

Abstract
Efferent brain-stem neurons release acetylcholine to desensitize cochlear hair cells and can protect the inner ear from acoustic trauma. That protection is absent from knockout mice lacking efferent inhibition and is stronger in mice with a gain-of-function point mutation of the hair cell-specific nicotinic acetylcholine receptor. The present work uses viral transduction of gain-of-function receptors to restore acoustic prophylaxis to the knockout mice. Widespread postsynaptic expression of the transgene was visualized in excised tissue with a fluorophore-conjugated peptide toxin that binds selectively to hair cell acetylcholine receptors. Viral transduction into efferent knockout mice reduced the temporary hearing loss measured 1 day post acoustic trauma. The acoustic evoked-response waveform (auditory brain-stem response) recovered more rapidly in treated mice than in control mice. Thus, both cochlear amplification by outer hair cells (threshold shift) and afferent signaling (evoked-response amplitude) in knockout mice were protected by viral transduction of hair cell acetylcholine receptors. Gene therapy to strengthen efferent cochlear feedback could be complementary to existing and future therapies to prevent hearing loss, including ear coverings, hearing aids, single-gene repair, or small-molecule therapies.
Keywords: cochlea, acoustic trauma, efferent inhibition, gain-of-function nAChRs, AAV, gene therapy
Graphical abstract
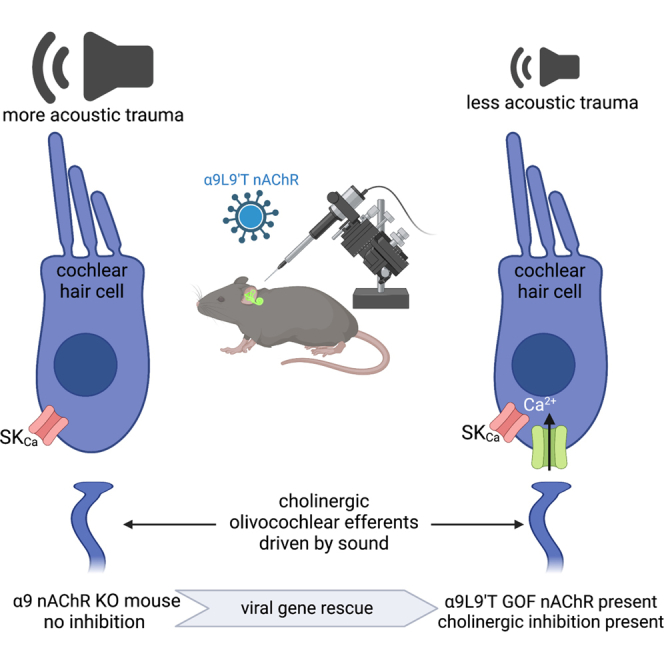
Cholinergic efferent neurons inhibit cochlear hair cells to regulate sensitivity. Consequently, mutant mice lacking the hair cell acetylcholine receptor are more sensitive to acoustic trauma. The authors use viral gene therapy to express the missing receptor, restoring a degree of acoustic protection.
Introduction
Age-related hearing loss (presbycusis) afflicts one in three people ages 65–74, rising to nearly half of those over 75. Early-onset presbycusis runs in families and is exacerbated by loud noise exposure (https://www.nidcd.nih.gov/health/statistics/hearing-charts-tables). Given the years-long onset and obligatory exposure in the workplace, military, or other environments, therapeutic options are limited to the use of protective ear coverings. The present work explores the possibility that cholinergic inhibition by efferent neurons could be leveraged to enhance protection of the inner ear.
Mechanosensory hair cells of vertebrates are subject to cholinergic inhibition by efferent brain-stem neurons.1,2 Cholinergic olivocochlear efferents are driven by sound3,4 to inhibit outer hair cells (OHCs) and provide gain control of the mature cochlea. Efferent feedback is thought to extend the dynamic range, reduce noise masking, enhance selective attention, and contribute to perceptual learning (reviewed in Guinnan,5 Fuchs and Lauer,6 and Lauer et al.7) In addition, transient efferent inhibition of early postnatal inner hair cells (IHCs) may contribute to functional maturation of the auditory pathway.8,9,10,11
Beyond these actions, animal studies support a role for olivocochlear inhibition in protection from acoustic trauma12 in which susceptibility to noise damage correlates inversely with expression of the α9 nicotinic acetylcholine receptor (Chrna9) subunit.13,14 Direct support has been provided by mouse models in which either the α9 or the α10 (Chrna10) subunit of the hair cell’s nAChR was knocked out15 or altered to produce a gain-of-function nAChR (α9L9′T). Noise-induced and age-related hearing loss is exacerbated in α9-knockout mice, but mitigated in the α9L9′T gain-of-function-knockin mice.16,17 The protective effects of efferent feedback make it a promising target for therapeutic intervention. For example, positive allosteric modulators of the hair cell’s nAChR could provide a drug regimen to protect from hearing loss.18 Another strategy is to employ gene therapy to convert hair cell nAChRs to the gain-of-function phenotype to preserve residual hearing. The present work describes viral transduction for genetic rescue of “efferent-knockout” mice as a proof of principle for generalized acoustic prophylaxis.
Gene therapy for sensory loss broke ground with application to retinitis pigmentosa, reviewed in Delmaghani and El-Amraoui.19 Efforts to repair the inner ear have focused largely on the rescue of single-gene mutations in mouse models,19,20 with some examples including vesicular glutamate transporters,21 the presynaptic release protein otoferlin,22,23 and mechanotransduction channels,24 reviewed in Lustig and Akil20 and Ahmed et al.25 These strategies aim to correct specific, monogenic causes of congenital hearing loss. Progress is beginning for “generic” noise-induced26 and age-related hearing loss, with presbycusis being by far the most prevalent pathology in industrialized societies (https://www.nidcd.nih.gov/health/age-related-hearing-loss). Environmental noise exposure can hasten the onset and increase the severity of presbycusis.27,28 Thus, strengthening the naturally occurring sound-evoked feedback inhibition will provide enhanced automatic gain control, matching the level of protection to the level of threat. Therefore, the present study pursues a genetic “gain-of-function” strategy using viral transduction to repair the inner ear’s own protective feedback and thereby reduce acoustic trauma. An additional benefit of this strategy is that it could apply to any pathology that is exacerbated by noise and so would be complementary to other therapies. This could have particular benefit for those at risk of early-onset, activity-dependent hearing loss.
Results
Imaging virally transduced nAChRs with Cy3-RgIA-5727 (α-conotoxin)
The efficacy of posterior semicircular canal injection with adeno-associated virus AAV2.7m8 was determined initially by cochlear expression of green fluorescent protein (GFP) carried by the virus. Injection at postnatal days 0–2 (P0–P2) produced widespread expression of GFP throughout the cochlea at P19–P21 (Figures 1C and 1D). GFP expression was seen in IHCs and OHCs and in supporting cells that surround the IHCs, as reported previously.29
Figure 1.

Viral transduction
(A) AAV2.7m8.CAG.mChrna9.IRES.eGFP.bGH was synthesized to carry either green fluorescent protein or the gain-of-function α9L9′T nAChR. (B) Virus was injected into the left posterior semicircular canal at P0–P2. Auditory brain-stem responses (ABRs) were recorded at 4 weeks, followed by exposure to loud sound 2 days later, followed by ABRs 1 and 14 days post noise exposure. The injection protocol was validated by examining GFP expression. (C) Middle turn of a P21 left cochlea. GFP fluorescence was observed in both inner and outer hair cells and pillar cells throughout the cochlea. (D) Enlargement of boxed region from (C) showing GFP fluorescence in hair cells and supporting cells.
Viral transduction of α9L9′T into α9-null mice was assessed by labeling hair cell nAChRs with Cy3-conjugated RgIA-5727. RgIA is a modified form of an α peptide isolated from cone snail venom that binds to and blocks α9-containing nAChRs with very high affinity.30 RgIA conjugated to the fluorophore Cy3 (Cy3-RgIA-5727) was shown to bind irreversibly at hair cell postsynaptic densities opposite presynaptic efferent terminals.31 In the present work, Cy3-RgIA-5727 labeled OHCs of α9-null mice after viral transduction in a pattern consistent with the known efferent innervation of cochlear tissue (Figure 2). When hair cells were visualized by loading with the permeant fluorescent dye FM4-64, bright, circumscribed, Cy3 puncta were observed at the base of OHCs in 3-week-old mouse cochleas (Figure 2). Fluorescent puncta were also seen near the cuticular plate, where efferent synaptic contacts have been described in electron micrographs32,33,34,35 and demonstrated by hemagglutinin (HA)-tagged nAChR expression.36 In addition to labeled synaptic puncta, a nonspecific red Cy3 signal combines with green FM4-64 to produce yellow or orange OHC and IHC stereociliary bundles. The hydrophobic Cy3 moiety sticks to membranes and becomes most evident where multiple layers of membrane overlap. This also occurs in the tightly packed Kölliker’s organ present in the neonatal cochlea.36 The specificity of Cy3-RgIA-5727 for α9-containing nAChRs is confirmed by the lack of synaptic labeling in negative control α9-null cochlear tissue (Figure 2A1 inset, reproduced from Fisher et al.31 with permission). Note that OHC bundles remain yellow, and nonspecific Cy3 label is seen medial to the IHCs. Following virus injection at P0–P2, expression of α9L9′T as shown by Cy3-RgIA-5727 label in live tissue explants increased from 2 to 3 weeks. By 3 weeks postinjection 81 Cy3 puncta associated with 57 OHCs were counted in one middle cochlear segment and 43 Cy3 puncta on 47 OHCs in a second mid-cochlear segment from a different mouse. A few small, scattered Cy3 puncta were found medial to IHCs in these tissues, but younger cochleas (when IHCs have numerous efferent synapses) were not examined.
Figure 2.

Expression of virally transduced α9L9′T in α9-null cochleas
Viral injection at P0–P2 was followed by labeling of acutely isolated live tissue with Cy3-RgIA-5727 conopeptide at P21. (A) A 3D projection looking along the organ of Corti. (A1) A yz projection from a negative control (no viral injection) α9-null cochlea (reprinted with permission from Fisher et al.31). (B) Left dashed arrow indicates this yz projection from (A). (C) Right dashed arrow indicates this xz projection from (A). Cy3 puncta (red) were found at the synaptic pole of every outer hair cell in the middle turn of an excised cochlea. Puncta also were found near the cuticular surface of outer hair cells (triangles), where efferent synapses have been reported. Cy3 puncta were seen rarely near inner hair cells at this age. Hair cells in live tissue were visualized by accumulation of FM4-64 vital dye.
Noise-induced hearing loss
Acoustic trauma causes hearing loss that is greater in α9-null mice than in wild-type littermates, but is reduced in α9L9′T mice compared with wild type or α9 null.16,17,37 An acoustic trauma protocol was developed to determine if the acoustic vulnerability of α9-null mice could be “rescued” by viral transduction of α9L9′T DNA. It is not possible to know a priori how effective any individual virus injection might be. Thus, to reveal even partial protection, the trauma protocol was designed to produce a temporary threshold shift (20–30 dB across tested frequencies). Mice were exposed to 90 dB noise (2–20 kHz) for 2 h. Auditory brain-stem response (ABR) thresholds 1 and 14 days post trauma were compared with baseline thresholds collected prior to acoustic trauma. It was noted previously37 that transgenic mice expressing the α9L9′T gain-of-function nAChR have baseline thresholds 5–10 dB higher than those of wild-type littermates. In that previous study this was attributed to enhanced efferent feedback activated by the test sounds and was reversed by blocking hair cell nAChRs. Mice injected with the α9L9′T-containing virus (combined with GFP-expressing virus) had ABR thresholds averaging 10 dB higher than those of uninjected mice or those given control injections with GFP-expressing virus only (Figure 3A). There were significant differences in threshold between α9L9′T-injected mice and uninjected, or GFP-injected mice, at all but 12 kHz (multiple unpaired t -tests with Welch correction). Mixed-effects linear model testing (restricted maximum likelihood [REML]) found that thresholds across all frequencies (including clicks) were significantly elevated in α9L9′T-injected compared with uninjected mice (p < 0.0001, F (1, 138) = 50.09) and likewise for α9L9′T-injected mice compared with GFP-injected mice (p < 0.0001, F (1, 54) = 40.41). Pretrauma thresholds did not differ between uninjected and GFP-injected mice, demonstrating that the surgery, injection procedure, viral infection, and GFP expression per se were not damaging.
Figure 3.

Noise-induced hearing loss in α9-null mice and impact of virus injection
(A) ABR threshold prior to acoustic trauma. Mean and SEM for C57BL/6J α9-null mice injected with α9L9′T virus plus GFP virus (magenta, n = 31). Mean and SEM for uninjected mice (black, n = 24). Mean and SEM for GFP (only)-injected mice (blue, n = 10). Blue asterisks compare α9L9′T plus GFP-injected with GFP only-injected. Black asterisks compare α9L9′T-injected to uninjected. (B) One day post-acoustic-trauma thresholds did not differ among all three cohorts of mice. (C) Fourteen days after trauma, thresholds recovered, with α9L9′T-injected mice having significantly higher thresholds than either set of control mice. (D) Mean (±SEM) ABR threshold for 24 uninjected α9-null C57BL/6J mice. Black circles, before; black squares, 1 day post; gray line and triangles, 14 days post. (E) Mean (±SEM) ABR threshold for 10 α9-null C57BL/6J mice injected with GFP virus only. Blue circles, before; cyan squares, 1 day post; pale blue line and triangles, 14 days post. (F) Mean (±SEM) ABR threshold for 31 α9-null C57BL/6J mice injected with α9L9′T virus plus GFP virus. Magenta circles, before; pink squares, 1 day post; light pink line and triangles, 14 days post trauma. Statistical significance from multiple unpaired t tests with Welch correction, ∗p < 0.05, ∗∗p < 0.01, ∗∗∗p < 0.001.
At 1 day post acoustic trauma, there were no significant differences in ABR threshold among the three mouse cohorts (Figure 3B). Thresholds at 14 days post acoustic trauma were again significantly different between α9L9′T-injected mice and the others (Figure 3C), returning to the baseline condition. Mixed effects linear model testing (REML) of the 14 day post-trauma thresholds in α9L9′T-injected was compared with that of uninjected mice (p < 0.0001, F (1, 126) = 25.09) and likewise for α9L9′T-injected mice compared with that of GFP-injected mice (p = 0.0004, F (1, 269) = 12.97). Fourteen days post trauma, the original differential sensitivity was re-established.
To probe these results further, each cohort of mice was examined independently for the impact of acoustic trauma at 1 and 14 days post trauma. In the α9-null mice that received no virus injection (Figure 3D), the acoustic trauma protocol produced a significant threshold elevation in click and pure-tone thresholds from 8 to 32 kHz 1 day after exposure (click 13.8 ΔdB, 8 kHz 17.2 ΔdB, 12 kHz 14.8 ΔdB, 16 Hz 21.9 ΔdB, 24 kHz 22.7 ΔdB, 32 kHz 28.5 ΔdB, p < 0.001 for all frequencies, Welch correction for multiple unpaired t tests). After 14 days, ABR threshold returned to near baseline; thus, it was a temporary threshold shift. Mice injected with the GFP-containing virus only (Figure 3E) experienced threshold shifts at 1 day post trauma, similar to those in the uninjected mice, with recovery at 14 days (1 day: click 15.8 ΔdB, 8 kHz 24.7 ΔdB, 12 kHz 22.5 ΔdB, 16 kHz 20.2 ΔdB, 24 kHz 28.5 ΔdB, 32 kHz 30.8 ΔdB; p < 0.01 or 0.001 for comparisons at each pure-tone frequency [clicks p < 0.05], Welch correction for multiple unpaired t tests.) In contrast, mice that received a unilateral injection of the α9-containing virus (plus the GFP-containing virus) had no, or smaller, shifts at all frequencies 1 day post trauma, with significant hearing loss only at mid- and upper frequencies (Figure 3F). Threshold shifts at 1 day were click 8.7 dB, 8 kHz 8.9 dB, 12 kHz 7.4 dB, 16 kHz 12.1 dB, 24 kHz 16.8 dB, 32 kHz 19.5 dB; significant for 16, 24, and 32 kHz only.
Mixed-effects linear model testing (REML) was run to compare threshold shifts at 1 day post trauma between experimental conditions and across frequencies. The 1-day threshold shifts in mice injected with α9-containing virus (plus GFP-containing virus) were significantly less than those for control mice that received no injection (p = 0.0013, F (1, 30.00) = 12.67). A significant difference in threshold shift also was found comparing α9L9′T (plus GFP-containing virus)-injected mice with those injected with the GFP-containing virus only (p < 0.0001, F (1, 231) = 28.28). Thus, injection of an AAV vector expressing the gain-of-function α9L9′T nAChR subunit specifically reduced the temporary threshold shift caused by acoustic trauma in α9-null mice.
ABR waveforms
A change in ABR threshold could be due to degraded mechanotransduction, reduced OHC function, altered endolymph, etc. Damage also can occur at the synaptic contacts between IHCs and type I afferents38,39 without necessarily altering acoustic thresholds. This deficit can be revealed by measuring the amplitude and latency of wave 1 of the ABR. Acoustic trauma reduces the maximum amplitude, indicating fewer type I afferents contributing to the initial compound action potential, either from reduced numbers or from a spread in latencies. Previous studies of α9L9′T transgenic knockin mice found reduced amplitude wave 1 of the ABR compared with wild-type or α9-knockout mice16 that was unchanged after acoustic trauma. Likewise, in the present work, the baseline (pretrauma) amplitude of wave 1 of the click-evoked ABR was significantly smaller in mice injected with α9-containing virus (plus GFP virus) compared with mice that were not injected, as well as that of mice injected with GFP-containing virus only (Figure 4A). Two-way ANOVA of ABR wave 1 for α9L9′T-injected versus uninjected gave p < 0.0001, F (1, 298) = 33.38 and likewise, for α9L9′T-injected versus GFP-injected, p < 0.0001, F (1, 269) = 51.93. ABR wave 1 of GFP-injected mice versus that of uninjected mice was not significantly different across frequencies (p = 0.2578, F (1, 245) = 1.286). At 14 days after acoustic trauma there were no significant differences at each frequency among all cohorts (multiple unpaired t tests with Welch correction) (Figure 4B). Further insight is obtained by examining each cohort separately.
Figure 4.

ABR wave 1 amplitudes for saturating loud tones
(A) Baseline wave 1 amplitudes prior to acoustic trauma: average (±SEM) for uninjected (black, n = 24), GFP virus only-injected (blue, n = 10), and α9L9′T-containing virus plus GFP-containing virus-injected (magenta, n = 31). Multiple unpaired t tests with Welch correction, ∗p < 0.05, ∗∗p < 0.01, ∗∗∗p < 0.001, blue asterisks for α9 versus GFP, black for α9 versus uninjected. (B) Wave 1 amplitude 14 days post trauma; no significant differences by unpaired t test with Welch correction at each frequency. (C) The ratio of wave 1 amplitude at day 14 (averaged) over the pretrauma value (averaged). Wave 1 amplitude recovered more in α9L9′T-injected mice. (D) Average (±SEM) of wave 1 amplitude in 24 uninjected mice. Wave 1 amplitude fell by ∼50% 1 day post trauma and remained unchanged 14 days later. (E) Wave 1 amplitude of 10 GFP-injected mice before and 1 and 14 days post trauma. Wave 1 amplitude fell by ∼50% 1 day post trauma and remained unchanged 14 days later. (F) Wave 1 amplitude of 31 α9L9′T-injected mice before and 1 and 14 days post trauma. Amplitude declined ∼50% at day 1 but showed no significant difference from pretrauma control at 14 days. Asterisks for pretrauma and 1 day post comparison. Multiple unpaired t tests with Welch correction, ∗p < 0.05, ∗∗p < 0.01, ∗∗∗p < 0.001. (G) Click-evoked ABR from exemplar control mouse (no injection) before (black) and 14 days post (gray) acoustic trauma. (H) Click-evoked ABR from exemplar GFP-injected mouse before (dark blue) and 14 days post trauma (light blue). (I) Click-evoked ABR from α9L9′T-injected mouse before (purple) and 14 days post trauma (pink).
One day after acoustic trauma, both groups of control mice (uninjected and GFP injected) displayed a significant reduction in the amplitude of wave 1 of the ABR evoked by the loudest clicks and pure tones from 8 to 32 kHz (Figures 4D and 4E). Wave 1 amplitude remained smaller 14 days after acoustic trauma, indicating that this deficit did not recover (unlike elevated ABR threshold). Wave 1 of the ABR of mice that received the α9L9′T-containing virus also was reduced after acoustic trauma but recovered substantially 14 days later (Figure 4F) (mixed effects model [REML] for day 14 compared with day 1 amplitude; p = 0.0019, F (1, 150) = 9.997). Fourteen days post trauma significant differences again were found between α9L9′T-injected and control mice. This is evident in the ratio of wave 1 amplitude at 14 days to that before trauma (Figure 4C): near 1.0 for α9L9′T-injected mice but approximately 0.5 for the controls. A mixed-effects model (REML) for the α9L9′T-injected mice showed that the ratio of wave 1 amplitudes at 14 days to the pretrauma value was significantly larger than that of the GFP-injected mice (p < 0.0001, F (1, 261) = 20.40) and that of the uninjected mice (p = 0.0116, F (1, 261) = 6.469). Thus, mice in all conditions underwent significant wave 1 amplitude loss 1 day post acoustic trauma. However, the α9L9′T-injected mice significantly recovered this parameter, while both sets of control mice did not.
The latency to wave 1 of the ABR is another indicator of afferent excitation. Longer latencies suggest disruption in coordinate firing or otherwise delayed conduction. This disruption could be pre- or postsynaptic in origin. ABR wave 1 latency was significantly greater in α9L9′T-injected mice than in uninjected or GFP-injected mice (Figure 5A). Fourteen days post acoustic trauma wave 1 latency remained significantly longer in α9L9′T-injected than in both sets of control mice (Figure 5B). Wave 1 latency was significantly greater 1 day after acoustic trauma in control cohorts, uninjected control mice (Figure 5C) and GFP-injected mice (Figure 5D). In contrast, ABR wave 1 latencies increased only at 24 and 32 kHz in α9L9′T-injected mice (Figure 5E), suggesting better preservation of function. However, direct comparison of cohorts 1 day post trauma showed no significant differences in the extent of latency shift across frequency among all groups (Figure 5F). Latencies recovered to pretrauma control values at 14 days in all groups.
Figure 5.

ABR wave 1 latencies
(A) Average (±SEM) for ABR wave 1 latency prior to acoustic trauma in uninjected α9-null (black), GFP virus only-injected (blue), and α9L9′T-containing virus plus GFP-containing virus-injected (magenta). Cyan asterisks for α9L9′T versus GFP animals, black asterisks for α9L9′T versus uninjected animals. (B) ABR wave 1 latencies 14 days post trauma. (C) ABR wave 1 latency for uninjected α9-null mice, before and 1 and 14 days post trauma. (D) ABR wave 1 latency for GFP-injected α9-null mice, before and 1 and 14 days post trauma. (E) ABR wave 1 latency for α9L9′T-injected α9-null mice, before and 1 and 14 days post trauma. (F) Average (±SEM) latency shift at 1 day post trauma in all cohorts. Multiple t tests with Welch correction, ∗p < 0.05, ∗∗p < 0.01, ∗∗∗p < 0.001.
Ribbon synapses of IHCs
Changes in wave 1 of the ABR have been correlated with the loss of synaptic contacts between IHCs and type I afferents. Thus, ribbon synapses were counted in IHCs of α9-knockout mice with or without acoustic trauma (2 h exposure to 90 dB broadband sound), with or without viral transduction (either α9L9′T virus plus GFP virus or GFP virus alone, the latter serving as the injection control condition). This generated six experimental groups for analysis: (1) no virus injection, no acoustic trauma; (2) no virus injection plus acoustic trauma; (3) GFP viral injection, no acoustic trauma; (4) GFP virus injection plus acoustic trauma; (5) α9L9′T and GFP virus injection, no acoustic trauma; and (6) α9L9′T and GFP virus plus acoustic trauma. Synaptic immunohistology was carried out 14 or 15 days after acoustic trauma (P45–P46). Each cochlea was divided into segments approximately 100 μm in length (containing on average 10 IHCs and 30 OHCs), at locations corresponding to center frequencies of 8, 12, 16, 24, and 32 kHz (based on the standardized mouse cochlear frequency map40). CtBP2 (ribbons), GluA2/3 (postsynaptic AMPAR clusters), ribbons colocalized with GluA2/3 puncta, and synaptophysin (efferent terminals) immunopuncta were counted in confocal z stacks. Counts were conducted repeatedly using ImageJ or Imaris by blinded observers and consolidated post hoc before identifying experimental condition. Ribbons per IHC ranged from 6 to 18 depending on cochlear position, trending to highest numbers at the 16 kHz position in control cochleas as previously reported.16 Immunolabel for GluA2/3 provided the lowest signal to noise. Therefore, the better-labeled ribbons were first demarcated in each tissue sample, then the GluA2/3 channel was examined for corresponding immunopuncta. In some cases, merged colors (green plus red to produce yellow) also were used. Overall, 5% of ribbons could not be associated with GluA2/3.
Synapses per IHC ranged from 6 to 18 depending on cochlear position, tending to highest numbers at the 16 or 24 kHz positions in control cochleas. IHCs from α9L9′T-injected cochleas had fewer synapses, with a peak at 12 kHz but a flatter tonotopic distribution overall (Figure 6B). Synapse counts in IHCs of α9L9′T-injected cochleas differed from controls (p = 0.008, F (2, 60) = 5.232), but the interaction with frequency failed significance (p = 0.699, F (8, 60) = 0.690). Following acoustic trauma (Figure 6C), α9L9′T-injected IHCs still had fewer synapses than controls (p = 0.0008, F (2, 64)) and the interaction with frequency remained nonsignificant (p = 0.359, F (8, 64) = 1.124). The effect of acoustic trauma on synapse numbers within each cohort revealed no loss, with the exception of the 32 kHz position in GFP-injected cochleas. There was no synapse loss at 32 kHz in the α9L9′T-injected cochleas, perhaps corresponding with the better recovery of wave 1 (Figure 4F). Overall, however, these data on synapse number do not correlate with the sustained drop in amplitude of wave 1 of the ABR for all frequencies in control or experimental cochleas 14 days after acoustic trauma.
Figure 6.

Afferent synapses of inner hair cells (mean ± SEM)
Antibodies to CtBP2 and GluA2/3 were used to label pre- and postsynaptic specializations of inner hair cells before and 14 days post acoustic trauma. Colocalized immunopuncta were identified in confocal z stacks. (A) Mean number of inner hair cells per condition. Total IHC numbers ranged from 165 to 476 for 4 to12 tissue samples in different groups. (B) Synapse counts prior to acoustic trauma. Inner hair cells of mice injected with α9-containing virus (in combination with GFP-containing virus) tended to have slightly fewer afferent synapses, but with a significant difference only at 16 kHz from mice receiving no injection; ∗p < 0.05 (multiple t tests with Welch correction). (C) Two weeks post acoustic trauma, inner hair cells from cochleas of α9L9′T plus GFP-injected and GFP only-injected mice had significantly fewer afferent synapses than uninjected mice at 8 and 24 kHz positions (∗p < 0.05, multiple t tests with Welch correction). Linear mixed model showed no significant difference among all cohorts as a function of frequency or condition. There were no significant differences between α9L9′T plus GFP-injected and GFP only-injected mice. (D) The number of afferent synapses did not differ significantly 2 weeks post trauma in inner hair cells of uninjected mice at any cochlear location. (E) The number of afferent synapses differed significantly at the 32 kHz location 2 weeks post trauma in inner hair cells of GFP-injected mice; ∗∗p < 0.01 (multiple t test with Welch correction). (F) The number of afferent synapses did not differ significantly at any cochlear location 2 weeks post trauma in inner hair cells of α9L9′T-injected mice. (G) Maximum intensity projection (MIP) of the 16 kHz region in a control (uninjected) mouse. (H) MIP of the 16 kHz region in a GFP-injected mouse. (I) MIP of the 16 kHz region in an α9L9′T-injected mouse. Inner hair cell nuclei labeled red by CtBP2 immunolabel. Correspondence of CtBP2 (red) and GluA2 (green) immunolabel results in yellow synaptic loci.
Afferent and efferent synapses of OHCs
In previous work it was found that acoustic trauma had opposing effects on ribbon synapses of IHCs and OHCs, decreasing those of IHCs but increasing those of OHCs.41 Thus, reduced acoustic trauma in α9L9′T-injected mice might be expected to prevent ribbon augmentation in OHCs. There were no significant differences in numbers of OHCs among all cohorts (Figure 7A). However, there was some effect of the α9L9′T treatment on OHC ribbons. In uninjected and GFP-injected cochleas there was a falling gradient of ribbons per outer hair as one moved from lower- to higher-frequency positions in the cochlea cells (i.e., more in apical OHCs, fewer in basal OHCs). In contrast, α9L9′T-injected cochleas had equal numbers of OHC ribbons at all locations (Figure 7B). After acoustic trauma these differential patterns were more pronounced, with α9L9′T-injected cochleas developing a tonotopic gradient that nearly reversed the control pattern (Figure 7C). Linear mixed-model testing showed that the effect of noise differed significantly by condition (p = 0.0047, F (2, 60) = 5.86) and frequency (p = 0.0059, F (4, 60) = 4.025), reflecting these differing tonotopic patterns of OHC ribbons.
Figure 7.

Ribbons in outer hair cells
(A) The average number of OHCs in each experimental cohort. Number of samples ranged from 4 to 12, number of OHCs ranged from 485 to 1,230. (B) Ribbon counts in outer hair cells at five frequency loci in cochleas of uninjected, GFP only-injected, and α9L9′T plus GFP-injected mice counted in confocal z stacks (mean ± SEM). Ribbons were significantly fewer (∗p < 0.05) at 12 and 16 kHz in α9L9′T-injected and higher at 32 kHz versus uninjected cochleas (multiple t tests with Welch correction, blue asterisks for α9L9′T versus GFP injected, black asterisks for α9L9′T versus uninjected). (C) Ribbons were significantly fewer (∗p < 0.05, ∗∗p < 0.01) at 8 and 16 kHz in α9L9′T versus controls and significantly higher at 32 kHz versus GFP-injected cochleas (multiple t tests with Welch correction, cyan asterisks for α9L9′T versus GFP injected, black asterisks for α9L9′T versus uninjected). (D) Ribbon counts in outer hair cells of uninjected cochleas were significantly higher 14 days after trauma at 24 kHz (multiple t tests with Welch correction, ∗p < 0.05). (E) Ribbon counts in outer hair cells of GFP-injected cochleas. (F) Ribbon counts in outer hair cells of α9L9′T-injected cochleas.
While between-group comparisons were significant for the effect of noise, tonotopic gradients dominated within group ribbon counts. For uninjected cochleas (Figure 7D), two-way ANOVA showed that frequency (cochlear location) accounted for 42% of the total variance (p < 0.0001, F (4, 67) = 16.77). But, pre- versus post-noise exposure was not significant (p = 0.1173, F = 2.517). For GFP-injected cochleas, two-way ANOVA (Figure 7E) showed that frequency (cochlear location) accounted for 46.54% of the total variance, with p = 0.0002, F (4, 31) = 7.95. Synapse counts pre- versus post-noise exposure were not significantly different (p = 0.429, F = 0.64). The pattern of ribbon distribution for α9L9′T-injected mice differed from the control tonotopic pattern both pre- and post-acoustic trauma (Figure 7F). Ribbon counts did not rise to statistical significance as a function of either acoustic trauma or frequency. Two-way ANOVA for α9L9′T-injected cochleas showed that frequency (cochlear location) accounted for only 9.9% of the total variance, with p = 0.475, F (4, 30) = 0.90, and no significant difference after noise exposure (p = 0.699, F (1, 30) = 0.1524). Thus, while α9L9′T-injected cochleas lost the control tonotopic pattern of OHC ribbon expression, noise exposure, as for the control cochleas, did not alter that pattern significantly.
Efferent synapses on outer hair cells
Previous studies have described quantitative differences in efferent innervation density of OHCs in α9-transgenic mouse cochleas, with total presynaptic terminal volume (sum of all contacts per OHC) smaller on α9-null OHCs and larger on OHCs of α9L9′T gain-of-function mice.42 This issue and the impact of noise were examined by quantifying efferent synaptic volumes on OHCs in cochlear tissue of control and α9L9′T-injected mice with and without acoustic trauma. Efferent synaptic volumes on individual OHCs ranged between 10 and 40 μm3 with average values between 15 and 25 μm3 (Figure 8B), with a trend to lower values at the 32 kHz position. There were no significant differences among all cohorts before noise exposure (two-way ANOVA) (Figure 8B). Average values were significantly higher in both sets of virus-injected cochleas from noise-exposed mice, compared with uninjected (Figure 8C; as a function of both frequency, p = 0.0097, and condition, p < 0.0001, two-way ANOVA). Examination of efferent synaptic volumes in each cohort independently found no effect of noise exposure on efferent volumes of OHCs in uninjected cochleas (Figure 8D) and a significant difference only at 12 kHz for GFP-injected cochleas (Figure 8E). However, efferent synaptic volumes were significantly larger in α9L9′T-injected cochleas after noise exposure (Figure 8F; effect of noise p = 0.0004, two-way ANOVA, F (1, 31) = 15.94).
Figure 8.

Efferent synapses on outer hair cells
(A) The average number of OHCs in each experimental cohort, no significant differences. N, number of samples ranged from 4 to 12; n, number of OHCs ranged from 485 to 1,230 (repeated from Figure 7). (B) Efferent synaptic volumes per OHC without acoustic trauma. (C) Efferent synaptic volumes 14 days after acoustic trauma. (D) Synaptic volumes in uninjected cochleas with and without noise exposure. (E) Synaptic volumes in GFP only-injected cochleas with and without noise exposure. (F) Synaptic volumes in α9L9′T plus GFP-injected cochleas with and without noise exposure. Multiple t tests with Welch correction, ∗p < 0.05, ∗∗p < 0.01, ∗∗∗p < 0.001. (G) Maximum intensity projection (MIP) of three rows of outer hair cells (OHCs) in the 16 kHz region of a control (uninjected) mouse. (H) MIP of three rows of OHCs in the 16 kHz region of a GFP-injected mouse. (I) MIP of three rows of OHCs in the 16 kHz region of an α9L9′T-injected mouse. Efferent terminals were immunolabeled for synaptophysin (yellow), OHC ribbons were labeled for CtBP2 (red) in (G), (H), and (I).
Discussion
There is compelling evidence that inhibitory efferent feedback can protect the cochlea from acoustic damage.13,42,43,44 This includes genetic alteration of the hair cell nAChR,16,17,37 motivating the desire to exploit this mechanism therapeutically.18,45 The present work explores a “gene rescue” strategy using viral transduction of the gain-of-function α9L9′T to enhance acoustic protection in α9-null mice. The AAV2.7m8 virus was shown previously to transduce cochlear hair cells efficiently with GFP,29 as replicated here. Virally mediated expression of α9L9′T at synaptic sites in α9-null hair cells 3 weeks post injection was demonstrated in the present work by labeling with Cy3-RgIA-5727, a fluorophore-conjugated modification of a Conus venom peptide that is a high-affinity, slowly reversible antagonist of α9-containing nAChRs.30,31
Does gene rescue of efferent feedback preserve sensitivity?
Surgical introduction of virus was not damaging per se, as shown by equivalent hearing in mice receiving a GFP-containing virus compared with uninjected mice. Rather, injection of α9L9′T-containing virus specifically altered ABR thresholds and sensitivity to acoustic trauma. The acoustic protection was not as effective as in the transgenic α9L9′T-knockin, where expression is guaranteed to be universal. But, injection with α9L9′T-containing virus did mitigate the usual pattern of noise-induced threshold shifts,46 completely preserving ABR threshold at lower frequencies and reducing the loss at higher frequencies. Whether complete prophylaxis with preservation of hearing might be achieved by further technical improvement is a continued goal of this work.
A counter argument proposes that α9L9′T-transduced mice were simply less sensitive to sound, so the smaller threshold shift results from a higher initial baseline. In other words, α9L9′T transduction is equivalent to putting in ear plugs. However, efferent feedback is distinctly different from ear plugging. Efferent neurons have sensitive, V-shaped tuning curves to sound,3,4 providing protection that is both intensity and frequency tuned. As sound gets louder, efferents fire more frequently, transmitter release facilitates,47 and suppression gets stronger and is directed specifically against the frequency content of that sound. Additional support is derived from studies on α9L9′T-knockin mice.48 Efferent synapses in these mice have a significantly lower resting probability of release than in wild type, but facilitate strongly with repetitive activation. This is thought to be a form of homeostatic plasticity to compensate for the stronger postsynaptic effect of α9L9′T nAChRs. The benefit for hearing is that efferent activity will be less effective near threshold (than in wild type) and even more effective for louder sounds. It is conceivable that such homeostatic adjustment has not yet occurred by the time of testing in the present work, so longer-term studies will be necessary to determine if synaptic plasticity also arises in virally transduced mice. Ultimately, more complex listening tasks will be necessary to determine whether enhanced efferent feedback preserves hearing discrimination, as opposed to click and pure-tone thresholds in quiet. Furthermore, the ultimate utility of engineering medial olivocochlear (MOC) feedback to preserve hearing needs to be examined in wild-type mice, rather than in α9 knockouts. To these ends, future work will use a viral construct containing an HA-tagged version of α9L9′T36 so that the transduced subunit can be visualized throughout the lifespan. Another strategy to consider is simple overexpression of wild-type α9 nAChRs, as found previously in a transgenic mouse line.13 This could be implemented using HA tagging to distinguish foreign from native nAChRs.
Does gene rescue preserve the ABR waveform and alter synapse numbers?
Following acoustic trauma, all cohorts of mice experienced a 50% drop in wave 1 ABR amplitude that recovered 14 days later only in the α9L9′T-injected mice. This result, and the recovery of ABR threshold in these same animals, suggests that this type of acoustic trauma caused a temporary threshold shift, but lasting “hidden hearing loss” only in the control animals (i.e., reduced numbers of responding type I afferents for a saturating loud sound). Recovery of wave 1 amplitude in α9L9′T-injected mice suggests that synaptopathy also was mitigated. Wave 1 latency measurements partially followed this pattern, with a smaller increase in latency for α9L9′T-injected mice than in either control cohort. However, in contrast to wave 1 amplitude that remained suppressed 14 days post trauma, latencies recovered completely in all cohorts of mice.
IHC synapse counts 14 days post trauma did not cohere completely with the ABR waveform changes. In uninjected and GFP-injected cochleas acoustic trauma reduced synapse numbers at 32 kHz only, while wave 1 amplitude remained suppressed for all frequencies. ABR wave 1 amplitude was smaller in α9L9′T-injected mice than in control mice, and IHCs had significantly fewer synapses than control mice. This may suggest that early postnatal injection of α9L9′T alters the developmental interplay of afferent synapses with efferent synapses present on IHCs in the first 2 postnatal weeks.
There is a qualitative suggestion that the tonotopic distribution of ribbon synapses of IHCs was altered in α9L9′T-injected cochleas, appearing to be flatter than the peaked distribution of control cochleas. Alteration of tonotopic distribution was more obvious for ribbons in OHCs. Uninjected and GFP-injected cochleas have a declining gradient of ribbon numbers, moving from lower- to higher-frequency positions. This tonotopic gradient is not present, and may reverse after trauma, in α9-injected cochleas. Thus, the difference in ribbon counts for OHCs at low-frequency positions was even greater among control cohorts after trauma than in the α9L9′T-injected mice, perhaps because the α9L9′T-injected mice were better protected and so were not driven to increase OHC ribbon synapses as reported previously for more severe acoustic trauma.41
Previous work showed that damaging sound reduced efferent innervation.49 In the present work, efferent synapses on OHCs also showed some influence of the α9L9′T virus. After acoustic trauma, α9-injected cochleas had significantly larger efferent synaptic contacts. Neither uninjected nor GFP-injected cochleas showed significant differences after trauma.
Comparison with transgenic mice
How similar was viral transduction to genetic knockin of α9L9′T? Baseline thresholds in the virally transduced mice were on average 10 dB higher than those of control mice. Thresholds of transgenic α9L9′T knockins were 5–10 dB higher than in wild type.16,17 The knockin mice were completely protected from acoustic trauma, whereas viral transduction was only partially protective, suggesting less efficient expression compared with the knockin. Work on α9L9′T-knockin mice showed that IHC synaptopathy (hidden hearing loss) also was ameliorated compared with knockout or wild-type mice. The amplitude of wave 1 was irreversibly reduced by trauma in wild-type and α9-knockout mice, but unchanged in the transgenic α9L9′T knockins. In the present work, ABR wave 1 amplitude was reduced by trauma in all cohorts but recovered only in those mice virally transduced with α9L9′T. Thus, viral expression of α9L9′T has some benefit for synaptopathy as well. However, caution is required, since wave 1 amplitude was initially smaller and latencies longer for α9L9′T-injected mice compared with uninjected or GFP-injected mice. There were no significant differences in the effects of trauma on wave 1 latency among these three cohorts, as reported previously for the transgenic mice.
In α9L9′T-transgenic mice, IHCs maintain and actually increase synaptic counts after acoustic trauma, whereas wild-type and knockout IHCs lose synapses.16 The present study did not replicate this result. Rather, reduction of ABR wave 1 amplitude was not reflected in a comparable loss of IHC synapses in any of the cohorts of mice. The viral injection strategy does avoid possible developmental effects that could occur in the α9L9′T transgenic knockin mouse.
Conclusions
The next steps will be to assess the impact of α9L9′T viral transduction in wild-type mice. Behavioral thresholds need to be tested in more complex acoustic environments to learn if acoustic protection after viral transduction improves discriminative hearing.50 The longevity of transfected DNA needs to be explored further. Previous studies found continued expression of GFP for months.29 However, benefit for humans will require expression for decades. Finally, while there is no evidence for altered vestibular function, α9-containing nAChRs do mediate inhibition of type II vestibular hair cells, the consequences of which should be addressed.
While important questions remain, the present results constitute a proof of principle to motivate further exploration for therapeutic application. Age-related hearing loss is worsened by genetic variants that associate with earlier onset and is exacerbated by noise exposure.51 Genetic markers and family history of hearing loss can identify an initial population likely to benefit most from enhanced efferent protection. Such a strategy could provide still more general benefits. Long-lasting enhancement of efferent protection could play a role wherever acoustic exposure exacerbates hearing loss, whether genetic, environmental, or of undefined etiology. It could be complementary to gene replacement for monogenic deafness. Long-lasting, virally mediated α9L9′T expression could reduce the dosage or frequency of small-molecule therapies (e.g., prophylaxis for ototoxic medications or positive allosteric modulators of the nAChR). Ultimately, one could imagine efferent upregulation as generally recommended for amelioration of age-related hearing loss, especially for those with unavoidable exposure to damaging levels of sound in the workplace or military service. A compelling argument for this strategy is that efferent activity is itself regulated by the acoustic environment. Thus, genetic enhancement of efferent inhibition leverages an intrinsic protective mechanism of the inner ear. An interesting associated question is whether enhanced efferent feedback also might benefit gain-of-function pathologies such as hyperacusis or tinnitus.
Materials and methods
Study design
α9-null mice were injected with virus via the posterior semicircular canal. Typically, 1 μL of viral solution plus Fast Green dye was injected into early postnatal (P0–P2) mice. Hearing tests were conducted 3 weeks later (Figure 1B), followed by acoustic overexposure and subsequent hearing tests 1 day and 14 days later. The efficacy of viral treatment was not known a priori, but prior studies of transgenic mice suggested that we could expect ∼10 dB shift in baseline threshold and ∼30 dB difference in threshold shift after acoustic overexposure as maximal effects. Since viral transduction might be less comprehensive than expression in genetically modified mice, less stringent sound exposure was used, and thus smaller experimental effects were obtained. Hearing tests were conducted on 31 α9 plus GFP-injected mice, 24 uninjected mice, and 10 mice injected with GFP virus only. Synaptic immunolabeling was carried out on 4–12 mice for each condition, including hundreds of IHCs and OHCs.
The objectives of this research were to determine if viral transduction could provide synaptic expression of nAChRs in cochlear hair cells. This was confirmed by histology. Virus-injected animals were tested for hearing (ABR thresholds) prior to and following acoustic trauma (Figure 1B). The hypothesis to be tested was that viral transduction of nAChRs in cochlear hair cells would provide protection against acoustic trauma. Immunohistology of cochlear innervation was carried out to determine if functional changes could be correlated with changes in synaptic contacts. All experiments were carried out on α9-null (knockout) mice.
This was an admixture of controlled laboratory experiment and observational study. Treatments included minor surgery to enable viral injections into the posterior semicircular canal, followed at various times later by hearing tests and the effects of loud sound exposure. Details of measurement techniques are given in the following sections.
Mice were purpose bred for use in this study. Both male and female mouse pups were utilized, otherwise no randomization was employed.
Mouse breeding was carried out independent of experimentalists. The experimental condition was whether or not virus was injected and which type of virus. Following that treatment, the animals were coded. Investigators who made assessments, made measurements, and quantified results were given only the anonymized code. Measurements of ABR recordings and synaptic counts in immunolabeled tissue were carried out by two independent, blinded observers.
α9L9′T AAV2.7m8
The α9L9′T sequence was inserted into AAV2.7m8 (obtained from Addgene, University of California, Berkeley, MTA no. 486064) (Figure 1A). This modified form of the AAV2 virus was designed by the Bennet laboratory at the University of Pennsylvania.52 It contains a 10-amino-acid peptide inserted at position 588 of the AAV2 capsid protein sequence. AAV2.7m8 was shown previously to provide efficient transduction of inner and outer cochlear hair cells with GFP.29 Control experiments in the present study were conducted with that same GFP-containing AAV2.7m8 (9.75 × 1012 GC/mL). Mouse α9L9′T cDNA was incorporated into a plasmid (Genewiz, South Plainfield, NJ 07080) and submitted to the Penn Vector Core (Gene Therapy Program, University of Pennsylvania School of Medicine) for incorporation into the AAV2.7m8 backbone. The resulting viral vector, (AAV2.7m8.CAG.nAChRα9L9′T.bGH) was provided for use at 3.26 × 1013 viral copies/mL. This was subdivided into 100 μL aliquots and stored at −80°C until use. Each 100 μL aliquot was stored at 4°C (i.e., not refrozen) until exhausted.
Mice (α9 nulls and wild type)
Care and housing of animals was in accordance with institutional guidelines as specified in Johns Hopkins IACUC protocol MO19M478. α9-null (homozygous knockout) mice have qualitatively normal efferent innervation, but no cochlear inhibition.53 In the altered gene the ligand-binding site, the first and second transmembrane domains, and a portion of the third transmembrane domain of the α9 coding region were excised. The original mice (129S6 background) were subsequently backcrossed onto C57BL/6J for more than 10 generations and breeding colonies maintained in the Johns Hopkins Research Animal Resource.
Mice (C57BL/6J; RRID: IMSR_JAX:000664) were purchased from The Jackson Laboratory, and α9 knockouts (C57 background) were bred and maintained in the Johns Hopkins University School of Medicine Research Animal Resource facility. Mice were placed on a 12-h light-dark cycle, fed an autoclaved Teklad diet, and housed in cages with automatic water and filtered air. All experiments were carried out under protocols approved by the Institutional Animal Care and Use Committee protocol #MO19M478. Male and female mice were used in all experiments.
Injection protocol
Virus (1.1 μL at 1013 GC/mL) was injected into the posterior semicircular canal using a micropipette pulled from borosilicate glass to produce a tip opening of 10–40 μm (Kwik-Fil 1B100-4, World Precision Instruments, Sarasota, FL). The α9L9′T-containing virus was always injected together with the GFP-containing virus (i.e., two independent viruses) in a 1:1 ratio. The GFP virus (∼1013 GC/mL) was included initially with the α9 virus injection to assess the degree of viral infection as indicated by GFP label (in the absence of reliable nAChR antibodies). Since conopeptide labeling for α9L9′T nAChRs proved successful (see below), GFP injection was not necessary, but was continued for consistency. To control for the impact of surgery and virus injection per se, a cohort of mice were injected with GFP-containing virus only. Mouse pups (0–2 days of age, P0–P2) were anesthetized by cooling. Under a stereo dissecting scope, a small dorsal-ventral postauricular incision was made through skin and muscle to expose the posterior semicircular canal as a darker tube surrounded by whiter tissue. The injection pipette was used to penetrate through the soft bone in young animals. Repeated, brief pressure pulses were used to inject the viral aliquot over approximately 40 s. The pipette was removed, the skin was sutured together over the incision, and the animals were left to recover on a warming pad. The wound site was disinfected with betadine and the entire procedure carried out under sterile conditions.
Fluorophore-conjugated conotoxin peptide (Cy3-RgIA-5727) labeling of cochlear nAChRs
The fluorophore-conjugated modified conopeptide Cy3-RgIA-5727 labels hair cell nAChRs in unfixed tissue.31 Thus, live cochlear tissue (divided into apical, middle, and basal turns) from control and virus-injected mice was dissected from the isolated otic capsule and secured with a thin spring clip fashioned from minuten nadeln cemented to a glass coverslip. The live tissue was exposed to 10–250 nM Cy3-RgIA-5727 in artificial perilymph (5.8 mM KCl, 144 mM NaCl, 1.3 mM CaCl2, 0.9 mM MgCl2, 0.7 mM NaH2PO4, 5.6 mM D-glucose, and 10 mM HEPES [300 mOsm, pH 7.4, adjusted with NaOH]) for 15 min. The vital dye FM4-64 (10–50 μM) loads into hair cells through open mechanotransduction channels.54 The labeled tissue was examined on a Nikon A1R-MP upright confocal microscope and z-stack image series were acquired using Nikon Nis Element software. Three-dimensional videos were made with the Movie Maker tool in Nis Element software. Image analysis was conducted by a blinded observer in ImageJ/FIJI, in Imaris, or using a virtual reality viewing system (Syglass, Istovisio). A second blinded observer re-counted the images for comparison.
Acoustic trauma protocol
C57BL/6J mice were transferred to a low-noise satellite housing facility from the day before noise exposure through the week after noise exposure until the endpoint for histology. Due to the susceptibility of C57BL/6J mice to age-related hearing loss,55 the effect of acoustic trauma was examined before mice were 7 weeks of age. Awake, unrestrained mice were exposed to a 90 dB SPL white noise band (2–20 kHz) for 2 h to produce a temporary threshold shift of 20–30 dB. Mice were exposed to noise in a reverberant sound-attenuating chamber (58 × 40 × 30 cm; width, depth, height) with three overhead, Promaster TW47 1200 W dome tweeter speakers that produced maximum energy in the sound spectrum from 2 to 16 kHz. Speakers were approximately 25 cm above the heads of the mice. Broadband noise was generated by two JKT tone and noise generators (KV2 Audio, Czech Republic) powered by Neewer nw-100 phantom power sources. The noise generators were connected to two Crown Drivecore XLS2502 amplifiers: one driving the two peripheral speakers in Y input mode, the other driving a central speaker in bridge mode. The sound spectra and decibel level were tested in each setup using a Larson-Davis LXT sound level meter with a ½-inch free-field microphone. Care was taken to measure the sound level at the position of the head of the experimental animals. When not being studied, mice were housed in a “quiet” room where average noise levels (third octave band levels) were below 40 dB SPL.
Auditory brain-stem response measurements
The ABR system (Tucker-Davis Technologies), procedures, and quantification software used for this study have been previously described.56,57 Mice were anesthetized with an intraperitoneal injection of 0.1 cc per 20 g body weight of a mixture of ketamine (100 mg/kg) and xylazine (1 mg/kg in phosphate-buffered saline [PBS]). The animals were placed on a gauze-covered heating pad in a sound-attenuating chamber lined with Sonex acoustic foam panels and their eyes were swabbed with petrolatum-based ophthalmic ointment to prevent corneal ulcers. Subdermal platinum electrodes were placed at the level of the vertex of the skull (noninverting), behind the ventral edge of the left pinna (inverting), and at the base of the tail (ground). Clicks or pure-tone stimuli (512 repetitions 10 to 90 dB in 10 dB steps, 21 stimuli/s) were used to generate averaged ABR waveforms. The duration of the tonal stimulus was 5 ms, with a 1 ms rise and fall time. A TDT MF1 free-field speaker was used to present the stimuli 10 cm from the mouse pinna. The ABR threshold was defined with custom MATLAB software (B.J. May) by calculating the averaged peak-to-peak voltage during a 5 ms interval, beginning 1 ms after the onset of the stimulus, compared with the averaged peak-to-peak voltage in a 5 ms window 20 ms after the stimulus (reflecting the baseline physiological noise level). The threshold was determined by interpolating the stimulus level where the peak-to-peak response was greater than 2 standard deviations above the baseline noise. Each threshold determination was confirmed by visual inspection of averaged ABR waveforms.
Synaptic immunolabeling
Two weeks after acoustic trauma (P45–P46) the mice were euthanized by isoflurane anesthesia and decapitation. Otic capsules were removed from the temporal bone and perfused with 4% paraformaldehyde (PFA) in PBS through the oval and round windows. All samples were postfixed in fresh fixative solution for 30 min at room temperature (RT) and rinsed thoroughly with PBS, and the cochlear epithelium was microdissected. After removal of Reissner’s and tectorial membranes, the tissue was permeabilized in PBS containing 0.5% Triton X-100 and 10% normal donkey serum and for 1 to 2 h at RT. A mixture of primary antibodies was applied to the tissue samples and then incubated overnight at RT or at 4°C. Following several rinses in PBS, tissue samples were transferred to Alexa Fluor-conjugated secondary antibody (Life Technologies, 1:1,000 dilution) solution supplemented with the nuclear dye DAPI (1:5,000) and then incubated for 1 to 2 h at RT. The samples were rinsed several times in PBS and mounted in FluorSave antifade mounting medium (CalBiochem, San Diego, CA) using grade 1 coverslips recommended for confocal microscopy. Primary antibodies used in this study included mouse IgG1 anti-CtBP2 (clone 16; BD Biosciences, AB_39943), rabbit anti-GluAR2/3 (Millipore, AB1506), and rabbit anti-synaptophysin (Chemicon, AB9272). Images were acquired on an LSM700 confocal microscope (Zeiss Axio Imager Z2) using 40× NA 1.3 and 63× NA 1.4 oil immersion objectives. Images were analyzed using ImageJ/Fiji (RRID: SCR_002285) or Imaris (RRID: SCR_007370).
Statistics and rigor
Pairwise comparisons were tested for significance using two-tailed Student’s t test for unpaired samples, with Welch’s correction for multiple comparisons. Group analyses were evaluated using two-way ANOVA or mixed effects linear model testing (REML). Data analysis (determining ABR threshold, counting positive synaptic labeling) was carried out by two independent observers, each blinded to experimental condition. Controls for virus injection included no injection (so also no surgery) and animals receiving an injection of GFP-encoding virus only. Negative controls for antibody labeling of synapses included uninjected mice, as well as the opposite (uninjected) ear of virus-treated mice. The specific labeling of α9-containing nAChRs by Cy3-RgIA-5727 was validated in previous work,31 which included labeling of wild-type and α9-knockout cochlear tissue, as well as wild-type tissue previously absorbed with unlabeled RgIA-5727.
Data availability
Internet access to original recordings and images will be provided upon request.
Acknowledgments
We thank F. Chakir for outstanding technical support, J.M. McIntosh for Cy3-RgIA-5727 Conus peptide, B.J. May for the ABR threshold app, and A. Goldring for developing the viral injection protocol in our laboratory. Funding was provided by the National Institute on Deafness and Other Communication Disorders, grants R01 DC001508 (P.A.F., A.B.E.) and R01DC017620 (A.L.), and the David M. Rubenstein Fund for Hearing Research and Professorship (P.A.F.).
Author contributions
Experiments were designed by Y.Z., H.H., P.F.V.Y., M.B.W., A.B.E., and P.A.F. Methodology was perfected by Y.Z., H.H., P.F.V.Y., M.B.W., W.C., A.B.E., and A.L. Experimental work and visualization were conducted by Y.Z., H.H., P.F.V.Y., M.B.W., and P.A.F. Funding was acquired by P.A.F., A.B.E., and A.L. The project was administered by P.A.F. and H.H. Supervision was conducted by P.A.F. and A.L. P.A.F. wrote the original draft. Manuscript review and editing were completed by Y.Z., H.H., P.F.V.Y., M.B.W., A.B.E., W.C., A.L., and P.A.F.
Declaration of interests
The authors declare no competing or other financial interests.
References
- 1.Galambos R. Suppression of auditory nerve activity by stimulation of efferent fibers to cochlea. J. Neurophysiol. 1956;19:424–437. doi: 10.1152/jn.1956.19.5.424. [DOI] [PubMed] [Google Scholar]
- 2.Klinke R., Galley N. Efferent innervation of vestibular and auditory receptors. Physiol. Rev. 1974;54:316–357. doi: 10.1152/physrev.1974.54.2.316. [DOI] [PubMed] [Google Scholar]
- 3.Robertson D., Gummer M. Physiological and morphological characterization of efferent neurones in the Guinea pig cochlea. Hear. Res. 1985;20:63–77. doi: 10.1016/0378-5955(85)90059-0. [DOI] [PubMed] [Google Scholar]
- 4.Liberman M.C., Brown M.C. Physiology and anatomy of single olivocochlear neurons in the cat. Hear. Res. 1986;24:17–36. doi: 10.1016/0378-5955(86)90003-1. [DOI] [PubMed] [Google Scholar]
- 5.Guinan J.J., Jr. Cochlear efferent innervation and function. Curr. Opin. Otolaryngol. Head Neck Surg. 2010;18:447–453. doi: 10.1097/MOO.0b013e32833e05d6. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 6.Fuchs P.A., Lauer A.M. Efferent inhibition of the cochlea. Cold Spring Harb. Perspect. Med. 2019;9 doi: 10.1101/cshperspect.a033530. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 7.Lauer A.M., Jimenez S.V., Delano P.H. Olivocochlear efferent effects on perception and behavior. Hear. Res. 2022;419 doi: 10.1016/j.heares.2021.108207. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 8.Nouvian R., Eybalin M., Puel J.L. Cochlear efferents in developing adult and pathological conditions. Cell Tissue Res. 2015;361:301–309. doi: 10.1007/s00441-015-2158-z. [DOI] [PubMed] [Google Scholar]
- 9.Frank M.M., Goodrich L.V. Talking back: development of the olivocochlear efferent system. Wiley Interdiscip. Rev. Dev. Biol. 2018;7:e324. doi: 10.1002/wdev.324. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 10.Di Guilmi M.N., Boero L.E., Castagna V.C., Rodríguez-Contreras A., Wedemeyer C., Gómez-Casati M.E., Elgoyhen A.B. Strengthening of the efferent olivocochlear system leads to synaptic dysfunction and tonotopy disruption of a central auditory nucleus. J. Neurosci. 2019;39:7037–7048. doi: 10.1523/JNEUROSCI.2536-18.2019. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 11.Wang Y., Sanghvi M., Gribizis A., Zhang Y., Song L., Morley B., Barson D.G., Santos-Sacchi J., Navaratnam D., Crair M. Efferent feedback controls bilateral auditory spontaneous activity. Nat. Commun. 2021;12:2449. doi: 10.1038/s41467-021-22796-8. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 12.Rajan R., Johnstone B.M. Electrical stimulation of cochlear efferents at the round window reduces auditory desensitization in Guinea pigs. I. Dependence on electrical stimulation parameters. Hear. Res. 1988;36:53–73. doi: 10.1016/0378-5955(88)90137-2. [DOI] [PubMed] [Google Scholar]
- 13.Maison S.F., Luebke A.E., Liberman M.C., Zuo J. Efferent protection from acoustic injury is mediated via alpha9 nicotinic acetylcholine receptors on outer hair cells. J. Neurosci. 2002;22:10838–10846. doi: 10.1523/JNEUROSCI.22-24-10838.2002. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 14.Luebke A.E., Foster P.K. Variation in inter-animal susceptibility to noise damage is associated with alpha 9 acetylcholine receptor subunit expression level. J. Neurosci. 2002;22:4241–4247. doi: 10.1523/JNEUROSCI.22-10-04241.2002. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 15.Vetter D.E., Katz E., Maison S.F., Taranda J., Turcan S., Ballestero J., Liberman M.C., Elgoyhen A.B., Boulter J. The alpha10 nicotinic acetylcholine receptor subunit is required for normal synaptic function and integrity of the olivocochlear system. Proc. Natl. Acad. Sci. USA. 2007;104:20594–20599. doi: 10.1073/pnas.0708545105. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 16.Boero L.E., Castagna V.C., Di Guilmi M.N., Goutman J.D., Elgoyhen A.B., Gómez-Casati M.E. Enhancement of the medial olivocochlear system prevents hidden hearing loss. J. Neurosci. 2018;38:7440–7451. doi: 10.1523/JNEUROSCI.0363-18.2018. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 17.Boero L.E., Castagna V.C., Terreros G., Moglie M.J., Silva S., Maass J.C., Fuchs P.A., Delano P.H., Elgoyhen A.B., Gómez-Casati M.E. Preventing presbycusis in mice with enhanced medial olivocochlear feedback. Proc. Natl. Acad. Sci. USA. 2020;117:11811–11819. doi: 10.1073/pnas.2000760117. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 18.Elgoyhen A.B., Katz E., Fuchs P.A. The nicotinic receptor of cochlear hair cells: a possible pharmacotherapeutic target? Biochem. Pharmacol. 2009;78:712–719. doi: 10.1016/j.bcp.2009.05.023. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 19.Delmaghani S., El-Amraoui A. Inner ear gene therapies take off: current promises and future challenges. J. Clin. Med. 2020;9 doi: 10.3390/jcm9072309. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 20.Lustig L., Akil O. Cochlear gene therapy. Cold Spring Harb. Perspect. Med. 2019;9 doi: 10.1101/cshperspect.a033191. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 21.Akil O., Seal R.P., Burke K., Wang C., Alemi A., During M., Edwards R.H., Lustig L.R. Restoration of hearing in the VGLUT3 knockout mouse using virally mediated gene therapy. Neuron. 2012;75:283–293. doi: 10.1016/j.neuron.2012.05.019. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 22.Akil O., Dyka F., Calvet C., Emptoz A., Lahlou G., Nouaille S., Boutet de Monvel J., Hardelin J.P., Hauswirth W.W., Avan P., et al. Dual AAV-mediated gene therapy restores hearing in a DFNB9 mouse model. Proc. Natl. Acad. Sci. USA. 2019;116:4496–4501. doi: 10.1073/pnas.1817537116. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 23.Al-Moyed H., Cepeda A.P., Jung S., Moser T., Kügler S., Reisinger E. A dual-AAV approach restores fast exocytosis and partially rescues auditory function in deaf otoferlin knock-out mice. EMBO Mol. Med. 2019;11 doi: 10.15252/emmm.201809396. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 24.Wu J., Solanes P., Nist-Lund C., Spataro S., Shubina-Oleinik O., Marcovich I., Goldberg H., Schneider B.L., Holt J.R. Single and dual vector gene therapy with AAV9-PHP.B rescues hearing in Tmc1 mutant mice. Mol. Ther. 2020 doi: 10.1016/j.ymthe.2020.11.016. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 25.Ahmed H., Shubina-Oleinik O., Holt J.R. Emerging gene therapies for genetic hearing loss. J. Assoc. Res. Otolaryngol. 2017;18:649–670. doi: 10.1007/s10162-017-0634-8. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 26.Hashimoto K., Hickman T.T., Suzuki J., Ji L., Kohrman D.C., Corfas G., Liberman M.C. Protection from noise-induced cochlear synaptopathy by virally mediated overexpression of NT3. Sci. Rep. 2019;9 doi: 10.1038/s41598-019-51724-6. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 27.Gates G.A., Schmid P., Kujawa S.G., Nam B., D'Agostino R. Longitudinal threshold changes in older men with audiometric notches. Hear. Res. 2000;141:220–228. doi: 10.1016/s0378-5955(99)00223-3. [DOI] [PubMed] [Google Scholar]
- 28.Wang J., Puel J.L. Presbycusis: an update on cochlear mechanisms and therapies. J. Clin. Med. 2020;9 doi: 10.3390/jcm9010218. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 29.Isgrig K., McDougald D.S., Zhu J., Wang H.J., Bennett J., Chien W.W. AAV2.7m8 is a powerful viral vector for inner ear gene therapy. Nat. Commun. 2019;10:427. doi: 10.1038/s41467-018-08243-1. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 30.Ellison M., Haberlandt C., Gomez-Casati M.E., Watkins M., Elgoyhen A.B., McIntosh J.M., Olivera B.M. Alpha-RgIA: a novel conotoxin that specifically and potently blocks the alpha9alpha10 nAChR. Biochemistry. 2006;45:1511–1517. doi: 10.1021/bi0520129. [DOI] [PubMed] [Google Scholar]
- 31.Fisher F., Zhang Y., Vincent P.F.Y., Gajewiak J., Gordon T.J., Glowatzki E., Fuchs P.A., McIntosh J.M. Cy3-RgIA-5727 labels and inhibits alpha9-containing nAChRs of cochlear hair cells. Front. Cell. Neurosci. 2021;15 doi: 10.3389/fncel.2021.697560. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 32.Brown M.C. Morphology of labeled efferent fibers in the Guinea pig cochlea. J. Comp. Neurol. 1987;260:605–618. doi: 10.1002/cne.902600412. [DOI] [PubMed] [Google Scholar]
- 33.Liberman M.C., Dodds L.W., Pierce S. Afferent and efferent innervation of the cat cochlea: quantitative analysis with light and electron microscopy. J. Comp. Neurol. 1990;301:443–460. doi: 10.1002/cne.903010309. [DOI] [PubMed] [Google Scholar]
- 34.Nadol J.B., Jr., Burgess B.J. Supranuclear efferent synapses on outer hair cells and Deiters' cells in the human organ of Corti. Hear. Res. 1994;81:49–56. doi: 10.1016/0378-5955(94)90152-x. [DOI] [PubMed] [Google Scholar]
- 35.Sato M., Henson M.M., Smith D.W. Synaptic specializations associated with the outer hair cells of the Japanese macaque. Hear. Res. 1997;108:46–54. doi: 10.1016/s0378-5955(97)00049-x. [DOI] [PubMed] [Google Scholar]
- 36.Vyas P., Wood M.B., Zhang Y., Goldring A.C., Chakir F.Z., Fuchs P.A., Hiel H. Characterization of HA-tagged alpha9 and alpha10 nAChRs in the mouse cochlea. Sci. Rep. 2020;10 doi: 10.1038/s41598-020-78380-5. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 37.Taranda J., Maison S.F., Ballestero J.A., Katz E., Savino J., Vetter D.E., Boulter J., Liberman M.C., Fuchs P.A., Elgoyhen A.B. A point mutation in the hair cell nicotinic cholinergic receptor prolongs cochlear inhibition and enhances noise protection. PLoS Biol. 2009;7:e18. doi: 10.1371/journal.pbio.1000018. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 38.Stamataki S., Francis H.W., Lehar M., May B.J., Ryugo D.K. Synaptic alterations at inner hair cells precede spiral ganglion cell loss in aging C57BL/6J mice. Hear. Res. 2006;221:104–118. doi: 10.1016/j.heares.2006.07.014. [DOI] [PubMed] [Google Scholar]
- 39.Liberman M.C., Kujawa S.G. Cochlear synaptopathy in acquired sensorineural hearing loss: manifestations and mechanisms. Hear. Res. 2017;349:138–147. doi: 10.1016/j.heares.2017.01.003. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 40.Müller M., von Hünerbein K., Hoidis S., Smolders J.W.T. A physiological place-frequency map of the cochlea in the CBA/J mouse. Hear. Res. 2005;202:63–73. doi: 10.1016/j.heares.2004.08.011. [DOI] [PubMed] [Google Scholar]
- 41.Wood M.B., Nowak N., Mull K., Goldring A., Lehar M., Fuchs P.A. Acoustic trauma increases ribbon number and size in outer hair cells of the mouse cochlea. J. Assoc. Res. Otolaryngol. 2021;22:19–31. doi: 10.1007/s10162-020-00777-w. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 42.Maison S.F., Usubuchi H., Liberman M.C. Efferent feedback minimizes cochlear neuropathy from moderate noise exposure. J. Neurosci. 2013;33:5542–5552. doi: 10.1523/JNEUROSCI.5027-12.2013. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 43.Liberman M.C., Liberman L.D., Maison S.F. Efferent feedback slows cochlear aging. J. Neurosci. 2014;34:4599–4607. doi: 10.1523/JNEUROSCI.4923-13.2014. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 44.Fuente A. The olivocochlear system and protection from acoustic trauma: a mini literature review. Front. Syst. Neurosci. 2015;9:94. doi: 10.3389/fnsys.2015.00094. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 45.Elgoyhen A.B. The alpha9alpha10 nicotinic acetylcholine receptor: a compelling drug target for hearing loss? Expert Opin. Ther. Targets. 2022;26:291–302. doi: 10.1080/14728222.2022.2047931. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 46.Hultcrantz M., Li H.S. Inner ear morphology in CBA/Ca and C57BL/6J mice in relationship to noise, age and phenotype. Eur. Arch. Oto-Rhino-Laryngol. 1993;250:257–264. doi: 10.1007/BF00186222. [DOI] [PubMed] [Google Scholar]
- 47.Ballestero J., Zorrilla de San Martín J., Goutman J., Elgoyhen A.B., Fuchs P.A., Katz E. Short-term synaptic plasticity regulates the level of olivocochlear inhibition to auditory hair cells. J. Neurosci. 2011;31:14763–14774. doi: 10.1523/JNEUROSCI.6788-10.2011. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 48.Wedemeyer C., Vattino L.G., Moglie M.J., Ballestero J., Maison S.F., Di Guilmi M.N., Taranda J., Liberman M.C., Fuchs P.A., Katz E., Elgoyhen A.B. A gain-of-function mutation in the alpha9 nicotinic acetylcholine receptor alters medial olivocochlear efferent short-term synaptic plasticity. J. Neurosci. 2018;38:3939–3954. doi: 10.1523/JNEUROSCI.2528-17.2018. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 49.Canlon B., Fransson A., Viberg A. Medial olivocochlear efferent terminals are protected by sound conditioning. Brain Res. 1999;850:253–260. doi: 10.1016/s0006-8993(99)02091-0. [DOI] [PubMed] [Google Scholar]
- 50.Prosen C.A., Bath K.G., Vetter D.E., May B.J. Behavioral assessments of auditory sensitivity in transgenic mice. J. Neurosci. Methods. 2000;97:59–67. doi: 10.1016/s0165-0270(00)00169-2. [DOI] [PubMed] [Google Scholar]
- 51.Jimenez J.E., Nourbakhsh A., Colbert B., Mittal R., Yan D., Green C.L., Nisenbaum E., Liu G., Bencie N., Rudman J., et al. Diagnostic and therapeutic applications of genomic medicine in progressive, late-onset, nonsyndromic sensorineural hearing loss. Gene. 2020;747 doi: 10.1016/j.gene.2020.144677. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 52.Dalkara D., Byrne L.C., Klimczak R.R., Visel M., Yin L., Merigan W.H., Flannery J.G., Schaffer D.V. In vivo-directed evolution of a new adeno-associated virus for therapeutic outer retinal gene delivery from the vitreous. Sci. Transl. Med. 2013;5 doi: 10.1126/scitranslmed.3005708. [DOI] [PubMed] [Google Scholar]
- 53.Vetter D.E., Liberman M.C., Mann J., Barhanin J., Boulter J., Brown M.C., Saffiote-Kolman J., Heinemann S.F., Elgoyhen A.B. Role of alpha9 nicotinic ACh receptor subunits in the development and function of cochlear efferent innervation. Neuron. 1999;23:93–103. doi: 10.1016/s0896-6273(00)80756-4. [DOI] [PubMed] [Google Scholar]
- 54.Roux I., Wersinger E., McIntosh J.M., Fuchs P.A., Glowatzki E. Onset of cholinergic efferent synaptic function in sensory hair cells of the rat cochlea. J. Neurosci. 2011;31:15092–15101. doi: 10.1523/JNEUROSCI.2743-11.2011. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 55.Johnson K.R., Erway L.C., Cook S.A., Willott J.F., Zheng Q.Y. A major gene affecting age-related hearing loss in C57BL/6J mice. Hear. Res. 1997;114:83–92. doi: 10.1016/s0378-5955(97)00155-x. [DOI] [PubMed] [Google Scholar]
- 56.Lauer A.M., May B.J. The medial olivocochlear system attenuates the developmental impact of early noise exposure. J. Assoc. Res. Otolaryngol. 2011;12:329–343. doi: 10.1007/s10162-011-0262-7. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 57.Lina I.A., Lauer A.M. Rapid measurement of auditory filter shape in mice using the auditory brainstem response and notched noise. Hear. Res. 2013;298:73–79. doi: 10.1016/j.heares.2013.01.002. [DOI] [PMC free article] [PubMed] [Google Scholar]
Associated Data
This section collects any data citations, data availability statements, or supplementary materials included in this article.
Data Availability Statement
Internet access to original recordings and images will be provided upon request.