CD41), all of which were repressed by RUNX1C in RNA-seq experiments (Figure 5F; supplemental Figure 6F-H). These data are consistent with our comparative transcriptomic analyses in Gata1s-FLCs that uncovered that the activation of MYC and E2F targets by GATA1s and RUNX1A results in a synergistic induction of proliferation and an immature phenotype, whereas the opposite was true for RUNX1C (Figure 3C-E).
Taken together, our proteomic and genomic analyses imply that RUNX1A interferes with normal RUNX1C function by displacing it from target gene promoters. Instead, RUNX1A interacts with the MYC cofactor MAX and induces an MYC- and E2F-driven expression program. The concerted alteration of normal megakaryocytic progenitor gene expression programs through GATA1s and RUNX1A leads to malignant transformation in these cells via the upregulation of oncogenic gene expression programs and the perturbation of RUNX1C-regulated differentiation programs, respectively.
Targeting MYC:MAX dimerization as a therapeutic approach for ML-DS
Finally, we investigated the role of the MYC cofactor MAX, which we hypothesized to be central in the synergy between GATA1s and RUNX1A, during leukemia pathogenesis. Short hairpin RNA–mediated knock down and sgRNA-mediated knock out of MAX inhibited the growth of CMK cells in vitro (supplemental Figure 7A-B). These findings were confirmed in murine RUNX1A Gata1s-FLCs and in ML-DS blasts derived from 2 patients (supplemental Figure 7C-F). Normal CD34+ HSPCs showed only a mild growth impairment upon MAX depletion (supplemental Figure 7G-H). Importantly, pharmacological disruption of MYC:MAX dimerization using the MYC inhibitor MYCi36149 caused apoptotic cell death in RUNX1A Gata1s-FLCs and in CMK and K562 cells, which are MYC-dependent (Figure 1B), in a dose-dependent manner (Figure 6A; supplemental Figure 8A-B). Accordingly, MYCi361 also induced apoptosis and partial differentiation in ML-DS blasts derived from 3 patients, with median lethal concentration (50% inhibitory concentration [IC50]) values of 2.09 μM, 2.63 μM, and 3.82 μM, respectively (Figure 6B; supplemental Figure 8C). Similarly, blasts derived from patients with non-DS acute megakaryoblastic leukemia (AMKL) or KMT2Ar, 2 other AML subgroups that have elevated RUNX1A:RUNX1C ratios (supplemental Figure 1H-K) also showed good responses with partial differentiation and median IC50 values between 3.9 and 4.32 μM and from 3.9 to 5.88 μM, respectively (supplemental Figure 8D-H). Normal CD34+ HSPCs from 2 donors had IC50 values of 7.291 μM and 7.168 μM, respectively (supplemental Figure 8I-K), outlining a therapeutic window. Lastly, we evaluated MAX knockdown in vivo through fluorescence-based competitive transplantation assays using patient-derived xenografts from patients with ML-DS and AMKL (Figure 6C). In both cases, MAX-depleted leukemic blasts were significantly diminished in the bone marrow of recipient mice at the experimental end point (Figure 6D).
Figure 6.

Interfering with MYC:MAX dimerization as a therapeutic strategy. Dose response curves for MYC:MAX dimerization inhibitor MYCi361 in Gata1s-FLC cells (A, left) and in ML-DS blasts derived from 3 patients (B, left) 24 hours after treatment in vitro. Corresponding IC50 values are depicted below the graphs. Bar graphs showing the percentage of annexin V+ cells after treatment with the indicated doses of MYCi361 compared with the dimethyl sulfoxide control in Gata1s-FLCs (A, right) and in ML-DS blasts (B, right); data represented as mean ± SD (n = 3, 1-way ANOVA). (C) Experimental setup for evaluating MAX dependence in ML-DS and AMKL cells in vivo. ML-DS or AMKL blasts derived from 2 patients were transduced with short hairpin MAX (shMAX) (GFP+) or short hairpin control (shCtrl) (GFP+) and mixed 1:1 with shCtrl-transduced blasts (dTomato+), before transplantation into sublethally irradiated recipient mice. (D) Ratio of GFP+ to dTomato+ cells in the bone marrow of mice euthanized 4 to 8 weeks after transplantation (n = 5, Kruskal-Wallis test).
Thus, we have demonstrated that the MYC cofactor MAX is key to leukemic transformation mediated by RUNX1A and GATA1s, and that it can be therapeutically exploited.
Discussion
Aneuploidies, and in particular, partial or complete amplifications of chromosome 21, are frequent alterations in leukemia; however, their underlying pathomechanisms remain enigmatic.3 In this study, we leveraged the model system of DS-associated TAM and ML-DS to interrogate oncogenic factors on chromosome 21 for their roles in AML pathogenesis. A CRISPR-Cas9 screen of chromosome 21 genes unexpectedly revealed RUNX1 as an ML-DS dependency in cell lines and patient-derived blasts in vitro and in vivo, despite former controversy regarding its role in TAM and ML-DS.3,24 Through detailed functional validation, we discovered that RUNX1 isoform disequilibrium in the form of RUNX1A bias, rather than RUNX1 gene dosage, is key to trisomy 21–associated leukemogenesis. By dissecting the consequences of RUNX1 disequilibrium in TAM/ML-DS, we showed that RUNX1A acts in concert with pathognomonic GATA1s mutations in this context, thereby blocking megakaryocytic differentiation and accelerating progenitor proliferation, effects that were reversed upon restoring the RUNX1A:RUNX1C equilibrium in murine models and patient-derived blasts in vitro and in vivo. These findings are in line with the known function of RUNX1C during adult hematopoiesis,50, 51, 52 in which it regulates megakaryopoiesis by controlling the proliferation and survival of committed progenitors.53 Thus, through the systematic interrogation of chromosome 21–encoded coding genes, our work contributes to understanding the synergy between trisomy 21 and GATA1 mutations in ML-DS leukemogenesis. We suggest a model in which RUNX1A:RUNX1C disequilibrium in TAM and ML-DS is a consequence of the combination of trisomy 21 and GATA1s mutations in fetal HSPCs. Our findings have further implications for other types of leukemia with numerical or structural alterations of chromosome 21 and/or RUNX1 isoform disequilibrium. Indeed, we also observed elevated RUNX1A:RUNX1C ratios in non-DS AMKL, KMT2A-rearranged AML, and AML with complex karyotype (supplemental Figure 1E-K). The exact mechanism leading to RUNX1A:RUNX1C dysregulation in trisomy 21 HSPCs remains an open question. One hypothesis is that altered transcriptional regulation by GATA1s and elevated levels of the Hsa21-encoded splicing factor U2AF1 may combine to drive RUNX1 isoform imbalance in TAM and ML-DS (supplemental Figure 2); different mechanisms or mutations in splicing factors may be responsible in other subtypes of AML or myelodysplastic syndromes.54 In addition, RUNX1A might further enhance the oncogenic effect of miR-125b, another well-characterized oncogenic driver on chromosome 21.16
Mechanistically, we showed that the altered protein interaction network of RUNX1A (compared with RUNX1B/C) underlies its oncogenic function. RUNX1A fails to interact with GATA1 and GATA1s to control megakaryopoiesis.46 Our proteomics analyses point toward RUNX1A’s increased affinity for the MYC cofactor MAX. We propose a model in which RUNX1A, in complex with MAX, displaces RUNX1C from its endogenous binding sites where it normally recruits GATA1. Thus, RUNX1A perturbs RUNX1C:GATA1 regulated gene expression and activates MYC:MAX and E2F target genes, leading to a megakaryocytic differentiation block and acceleration of progenitor proliferation. This model would help explain the synergistic oncogenic effect of RUNX1A and GATA1s; GATA1s not only fails to repress RUNX1A but also E2F and MYC target genes during megakaryopoiesis,18 suggesting a double hit on these pathways in fetal trisomy 21 HSPCs. The central role of MAX in the RUNX1A-induced phenotype was validated by genetically interfering with MAX expression. Importantly, the dependency of ML-DS blasts on intact MAX represents a vulnerability that can be therapeutically exploited, as we demonstrated by using the MYC:MAX dimerization inhibitor MYCi36149 to eradicate blasts from patients with ML-DS.
Although previous studies have reported the oncogenic and overriding negative effects that RUNX1A exerts on RUNX1B,54, 55, 56, 57, 58 our findings are unexpected and have general implications for basic oncology research. First, this study demonstrates that species-specific differences in isoform expression must be considered when interpreting species-specific or divergent phenotypes in mouse models. RUNX1A is a primate-specific isoform of RUNX1,31 which may explain the inability of DS mouse models32,59 to recapitulate the human TAM/ML-DS phenotype or the interplay of GATA1s and trisomy 21 seen in human induced pluripotent stem cells.3,11,19,60 Second, this study underlines the importance of accounting for all isoforms when studying the oncogenic effects of a given gene and emphasizes the importance of alternative splicing in the pathogenesis of cancer. Various mutations in splicing factors have been identified in AML and MDS genomes.61 Whether these mutations are causative for the increased RUNX1A:RUNX1B/C ratio observed in MDS54 will need to be investigated in the future.
Overall, our study provides a general framework for interrogating the contribution of aneuploidy in oncogenesis. Given the current state of the field, in which we have a near-complete map of the cytogenetic, mutational, and epigenetic landscape of cancer, these insights will be crucial for understanding the complex cooperation between common fusion oncogenes, recurrently mutated genes, and larger amplifications or deletions during disease initiation and progression. As we have illustrated with the identification of MAX as a RUNX1A cofactor and its pharmacological inhibition, this knowledge can have direct therapeutic implications.
Conflict-of-interest disclosure: D.R. has advisory roles for Celgene Corporation, Novartis, bluebird bio, and Janssen and receives research funding from CLS Behring and Roche. J.-H.K. has advisory roles for bluebird bio, Novartis, Roche, and Jazz Pharmaceuticals. M.-L.Y. is partially employed by Alacris Theranostics. The remaining authors declare no competing financial interests.
Acknowledgments
The authors thank D. Trono of École Polytechnique Fédérale de Lausanne, Lausanne, Switzerland, for kindly providing pMD2.G (Addgene plasmid 12259) and psPAX2 (Addgene plasmid 12260) and D. E. Zhang for sharing the RUNX1A complementary DNA.
This work was supported by grants from the German Research Foundation (DFG; KL-2374/1-3) (J.-H.K.), BMBF (01GM1911D myPred) (J.-H.K.), the Deutsche Krebshilfe (DKH; #109251 and #110806) (J.-H.K.), the European Research Council under the European Union’s Horizon 2020 research and innovation programme (grant agreement number 714226) (J.-H.K.) and the DKH (#111743) (D.H.). S.G., M.L., F.J.S., S.-K.K., and L.S. were supported by the Hannover Biomedical Research School. J.-H.K. is a recipient of the St. Baldrick’s Robert Arceci Innovations Award.
Figures and the visual abstract were created with BioRender.com.
Authorship
Contribution: S.G. and D.B.-H. performed experiments, analyzed and interpreted data, and revised the manuscript; H.I., R.B., O.A.-V., L.V., A.-L.S., S.L., M.L., C.I., L.S, F.J.S., and S.-K.K. performed experiments and analyzed data; E.R., K.S., M.G., S.M., R.W., and M.N. performed bioinformatics analysis and data interpretation and revised the manuscript; A.S., S.H., and M.-L.Y. supervised the analyses and revised the manuscript; D.R. provided patient material and data; and D.H. and J.-H.K. designed the study, analyzed and interpreted data, wrote the manuscript, and academically drove the project.
Footnotes
The RNA sequencing gene expression data reported in this article have been deposited in the European Nucleotide Archive (accession numbers ERS6071576-ERS6071595). The CUT&RUN data reported in this article have been deposited in the European Nucleotide Archive (accession numbers ERS5956460-ERS5956467). The mass spectrometry proteomics data reported in this article have been deposited in the ProteomeXchange Consortium via the PRIDE partner repository (data set identifier PXD030616 and 10.6019/PXD030616).
Data will be available immediately following publication, no end date, and are available on request from the corresponding authors, Jan-Henning Klusmann (jan-henning.klusmann@kgu.de) and Dirk Heckl (dirk.heckl@uk-halle.de).
The online version of this article contains a data supplement.
There is a Blood Commentary on this article in this issue.
The publication costs of this article were defrayed in part by page charge payment. Therefore, and solely to indicate this fact, this article is hereby marked “advertisement” in accordance with 18 USC section 1734.
Contributor Information
Dirk Heckl, Email: dirk.heckl@uk-halle.de.
Jan-Henning Klusmann, Email: jan-henning.klusmann@kgu.de.
Supplementary Material
References
- 1.Cancer Genome Atlas Research N. Ley TJ, Miller C, et al. Genomic and epigenomic landscapes of adult de novo acute myeloid leukemia. N Engl J Med. 2013;368(22):2059–2074. doi: 10.1056/NEJMoa1301689. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 2.Bolouri H, Farrar JE, Triche T, Jr., et al. The molecular landscape of pediatric acute myeloid leukemia reveals recurrent structural alterations and age-specific mutational interactions. Nat Med. 2018;24(1):103–112. doi: 10.1038/nm.4439. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 3.Maclean GA, Menne TF, Guo G, et al. Altered hematopoiesis in trisomy 21 as revealed through in vitro differentiation of isogenic human pluripotent cells. Proc Natl Acad Sci U S A. 2012;109(43):17567–17572. doi: 10.1073/pnas.1215468109. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 4.Mitelman F, Heim S, Mandahl N. Trisomy 21 in neoplastic cells. Am J Med Genet Suppl. 1990;7:262–266. doi: 10.1002/ajmg.1320370752. [DOI] [PubMed] [Google Scholar]
- 5.Duijf PH, Schultz N, Benezra R. Cancer cells preferentially lose small chromosomes. Int J Cancer. 2013;132(10):2316–2326. doi: 10.1002/ijc.27924. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 6.Wechsler J, Greene M, McDevitt MA, et al. Acquired mutations in GATA1 in the megakaryoblastic leukemia of Down syndrome. Nat Genet. 2002;32(1):148–152. doi: 10.1038/ng955. [DOI] [PubMed] [Google Scholar]
- 7.Labuhn M, Perkins K, Matzk S, et al. Mechanisms of progression of myeloid preleukemia to transformed myeloid leukemia in children with Down syndrome. Cancer Cell. 2019;36(2):123–138.e110. doi: 10.1016/j.ccell.2019.06.007. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 8.Yoshida K, Toki T, Okuno Y, et al. The landscape of somatic mutations in Down syndrome-related myeloid disorders. Nat Genet. 2013;45(11):1293–1299. doi: 10.1038/ng.2759. [DOI] [PubMed] [Google Scholar]
- 9.Gurbuxani S, Vyas P, Crispino JD. Recent insights into the mechanisms of myeloid leukemogenesis in Down syndrome. Blood. 2004;103(2):399–406. doi: 10.1182/blood-2003-05-1556. [DOI] [PubMed] [Google Scholar]
- 10.Banno K, Omori S, Hirata K, et al. Systematic cellular disease models reveal synergistic interaction of trisomy 21 and GATA1 mutations in hematopoietic abnormalities. Cell Rep. 2016;15(6):1228–1241. doi: 10.1016/j.celrep.2016.04.031. [DOI] [PubMed] [Google Scholar]
- 11.Roy A, Cowan G, Mead AJ, et al. Perturbation of fetal liver hematopoietic stem and progenitor cell development by trisomy 21. Proc Natl Acad Sci U S A. 2012;109(43):17579–17584. doi: 10.1073/pnas.1211405109. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 12.Ng AP, Hyland CD, Metcalf D, et al. Trisomy of Erg is required for myeloproliferation in a mouse model of Down syndrome. Blood. 2010;115(19):3966–3969. doi: 10.1182/blood-2009-09-242107. [DOI] [PubMed] [Google Scholar]
- 13.Birger Y, Goldberg L, Chlon TM, et al. Perturbation of fetal hematopoiesis in a mouse model of Down syndrome's transient myeloproliferative disorder. Blood. 2013;122(6):988–998. doi: 10.1182/blood-2012-10-460998. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 14.Malinge S, Bliss-Moreau M, Kirsammer G, et al. Increased dosage of the chromosome 21 ortholog Dyrk1a promotes megakaryoblastic leukemia in a murine model of Down syndrome. J Clin Invest. 2012;122(3):948–962. doi: 10.1172/JCI60455. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 15.Volk A, Liang K, Suraneni P, et al. A CHAF1B-dependent molecular switch in hematopoiesis and leukemia pathogenesis. Cancer Cell. 2018;34(5):707–723.e707. doi: 10.1016/j.ccell.2018.10.004. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 16.Alejo-Valle O, Weigert K, Bhayadia R, et al. The megakaryocytic transcription factor ARID3A suppresses leukemia pathogenesis. Blood. 2022;139(5):651–665. doi: 10.1182/blood.2021012231. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 17.Wagenblast E, Araujo J, Gan OI, et al. Mapping the cellular origin and early evolution of leukemia in Down syndrome. Science. 2021;373(6551) doi: 10.1126/science.abf6202. [DOI] [PubMed] [Google Scholar]
- 18.Klusmann JH, Godinho FJ, Heitmann K, et al. Developmental stage-specific interplay of GATA1 and IGF signaling in fetal megakaryopoiesis and leukemogenesis. Genes Dev. 2010;24(15):1659–1672. doi: 10.1101/gad.1903410. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 19.Chou ST, Byrska-Bishop M, Tober JM, et al. Trisomy 21-associated defects in human primitive hematopoiesis revealed through induced pluripotent stem cells. Proc Natl Acad Sci U S A. 2012;109(43):17573–17578. doi: 10.1073/pnas.1211175109. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 20.Grimm J, Heckl D, Klusmann JH. Molecular mechanisms of the genetic predisposition to acute megakaryoblastic leukemia in infants with Down syndrome. Front Oncol. 2021;11 doi: 10.3389/fonc.2021.636633. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 21.Okuda T, van Deursen J, Hiebert SW, Grosveld G, Downing JR. AML1, the target of multiple chromosomal translocations in human leukemia, is essential for normal fetal liver hematopoiesis. Cell. 1996;84(2):321–330. doi: 10.1016/s0092-8674(00)80986-1. [DOI] [PubMed] [Google Scholar]
- 22.Wang Q, Stacy T, Binder M, Marin-Padilla M, Sharpe AH, Speck NA. Disruption of the Cbfa2 gene causes necrosis and hemorrhaging in the central nervous system and blocks definitive hematopoiesis. Proc Natl Acad Sci U S A. 1996;93(8):3444–3449. doi: 10.1073/pnas.93.8.3444. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 23.North TE, de Bruijn MF, Stacy T, et al. Runx1 expression marks long-term repopulating hematopoietic stem cells in the midgestation mouse embryo. Immunity. 2002;16(5):661–672. doi: 10.1016/s1074-7613(02)00296-0. [DOI] [PubMed] [Google Scholar]
- 24.Kirsammer G, Jilani S, Liu H, et al. Highly penetrant myeloproliferative disease in the Ts65Dn mouse model of Down syndrome. Blood. 2008;111(2):767–775. doi: 10.1182/blood-2007-04-085670. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 25.Liu X, Zhang Q, Zhang DE, et al. Overexpression of an isoform of AML1 in acute leukemia and its potential role in leukemogenesis. Leukemia. 2009;23(4):739–745. doi: 10.1038/leu.2008.350. [DOI] [PubMed] [Google Scholar]
- 26.Bourquin JP, Subramanian A, Langebrake C, et al. Identification of distinct molecular phenotypes in acute megakaryoblastic leukemia by gene expression profiling. Proc Natl Acad Sci U S A. 2006;103(9):3339–3344. doi: 10.1073/pnas.0511150103. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 27.Ghozi MC, Bernstein Y, Negreanu V, Levanon D, Groner Y. Expression of the human acute myeloid leukemia gene AML1 is regulated by two promoter regions. Proc Natl Acad Sci U S A. 1996;93(5):1935–1940. doi: 10.1073/pnas.93.5.1935. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 28.Fujita Y, Nishimura M, Taniwaki M, Abe T, Okuda T. Identification of an alternatively spliced form of the mouse AML1/RUNX1 gene transcript AML1c and its expression in early hematopoietic development. Biochem Biophys Res Commun. 2001;281(5):1248–1255. doi: 10.1006/bbrc.2001.4513. [DOI] [PubMed] [Google Scholar]
- 29.Sroczynska P, Lancrin C, Kouskoff V, Lacaud G. The differential activities of Runx1 promoters define milestones during embryonic hematopoiesis. Blood. 2009;114(26):5279–5289. doi: 10.1182/blood-2009-05-222307. [DOI] [PubMed] [Google Scholar]
- 30.Challen GA, Goodell MA. Runx1 isoforms show differential expression patterns during hematopoietic development but have similar functional effects in adult hematopoietic stem cells. Exp Hematol. 2010;38(5):403–416. doi: 10.1016/j.exphem.2010.02.011. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 31.Komeno Y, Yan M, Matsuura S, et al. Runx1 exon 6-related alternative splicing isoforms differentially regulate hematopoiesis in mice. Blood. 2014;123(24):3760–3769. doi: 10.1182/blood-2013-08-521252. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 32.Li Z, Godinho FJ, Klusmann JH, Garriga-Canut M, Yu C, Orkin SH. Developmental stage-selective effect of somatically mutated leukemogenic transcription factor GATA1. Nat Genet. 2005;37(6):613–619. doi: 10.1038/ng1566. [DOI] [PubMed] [Google Scholar]
- 33.Ling T, Birger Y, Stankiewicz MJ, et al. Chromatin occupancy and epigenetic analysis reveal new insights into the function of the GATA1 N terminus in erythropoiesis. Blood. 2019;134(19):1619–1631. doi: 10.1182/blood.2019001234. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 34.Birdwell C, Fiskus W, Kadia TM, DiNardo CD, Mill CP, Bhalla KN. EVI1 dysregulation: impact on biology and therapy of myeloid malignancies. Blood Cancer J. 2021;11(3):64. doi: 10.1038/s41408-021-00457-9. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 35.Schwarzer A, Emmrich S, Schmidt F, et al. The non-coding RNA landscape of human hematopoiesis and leukemia. Nat Commun. 2017;8(1):218. doi: 10.1038/s41467-017-00212-4. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 36.Emmrich S, Rasche M, Schoning J, et al. miR-99a/100∼125b tricistrons regulate hematopoietic stem and progenitor cell homeostasis by shifting the balance between TGFβ and Wnt signaling. Genes Dev. 2014;28(8):858–874. doi: 10.1101/gad.233791.113. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 37.Grinev VV, Barneh F, Ilyushonak IM, et al. RUNX1/RUNX1T1 mediates alternative splicing and reorganises the transcriptional landscape in leukemia. Nat Commun. 2021;12(1):520. doi: 10.1038/s41467-020-20848-z. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 38.Arkoun B, Robert E, Boudia F, et al. Stepwise GATA1 and SMC3 mutations alter megakaryocyte differentiation in a Down syndrome leukemia model. J Clin Invest. 2022;132(14) doi: 10.1172/JCI156290. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 39.Jutzi JS, Bogeska R, Nikoloski G, et al. MPN patients harbor recurrent truncating mutations in transcription factor NF-E2. J Exp Med. 2013;210(5):1003–1019. doi: 10.1084/jem.20120521. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 40.Marcault C, Zhao LP, Maslah N, et al. Impact of NFE2 mutations on AML transformation and overall survival in patients with myeloproliferative neoplasms. Blood. 2021;138(21):2142–2148. doi: 10.1182/blood.2020010402. [DOI] [PubMed] [Google Scholar]
- 41.Elagib KE, Racke FK, Mogass M, Khetawat R, Delehanty LL, Goldfarb AN. RUNX1 and GATA-1 coexpression and cooperation in megakaryocytic differentiation. Blood. 2003;101(11):4333–4341. doi: 10.1182/blood-2002-09-2708. [DOI] [PubMed] [Google Scholar]
- 42.Alford KA, Reinhardt K, Garnett C, et al. Analysis of GATA1 mutations in Down syndrome transient myeloproliferative disorder and myeloid leukemia. Blood. 2011;118(8):2222–2238. doi: 10.1182/blood-2011-03-342774. [DOI] [PubMed] [Google Scholar]
- 43.Kanezaki R, Toki T, Terui K, et al. Down syndrome and GATA1 mutations in transient abnormal myeloproliferative disorder: mutation classes correlate with progression to myeloid leukemia. Blood. 2010;116(22):4631–4638. doi: 10.1182/blood-2010-05-282426. [DOI] [PubMed] [Google Scholar]
- 44.Crossley M, Merika M, Orkin SH. Self-association of the erythroid transcription factor GATA-1 mediated by its zinc finger domains. Mol Cell Biol. 1995;15(5):2448–2456. doi: 10.1128/mcb.15.5.2448. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 45.Shimizu R, Trainor CD, Nishikawa K, Kobayashi M, Ohneda K, Yamamoto M. GATA-1 self-association controls erythroid development in vivo. J Biol Chem. 2007;282(21):15862–15871. doi: 10.1074/jbc.M701936200. [DOI] [PubMed] [Google Scholar]
- 46.Xu G, Kanezaki R, Toki T, et al. Physical association of the patient-specific GATA1 mutants with RUNX1 in acute megakaryoblastic leukemia accompanying Down syndrome. Leukemia. 2006;20(6):1002–1008. doi: 10.1038/sj.leu.2404223. [DOI] [PubMed] [Google Scholar]
- 47.Meers MP, Tenenbaum D, Henikoff S. Peak calling by Sparse Enrichment Analysis for CUT&RUN chromatin profiling. Epigenet Chromatin. 2019;12(1):42. doi: 10.1186/s13072-019-0287-4. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 48.Tijssen MR, Cvejic A, Joshi A, et al. Genome-wide analysis of simultaneous GATA1/2, RUNX1, FLI1, and SCL binding in megakaryocytes identifies hematopoietic regulators. Dev Cell. 2011;20(5):597–609. doi: 10.1016/j.devcel.2011.04.008. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 49.Han H, Jain AD, Truica MI, et al. Small-molecule MYC inhibitors suppress tumor growth and enhance immunotherapy. Cancer Cell. 2019;36(5):483–497.e415. doi: 10.1016/j.ccell.2019.10.001. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 50.Ichikawa M, Asai T, Saito T, et al. AML-1 is required for megakaryocytic maturation and lymphocytic differentiation, but not for maintenance of hematopoietic stem cells in adult hematopoiesis. Nat Med. 2004;10(3):299–304. doi: 10.1038/nm997. [DOI] [PubMed] [Google Scholar]
- 51.Growney JD, Shigematsu H, Li Z, et al. Loss of Runx1 perturbs adult hematopoiesis and is associated with a myeloproliferative phenotype. Blood. 2005;106(2):494–504. doi: 10.1182/blood-2004-08-3280. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 52.Senserrich J, Batsivari A, Rybtsov S, et al. Analysis of Runx1 Using induced gene ablation reveals its essential role in pre-liver HSC development and limitations of an in vivo approach. Stem Cell Rep. 2018;11(3):784–794. doi: 10.1016/j.stemcr.2018.08.004. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 53.Draper JE, Sroczynska P, Leong HS, et al. Mouse RUNX1C regulates premegakaryocytic/erythroid output and maintains survival of megakaryocyte progenitors. Blood. 2017;130(3):271–284. doi: 10.1182/blood-2016-06-723635. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 54.Sakurai H, Harada Y, Ogata Y, et al. Overexpression of RUNX1 short isoform has an important role in the development of myelodysplastic/myeloproliferative neoplasms. Blood Adv. 2017;1(18):1382–1386. doi: 10.1182/bloodadvances.2016002725. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 55.Tanaka T, Tanaka K, Ogawa S, et al. An acute myeloid leukemia gene, AML1, regulates hemopoietic myeloid cell differentiation and transcriptional activation antagonistically by two alternative spliced forms. EMBO J. 1995;14(2):341–350. doi: 10.1002/j.1460-2075.1995.tb07008.x. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 56.Bertrand-Philippe M, Ruddell RG, Arthur MJ, Thomas J, Mungalsingh N, Mann DA. Regulation of tissue inhibitor of metalloproteinase 1 gene transcription by RUNX1 and RUNX2. J Biol Chem. 2004;279(23):24530–24539. doi: 10.1074/jbc.M311804200. [DOI] [PubMed] [Google Scholar]
- 57.Guo H, Ma O, Speck NA, Friedman AD. Runx1 deletion or dominant inhibition reduces Cebpa transcription via conserved promoter and distal enhancer sites to favor monopoiesis over granulopoiesis. Blood. 2012;119(19):4408–4418. doi: 10.1182/blood-2011-12-397091. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 58.Ran D, Shia WJ, Lo MC, et al. RUNX1a enhances hematopoietic lineage commitment from human embryonic stem cells and inducible pluripotent stem cells. Blood. 2013;121(15):2882–2890. doi: 10.1182/blood-2012-08-451641. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 59.Alford KA, Slender A, Vanes L, et al. Perturbed hematopoiesis in the Tc1 mouse model of Down syndrome. Blood. 2010;115(14):2928–2937. doi: 10.1182/blood-2009-06-227629. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 60.Byrska-Bishop M, VanDorn D, Campbell AE, et al. Pluripotent stem cells reveal erythroid-specific activities of the GATA1 N-terminus. J Clin Invest. 2015;125(3):993–1005. doi: 10.1172/JCI75714. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 61.Makishima H, Visconte V, Sakaguchi H, et al. Mutations in the spliceosome machinery, a novel and ubiquitous pathway in leukemogenesis. Blood. 2012;119(14):3203–3210. doi: 10.1182/blood-2011-12-399774. [DOI] [PMC free article] [PubMed] [Google Scholar]
Associated Data
This section collects any data citations, data availability statements, or supplementary materials included in this article.