

Abstract
Background
Behavioural variant fronto-temporal dementia (bvFTD) is characterised by a progressive change in personality in association with atrophy of the frontal and temporal lobes. Whilst language impairment has been described in people with bvFTD, little is currently known about the extent or type of linguistic difficulties that occur, particularly in the genetic forms.
Methods
Participants with genetic bvFTD along with healthy controls were recruited from the international multicentre Genetic FTD Initiative (GENFI). Linguistic symptoms were assessed using items from the Progressive Aphasia Severity Scale (PASS). Additionally, participants undertook the Boston Naming Test (BNT), modified Camel and Cactus Test (mCCT) and a category fluency test. Participants underwent a 3T volumetric T1-weighted MRI, with language network regional brain volumes measured and compared between the genetic groups and controls.
Results
76% of the genetic bvFTD cohort had impairment in at least one language symptom: 83% C9orf72, 80% MAPT and 56% GRN mutation carriers. All three genetic groups had significantly impaired functional communication, decreased fluency, and impaired sentence comprehension. C9orf72 mutation carriers also had significantly impaired articulation and word retrieval as well as dysgraphia whilst the MAPT mutation group also had impaired word retrieval and single word comprehension. All three groups had difficulties with naming, semantic knowledge and verbal fluency. Atrophy in key left perisylvian language regions differed between the groups, with generalised involvement in the C9orf72 group and more focal temporal and insula involvement in the other groups. Correlates of language symptoms and test scores also differed between the groups.
Conclusions
Language deficits exist in a substantial proportion of people with familial bvFTD across all three genetic groups. Significant atrophy is seen in the dominant perisylvian language areas and correlates with language impairments within each of the genetic groups. Improved understanding of the language phenotype in the main genetic bvFTD subtypes will be helpful in future studies, particularly in clinical trials where accurate stratification and monitoring of disease progression is required.
Supplementary Information
The online version contains supplementary material available at 10.1007/s00415-022-11512-1.
Keywords: Frontotemporal dementia, Genetics, Language, Tau, Progranulin, C9orf72
Background
Frontotemporal dementia (FTD) is a neurodegenerative disorder affecting particularly those under the age of 65 [1]. The most common presentation is the behavioural variant (bvFTD) [2], which is characterised by a progressive change in personality including loss of inhibitory control, apathy, obsessive–compulsive behaviour, reduced empathy and altered dietary preferences [3]. The main cognitive domains affected in bvFTD are executive function and social cognition [3, 4]. However, impairment in other domains has been described including episodic memory [4, 5] and language [4, 6–8].
Language difficulties are the presenting symptoms of the primary progressive aphasias (PPA), another form of FTD, of which there are three main subtypes: nonfluent variant (nfvPPA), where there are difficulties with grammar and/or speech apraxia, semantic variant (svPPA), where there is impaired naming and word comprehension, and logopenic variant (lvPPA), in which there are word retrieval problems [4, 9]. Of note, a number of patients do not fit criteria for any of the three core syndromes, often called PPA-unclassified, atypical PPA, or PPA-not otherwise specified (PPA-NOS) [10–12]. Features of each of the language variants have been described in people with bvFTD, but few studies have been performed to investigate the type or extent of linguistic impairment in bvFTD, and little is known about whether there are particular clinico-pathological associations for any of the symptoms [8, 13, 14].
Around a third of individuals with FTD have an autosomal dominant inheritance [15], with mutations in progranulin (GRN), microtubule-associated protein tau (MAPT) and chromosome 9 open reading frame 72 (C9orf72) being the most common causes [16]. BvFTD is the most frequent phenotype in all of the genetic forms of FTD with only a minority having PPA. However, like in sporadic disease, language problems have been described in people with genetic bvFTD [7, 17], albeit with little known so far about the exact features and whether differences exist between individuals within the main genetic groups (GRN, MAPT and C9orf72). Such information is important, not only to understand the disease better during the symptomatic period of the disorder, but also to further our knowledge of what symptoms to expect (and measure) in the prodromal stage of FTD. This would hopefully allow for improved stratification in clinical trials of disease-modifying treatments as well as more accurate measurement of treatment response.
This study therefore aims to explore the language phenotype of genetic bvFTD within the Genetic FTD Initiative (GENFI) cohort by investigating the linguistic features of the different genetic forms of FTD, including in relation to structural imaging measures.
Methods
Participants
Participants were recruited from the fifth data freeze of the GENFI study between 20 January 2012 and 30 May 2019, including sites in the UK, Canada, Belgium, France, Germany, Italy, the Netherlands, Portugal, Spain and Sweden. All aspects of the study were approved by local ethics committees, and written informed consent was obtained from all participants.
Participants underwent a standardised clinical assessment including a clinical history and neurological examination, neuro-psychometric assessment, the Mini-Mental State Examination (MMSE), and the CDR® plus NACC FTLD [18]. The CDR® plus NACC FTLD was used to classify mutation carriers as asymptomatic (global score of 0), prodromal (score 0.5) or symptomatic (score ≥ 1). To investigate the features of bvFTD, we reviewed all symptomatic mutation carriers recruited in the study and excluded those severely affected (CDR® plus NACC FTLD score of 3 i.e. only included those with a score of 1 or 2). In total, 43 participants met consensus diagnostic criteria for bvFTD [3]: 24 with C9orf72 expansions, 9 with GRN mutations and 10 with MAPT mutations. A comparison group of 100 healthy controls from the GENFI cohort (i.e. family members who did not carry a genetic mutation) was included, matched with the overall bvFTD group on age, sex, and years of education. Demographics are shown in Table 1.
Table 1.
Demographics, clinical scores, severity of linguistic symptoms, cognitive task data and regional brain volumes for the bvFTD groups and healthy controls
| Controls | bvFTD | |||
|---|---|---|---|---|
| C9orf72 | GRN | MAPT | ||
| Number of participants | 100 | 24 | 9 | 10 |
| % Male | 45 | 67 | 44 | 80 |
| % Right-handed | 95 | 96 | 100 | 70 |
| Age (years) | 60.2 (7.1) | 62.3 (7.9) | 67.2 (7.4) | 59.1 (7.7) |
| Education (years) | 13.5 (3.1) | 14.5 (3.6) | 12.0 (3.2) | 13.4 (3.5) |
| MMSE | 29.1 (1.2) | 26.0 (3.4) | 23.9 (5.1) | 24.4 (5.0) |
| CDR® plus NACC FTLD Global score | 0.1 (0.2) | 1.6 (0.5) | 1.7 (0.5) | 1.8 (0.4) |
| CDR® plus NACC FTLD Sum of Boxes | 0.3 (0.6) | 8.5 (3.9) | 8.0 (3.5) | 9.0 (3.1) |
| Progressive Aphasia Severity Scale Sum of Boxes | 0.1 (0.4) | 2.7 (2.6) | 2.2 (2.5) | 3.3 (2.6) |
| Linguistic symptoms | ||||
| Impaired articulation | 0.07 (0.18) | 0.27 (0.53) | 0.06 (0.17) | 0.10 (0.21) |
| Decreased fluency | 0.00 (0.00) | 0.44 (0.61) | 0.39 (0.36) | 0.55 (0.50) |
| Impaired grammar/syntax | 0.00 (0.00) | 0.06 (0.17) | 0.11 (0.33) | 0.15 (0.34) |
| Impaired word retrieval | 0.08 (0.21) | 0.48 (0.56) | 0.44 (0.68) | 0.80 (0.59) |
| Impaired speech repetition | 0.01 (0.05) | 0.04 (0.14) | 0.06 (0.17) | 0.00 (0.00) |
| Impaired sentence comprehension | 0.00 (0.00) | 0.38 (0.61) | 0.22 (0.36) | 0.45 (0.69) |
| Impaired single word comprehension | 0.01 (0.05) | 0.06 (0.17) | 0.00 (0.00) | 0.40 (0.66)ab |
| Dyslexia | 0.02 (0.12) | 0.08 (0.41) | 0.11 (0.33) | 0.15 (0.24) |
| Dysgraphia | 0.01 (0.05) | 0.17 (0.32) | 0.22 (0.44) | 0.15 (0.34) |
| Impaired functional communication | 0.03 (0.13) | 0.73 (0.83) | 0.56 (0.73) | 0.55 (0.44) |
| Cognitive tasks | ||||
| Boston Naming Test (/30) | 27.8 (2.0) | 23.9 (4.0) | 24.3 (4.9) | 17.0 (7.7)ab |
| Modified Camel and Cactus Test (/32) | 30.0 (1.5) | 24.7 (5.2) | 25.1 (4.3) | 24.5 (5.9) |
| Category Fluency (max in 60 s) | 23.3 (5.6) | 13.0 (6.2) | 12.4 (6.6) | 12.7 (5.2) |
| Regional left hemisphere brain volumes (as a % of TIV) | ||||
| Inferior frontal gyrus | 0.56 (0.06) | 0.47 (0.09)c | 0.47 (0.08) | 0.55 (0.06) |
| Insula | 0.36 (0.03) | 0.28 (0.04) | 0.30 (0.02) | 0.26 (0.05)b |
| Motor cortex | 1.36 (0.12) | 1.21 (0.13)c | 1.22 (0.07) | 1.33 (0.12) |
| Temporal pole | 0.50 (0.06) | 0.43 (0.08) | 0.46 (0.07) | 0.35 (0.09)ab |
| Superior temporal gyrus | 0.48 (0.05) | 0.41 (0.04) | 0.42 (0.05) | 0.41 (0.04) |
| Supratemporal region | 0.40 (0.04) | 0.37 (0.04) | 0.35 (0.05) | 0.37 (0.04) |
| Angular gyrus | 0.50 (0.06) | 0.43 (0.05)c | 0.49 (0.08) | 0.50 (0.08) |
Data are shown as mean (standard deviation). Bold items are significantly different to controls
bvFTD behavioural variant fronto-temporal dementia; TIV total intracranial volume
asignificantly impaired compared to C9orf72 mutation carriers
bsignificantly impaired compared to GRN mutation carriers
csignificantly impaired compared to MAPT mutation carriers
Language symptom assessment
Language was assessed by a clinician using the GENFI linguistic symptom scale, which is based on the Progressive Aphasia Severity Scale (PASS) [19]. This contains ten language symptoms scored as per a clinical dementia rating scale i.e. 0 = asymptomatic, 0.5 = questionable/very mild, 1 = mild, 2 = moderate and 3 = severe: impaired articulation, decreased fluency, impaired grammar/syntax, impaired word retrieval, impaired speech repetition, impaired sentence comprehension, impaired single word comprehension, dyslexia, dysgraphia, and impaired functional communication.
Linguistic and non-linguistic cognitive assessment
Within the GENFI neuropsychology battery, the 30-item version of the Boston Naming Test [20, 21] (BNT), the modified Camel and Cactus Test [22] (mCCT) and category fluency (animals) were the linguistic measures used.
The rest of the GENFI neuropsychology battery includes tests of attention and executive function including the Trail Making Test parts A and B (TMTA and TMTB), D-KEFS Colour-Word Inference Test (CWIT), WAIS-R Digit Symbol test, and WMS-R Digit Span Forwards (DSF) and Backward (DSB) as well as tests of visuospatial skills (WASI Block Design), episodic memory (the Free and Cued Selective Reminding Test, FCSRT) and social cognition (mini-Social Cognition and Emotion Assessment, mini-SEA, which includes a Faux Pas test of theory of mind and a Facial Emotion Recognition Test).
Imaging
One hundred and twenty nine participants had a 3T volumetric T1-weighted magnetic resonance imaging (MRI) scan (46 Siemens Prisma, 18 Siemens Trio, 17 Siemens Skyra, 47 Philips Achieva, 1 GE Signa HD) of sufficient quality to be analysed: 37 patients with bvFTD (20 with C9orf72, 8 with GRN, and 9 with MAPT mutations) and 92 controls. Those without scans had either not been scanned due to contraindications or had a poor quality scan due to movement or other artefacts.
Volumetric MRI scans were first bias field-corrected and whole brain parcellated using the geodesic information flow (GIF) algorithm [23], which is based on atlas propagation and label fusion. We focussed on key language regions that were present in the GIF parcellation atlas, calculating grey matter volumes of the cortex for seven left hemisphere perisylvian regions (Fig. 1a): inferior frontal gyrus, insula, motor cortex, temporal pole, superior temporal gyrus, supratemporal region, and angular gyrus. All measures were expressed as a percentage of total intracranial volume (TIV) computed with SPM12 v6470 (Statistical Parametric Mapping, Wellcome Trust Centre for Neuroimaging, London, UK) running under Matlab R2014b (Math Works, Natick, MA, USA) [24].
Fig. 1.

a Left perisylvian regions included in the MR imaging analysis are shown in this artificial representation of the lateral surface of the brain, with the insula and supratemporal region shown in darker blue to represent that they are deeper structures within the sylvian fissure, and b region of interest volumes in each genetic group as a percentage of mean control volume: IFG inferior frontal gyrus; INS insula; MOT motor cortex; TP temporal pole; STG superior temporal gyrus; STR supratemporal region; ANG angular gyrus. The darkest colours represent areas of lowest brain volume as per the key
Statistical analysis
All statistical analyses were performed using Stata/MP 16.1. Statistical tests of normality were performed using the Shapiro–Wilk test. Demographics were compared between groups using either linear regression (age and education) or a chi-squared test (sex). Linear regressions adjusting for age and sex were used to compare the MMSE, CDR® plus NACC FTLD and PASS scores as well as the cognitive tasks and regional brain volumes between groups. Individual linguistic symptoms were compared in each disease group versus controls using linear regressions adjusting for age and sex, and 95% bias-corrected bootstrapped confidence intervals with 2000 repetitions (as there was minimal variation from zero in severity scores for the control group), and between genetic groups using an ordinal logistic regression adjusting for age and sex. Comparison between language-associated brain regions and both individual language symptoms and linguistic tasks was performed using Spearman rank correlations.
Results
Demographics
No significant differences were seen between the groups in years of education, but the GRN mutation carriers were significantly older than controls (p = 0.007) and MAPT mutation carriers (p = 0.020), and the MAPT mutation group had more males than controls (Chi2 = 4.46, p = 0.035) (Table 1).
Disease severity
The MMSE and CDR® plus NACC FTLD Sum of Boxes scores were significantly different to controls in each genetic group, but there were no significant differences between the genetic groups (Table 1).
Language symptoms
76% of the total bvFTD cohort had impairment in at least one language symptom (33 out of 43 participants): 83% of the C9orf72 group, 56% of the GRN group and 80% of the MAPT group. In comparison, only 17% of the controls showed any impairment (Table 1, Fig. 2). However, a significant number of patients in the MAPT and C9orf72 groups only had only one or two language symptoms, with a similar number of cases showing 3 or more language symptoms across all three groups (Fig. 3).
Fig. 2.

The percentage of participants in each of the groups who score 0 = absent, 0.5 = very mild/questionable, 1 = mild, 2 = moderate, or 3 = severe for each linguistic symptom. Values along the x-axis represent the frequency (%) with which the symptom is present in any severity category (0.5–3). An asterisk above the bar indicates that the symptom severity is significantly greater than controls
Fig. 3.
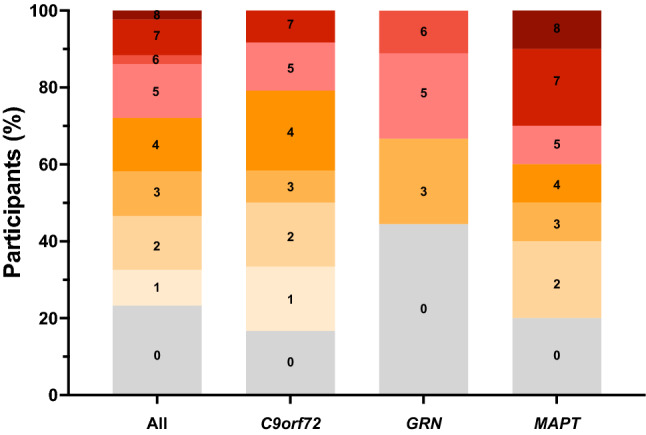
The percentage of participants with a particular number of language symptoms present, including all mutation carriers and each of the mutation groups
All three groups had significantly impaired functional communication compared to controls: severity mean 0.73 (standard deviation 0.83), frequency 58% in the C9orf72 expansion carriers, 0.56 (0.73), 44% in the GRN mutation carriers and 0.55 (0.44), 70% in the MAPT mutation carriers. Similarly, all groups had significantly decreased fluency (0.44 (0.61), 46% in the C9orf72 group, 0.39 (0.36), 56% in the GRN group and 0.55 (0.50), 60% in the MAPT group) and impaired sentence comprehension (0.38 (0.61), 29% in the C9orf72 group, 0.22 (0.36), 33% in the GRN group and 0.45 (0.69), 40% in the MAPT group) compared to controls.
However, the pattern of linguistic symptomatology otherwise varied across the genetic groups. For the C9orf72 group, impaired word retrieval (0.48 (0.56), 50%), impaired articulation (0.27 (0.53), 25%) and dysgraphia (0.17 (0.32), 14%) were significantly different to controls, whilst for the MAPT group, impaired word retrieval (0.80 (0.59), 80%) was significantly different to controls, and impaired single word comprehension (0.40 (0.66), 40%) was significantly different to both the other two genetic groups. No other symptoms were significantly different to controls or the other groups in the GRN mutation carriers.
Cognitive assessment
The C9orf72 and MAPT groups scored significantly lower than controls on the BNT (p < 0.001, Table 1), with a trend to a lower score in the GRN group (p = 0.060). The MAPT mutation carriers scored the lowest of the three groups, significantly lower than the C9orf72 and GRN mutation carrier groups (p = 0.006 and p = 0.008 respectively). All three groups scored significantly lower than controls on the mCCT (C9orf72, p < 0.001; GRN, p = 0.005, MAPT, p = 0.009), and on category fluency (p < 0.001 for all three groups).
As expected, in the other cognitive tasks, all three genetic bvFTD groups showed evidence of executive dysfunction and impaired social cognition (Supplementary Table 1, Additional File 1). Additionally, significantly lower scores on the FCSRT and Block Design were seen in all three groups compared with controls, with DSF also significantly impaired in the C9orf72 group (Supplementary Table 1, Additional File 1).
Imaging analysis
All the left hemisphere regional brain volumes were significantly reduced compared to controls in the C9orf72 expansion carriers (Table 1, Fig. 1b): insula (78% of mean control volume, p < 0.001), inferior frontal gyrus (84%, p = 0.002), superior temporal gyrus (85%, p < 0.001), temporal pole (86%, p < 0.001), angular gyrus (86%, p < 0.001), motor cortex (89%, p < 0.001) and supratemporal region (93%, p = 0.005). Inferior frontal gyrus, motor cortex and angular gyrus were significantly lower in volume compared to MAPT mutation carriers (p = 0.027, 0.012, and 0.005 respectively).
The GRN group had significantly reduced volume compared to controls in the insula (83%, p < 0.001), superior temporal gyrus (88%, p = 0.005), supratemporal region (88%, p = 0.041), and motor cortex (90%, p = 0.003), with a trend to a decreased volume in the inferior frontal gyrus (84%, p = 0.051) (Table 1, Fig. 1b).
The temporal pole was the most atrophied region in the MAPT mutation carriers (70%, p < 0.001) significantly lower than controls and the other mutation groups. The insula (72%, p < 0.001), superior temporal gyrus (85%, p < 0.001) and supratemporal region (93%, p = 0.043) were also significantly lower in volume compared with controls (Table 1, Fig. 1b).
For the linguistic symptoms, in the C9orf72 group, impaired articulation significantly negatively correlated with volume of the inferior frontal gyrus (r = − 0.64, p = 0.002) and motor cortex (r = − 0.59, p = 0.008) whilst decreased fluency also negatively correlated with motor cortex volume (r = − 0.65, p = 0.002) as well as insula volume (r = − 0.54, p = 0.016) (Supplementary Table 2, Additional File 2). Impaired word retrieval negatively correlated with motor cortex volume (r = − 0.51, p = 0.025), and impaired functional communication negatively correlated with insula volume (r = − 0.54, p = 0.009). In the GRN group, impaired sentence comprehension negatively correlated with the volume of temporal regions: temporal pole (r = − 0.73, p = 0.040); superior temporal gyrus (r = − 0.78, p = 0.021), and supratemporal region (r = − 0.73, p = 0.040). Finally, in the MAPT group, decreased fluency (r = − 0.87, p = 0.012), impaired single word comprehension (r = − 0.79, p = 0.034), and dyslexia (r = − 0.79, p = 0.034) all negatively correlated with inferior frontal gyrus volume, whilst impaired sentence comprehension negatively correlated with angular gyrus volume (r = − 0.80, p = 0.030).
In the C9orf72 group, there was a strong positive correlation between scores on the linguistic cognitive tasks and left inferior frontal gyrus, left insula and left angular gyrus volumes (Table 2): BNT (inferior frontal gyrus r = 0.50, p = 0.029; angular gyrus r = 0.77, p = < 0.001); mCCT (inferior frontal gyrus r = 0.47, p = 0.044; insula r = 0.61, p = 0.005; angular gyrus r = 0.47, p = 0.043); CF (inferior frontal gyrus r = 0.67, p = 0.002; insula r = 0.50, p = 0.029). In the GRN group, there was a significant positive correlation between BNT score and temporal volumes: superior temporal gyrus r = 0.82, p = 0.013; supratemporal region r = 0.80, p = 0.018. Although there were no significant correlations of mCCT score with volumes, CF score was correlated with left insula and angular gyrus volumes (r = 0.86, p = 0.007; r = 0.74, p = 0.038 respectively). In MAPT mutation carriers, a strong positive correlation was seen between mCCT score and left superior temporal gyrus volume (r = 0.93, p = 0.003), but no significant correlations were seen for the BNT or CF scores.
Table 2.
Correlations between the linguistic cognitive tasks and left hemisphere regional brain volumes in each genetic group
| BNT | mCCT | CF | ||
|---|---|---|---|---|
| Inferior frontal gyrus | C9orf72 | 0.50 | 0.47 | 0.67 |
| GRN | − 0.19 | 0.00 | 0.58 | |
| MAPT | − 0.14 | − 0.11 | 0.16 | |
| Insula | C9orf72 | 0.35 | 0.61 | 0.50 |
| GRN | 0.16 | 0.21 | 0.86 | |
| MAPT | 0.63 | 0.48 | 0.34 | |
| Motor cortex | C9orf72 | 0.12 | 0.40 | 0.37 |
| GRN | 0.35 | 0.31 | 0.45 | |
| MAPT | − 0.34 | − 0.07 | 0.41 | |
| Temporal pole | C9orf72 | 0.06 | 0.05 | − 0.20 |
| GRN | 0.43 | 0.38 | 0.46 | |
| MAPT | 0.41 | 0.15 | − 0.20 | |
| Superior temporal gyrus | C9orf72 | 0.08 | 0.33 | 0.11 |
| GRN | 0.82 | 0.48 | 0.17 | |
| MAPT | 0.45 | 0.93 | 0.56 | |
| Supratemporal region | C9orf72 | 0.26 | 0.14 | 0.17 |
| GRN | 0.80 | 0.14 | − 0.02 | |
| MAPT | 0.67 | 0.56 | 0.23 | |
| Angular gyrus | C9orf72 | 0.77 | 0.47 | 0.36 |
| GRN | 0.06 | 0.21 | 0.74 | |
| MAPT | − 0.74 | − 0.26 | − 0.05 |
Rho is shown for each correlation, with significant values shown in bold
BNT Boston naming test; mCCT modified Camel and Cactus Test; CF category fluency
Discussion
In this study, we have shown that language impairment is present in a substantial proportion of people with the genetic form of bvFTD. Furthermore, significant atrophy is seen in the dominant perisylvian language regions and correlates with linguistic features in each of the three genetic groups. Three symptoms were seen significantly more than controls in all three genetic groups: impaired functional communication, decreased fluency and impaired sentence comprehension. Each group also had problems with naming, semantic knowledge and verbal fluency. However, differences were also present, with the MAPT mutation group having difficulties with single word comprehension and the C9orf72 group having impaired articulation and dysgraphia. Similarly, whilst the three genetic groups had overlapping left insula atrophy, there were differences between them, with inferior frontal involvement particularly in the C9orf72 group, anterior temporal atrophy in MAPT mutation carriers, and superior temporal volume loss in the GRN mutation carriers.
All three groups had difficulties with impaired functional communication. Whilst this could be in part due to individual or combined linguistic deficits, previous studies have shown that people with bvFTD can be impaired in conversational discourse and the pragmatics of language [25, 26]. People with bvFTD can have difficulties with participating in communication in the first place [26], as well as how best to communicate effectively [25]. These deficits are likely to arise from a combination of executive function and social cognition deficits that are present here in each of the three genetic groups [27, 28].
Similarly, impaired sentence comprehension was seen in all three groups. Difficulties in this domain can be due to abnormal grammatical processing, but this was not significantly impaired in any of the groups within this cohort. Problems in sentence comprehension can also be due to the inability to hold the sentence in short-term memory, as is seen in people with lvPPA, with shorter sentences easier to comprehend than longer ones [29]. The C9orf72 mutation carriers had significantly shorter forwards digit span than controls (with a trend in GRN mutation carriers), but whilst this may have contributed to impaired sentence comprehension, this deficit in short-term memory was not as severe as that seen in lvPPA. Lastly, separate to grammar and short-term memory, prior research in FTD has suggested a role for decreased executive resources as a contributor to impaired sentence comprehension [30, 31], and certainly all three groups had significant executive dysfunction on testing here. It may well be therefore that a combination of executive dysfunction and impaired short-term memory leads to impaired sentence comprehension across the different groups.
Also seen in all three groups was decreased fluency. This can be caused by multiple underlying linguistic deficits including changes in articulation, grammar and word retrieval as well as an impairment of word generation or prosody [32]. The C9orf72 mutation group had significant articulatory deficits and both C9orf72 and MAPT mutation groups had word retrieval problems, but all three groups also had decreased verbal fluency scores compared to controls. Word generation difficulties are a major contributor to decreased verbal fluency scores in bvFTD [33], with associated executive dysfunction being an important cause [34]. Prosodic changes have also previously been noted in bvFTD with changes in different aspects of speech timing found in one study [32]. It is therefore likely that the decreased fluency noted on the linguistic symptom scale here is multifactorial in each group with contributions from different factors.
The C9orf72 mutation carriers had the most language impairment of any of the groups, with 83% having deficits in at least one symptom. As well as the difficulties above, 25% of patients were noted to have impaired articulation. It was not noted if this was due to apraxia of speech, dysarthria or other reasons in the symptom scales, but the score did correlate with atrophy in the inferior frontal gyrus and motor cortex. None of these carriers had an associated diagnosis of amyotrophic lateral sclerosis (ALS), but prior studies have shown the presence of motor deficits in people with FTD that do not meet criteria for ALS [35]. Correlation with inferior frontal cortex volume and/or the insula was seen also for decreased fluency and performance on the cognitive language tasks, suggesting that the impairments are not just down to motor speech deficits, but that there are true language-based speech production difficulties as well. The C9orf72 group had more widespread involvement of the perisylvian language areas in general, including more posterior regions like the angular gyrus. Interestingly, scores on the BNT and mCCT both correlated significantly with angular gyrus atrophy. This region forms part of what has been called Geschwind’s area and is associated with multiple language functions [36] including word retrieval and semantic processing as well as writing, which is also impaired in the C9orf72 mutation carrier group. It is therefore likely that the more extensive linguistic deficits seen in this group relate to the wider involvement of key linguistic regions in the brain as the disease progresses in addition to a contribution from executive dysfunction. As mentioned above, this latter cognitive deficit is likely to be a factor in a number of apparent language problems [34], including the decreased score in the mCCT, which has previously been shown to be related to the executive control of semantic knowledge [22].
80% of the MAPT mutation group had linguistic symptoms with single word comprehension being impaired more than the other two groups. The MAPT group also included individuals with the greatest number of language symptoms affected, and with the lowest naming scores of all three groups. The group also had impairments in the mCCT and in verbal fluency. Previous studies of MAPT-associated bvFTD have highlighted the presence of semantic impairment, a feature that can occur quite early in the disease process, even prodromally [22, 37, 38]. This is generally associated with atrophy of the anterior temporal lobes (the ‘semantic hub’), as is seen prominently here, with mCCT score correlating with temporal lobe atrophy. Interestingly, there was also a correlation of decreased fluency, impaired single word comprehension and dyslexia with inferior frontal gyrus volume in this group, suggesting more wider contributions to the language deficits.
The GRN-bvFTD had the least linguistic symptomatology of all the groups. This sits in contrast to GRN mutations being the commonest cause of PPA in genetic FTD, with usually either a nonfluent variant or a mixed (not otherwise specified) phenotype [12, 39]. It may be that many of the linguistic deficits seen here are related to the executive dysfunction found in the group, but in fact impaired sentence comprehension as well as BNT score correlated with temporal lobe volumes, suggesting a primary linguistic impairment, potentially of semantics within the GRN group.
An important question arises about what diagnosis should be given to people who have bvFTD but also early, prominent language deficits. The current diagnostic criteria are poorly equipped for situations like this e.g. to fulfil a diagnosis of PPA the ‘most prominent clinical feature’ must be ‘difficulty with language’ i.e. there is no current option to make a dual diagnosis of bvFTD with PPA (or even ‘secondary’ progressive aphasia). Future revisions of the diagnostic criteria should consider this issue.
Limitations
In general, despite a well-defined cohort in the GENFI study, once groups were stratified, the numbers that could be studied were much smaller. Further studies of larger genetic FTD cohorts to replicate these findings will be helpful. Another limitation was the limited availability of language cognitive tests within the GENFI battery. There is a lack of validated cross-language verbal linguistic tasks in general and, with multiple languages represented in the GENFI study, the cognitive battery has mostly non-verbal or already validated tasks. Future studies should therefore develop novel cross-language tasks with normative data that can be used in multinational studies like GENFI.
Conclusions
Despite a primary bvFTD diagnosis, language problems are present extensively in genetic FTD, with overlapping but distinct patterns of linguistic deficits and associated brain atrophy. Improved understanding of the relationship between bvFTD and its language phenotype will aid more focussed assessments and interpretations of data within FTD studies. This in turn will guide the future stratification of individuals within clinical trials as well as the monitoring of disease progression and treatment response.
Supplementary Information
Below is the link to the electronic supplementary material.
Acknowledgements
We thank the research participants and their families for their contribution to the study. Several authors of this publication are members of the European Reference Network for Rare Neurological Diseases—Project ID No 739510. The Dementia Research Centre is supported by Alzheimer’s Research UK, Alzheimer’s Society, Brain Research UK, and The Wolfson Foundation. This work was supported by the NIHR UCL/H Biomedical Research Centre, the Leonard Wolfson Experimental Neurology Centre (LWENC) Clinical Research Facility, and the UK Dementia Research Institute, which receives its funding from UK DRI Ltd, funded by the UK Medical Research Council, Alzheimer's Society and Alzheimer’s Research UK. This work was also supported by the JPND GENFI-PROX grant (2019-02248; to JDR, MO, BB, CG, JvS and MS. [latter via DLR/DFG 01ED2008B]). JDR is supported by the Miriam Marks Brain Research UK Senior Fellowship and has received funding from an MRC Clinician Scientist Fellowship (MR/M008525/1) and the NIHR Rare Disease Translational Research Collaboration (BRC149/NS/MH). This work was also supported by the MRC UK GENFI grant (MR/M023664/1), the Bluefield Project and the JPND GENFI-PROX grant (2019-02248). Several authors of this publication are members of the European Reference Network for Rare Neurological Diseases—Project ID No 739510. RC/CG are supported by a Frontotemporal Dementia Research Studentships in Memory of David Blechner funded through The National Brain Appeal (RCN 290173). MB is supported by a Fellowship award from the Alzheimer’s Society, UK (AS-JF-19a-004-517). MB’s work is also supported by the UK Dementia Research Institute which receives its funding from DRI Ltd, funded by the UK Medical Research Council, Alzheimer’s Society and Alzheimer’s Research UK. JCVS was supported by the Dioraphte Foundation grant 09-02-03-00, the Association for Frontotemporal Dementias Research Grant 2009, The Netherlands Organization for Scientific Research (NWO) grant HCMI 056-13-018, ZonMw Memorabel (Deltaplan Dementie, project number 733 051 042), Alzheimer Nederland and the Bluefield project. FM received funding from the Tau Consortium and the Center for Networked Biomedical Research on Neurodegenerative Disease (CIBERNED). RS-V is supported by an Alzheimer’s Research UK Clinical Research Training Fellowship (ARUK-CRF2017B-2), and has received funding from Fundació Marató de TV3, Spain (grant no. 20143810). CG received funding from JPND-Prefrontals VR Dnr 529-2014-7504, VR 2015-02926 and 2018-02754, the Swedish FTD Inititative-Schörling Foundation, Alzheimer Foundation, Brain Foundation and Stockholm County Council ALF. MM has received funding from a Canadian Institute of Health Research operating grant and the Weston Brain Institute and Ontario Brain Institute. JBR has received funding from the Welcome Trust (103838) and is supported by the Cambridge University Centre for Frontotemporal Dementia, the Medical Research Council (SUAG/051 G101400) and the National Institute for Health Research (NIHR) Cambridge Biomedical Research Centre (BRC-1215-20014). EF has received funding from a CIHR grant #327387. DG received support from the EU Joint Programme—Neurodegenerative Disease Research (JPND) and the Italian Ministry of Health (PreFrontALS) grant 733051042. RV has received funding from the Mady Browaeys Fund for Research into Frontotemporal Dementia. MO has received funding from BMBF (FTLDc). JL received funding for this work by the Deutsche Forschungsgemeinschaft (DFG, German Research Foundation) under Germany’s Excellence Strategy within the framework of the Munich Cluster for Systems Neurology (EXC 2145 SyNergy—ID 390857198). Group authorship for the Genetic FTD Initiative (GENFI): Annabel Nelson: Department of Neurodegenerative Disease, Dementia Research Centre, UCL Queen Square Institute of Neurology, London, UK. David L Thomas: Neuroimaging Analysis Centre, Department of Brain Repair and Rehabilitation, UCL Institute of Neurology, Queen Square, London, UK. Emily Todd: Department of Neurodegenerative Disease, Dementia Research Centre, UCL Queen Square Institute of Neurology, London, UK. Hanya Benotmane: UK Dementia Research Institute at University College London, UCL Queen Square Institute of Neurology, London, UK. Jennifer Nicholas: Department of Medical Statistics, London School of Hygiene and Tropical Medicine, London, UK. Rachelle Shafei: Department of Neurodegenerative Disease, Dementia Research Centre, UCL Queen Square Institute of Neurology, London, UK. Carolyn Timberlake: Department of Clinical Neurosciences, University of Cambridge, Cambridge, UK. Thomas Cope: Department of Clinical Neuroscience, University of Cambridge, Cambridge, UK. Timothy Rittman: Department of Clinical Neurosciences, University of Cambridge, Cambridge, UK. Alberto Benussi: Centre for Neurodegenerative Disorders, Department of Clinical and Experimental Sciences, University of Brescia, Brescia, Italy. Enrico Premi: Stroke Unit, ASST Brescia Hospital, Brescia, Italy. Roberto Gasparotti: Neuroradiology Unit, University of Brescia, Brescia, Italy. Silvana Archetti: Biotechnology Laboratory, Department of Diagnostics, ASST Brescia Hospital, Brescia, Italy. Stefano Gazzina: Neurology, ASST Brescia Hospital, Brescia, Italy. Valentina Cantoni: Centre for Neurodegenerative Disorders, Department of Clinical and Experimental Sciences, University of Brescia, Brescia, Italy. Andrea Arighi: Fondazione IRCCS Ca’ Granda Ospedale Maggiore Policlinico, Neurodegenerative Diseases Unit, Milan, Italy; University of Milan, Centro Dino Ferrari, Milan, Italy. Chiara Fenoglio: Fondazione IRCCS Ca’ Granda Ospedale Maggiore Policlinico, Neurodegenerative Diseases Unit, Milan, Italy; University of Milan, Centro Dino Ferrari, Milan, Italy. Elio Scarpini: Fondazione IRCCS Ca’ Granda Ospedale Maggiore Policlinico, Neurodegenerative Diseases Unit, Milan, Italy; University of Milan, Centro Dino Ferrari, Milan, Italy. Giorgio Fumagalli: Fondazione IRCCS Ca’ Granda Ospedale Maggiore Policlinico, Neurodegenerative Diseases Unit, Milan, Italy; University of Milan, Centro Dino Ferrari, Milan, Italy. Vittoria Borracci: Fondazione IRCCS Ca’ Granda Ospedale Maggiore Policlinico, Neurodegenerative Diseases Unit, Milan, Italy; University of Milan, Centro Dino Ferrari, Milan, Italy. Giacomina Rossi: Fondazione IRCCS Istituto Neurologico Carlo Besta, Milano, Italy. Giorgio Giaccone: Fondazione IRCCS Istituto Neurologico Carlo Besta, Milano, Italy. Giuseppe Di Fede: Fondazione IRCCS Istituto Neurologico Carlo Besta, Milano, Italy. Paola Caroppo: Fondazione IRCCS Istituto Neurologico Carlo Besta, Milano, Italy. Pietro Tiraboschi: Fondazione IRCCS Istituto Neurologico Carlo Besta, Milano, Italy. Sara Prioni: Fondazione IRCCS Istituto Neurologico Carlo Besta, Milano, Italy. Veronica Redaelli: Fondazione IRCCS Istituto Neurologico Carlo Besta, Milano, Italy. David Tang-Wai: The University Health Network, Krembil Research Institute, Toronto, Canada. Ekaterina Rogaeva: Tanz Centre for Research in Neurodegenerative Diseases, University of Toronto, Toronto, Canada. Miguel Castelo-Branco: Faculty of Medicine, University of Coimbra, Coimbra, Portugal. Morris Freedman: Baycrest Health Sciences, Rotman Research Institute, University of Toronto, Toronto, Canada. Ron Keren: The University Health Network, Toronto Rehabilitation Institute, Toronto, Canada. Sandra Black: Sunnybrook Health Sciences Centre, Sunnybrook Research Institute, University of Toronto, Toronto, Canada. Sara Mitchell: Sunnybrook Health Sciences Centre, Sunnybrook Research Institute, University of Toronto, Toronto, Canada. Christen Shoesmith: Department of Clinical Neurological Sciences, University of Western Ontario, London, Ontario, Canada. Robart Bartha: Department of Medical Biophysics, The University of Western Ontario, London, Ontario, Canada; Centre for Functional and Metabolic Mapping, Robarts Research Institute, The University of Western Ontario, London, Ontario, Canada. Rosa Rademakers: Center for Molecular Neurology, University of Antwerp. Jackie Poos: Department of Neurology, Erasmus Medical Center, Rotterdam, Netherlands. Janne M. Papma: Department of Neurology, Erasmus Medical Center, Rotterdam, Netherlands. Lucia Giannini: Department of Neurology, Erasmus Medical Center, Rotterdam, Netherlands. Rick van Minkelen: Department of Clinical Genetics, Erasmus Medical Center, Rotterdam, Netherlands. Yolande Pijnenburg: Amsterdam University Medical Centre, Amsterdam VUmc, Amsterdam, Netherlands. Benedetta Nacmias: Department of Neuroscience, Psychology, Drug Research and Child Health, University of Florence, Florence, Italy. Camilla Ferrari: Department of Neuroscience, Psychology, Drug Research and Child Health, University of Florence, Florence, Italy. Cristina Polito: Department of Biomedical, Experimental and Clinical Sciences “Mario Serio”, Nuclear Medicine Unit, University of Florence, Florence, Italy. Gemma Lombardi: Department of Neuroscience, Psychology, Drug Research and Child Health, University of Florence, Florence, Italy. Valentina Bessi: Department of Neuroscience, Psychology, Drug Research and Child Health, University of Florence, Florence, Italy. Michele Veldsman: Nuffield Department of Clinical Neurosciences, Medical Sciences Division, University of Oxford, Oxford, UK. Christin Andersson: Department of Clinical Neuroscience, Karolinska Institutet, Stockholm, Sweden. Hakan Thonberg: Center for Alzheimer Research, Division of Neurogeriatrics, Karolinska Institutet, Stockholm, Sweden. Linn Öijerstedt: Center for Alzheimer Research, Division of Neurogeriatrics, Department of Neurobiology, Care Sciences and Society, Bioclinicum, Karolinska Institutet, Solna, Sweden; Unit for Hereditary Dementias, Theme Aging, Karolinska University Hospital, Solna, Sweden. Vesna Jelic: Division of Clinical Geriatrics, Karolinska Institutet, Stockholm, Sweden. Paul Thompson: Division of Neuroscience and Experimental Psychology, Wolfson Molecular Imaging Centre, University of Manchester, Manchester, UK. Tobias Langheinrich: Division of Neuroscience and Experimental Psychology, Wolfson Molecular Imaging Centre, University of Manchester, Manchester, UK; Manchester Centre for Clinical Neurosciences, Department of Neurology, Salford Royal NHS Foundation Trust, Manchester, UK. Albert Lladó: Alzheimer’s disease and Other Cognitive Disorders Unit, Neurology Service, Hospital Clínic, Barcelona, Spain. Anna Antonell: Alzheimer’s disease and Other Cognitive Disorders Unit, Neurology Service, Hospital Clínic, Barcelona, Spain. Jaume Olives: Alzheimer’s disease and Other Cognitive Disorders Unit, Neurology Service, Hospital Clínic, Barcelona, Spain. Mircea Balasa: Alzheimer’s disease and Other Cognitive Disorders Unit, Neurology Service, Hospital Clínic, Barcelona, Spain. Nuria Bargalló: Imaging Diagnostic Center, Hospital Clínic, Barcelona, Spain. Sergi Borrego-Ecija: Alzheimer’s disease and Other Cognitive Disorders Unit, Neurology Service, Hospital Clínic, Barcelona, Spain. Ana Verdelho: Department of Neurosciences and Mental Health, Centro Hospitalar Lisboa Norte—Hospital de Santa Maria & Faculty of Medicine, University of Lisbon, Lisbon, Portugal. Carolina Maruta: Laboratory of Language Research, Centro de Estudos Egas Moniz, Faculty of Medicine, University of Lisbon, Lisbon, Portugal. Catarina B. Ferreira: Laboratory of Neurosciences, Faculty of Medicine, University of Lisbon, Lisbon, Portugal. Gabriel Miltenberger: Faculty of Medicine, University of Lisbon, Lisbon, Portugal. Frederico Simões do Couto: Faculdade de Medicina, Universidade Católica Portuguesa. Alazne Gabilondo: Cognitive Disorders Unit, Department of Neurology, Donostia University Hospital, San Sebastian, Gipuzkoa, Spain; Neuroscience Area, Biodonostia Health Research Insitute, San Sebastian, Gipuzkoa, Spain. Ana Gorostidi: Neuroscience Area, Biodonostia Health Research Insitute, San Sebastian, Gipuzkoa, Spain. Jorge Villanua: OSATEK, University of Donostia, San Sebastian, Gipuzkoa, Spain. Marta Cañada: CITA Alzheimer, San Sebastian, Gipuzkoa, Spain. Mikel Tainta: Neuroscience Area, Biodonostia Health Research Insitute, San Sebastian, Gipuzkoa, Spain. Miren Zulaica: Neuroscience Area, Biodonostia Health Research Insitute, San Sebastian, Gipuzkoa, Spain. Myriam Barandiaran: Cognitive Disorders Unit, Department of Neurology, Donostia University Hospital, San Sebastian, Gipuzkoa, Spain; Neuroscience Area, Biodonostia Health Research Insitute, San Sebastian, Gipuzkoa, Spain. Patricia Alves: Neuroscience Area, Biodonostia Health Research Insitute, San Sebastian, Gipuzkoa, Spain; Department of Educational Psychology and Psychobiology, Faculty of Education, International University of La Rioja, Logroño, Spain. Benjamin Bender: Department of Diagnostic and Interventional Neuroradiology, University of Tübingen, Tübingen, Germany. Carlo Wilke: Department of Neurodegenerative Diseases, Hertie-Institute for Clinical Brain Research and Center of Neurology, University of Tübingen, Tübingen, Germany; Center for Neurodegenerative Diseases (DZNE), Tübingen, Germany. Lisa Graf: Department of Neurodegenerative Diseases, Hertie-Institute for Clinical Brain Research and Center of Neurology, University of Tübingen, Tübingen, Germany. Annick Vogels: Department of Human Genetics, KU Leuven, Leuven, Belgium. Mathieu Vandenbulcke: Geriatric Psychiatry Service, University Hospitals Leuven, Belgium; Neuropsychiatry, Department of Neurosciences, KU Leuven, Leuven, Belgium. Philip Van Damme: Neurology Service, University Hospitals Leuven, Belgium; Laboratory for Neurobiology, VIB-KU Leuven Centre for Brain Research, Leuven, Belgium. Rose Bruffaerts: Department of Biomedical Sciences, University of Antwerp, Antwerp, Belgium; Biomedical Research Institute, Hasselt University, 3500 Hasselt, Belgium. Koen Poesen: Laboratory for Molecular Neurobiomarker Research, KU Leuven, Leuven, Belgium. Pedro Rosa-Neto: Translational Neuroimaging Laboratory, McGill Centre for Studies in Aging, McGill University, Montreal, Québec, Canada. Serge Gauthier: Alzheimer Disease Research Unit, McGill Centre for Studies in Aging, Department of Neurology & Neurosurgery, McGill University, Montreal, Québec, Canada. Agnès Camuzat: Sorbonne Université, Paris Brain Institute—Institut du Cerveau—ICM, Inserm U1127, CNRS UMR 7225, AP-HP—Hôpital Pitié-Salpêtrière, Paris, France. Alexis Brice: Sorbonne Université, Paris Brain Institute—Institut du Cerveau—ICM, Inserm U1127, CNRS UMR 7225, AP-HP—Hôpital Pitié-Salpêtrière, Paris, France; Reference Network for Rare Neurological Diseases (ERN-RND). Anne Bertrand: Sorbonne Université, Paris Brain Institute—Institut du Cerveau—ICM, Inserm U1127, CNRS UMR 7225, AP-HP—Hôpital Pitié-Salpêtrière, Paris, France; Inria, Aramis project-team, F-75013, Paris, France; Centre pour l'Acquisition et le Traitement des Images, Institut du Cerveau et la Moelle, Paris, France. Aurélie Funkiewiez: Centre de référence des démences rares ou précoces, IM2A, Département de Neurologie, AP-HP—Hôpital Pitié-Salpêtrière, Paris, France; Sorbonne Université, Paris Brain Institute – Institut du Cerveau—ICM, Inserm U1127, CNRS UMR 7225, AP-HP—Hôpital Pitié-Salpêtrière, Paris, France. Daisy Rinaldi: Centre de référence des démences rares ou précoces, IM2A, Département de Neurologie, AP-HP—Hôpital Pitié-Salpêtrière, Paris, France; Sorbonne Université, Paris Brain Institute—Institut du Cerveau—ICM, Inserm U1127, CNRS UMR 7225, AP-HP—Hôpital Pitié-Salpêtrière, Paris, France; Département de Neurologie, AP-HP—Hôpital Pitié-Salpêtrière, Paris, France. Dario Saracino: Sorbonne Université, Paris Brain Institute – Institut du Cerveau—ICM, Inserm U1127, CNRS UMR 7225, AP-HP—Hôpital Pitié-Salpêtrière, Paris, France; Inria, Aramis project-team, F-75013, Paris, France; Centre de référence des démences rares ou précoces, IM2A, Département de Neurologie, AP-HP—Hôpital Pitié-Salpêtrière, Paris, France. Olivier Colliot: Sorbonne Université, Paris Brain Institute—Institut du Cerveau—ICM, Inserm U1127, CNRS UMR 7225, AP-HP—Hôpital Pitié-Salpêtrière, Paris, France; Inria, Aramis project-team, F-75013, Paris, France; Centre pour l'Acquisition et le Traitement des Images, Institut du Cerveau et la Moelle, Paris, France. Sabrina Sayah: Sorbonne Université, Paris Brain Institute—Institut du Cerveau—ICM, Inserm U1127, CNRS UMR 7225, AP-HP—Hôpital Pitié-Salpêtrière, Paris, France. Catharina Prix: Neurologische Klinik, Ludwig-Maximilians-Universität München, Munich, Germany. Elisabeth Wlasich: Neurologische Klinik, Ludwig-Maximilians-Universität München, Munich, Germany. Olivia Wagemann: Neurologische Klinik, Ludwig-Maximilians-Universität München, Munich, Germany. Sandra Loosli: Neurologische Klinik, Ludwig-Maximilians-Universität München, Munich, Germany. Sonja Schönecker: Neurologische Klinik, Ludwig-Maximilians-Universität München, Munich, Germany. Tobias Hoegen: Neurologische Klinik, Ludwig-Maximilians-Universität München, Munich, Germany. Jolina Lombardi: Department of Neurology, University of Ulm, Ulm. Sarah Anderl-Straub: Department of Neurology, University of Ulm, Ulm, Germany. Adeline Rollin: CHU, CNR-MAJ, Labex Distalz, LiCEND Lille, France. Gregory Kuchcinski: Univ Lille, France; Inserm 1172, Lille, France; CHU, CNR-MAJ, Labex Distalz, LiCEND Lille, France. Maxime Bertoux: Inserm 1172, Lille, France; CHU, CNR-MAJ, Labex Distalz, LiCEND Lille, France. Thibaud Lebouvier: Univ Lille, France; Inserm 1172, Lille, France; CHU, CNR-MAJ, Labex Distalz, LiCEND Lille, France. Vincent Deramecourt: Univ Lille, France; Inserm 1172, Lille, France; CHU, CNR-MAJ, Labex Distalz, LiCEND Lille, France. Beatriz Santiago: Neurology Department, Centro Hospitalar e Universitario de Coimbra, Coimbra, Portugal. Diana Duro: Faculty of Medicine, University of Coimbra, Coimbra, Portugal. Maria João Leitão: Centre of Neurosciences and Cell Biology, Universidade de Coimbra, Coimbra, Portugal. Maria Rosario Almeida: Faculty of Medicine, University of Coimbra, Coimbra, Portugal. Miguel Tábuas-Pereira: Neurology Department, Centro Hospitalar e Universitario de Coimbra, Coimbra, Portugal. Sónia Afonso: Instituto Ciencias Nucleares Aplicadas a Saude, Universidade de Coimbra, Coimbra, Portugal.
Abbreviations
- ALS
Amyotrophic lateral sclerosis
- bvFTD
Behavioural variant fronto-temporal dementia
- BNT
Boston naming test
- C9orf72
Chromosome 9 open reading frame 72
- CWIT
D-KEFS colour-word interference test
- FCSRT
Free and cued selective reminding test
- FTD
Frontotemporal dementia
- GENFI
Genetic frontotemporal dementia initiative
- GIF
Geodesic information flow
- lvPPA
Logopenic variant primary progressive aphasia
- MRI
Magnetic resonance imaging
- MAPT
Microtubule-associated protein tau
- MMSE
Mini-mental state examination
- mini-SEA
Mini-social cognition and emotion assessment
- mCCT
Modified camel and cactus test
- nfvPPA
Non-fluent variant primary progressive aphasia
- PPA
Primary progressive aphasia
- GRN
Progranulin
- PASS
Progressive aphasia severity scale
- svPPA
Semantic variant primary progressive aphasia
- TIV
Total intracranial volume
- TMT
Trail making test
- DSF/DSB
WMS-R digit span forward/backward
Author contributions
All authors contributed to the study conception and design. Material preparation, data manipulation and analysis were performed by KS, LLR and JDR. AMM contributed to the analysis of the data. The first draft of the manuscript was written by KS, LLR and JDR and all authors commented on previous versions of the manuscript. All authors read and approved the final manuscript.
Availability of data and materials
The datasets used and/or analysed during the current study are available from the corresponding author on reasonable request.
Declarations
Conflicts of interest
The authors declare that they have no conflict of interest.
Ethical approval and consent to participate
All GENFI sites had local ethical approval for the study, and all participants gave written informed consent.
Footnotes
The list of consortium authors are mentioned in Acknowledgements.
Jonathan D. Rohrer and Lucy L. Russell are joint senior authors.
Contributor Information
Lucy L. Russell, Email: l.russell@ucl.ac.uk
On Behalf of the Genetic FTD Initiative (GENFI):
Annabel Nelson, David L. Thomas, Emily Todd, Hanya Benotmane, Jennifer Nicholas, Rachelle Shafei, Carolyn Timberlake, Thomas Cope, Timothy Rittman, Alberto Benussi, Enrico Premi, Roberto Gasparotti, Silvana Archetti, Stefano Gazzina, Valentina Cantoni, Andrea Arighi, Chiara Fenoglio, Elio Scarpini, Giorgio Fumagalli, Vittoria Borracci, Giacomina Rossi, Giorgio Giaccone, Giuseppe Di Fede, Paola Caroppo, Pietro Tiraboschi, Sara Prioni, Veronica Redaelli, David Tang-Wai, Ekaterina Rogaeva, Miguel Castelo-Branco, Morris Freedman, Ron Keren, Sandra Black, Sara Mitchell, Christen Shoesmith, Robart Bartha, Rosa Rademakers, Jackie Poos, Janne M. Papma, Lucia Giannini, Rick van Minkelen, Yolande Pijnenburg, Benedetta Nacmias, Camilla Ferrari, Cristina Polito, Gemma Lombardi, Valentina Bessi, Michele Veldsman, Christin Andersson, Hakan Thonberg, Linn Öijerstedt, Vesna Jelic, Paul Thompson, Tobias Langheinrich, Albert Lladó, Anna Antonell, Jaume Olives, Mircea Balasa, Nuria Bargalló, Sergi Borrego-Ecija, Ana Verdelho, Carolina Maruta, Catarina B. Ferreira, Gabriel Miltenberger, Frederico Simões do Couto, Alazne Gabilondo, Ana Gorostidi, Jorge Villanua, Marta Cañada, Mikel Tainta, Miren Zulaica, Myriam Barandiaran, Patricia Alves, Benjamin Bender, Carlo Wilke, Lisa Graf, Annick Vogels, Mathieu Vandenbulcke, Philip Van Damme, Rose Bruffaerts, Koen Poesen, Pedro Rosa-Neto, Serge Gauthier, Agnès Camuzat, Alexis Brice, Anne Bertrand, Aurélie Funkiewiez, Daisy Rinaldi, Dario Saracino, Olivier Colliot, Sabrina Sayah, Catharina Prix, Elisabeth Wlasich, Olivia Wagemann, Sandra Loosli, Sonja Schönecker, Tobias Hoegen, Jolina Lombardi, Sarah Anderl-Straub, Adeline Rollin, Gregory Kuchcinski, Maxime Bertoux, Thibaud Lebouvier, Vincent Deramecourt, Beatriz Santiago, Diana Duro, Maria João Leitão, Maria Rosario Almeida, Miguel Tábuas-Pereira, and Sónia Afonso
References
- 1.Hodges J, et al. Survival in frontotemporal dementia. Neurology. 2003;61(3):349–354. doi: 10.1212/01.WNL.0000078928.20107.52. [DOI] [PubMed] [Google Scholar]
- 2.Hogan DB, et al. The prevalence and incidence of frontotemporal dementia: a systematic review. Can J Neurol Sci. 2016;43(S1):S96–S109. doi: 10.1017/cjn.2016.25. [DOI] [PubMed] [Google Scholar]
- 3.Rascovsky K, et al. Sensitivity of revised diagnostic criteria for the behavioural variant of frontotemporal dementia. Brain. 2011;134(9):2456–2477. doi: 10.1093/brain/awr179. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 4.Neary D, et al. Frontotemporal lobar degeneration: a consensus on clinical diagnostic criteria. Neurology. 1998;51(6):1546–1554. doi: 10.1212/WNL.51.6.1546. [DOI] [PubMed] [Google Scholar]
- 5.Hornberger M, et al. How preserved is episodic memory in behavioral variant frontotemporal dementia? Neurology. 2010;74(6):472–479. doi: 10.1212/WNL.0b013e3181cef85d. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 6.Cheran G, et al. Cognitive indicators of preclinical behavioral variant frontotemporal dementia in MAPT carriers. J Int Neuropsychol Soc. 2019;25(2):184–194. doi: 10.1017/S1355617718001005. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 7.Hardy CJD, et al. The language profile of behavioral variant frontotemporal dementia. J Alzheimers Dis. 2016;50(2):359–371. doi: 10.3233/JAD-150806. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 8.Saxon JA, et al. Examining the language and behavioural profile in FTD and ALS-FTD. J Neurol Neurosurg Psychiatry. 2017;88(8):675–680. doi: 10.1136/jnnp-2017-315667. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 9.Gorno-Tempini ML, et al. Cognition and anatomy in three variants of primary progressive aphasia. Annal Neurol. 2004;55(3):335–346. doi: 10.1002/ana.10825. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 10.Foiani MS, et al. Plasma tau is increased in frontotemporal dementia. J Neurol Neurosurg Psychiatry. 2018;89(8):804–807. doi: 10.1136/jnnp-2017-317260. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 11.Harris JM, et al. Classification and pathology of primary progressive aphasia. Neurology. 2013;81(21):1832–1839. doi: 10.1212/01.wnl.0000436070.28137.7b. [DOI] [PubMed] [Google Scholar]
- 12.Sajjadi SA, et al. Primary progressive aphasia: a tale of two syndromes and the rest. Neurology. 2012;78(21):1670–1677. doi: 10.1212/WNL.0b013e3182574f79. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 13.Geraudie A, et al. Speech and language impairments in behavioral variant frontotemporal dementia: a systematic review. Neurosci Biobehav Rev. 2021;131:1076–1095. doi: 10.1016/j.neubiorev.2021.10.015. [DOI] [PubMed] [Google Scholar]
- 14.Staffaroni AM, et al. Uniform data set language measures for bvFTD and PPA diagnosis and monitoring. Alzheimers Dement (Amst) 2021;13(1):e12148. doi: 10.1002/dad2.12148. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 15.Rohrer J, et al. The heritability and genetics of frontotemporal lobar degeneration. Neurology. 2009;73(18):1451–1456. doi: 10.1212/WNL.0b013e3181bf997a. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 16.Warren JD, Rohrer JD, Rossor MN (2013) Clinical review. Frontotemporal dementia. BMJ, 347, f4827. Members Copy, Not for Commercial Sale [DOI] [PMC free article] [PubMed]
- 17.Garrard P, Carroll E. Presymptomatic semantic impairment in a case of fronto–temporal lobar degeneration associated with the+ 16 mutation in MAPT. Neurocase. 2005;11(5):371–383. doi: 10.1080/13554790500205421. [DOI] [PubMed] [Google Scholar]
- 18.Miyagawa T, et al. Use of the CDR® plus NACC FTLD in mild FTLD: data from the ARTFL/LEFFTDS consortium. Alzheimers Dement. 2020;16(1):79–90. doi: 10.1016/j.jalz.2019.05.013. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 19.Sapolsky D, Domoto-Reilly K, Dickerson BC. Use of the progressive aphasia severity scale (PASS) in monitoring speech and language status in PPA. Aphasiology. 2014;28(8–9):993–1003. doi: 10.1080/02687038.2014.931563. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 20.Graves R, et al. Boston naming test short forms: a comparison of previous forms with new item response theory based forms. J Clin Exp Neuropsychol. 2004;26(7):891–902. doi: 10.1080/13803390490510716. [DOI] [PubMed] [Google Scholar]
- 21.Mack WJ, et al. Boston naming test: shortened versions for use in Alzheimer’s disease. J Gerontol. 1992;47(3):P154–P158. doi: 10.1093/geronj/47.3.P154. [DOI] [PubMed] [Google Scholar]
- 22.Moore K, et al. A modified Camel and Cactus Test detects presymptomatic semantic impairment in genetic frontotemporal dementia within the GENFI cohort. Appl Neuropsychol. 2021;29:1–8. doi: 10.1080/23279095.2020.1716357. [DOI] [PubMed] [Google Scholar]
- 23.Cardoso MJ, et al. Geodesic information flows: spatially-variant graphs and their application to segmentation and fusion. IEEE Trans Med Imaging. 2015;34(9):1976–1988. doi: 10.1109/TMI.2015.2418298. [DOI] [PubMed] [Google Scholar]
- 24.Malone IB, et al. Accurate automatic estimation of total intracranial volume: a nuisance variable with less nuisance. Neuroimage. 2015;104:366–372. doi: 10.1016/j.neuroimage.2014.09.034. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 25.Healey ML, et al. Getting on the same page: the neural basis for social coordination deficits in behavioral variant frontotemporal degeneration. Neuropsychologia. 2015;69:56–66. doi: 10.1016/j.neuropsychologia.2015.01.028. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 26.Rousseaux M, et al. An analysis of communication in conversation in patients with dementia. Neuropsychologia. 2010;48(13):3884–3890. doi: 10.1016/j.neuropsychologia.2010.09.026. [DOI] [PubMed] [Google Scholar]
- 27.Healey M, et al. More than words: extra-Sylvian neuroanatomic networks support indirect speech act comprehension and discourse in behavioral variant frontotemporal dementia. Front Hum Neurosci. 2020;14:598131. doi: 10.3389/fnhum.2020.598131. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 28.Luzzi S, et al. The neural bases of discourse semantic and pragmatic deficits in patients with frontotemporal dementia and Alzheimer’s disease. Cortex. 2020;128:174–191. doi: 10.1016/j.cortex.2020.03.012. [DOI] [PubMed] [Google Scholar]
- 29.Gorno-Tempini ML, et al. The logopenic/phonological variant of primary progressive aphasia. Neurology. 2008;71(16):1227–1234. doi: 10.1212/01.wnl.0000320506.79811.da. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 30.Grossman M, Rhee J, Moore P. Sentence processing in frontotemporal dementia. Cortex. 2005;41(6):764–777. doi: 10.1016/S0010-9452(08)70295-8. [DOI] [PubMed] [Google Scholar]
- 31.Peelle JE, et al. Sentence comprehension and voxel-based morphometry in progressive nonfluent aphasia, semantic dementia, and nonaphasic frontotemporal dementia. J Neurolinguistics. 2008;21(5):418–432. doi: 10.1016/j.jneuroling.2008.01.004. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 32.Vogel AP, et al. Motor speech signature of behavioral variant frontotemporal dementia: refining the phenotype. Neurology. 2017;89(8):837–844. doi: 10.1212/WNL.0000000000004248. [DOI] [PubMed] [Google Scholar]
- 33.Laisney M, et al. The underlying mechanisms of verbal fluency deficit in frontotemporal dementia and semantic dementia. J Neurol. 2009;256(7):1083–1094. doi: 10.1007/s00415-009-5073-y. [DOI] [PubMed] [Google Scholar]
- 34.Snowden JS, et al. Distinct clinical and pathological characteristics of frontotemporal dementia associated with C 9ORF72 mutations. Brain. 2012;135(3):693–708. doi: 10.1093/brain/awr355. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 35.Burrell JR, et al. Motor neuron dysfunction in frontotemporal dementia. Brain. 2011;134(9):2582–2594. doi: 10.1093/brain/awr195. [DOI] [PubMed] [Google Scholar]
- 36.Seghier ML. The angular gyrus: multiple functions and multiple subdivisions. Neuroscientist. 2013;19(1):43–61. doi: 10.1177/1073858412440596. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 37.Pickering-Brown SM, et al. Frequency and clinical characteristics of progranulin mutation carriers in the Manchester frontotemporal lobar degeneration cohort: comparison with patients with MAPT and no known mutations. Brain. 2008;131(3):721–731. doi: 10.1093/brain/awm331. [DOI] [PubMed] [Google Scholar]
- 38.Pickering-Brown S, et al. Inherited frontotemporal dementia in nine British families associated with intronic mutations in the tau gene. Brain. 2002;125(4):732–751. doi: 10.1093/brain/awf069. [DOI] [PubMed] [Google Scholar]
- 39.Harris JM, et al. Sensitivity and specificity of FTDC criteria for behavioral variant frontotemporal dementia. Neurology. 2013;80(20):1881–1887. doi: 10.1212/WNL.0b013e318292a342. [DOI] [PubMed] [Google Scholar]
Associated Data
This section collects any data citations, data availability statements, or supplementary materials included in this article.
Supplementary Materials
Data Availability Statement
The datasets used and/or analysed during the current study are available from the corresponding author on reasonable request.