

Abstract
Venous leg ulcers, diabetic foot ulcers, and pressure ulcers are complex chronic wounds with multifactorial etiologies that are associated with high patient morbidity and mortality. Despite considerable progress in deciphering the pathologies of chronic wounds using “omics” approaches, considerable gaps in knowledge remain, and current therapies are often not efficacious. We provide a comprehensive overview of current understanding of the molecular mechanisms that impair healing and current knowledge on cell-specific dysregulation including keratinocytes, fibroblasts, immune cells, endothelial cells and their contributions to impaired reepithelialization, inflammation, angiogenesis, and tissue remodeling that characterize chronic wounds. We also provide a rationale for further elucidation of ulcer-specific pathologic processes that can be therapeutically targeted to shift chronic nonhealing to acute healing wounds.
Chronic nonhealing wounds are the result of a failure of the tissue repair process to reestablish structurally and functionally intact cutaneous barrier and are frequently associated with underlying conditions that include vascular disease, diabetes, and aging (Eming et al. 2014). Although differing in etiology, chronic wounds—including venous leg ulcers (VLUs), diabetic foot ulcers (DFUs), and pressure ulcers (PUs)—share an immense negative impact due to high incidences, associated complications, and frequent recurrences (Armstrong et al. 2017; Petersen et al. 2020). In addition, chronic wounds are reaching epidemic proportions worldwide. It has been estimated that up to 2% of the world population will develop a chronic wound, with 6.5 million patients affected in the United States alone (Richmond et al. 2013; Eming et al. 2014; Schneider et al. 2021). Treatment of chronic wounds has become a tremendous economic burden incurring costs of approximately $20–$25 billion annually in the United States (Schneider et al. 2021). Despite the high mortality rate and severity of chronic wounds, the mechanisms that result in their development remain partially understood, and available treatment options are often not fully efficacious in restoring healing. Several factors have played a role in limiting research on chronic wounds and hindered the development of new therapies, including a lack of animal models that fully recapitulate the human condition and complexity of a heterogeneous patient population with multifactorial ulcer etiologies (Gordilloet al. 2013; Eming et al. 2014; Elliot et al. 2018; Pastar et al. 2018). Recent advances in “omics” approaches using human tissue and various technologies including RNA sequencing (RNA-seq), single-cell RNA-seq, spatial transcriptomics, proteomics, metabolomics, and microbiome/metagenomics have deepened broad understanding of chronic wound pathology at the cellular, molecular, pathway levels, and host–pathogen interfaces (Pastar et al. 2012; Gardner et al. 2013; Fadini et al. 2014; Kalan et al. 2016; Loesche et al. 2017; Stone et al. 2017, 2020b; Ramirez et al. 2018; Cavassan et al. 2019; Kalan et al. 2019; Januszyk et al. 2020; Sawaya et al. 2020; Theocharidis et al. 2020, 2022). These technologies together with accessibility of patients’ biomaterials, including ulcer debridement tissue, wound fluid, and swabs, provide the opportunity to concurrently analyze multiple deregulated targets and thereby identify pathways responsible for impaired healing in patient samples (Fig. 1). Furthermore, comparative “omics” analyses have been successfully employed to elucidate unique features that distinguish chronic from physiologically healing wounds, leading to identification of specific molecular signatures for nonhealing ulcers and identification of novel targets for therapeutic intervention (Ramirez et al. 2018; Sawaya et al. 2020; Stone et al. 2020b; Theocharidis et al. 2022). Here, we outline the current knowledge and the most recent findings focused on deciphering wound healing impairment at the molecular and cellular levels in patients affected with chronic wounds. These mechanisms involve the major structural and resident skin cells: keratinocytes, immune cells, fibroblasts, and endothelial cells. Their roles in healing inhibition are reviewed, providing a rationale for approaching future development of advanced therapeutics.
Figure 1.
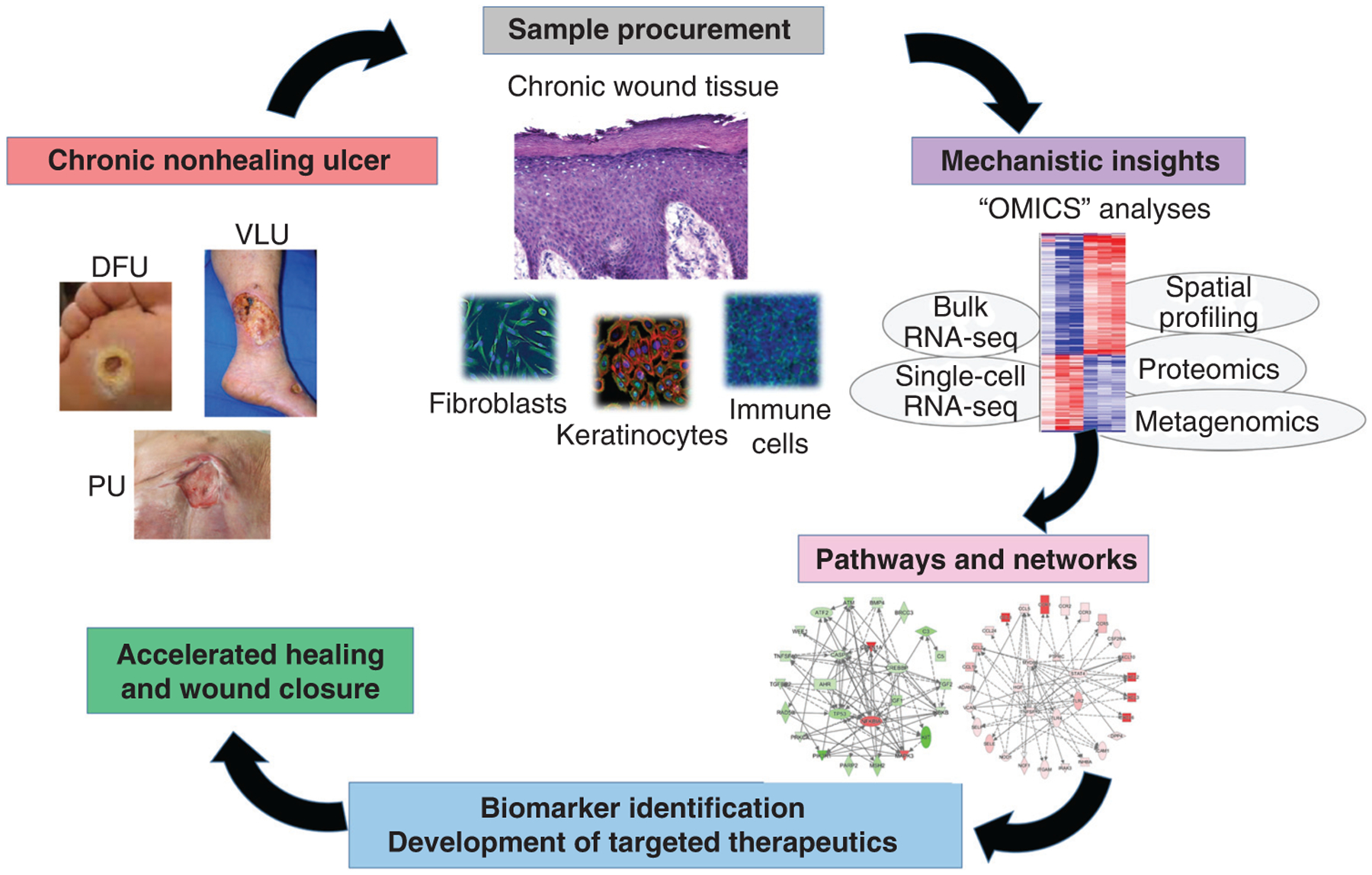
“Omics” approaches to therapeutic targeting of chronic ulcers. Comparative “omics” and bioinformatics analyses can be used to decipher pathophysiologic mechanisms that drive chronic wound pathology and to identify biomarkers and novel therapies likely to heal chronic wounds. (DFU) Diabetic foot ulcer, (VLU) venous leg ulcer, (PU) pressure ulcer.
DEREGULATION OF KERATINOCYTE DIFFERENTIATION, MIGRATION, AND PROLIFERATION—HALLMARKS OF THE NONHEALING WOUND PHENOTYPE
Keratinocytes, a major cellular component of the epidermis, play several critical roles in the wound healing process and are among the first responders to injury (Pastar et al. 2008, 2014). Once the skin is wounded, keratinocytes undergo activation, with the goal to migrate, proliferate, and differentiate within the wound bed and ultimately restore the epidermal barrier (Tomic-Canic et al. 1998; Freedberg et al. 2001; Pastar et al. 2014). Keratinocytes secrete a plethora of cytokines and growth factors; these, in turn, recruit and coordinate other cell types involved in wound healing, stimulate matrix formation, and promote angiogenesis (Pastar et al. 2008; Barrientos et al. 2014). In contrast to tightly controlled keratinocyte activation during physiological wound healing, the reepithelialization process is ineffective in chronic ulcers due to keratinocyte hyperproliferation, poor migration, and deregulated differentiation (Table 1). Contrary to normal skin, where proliferating keratinocytes reside exclusively in the basal layer (Morasso and Tomic-Canic 2005), hyperproliferative keratinocytes in chronic wounds continue to divide throughout basal and suprabasal layers of the epidermis (Stojadinovic et al. 2005). One of the first studies using gene expression profiling of chronic wound tissue pinpointed incomplete activation and differentiation of keratinocytes and their contribution to parakeratosis and hyper-keratosis (Stojadinovic et al. 2008). Expression of early keratinocyte differentiation markers, keratins 1 and 10 (K1 and K10), was suppressed at the nonhealing edge of VLU, unlike the late differentiation markers involving involucrin, transglutaminase 1, and subset of small proline-rich proteins (SPRR1A, SPRR1B, SPRR2B, SPRR3), which were up-regulated in chronic VLU (Stojadinovic et al. 2008). This dysregulation of the differentiation process points toward impaired barrier, despite the formation of hyper-keratotic cornified layer. Interestingly, keratinocytes at the nonhealing edge of VLU exhibit a gene signature that reflects high mitotic activity, with several crucial checkpoint control proteins down-regulated (such as retinoblastoma [Rb], p107, and p130), and with up-regulation of cell-cycle genes (cyclin B1, cyclin D2, cyclin A2, cyclin F, cyclin M4, and cell division cycle 2). Deregulated expression of cell-cycle-associated genes implies a loss of cell-cycle control, and together with the suppression of genes involved in regulation of an epidermal stem cells niche (Stojadinovic et al. 2014), contributes to the epidermal hyperproliferation evident at the VLU wound edge (Stojadinovic et al. 2008).
Table 1.
Overview of representative findings focused on understanding cellular responses involved in reepithelialization in acute and chronic wounds (CWs)
| Processes and cells | Acute wound | Chronic wound | Type of CWs | References |
|---|---|---|---|---|
| Epithelialization | Keratinocyte activation, regulated migration, proliferation, and differentiation | Deregulation of proliferation, differentiation, and migration | VLU DFU PU |
Eming et al. 2014; Pastar et al. 2014 |
| Keratinocyte proliferation | Tightly regulated during acute wound healing; proliferating keratinocytes reside in the basal layer | Hyperproliferative keratinocytes in the upper epidermal layers due to nuclear presence of β-catenin and c-myc | DFU VLU |
Morasso and Tomic-Canic 2005; Stojadinovic et al. 2005, 2008, 2014; Jozic et al. 2017; Stone et al. 2017 |
| Keratinocyte migration | Stimulated epidermal migration and K6/16 activation | Inhibition of keratinocyte migration though multiple signaling pathways including GR and WNT/β- catenin | DFU VLU |
Stojadinovic et al. 2005; Vukelic et al. 2010; Jozic et al. 2017, 2019, 2021; Sawaya et al. 2018; Marjanovic et al. 2022 |
| Keratinocyte differentiation | Activated during barrier restoration | Suppression of keratinocyte differentiation markers (K1 and K10) | VLU | Stojadinovic et al. 2008 |
| Epidermal stem cells (ESCs) | ESCs contribute to epidermal homeostasis and wound closure | Deregulation and deprivation of ESC niche | VLU | Morasso and Tomic-Canic 2005; Stojadinovic et al. 2014 |
| Growth factors signaling | Coordinated action of growth factors and receptors | Dysfunctional EGF and TGF-β signaling | DFU VLU PU |
Brem et al. 2007; Barrientos et al. 2008; Pastar et al, 2010; Jozic et al. 2019 |
| Keratinocyte immunity | Tightly regulated TLR response and production of proinflammatory cytokines and chemokines | Deregulation due to nuclear TLR3, suppression of PDL1, accumulated DNA breaks, and enriched MHC-II | DFU VLU PU |
Pastar et al. 2014; Ramirez et al. 2018; Li et al. 2021, 2022a; Kuai et al. 2022 |
| Antimicrobial response | Increased levels of AMP contribute to response against extracellular and intracellular pathogens | Increased levels of AMPs against extracellular bacteria, suppression of P-2 against intracellular pathogens | DFU | Thorey et al. 2001; Dressel et al. 2010; Sawaya et al. 2020; Pastar et al. 2021c |
(VLU) Venous leg ulcer, (DFU) diabetic foot ulcer, (PU) pressure ulcer, (GR) glucocorticoid receptor, (EGF) epidermal growth factor,
(TGF-β) transforming growth factor β, (TLR) Toll-like receptor, (MHC-II) major histocompatibility complex class II, (AMP) antimicrobial peptide.
The hyperproliferative and nonmigratory phenotype of keratinocytes at the nonhealing edge is also characterized by nuclear presence and overexpression of β-catenin and protooncogene c-myc (Waikelet al. 2001; Stojadinovic et al. 2005, 2014; Lindley et al. 2016; Stone et al. 2017). In contrast to membranous and cytoplasmic localization in normal skin, β-catenin is nuclear in keratinocytes at the nonhealing wound edge, leading to inhibition of keratinocyte migration (Stojadinovic et al. 2005, 2014; Stone et al. 2017). β-Catenin-mediated inhibition of healing is driven by blockade of the epidermal growth factor (EGF) response, inducing c-myc expression and suppression of indispensable cytoskeletal components for epidermal migration K6/K16 through cytoplasmic glucocorticoid receptor (GR) activation (Stojadinovic et al. 2005).
Molecular mechanisms of β-catenin activation in chronic wound keratinocytes also involve membranous glucocorticoid receptor (mbGR) (Jozic et al. 2017). While glucocorticoids are known as potent inhibitors of reepithelialization acting through the phosphorylation and nuclearization of GRs (Vukelic et al. 2010; Jozic et al. 2017), mbGR suppresses keratinocyte migration and wound healing by activating a Wnt-like phospholipase/protein kinase C signaling cascade leading to atypical activation and nuclear localization of β-catenin and c-myc (Jozic et al. 2017). Cumulatively, these findings generated from patients’ tissue resulted in the current clinical trial evaluating nuclear c-myc and p-GR as predictive and diagnostic tissue biomarkers for healing outcomes in DFUs (ClinicalTrials.gov 2020). Of note, the inhibitory effects of glucocorticoids can be therapeutically reversed by topically applied statins, which inhibit epidermal cortisol synthesis and subsequent activation of GRs (Sawaya et al. 2018). In addition, topical mevastatin-induced expression of noncoding RNA Gas5 resulted in suppression of c-myc and accelerated wound closure (Sawaya et al. 2018). Recent work has identified an additional mechanism of anti-healing activity of glucocorticoids via interaction of GR with Caveolin 1 (Cav1), a key scaffolding component of plasma membrane caveolae. Cav1 interacts with and sequesters mbGR and EGFR in keratinocytes to inhibit migration and wound closure in both DFU and VLU (Jozic et al. 2019). Cav1 is significantly up-regulated in nonhealing ulcers, in contrast to physiological healing wounds where it is markedly suppressed (Jozic et al. 2019). The Cav1-mediated sequestration of EGFR together with its mislocalization from membrane to cytoplasm (Brem et al. 2007; Sawaya et al. 2019) can account for diminished EGF signaling in VLUs and DFUs, and lack of response to topically applied EGF (Falanga et al. 1992). Jozic et al. (2021) have also shown that Cav1 induction in keratinocytes at the nonhealing wound edge keeps the cytoskeletal machinery involved in cell migration in the stalled state through deregulation of the cytoskeleton components including members of Rho family of GTPases, Cdc42, and RhoA and their inactivator ArhGAP35, while pharmacological inhibition or genetic depletion of Cav1 accelerated wound closure.
Up-regulation of the pro-oncogene c-myc and activation of WNT/β-catenin signaling is also a common event leading to the development of many cancers and can contribute to the known predisposition of chronic wounds to tumor formation (Gabay et al. 2014; Zhan et al. 2017). Interestingly, a recent study identified up-regulation of specific post-transcriptional gene expression regulator microRNA (miR), miR193b-3p, as a tumor-suppressive mechanism in DFUs via down-regulation of Kirsten rat sarcoma viral oncogene (KRAS) signaling (Marjanovic et al. 2022). While miR193b-3p contributes to low tumor incidence in DFUs, it also acts as a master inhibitor of cellular migration and epithelialization by disrupting stress fiber formation through suppression of RhoA activity. Up-regulation of miR193b-3p was a unique feature of DFUs and not regulated in VLUs, which develop squamous cell carcinomas far more frequently (Trent and Kirsner 2003; Marjanovic et al. 2022).
Another feature of the DFU keratinocytes is their decreased DNA damage repair response. Ramirez et al. showed accumulated DNA breaks in DFU keratinocytes as a result of suppression of the genes involved in DNA repair (MutS Homolog 2-MSH2, Double Strand Break Repair Protein-RAD50, WEE1 G2 Checkpoint Kinase, Tumor Protein P53, and Receptor Tyrosine Kinase-KIT) (Ramirez et al. 2018). The suppression of DNA repair is driven by miR15b-5p, induced by common DFU pathogen Staphylococcus aureus, which targets genes involved in the DNA damage response (Ramirez et al. 2018). Additional miRs also contribute to the nonhealing phenotype of chronic wounds (detailed in Li et al. 2022b).
Lack of keratinocyte migration in conjunction with the persistent low-grade inflammation in DFUs may be partially attributed to deregulated expression of immune inhibitory receptor ligand Programmed death-ligand 1 (PDL1) (Kuai et al. 2022). Transmembrane protein PDL1 is suppressed in DFU epidermis when compared to normal skin, contributing to delayed wound closure (Kuai et al. 2022). Moreover, treatment of murine diabetic wounds with PDL1 resulted in improved reepithelialization and modulation of prolonged inflammation (Kuai et al. 2022).
DEREGULATION OF KERATINOCYTE IMMUNE FUNCTIONS
A less-studied role of keratinocytes in chronic wounds is their immune functions. Keratinocytes communicate with immune cells through numerous cytokines, chemokines, and extracellular vesicles (Pastar et al. 2014; McBride et al. 2017). Epidermal keratinocytes directly interact with T cells via antigen presentation and produce antimicrobial peptides (AMPs) that inhibit the growth of wound pathogens (Piipponen et al. 2020). However, keratinocyte immune functions are deregulated in chronic wounds. Keratinocytes from VLUs, DFUs, and PUs are characterized by increased levels and nuclear localization of Toll-like receptor 3 (TLR3) (Li et al. 2021). Furthermore, major histocompatibility complex class II (MHC-II)-expressing subgroup of keratinocytes is significantly enriched in PUs when compared to either acute wounds or normal skin (Li et al. 2022a). This is attributed to IFN-γ in the wound fluid from PUs, resulting in induction of MHC-II in keratinocytes, which in turn attenuates autologous T cell activation (Li et al. 2022a). AMPs, human β defensin 2 (hBD-2), psoriasin, S100A8, and S100A9 are induced in the epidermis of chronic VLUs compared to skin from healthy individuals (Thorey et al. 2001; Dressel et al. 2010), whereas S100A7 and DEFB4A/DEFB4B are up-regulated in both DFUs and acute human wounds (Sawaya et al. 2020).
In contrast to up-regulation of AMPs targeting extracellular bacteria in chronic wounds, recent studies have identified suppression of a unique antimicrobial effector Perforin 2 (P-2), targeting intracellular pathogens in chronic DFUs (Pastar et al. 2021c). While the intracellular life cycle of S. aureus is not completely understood, P-2 is shown to be essential for control and elimination of intracellular bacteria in both professional and nonprofessional phagocytic skin cells and is induced during physiological wound healing (Strbo et al. 2019). Moreover, pathogenic S. aureus can invade and persist inside the keratinocytes to contribute to wound chronicity (Pastar et al. 2021c). P-2 was found suppressed in DFU epidermis, which was associated with accumulation of intracellular S. aureus, induction of AIM-2 inflammasome, and pyroptosis that collectively contributed to healing inhibition and unresolved inflammation in DFUs (Pastar et al. 2021c). Unlike pathogenic S. aureus responsible for P-2 suppression (Strbo et al. 2019), commensal Staphylococcus epidermidis was able to induce P-2 in keratinocytes and γδ T cells (Pastar et al. 2020), suggesting potential of therapeutic targeting of P-2 to prevent or reverse persistent intracellular chronic wound infections. Taken together, these studies underscore the importance of intracellular bacterial niche in chronic wound keratinocytes and their contribution to inhibition of wound healing in addition to the well described role of the extracellular microbiome (see White and Grice 2022).
While specific processes of hyperproliferation and impaired migration and differentiation, together with dysregulation of associated pathways and regulators are characteristics of all types of chronic wounds, the recent findings on up-regulation of tumor suppressor miR-193b-3p unique for DFUs, also suggest that pathology of keratinocytes is ulcer-type specific (Marjanovic et al. 2022). Future use of single-cell and spatial “omics” methods is required to shed more light on pathways and therapeutic approaches targeting keratinocytes and reepithelialization in specific types of chronic wounds.
INFLAMMATION IN CHRONIC WOUNDS—A FRIEND, A FOE AND MORE
One key characteristic of chronic nonhealing wounds is unresolved inflammation. It has been postulated that the chronic wound is “stuck in a chronic inflammatory state” that does not progress. Recent studies, however, contributed to a paradigm shift, showing that imbalance of proinflammatory and anti-inflammatory responses along with impaired function, and signaling of inflammatory cells traps the wound in a dysregulated inflammatory state that fails to reach levels seen in acute wounds. In turn, this ineffective inflammatory state fails to stimulate progression of healing to reepithelization and remodeling phases and promote successful closure (Eming et al. 2014; Stone et al. 2017; Ramirez et al. 2018; Theocharidis et al. 2022). Comparative transcriptomic analyses of chronic wounds and acute physiological wounds have demonstrated suboptimal levels of inflammatory signaling in nonhealing VLUs and DFUs (Stone et al. 2017; Sawaya et al. 2019, 2020). Promoting inflammatory profile to the one resembling an acute wound has been demonstrated to clinically correlate with reversal from a nonhealing to healing phenotype in VLUs as a response to treatment with a bioengineered human skin equivalent (Stone et al. 2017). Below we review current knowledge of this dysregulated inflammatory process by delineating the role of each immune cell and their contribution to the overall chronic wound pathogenesis (Table 2).
Table 2.
Summary of representative findings from studies focusing on understanding the role of inflammation and its cellular components in acute and chronic wounds (CWs)
| Processes and cells | Acute wound | Chronic wound | Type of CW | References |
|---|---|---|---|---|
| Inflammation | High levels of controlled inflammatory cell activity | Low, persistent level not reaching levels of acute wounds | DFU VLU |
Stone et al. 2017, 2020b; Sawaya et al. 2020; Theocharidis et al. 2022 |
| Neutrophils | Present during early inflammatory phase; low levels of controlled NETosis | Increased NETosis, neutrophil numbers decreased at ulcer edge and increased in ulcer bed | DFU VLU PU |
Diegelmann 2003; Liu et al. 2019; Sawaya et al. 2020, 2022; Yang et al. 2020; Theocharidis et al. 2022 |
| Macrophages | Controlled M1-M2 transition assures resolution of inflammation | Increased level of M1 macrophages associated with healing ulcers | DFU | Theocharidis et al. 2022 |
| Mast cells | Controlled degranulation in early phases; low numbers | Increased numbers at the wound edge and degranulation promoting ECM degradation | VLU DFU |
Huttunen et al. 2000; Abd-El-Aleem et al. 2005; Theocharidis et al. 2022 |
| T cells | T cell recruitment with high CD4+ proportion; epidermal T cell activation | Shift in T cell populations; increased naive T cell proportion correlates with healing DFUs; higher levels of natural killer T (NKT) cells in nonhealing DFUs; decreased epidermal T cell activation | DFU VLU |
Toulon et al. 2009, Stone et al. 2017; Moura et al. 2019; Theocharidis et al. 2022 |
| Langerhans cells | Increased number during early inflammatory phase | Decreased number associated with nonhealing outcomes | DFU | Stojadinovic et al. 2013 |
(DFU) Diabetic foot ulcer, (VLU) venous leg ulcer, (PU) pressure ulcer.
One of the first responders to the injury site are mast cells. Mast cell recruitment is facilitated by keratinocyte-released mediators (Trautmann et al. 2000), and once present, they contribute to clot stabilization, endothelial permeability, vasodilation, neoangiogenesis, and fibrogenesis. Mast cells degranulate and release vasoactive and proinflammatory mediators including histamine, tumor necrosis factor α (TNF-α), interleukin 8 (IL-8), and proteases that facilitate the influx of neutrophils and other immune cells to the wound site. They also stimulate proinflammatory mediator production by keratinocytes and fibroblasts (Kohda et al. 2002; Giustizieri et al. 2004; Nishida et al. 2019), and are essential to bacterial killing in infected wounds (Zimmermann et al. 2019).
However, studies have observed differential mast cell mediators and location in chronic VLUs as compared with normal skin and acute wounds, suggesting that their altered function contributes to impaired wound healing. Immunohistochemistry of perilesional VLU skin identified increased mast cells and TNF-α and tryptase expression, and identified increased chymase expression, a protease that degrades the extracellular matrix and damages tissue in the VLU bed (Huttunen et al. 2000; Abd-El-Aleem et al. 2005). At elevated concentrations in vitro, mast cells and their mediators inhibit keratinocyte activity and epithelial outgrowth (Huttunen et al. 2001), which may also contribute to delayed wound healing in vivo. Increased degranulation of mast cells also contributes to abnormal wound healing in DFUs (Theocharidis et al. 2022). Patients with diabetes have increased numbers of degranulated mast cells even in unwounded skin (Dong et al. 2020; Tellechea et al. 2020). Pretreatment with a mast cell degranulation inhibitor attenuated wound-healing impairment in a diabetic mouse model and shifted macrophages to a regenerative M2 phenotype (Tellechea et al. 2016, 2020). Further studies are needed to more specifically define mast cell contributions to prolonged inflammation and impaired wound healing in VLUs and PUs.
A specialized subset of epidermal dendritic cells, Langerhans cells (LCs), represents a potent group of antigen-presenting cells known as the first-line defenders in skin (Gallo and Gallucci 2013). LCs are an important immune component during the early phase of physiological acute wound healing (Gallo and Gallucci 2013; Stojadinovic et al. 2013). Decreased numbers of LCs are associated with nonhealing DFUs, while the epidermis of healing DFUs has been shown to be repopulated with higher numbers of LCs (Stojadinovic et al. 2013).
During the early inflammatory phase, neutrophils play an important role in the first-line defense of clearing foreign debris, necrotic wound tissue, and bacteria, and promote inflammation via cytokines and chemokines including TNF-α, IL-1β, IL-6, and C-X-C motif chemokine ligand (CXCL8) (Pittman and Kubes 2013). The levels of neutrophils in chronic wounds are previously thought to be elevated compared to acute wounds, as demonstrated by increased neutrophil infiltration and its marker myeloperoxidase (MPO) in chronic VLUs and PUs (Diegelmann 2003; Eming et al. 2010). However, further studies showed that neutrophil presence varies based on wound etiology and location, with elevated levels in the wound bed due to prolonged inflammation attenuating neutrophil apoptosis or phagocytosis (Theocharidis et al. 2022) and decreased levels at the wound edge reflecting impaired migratory ability of immune cells (Sawaya et al. 2020), or increased cell death from release of neutrophil extracellular traps (NETs) (Wong et al. 2015; Sawaya et al. 2022). NETs, which are web-like matrices lined with neutrophil-produced cytotoxic proteins, function to either directly kill or facilitate phagocytosis of extracellular pathogens. However, NET release, known as NETosis, can damage the surrounding tissue and contribute to prolonged inflammation (Fadini et al. 2016). At the DFU ulcer edge, decreased neutrophil recruitment has been linked to deregulation of transcriptional networks involving Forkhead Box M1 (FOXM1) and Signal Transducer and Activator of Transcription 3 (STAT3), which activate and promote survival of immune cells (Sawaya et al. 2020). Suppression of triggering receptor expressed on myeloid cells 1 (TREM1), an upstream regulator of FOXM1 and negative regulator of NET formation, was found to be responsible for dysfunctional neutrophils, accumulated NETs, and increased reactive oxygen species (ROS) in DFUs (AP Sawaya et al. in prep.). S. aureus biofilms have been shown to evade neutrophil-mediated killing by eliciting NETosis that is ineffective at clearing biofilm (Bhattacharya et al. 2018), suggesting that wound infection modulates NET function, and increased NETosis does not necessarily translate to antimicrobial benefits. In diabetes, neutrophils are primed to undergo NETosis (Wong et al. 2015), and NET overproduction has been reported in DFU patients as correlating with infection and wound severity (Liu et al. 2019; Yang et al. 2020). While the deregulation of neutrophils and the role of NETosis have been characterized in DFUs, they have not yet been well-studied in other types of chronic ulcers.
As monocytes migrate to the wound and mature into macrophages, they play a key role in both initiation and resolution of the inflammatory response by clearing apoptotic neutrophils and restoring tissue integrity for wound closure. In physiological wound healing, the transition from the inflammatory to proliferative phase is facilitated by a shift in macrophage polarization from classically activated M1 phenotype, characterized by proinflammatory cytokines, to alternatively activated M2 phenotype, characterized by markers of inflammatory resolution and initiation of tissue repair (Lawrence and Natoli 2011; Murray et al. 2014; Nassiri et al. 2015). However, in-depth transcriptomic studies identified multiple unique subtypes of macrophages confirming a broad spectrum of macrophage activation programs beyond the initial concept of polarization (M1 versus M2) (Xue et al. 2014), indicating complexity of macrophage phenotypes in wound healing. Studies focusing on M1-M2 polarization have shown that transition to the M2 phenotype can be induced by efferocytosis, a process in which macrophages phagocytose apoptotic neutrophils (Elliott et al. 2017), as well as by various signals from regulatory T cells and epigenetic modifications (Nosbaum et al. 2016; Pastar et al. 2021b). Until recently, the accepted paradigm proposed that in nonhealing wounds, overexpression of proinflammatory cytokines and impaired clearance of apoptotic neutrophils led to deregulated polarization and activation of wound macrophages with a predominant M1 phenotype (Nassiri et al. 2015). However, more recent studies employing single-cell transcriptomics in tissue from clinically monitored DFU patients identified increased numbers and M1 macrophage polarization in the healing DFUs, in contrast to decreased M1 macrophages in nonhealing DFUs (Theocharidis et al. 2022). It is important to note that excessive activity of M2 macrophages in wound healing may lead to fibrotic scarring via activation of the Wnt/β-catenin pathway (Gay et al. 2020). Taken together, balanced regulation of M1-M2 polarization is indispensable for proper wound healing, although what constitutes a successful macrophage wound healing response likely varies by ulcer subtype.
The role of adaptive immunity in chronic wounds has not been extensively investigated; however, preliminary evidence suggests T cells also take part in maintaining a dysfunctional proinflammatory profile for nonhealing ulcers (Theocharidis et al. 2022). Processes of T cell differentiation and migration are diminished at the wound edge of nonhealing VLUs (Stone et al. 2017), resulting in fewer numbers of T cells in chronic wounds in an impaired or unresponsive state, unable to secrete factors such as insulin growth factor 1 (IGF-1) and IL-2 that are produced in a normal wound healing response (Toulon et al. 2009; Xu et al. 2017). Studies have also demonstrated that infection can induce strong T cell stimulation that reduces T cell receptor (TCR) repertoire diversity, thereby affecting the immune response to subsequent infections by different pathogens. Patients with diabetes have decreased serum levels of naive T cells and increased levels of effector T cells, and effector T cells accumulate and display a significantly reduced TCR-β repertoire diversity in the serum (Moura et al. 2017, 2019) and tissue (Loots et al. 1998) of patients affected with DFUs and VLUs, respectively. Effector T cells are major producers of interferon γ (IFN-γ) and TNF-α inflammatory cytokines that in turn promote naive T cell activation and differentiation (Moura et al. 2017; Mehta et al. 2018). The ratio of T cell phenotype (effector/naive) and level of TCR repertoire diversity in serum have been proposed as predictive biomarkers of DFU healing outcome (Moura et al. 2019). A recent study using single-cell transcriptomics identified higher proportions of naive and early differentiated progenitor T-lymphocytes in healing DFUs, while nonhealing DFUs had higher proportions of cytotoxic natural killer T (NKT) cells, indicating a shift in T cell subpopulations that correlates with healing outcomes (Theocharidis et al. 2022). Whereas the “omics” technologies have deepened understanding of specific inflammatory cell subtypes in chronic DFU pathology, future longitudinal studies that profile tissues from expanded cohorts of all types of chronic wounds are required to elucidate dynamic changes in the inflammatory cell milieu and their contributions to clinical outcomes and response to therapies in patients.
ALTERED FIBROBLAST FUNCTION IN CHRONIC WOUNDS
Fibroblasts are the main manufacturers of the extracellular matrix (ECM) and granulation tissue during physiological wound healing. The transformation from quiescent dermal fibroblast to activated myofibroblast is crucial for successful wound healing and contraction, recruitment of immune cells, and angiogenesis (Li and Wang 2011; Hinz 2016; Arif et al. 2021). Acute wound fibroblasts are astoundingly heterogeneous in terms of their developmental origin, location in tissue, gene expression, and ECM secretion as also detailed in this collection (please see Ganier et al. 2022 and Jiang et al. 2022), and the most recent findings identified fibroblast heterogeneity in chronic wounds as well (Theocharidis et al. 2022).
Although VLUs, DFUs, and PUs differ by etiology, fibroblast dysfunction and phenotypic cellular aging remain unifying themes (Table 3; Brem et al. 2008; Eming et al. 2014). Fibroblasts isolated from nonhealing ulcers demonstrate severely attenuated migratory capacity, mitogenic response, ECM deposition and proliferation, and increased apoptosis (Ågren et al. 1999; Brem et al. 2008; Liang et al. 2016; Maione et al. 2016). Aged wound fibroblasts divide and migrate less, produce less ECM, and demonstrate up-regulated MMP activity compared to their younger counterparts, which leads to insufficient tension and granulation tissue formation in the wound bed (Ashcroft et al. 1997; Fujiwara et al. 2019; Mahmoudi et al. 2019; Ding et al. 2021). Premature aging of PU fibroblasts is attributed to high oxidative stress caused by a deficiency of the chemokines necessary to overcome the persistent inflammation (Wall et al. 2008; Fujiwara et al. 2019).
Table 3.
Overview of representative findings from studies focused on understanding cellular responses involved in extracellular matrix (ECM) formation and angiogenesis in acute and chronic wounds (CWs)
| Processes and cells | Acute wound | Chronic wound | Type of CW | References |
|---|---|---|---|---|
| ECM formation and fibroblasts | ||||
| Fibroblast proliferation and migration | Similar replicative capacity as fibroblasts isolated from healthy intact skin | Decreased proliferation, migration, decreased mitogenic response, increased senescence | DFU VLU |
Ågren et al. 1999; Brem et al. 2008; Berberich et al. 2020 |
| MMPs and TIMPs | Small initial increase, with resolution of ECM remodeling | Significantly greater number of proteases in chronic wounds, low levels of TIMPs | DFU VLU PU |
Grinnell and Zhu 1996; Cook et al. 2000; Liu et al. 2009; Singh et al. 2014; Lindley et al. 2016; Jindatanmanusan et al. 2018; Trøstrup et al. 2018; Stacey et al. 2019 |
| Growth factors | Coordinated action of growth factors and receptors | Increased TGF-β and IGF; EGF degradation; dysfunctional TGF-β | DFU VLU PU |
Barrientos et al. 2008; Brem et al. 2008; Berberich et al. 2020 |
| Lysosomes | N/A | Accumulation of lysosomal proteins | VLU | Berberich et al. 2020 |
| Fibrosis | Minimal, tightly regulated ECM remodeling | Enriched fibrotic and fibrogenic pathways and associated gene networks | VLU | Stone et al. 2020a,b |
| Fibronectin (FN), chondroitin sulfate (CS), and tenascin (TN) | Abundant expression early in healing process, return to nonwounded levels by 12 mo, stable FN | Prolonged (>12 mo) presence of FN, CS, and TN, increased degradation of FN | DFU VLU |
Rao et al. 1995; Loots et al. 1998; Stone et al. 2020b |
| Angiogenesis | ||||
| Angiogenesis stimulators/inhibitors | Increased angiogenic stimulators and decreased antiangiogenic proteins | Increased antiangiogenic proteins (annexin V) in chronic wounds and decreased or rapidly degraded angiogenic stimulators (VEGF) | DFU | Lauer et al. 2000; Romagnani et al. 2004; Edsberg et al. 2012; Krisp et al. 2013; Singh et al. 2017, 2022 |
| Bone marrow–derived stem cells and endothelial progenitors | Recruitment of stem cells and progenitors in response to wounding | Impaired recruitment of bone marrow–derived stem cells and endothelial progenitors | DFU VLU |
Gallagher et al. 2007; Liu and Velazquez 2008; Thom et al. 2016; Singh et al. 2022 |
| Pericapillary fibrin cuffs | Tortuous capillaries and fibrin cuffs absent | Extravasation of macromolecules and possible trapping of TGF-β | VLU | Leu et al. 1995 |
(DFU) Diabetic foot ulcer, (VLU) venous leg ulcer, (PU) pressure ulcer, (TIMP) tissue inhibitor of matrix metalloproteinase.
Related to aging, fibroblast senescence is also implicated in wound chronicity (Liang et al. 2016; Berlanga-Acosta et al. 2020; Wang and Shi 2020). Fibroblast senescence is beneficial in early phases of acute wound healing and in curbing fibrosis at healing resolution (Brockman et al. 1970; Jun and Lau 2010a,b; Pauty et al. 2021). However, inappropriate fibroblast senescence in VLUs and DFUs is associated with inability to heal (Stanley and Osler 2001; Harding et al. 2005; Wilkinson et al. 2019). Fibroblasts isolated from PUs also demonstrate increased replicative senescence with elevated expressions of plasmin, plasminogen activator inhibitor-1 (PAI-1), and TGF-β (Vande Berg et al. 2005), which is in line with findings that fibroblast senescence is induced by the deposition of a “senescence-promoting” ECM, including PAI-1 (Hiebert et al. 2018).
Chronic hypoxia is a known contributor to wound chronicity (Sheffield 1998; Eisenbud 2012). Hypoxia in the early stages of wound healing is beneficial in that it induces myofibroblast expression of genes such as transforming growth factor β (TGF-β) and collagen A1 (COLA1) (Falanga et al. 2002) and stimulates recruitment of peripheral stem cells to the wound (Brem and Tomic-Canic 2007; Gallagher et al. 2007). Increased levels of blood-borne stem cells and their intracellular hypoxia inducible factors (HIFs) are positively correlated with healing status (Thom et al. 2016). However, hypoxia impairs hydroxylation of proline and lysine during collagen synthesis, and consequently the maturation of procollagen and prolyl hydroxylase activity (Gordillo and Sen 2003; Tandara and Mustoe 2004; Thackham et al. 2008; Sen 2009; Chambers and Leaper 2011). As such, chronic hypoxia negatively affects fibroblast growth, activity, and TGF-β and EGF receptor expression (Falanga et al. 1994; Tandara and Mustoe 2004; Mustoe et al. 2006; Eisenbud 2012). However, restoring oxygen levels with hyperbaric therapy can result in increased growth and activation of myofibroblasts and increased granulation tissue deposition (Zhao et al. 1994; Kranke et al. 2015).
An altered wound microenvironment acts as another driver of fibroblast dysfunction and chronic wound formation. Hyperglycemia impairs proliferation, migration, and differentiation of DFU fibroblasts (DFUFs) and accelerates their senescence (Bian et al. 2020; Wan et al. 2021). Fibroblasts isolated from nonulcerated diabetic skin are strikingly similar to fibroblasts isolated from healthy foot skin in terms of their function and gene expression profiles, implying that hyperglycemia itself does not negatively impact fibroblasts prior to wounding (Ramirez et al. 2015). However, hyperglycemia induces a metabolic epigenetic memorys witch in fibroblasts that is maintained even when reexposed to normal glucose levels and may prime diabetic skin to ulcer formation (Park et al. 2014a). A unique population of fibroblasts has been identified in the ulcer beds of the DFU patients with healing wounds and is characterized by overexpression of matrix remodeling and inflammatory response genes (Theocharidis et al. 2022). In addition, fibroblast plasticity is dysregulated in chronic wounds due to aberrant activation of Notch1 signaling, suggesting that different subpopulations of fibroblasts may be responsible for the failure to achieve healing (Shao et al. 2020). Proteomic analyses of chronic wound fibroblasts identified two major groups of dysregulated processes. First, dysfunctional lysosomal capacity and protein turnover, which is commonly dysregulated in age-associated diseases, contributes to impaired cell motility and proliferation (Cellerino and Ori 2017; Berberich et al. 2020). Second, altered TGF-β activity causes aberrant myofibroblast contraction (Berberich et al. 2020). Dysregulation of TGF-β pathway and unresponsiveness of its receptor has been documented in all types of chronic wounds (Kim et al. 2003; Dalton et al. 2007; Pastar et al. 2010; Zhang et al. 2016; Brunner et al. 2021). Additionally, chronic wound fibroblasts have a diminished mitogenic response to growth factors found in the wound bed including platelet-derived growth factor BB (PDGF-BB), fibroblast growth factor (FGF), and EGF (Stanley et al. 1997; Ågren et al. 1999). Improper gap junction formation and subsequent pathologic spread of various apoptotic and inflammatory signals in chronic wounds is attributed to dermal up-regulation of connexins (Sutcliffe et al. 2015).
Reprogramming DFUFs has been explored as a promising therapeutic option for chronic wounds. In a series of studies, DFUFs from non-healing DFUs were first reprogrammed into induced pluripotent stem cells (iPSCs) to reacquire wound healing function and then reprogrammed back into fibroblasts (Takahashi et al. 2007; Gerami-Naini et al. 2016; Kashpur et al. 2019). Post-reprogrammed DFUFs reversed the impaired healing capacity seen in the original DFUFs, which was attributed to miR-mediated epigenetic mechanisms (Kashpur et al. 2019; Pastar et al. 2021a) revealing potential of iPSC-based therapeutic approach for chronic wounds.
IMPAIRED ANGIOGENESIS IN CHRONIC WOUNDS
Angiogenesis and vasculogenesis are integral components of wound healing, which function to increase immune cell recruitment, supply oxygen, and nutrientsto the hypermetabolic regenerating wound, as well as dispose of toxic metabolites and waste products (Eming et al. 2007). Impaired angiogenesis is associated with a poor nutritional supply state and tissue hypoxia that ultimately induces cellular death in chronic wounds (Table 3; Biswas et al. 2010; Eming et al. 2010; Krisp et al. 2013). Dysregulation of these pathways is implicated in chronic wound formation and in their failure to heal (Brem and Tomic-Canic 2007; Costa and Soares 2013; Okonkwo and DiPietro 2017).
Antiangiogenic proteins, such as myeloperoxidase, are expressed at higher levels in DFU versus acute physiological wounds. Conversely, angiogenic stimulators, including superoxide dismutase and angiogenin, exhibit decreased expression in DFUs (Krisp et al. 2013; Singh et al. 2022). Ischemia-induced microvascular endothelial cell damage contributes to slowed angiogenesis in VLUs, and hyperglycemia causes alterations in structure and function of both micro and macro vessels, in part through miR-200b induction (Leu et al. 1995; Chan et al. 2012; Singh et al. 2017; Haspula et al. 2019). Additionally, chronic wounds display an aberrant cleavage and destruction of growth factors and their receptors by increased protease activity, major factors include VEGF in DFUs and chemokine ligand 9in Pus (Lauer et al. 2000; Romagnani et al. 2004; Edsberg et al. 2012; Singh et al. 2017).
A major consequence of impaired angiogenesis in chronic wounds is inability to recruit pro-healing stem cells, including bone marrow–derived cells and endothelial progenitor cells (EPCs), to the site of injury (Gallagher et al. 2007; Thom et al. 2016; Singh et al. 2022). This recruitment must be synchronized by a repertoire of chemokines, which are exhausted in conditions that have compromised healing responses including human aging and diabetes (Gallagher et al. 2007; Barrientos et al. 2008; Pastar et al. 2010; Thom et al. 2016; Singh et al. 2022). Aberrant EPC behavior, cell numbers, recruitment, and transition to a proinflammatory state have been well documented in diabetic patients (Gallagher et al. 2007; Liu and Velazquez 2008). Common causes of EPC dysfunction include hyperglycemia, increased oxidative stress, chronic inflammation, and activation of NADPH oxidase (Drela et al. 2012; Kim et al. 2012; Yu et al. 2016), and similar themes of EPC dysfunction have been confirmed in murine models of diabetic wound healing (Gallagher et al. 2007; Albiero et al. 2011; Singh et al. 2022). Genetic studies have highlighted a nitric oxide synthase 1 adaptor protein (NOS1AP) variation in the population that is associated with impaired wound healing and attenuated stem cell mobilization in DFU patients (Margolis et al. 2017). Consequently, hyperbaric oxygen therapy is beneficial to wound healing by promoting EPC mobilization and recruitment in addition to its benefits in reversing hypoxia, as discussed above (Thom et al. 2011; Huang et al. 2020). A better understanding of the mechanisms that result in dysfunction of angiogenesis is of vital importance in the wound healing field especially with an increase in microvascular complications that accompanies the aging population affected with wound healing disorders.
IMBALANCE OF EXTRACELLULAR MATRIX (ECM) COMPONENTS IN CHRONIC WOUNDS
Chronic wounds are characterized by a dysfunctional ECM and altered protease activity (Monika et al. 2021). Matrix metalloproteinases (MMPs), and their regulators, tissue inhibitor of matrix metalloproteinase (TIMP), are enzymes that play an integral role in all stages of wound healing (McCarty and Percival 2013; Michopoulou and Rousselle 2015). While the tight control of MMP dynamics is crucial for proper wound healing, their perturbed activities including chronically elevated levels of MMPs, reduced levels of TIMPs, or altered ratios of the two, lead to impaired remodeling response and failure of wounds to heal (Cook et al. 2000; Lindley et al. 2016). Elevated MMP9 and a high MMP9/TIMP1 ratio has been proposed as a predictor of nonhealing ulcers (Liu et al. 2009; Singh et al. 2014; Jindatanmanusan et al. 2018; Trøstrup et al. 2018; Jones et al. 2019), whereas a high MMP1/TIMP1 ratio is associated with improved healing outcomes (Muller et al. 2008). MMP13 and granulocyte-macrophage colony-stimulating factor (GM-CSF) levels in wound fluid may be an important healing status biomarker specific for VLUs (Stacey et al. 2019). Interestingly, adding MMP inhibitors to chronic wound fluid only partially inhibits proteolytic activity, demonstrating that non-MMP proteinases also contribute to ECM destruction in chronic wounds (Trøstrup et al. 2018). In addition to increased collagen-matrix degradation, the chronic wound environment is characterized by up-regulated serine proteases that degrade important components of the ECM including fibronectin, dermatopontin, and important growth factors and receptors necessary for ECM remodeling and cell growth (Rao et al. 1995; Wlaschek et al. 1997; Lobmann et al. 2002; Barrientos et al. 2008; Buchstein et al. 2009; Eming et al. 2014; Krishnaswamy et al. 2014). For example, fibronectin (FN) mRNA expressed by fibroblasts is increased in chronic wounds; however, FN-rich ECMs are thin, defective, and unstable, which is likely due to increased ECM turnover caused by increased levels of elastase serine protease present in chronic wounds (Grinnell and Zhu 1996; Yager et al. 1997; Maione et al. 2016). In addition, lack of FN cell-surface receptor on keratinocytes prevents migration of epidermal keratinocytes in chronic wounds despite higher levels of FN expressed by fibroblasts (Ongenae et al. 2000). Human neutrophil elastase is increased in chronic wound tissue including chronic VLUs, PUs, and DFUs (Rogers et al. 1995; Eming et al. 2010; Wiegand et al. 2010). Increased neutrophil elastase in DFUs is also associated with infection and worsening wound severity (Fadini et al. 2016).
Chronic wounds, specifically VLUs, have also shown evidence of marked tissue fibrosis (Blumberg et al. 2012; Stone et al. 2020a,b). In nonhealing VLUs, prolonged ineffective inflammation has been associated with a sustained remodeling response and the development of a fibrotic ulcer (Stone et al. 2020b). Gene expression profiling of chronic nonhealing VLUs revealed enrichment of collagens and secreted matricellular proteins in combination with up-regulation of tenascin (TNC), osteopontin (SPP1), FN1, connective tissue and hepatocyte growth factors (CTGFs, HGFs), and plasminogen activator inhibitor 1 PAI-1 (Stone et al. 2020b). Profibrotic TGF-β signaling was also found enriched and activated in the ulcer bed, thus confirming the chronic VLU as a fibrotic condition. Further studies showed bioengineered bilayered cellular construct (BLCC) application decreased TGF-β1-induced gene expression with a concurrent increase in expression of the TGF-β inhibitor decorin. BLCC application also resulted in increased expression of MMP-8 and levels of MMP-activating zinc, which stimulated antifibrotic remodeling. Ultimately, these studies indicated a novel treatment approach of chronic wounds, preventing fibrosis as well as a new classification of VLU as a fibrotic disease (Stone et al. 2020a,b).
Together, these reports underscore the complex, temporospatial nature of ECM turnover within the wound microenvironment. They highlight the need for continued exploration into ECM dynamics in chronic wounds, reversal of fibrosis, the inhibition of proteases, and pathways that lead to their activation to develop effective therapies (McCarty and Percival 2013).
CONCLUSIONS AND FUTURE DIRECTIONS
Despite major recent advances in understanding pathophysiology of chronic wounds and molecular mechanisms of wound healing impairment, treatment options remain limited with only a handful of products FDA approved for efficacy. However, over 50% of DFUs and over 70% of VLUs fail to heal with standard of care therapies (Eming et al. 2014), and there is no efficacious therapy approved for PU treatment, emphasizing the urgent need for the development of novel therapeutic modalities.
A lack of validated preclinical models is one of the major challenges in the chronic wound healing field that hinders translation of therapies to clinical practice (Gordillo et al. 2013; Eming et al. 2014; Elliot et al. 2018; Pastar et al. 2018). Preclinical animal testing is required for preclinical evaluation and regulatory approval of novel therapeutics; their low concordance rate with complex human chronic wounds represents a significant impediment to development of products with clinical efficacies. For example, numerous studies using various growth factors as chronic wound therapeutics were considered promising based on the preclinical data but have failed to show efficacy in clinical trials (Falanga et al. 1992; Barrientos et al. 2008; Yamakawa and Hayashida 2019). Intrinsic suppression of EGF signaling, including mislocalization of the EGF receptor (Brem et al. 2007; Jozic et al. 2019; Sawaya et al. 2019), and suppression of TGF-β receptors (Pastar et al. 2010) found only in human tissue but not in animal models, together with a harsh proteolytic chronic wound environment, have been identified as major factors responsible for inadequate translation of EGF and TGF-β to clinical practice (Eming et al. 2014; Eming and Tomic-Canic 2017). Further to the point, even when tested post-FDA approval, topical PDGF showed that its effect was dependent on the animal model used (Park et al. 2014b).
Another impediment to more approved therapies is limitation of a single primary end point, complete closure. Current ongoing initiatives identified multiple new measurable wound care end points and supporting evidence from both clinical practice and patient-reported outcomes (Driver et al. 2017, 2019; Gould et al. 2021). These findings provide new insights and a potential roadmap for future acceptance of multiple end points that would accelerate the process of approval and potentially bring more therapies to patients who desperately need them.
The most recent “omics” work including current and future data resources generated from the human biomaterials combined with new bioinformatics tools provide better insights and “higher resolution” understanding of pathophysiology for disease target-based repositioning methods (Sawaya et al. 2018, 2019). Expanded analyses of existing transcriptional profiling or single-cell expression data can highlight the relationships between chronic wound signatures and gene expression profiles associated with existing drugs, identifying new therapeutic strategies (Subramanian et al. 2017). Whereas computational drug methods using expression data sets have already been applied to repurpose existing FDA-approved drugs in a limited number of cutaneous diseases (Nyström et al. 2015; Mirza et al. 2017; Lee et al. 2022), similar approaches have not been fully explored for chronic wounds.
The following recent series of studies can be extrapolated in future investigations to elucidate the mechanisms of existing or new therapies. The bilayered human skin equivalent (BLCC) is the only FDA-approved therapy that has demonstrated efficacy in healing chronic VLUs (Falanga et al. 1998; ClinicalTrials.gov 2011; Stone et al. 2017) and a clinical trial was designed to understand the mechanisms of action of the BLCC in promoting healing of chronic wounds by exploring the transcriptional profiles of ulcer tissue from patients that were treated with BLCC versus those who were treated with standard of care. The findings unveiled specific biologic processes through which BLCC application converts chronic non-healing VLUs into an acute healing phenotype, including the role of stimulated inflammatory response and reversal of fibrosis (Stone et al. 2017, 2020b). Importantly, these processes can serve as the basis for future studies to find much needed novel therapies for chronic ulcers. Similar approaches can be integrated into clinical trials to elucidate the mechanisms of the chronic wound healing response and most importantly to ultimately improve the lives of patients suffering from chronic wounds.
Complexity of pathophysiology of chronic wounds mandates multifaceted approach coupling diagnostics and monitoring tools with combinatorial targeted therapies. One can envision treatment that targets multiple cellular dysfunctions (described above in detail) in a precise timely manner only to wounds that would benefit from such combinatorial treatment. Future longitudinal studies using “omics” approaches will yield identification of biomarkers or predictors of healing outcomes, monitors of progression during the treatments, predictors of infection, or detection of a biofilm, all of which could help steer clinical decision making (Lindley et al. 2016). Adapting precision medicine strategies to this complex clinical problem, including development of biomarkers, will facilitate better personalized treatment approach allowing for targeting clinical interventions and therapies to individual wound and patient with maximum efficacy.
ACKNOWLEDGMENTS
We are grateful to patients, past and current members of our collaborative clinical and research teams, as well as our colleagues in the field for the inspiration. M.T.-C. acknowledges funding by the National Institutes of Health (NIH) (R01NR015649; U01DK119085; R01NR01388; RC1DK086364 to M.T.-C.), NIH Bench-to-Bedside award, and University of Miami SAC 2013-19.
COMPETING INTEREST STATEMENT
Dr. Pastar’s research is in part supported by Vomaris and Next Science. Dr. Tomic-Canic’s research is partly supported by Organogensis and she serves on an advisory board of Molnlycke.
Footnotes
Additional Perspectives on Wound Healing: From Bench to Bedside available at www.cshperspectives.org
REFERENCES
*Reference is also in this subject collection.
- Abd-El-Aleem SA, Morgan C, Ferguson MW, McCollum CN, Ireland GW. 2005. Spatial distribution of mast cells in chronic venous leg ulcers. Eur J Histochem 49: 265–272. [PubMed] [Google Scholar]
- Ågren MS, Steenfos HH, Dabelsteen S, Hansen JB, Dabelsteen E. 1999. Proliferation and mitogenic response to PDGF-BB of fibroblasts isolated from chronic venous leg ulcers is ulcer-age dependent. J Invest Dermatol 112: 463–469. doi: 10.1046/j.1523-1747.1999.00549.x [DOI] [PubMed] [Google Scholar]
- Albiero M, Menegazzo L, Boscaro E, Agostini C, Avogaro A, Fadini GP. 2011. Defective recruitment, survival and proliferation of bone marrow-derived progenitor cells at sites of delayed diabetic wound healing in mice. Diabetologia 54: 945–953. doi: 10.1007/s00125-010-2007-2 [DOI] [PubMed] [Google Scholar]
- Arif S, Attiogbe E, Moulin VJ. 2021. Granulation tissue myofibroblasts during normal and pathological skin healing: the interaction between their secretome and the microenvironment. Wound Repair Regen 29: 563–572. doi: 10.1111/wrr.12919 [DOI] [PubMed] [Google Scholar]
- Armstrong DG, Boulton AJM, Bus SA. 2017. Diabetic foot ulcers and their recurrence. N Engl J Med 376: 2367–2375. doi: 10.1056/NEJMra1615439 [DOI] [PubMed] [Google Scholar]
- Ashcroft GS, Horan MA, Ferguson MW. 1997. Aging is associated with reduced deposition of specific extracellular matrix components, an upregulation of angiogenesis, and an altered inflammatory response in a murine incisional wound healing model. J Invest Dermatol 108: 430–437. doi: 10.1111/1523-1747.ep12289705 [DOI] [PubMed] [Google Scholar]
- Barrientos S, Stojadinovic O, Golinko MS, Brem H, Tomic-Canic M. 2008. Growth factors and cytokines in wound healing. Wound Repair Regen 16: 585–601. doi: 10.1111/j.1524-475X.2008.00410.x [DOI] [PubMed] [Google Scholar]
- Barrientos S, Brem H, Stojadinovic O, Tomic-Canic M. 2014. Clinical application of growth factors and cytokines in wound healing. Wound Repair Regen 22: 569–578. doi: 10.1111/wrr.12205 [DOI] [PMC free article] [PubMed] [Google Scholar]
- Berberich B, Thriene K, Gretzmeier C, Kühl T, Bayer H, Athanasiou I, Rafei-Shamsabadi DA, Bruckner-Tuderman L, Nyström A, Kiritsi D, et al. 2020. Proteomic profiling of fibroblasts isolated from chronic wounds identifies disease-relevant signaling pathways. J Invest Dermatol 140: 2280–2290.e4. doi: 10.1016/j.jid.2020.02.040 [DOI] [PubMed] [Google Scholar]
- Berlanga-Acosta JA, Guillén-Nieto GE, Rodríguez-Rodríguez N, Mendoza-Mari Y, Bringas-Vega ML, Berlanga-Saez JO, García Del Barco Herrera D, Martinez-Jimenez I, Hernandez-Gutierrez S, Valdés-Sosa PA. 2020. Cellular senescence as the pathogenic hub of diabetes-related wound chronicity. Front Endocrinol (Lausanne) 11: 573032. doi: 10.3389/fendo.2020.573032 [DOI] [PMC free article] [PubMed] [Google Scholar]
- Bhattacharya M, Berends ETM, Chan R, Schwab E, Roy S, Sen CK, Torres VJ, Wozniak DJ. 2018. Staphylococcus aureus biofilms release leukocidins to elicit extracellular trap formation and evade neutrophil-mediated killing. Proc Natl Acad Sci 115: 7416–7421. doi: 10.1073/pnas.1721949115 [DOI] [PMC free article] [PubMed] [Google Scholar]
- Bian X, Li B, Yang J, Ma K, Sun M, Zhang C, Fu X. 2020. Regenerative and protective effects of dMSC-sEVs on high-glucose-induced senescent fibroblasts by suppressing RAGE pathway and activating Smad pathway. Stem Cell Res Ther 11: 166. doi: 10.1186/s13287-020-01681-z [DOI] [PMC free article] [PubMed] [Google Scholar]
- Biswas S, Roy S, Banerjee J, Hussain SR, Khanna S, Meenakshisundaram G, Kuppusamy P, Friedman A, Sen CK. 2010. Hypoxia inducible microRNA 210 attenuates keratinocyte proliferation and impairs closure in a murine model of ischemic wounds. Proc Natl Acad Sci 107: 6976–6981. doi: 10.1073/pnas.1001653107 [DOI] [PMC free article] [PubMed] [Google Scholar]
- Blumberg SN, Maggi J, Melamed J, Golinko M, Ross F, Chen W. 2012. A histopathologic basis for surgical debridement to promote healing of venous ulcers. J Am Coll Surg 215: 751–757. doi: 10.1016/j.jamcollsurg.2012.08.008 [DOI] [PubMed] [Google Scholar]
- Brem H, Tomic-Canic M. 2007. Cellular and molecular basis of wound healing in diabetes. J Clin Invest 117: 1219–1222. doi: 10.1172/JCI32169 [DOI] [PMC free article] [PubMed] [Google Scholar]
- Brem H, Stojadinovic O, Diegelmann RF, Entero H, Lee B, Pastar I, Golinko M, Rosenberg H, Tomic-Canic M. 2007. Molecular markers in patients with chronic wounds to guide surgical debridement. Mol Med 13: 30–39. doi: 10.2119/2006-00054.Brem [DOI] [PMC free article] [PubMed] [Google Scholar]
- Brem H, Golinko MS, Stojadinovic O, Kodra A, Diegelmann RF, Vukelic S, Entero H, Coppock DL, Tomic-Canic M. 2008. Primary cultured fibroblasts derived from patients with chronic wounds: a methodology to produce human cell lines and test putative growth factor therapy such as GMCSF. J Transl Med 6: 75. doi: 10.1186/1479-5876-6-75 [DOI] [PMC free article] [PubMed] [Google Scholar]
- Brockman SJ, Dirks D, Eviatar A, Goodhill V, Wilber L. 1970. Frontal bone conduction, impedance, and myringomanometry in the diagnosis of conductive lesions. Trans Pac Coast Otoophthalmol Soc Annu Meet 51: 65–85. [PubMed] [Google Scholar]
- Brunner G, Roux M, Böhm V, Meiners T. 2021. Cellular and molecular changes that predispose skin in chronic spinal cord injury to pressure ulcer formation. Int Wound J 18: 728–737. doi: 10.1111/iwj.13575 [DOI] [PMC free article] [PubMed] [Google Scholar]
- Buchstein N, Hoffmann D, Smola H, Lang S, Paulsson M, Niemann C, Krieg T, Eming SA. 2009. Alternative proteolytic processing of hepatocyte growth factor during wound repair. Am J Pathol 174: 2116–2128. doi: 10.2353/ajpath.2009.080597 [DOI] [PMC free article] [PubMed] [Google Scholar]
- Cavassan NRV, Camargo CC, de Pontes LG, Barraviera B, Ferreira RS, Miot HA, Abbade LPF, Dos Santos LD. 2019. Correlation between chronic venous ulcer exudate proteins and clinical profile: a cross-sectional study. J Proteomics 192: 280–290. doi: 10.1016/j.jprot.2018.09.009 [DOI] [PubMed] [Google Scholar]
- Cellerino A, Ori A. 2017. What have we learned on aging from omics studies? Semin Cell Dev Biol 70: 177–189. doi: 10.1016/j.semcdb.2017.06.012 [DOI] [PubMed] [Google Scholar]
- Chambers AC, Leaper DJ. 2011. Role of oxygen in wound healing: a review of evidence. J Wound Care 20: 160–164. doi: 10.12968/jowc.2011.20.4.160 [DOI] [PubMed] [Google Scholar]
- Chan YC, Roy S, Khanna S, Sen CK. 2012. Downregulation of endothelial microRNA-200b supports cutaneous wound angiogenesis by desilencing GATA binding protein 2 and vascular endothelial growth factor receptor 2. Arterioscler Thromb Vasc Biol 32: 1372–1382. doi: 10.1161/ATVBAHA.112.248583 [DOI] [PMC free article] [PubMed] [Google Scholar]
- ClinicalTrials.gov. 2011. A post marketing study of apligraf in non-healing wounds of subjects with venous leg ulcers. https://ClinicalTrials.gov/show/NCT01327937.2/11/22
- ClinicalTrials.gov. 2020. C-myc biomarker study for diabetic foot ulcers. https://ClinicalTrials.gov/show/NCT04591691.2/17/2022
- Cook H, Davies KJ, Harding KG, Thomas DW. 2000. Defective extracellular matrix reorganization by chronic wound fibroblasts is associated with alterations in TIMP-1, TIMP-2, and MMP-2 activity. J Invest Dermatol 115: 225–233. doi: 10.1046/j.1523-1747.2000.00044.x [DOI] [PubMed] [Google Scholar]
- Costa PZ, Soares R. 2013. Neovascularization in diabetes and its complications. Unraveling the angiogenic paradox. Life Sci 92: 1037–1045. doi: 10.1016/j.lfs.2013.04.001 [DOI] [PubMed] [Google Scholar]
- Dalton SJ, Whiting CV, Bailey JR, Mitchell DC, Tarlton JF. 2007. Mechanisms of chronic skin ulceration linking lactate, transforming growth factor-β, vascular endothelial growth factor, collagen remodeling, collagen stability, and defective angiogenesis. J Invest Dermatol 127: 958–968. doi: 10.1038/sj.jid.5700651 [DOI] [PubMed] [Google Scholar]
- Diegelmann RF. 2003. Excessive neutrophils characterize chronic pressure ulcers. Wound Repair Regen 11: 490–495. doi: 10.1046/j.1524-475X.2003.11617.x [DOI] [PubMed] [Google Scholar]
- Ding X, Kakanj P, Leptin M, Eming SA. 2021. Regulation of the wound healing response during aging. J Invest Dermatol 141: 1063–1070. doi: 10.1016/j.jid.2020.11.014 [DOI] [PubMed] [Google Scholar]
- Dong J, Chen L, Zhang Y, Jayaswal N, Mezghani I, Zhang W, Veves A. 2020. Mast cells in diabetes and diabetic wound healing. Adv Ther 37: 4519–4537. doi: 10.1007/s12325-020-01499-4 [DOI] [PMC free article] [PubMed] [Google Scholar]
- Drela E, Stankowska K, Kulwas A, Rosc D. 2012. Endothelial progenitor cells in diabetic foot syndrome. Adv Clin Exp Med 21: 249–254. [PubMed] [Google Scholar]
- Dressel S, Harder J, Cordes J, Wittersheim M, Meyer-Hoffert U, Sunderkötter C, Gläser R. 2010. Differential expression of antimicrobial peptides in margins of chronic wounds. Exp Dermatol 19: 628–632. doi: 10.1111/j.1600-0625.2009.01030.x [DOI] [PubMed] [Google Scholar]
- Driver VR, Gould LJ, Dotson P, Gibbons GW, Li WW, Ennis WJ, Kirsner RS, Eaglstein WH, Bolton LL, Carter MJ. 2017. Identification and content validation of wound therapy clinical endpoints relevant to clinical practice and patient values for FDA approval. Part 1: Survey of the wound care community. Wound Repair Regen 25: 454–465. doi: 10.1111/wrr.12533 [DOI] [PubMed] [Google Scholar]
- Driver VR, Gould LJ, Dotson P, Allen LL, Carter MJ, Bolton LL. 2019. Evidence supporting wound care end points relevant to clinical practice and patients’ lives. Part 2: Literature survey. Wound Repair Regen 27: 80–89. doi: 10.1111/wrr.12676 [DOI] [PubMed] [Google Scholar]
- Edsberg LE, Wyffels JT, Brogan MS, Fries KM. 2012. Analysis of the proteomic profile of chronic pressure ulcers. Wound Repair Regen 20: 378–401. doi: 10.1111/j.1524-475X.2012.00791.x [DOI] [PubMed] [Google Scholar]
- Eisenbud DE. 2012. Oxygen in wound healing: nutrient, antibiotic, signaling molecule, and therapeutic agent. Clin Plast Surg 39: 293–310. doi: 10.1016/j.cps.2012.05.001 [DOI] [PubMed] [Google Scholar]
- Elliot S, Wikramanayake TC, Jozic I, Tomic-Canic M. 2018. A modeling conundrum: murine models for cutaneous wound healing. J Invest Dermatol 138: 736–740. doi: 10.1016/j.jid.2017.12.001 [DOI] [PubMed] [Google Scholar]
- Elliott MR, Koster KM, Murphy PS. 2017. Efferocytosis signaling in the regulation of macrophage inflammatory responses. J Immunol 198: 1387–1394. doi: 10.4049/jimmunol.1601520 [DOI] [PMC free article] [PubMed] [Google Scholar]
- Eming SA, Tomic-Canic M. 2017. Updates in wound healing: mechanisms and translation. Exp Dermatol 26: 97–98. doi: 10.1111/exd.13281 [DOI] [PubMed] [Google Scholar]
- Eming SA, Brachvogel B, Odorisio T, Koch M. 2007. Regulation of angiogenesis: wound healing as a model. Prog Histochem Cytochem 42: 115–170. doi: 10.1016/j.proghi.2007.06.001 [DOI] [PubMed] [Google Scholar]
- Eming SA, Koch M, Krieger A, Brachvogel B, Kreft S, Bruckner-Tuderman L, Krieg T, Shannon JD, Fox JW. 2010. Differential proteomic analysis distinguishes tissue repair biomarker signatures in wound exudates obtained from normal healing and chronic wounds. J Proteome Res 9: 4758–4766. doi: 10.1021/pr100456d [DOI] [PubMed] [Google Scholar]
- Eming SA, Martin P, Tomic-Canic M. 2014. Wound repair and regeneration: mechanisms, signaling, and translation. Sci Transl Med 6: 265sr266. doi: 10.1126/scitranslmed.3009337 [DOI] [PMC free article] [PubMed] [Google Scholar]
- Fadini GP, Albiero M, Millioni R, Poncina N, Rigato M, Scotton R, Boscari F, Brocco E, Arrigoni G, Villano G, et al. 2014. The molecular signature of impaired diabetic wound healing identifies serpinB3 as a healing biomarker. Diabetologia 57: 1947–1956. doi: 10.1007/s00125-014-3300-2 [DOI] [PubMed] [Google Scholar]
- Fadini GP, Menegazzo L, Rigato M, Scattolini V, Poncina N, Bruttocao A, Ciciliot S, Mammano F, Ciubotaru CD, Brocco E, et al. 2016. NETosis delays diabetic wound healing in mice and humans. Diabetes 65: 1061–1071. doi: 10.2337/db15-0863 [DOI] [PubMed] [Google Scholar]
- Falanga V, Eaglstein WH, Bucalo B, Katz MH, Harris B, Carson P. 1992. Topical use of human recombinant epidermal growth factor (h-EGF) in venous ulcers. J Dermatol Surg Oncol 18: 604–606. doi: 10.1111/j.1524-4725.1992.tb03514.x [DOI] [PubMed] [Google Scholar]
- Falanga V, Takagi H, Ceballos PI, Pardes JB. 1994. Low oxygen tension decreases receptor binding of peptide growth factors in dermal fibroblast cultures. Exp Cell Res 213: 80–84. doi: 10.1006/excr.1994.1175 [DOI] [PubMed] [Google Scholar]
- Falanga V, Margolis D, Alvarez O, Auletta M, Maggiacomo F, Altman M, Jensen J, Sabolinski M, Hardin-Young J. 1998. Rapid healing of venous ulcers and lack of clinical rejection with an allogeneic cultured human skin equivalent. Human skin equivalent investigators group. Arch Dermatol 134: 293–300. doi: 10.1001/archderm.134.3.293 [DOI] [PubMed] [Google Scholar]
- Falanga V, Zhou L, Yufit T. 2002. Low oxygen tension stimulates collagen synthesis and COL1A1 transcription through the action of TGF-β1. J Cell Physiol 191: 42–50. doi: 10.1002/jcp.10065 [DOI] [PubMed] [Google Scholar]
- Freedberg IM, Tomic-Canic M, Komine M, Blumenberg M. 2001. Keratins and the keratinocyte activation cycle. J Invest Dermatol 116: 633–640. doi: 10.1046/j.1523-1747.2001.01327.x [DOI] [PubMed] [Google Scholar]
- Fujiwara T, Dohi T, Maan ZN, Rustad KC, Kwon SH, Padmanabhan J, Whittam AJ, Suga H, Duscher D, Rodrigues M, et al. 2019. Age-associated intracellular superoxide dismutase deficiency potentiates dermal fibroblast dysfunction during wound healing. Exp Dermatol 28: 485–492. doi: 10.1111/exd.13404 [DOI] [PubMed] [Google Scholar]
- Gabay M, Li Y, Felsher DW. 2014. MYC activation is a hallmark of cancer initiation and maintenance. Cold Spring Harb Perspect Med 4: a014241. doi: 10.1101/cshperspect.a014241 [DOI] [PMC free article] [PubMed] [Google Scholar]
- Gallagher KA, Liu ZJ, Xiao M, Chen H, Goldstein LJ, Buerk DG, Nedeau A, Thom SR, Velazquez OC. 2007. Diabetic impairments in NO-mediated endothelial progenitor cell mobilization and homing are reversed by hyperoxia and SDF-1α. J Clin Invest 117: 1249–1259. doi: 10.1172/JCI29710 [DOI] [PMC free article] [PubMed] [Google Scholar]
- Gallo PM, Gallucci S. 2013. The dendritic cell response to classic, emerging, and homeostatic danger signals. Implications for autoimmunity. Front Immunol 4: 138. doi: 10.3389/fimmu.2013.00138 [DOI] [PMC free article] [PubMed] [Google Scholar]
- *.Ganier C, Rognoni E, Goss G, Lynch M, Watt FM. 2022. Fibroblast heterogeneity in healthy and wounded skin. Cold Spring Harb Perspect Biol doi: 10.1101/cshperspect.a041238 [DOI] [PMC free article] [PubMed] [Google Scholar]
- Gardner SE, Hillis SL, Heilmann K, Segre JA, Grice EA. 2013. The neuropathic diabetic foot ulcer microbiome is associated with clinical factors. Diabetes 62: 923–930. doi: 10.2337/db12-0771 [DOI] [PMC free article] [PubMed] [Google Scholar]
- Gay D, Ghinatti G, Guerrero-Juarez CF, Ferrer RA, Ferri F, Lim CH, Murakami S, Gault N, Barroca V, Rombeau I, et al. 2020. Phagocytosis of Wnt inhibitor SFRP4 by late wound macrophages drives chronic Wnt activity for fibrotic skin healing. Sci Adv 6: eaay3704. doi: 10.1126/sciadv.aay3704 [DOI] [PMC free article] [PubMed] [Google Scholar]
- Gerami-Naini B, Smith A, Maione AG, Kashpur O, Carpinito G, Veves A, Mooney DJ, Garlick JA. 2016. Generation of induced pluripotent stem cells from diabetic foot ulcer fibroblasts using a nonintegrative Sendai virus. Cell Reprogram 18: 214–223. doi: 10.1089/cell.2015.0087 [DOI] [PMC free article] [PubMed] [Google Scholar]
- Giustizieri ML, Albanesi C, Fluhr J, Gisondi P, Norgauer J, Girolomoni G. 2004. H1 histamine receptor mediates inflammatory responses in human keratinocytes. J Allergy Clin Immunol 114: 1176–1182. doi: 10.1016/j.jaci.2004.07.054 [DOI] [PubMed] [Google Scholar]
- Gordillo GM, Sen CK. 2003. Revisiting the essential role of oxygen in wound healing. Am J Surg 186: 259–263. doi: 10.1016/S0002-9610(03)00211-3 [DOI] [PubMed] [Google Scholar]
- Gordillo GM, Bernatchez SF, Diegelmann R, Di Pietro LA, Eriksson E, Hinz B, Hopf HW, Kirsner R, Liu P, Parnell LK, et al. 2013. Preclinical models of wound healing: is man the model? proceedings of the wound healing society symposium. Adv Wound Care (New Rochelle) 2: 1–4. doi: 10.1089/wound.2012.0367 [DOI] [PMC free article] [PubMed] [Google Scholar]
- Gould LJ, Liu J, Wan R, Carter MJ, Dotson MP, Driver VR. 2021. Evidence supporting wound care end points relevant to clinical practice and patients’ lives. Part 3: The patient survey. Wound Repair Regen 29: 60–69. doi: 10.1111/wrr.12872 [DOI] [PubMed] [Google Scholar]
- Grinnell F, Zhu M. 1996. Fibronectin degradation in chronic wounds depends on the relative levels of elastase, α1-proteinase inhibitor, and α2-macroglobulin. J Invest Dermatol 106: 335–341. doi: 10.1111/1523-1747.ep12342990 [DOI] [PubMed] [Google Scholar]
- Harding KG, Moore K, Phillips TJ. 2005. Wound chronicity and fibroblast senescence–implications for treatment. Int Wound J 2: 364–368. doi: 10.1111/j.1742-4801.2005.00149.x [DOI] [PMC free article] [PubMed] [Google Scholar]
- Haspula D, Vallejos AK, Moore TM, Tomar N, Dash RK, Hoffmann BR. 2019. Influence of a hyperglycemic microenvironment on a diabetic versus healthy rat vascular endothelium reveals distinguishable mechanistic and phenotypic responses. Front Physiol 10: 558. doi: 10.3389/fphys.2019.00558 [DOI] [PMC free article] [PubMed] [Google Scholar]
- Hiebert P, Wietecha MS, Cangkrama M, Haertel E, Mavrogonatou E, Stumpe M, Steenbock H, Grossi S, Beer HD, Angel P, et al. 2018. Nrf2-mediated fibroblast reprogramming drives cellular senescence by targeting the matrisome. Dev Cell 46: 145–161.e10. doi: 10.1016/j.devcel.2018.06.012 [DOI] [PubMed] [Google Scholar]
- Hinz B 2016. The role of myofibroblasts in wound healing. Curr Res Transl Med 64: 171–177. doi: 10.1016/j.retram.2016.09.003 [DOI] [PubMed] [Google Scholar]
- Huang X, Liang P, Jiang B, Zhang P, Yu W, Duan M, Guo L, Cui X, Huang M, Huang X. 2020. Hyperbaric oxygen potentiates diabetic wound healing by promoting fibroblast cell proliferation and endothelial cell angiogenesis. Life Sci 259: 118246. doi: 10.1016/j.lfs.2020.118246 [DOI] [PubMed] [Google Scholar]
- Huttunen M, Aalto ML, Harvima RJ, Horsmanheimo M, Harvima IT. 2000. Alterations in mast cells showing tryptase and chymase activity in epithelializating and chronic wounds. Exp Dermatol 9: 258–265. doi: 10.1034/j.1600-0625.2000.009004258.x [DOI] [PubMed] [Google Scholar]
- Huttunen M, Hyttinen M, Nilsson G, Butterfield JH, Horsmanheimo M, Harvima IT. 2001. Inhibition of keratinocyte growth in cell culture and whole skin culture by mast cell mediators. Exp Dermatol 10: 184–192. doi: 10.1034/j.1600-0625.2001.010003184.x [DOI] [PubMed] [Google Scholar]
- Januszyk M, Chen K, Henn D, Foster DS, Borrelli MR, Bonham CA, Sivaraj D, Wagh D, Longaker MT, Wan DC, et al. 2020. Characterization of diabetic and non-diabetic foot ulcers using single-cell RNA-sequencing. Micromachines (Basel) 11: 815. doi: 10.3390/mi11090815 [DOI] [PMC free article] [PubMed] [Google Scholar]
- *.Jiang D, Guo R, Machens HG, Rinkevich Y. 2022. Diversity of fibroblasts and their roles in wound healing. Cold Spring Harb Perspect Biol doi: 10.1101/cshperspect.a041222 [DOI] [PMC free article] [PubMed] [Google Scholar]
- Jindatanmanusan P, Luanraksa S, Boonsiri T, Nimmanon T, Arnutti P. 2018. Wound fluid matrix metalloproteinase-9 as a potential predictive marker for the poor healing outcome in diabetic foot ulcers. Patholog Res Int 2018: 1631325. doi: 10.1155/2018/1631325 [DOI] [PMC free article] [PubMed] [Google Scholar]
- Jones JI, Nguyen TT, Peng Z, Chang M. 2019. Targeting MMP-9 in diabetic foot ulcers. Pharmaceuticals (Basel) 12: 79. doi: 10.3390/ph12020079 [DOI] [PMC free article] [PubMed] [Google Scholar]
- Jozic I, Vukelic S, Stojadinovic O, Liang L, Ramirez HA, Pastar I, Tomic Canic M. 2017. Stress signals, mediated by membranous glucocorticoid receptor, activate PLC/PKC/GSK-3β/β-catenin pathway to inhibit wound closure. J Invest Dermatol 137: 1144–1154. doi: 10.1016/j.jid.2016.11.036 [DOI] [PMC free article] [PubMed] [Google Scholar]
- Jozic I, Sawaya AP, Pastar I, Head CR, Wong LL, Glinos GD, Wikramanayake TC, Brem H, Kirsner RS, Tomic-Canic M. 2019. Pharmacological and genetic inhibition of caveolin-1 promotes epithelialization and wound closure. Mol Ther 27: 1992–2004. doi: 10.1016/j.ymthe.2019.07.016 [DOI] [PMC free article] [PubMed] [Google Scholar]
- Jozic I, Abujamra BA, Elliott MH, Wikramanayake TC, Marjanovic J, Stone RC, Head CR, Pastar I, Kirsner RS, Andreopoulos FM, et al. 2021. Glucocorticoid-mediated induction of caveolin-1 disrupts cytoskeletal organization, inhibits cell migration and re-epithelialization of non-healing wounds. Commun Biol 4: 757. doi: 10.1038/s42003-021-02298-5 [DOI] [PMC free article] [PubMed] [Google Scholar]
- Jun JI, Lau LF. 2010a. Cellular senescence controls fibrosis in wound healing. Aging (Albany NY) 2: 627–631. doi: 10.18632/aging.100201 [DOI] [PMC free article] [PubMed] [Google Scholar]
- Jun JI, Lau LF. 2010b. The matricellular protein CCN1 induces fibroblast senescence and restricts fibrosis in cutaneous wound healing. Nat Cell Biol 12: 676–685. doi: 10.1038/ncb2070 [DOI] [PMC free article] [PubMed] [Google Scholar]
- Kalan L, Loesche M, Hodkinson BP, Heilmann K, Ruthel G, Gardner SE, Grice EA. 2016. Redefining the chronic-wound microbiome: fungal communities are prevalent, dynamic, and associated with delayed healing. MBio 7: e01058–16. doi: 10.1128/mBio.01058-16 [DOI] [PMC free article] [PubMed] [Google Scholar]
- Kalan LR, Meisel JS, Loesche MA, Horwinski J, Soaita I, Chen X, Uberoi A, Gardner SE, Grice EA. 2019. Strain- and species-level variation in the microbiome of diabetic wounds is associated with clinical outcomes and therapeutic efficacy. Cell Host Microbe 25: 641–655.e5. doi: 10.1016/j.chom.2019.03.006 [DOI] [PMC free article] [PubMed] [Google Scholar]
- Kashpur O, Smith A, Gerami-Naini B, Maione AG, Calabrese R, Tellechea A, Theocharidis G, Liang L, Pastar I, Tomic-Canic M, et al. 2019. Differentiation of diabetic foot ulcer-derived induced pluripotent stem cells reveals distinct cellular and tissue phenotypes. FASEB J 33: 1262–1277. doi: 10.1096/fj.201801059 [DOI] [PMC free article] [PubMed] [Google Scholar]
- Kim BC, Kim HT, Park SH, Cha JS, Yufit T, Kim SJ, Falanga V. 2003. Fibroblasts from chronic wounds show altered TGF-β-signaling and decreased TGF-β type II receptor expression. J Cell Physiol 195: 331–336. doi: 10.1002/jcp.10301 [DOI] [PubMed] [Google Scholar]
- Kim KA, Shin YJ, Kim JH, Lee H, Noh SY, Jang SH, Bae ON. 2012. Dysfunction of endothelial progenitor cells under diabetic conditions and its underlying mechanisms. Arch Pharm Res 35: 223–234. doi: 10.1007/s12272-012-0203-y [DOI] [PubMed] [Google Scholar]
- Kohda F, Koga T, Uchi H, Urabe K, Furue M. 2002. Hista-mine-induced IL-6 and IL-8 production are differentially modulated by IFN-γ and IL-4 in human keratinocytes. J Dermatol Sci 28: 34–41. doi: 10.1016/S0923-1811(01)00147-5 [DOI] [PubMed] [Google Scholar]
- Kranke P, Bennett MH, Martyn-St James M, Schnabel A, Debus SE, Weibel S. 2015. Hyperbaric oxygen therapy for chronic wounds. Cochrane Database Syst Rev 2015: CD004123. doi: 10.1002/14651858.CD004123.pub4 [DOI] [PMC free article] [PubMed] [Google Scholar]
- Krishnaswamy VR, Manikandan M, Munirajan AK, Vijayaraghavan D, Korrapati PS. 2014. Expression and integrity of dermatopontin in chronic cutaneous wounds: a crucial factor in impaired wound healing. Cell Tissue Res 358: 833–841. doi: 10.1007/s00441-014-2000-z [DOI] [PubMed] [Google Scholar]
- Krisp C, Jacobsen F, McKay MJ, Molloy MP, Steinstraesser L, Wolters DA. 2013. Proteome analysis reveals antiangiogenic environments in chronic wounds of diabetes mellitus type 2 patients. Proteomics 13: 2670–2681. doi: 10.1002/pmic.201200502 [DOI] [PubMed] [Google Scholar]
- Kuai L, Xiang YW, Chen QL, Ru Y, Yin SY, Li W, Jiang JS, Luo Y, Song JK, Lu B, et al. 2022. PD-L1 triggered by binding eIF3I contributes to the amelioration of diabetes-associated wound healing defects by regulating IRS4. J Invest Dermatol 142: 220–231.e8. doi: 10.1016/j.jid.2021.06.028 [DOI] [PubMed] [Google Scholar]
- Lauer G, Sollberg S, Cole M, Flamme I, Stürzebecher J, Mann K, Krieg T, Eming SA. 2000. Expression and proteolysis of vascular endothelial growth factor is increased in chronic wounds. J Invest Dermatol 115: 12–18. doi: 10.1046/j.1523-1747.2000.00036.x [DOI] [PubMed] [Google Scholar]
- Lawrence T, Natoli G. 2011. Transcriptional regulation of macrophage polarization: enabling diversity with identity. Nat Rev Immunol 11: 750–761. doi: 10.1038/nri3088 [DOI] [PubMed] [Google Scholar]
- Lee GH, Lekwuttikarn R, Tafoya E, Martin M, Sarin KY, Teng JM. 2022. Transcriptomic repositioning analysis identifies mTOR inhibitor as potential therapy for epidermolysis bullosa simplex. J Invest Dermatol 142: 382–389. doi: 10.1016/j.jid.2021.07.170 [DOI] [PubMed] [Google Scholar]
- Leu AJ, Leu HJ, Franzeck UK, Bollinger A. 1995. Microvascular changes in chronic venous insufficiency—a review. Cardiovasc Surg 3: 237–245. doi: 10.1016/0967-2109(95)93871-L [DOI] [PubMed] [Google Scholar]
- Li B, Wang JH. 2011. Fibroblasts and myofibroblasts in wound healing: force generation and measurement. J Tissue Viability 20: 108–120. doi: 10.1016/j.jtv.2009.11.004 [DOI] [PMC free article] [PubMed] [Google Scholar]
- Li D, Peng H, Qu L, Sommar P, Wang A, Chu T, Li X, Bi X, Liu Q, Gallais Sérézal I, et al. 2021. miR-19a/b and miR-20a promote wound healing by regulating the inflammatory response of keratinocytes. J Invest Dermatol 141: 659–671. doi: 10.1016/j.jid.2020.06.037 [DOI] [PubMed] [Google Scholar]
- Li D, Cheng S, Pei Y, Sommar P, Kärner J, Herter EK, Toma MA, Zhang L, Pham K, Cheung YT, et al. 2022a. Single-cell analysis reveals major histocompatibility complex II–expressing keratinocytes in pressure ulcers with worse healing outcomes. J Invest Dermatol 142: 705–716. doi: 10.1016/j.jid.2021.07.176 [DOI] [PubMed] [Google Scholar]
- *.Li D, Niu G, Landén NX. 2022b. Beyond the code: noncoding RNAs in skin wound healing. Cold Spring Harb Perspect Biol doi: 10.1101/cshperspect.a041230 [DOI] [PMC free article] [PubMed] [Google Scholar]
- Liang L, Stone RC, Stojadinovic O, Ramirez H, Pastar I, Maione AG, Smith A, Yanez V, Veves A, Kirsner RS, et al. 2016. Integrative analysis of miRNA and mRNA paired expression profiling of primary fibroblast derived from diabetic foot ulcers reveals multiple impaired cellular functions. Wound Repair Regen 24: 943–953. doi: 10.1111/wrr.12470 [DOI] [PMC free article] [PubMed] [Google Scholar]
- Lindley LE, Stojadinovic O, Pastar I, Tomic-Canic M. 2016. Biology and biomarkers for wound healing. Plast Reconstr Surg 138: 18S–28S. doi: 10.1097/PRS.0000000000002682 [DOI] [PMC free article] [PubMed] [Google Scholar]
- Liu ZJ, Velazquez OC. 2008. Hyperoxia, endothelial progenitor cell mobilization, and diabetic wound healing. Anti-oxid Redox Signal 10: 1869–1882. doi: 10.1089/ars.2008.2121 [DOI] [PMC free article] [PubMed] [Google Scholar]
- Liu Y, Min D, Bolton T, Nubé V, Twigg SM, Yue DK, McLennan SV. 2009. Increased matrix metalloproteinase-9 predicts poor wound healing in diabetic foot ulcers. Diabetes Care 32: 117–119. doi: 10.2337/dc08-0763 [DOI] [PMC free article] [PubMed] [Google Scholar]
- Liu D, Yang P, Gao M, Yu T, Shi Y, Zhang M, Yao M, Liu Y, Zhang X. 2019. NLRP3 activation induced by neutrophil extracellular traps sustains inflammatory response in the diabetic wound. Clin Sci (Lond) 133: 565–582. doi: 10.1042/CS20180600 [DOI] [PubMed] [Google Scholar]
- Lobmann R, Ambrosch A, Schultz G, Waldmann K, Schiweck S, Lehnert H. 2002. Expression of matrix-metalloproteinases and their inhibitors in the wounds of diabetic and non-diabetic patients. Diabetologia 45: 1011–1016. doi: 10.1007/s00125-002-0868-8 [DOI] [PubMed] [Google Scholar]
- Loesche M, Gardner SE, Kalan L, Horwinski J, Zheng Q, Hodkinson BP, Tyldsley AS, Franciscus CL, Hillis SL, Mehta S, et al. 2017. Temporal stability in chronic wound microbiota is associated with poor healing. J Invest Dermatol 137: 237–244. doi: 10.1016/j.jid.2016.08.009 [DOI] [PMC free article] [PubMed] [Google Scholar]
- Loots MA, Lamme EN, Zeegelaar J, Mekkes JR, Bos JD, Middelkoop E. 1998. Differences in cellular infiltrate and extracellular matrix of chronic diabetic and venous ulcers versus acute wounds. J Invest Dermatol 111: 850–857. [DOI] [PubMed] [Google Scholar]
- Mahmoudi S, Mancini E, Xu L, Moore A, Jahanbani F, Hebestreit K, Srinivasan R, Li X, Devarajan K, Prélot L, et al. 2019. Heterogeneity in old fibroblasts is linked to variability in reprogramming and wound healing. Nature 574: 553–558. doi: 10.1038/s41586-019-1658-5 [DOI] [PMC free article] [PubMed] [Google Scholar]
- Maione AG, Smith A, Kashpur O, Yanez V, Knight E, Mooney DJ, Veves A, Tomic-Canic M, Garlick JA. 2016. Altered ECM deposition by diabetic foot ulcer-derived fibroblasts implicates fibronectin in chronic wound repair. Wound Repair Regen 24: 630–643. doi: 10.1111/wrr.12437 [DOI] [PMC free article] [PubMed] [Google Scholar]
- Margolis DJ, Hampton M, Hoffstad O, Mala DS, Mirza Z, Woltereck D, Shannon S, Troiano MA, Mitra N, Yang M, et al. 2017. NOS1AP genetic variation is associated with impaired healing of diabetic foot ulcers and diminished response to healing of circulating stem/progenitor cells. Wound Repair Regen 25: 733–736. doi: 10.1111/wrr.12564 [DOI] [PMC free article] [PubMed] [Google Scholar]
- Marjanovic J, Ramirez HA, Jozic I, Stone RC, Wikramanayake TC, Head CR, Abdo Abujamra B, Ojeh N, Kirsner RS, Lev-Tov H, et al. 2022. Dichotomous role of miR193b-3p in diabetic foot ulcers maintains inhibition of healing and suppression of tumor formation. Sci Transl Med 14: eabg8397. doi: 10.1126/scitranslmed.abg8397 [DOI] [PMC free article] [PubMed] [Google Scholar]
- McBride JD, Rodriguez-Menocal L, Badiavas EV. 2017. Extracellular vesicles as biomarkers and therapeutics in dermatology: a focus on exosomes. J Invest Dermatol 137: 1622–1629. doi: 10.1016/j.jid.2017.04.021 [DOI] [PubMed] [Google Scholar]
- McCarty SM, Percival SL. 2013. Proteases and delayed wound healing. Adv Wound Care (New Rochelle) 2: 438–447. doi: 10.1089/wound.2012.0370 [DOI] [PMC free article] [PubMed] [Google Scholar]
- Mehta AK, Gracias DT, Croft M. 2018. TNF activity and T cells. Cytokine 101: 14–18. doi: 10.1016/j.cyto.2016.08.003 [DOI] [PMC free article] [PubMed] [Google Scholar]
- Michopoulou A, Rousselle P. 2015. How do epidermal matrix metalloproteinases support re-epithelialization during skin healing? Eur J Dermatol 25: 33–42. doi: 10.1684/ejd.2015.2553 [DOI] [PubMed] [Google Scholar]
- Mirza AN, Fry MA, Urman NM, Atwood SX, Roffey J, Ott GR, Chen B, Lee A, Brown AS, Aasi SZ, et al. 2017. Combined inhibition of atypical PKC and histone deacetylase 1 is cooperative in basal cell carcinoma treatment. JCI Insight 2: e97071. doi: 10.1172/jci.insight.97071 [DOI] [PMC free article] [PubMed] [Google Scholar]
- Monika P, Waiker PV, Chandraprabha MN, Rangarajan A, Murthy KNC. 2021. Myofibroblast progeny in wound biology and wound healing studies. Wound Repair Regen 29: 531–547. doi: 10.1111/wrr.12937 [DOI] [PubMed] [Google Scholar]
- Morasso MI, Tomic-Canic M. 2005. Epidermal stem cells: the cradle of epidermal determination, differentiation and wound healing. Biol Cell 97: 173–183. doi: 10.1042/BC20040098 [DOI] [PMC free article] [PubMed] [Google Scholar]
- Moura J, Rodrigues J, Gonçalves M, Amaral C, Lima M, Carvalho E. 2017. Impaired T-cell differentiation in diabetic foot ulceration. Cell Mol Immunol 14: 758–769. doi: 10.1038/cmi.2015.116 [DOI] [PMC free article] [PubMed] [Google Scholar]
- Moura J, Rodrigues J, Gonçalves M, Amaral C, Lima M, Carvalho E. 2019. Imbalance in T-cell differentiation as a biomarker of chronic diabetic foot ulceration. Cell Mol Immunol 16: 833–834. doi: 10.1038/s41423-019-0233-1 [DOI] [PMC free article] [PubMed] [Google Scholar]
- Muller M, Trocme C, Lardy B, Morel F, Halimi S, Benhamou PY. 2008. Matrix metalloproteinases and diabetic foot ulcers: the ratio of MMP-1 to TIMP-1 is a predictor of wound healing. Diabet Med 25: 419–426. doi: 10.1111/j.1464-5491.2008.02414.x [DOI] [PMC free article] [PubMed] [Google Scholar]
- Murray PJ, Allen JE, Biswas SK, Fisher EA, Gilroy DW, Goerdt S, Gordon S, Hamilton JA, Ivashkiv LB, Lawrence T, et al. 2014. Macrophage activation and polarization: nomenclature and experimental guidelines. Immunity 41: 14–20. doi: 10.1016/j.immuni.2014.06.008 [DOI] [PMC free article] [PubMed] [Google Scholar]
- Mustoe TA, O’Shaughnessy K, Kloeters O. 2006. Chronic wound pathogenesis and current treatment strategies: a unifying hypothesis. Plast Reconstr Surg 117: 35S–41S. doi: 10.1097/01.prs.0000225431.63010.1b [DOI] [PubMed] [Google Scholar]
- Nassiri S, Zakeri I, Weingarten MS, Spiller KL. 2015. Relative expression of proinflammatory and antiinflammatory genes reveals differences between healing and nonhealing human chronic diabetic foot ulcers. J Invest Dermatol 135: 1700–1703. doi: 10.1038/jid.2015.30 [DOI] [PubMed] [Google Scholar]
- Nishida K, Hasegawa A, Yamasaki S, Uchida R, Ohashi W, Kurashima Y, Kunisawa J, Kimura S, Iwanaga T, Watarai H, et al. 2019. Mast cells play role in wound healing through the ZnT2/GPR39/IL-6 axis. Sci Rep 9: 10842. doi: 10.1038/s41598-019-47132-5 [DOI] [PMC free article] [PubMed] [Google Scholar]
- Nosbaum A, Prevel N, Truong HA, Mehta P, Ettinger M, Scharschmidt TC, Ali NH, Pauli ML, Abbas AK, Rosenblum MD. 2016. Cutting edge: regulatory T cells facilitate cutaneous wound healing. J Immunol 196: 2010–2014. doi: 10.4049/jimmunol.1502139 [DOI] [PMC free article] [PubMed] [Google Scholar]
- Nyström A, Thriene K, Mittapalli V, Kern JS, Kiritsi D, Dengjel J, Bruckner-Tuderman L. 2015. Losartan ameliorates dystrophic epidermolysis bullosa and uncovers new disease mechanisms. EMBO Mol Med 7: 1211–1228. doi: 10.15252/emmm.201505061 [DOI] [PMC free article] [PubMed] [Google Scholar]
- Okonkwo UA, DiPietro LA. 2017. Diabetes and wound angiogenesis. Int J Mol Sci 18: 1419. doi: 10.3390/ijms18071419 [DOI] [PMC free article] [PubMed] [Google Scholar]
- Ongenae KC, Phillips TJ, Park HY. 2000. Level of fibronectin mRNA is markedly increased in human chronic wounds. Dermatol Surg 26: 447–451. doi: 10.1046/j.1524-4725.2000.99281.x [DOI] [PubMed] [Google Scholar]
- Park LK, Maione AG, Smith A, Gerami-Naini B, Iyer LK, Mooney DJ, Veves A, Garlick JA. 2014a. Genome-wide DNA methylation analysis identifies a metabolic memory profile in patient-derived diabetic foot ulcer fibroblasts. Epigenetics 9: 1339–1349. doi: 10.4161/15592294.2014.967584 [DOI] [PMC free article] [PubMed] [Google Scholar]
- Park SA, Raghunathan VK, Shah NM, Teixeira L, Motta MJ, Covert J, Dubielzig R, Schurr M, Isseroff RR, Abbott NL, et al. 2014b. PDGF-BB does not accelerate healing in diabetic mice with splinted skin wounds. PLoS ONE 9: e104447. doi: 10.1371/journal.pone.0104447 [DOI] [PMC free article] [PubMed] [Google Scholar]
- Pastar I, Stojadinovic O, Tomic-Canic M. 2008. Role of keratinocytes in healing of chronic wounds. Surg Technol Int 17: 105–112. [PubMed] [Google Scholar]
- Pastar I, Stojadinovic O, Krzyzanowska A, Barrientos S, Stuelten C, Zimmerman K, Blumenberg M, Brem H, Tomic-Canic M. 2010. Attenuation of the transforming growth factor β-signaling pathway in chronic venous ulcers. Mol Med 16: 92–101. doi: 10.2119/molmed.2009.00149 [DOI] [PMC free article] [PubMed] [Google Scholar]
- Pastar I, Khan AA, Stojadinovic O, Lebrun EA, Medina MC, Brem H, Kirsner RS, Jimenez JJ, Leslie C, Tomic-Canic M. 2012. Induction of specific microRNAs inhibits cutaneous wound healing. J Biol Chem 287: 29324–29335. doi: 10.1074/jbc.M112.382135 [DOI] [PMC free article] [PubMed] [Google Scholar]
- Pastar I, Stojadinovic O, Yin NC, Ramirez H, Nusbaum AG, Sawaya A, Patel SB, Khalid L, Isseroff RR, Tomic-Canic M. 2014. Epithelialization in wound healing: a comprehensive review. Adv Wound Care (New Rochelle) 3: 445–464. doi: 10.1089/wound.2013.0473 [DOI] [PMC free article] [PubMed] [Google Scholar]
- Pastar I, Wong LL, Egger AN, Tomic-Canic M. 2018. Descriptive vs mechanistic scientific approach to study wound healing and its inhibition: is there a value of translational research involving human subjects? Exp Dermatol 27: 551–562. doi: 10.1111/exd.13663 [DOI] [PMC free article] [PubMed] [Google Scholar]
- Pastar I, O’Neill K, Padula L, Head CR, Burgess JL, Chen V, Garcia D, Stojadinovic O, Hower S, Plano GV, et al. 2020. Staphylococcus epidermidis boosts innate immune response by activation of gamma delta T cells and induction of perforin-2 in human skin. Front Immunol 11: 550946. doi: 10.3389/fimmu.2020.550946 [DOI] [PMC free article] [PubMed] [Google Scholar]
- Pastar I, Marjanovic J, Liang L, Stone RC, Kashpur O, Jozic I, Head CR, Smith A, Gerami-Naini B, Garlick JA, et al. 2021a. Cellular reprogramming of diabetic foot ulcer fibroblasts triggers pro-healing miRNA-mediated epigenetic signature. Exp Dermatol 30: 1065–1072. doi: 10.1111/exd.14405 [DOI] [PubMed] [Google Scholar]
- Pastar I, Marjanovic J, Stone RC, Chen V, Burgess JL, Mervis JS, Tomic-Canic M. 2021b. Epigenetic regulation of cellular functions in wound healing. Exp Dermatol 30: 1073–1089.doi: 10.1111/exd.14325 [DOI] [PMC free article] [PubMed] [Google Scholar]
- Pastar I, Sawaya AP, Marjanovic J, Burgess JL, Strbo N, Rivas KE, Wikramanayake TC, Head CR, Stone RC, Jozic I, et al. 2021c. Intracellular Staphylococcus aureus triggers pyroptosis and contributes to inhibition of healing due to perforin-2 suppression. J Clin Invest 131: e133727. doi: 10.1172/JCI133727 [DOI] [PMC free article] [PubMed] [Google Scholar]
- Pauty J, Nakano S, Usuba R, Nakajima T, Johmura Y, Omori S, Sakamoto N, Kikuchi A, Nakanishi M, Matsunaga YT. 2021. A 3D tissue model-on-a-chip for studying the effects of human senescent fibroblasts on blood vessels. Biomater Sci 9: 199–211. doi: 10.1039/D0BM01297A [DOI] [PubMed] [Google Scholar]
- Petersen BJ, Rothenberg GM, Lakhani PJ, Zhou M, Linders DR, Bloom JD, Wood KA, Armstrong DG. 2020. Ulcer metastasis? anatomical locations of recurrence for patients in diabetic foot remission. J Foot Ankle Res 13: 1. doi: 10.1186/s13047-020-0369-3 [DOI] [PMC free article] [PubMed] [Google Scholar]
- Piipponen M, Li D, Landén NX. 2020. The immune functions of keratinocytes in skin wound healing. Int J Mol Sci 21: 8790. doi: 10.3390/ijms21228790 [DOI] [PMC free article] [PubMed] [Google Scholar]
- Pittman K, Kubes P. 2013. Damage-associated molecular patterns control neutrophil recruitment. J Innate Immun 5: 315–323. doi: 10.1159/000347132 [DOI] [PMC free article] [PubMed] [Google Scholar]
- Ramirez HA, Liang L, Pastar I, Rosa AM, Stojadinovic O, Zwick TG, Kirsner RS, Maione AG, Garlick JA, Tomic-Canic M. 2015. Comparative genomic, microRNA, and tissue analyses reveal subtle differences between non-diabetic and diabetic foot skin. PLoS ONE 10: e0137133. doi: 10.1371/journal.pone.0137133 [DOI] [PMC free article] [PubMed] [Google Scholar]
- Ramirez HA, Pastar I, Jozic I, Stojadinovic O, Stone RC, Ojeh N, Gil J, Davis SC, Kirsner RS, Tomic-Canic M. 2018. Staphylococcus aureus triggers induction of miR-15B-5P to diminish DNA repair and deregulate inflammatory response in diabetic foot ulcers. J Invest Dermatol 138: 1187–1196. doi: 10.1016/j.jid.2017.11.038 [DOI] [PMC free article] [PubMed] [Google Scholar]
- Rao CN, Ladin DA, Liu YY, Chilukuri K, Hou ZZ, Woodley DT. 1995. α1-antitrypsin is degraded and non-functional in chronic wounds but intact and functional in acute wounds: the inhibitor protects fibronectin from degradation by chronic wound fluid enzymes. J Invest Dermatol 105: 572–578. doi: 10.1111/1523-1747.ep12323503 [DOI] [PubMed] [Google Scholar]
- Richmond NA, Lamel SA, Davidson JM, Martins-Green M, Sen CK, Tomic-Canic M, Vivas AC, Braun LR, Kirsner RS. 2013. US-national institutes of health-funded research for cutaneous wounds in 2012. Wound Repair Regen 21: 789–792. doi: 10.1111/wrr.12099 [DOI] [PubMed] [Google Scholar]
- Rogers AA, Burnett S, Moore JC, Shakespeare PG, Chen WY. 1995. Involvement of proteolytic enzymes—plasminogen activators and matrix metalloproteinases—in the pathophysiology of pressure ulcers. Wound Repair Regen 3: 273–283. doi: 10.1046/j.1524-475X.1995.30307.x [DOI] [PubMed] [Google Scholar]
- Romagnani P, Lasagni L, Annunziato F, Serio M, Romagnani S. 2004. CXC chemokines: the regulatory link between inflammation and angiogenesis. Trends Immunol 25: 201–209. doi: 10.1016/j.it.2004.02.006 [DOI] [PubMed] [Google Scholar]
- Sawaya AP, Pastar I, Stojadinovic O, Lazovic S, Davis SC, Gil J, Kirsner RS, Tomic-Canic M. 2018. Topical mevastatin promotes wound healing by inhibiting the transcription factor c-Myc via the glucocorticoid receptor and the long non-coding RNA Gas5. J Biol Chem 293: 1439–1449. doi: 10.1074/jbc.M117.811240 [DOI] [PMC free article] [PubMed] [Google Scholar]
- Sawaya AP, Jozic I, Stone RC, Pastar I, Egger AN, Stojadinovic O, Glinos GD, Kirsner RS, Tomic-Canic M. 2019. Mevastatin promotes healing by targeting caveolin-1 to restore EGFR signaling. JCI Insight 4: e129320. doi: 10.1172/jci.insight.129320 [DOI] [PMC free article] [PubMed] [Google Scholar]
- Sawaya AP, Stone RC, Brooks SR, Pastar I, Jozic I, Hasneen K, O’Neill K, Mehdizadeh S, Head CR, Strbo N, et al. 2020. Deregulated immune cell recruitment orchestrated by FOXM1 impairs human diabetic wound healing. Nat Commun 11: 4678. doi: 10.1038/s41467-020-18276-0 [DOI] [PMC free article] [PubMed] [Google Scholar]
- Sawaya AP, Stone RC, Mehdizadeh S, Pastar I, Worrell S, Balukoff NC, Kaplan MJ, Tomic-Canic M, Morasso MI. 2022. FOXM1 network in association with TREM1 suppression regulates NET formation in diabetic foot ulcers. EMBO Rep 23: e54558. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Schneider C, Stratman S, Kirsner RS. 2021. Lower extremity ulcers. Med Clin North Am 105: 663–679. doi: 10.1016/j.mcna.2021.04.006 [DOI] [PubMed] [Google Scholar]
- Sen CK. 2009. Wound healing essentials: let there be oxygen. Wound Repair Regen 17: 1–18. doi: 10.1111/j.1524-475X.2008.00436.x [DOI] [PMC free article] [PubMed] [Google Scholar]
- Shao H, Li Y, Pastar I, Xiao M, Prokupets R, Liu S, Yu K, Vazquez-Padron RI, Tomic-Canic M, Velazquez OC, et al. 2020. Notch1 signaling determines the plasticity and function of fibroblasts in diabetic wounds. Life Sci Alliance 3: e20200076. doi: 10.26508/lsa.202000769 [DOI] [PMC free article] [PubMed] [Google Scholar]
- Sheffield PJ. 1998. Measuring tissue oxygen tension: a review. Undersea Hyperb Med 25: 179–188. [PubMed] [Google Scholar]
- Singh K, Agrawal NK, Gupta SK, Mohan G, Chaturvedi S, Singh K. 2014. Differential expression of matrix metal-loproteinase-9 gene in wounds of type 2 diabetes mellitus cases with susceptible −1562C>T genotypes and wound severity. Int J Low Extrem Wounds 13: 94–102. doi: 10.1177/1534734614534980 [DOI] [PubMed] [Google Scholar]
- Singh K, Pal D, Sinha M, Ghatak S, Gnyawali SC, Khanna S, Roy S, Sen CK. 2017. Epigenetic modification of micro-RNA-200b contributes to diabetic vasculopathy. Mol Ther 25: 2689–2704. doi: 10.1016/j.ymthe.2017.09.009 [DOI] [PMC free article] [PubMed] [Google Scholar]
- Singh K, Maity P, Koroma AK, Basu A, Pandey RK, Vander Beken S, Haas P, Krug L, Hainzl A, Sindrilaru A, et al. 2022. Angiogenin released from ABCB5+ stromal precursors improves healing of diabetic wounds by promoting angiogenesis. J Invest Dermatol 142: 1725–1736.e10. doi: 10.1016/j.jid.2021.10.026 [DOI] [PMC free article] [PubMed] [Google Scholar]
- Stacey MC, Phillips SA, Farrokhyar F, Swaine JM. 2019. Evaluation of wound fluid biomarkers to determine healing in adults with venous leg ulcers: a prospective study. Wound Repair Regen 27: 509–518. doi: 10.1111/wrr.12723 [DOI] [PubMed] [Google Scholar]
- Stanley A, Osler T. 2001. Senescence and the healing rates of venous ulcers. J Vasc Surg 33: 1206–1211. doi: 10.1067/mva.2001.115379 [DOI] [PubMed] [Google Scholar]
- Stanley AC, Park HY, Phillips TJ, Russakovsky V, Menzoian JO. 1997. Reduced growth of dermal fibroblasts from chronic venous ulcers can be stimulated with growth factors. J Vasc Surg 26: 994–999; discussion 999–1001. doi: 10.1016/S0741-5214(97)70012-0 [DOI] [PubMed] [Google Scholar]
- Stojadinovic O, Brem H, Vouthounis C, Lee B, Fallon J, Stallcup M, Merchant A, Galiano RD, Tomic-Canic M. 2005. Molecular pathogenesis of chronic wounds: the role of β-catenin and c-myc in the inhibition of epithelialization and wound healing. Am J Pathol 167: 59–69. doi: 10.1016/S0002-9440(10)62953-7 [DOI] [PMC free article] [PubMed] [Google Scholar]
- Stojadinovic O, Pastar I, Vukelic S, Mahoney MG, Brennan D, Krzyzanowska A, Golinko M, Brem H, Tomic-Canic M. 2008. Deregulation of keratinocyte differentiation and activation: a hallmark of venous ulcers. J Cell Mol Med 12: 2675–2690. doi: 10.1111/j.1582-4934.2008.00321.x [DOI] [PMC free article] [PubMed] [Google Scholar]
- Stojadinovic O, Yin N, Lehmann J, Pastar I, Kirsner RS, Tomic-Canic M. 2013. Increased number of Langerhans cells in the epidermis of diabetic foot ulcers correlates with healing outcome. Immunol Res 57: 222–228. doi: 10.1007/s12026-013-8474-z [DOI] [PMC free article] [PubMed] [Google Scholar]
- Stojadinovic O, Pastar I, Nusbaum AG, Vukelic S, Krzyzanowska A, Tomic-Canic M. 2014. Deregulation of epidermal stem cell niche contributes to pathogenesis of non-healing venous ulcers. Wound Repair Regen 22: 220–227. doi: 10.1111/wrr.12142 [DOI] [PMC free article] [PubMed] [Google Scholar]
- Stone RC, Stojadinovic O, Rosa AM, Ramirez HA, Badiavas E, Blumenberg M, Tomic-Canic M. 2017. A bioengineered living cell construct activates an acute wound healing response in venous leg ulcers. Sci Transl Med 9: eaaf8611. doi: 10.1126/scitranslmed.aaf8611 [DOI] [PMC free article] [PubMed] [Google Scholar]
- Stone RC, Chen V, Burgess J, Pannu S, Tomic-Canic M. 2020a. Genomics of human fibrotic diseases: disordered wound healing response. Int J Mol Sci 21: 8590. doi: 10.3390/ijms21228590 [DOI] [PMC free article] [PubMed] [Google Scholar]
- Stone RC, Stojadinovic O, Sawaya AP, Glinos GD, Lindley LE, Pastar I, Badiavas E, Tomic-Canic M. 2020b. A bioengineered living cell construct activates metallothionein/zinc/MMP8 and inhibits TGFβ to stimulate remodeling of fibrotic venous leg ulcers. Wound Repair Regen 28: 164–176. doi: 10.1111/wrr.12778 [DOI] [PMC free article] [PubMed] [Google Scholar]
- Strbo N, Pastar I, Romero L, Chen V, Vujanac M, Sawaya AP, Jozic I, Ferreira ADF, Wong LL, Head C, et al. 2019. Single cell analyses reveal specific distribution of anti-bacterial molecule perforin-2 in human skin and its modulation by wounding and Staphylococcus aureus infection. Exp Dermatol 28: 225–232. doi: 10.1111/exd.13870 [DOI] [PMC free article] [PubMed] [Google Scholar]
- Subramanian A, Narayan R, Corsello SM, Peck DD, Natoli TE, Lu X, Gould J, Davis JF, Tubelli AA, Asiedu JK, et al. 2017. A next generation connectivity map: L1000 platform and the first 1,000,000 profiles. Cell 171: 1437–1452.e17. doi: 10.1016/j.cell.2017.10.049 [DOI] [PMC free article] [PubMed] [Google Scholar]
- Sutcliffe JE, Chin KY, Thrasivoulou C, Serena TE, O’Neil S, Hu R, White AM, Madden L, Richards T, Phillips AR, et al. 2015. Abnormal connexin expression in human chronic wounds. Br J Dermatol 173: 1205–1215. doi: 10.1111/bjd.14064 [DOI] [PubMed] [Google Scholar]
- Takahashi K, Tanabe K, Ohnuki M, Narita M, Ichisaka T, Tomoda K, Yamanaka S. 2007. Induction of pluripotent stem cells from adult human fibroblasts by defined factors. Cell 131: 861–872. doi: 10.1016/j.cell.2007.11.019 [DOI] [PubMed] [Google Scholar]
- Tandara AA, Mustoe TA. 2004. Oxygen in wound healing—more than a nutrient. World J Surg 28: 294–300. doi: 10.1007/s00268-003-7400-2 [DOI] [PubMed] [Google Scholar]
- Tellechea A, Leal EC, Kafanas A, Auster ME, Kuchibhotla S, Ostrovsky Y, Tecilazich F, Baltzis D, Zheng Y, Carvalho E, et al. 2016. Mast cells regulate wound healing in diabetes. Diabetes 65: 2006–2019. doi: 10.2337/db15-0340 [DOI] [PMC free article] [PubMed] [Google Scholar]
- Tellechea A, Bai S, Dangwal S, Theocharidis G, Nagai M, Koerner S, Cheong JE, Bhasin S, Shih TY, Zheng Y, et al. 2020. Topical application of a mast cell stabilizer improves impaired diabetic wound healing. J Invest Dermatol 140: 901–911.e11. doi: 10.1016/j.jid.2019.08.449 [DOI] [PubMed] [Google Scholar]
- Thackham JA, McElwain DL, Long RJ. 2008. The use of hyperbaric oxygen therapy to treat chronic wounds: a review. Wound Repair Regen 16: 321–330. doi: 10.1111/j.1524-475X.2008.00372.x [DOI] [PubMed] [Google Scholar]
- Theocharidis G, Baltzis D, Roustit M, Tellechea A, Dangwal S, Khetani RS, Shu B, Zhao W, Fu J, Bhasin S, et al. 2020. Integrated skin transcriptomics and serum multiplex as-says reveal novel mechanisms of wound healing in diabetic foot ulcers. Diabetes 69: 2157–2169. doi: 10.2337/db20-0188 [DOI] [PMC free article] [PubMed] [Google Scholar]
- Theocharidis G, Thomas BE, Sarkar D, Mumme HL, Pilcher WJR, Dwivedi B, Sandoval-Schaefer T, Sîrbulescu RF, Kafanas A, Mezghani I, et al. 2022. Single cell transcriptomic landscape of diabetic foot ulcers. Nat Commun 13: 181. doi: 10.1038/s41467-021-27801-8 [DOI] [PMC free article] [PubMed] [Google Scholar]
- Thom SR, Milovanova TN, Yang M, Bhopale VM, Sorokina EM, Uzun G, Malay DS, Troiano MA, Hardy KR, Lambert DS, et al. 2011. Vasculogenic stem cell mobilization and wound recruitment in diabetic patients: increased cell number and intracellular regulatory protein content associated with hyperbaric oxygen therapy. Wound Repair Regen 19: 149–161. doi: 10.1111/j.1524-475X.2010.00660.x [DOI] [PMC free article] [PubMed] [Google Scholar]
- Thom SR, Hampton M, Troiano MA, Mirza Z, Malay DS, Shannon S, Jennato NB, Donohue CM, Hoffstad O, Woltereck D, et al. 2016. Measurements of CD34+/CD45– dim stem cells predict healing of diabetic neuropathic wounds. Diabetes 65: 486–497. doi: 10.2337/db15-0517 [DOI] [PMC free article] [PubMed] [Google Scholar]
- Thorey IS, Roth J, Regenbogen J, Halle JP, Bittner M, Vogl T, Kaesler S, Bugnon P, Reitmaier B, Durka S, et al. 2001. The Ca2+-binding proteins S100A8 and S100A9 are encoded by novel injury-regulated genes. J Biol Chem 276: 35818–35825. doi: 10.1074/jbc.M104871200 [DOI] [PubMed] [Google Scholar]
- Tomic-Canic M, Komine M, Freedberg IM, Blumenberg M. 1998. Epidermal signal transduction and transcription factor activation in activated keratinocytes. J Dermatol Sci 17: 167–181. doi: 10.1016/S0923-1811(98)00016-4 [DOI] [PubMed] [Google Scholar]
- Toulon A, Breton L, Taylor KR, Tenenhaus M, Bhavsar D, Lanigan C, Rudolph R, Jameson J, Havran WL. 2009. A role for human skin-resident T cells in wound healing. J Exp Med 206: 743–750. doi: 10.1084/jem.20081787 [DOI] [PMC free article] [PubMed] [Google Scholar]
- Trautmann A, Toksoy A, Engelhardt E, Brocker EB, Gillitzer R. 2000. Mast cell involvement in normal human skin wound healing: expression of monocyte chemoattractant protein-1 is correlated with recruitment of mast cells which synthesize interleukin-4 in vivo. J Pathol 190: 100–106. doi: [DOI] [PubMed] [Google Scholar]
- Trent JT, Kirsner RS. 2003. Wounds and malignancy. Adv Skin Wound Care 16: 31–34. doi: 10.1097/00129334-200301000-00014 [DOI] [PubMed] [Google Scholar]
- Trøstrup H, Holstein P, Karlsmark T, Moser C, Ågren MS. 2018. Uncontrolled gelatin degradation in non-healing chronic wounds. J Wound Care 27: 724–734. doi: 10.12968/jowc.2018.27.11.724 [DOI] [PubMed] [Google Scholar]
- Vande Berg JS, Rose MA, Haywood-Reid PL, Rudolph R, Payne WG, Robson MC. 2005. Cultured pressure ulcer fibroblasts show replicative senescence with elevated production of plasmin, plasminogen activator inhibitor-1, and transforming growth factor-β1. Wound Repair Regen 13: 76–83. doi: 10.1111/j.1067-1927.2005.130110.x [DOI] [PubMed] [Google Scholar]
- Vukelic S, Stojadinovic O, Pastar I, Vouthounis C, Krzyzanowska A, Das S, Samuels HH, Tomic-Canic M. 2010. Farnesyl pyrophosphate inhibits epithelialization and wound healing through the glucocorticoid receptor. J Biol Chem 285: 1980–1988. doi: 10.1074/jbc.M109.016741 [DOI] [PMC free article] [PubMed] [Google Scholar]
- Waikel RL, Kawachi Y, Waikel PA, Wang XJ, Roop DR. 2001. Deregulated expression of c-Myc depletes epidermal stem cells. Nat Genet 28: 165–168. doi: 10.1038/88889 [DOI] [PubMed] [Google Scholar]
- Wall IB, Moseley R, Baird DM, Kipling D, Giles P, Laffafian I, Price PE, Thomas DW, Stephens P. 2008. Fibroblast dysfunction is a key factor in the non-healing of chronic venous leg ulcers. J Invest Dermatol 128: 2526–2540. doi: 10.1038/jid.2008.114 [DOI] [PubMed] [Google Scholar]
- Wan R, Weissman JP, Grundman K, Lang L, Grybowski DJ, Galiano RD. 2021. Diabetic wound healing: the impact of diabetes on myofibroblast activity and its potential therapeutic treatments. Wound Repair Regen 29: 573–581. doi: 10.1111/wrr.12954 [DOI] [PubMed] [Google Scholar]
- Wang Z, Shi C. 2020. Cellular senescence is a promising target for chronic wounds: a comprehensive review. Burns Trauma 8: tkaa021. doi: 10.1093/burnst/tkaa021 [DOI] [PMC free article] [PubMed] [Google Scholar]
- *.White EK, Grice EA. 2022. The wound microbiome. Cold Spring Harb Perspect Biol doi: 10.1101/cshperspect.a041218 [DOI] [PMC free article] [PubMed] [Google Scholar]
- Wiegand C, Schönfelder U, Abel M, Ruth P, Kaatz M, Hipler UC. 2010. Protease and pro-inflammatory cytokine concentrations are elevated in chronic compared to acute wounds and can be modulated by collagen type I in vitro. Arch Dermatol Res 302: 419–428. doi: 10.1007/s00403-009-1011-1 [DOI] [PubMed] [Google Scholar]
- Wilkinson HN, Clowes C, Banyard KL, Matteuci P, Mace KA, Hardman MJ. 2019. Elevated local senescence in diabetic wound healing is linked to pathological repair via CXCR2. J Invest Dermatol 139: 1171–1181.e6. doi: 10.1016/j.jid.2019.01.005 [DOI] [PubMed] [Google Scholar]
- Wlaschek M, Peus D, Achterberg V, Meyer-Ingold W, Scharffetter-Kochanek K. 1997. Protease inhibitors protect growth factor activity in chronic wounds. Br J Dermatol 137: 646–663. doi: 10.1111/j.1365-2133.1997.tb03804.x [DOI] [PubMed] [Google Scholar]
- Wong SL, Demers M, Martinod K, Gallant M, Wang Y, Goldfine AB, Kahn CR, Wagner DD. 2015. Diabetes primes neutrophils to undergo NETosis, which impairs wound healing. Nat Med 21: 815–819. doi: 10.1038/nm.3887 [DOI] [PMC free article] [PubMed] [Google Scholar]
- Xu P, Fu X, Xiao N, Guo Y, Pei Q, Peng Y, Zhang Y, Yao M. 2017. Involvements of γδT lymphocytes in acute and chronic skin wound repair. Inflammation 40: 1416–1427. doi: 10.1007/s10753-017-0585-6 [DOI] [PubMed] [Google Scholar]
- Xue J, Schmidt SV, Sander J, Draffehn A, Krebs W, Quester I, De Nardo D, Gohel TD, Emde M, Schmidleithner L, et al. 2014. Transcriptome-based network analysis reveals a spectrum model of human macrophage activation. Immunity 40: 274–288. doi: 10.1016/j.immuni.2014.01.006 [DOI] [PMC free article] [PubMed] [Google Scholar]
- Yager DR, Chen SM, Ward SI, Olutoye OO, Diegelmann RF, Kelman Cohen I. 1997. Ability of chronic wound fluids to degrade peptide growth factors is associated with increased levels of elastase activity and diminished levels of proteinase inhibitors. Wound Repair Regen 5: 23–32. doi: 10.1046/j.1524-475X.1997.50108.x [DOI] [PubMed] [Google Scholar]
- Yamakawa S, Hayashida K. 2019. Advances in surgical applications of growth factors for wound healing. Burns Trauma 7: 10. doi: 10.1186/s41038-019-0148-1 [DOI] [PMC free article] [PubMed] [Google Scholar]
- Yang S, Gu Z, Lu C, Zhang T, Guo X, Xue G, Zhang L. 2020. Neutrophil extracellular traps are markers of wound healing impairment in patients with diabetic foot ulcers treated in a multidisciplinary setting. Adv Wound Care (New Rochelle) 9: 16–27. doi: 10.1089/wound.2019.0943 [DOI] [PMC free article] [PubMed] [Google Scholar]
- Yu CG, Zhang N, Yuan SS, Ma Y, Yang LY, Feng YM, Zhao D. 2016. Endothelial progenitor cells in diabetic microvascular complications: friends or foes? Stem Cells Int 2016: 1803989. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Zhan T, Rindtorff N, Boutros M. 2017. Wnt signaling in cancer. Oncogene 36: 1461–1473. doi: 10.1038/onc.2016.304 [DOI] [PMC free article] [PubMed] [Google Scholar]
- Zhang F, Ren Y, Liu P, Ren Y, Wang D. 2016. Expression of TGF-β1 and miRNA-145 in patients with diabetic foot ulcers. Exp Ther Med 11: 2011–2014. doi: 10.3892/etm.2016.3123 [DOI] [PMC free article] [PubMed] [Google Scholar]
- Zhao LL, Davidson JD, Wee SC, Roth SI, Mustoe TA. 1994. Effect of hyperbaric oxygen and growth factors on rabbit ear ischemic ulcers. Arch Surg 129: 1043–1049. doi: 10.1001/archsurg.1994.01420340057010 [DOI] [PubMed] [Google Scholar]
- Zimmermann C, Troeltzsch D, Giménez-Rivera VA, Galli SJ, Metz M, Maurer M, Siebenhaar F. 2019. Mast cells are critical for controlling the bacterial burden and the healing of infected wounds. Proc Natl Acad Sci 116: 20500–20504. doi: 10.1073/pnas.1908816116 [DOI] [PMC free article] [PubMed] [Google Scholar]