

Abstract
Quinolinic acid (QA) is a key intermediate of nicotinic acid (Niacin) which is an essential human nutrient and widely used in food and pharmaceutical industries. In this study, a quinolinic acid producer was constructed by employing comprehensive engineering strategies. Firstly, the quinolinic acid accumulation was improved by deactivation of NadC (to block the consumption pathway), NadR (to eliminate the repression of L-aspartate oxidase and quinolinate synthase), and PtsG (to slow down the glucose utilization rate and achieve a more balanced metabolism, to increase the availability of the precursor phosphoenolpyruvate). Further modifications to enhance quinolinic acid production were investigated by increasing the oxaloacetate pool through overproduction of phosphoenolpyruvate carboxylase and deactivation of acetate-producing pathway enzymes. Moreover, quinolinic acid production was accelerated by assembling NadB and NadA as a enzyme complex with the help of peptide-peptide interaction peptides RIAD and RIDD, which resulted in up to 3.7 g/L quinolinic acid being produced from 40 g/L glucose in shake-flask cultures. A quinolinic acid producer was constructed in this study, and these results lay a foundation for further engineering of microbial cell factories to efficiently produce quinolinic acid and subsequently converting this product to nicotinic acid for industrial applications.
Keywords: quinolinic acid, nicotinic acid, peptide-peptide interaction, enzyme complex, E. coli, metabolic engineering
1. Introduction
Quinolinic acid (QA), known as pyridine-2,3-dicarboxylic acid, is a dicarboxylic acid with a pyridine backbone, and it is a key precursor to nicotinic acid and nicotine in nature (Andreoli et al., 1963). Nicotinic acid, also known as niacin or vitamin B3, is one of the water-soluble B complex vitamins, and it commonly exists in living cells. In general, nicotinic acid is present mainly in the form of nicotinic acid amide coenzyme [Nicotinamide Adenine Dinucleotide (NAD+) or Nicotinamide Adenine Dinucleotide Phosphate (NADP+)] in vivo and it acts as a cofactor for a large and diverse number of cellular oxidative-reductive reactions. Nicotinic acid is an essential human nutrient as deficiency of nicotinic acid may cause the disease pellagra and neuropathies (Crook, 2014; Hammond et al., 2013). Consequently, nicotinic acid is widely used in the food and pharmaceutical industries. Nicotinic acid was mainly produced by chemical synthesis through oxidation of β-picoline (Andrushkevich and Ovchinnikova, 2012; Shishido et al., 2003); however, these processes result in considerable amounts of toxic wastes, which require great expense for proper disposal. Moreover, these methods use petroleum-based non-renewable chemicals as the precursor, which is affected by concerns about climate change and unpredictable oil refining prices. To overcome these concerns, production of fuels, pharmaceuticals, or bulk and fine chemicals from renewable feedstocks using microorganisms has arisen as an attractive alternative (Keasling, 2010; Lee et al., 2012; Woolston et al., 2013), especially given advancements in metabolic engineering, however, there is little published literature directed to the over-production of nicotinic acid or even quinolinic acid in microorganisms (Kim et al., 2016).
Early work on the enzymes and genes related to quinolinic acid formation from aspartate has been described (Chandler and Gholson, 1972; Flachmann et al., 1988; Griffith et al., 1975). Quinolinic acid, NAD+, Coenzyme A, and commercially important amino acids such as threonine and lysine (Becker et al., 2011; Ning et al., 2016; Park and Lee, 2010; Zhao et al., 2020) are generated via aspartate as a key precursor. Metabolic engineering at the oxaloacetate and aspartate nodes is important to give a high level of these key intermediates, as well as to provide high and specific conversion of aspartate to quinolinic acid. We have previously engineered cells to produce compounds derived from the oxaloacetate branch of the TCA cycle and have developed useful designs for metabolic networks to product compounds around this node (Cox et al., 2006; Martinez et al., 2018; Sánchez et al., 2005; Zhu et al., 2020). Likewise, we have investigated computational methods for finding novel pathways to relevant OAA derived compounds (Heath et al., 2010; Kim et al., 2020). There are two major NAD de novo biosynthetic pathways used for quinolinate production in vivo: NAD de novo biosynthesis I (from aspartate) in most prokaryotes and NAD de novo biosynthesis II (from tryptophan), also known as kynurenine pathway, in eukaryotes (Begley et al., 2001; Panozzo et al., 2002). These two pathways converge at quinolinic acid, a key intermediate of NAD biosynthetic pathways, and subsequently use three common steps to synthesize NAD. Five enzymes are involved in the conversion of tryptophan to quinolinate in the kynurenine pathway. In comparison, only two enzymes, L-aspartate oxidase (NadB) and quinolinate synthase (NadA), are needed for quinolinate production aerobically from aspartate (Magni et al., 1999). L-aspartate oxidase (NadB), is a monomer of 60 kDa flavoenzyme containing 1 mol of non-covalently bound FAD/mol protein, and it catalyzes the conversion of L-aspartate to 2-iminosuccinate (Mortarino et al., 1996; Nasu et al., 1982). Quinolinate synthase (NadA), which appears mainly as a dimeric protein of 80 kDa (Ollagnier-de Choudens et al., 2005), contains an oxygen-sensitive [4Fe-4S] cluster required to catalyze the condensation of 2-iminosuccinate with dihydroxyacetone phosphate (DHAP) to produce quinolinate (Ceciliani et al., 2000; Cicchillo et al., 2005; Ollagnier-de Choudens et al., 2005). We know that the concentration of NAD(H) and NADP(H) in vivo remains relatively stable, and the pool is maintained by a combination of gene expression regulation, feedback inhibition, and cofactor degradation [The NAD(P) recycling and salvage pathways] (Begley et al., 2001). The expression of NadB and NadA was reported to be repressed by a DNA binding transcriptional repressor NadR (Begley et al., 2001; Tritz and Chandler, 1973). Moreover, inhibition of aspartate oxidase activity was reported by the substrate aspartate, the product 2-iminosuccinate and also NAD+ (Mortarino et al., 1996; Seifert et al., 1990).
In this study, comprehensive engineering strategies were applied to E. coli K12 MG1655 to increase quinolinate production through the NAD de novo biosynthesis I (from aspartate) pathway. The approach included: blocking the consumption pathway, eliminating the transcriptional repression of NadA and NadB, decreasing the glucose consumption rate and increasing the availability of PEP followed by increasing the OAA pool for aspartate synthesis, and blocking the by-product acetate producing pathways, as well as improving the enzymatic efficiency by assembling NadA and NadB as a enzyme complex (Fig. 1). Finally, up to 3.7 g/L quinolinate was produced from 40 g/L glucose in shake-flask cultures of the improved strain. The results lay a foundation for further engineering of the strain for highly efficient production of quinolinate for industrial applications.
Fig. 1. Metabolic engineering of E. coli MG1655 for quinolinic acid production.
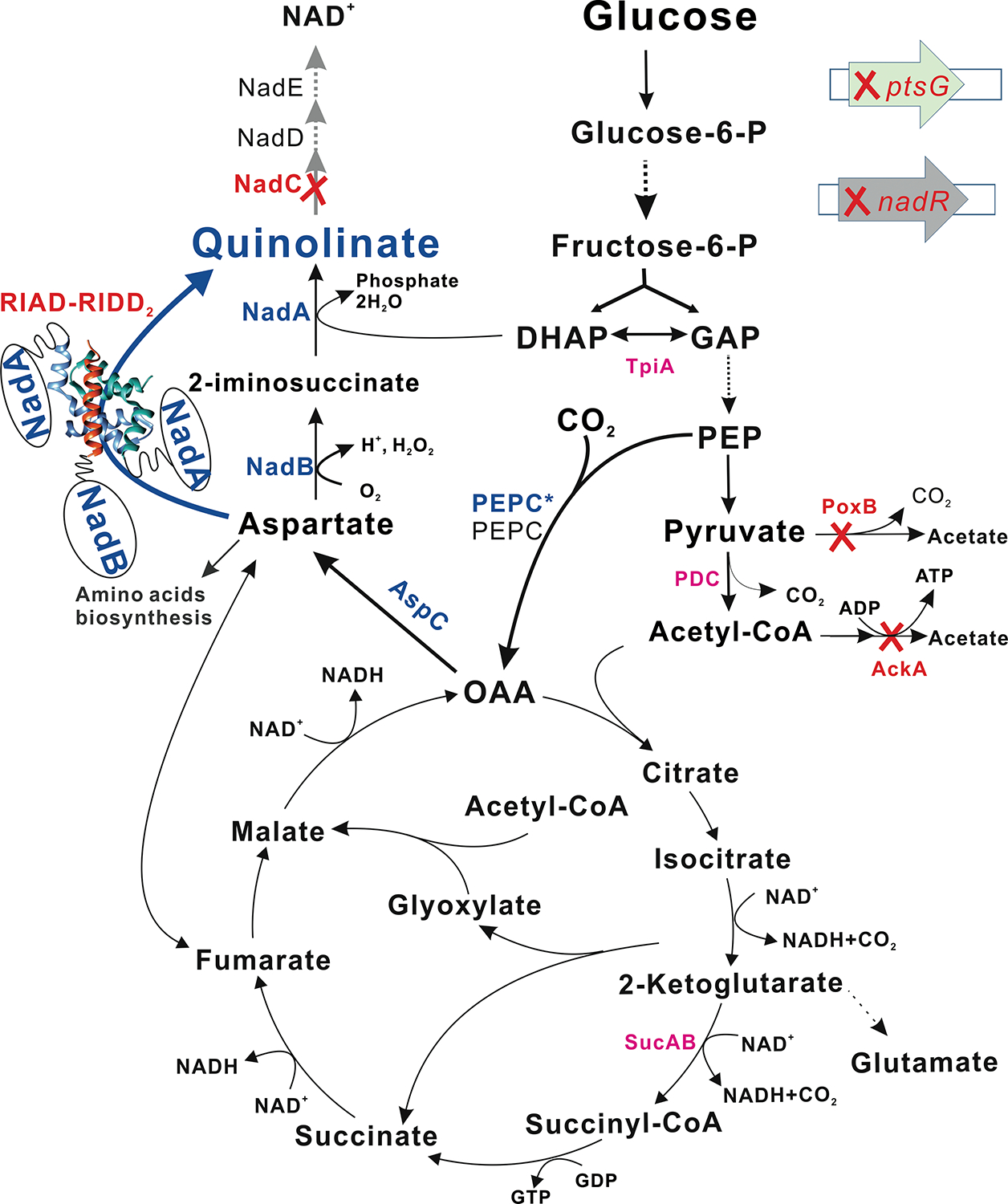
Quinolinic acid accumulation was enabled by deactivation of nadC (to block the consumption pathway), nadR (to eliminate the repression of L-aspartate oxidase NadB and quinolinate synthase NadA), and ptsG (to slow down the glucose consumption rate and achieve a more balanced metabolism, and to increase the availability of phosphoenolpyruvate). Quinolinic acid production was enhanced by increasing the oxaloacetate (OAA) pool through overexpression of phosphoenolpyruvate carboxylase (PEPC) and deactivation of the acetate-producing pathways (poxB, ackA). Quinolinic acid production was accelerated by assembling NadB and NadA as a enzyme complex with the help of peptide-peptide interaction peptides RIAD and RIDD (Carlson et al., 2006; Gold et al., 2006). PtsG: glucose specific PTS enzyme II subunit BC; NadR: DNA-binding transcriptional repressor; DHAP: dihydroxyacetone phosphate; GAP: glyceraldehyde 3-phosphate; TpiA: triose-phosphate isomerase; PDC: pyruvate dehydrogenase complex; poxB: pyruvate oxidase; AckA, acetate kinase; SucAB: 2-oxoglutarate dehydrogenase multi-enzyme complex subunit AB; AspC: aspartate aminotransferase; NadC: quinolinate phosphoribosyltransferase; NadD: nicotinate-mononucleotide adenylyltransferase; NadE: NAD synthetase; RIAD, an 18 amino acids peptide from the A kinase-anchoring proteins; RIDD2: RIDD dimer which contains 50 N-terminal residues of cAMP-dependent protein kinase for each peptide.
2. Materials and methods
2.1. Plasmids and strains
Plasmids and strains used in this study are listed in Table S1 and a brief description of the construction procedure is provided below.
The traditional digestion and ligation method was used for plasmid construction. NdeI-HindIII digested DNA fragment including lacI, pTrc promoter and multiple cloning sites (MCS) from pFZGNB16 (pTrc99a with a BglII site 193 bp upstream of the pTrc promoter) was ligated with a DNA fragment containing the kanamycin resistance gene and the origin of replication from pHL413Km (Thakker et al., 2011) to give pFZGNB33 (BglII-pTrc-MCS-Km-ori-lacI). Native genes, nadA, nadB and aspC were each individually amplified from E. coli MG1655 genomic DNA and inserted into pFZGNB33 to give pFZGNB34, pFZGNB35 and pFZGNB36, respectively. AspC, nadB and nadA gene fragments with ribosome binding sites (RBS) were combined in different orders to give pFZGNB37 to pFZGNB42 (Table S1). nadA from Bacillus subtilis (BsnadA) was amplified from Bacillus subtilis CB10 with an extra DNA fragment coding a HisTag at the C-terminal (BsnadA-HisTag) and inserted into pFZ33 to give pFZGNB43, and pTrc-nadB from pFZGNB35 was inserted into pFZGNB43 to give pFZGNB207. L-aspartate oxidase from Sulfolobus tokodaii (StnadB, GenBank: KC333624.1) was synthesized and sub-cloned into pTrc99a by Genscript (pTrc99a-StnadB). pTrc-StnadB was then inserted into pFZGNB34 to give pFZGNB189. The SpeI-HindIII digested pTrc-nadB PCR product from pFZGNB35 was inserted into XbaI-HindIII digested pFZGNB34 to give pFZGNB190. NadB from Pseudomonas putida KT2440 (PpnadB) was amplified from genomic DNA and inserted into pFZGNB33 to give pFZGNB183. pTrc-PpnadB from pFZGNB183 was inserted into pFZGNB34 to give pFZGNB193.
For NadA-NadB fusion protein constructions, DNA fragments encoding peptide linkers (GS)3, (GS)6 and (G4S)2 were introduced at the end of nadA or nadB without stop codon through PCR and combined with nadB or nadA to give pFZGNB153 to pFZGNB157 (Table S1). DNA fragments encoding peptides RIAD and RIDD with a linker (G4S)2G4CG were codon-optimized and synthesized by Synbio Technologies (Table S2). Overlap Extension PCR (OE-PCR) was then applied to combine the peptide encoding DNA fragment with nadA or nadB to give pFZGNB228 to pFZGNB231. pTrc-nadB-RIAD from pFZGNB229 and pTrc-RIAD-nadB from pFZGNB230 were inserted into pFZ228 and pFZGNB231 individually to give pFZGNB232 to pFZGNB235, respectively (Table S1).
N20 of pTargetF (Jiang et al., 2015) was replaced by a ptsG specific N20 (GTATCCGTACTGCCTATCGC) through inverse PCR to give pFZGNB44. The upstream and downstream homologous arms of ptsG were amplified from E. coli MG1655 genomic DNA and inserted between the EcoRI and HindIII sites of pFZGNB44 with an XbaI site between the two homologous arms to give pFZGNB46, which can be used for ptsG deletion. The same method was used to construct plasmids pFZGNB63, pFZGNB65, pFZGNB74, pFZGNB75 and pFZGNB76 for deactivation of tpiA, sucAB, lpd, ackA, and pta-ackA. pTrc-pepc was amplified from pKK313 (Wang et al., 1992) and inserted into pFZGNB46, pFZGNB75, and pFZGNB76 to give pFZGNB50, pFZGNB117, and pFZGNB118, which can be used to replace ptsG, ackA, pta-ackA with pTrc-pepc individually (Table S1).
E. coli K12 MG1655 was used as the parent strain in this study. Genes nadC and nadR were deleted using the lambda red recombination method (Datsenko and Wanner, 2000), CRISPR/Cas9 method was employed for deletion/replacement of the other selected genes (Jiang et al., 2015).
2.2. Fermentation of quinolinic acid from glucose
Freshly transformed strains were used for every batch fermentation. Ten single colonies were inoculated into 5 mL LB or quinolinic acid-producing medium with 50 mg/L kanamycin and cultured at 37 °C to form the inoculum seed culture. Fermentation was performed in 250-mL conical flasks that contain 1.5 g CaCO3. Fifty milliliters of quinolinic acid-producing medium supplemented with 50 mg/L kanamycin was added into each flask, and IPTG was added to a final concentration of 0.1 mM for induction. One percent of an overnight cell culture was inoculated into the fermentation medium. The cells were grown at 37 °C with shaking at 350 rpm unless otherwise stated. A sample of 1-mL of culture broth was withdrawn at designated intervals for product and metabolite analysis by HPLC. The previous reported medium (Kim et al., 2016) with higher concentration of Fe2+, Mn2+, and Zn2+ (increased from 5 mg/L to 10 mg/L) was used for fermentation, which contains 70 g/L of glucose, 17 g/L of (NH4)2SO4, 1 g/L of KH2PO4, 0.5 g/L of MgSO4 7 H2O, 2 g/L of yeast extract, 150 mg/L of methionine, 10 mg/L of FeSO4·7H2O, 10 mg/L of MnSO4·8H2O, and 10 mg/L of ZnSO4, unless otherwise specified.
2.3. SDS-PAGE analysis of soluable protein
To check the protein expression level of the targeted proteins, cells from 3mL cell culture was harvested ans stocked at −80 °C until further analysis. The cells were melt on ice and suspended in 0.8 mL of 50 mM Tris-Cl (pH 8.0), and 0.6 g glass beads was added. The cells were disrupted by disruptor Genie (2500 rpm) for 3 mins. Lyzed cells were then centrifuged at 13,000 rpm for 10 mins and the superanants were used for SDS-PAGE analysis.
2.4. Analytical methods
For analyzing the fermentation products and residual sugars and other metabolites, the samples were centrifuged at 13,000× g for 2 min and then the supernatant was filtered through a 0.2 μm syringe filter. The glucose, acetate, and quinolinate were quantified using the same method as previously described (Zhu et al., 2018). In brief, an HPLC system was equipped with a cation-exchange column Aminex HPX-87H (Bio-Rad, USA) and a differential refractive index detector RID-10A (Shimadzu, Japan) and UV detector at 210 nm. 2.5 mM H2SO4 served as the mobile phase running at 0.5 mL/min, and the column temperature was maintained at 55 °C. Authentic quinolinic acid (Sigma, St. Louis, MO) was used to generate a standard curve from 10 mg/L to 5 g/L, less than 10 mg/L is considered as not detectable (nd) under tested conditions.
3. Results and discussion
3.1. Engineering of E. coli MG1655 for quinolinic acid accumulation.
Quinolinic acid is an intermediate of the NAD+ de novo biosynthetic pathway under aerobic conditions (Fig. 1). The gene nadC of MG1655 was disrupted to block the consumption pathway and allow strain FZ700 to accumulate quinolinic acid. To increase expression of the NAD+ biosynthetic genes, the DNA-binding transcriptional repressor nadR (of nadA and nadB) was deactivated in FZ700 to give FZ703. Plasmid pFZGNB42 (pTrc-aspC-nadB-nadA) was introduced into the engineered strains to check their performance. The quinolinic acid accumulated in parent strain E. coli MG1655 is less than 43 mg/L under tested conditions, which is likely due to it being further converted to NAD+. 138 mg/L of quinolinic acid was accumulated in FZ700, and QA titer goes up to 544 mg/L in FZ703.
It’s worth to notice that there is more than 37 g/L acetate was accumulated in these strains, which has wasted most of the feedstock glucose. Deactivation of glucose specific transporter PtsG was reported to be able to reduce the acetate secretion significantly by slowing down the glucose consumption rate which results in a more balanced and efficient metabolism (De Anda et al., 2006; Gosset, 2005; Wong et al., 2008), and more PEP can be conserved and available for other metabolic pathways by decoupling glucose transportation from PEP-dependent phosphorylation (Chatterjee et al., 2001; Gosset, 2005; Liang et al., 2015; Lin et al., 2005). So, the glucose specific transporter PtsG was deactivated in FZ703 to give FZ723. Meanwhile, the S8D mutanted phosphoenolpyruvate carboxylase (PEPC) from sorghum (Wang et al., 1992) was also introduced to increase the OAA pool. This modification was accomplished by the replacement of ptsG with pTrc-pepc in FZ703 to give FZ734. After 1-day fermentation, up to 450 mg/L quinolinic acid was accumulated in FZ703, while only 283mg/L and 360 mg/L quinolinic acid was produced in FZ723 and FZ734 respectively. After 3-day fermentation, strains FZ723 and FZ734 catch up with the performance of strain FZ703. Finally, 627 mg/L and 740 mg/L quinolinic acid was accumulated in FZ723 and FZ734 respectively after 7-day fermentation, while, only 544 mg/L quinolinc acid accumulated in FZ703 (Table 1). However, there is a very high concentration of acetate was accumulated in FZ734 too (Table S2). The experiment clearly indicates that blocking the consumption pathway is necessary for quinolinic acid production, and deactivation of PtsG and introducing PEPC are helpful for quinolinic acid production. Strain FZ734 was used for further experiments.
Table 1.
Quinolinate production in engineered strains
| MG1655 | FZ700 | FZ703 | FZ723 | FZ734 | |
|---|---|---|---|---|---|
|
| |||||
| 1 day | 40.2 ± 1.6 | 97.1 ± 13.6 | 449.7 ± 24.3 | 282.6 ± 16.0 | 360.2 ± 29.2 |
| 3 day | 42.8 ± 3.5 | 138.2 ± 8.3 | 488.5 ± 23.4 | 500.8 ± 28.5 | 594.5 ± 13.7 |
| 5 day | 36.9 ± 1.8 | 133.8 ± 7.5 | 503.8 ± 26.4 | 544.0 ± 31.2 | 644.9 ± 16.4 |
| 7 day | 36.8 ± 2.1 | 135.4 ± 6.9 | 544.4 ± 20.4 | 627.3 ± 31.6 | 739.7 ± 16.9 |
|
| |||||
| nadC | + | − | − | − | − |
| nadR | + | + | − | − | − |
| ptsG | + | + | + | − | − |
| pTrc-pepc | − | − | − | − | + |
Plasmid pFZGNB42 (pTrc-aspC-nadB-nadA) was introduced into the tested strains to examine their performance, aerobic cultures were performed at 37 °C, 350 rpm. The numbers indicate quinolinate concentration (mg/L, average of three replicates with error bars indicating standard deviation).
Plasmid pFZGNB42 (pTrc-aspC-nadB-nadA) was introduced into the tested strains to check their performance, aerobic cultures were performed at 37 °C, 350 rpm. The numbers indicate quinolinate concentration (mg/L, average of three replicates with error bars indicating standard deviation).
3.2. Effect of varying the gene order of aspC, nadB, and nadA in expression constructs on quinolinic acid production
Plasmids containing the three genes for quinolinic acid production from OAA in different arrangements were constructed to give plasmids pFZGNB37 to pFZGNB42 (Fig. 2) and their performance on quinolinic acid production in FZ734 was evaluated. After 4 days of fermentation, more than 48 g/L of glucose was consumed in all tested strains. The culture of the strain where only aspC was overexpressed produced the lowest quinolinic acid titer (123 mg/L) compared to the results observed when nadA or nadB was overexpressed (Fig. 2). When all three genes were co-overexpressed in different gene orders on a plasmid, the quinolinic acid production was slightly improved with plasmid pFZGNB37 (pTrc-nadA-nadB-aspC), and the quinolinic acid titer increased to 404 mg/L with plasmid pFZGNB42 (pTrc-aspC-nadB-nadA). Unexpectedly, a higher quinolinic acid titer was observed in the strain bearing plasmid pFZGNB190, which only overexpressed nadA and nadB (pTrc-nadA+pTrc-nadB), where 667 mg/L quinolinic acid was accumulated (Fig. 2). This experiment suggested the level of AspC expressed from the native gene is already enough for these tested conditions.
Fig. 2. Quinolinic acid production in FZ734 harboring plasmids containing various gene orders of aspC, nadB, and nadA.
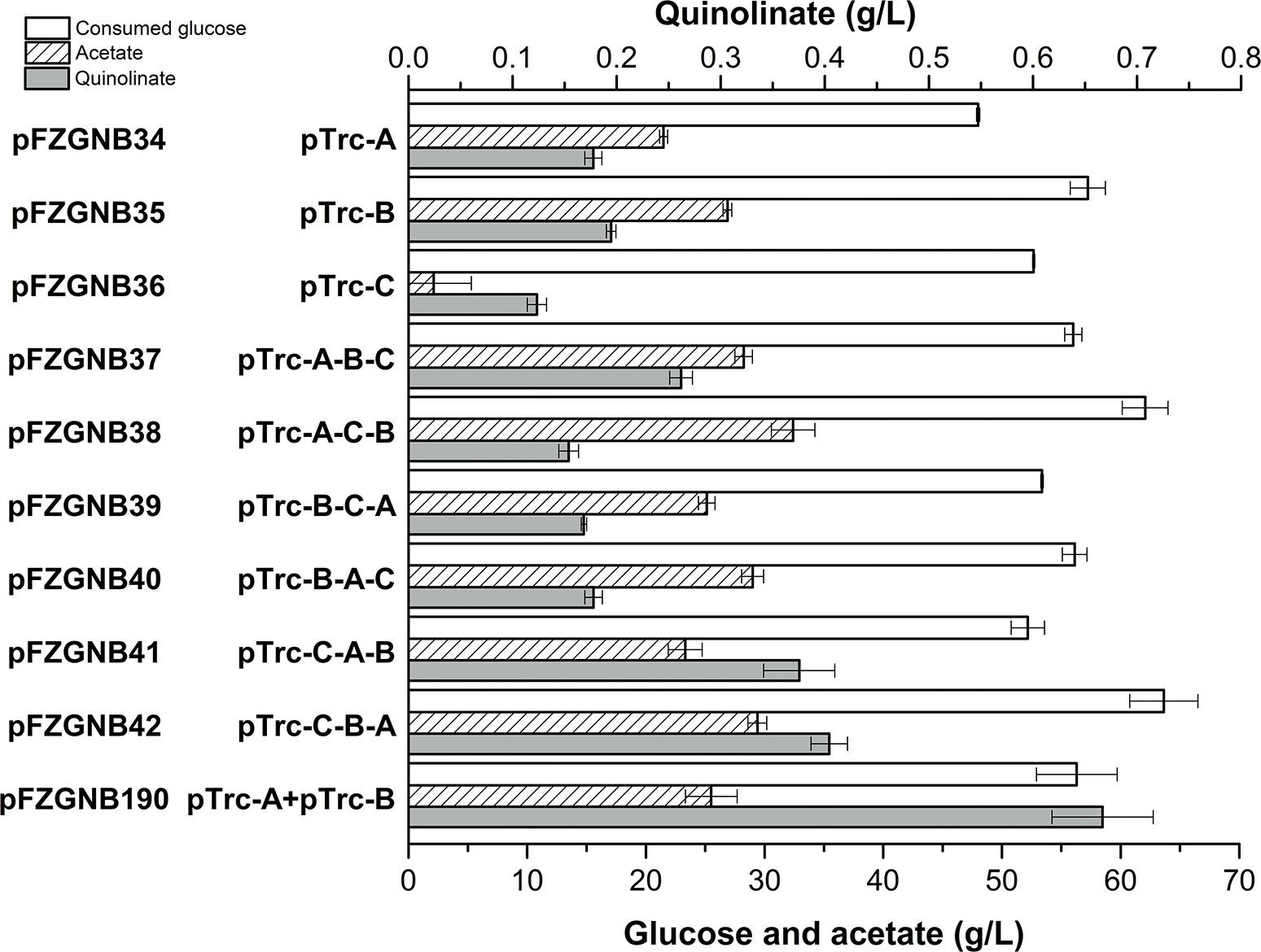
Aerobic cultures were performed at 37 °C, 350 rpm for 4 days. Values are the average of three replicates with error bars indicating standard deviation. A, B, and C indicate nadA, nadB, and aspC accordingly.
3.3. Examination of variants of nadA and nadB failed to improve quinolinate production
Although FZ734 with plasmid pFZGNB190 (pTrc-nadA+ pTrc-nadB) showed the best performance as described above in section 3.2, it was reported that protein NadB displays a substrate inactivation and its activity can be inhibited by substrate aspartate and NAD, which acts as a competitive inhibitor to FAD (Seifert et al., 1990). NadA was also reported to form inclusion bodies when it’s overexpressed (Ceciliani et al., 2000), which could diminish its in vivo activity. Screening enzymes from diverse sources has been used as an effective strategy to increase the productivity of heterologous pathways in specific hosts. So, some NadA and NadB variants from other species were employed and tested in FZ734 for comparison.
It was reported that the activity of NadB from Pseudomonas putida KT2440 (PpnadB) was not inhibited by aspartate when the concentration of aspartate are less than 50 mM. And the Km, kcat value of this enzyme was determined to be 2.26 mM, 10.6 s−1 against L-aspartate (Leese et al., 2013). NadB from Sulfolobus tokodaii (StNadB) was also reported to be less sensitive to NAD+ since it can bind the FAD cofactor tightly with a different structure (Bifulco et al., 2013; Sakuraba et al., 2008); moreover, it displays some other distinctive features, e.g., stable activity over a wide range of pH (from 7 to 10), and high thermostability (with a Tm higher than 79 °C), which would make it attractive for biotechnological applications. However, evaluation of constructs expressing these alternative genes under the same conditions described in Figure 2 revealed no improvement in quinolinic acid production (Fig. 3). This may be due to these enzymes having a lower affinity for aspartate, the Km value for L-aspartate of StNadB and PpNadB is 1.3 and 2.26 mM as reported (Bifulco et al., 2013; Leese et al., 2013), which is much higher than the Km value of E. coli NadB (0.048 mM). Another possible reason is the poor protein expression of these NadB variants (Fig. S1).
Fig. 3. The effect of nadA and nadB variants on quinolinic acid production in FZ734.
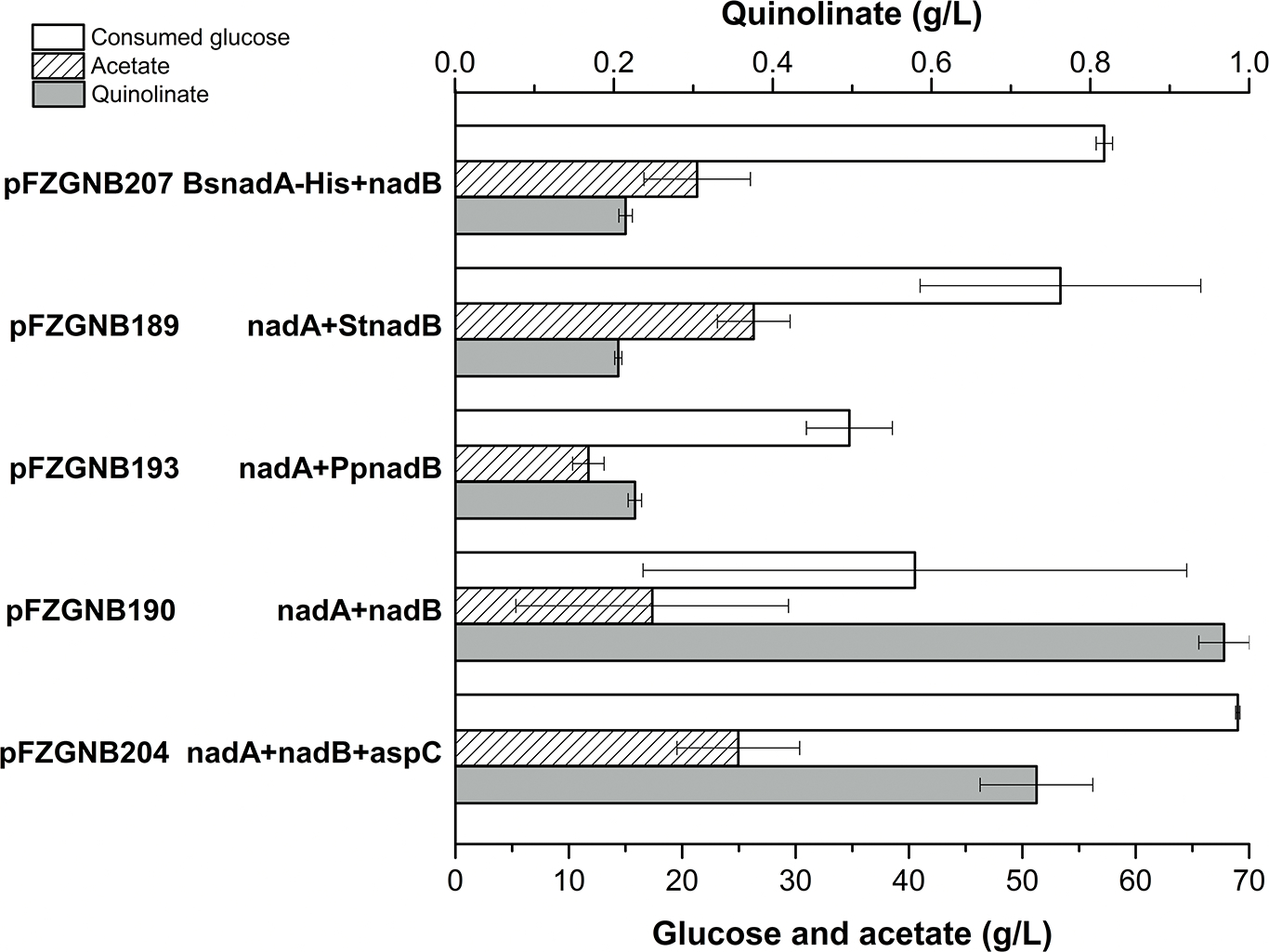
Aerobic cultures were performed at 37 °C, 350 rpm for 4 days. Proteins were overexpressed under the control of pTrc promoter in pFZGNB33. Values are the average of three replicates with error bars indicating standard deviation. BsnadA-HisTag indicates nadA from Bacillus subtilis with a His6-Tag at the C-terminal, StnadB indicates nadB from Sulfolobus tokodaii, and PpnadB indicates nadB from Pseudomonas putida KT2440.
NadA from Bacillus subtilis (BsNadA) had been successfully overexpressed with a His6 tag at its C-terminal (BsNadA-HisTag) in E. coli with a yield of 10 mg pure protein from 1 L culture (Marinoni et al., 2008). Unexpectedly, only 215 mg/L quinolinic acid was produced when the native nadA in pFZGNB190 was replaced with BsnadA-HisTag (Fig. 3), and no improvement on protein expression was observed in the tested conditions (Fig. S1). This may be due to the low activity of BsNadA-HisTag as previously reported (Marinoni et al., 2008).
Unfortunately, the performance of these strains with different NadA and NadB variants is not as good as the native E. coli enzymes NadA and NadB under the tested conditions in FZ734 (Fig. 3). The native aspC was also introduced into pFZGNB190 to give pFZGNB204; however, less quinolinic acid was accumulated in the strain with pFZGNB204 (Fig. 3), possibly due to the substrate inactivation of NadB, potentially caused by the overexpression of AspC.
3.4. Engineering of FZ734F to improve quinolinic acid production
Several manipulations were explored to enhance the carbon flux to quinolinate production, including increasing the substrate availability, downregulating the TCA cycle, and blocking the by-product acetate production. Triosephosphate isomerase (TpiA), which catalyzes the isomerization between glyceraldehyde 3-phosphate and dihydroxyacetone phosphate (DHAP), was deactivated to reserve DHAP for quinolinate biosynthesis. Meanwhile, we hypothesize that the low yield of quinolinate is due to too much carbon going through the TCA cycle, so sucAB and lpd were deactivated to down-regulate the carbon flux through TCA cycle. SucA and SucB are two subunits of the 2-oxoglutarate dehydrogenase multi-enzyme complex (OGDHC) that catalyzes the conversion of 2-oxoglutarate (2-ketoglutarate) to succinyl-CoA and CO2. Deactivation of sucAB could accumulate 2-oxoglutarate, which can be used for glutamate synthesis and will be beneficial for aspartate synthesis. Lipoamide dehydrogenase (Lpd) is the E3 component of three multicomponent enzyme complexes: pyruvate dehydrogenase, 2-oxoglutarate dehydrogenase complex, and the glycine cleavage system (Guest and Creaghan, 1972; Pettit and Reed, 1967; Steiert et al., 1990). Deactivation of lpd was reported to produce more pyruvate and L-glutamate under aerobic conditions (Li et al., 2006), as it affects the function of pyruvate dehydrogenase and 2-oxoglutarate dehydrogenase complex. However, the FZ738 (FZ734 ΔtpiA) or FZ742 (FZ734 Δlpd) strain displayed very poor cell growth (cell density was not measured due to the interfere of CaCO3) and failed to produce quinolinate (Fig. 4). Although the sucAB− strain can grow, unfortunately, it also failed to accumulate quinolinate. This may be due to the quinolinate production being an ATP consuming process, and it needs the ATP generated through the TCA cycle or other ATP producing pathway.
Fig. 4. Quinolinic acid production in engineered strains harboring plasmid pFZGNB190.
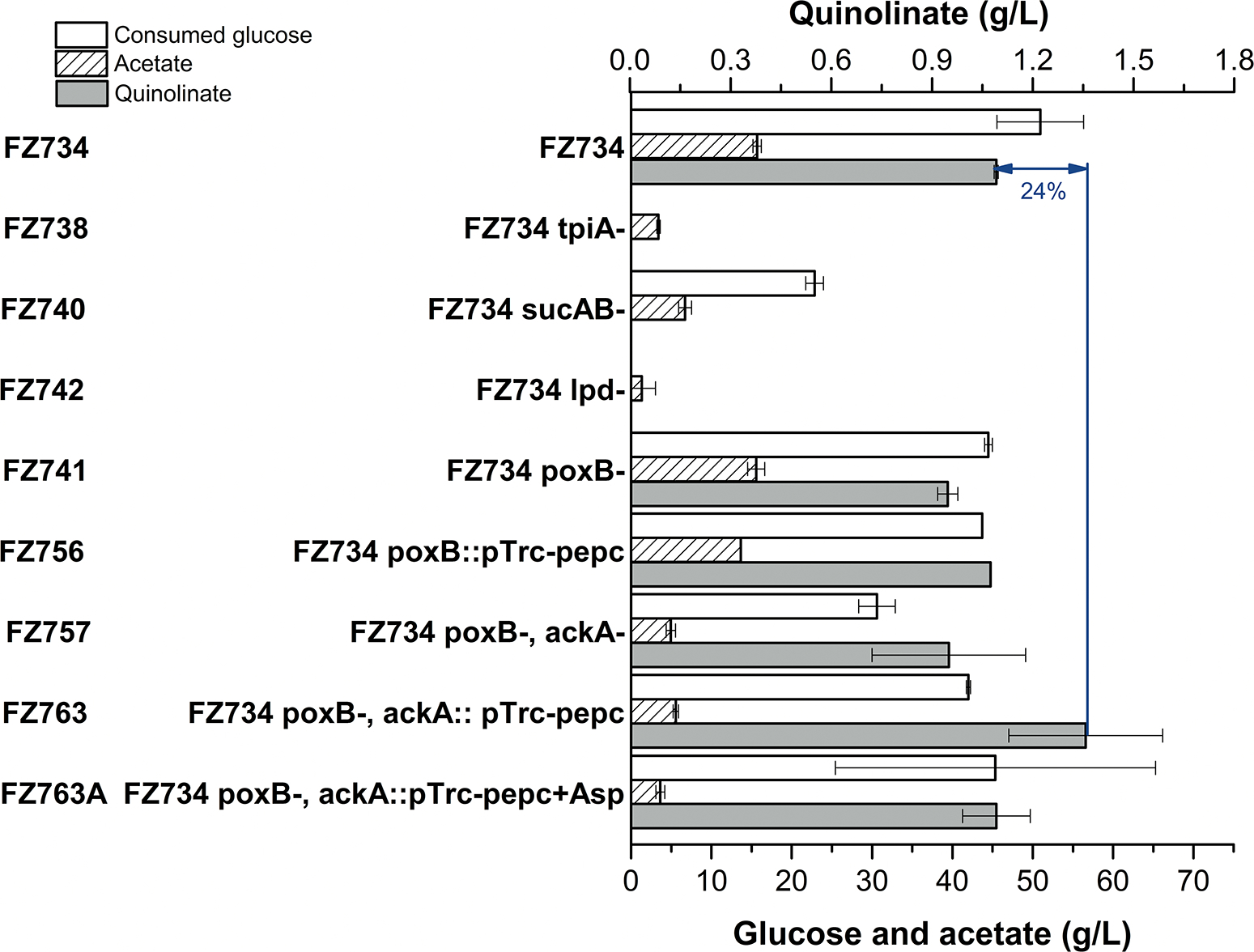
Aerobic cultures were performed at 37 °C, 350 rpm for 4 days. FZ763A indicates 5 g/L aspartate was added into the fermentation broth (+Asp). Values are the average of three replicates with error bars indicating standard deviation.
More than 20 g/L acetate accumulated in nearly in all tested strains, which may inhibit the cell growth and waste the substrate glucose, so the acetate producing pathways were selected as the next target for genetic modification. There are two predominant acetate producing pathways active aerobically in E. coli, they are the pyruvate oxidase (PoxB) and acetate kinase/phosphotransacetylase (AckA-Pta) pathways. Pyruvate oxidase (PoxB) is a peripheral membrane enzyme that catalyzes the oxidative decarboxylation of pyruvate to form acetate mainly functions at stationary phase, whereas the AckA-Pta catalyzed pathway, which mainly functions during the exponential phase, converts acetyl-CoA to acetate (Dittrich et al., 2005). When the fermentation was performed with strains harboring plasmid pFZGNB190, no difference was observed on acetate accumulation when only the PoxB was deactivated, while much less acetate was accumulated when both PoxB and AckA were deactivated (Fig. 4). However, there is 13% decrease in quinolinate titer in FZ757/pFZGNB190 due to 40% less glucose was consumed, which has two acetate producing pathways deactivated, compared with FZ734/pFZGNB190 (Fig. 4). Interestingly, improvement was observed when an extra copy of pTrc-pepc was introduced into FZ757 (FZ734 ΔpoxB, ΔackA) forming strain FZ763/pFZGNB190. The resulting strain yielded a 24.4% increase in quinolinic acid formation compared to FZ734/pFZGNB190, with up to 1358 mg/L being accumulated by consuming 42 g/L glucose (Fig. 4), it’s also worth to notice that there is more than 5.6 g/L acetate accumulated in FZ763/pFZGNB190 in 4-day fermentation (Fig. 4). However, when 5 g/L aspartate was added into the fermentation broth with strain FZ763/pFZGNB190, the quinolinate titer dropped to 1092 mg/L. The decrease is consistent with the result mentioned in section 3.3 (Fig. 3) that showed quinolinate titer dropped when AspC was overexpressed, indicating an imbalance in the synthesis and utilization of this intermediate.
Based on the strains’ performance, the strain FZ763/pFZGNB190 was further evaluated with lower glucose concentration (40 g/L). Interestingly, strain FZ763/pFZGNB190 fermentated with 40 g/L glucose has a higher quinoliate productivity based on 2-day samples, while the strain has comparable quinolinate titer after 4-day fermentation (Fig. S2). This may be due to the fermentation with 40 g/L glucose has much less metabolic stress and more suitable for chemicals production.
3.5. Improvement of quinolinate production by assembling NadA and NadB as an enzyme complex.
As we know, nature forms multi-enzyme complexes, membrane-containing/free organelles, or multidomain synthases to achieve high enzymatic efficiency, such as in tryptophan synthase, yeast fatty acid synthases, and polyketide synthase (Dutta et al., 2014; Lomakin et al., 2007). Synthetic multi-enzyme complexes have been developed to control the flux of metabolites and to improve product yield (Conrado et al., 2008; Quin et al., 2017). Using a fusion protein is one of the developed strategies to form synthetic multi-enzyme complexes, which can be easily constructed by genetically fusing two or more protein domains with a peptide linker, and it is also useful to improve the solubility of recombination proteins. While, no improvement was observed when NadA and NadB was fusioned with linkers (GS)3, (GS)6, or (G4S)2 (Fig. S3), although the performance of NadA-(GS)6-NadB (FZ763/pFZGNB157) is already comparable with the performance of separate proteins (FZ763/pFZGNB190). This may be due to the linker not being optimized, another possible reason is that the expression of the large fusion protein is not as good as the separate proteins (Fig. S4).
Synthetic multi-enzyme complexes can also be constructed by assembling the enzymes with a protein/peptide scaffold based on protein-protein/peptide interaction or even peptide-peptide interaction (Conrado et al., 2008; Quin et al., 2017). A dock-and-lock peptide interacting family of peptides RIAD and RIDD have been successfully applied for lycopene overproduction (Kang et al., 2019). RIAD, an 18 amino acid peptide which is from the A kinase-anchoring proteins, specifically binds to the RIDD dimer, which contains the first 50 N-terminal residues of cAMP-dependent protein kinase. Their small size, strong binding affinity, and fixed 1:2 binding stoichiometry ratio make them a suitable pair of protein tags for multi-enzyme assembly (Kang et al., 2019). So, NadB and NadA were then assembled as an enzyme complex by adding peptides RIAD and RIDD in this study. DNA fragments encoding peptides RIAD and RIDD with a linker (G4S)2G4CG were codon-optimized and synthesized by Synbio Technologies (Table S3). NadA appears as a dimeric protein and the dimeric structure is essential for its stability (Ollagnier-de Choudens et al., 2005), while NadB is a monomeric protein (Mattevi et al., 1999), so RIDD was combined with nadA and RIAD was combined with nadB both at N-terminal and C-terminal by Overlap Extension PCR (OE-PCR) to give pFZGNB228 to pFZGNB231. pTrc-nadB-RIAD from pFZGNB229, and pTrc-RIAD-nadB from pFZGNB230 was inserted into pFZ228 and pFZGNB231 individually to give pFZGNB232 to pFZGNB235 (Table S1). Since there is no significant improvement was observed in quinolinate titer when 70 g/L glucose was used compared to 40 g/L glucose, the strains were evaluated in QA producing medium with 40 g/L glucose. The strain FZ763/pFZGNB233 [RIDD-NadA (RIDD was added at the N-terminal of NadA with a G4SG4SG4CG linker) with RIAD-NadB] displayed a poor cell growth after two days fermentation, no quinolinate was observed (Fig. 5a), nearly no glucose was consumed (Fig. 5b), although comparable biomass was obtained after 4 days fermentation (cell density was not measured due to the present of CaCO3), it only produced 0.7 g/L quinolinate, which is 31% less than it produced in FZ763/pFZGNB190 (1028 mg/L, Fig. 5a). The other strains all have better performance than FZ763/pFZGNB190 (Fig. 5). After 2 days fermentation, FZ763/pFZGNB190 has already consumed all glucose, but only produced 1.0 g/L quinolinate. Strain FZ763/pFZGNB232 (RIDD-NadA with NadB-RIAD) has the highest quinolinate titer (2.8 g/L) due to more glucose was consumed (37.9 g/L) than other strains (except FZ763/pFZGNB190). Strain FZ763/pFZGNB234 (NadA-RIDD with NadB-RIAD) produced 2.6 g/L quinolinate from 29 g/L glucose with less acetate was accumulated (Fig. 5c). 37.4 g/L glucose was consumed in FZ763/pFZGNB235 (NadA-RIDD with RIAD-NadB), and 2.4 g/L quinolinate was accumulated. After 4 days fermentation, no significant improvement was observed in strain FZ763/pFZGNB190, FZ763/pFZGNB232 and FZ763/pFZGNB235 due to most of glucose was consumed within 2 days (Fig. 5a and 5b). Quinolinate titer in strain FZ763/pFZGNB234 was further increased to 3.7 g/L by consuming the residual glucose, which is 3.6-fold higher than FZ763/pFZGNB190 (Fig. 5) and more than 86.8 times higher than the parent strainMG1655/pFZGNB42 (Table 1 and Fig. 5a). Less acetate was accumulated in these strains, except strain FZ763/pFZGNB233, compared to strain FZ763/pFZGNB190 (Fig. 5c), which indicates a more balanced metabolism was achieved. Similar amount protein complexes were observed through a non-reducing SDS-PAGE analysis (Fig. S4), except FZ763/pFZGNB233, which indicates the different performance is not mainly due to the protein expression difference. The strains were also tested in QA producting medium with 70 g/L glucose as a carbon source, and there is no improvement on quinolinate titer was observed among most tested strains (Fig. S5). Interestingly, when the peptides were added to the N-terminal of NadA and NadB (RIDD-NadA with RIAD-NadB), the strain has the lowest quinolinate titer, which seems formed an unpreferred enzyme complex and leads to a metabolic burden; while it has the highest quinolinate titer when the peptides were added to the C-terminal (NadA-RIDD with NadB-RIAD), the improved titers may be due to the enzyme complex providing an efficient and exclusive catalytic cascade reaction with little free diffusion of unstable intermediates 2-iminosuccinate (Nasu et al., 1982), which may be need further structural studies to understand it. Another possible reason is the RIDD: RIAD complex has a ratio of 2:1 (Kang et al., 2019), which is beneficial for NadA dimer formation. Notably, the turnover frequency of E. coli NadB is only 0.267 s−1 (Tedeschi et al., 2010), which needs to be improved as it may limit the enzyme complex catalytic efficiency.
Fig. 5. The effect of NadA-NadB enzyme complex on quinolinic acid production in FZ763 with 40 g/L glucose.
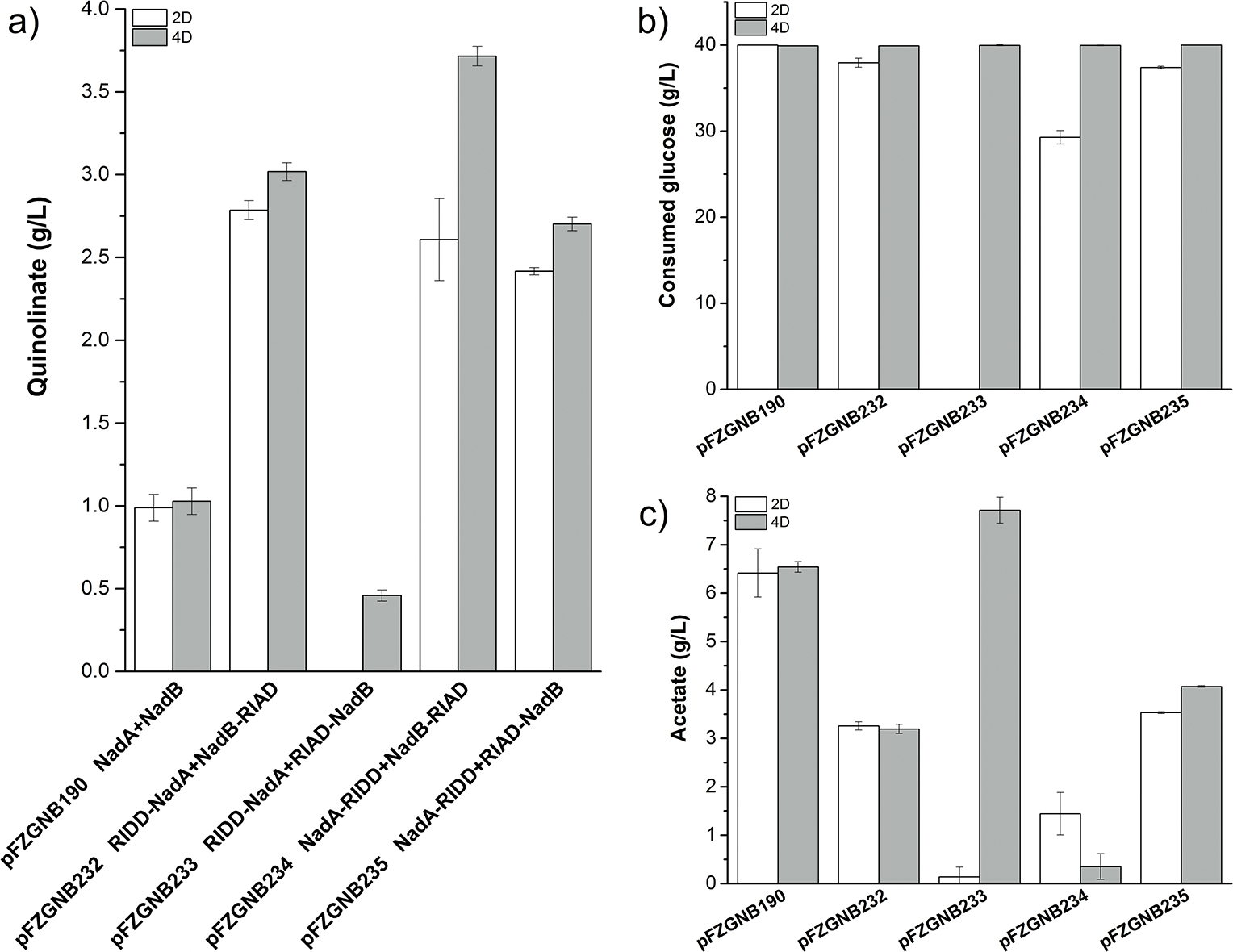
a) quinolinate titer, b) consumed glucose, c) accumulated acetate. Aerobic cultures were performed at 37 °C, 350 rpm, sampled at 2 days (2D) and 4 days (4D). NadA-RIDD and NadB-RIAD fusion proteins were over-expressed under the control of their pTrc promoter in pFZGNB227, NadA-NadB enzyme complex was assembled with the help of peptides, RIAD and RIDD. Values are the average of three replicates with error bars indicating standard deviation.
In conclusion, multiple engineering strategies were applied to increase the production of quinolinate. The quinolinate consumption pathway was blocked to enable the accumulation of quinolinate by deactivation of NadC, and quinolinate production was activated by knockout the repressor gene nadR. Then, the quinolinate production was enhanced by deactivation of glucose transporter gene ptsG to slow down the glucose consumption rate, to achieve a more balanced metabolism, and to improve the availability of PEP. Increasing the OAA pool through overexpression of PEPC also improved quinolinate production. Moreover, the acetate-producing pathways were deactivated as acetate is a major by-product in the engineered strain FZ734/pFZGNB190. Finally, quinolinate production was accelerated by assembling NadA and NadB as a enzyme complex with the help of peptide-peptide interaction peptides, RIAD and RIDD, and up to 3.7 g/L quinolinate was accumulated in FZ763/pFZGNB234 in shake-flask cultures. These results lay a foundation for further engineering of the strains to efficiently produce quinolinic acid or even nicotinic acid for industrial applications. Further improvement may be achieved by protein engineering of NadB to eliminate the feedback inhibition and improve the enzymatic activity, improving the expression of NadA, down-regulating the TCA cycle, optimizing the NadA-NadB enzyme complex, and assembling AspC, NadB, and NadA into a enzyme complex to allow more aspartate enter the reactions for quinolinate production before it diffuses into cytoplasmic environment.
Supplementary Material
Acknowledgment
The authors thank Dr. Charles Stewart, Department of Biosciences at Rice University, for kindly sharing Bacillus subtilis CB10 with us, and the authors thank Dr. Guoqiang Zhang at Jiangnan University (Wuxi, China) for helpful suggestions about fusion protein constructs. This work was supported by NSF DBI-1262491, NSF EAGER: DESYN-C3 CBET-1843556, and DOE BES DE-SC0014462. M. P. was supported by a training fellowship from the Gulf Coast Consortia (NLM Grant T15 LM007093).
Footnotes
Conflicts of interest
The authors declare no competing financial interests.
References
- Andreoli AJ, et al. , 1963. Quinolinic acid: a precursor to nicotinamide adenine dinucleotide in Escherichia coli. Biochemical and biophysical research communications. 12, 92. [DOI] [PubMed] [Google Scholar]
- Andrushkevich TV, Ovchinnikova EV, 2012. Gas Phase Catalytic Oxidation of β-Picoline to Nicotinic Acid: Catalysts, Mechanism and Reaction Kinetics. Catalysis Reviews. 54, 399–436. [Google Scholar]
- Becker J, et al. , 2011. From zero to hero—design-based systems metabolic engineering of Corynebacterium glutamicum for l-lysine production. Metabolic engineering. 13, 159–168. [DOI] [PubMed] [Google Scholar]
- Begley TP, et al. , 2001. The biosynthesis of nicotinamide adenine dinucleotides in bacteria. Vitamins & Hormones. 61, 103–119. [DOI] [PubMed] [Google Scholar]
- Bifulco D, et al. , 2013. A thermostable L-aspartate oxidase: a new tool for biotechnological applications. Applied microbiology and biotechnology. 97, 7285–7295. [DOI] [PubMed] [Google Scholar]
- Carlson CR, et al. , 2006. Delineation of type I protein kinase A-selective signaling events using an RI anchoring disruptor. Journal of Biological Chemistry. 281, 21535–21545. [DOI] [PubMed] [Google Scholar]
- Ceciliani F, et al. , 2000. Cloning, overexpression, and purification of Escherichia coli quinolinate synthetase. Protein expression and purification. 18, 64–70. [DOI] [PubMed] [Google Scholar]
- Chandler JL, Gholson R, 1972. De novo biosynthesis of nicotinamide adenine dinucleotide in Escherichia coli: excretion of quinolinic acid by mutants lacking quinolinate phosphoribosyl transferase. Journal of bacteriology. 111, 98–102. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Chatterjee R, et al. , 2001. Mutation of the ptsG gene results in increased production of succinate in fermentation of glucose byEscherichia coli. Applied and Environmental Microbiology. 67, 148–154. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Cicchillo RM, et al. , 2005. Escherichia coli quinolinate synthetase does indeed harbor a [4Fe-4S] cluster. Journal of the American Chemical Society. 127, 7310–7311. [DOI] [PubMed] [Google Scholar]
- Conrado RJ, et al. , 2008. Engineering the spatial organization of metabolic enzymes: mimicking nature’s synergy. Current opinion in biotechnology. 19, 492–499. [DOI] [PubMed] [Google Scholar]
- Cox SJ, et al. , 2006. Development of a metabolic network design and optimization framework incorporating implementation constraints: a succinate production case study. Metabolic engineering. 8, 46–57. [DOI] [PubMed] [Google Scholar]
- Crook MA, 2014. The importance of recognizing pellagra (niacin deficiency) as it still occurs. Nutrition. 30, 729. [DOI] [PubMed] [Google Scholar]
- Datsenko KA, Wanner BL, 2000. One-step inactivation of chromosomal genes in Escherichia coli K-12 using PCR products. Proceedings of the National Academy of Sciences. 97, 6640–6645. [DOI] [PMC free article] [PubMed] [Google Scholar]
- De Anda R, et al. , 2006. Replacement of the glucose phosphotransferase transport system by galactose permease reduces acetate accumulation and improves process performance of Escherichia coli for recombinant protein production without impairment of growth rate. Metabolic engineering. 8, 281–290. [DOI] [PubMed] [Google Scholar]
- Dittrich CR, et al. , 2005. Characterization of the acetate-producing pathways in Escherichia coli. Biotechnology progress. 21, 1062–1067. [DOI] [PubMed] [Google Scholar]
- Dutta S, et al. , 2014. Structure of a modular polyketide synthase. Nature. 510, 512–517. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Flachmann R, et al. , 1988. Molecular biology of pyridine nucleotide biosynthesis in Escherichia coli: cloning and characterization of quinolinate synthesis genes nadA and nadB. European journal of biochemistry. 175, 221–228. [DOI] [PubMed] [Google Scholar]
- Gold MG, et al. , 2006. Molecular basis of AKAP specificity for PKA regulatory subunits. Molecular cell. 24, 383–395. [DOI] [PubMed] [Google Scholar]
- Gosset G, 2005. Improvement of Escherichia coli production strains by modification of the phosphoenolpyruvate: sugar phosphotransferase system. Microbial cell factories. 4, 14. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Griffith GR, et al. , 1975. Studies on the de novo Biosynthesis of NAD in Escherichia coli: The Separation of the nadB Gene Product from the nadA Gene Product and Its Purification. European journal of biochemistry. 54, 239–245. [DOI] [PubMed] [Google Scholar]
- Guest J, Creaghan I, 1972. Lipoamide dehydrogenase mutants of Escherichia coli K 12. Biochemical Journal. 130, 8P. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Hammond N, et al. , 2013. Nutritional neuropathies. Neurologic clinics. 31, 477. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Heath AP, et al. , 2010. Finding metabolic pathways using atom tracking. Bioinformatics. 26, 1548–1555. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Jiang Y, et al. , 2015. Multigene editing in the Escherichia coli genome via the CRISPR-Cas9 system. Applied and environmental microbiology. 81, 2506–2514. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Kang W, et al. , 2019. Modular enzyme assembly for enhanced cascade biocatalysis and metabolic flux. Nature communications. 10, 1–11. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Keasling JD, 2010. Manufacturing molecules through metabolic engineering. Science. 330, 1355–1358. [DOI] [PubMed] [Google Scholar]
- Kim SM, et al. , 2020. Improving the organization and interactivity of metabolic pathfinding with precomputed pathways. BMC bioinformatics. 21, 1–22. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Kim SY, et al. , Method for the preparation of nicotinic acid. Google Patents, 2016. [Google Scholar]
- Lee JW, et al. , 2012. Systems metabolic engineering of microorganisms for natural and non-natural chemicals. Nature chemical biology. 8, 536. [DOI] [PubMed] [Google Scholar]
- Leese C, et al. , 2013. Cloning, expression, characterisation and mutational analysis of L-aspartate oxidase from Pseudomonas putida. Journal of Molecular Catalysis B: Enzymatic. 85, 17–22. [Google Scholar]
- Li M, et al. , 2006. Effect of lpdA gene knockout on the metabolism in Escherichia coli based on enzyme activities, intracellular metabolite concentrations and metabolic flux analysis by 13C-labeling experiments. Journal of biotechnology. 122, 254–266. [DOI] [PubMed] [Google Scholar]
- Liang Q, et al. , 2015. Comparison of individual component deletions in a glucose-specific phosphotransferase system revealed their different applications. Scientific reports. 5, 13200. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Lin H, et al. , 2005. Metabolic engineering of aerobic succinate production systems in Escherichia coli to improve process productivity and achieve the maximum theoretical succinate yield. Metabolic engineering. 7, 116–127. [DOI] [PubMed] [Google Scholar]
- Lomakin IB, et al. , 2007. The crystal structure of yeast fatty acid synthase, a cellular machine with eight active sites working together. Cell. 129, 319–332. [DOI] [PubMed] [Google Scholar]
- Magni G, et al. , 1999. Enzymology of Nad+ Synthesis. Advances in Enzymology and Related Areas of Molecular Biology: Mechanism of Enzyme Action, Part A. 73, 135–182. [DOI] [PubMed] [Google Scholar]
- Marinoni I, et al. , 2008. Characterization of l-aspartate oxidase and quinolinate synthase from Bacillus subtilis. The FEBS journal. 275, 5090–5107. [DOI] [PubMed] [Google Scholar]
- Martinez I, et al. , 2018. High yield production of four-carbon dicarboxylic acids by metabolically engineered Escherichia coli. Journal of industrial microbiology & biotechnology. 45, 53–60. [DOI] [PubMed] [Google Scholar]
- Mattevi A, et al. , 1999. Structure ofL-aspartate oxidase: implications for the succinate dehydrogenase/fumarate reductase oxidoreductase family. Structure. 7, 745–756. [DOI] [PubMed] [Google Scholar]
- Mortarino M, et al. , 1996. L-Aspartate Oxidase from Escherichia coli: I. Characterization of Coenzyme Binding and Product Inhibition. European journal of biochemistry. 239, 418–426. [DOI] [PubMed] [Google Scholar]
- Nasu S, et al. , 1982. L-Aspartate oxidase, a newly discovered enzyme of Escherichia coli, is the B protein of quinolinate synthetase. Journal of Biological Chemistry. 257, 626–632. [PubMed] [Google Scholar]
- Ning Y, et al. , 2016. Pathway construction and metabolic engineering for fermentative production of ectoine in Escherichia coli. Metabolic engineering. 36, 10–18. [DOI] [PubMed] [Google Scholar]
- Ollagnier-de Choudens S, et al. , 2005. Quinolinate synthetase, an iron–sulfur enzyme in NAD biosynthesis. FEBS letters. 579, 3737–3743. [DOI] [PubMed] [Google Scholar]
- Panozzo C, et al. , 2002. Aerobic and anaerobic NAD+ metabolism in Saccharomyces cerevisiae. FEBS letters. 517, 97–102. [DOI] [PubMed] [Google Scholar]
- Park JH, Lee SY, 2010. Metabolic pathways and fermentative production of L-aspartate family amino acids. Biotechnology journal. 5, 560–577. [DOI] [PubMed] [Google Scholar]
- Pettit FH, Reed LJ, 1967. Alpha-keto acid dehydrogenase complexes. 8. Comparison of dihydrolipoyl dehydrogenases from pyruvate and alpha-ketoglutarate dehydrogenase complexes of Escherichia coli. Proceedings of the National Academy of Sciences of the United States of America. 58, 1126. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Quin MB, et al. , 2017. Spatial organization of multi-enzyme biocatalytic cascades. Organic & biomolecular chemistry. 15, 4260–4271. [DOI] [PubMed] [Google Scholar]
- Sakuraba H, et al. , 2008. Structure of l-aspartate oxidase from the hyperthermophilic archaeon Sulfolobus tokodaii. Biochimica et Biophysica Acta (BBA)-Proteins and Proteomics. 1784, 563–571. [DOI] [PubMed] [Google Scholar]
- Sánchez AM, et al. , 2005. Novel pathway engineering design of the anaerobic central metabolic pathway in Escherichia coli to increase succinate yield and productivity. Metabolic engineering. 7, 229–239. [DOI] [PubMed] [Google Scholar]
- Seifert J, et al. , 1990. Expression of the E. coli nadB gene and characterization of the gene product L-aspartate oxidase. Biological chemistry Hoppe-Seyler. 371, 239–248. [PubMed] [Google Scholar]
- Shishido T, et al. , 2003. Vapor-phase oxidation of 3-picoline to nicotinic acid over Cr1-xAlxVO4 catalysts. Applied Catalysis A: General. 239, 287–296. [Google Scholar]
- Steiert P, et al. , 1990. The lpd gene product functions as the L protein in the Escherichia coli glycine cleavage enzyme system. Journal of bacteriology. 172, 6142–6144. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Tedeschi G, et al. , 2010. On the catalytic role of the active site residue E121 of E. coli L-aspartate oxidase. Biochimie. 92, 1335–1342. [DOI] [PubMed] [Google Scholar]
- Thakker C, et al. , 2011. Heterologous pyc gene expression under various natural and engineered promoters in Escherichia coli for improved succinate production. Journal of biotechnology. 155, 236–243. [DOI] [PubMed] [Google Scholar]
- Tritz GJ, Chandler JL, 1973. Recognition of a gene involved in the regulation of nicotinamide adenine dinucleotide biosynthesis. Journal of bacteriology. 114, 128–136. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Wang Y, et al. , 1992. Site-directed mutagenesis of the phosphorylatable serine (Ser8) in C4 phosphoenolpyruvate carboxylase from sorghum. The effect of negative charge at position 8. Journal of Biological Chemistry. 267, 16759–16762. [PubMed] [Google Scholar]
- Wong MS, et al. , 2008. Reduction of acetate accumulation in Escherichia coli cultures for increased recombinant protein production. Metabolic engineering. 10, 97–108. [DOI] [PubMed] [Google Scholar]
- Woolston BM, et al. , 2013. Metabolic engineering: past and future. Annual review of chemical and biomolecular engineering. 4, 259–288. [DOI] [PubMed] [Google Scholar]
- Zhao L, et al. , 2020. Expression regulation of multiple key genes to improve l-threonine in Escherichia coli. Microbial Cell Factories. 19, 1–23. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Zhu F, et al. , 2020. Improved succinate production from galactose-rich feedstocks by engineered Escherichia coli under anaerobic conditions. Biotechnology and Bioengineering. [DOI] [PubMed] [Google Scholar]
- Zhu F, et al. , 2018. Metabolic engineering of Escherichia coli to produce succinate from soybean hydrolysate under anaerobic conditions. Biotechnology and bioengineering. 115, 1743–1754. [DOI] [PubMed] [Google Scholar]
Associated Data
This section collects any data citations, data availability statements, or supplementary materials included in this article.