

Abstract
Recent technological and computational advances have made metagenomic assembly a viable approach to achieving high-resolution views of complex microbial communities. In previous benchmarking, short-read (SR) metagenomic assemblers had the highest accuracy, long-read (LR) assemblers generated the most contiguous sequences and hybrid (HY) assemblers balanced length and accuracy. However, no assessments have specifically compared the performance of these assemblers on low-abundance species, which include clinically relevant organisms in the gut. We generated semi-synthetic LR and SR datasets by spiking small and increasing amounts of Escherichia coli isolate reads into fecal metagenomes and, using different assemblers, examined E. coli contigs and the presence of antibiotic resistance genes (ARGs). For ARG assembly, although SR assemblers recovered more ARGs with high accuracy, even at low coverages, LR assemblies allowed for the placement of ARGs within longer, E. coli-specific contigs, thus pinpointing their taxonomic origin. HY assemblies identified resistance genes with high accuracy and had lower contiguity than LR assemblies. Each assembler type’s strengths were maintained even when our isolate was spiked in with a competing strain, which fragmented and reduced the accuracy of all assemblies. For strain characterization and determining gene context, LR assembly is optimal, while for base-accurate gene identification, SR assemblers outperform other options. HY assembly offers contiguity and base accuracy, but requires generating data on multiple platforms, and may suffer high misassembly rates when strain diversity exists. Our results highlight the trade-offs associated with each approach for recovering low-abundance taxa, and that the optimal approach is goal-dependent.
Keywords: metagenomic assembly, long reads, antibiotic resistance, plasmid assembly, low abundance, assembly benchmarking
Introduction
Metagenomic assembly has enabled the discovery of novel phyla and genera [1, 2]; identification of individual members in a microbiome, their functions and their genetic differences [3]; and detection of novel plasmids [4, 5] and antibiotic resistance mechanisms [6–8]. However, accurate reconstruction of complete individual genomes is challenging due to strain multiplicity, uneven read coverage between taxa, and homologous repeat regions [9]. These issues are particularly problematic for low-abundance species (<1% relative abundance), including clinically relevant organisms such as Escherichia coli [10, 11], which may contain antibiotic resistance genes (ARGs) located on mobile genetic elements (MGEs) and flanked by repeat and homologous regions [12, 13]. Short reads (SRs), e.g. 100–150 bp Illumina reads, provide minimal genomic context to allow assemblers to distinguish between homologous regions [14]. Relative to SR data, long reads (LRs) can overcome some of these issues by bridging repeat regions and providing enough genomic context to distinguish between related organisms [9, 15]. The minION sequencer from Oxford Nanopore Technology (ONT) is highly portable and inexpensive. However, ONT LRs are reported to have a higher base calling error rate than Illumina SRs (5–15% compared to <0.6%, respectively) [16, 17], which can result in assemblies with many indels, frameshifts and incorrect gene annotations [18]. Hybrid (HY) assembly, bringing together both SR and LR data, can balance the strengths of each data type by assembling contigs that are both long and accurate [19]. However, generating data on multiple sequencing platforms is unlikely to be cost-effective or feasible for many applications. As such, there is a clear need to fully explore the trade-offs associated with SR, LR and HY metagenomic assembly, including base accuracy, contiguity and completeness.
Previous benchmarking efforts have assessed the overall assembly quality of different metagenomic assemblers, as well as their ability to recover strains and genes of interest, such as ARGs [12, 13, 19–21]. Meyer et al. [20] used a synthetic metagenome with over 400 strains, including many same-species strains, and found that HY assemblers recovered high genome fractions of the greatest number of strains, but with a high number of misassemblies. Galata et al. [12], using a human fecal metagenome, found that, although LR assembly was associated with the greatest contiguity, SR and HY assemblies contained ARGs that were not present in those of LRs, likely due to erroneously generated indels. Brown et al. [13] found LR assemblies of wastewater to contain the most MGEs and ARGs on a contig, but, due to the high misassembly rate of LR assemblers, deemed these results unreliable and concluded that HY co-assembly of ARGs and MGEs was the most accurate. However, both Galata et al. and Brown et al. had no definitive catalog of ARG content for a given sample, so the calculation of sensitivity and specificity was not possible. Brown et al. [13] additionally compared misassembly rates for an in silico spiked-in isolate at various abundances in environmental metagenomes and found the HY assembly approach to produce the lowest misassembly rates. The merits of utilizing each data type for the assembly of low-abundance organisms and plasmids, which are of significant interest and potential threat in many contexts, remain unclear.
In this study, we focus on SR, LR and HY metagenomic assembly of low-abundance E. coli in human fecal samples. E. coli was selected due to its clinical relevance [11] and high prevalence, present in the guts of >90% of humans, but at a typical relative abundance of <1% [10, 11]. The E. coli genome is average size compared to other bacterial species’ genomes, typically includes a variety of plasmids, and shows remarkable diversity in strains—even from the same fecal sample—with a relatively modest overlap in gene content [22, 23]. We computationally spiked reads (LR and SR) of previously characterized E. coli isolates into fecal metagenomes across a range of coverage levels, reflecting realistic abundances in the human gut. To assess the effects of E. coli strain diversity, we performed this analysis in multiple metagenomic backgrounds with varying levels of pre-existing E. coli, as well as by spiking in E. coli strain pairs. We compared assemblies to the complete, closed assembly of the target E. coli genomes, and quantified quality of their plasmid and ARG content. Our results reveal general patterns in the utility of metagenomic assembly approaches for low-abundance species, genes and their genomic context, and highlight that optimal data generation for low-abundance species and ARGs is dependent on desired research outcome. For determining gene context, even in the presence of strain diversity, LR assembly is optimal. SR assemblies provide the best base-to-base accuracy for gene identification, and HY assemblies offer a balance of contiguity and accuracy, but with high misassembly rates in the presence of strain multiplicity and the associated costs of generating data on multiple platforms.
Results
To assess the relative performance of SR, LR and HY assembly to recover a low-abundance E. coli genome from a complex metagenome, we created semi-synthetic LR and SR metagenomic datasets with varying levels of spiked-in E. coli content. We selected a human fecal metagenome, previously sequenced on both Illumina and ONT platforms [12], that contained negligible levels of E. coli (B1; Table S1). Next, we computationally spiked in reads from one of three phylogenetically diverse, multidrug-resistant E. coli, (isolates I1, I2 and I3) previously sequenced using both ONT and Illumina technologies (Figure 1, Table 1, Table S2, Methods). The three isolates had complete ONT and Illumina HY-assembled genomes available, which we used to form the basis of ‘truth’ in our benchmarking experiments. Spike-in levels ranged from approximately 0.2% (1x coverage in 3 Gb-sized sample) to 10% (50x coverage) relative abundance. HY assemblers received these spike-in levels for each of the two data types.
Figure 1.

Semi-synthetic datasets for assessment of metagenomic assembly of low-abundance species. E. coli isolate reads were computationally added to metagenomic sequencing reads from a human fecal sample at 1x–50x spike-in coverage. SR, LR and HY assemblers were used to assemble the semi-synthetic datasets. We assessed the ability of each assembler to recover the ARGs within the well-characterized, spiked-in isolate.
Table 1.
Information on isolates
Rather than performing an exhaustive comparison of all available metagenomic assembly tools, we sought to compare metagenomic assembly based on data type, and thus, chose representative, widely used assemblers for SR, LR and HY assembly. For SR assembly, we used metaSPAdes [26], as well as MEGAHIT, a less computationally intensive, single-node assembler [27]. For LR assembly, we used metaFlye [28]. HY assembly can differ depending on the order in which read types are utilized; either (i) first building contigs using SRs, and then bridging these contigs with LRs (HY-SL), or (ii) generating contigs with LRs and then polishing these contigs to improve accuracy with SRs (HY-LS). Thus, for HY assemblies, we used OPERA-MS (HY-SL) [19] as well as metaFlye followed by Pilon [29] polishing (HY-LS).
HY assembly balanced contiguity and accuracy in E. coli assemblies
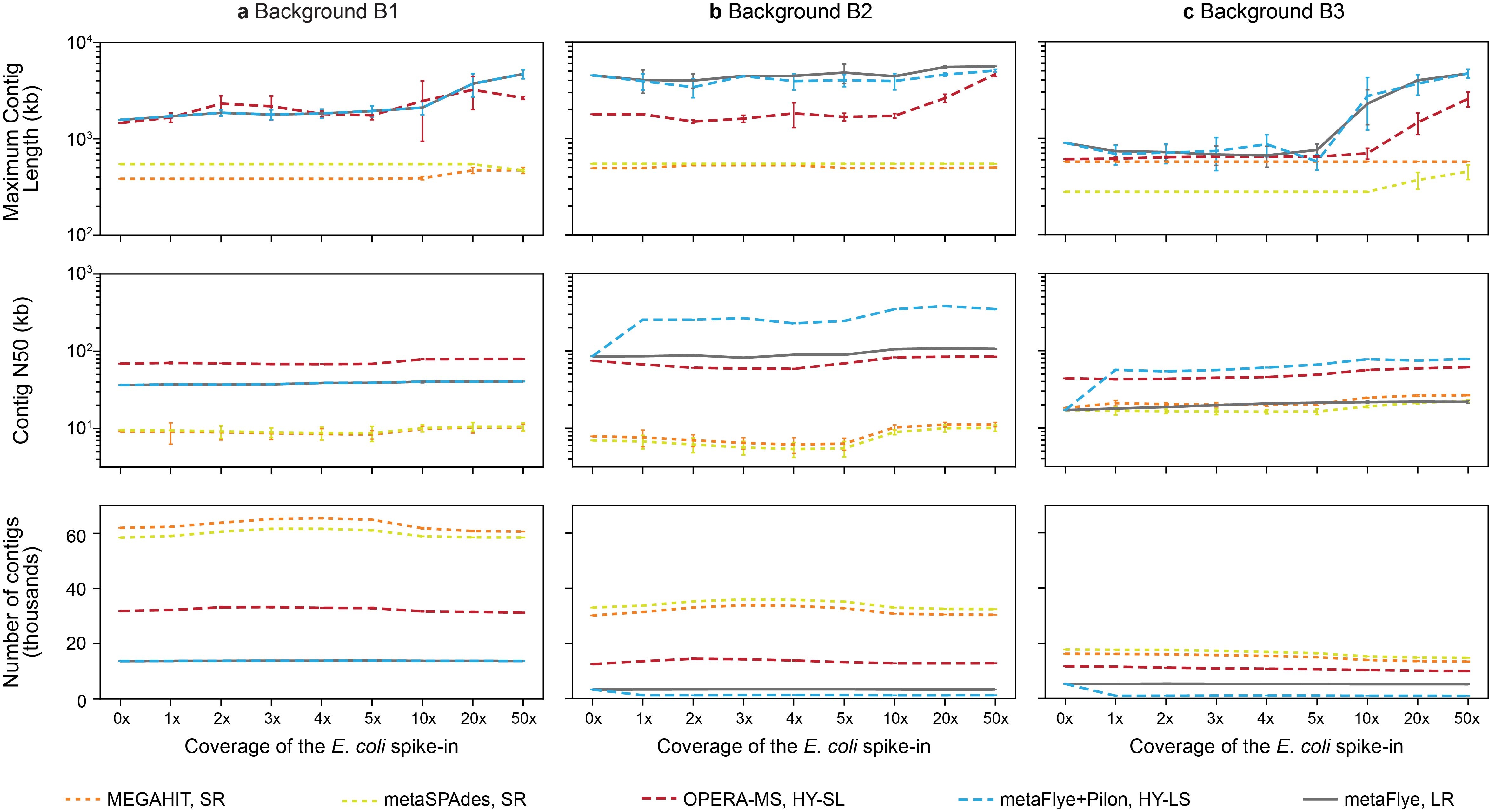
We first assessed the impact of the spiked-in E. coli on overall metagenome assembly metrics. As expected, the addition of the E. coli spike-in had little impact on the overall assembly of the B1 metagenome across all assembly approaches and spike-in levels, including the overall contig N50, maximum contig length and number of contigs (SI Figure S1a). Using metaQUAST [30] (Methods), we next assessed how well each assembly captured the spiked-in E. coli genome. As observed in previous work [12, 13], LR and HY metagenomic assemblies were the most contiguous by at least an order of magnitude at all coverage levels (Figure 2A). In fact, at ≥20x coverage, HY and LR assemblers recovered the entire (~5 Mb) E. coli chromosome in as little as 1–4 contigs. SR and HY-SL assemblies captured a greater proportion of the target genome than LR assemblies at ≤5x coverage (Figure 2B), and also offered higher base accuracy (Figure 2C). However, misassemblies (i.e. relocations and translocations) were elevated for MEGAHIT as well as for OPERA-MS (which uses MEGAHIT as its first assembly step) (Figure 2D). LR assembly generated large contigs with few misassemblies, but had relatively low accuracy and genome completeness, particularly at lower abundance. Polishing with short reads (HY-LS) improved accuracy, but required at least 10x coverage of both SRs and LRs to reach levels similar to SR assemblers (Figure 2C). OPERA-MS (HY-SL) had consistently higher identity and genome completeness than HY-LS (Figure 2A and C), though it generated more misassemblies across all coverages (Figure 2D).
Figure 2.

LR and HY assemblers generated more contiguous assemblies of E. coli in metagenomes. Assembly statistics for each coverage level, averaged across all three isolates individually spiked into the B1 background. (A) Percent of the E. coli genome present in the assembly, calculated by tabulating contigs >500 bp with alignment identity >95% to the spiked-in isolate’s reference genome (default parameters from metaQUAST). (B) NGA50 of E. coli isolate contigs in each assembly. Data are only shown at ≥3x coverage, where the sum of the reference contig alignments exceeded 50% of the reference genome length. (C) Percent identity of the longest alignment within a single contig to the E. coli isolate genome. (D) Number of misassemblies (including translocations and relocations) identified by metaQUAST. Error bars show +/- 1 S.D.
LR and HY assembly generated more contiguous plasmid assemblies
Plasmids frequently harbor ARGs and virulence factors, and can readily move between organisms, posing a threat to public health. They can also be a challenge to assemble given their often repetitive content. As such, we benchmarked the reconstruction of the 15 plasmids present across our three spike-in genomes on the B1 background using SR, LR and HY assembly.
We found that the same trends in contiguity and identity at low coverages observed for the whole E. coli genome also held true for plasmids (SI Figure S2A and C): SR assemblies were more fragmented, although accuracy was high. At low coverages, SR assemblies had gaps where transposases (i.e. transposable elements often found repeated throughout a genome) were located (Figure 3A and B; SI Figure S2A and B). OPERA-MS (HY-SL) had an improved ability to bridge transposons as coverage increased, but still left more gaps in the plasmid sequence than HY-LS (Figure 3C and D). All assemblers had comparable misassembly rates, mostly from translocations (SI Figure S2D). For each assembler, the degree of contiguity, misassembly and accuracy was consistent across all 15 predicted plasmids, ranging in length from 1.45 to 148 kbp (Table S2, SI Figure S2).
Figure 3.

metaFlye (with and without Pilon polishing) generated the most contiguous plasmid assemblies. Circular depictions of E. coli contigs in the single 150 kb plasmid present in the I3 isolate, assembled from spike-ins into the B1 background. Areas of correct assembly (blue) and misassemblies (red), as determined by metaQUAST, are shown for increasing spike-in coverages, ranging from 0x (innermost band) to 50x (outermost blue/red band). The outermost yellow and purple band indicates locations of ARGs (purple) and transposons (yellow) in the isolate genome. Assemblies are shown for (A) MEGAHIT (SR), (B) metaSPAdes (SR), (C) OPERA-MS (HY-SL), (D) metaFlye with Pilon polishing (HY-LS), (E) metaFlye (LR). Similar trends were seen across all 15 plasmids (SI Figure S2).
SR assembly captured more E. coli ARG sequence at low coverages; HY and LR captured more contiguous ARG sequences at all coverages
Given the importance of detecting ARGs in complex communities, we next aimed to assess how well ARGs could be identified from the different assemblies. We generated a ‘truth set’ of the 66–75 ARGs present in each isolate, identified using Resistance Gene Identifier [31] (RGI) (Methods). These genes were identified in each assembled metagenome using BLASTn [32] with a query set of the ARG sequences identified by RGI. ARGs from each metagenomic assembly were categorized based on their completeness, identity, and contiguity relative to the respective truth set (Figure 4).
Figure 4.

SR assemblers identified more E. coli isolate ARGs than LR and HY at coverages <5x. For each metagenomic assembly in the B1 background, the completeness of ARGs present in the E. coli isolate sequence is shown, averaged across spike-in experiments using the I1, I2 and I3 isolates, which contained 66, 70 and 75 ARGs, respectively. ARGs identified as ‘Strict’ or ‘Perfect’ hits using RGI are shown (Methods). (A) MEGAHIT; (B) metaSPAdes; (C) OPERA-MS; (D) metaFlye with Pilon polishing; (E) metaFlye.
At low abundance (1–5x), SR assembly outperformed LR and HY assembly, reconstructing the greatest number of ARGs with high sequence identity (Figure 4, SI Figure S3). At higher coverage levels, LR assembly contained full-length ARGs, although sequence identity often remained imperfect. This was considerably improved with SR Pilon polishing (HY-LS). Notably, ARGs were present in their entirety, or absent, using HY-LS and LR assembly, while the HY-SL approach, which begins with SR assembly followed by integration of LRs, also captured partial genes and led to fewer fragmented genes than present in SR assemblies.
HY and LR assemblers were most successful at recovering low-abundance ARGs with enough context to determine species specificity
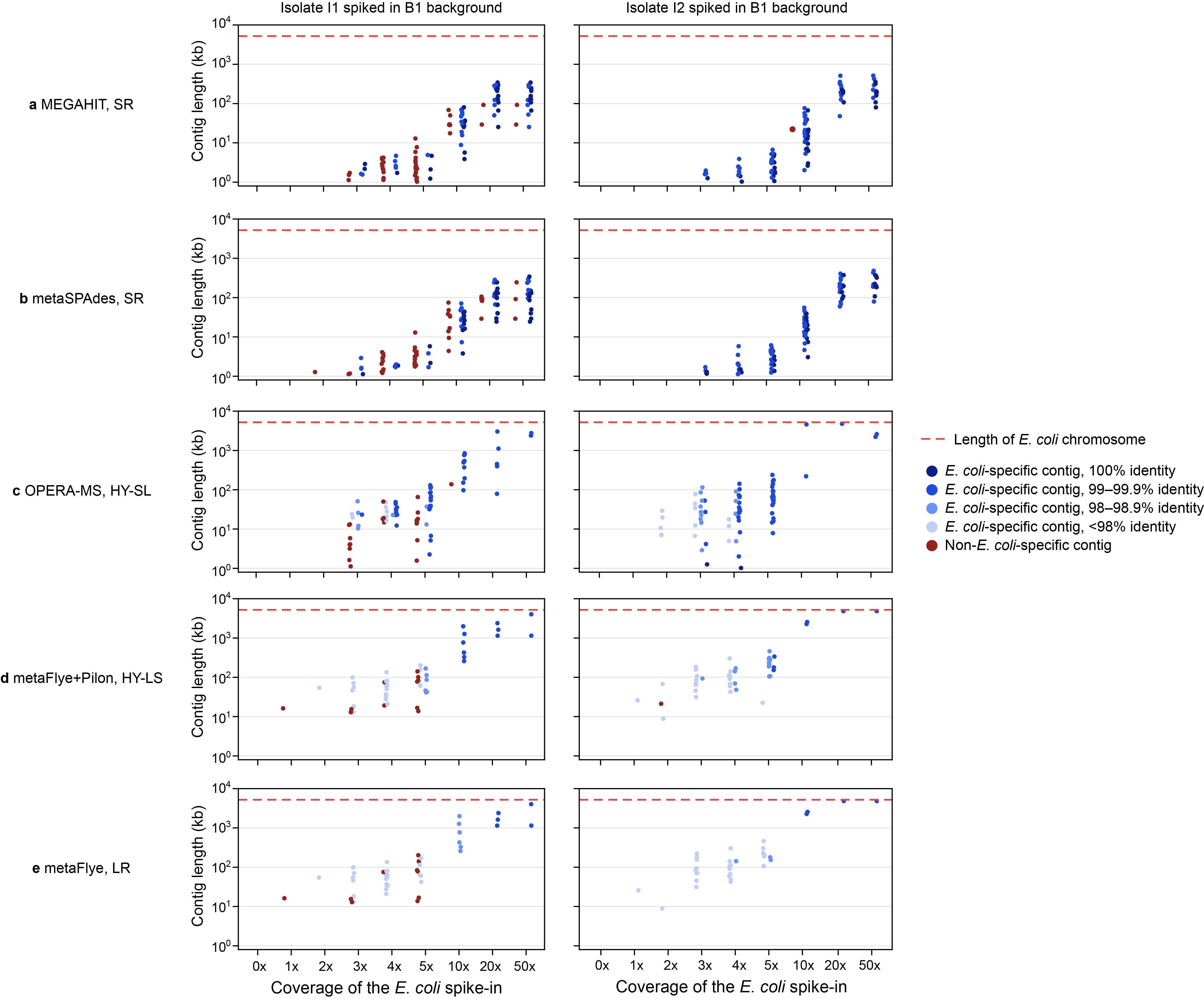
To determine the degree to which contigs containing ARGs could be correctly assigned to E. coli, we searched each contig (with >90% of their length aligning to the chromosome with >90% identity) containing a known chromosomally-encoded ARG against the Refseq database [33]. All ARG-containing contigs were classified as ‘E. coli-specific’ if the sole top hit was E. coli and were then evaluated for length and identity to the isolate (Figure 5; SI Figure S4).
Figure 5.

HY and LR assemblies contain long, species-specific contigs. Comparison of contig lengths for E. coli-specific versus non-E. coli-specific chromosome contigs for the I3 isolate spiked into the B1 background. Each E. coli contig >1 kb that contained chromosomal ARGs from the I3 isolate (>99% assembly length) is represented by a dot (blue = E. coli-specific; red = non-E. coli-specific). Contigs were considered species-specific if E. coli was the only top BLAST hit when searched against the Refseq database. The red dashed line shows the length of the I3 isolate’s chromosome. Data shown are for (A) MEGAHIT, (B) metaSPAdes, (C) OPERA-MS, (D) metaFlye with Pilon polishing, (E) metaFlye. Results for the I1 and I2 isolates spiked into the B1 background are shown in SI Figure S4.
Even at low coverages (≤5x), the HY and LR assemblies captured most (20–100%) chromosomal ARGs on E. coli-specific contigs (Figure 5). Contig length and E. coli specificity increased with spike-in coverage, and at coverage levels ≥10x, all ARGs were present on E. coli-specific contigs in HY and LR assemblies. While SR assembly was unable to achieve similar contiguity, the relatively high accuracy allowed for most shorter contigs (capturing 70–84% of ARGs from ≥10x) to be species-specific at high coverage levels.
E. coli strain diversity lowers the contiguity and accuracy of isolate and ARG assemblies, especially in HY assembly
While the above experiments were performed in a metagenomic background lacking E. coli to prevent interference from background strains, it is well known that many species, including E. coli, exist within communities containing multiple strains of varying relatedness and gene content [10, 11, 34]. Since metagenomic assemblers struggle with differentiating between different strains [19, 35], we sought to benchmark how the results of each assembly approach changed when resolving an isolate in the presence of other strains. Thus, we repeated the above experiments using two additional metagenomic backgrounds that contained native E. coli at relative abundances within ranges seen in healthy cohorts [36, 37], which we profiled at the strain level using StrainGE [38]: Background 2 (B2; containing one strain at 0.5% relative abundance, or 2.5x coverage) and Background 3 (B3; containing two strains at cumulatively 1.7% relative abundance or ~8.5x coverage) (Table S1). We estimated that the background E. coli strains had an average nucleotide identity (ANI) of 96.8–97% to the spiked-in isolates (Table S2, Methods). E. coli assemblies from these backgrounds were compared to those from equivalent E. coli spike-in levels in the B1 background with limited E. coli content. Overall, the presence of pre-existing strains reduced the accuracy of the spike-in genome assemblies, and increased fragmentation and misassembly rates, especially in background B3 (SI Figure S5B–D), which had the most background E. coli. We also observed a greater proportion of the target E. coli isolate aligning to assembled content at lower coverage levels, suggesting co-assembly of target and background E. coli sequence, or the assembly of both sequences on the same contig (SI Figure S5A). Similarly, the number of ARGs identified was generally higher, but with greater fragmentation and lower accuracy, due to co-assembly with background strains harboring similar genes (SI Figure S6).
To explore how metagenomic assembly approaches handle multiple strains more robustly, we generated semi-synthetic metagenomes containing the target strain I1 spiked into the metagenomic background B1, lacking native E. coli, with an additional competing E. coli isolate strain spiked in at the same abundance (Methods). We separately competed both I2 (97.3% ANI to I1) and I3 (99.7% ANI to I1) as competing strains with the I1 target to examine the impact of genetic similarity between the spiked-in genomes (Table 1; Table S2).
While the 2-fold higher E. coli content increased the fraction of the target genome aligning to assembled content, assemblies from spike-ins containing two strains at equal abundances were less accurate, more fragmented and incomplete (Figure 6A and Ci–iii) due to the co-assembly of the two strains. The SR assemblies did not disentangle homologous regions in most cases, resulting in reduced base accuracy relative to the target genome, even at high coverage. The reduction in identity was less pronounced for LR and HY assemblies due to the additional genomic context provided by LRs (Figure 6A and Ciii). Co-assembly of the two strains also resulted in considerably more misassemblies, especially for LR and HY assemblies at higher coverages (Figure 6A, and Ciii). Between the two SR assemblers considered here, MEGAHIT was associated with a greater number of misassemblies than metaSPAdes, particularly at high coverages (≥10x).
Figure 6.

For spike-ins with an equal abundance of two competing strains, assemblies were more fragmented and less accurate. Each column displays metrics for a different combination of isolate(s) spiked into background B1. (A) Isolate I1 only; (B) isolates I1 and I2 (99.7% ANI) at equal abundance; (C) isolates I1 and I3 (97% ANI) at equal abundance. Each row represents an assembly metric: (i) Percent isolate I1 assembled. The text on each graph reflects the maximum percent of the I1 genome assembled at 50x coverage out of all assemblers to show how strain multiplicity reduces genome completeness. (ii) Target E. coli (isolate I1) NGA50 (kb). (iii) Percent identity of the strain I1. (iv) The number of misassemblies in isolate I1’s assembly. Bottom right panel: Text indicates OPERA-MS misassembly values out-of-bounds.
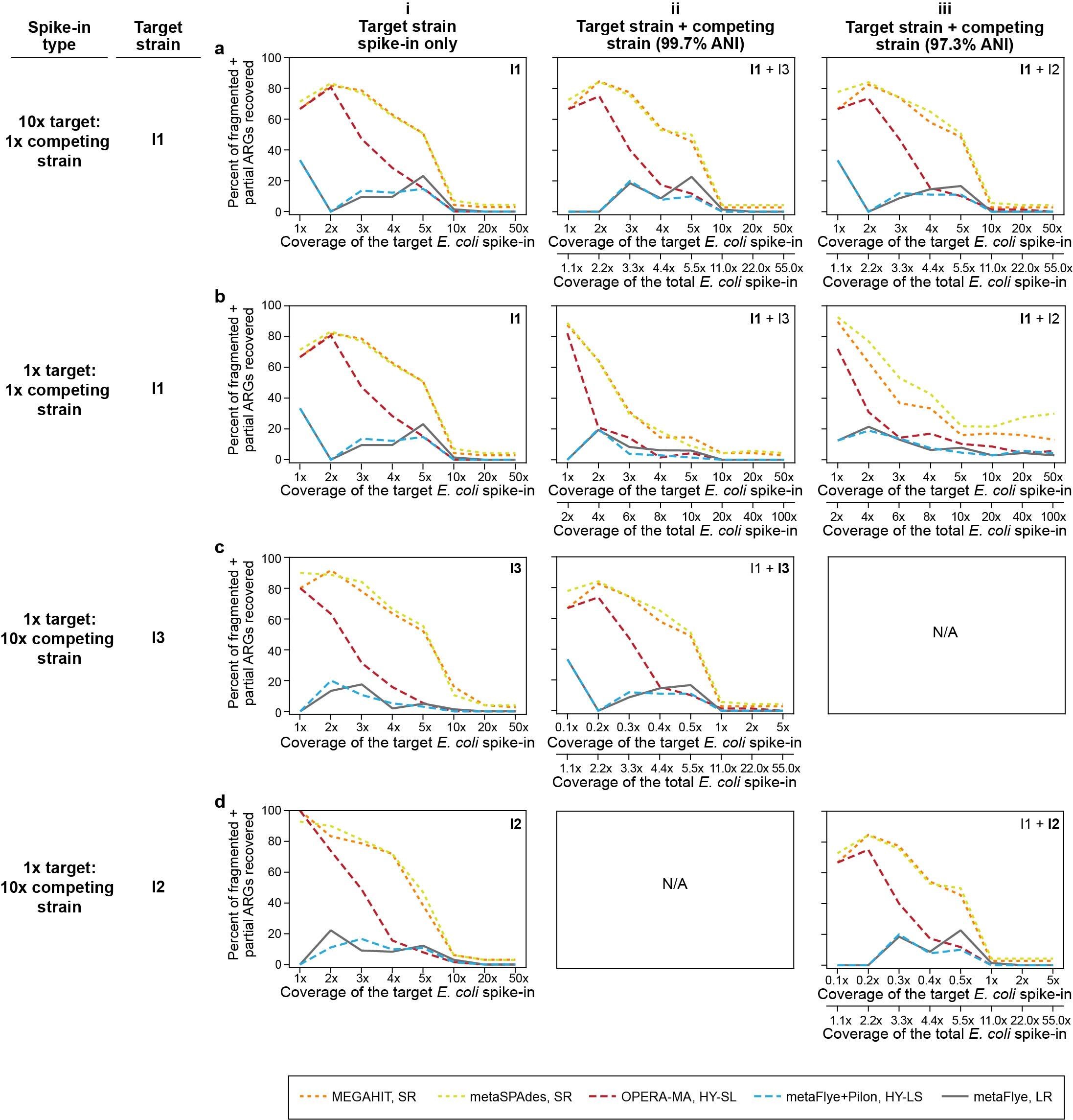
Spiking in a more similar competing strain (I3) had a lesser impact on the assembly of the target genome (I1) than a more distantly related competing strain (I2) due to the higher base-to-base similarity (Figure 6Bi–iv). For the I1–I3 spike-in, more of the common core E. coli genome was co-assembled at intermediate coverages without lowering accuracy for any assembler type. At high coverages, all assemblies were fragmented but more contiguous than the I1-I2 assemblies.
We also performed spike-ins using unequal ratios (1x target: 10x competing strain, or 10x target: 1x competing strain) for both the I1–I2 and I1–I3 pairs (SI Figure S7). A 1:10 minority competing strain caused only a slight change in metrics, including a slight increase in the number of misassemblies in the HY-SL assembly at intermediate coverages (SI Figure S7A-C). Unsurprisingly, when the competing strain was spiked in at 10-fold higher abundance than the target strain (SI Figure S7E and G), we saw a much larger degradation of assembly metrics compared to the degradation seen at other spike-in ratios, including greatly increased fragmentation of the target assembly. Recovery of minority strain I2 in a 1:10 spike-in with the more distantly related strain I1 resulted in the most fragmented and least accurate assemblies, highlighting the challenge of recovering minority strains, particularly in the presence of genomically divergent competing strains from the same species (SI Figure S7G).
In scenarios where we observed increased fragmentation and reduced accuracy of the E. coli assembly, we also observed a degradation in our ability to recover ARGs, particularly when the competing strain was less similar to the reference and spiked in at a higher ratio (SI Figure S8). The SR assembly and the HY approach starting with SR assembly (HY-SL) yielded more ARGs, but these were fragmented and less accurate (SI Figure S8).
Discussion
In this study, we systematically evaluated the ability of several approaches for metagenomic assembly to recover a target organism (E. coli) across a range of coverage levels, including at clinically relevant low abundances. We explored the impact of utilizing SR and LR sequence data, different assemblers, as well as the impact of competing strains from the same species on assembly. Our results highlight trade-offs between accuracy and contiguity, revealing that different approaches are better suited to different use cases, and providing insight into the capabilities and limitations of metagenomic assembly to characterize low-abundance organisms.
For low-abundance (≤5x) organisms, the optimal assembly approach depends on whether accuracy or genomic context is valued most. SR assemblies had the highest accuracy but provided limited genomic context, resulting in fragmented assemblies which frequently could not be confidently linked to the host species. Base accuracy was further reduced in the presence of multiple competing strains. Though this work is not meant to be an exhaustive assembler benchmarking, our comparison of two SR assemblies revealed that overall, metaSPAdes performed better than MEGAHIT, as its contigs contained less misassemblies. However, at high coverages (≥10x) and in the presence of a competing strain, metaSPAdes assembled much less of the target genome than MEGAHIT, perhaps because its algorithm assembled a consensus sequence between strains [26].
The LR assembler (metaFlye) provided insight into genome structure and species-specific context around genes of interest at the expense of base accuracy. While the LR assembler did not recover as much ARG content as the SR and HY assemblers at low coverages, most of its ARGs could be traced back to the host species. Non-E. coli-specific contigs contained homologous regions without enough accuracy or genomic context to be solely linked to E. coli.
In cases where both LR and SR data are available, HY assemblies, especially those from metaFlye with Pilon polishing, provided an exceptional balance of accuracy and contiguity. However, it should be noted that OPERA-MS (HY-SL) produced assemblies with a large number of misassemblies, especially in the presence of a competing strain, which may render its ARG genomic context unreliable. The misassemblies became an issue, especially at high coverages (≥10x), where OPERA-MS contigs contained >200 more misassemblies than that of all other assemblers. OPERA-MS’s misassembly rate was especially prominent at higher coverages in the multi-strain spike-in experiment, indicating it struggled at disambiguating different strains. Brown et al. [13] found OPERA-MS to have the lowest misassembly rate out of the assemblers we tested, a discrepancy with our results which may be due to the fact that we used stricter criteria to identify the reference genome in the assemblies. Additionally, if metaSPAdes were to be used instead of MEGAHIT as the first step in OPERA-MS assemblies, the HY-SL assembler may perform better.
While we do not anticipate considerable differences in assembler performance across other bacterial species, we only considered E. coli for our spike-in experiments. Although not explored in this paper, binning programs such as metaBAT [39] and CONCOCT [40] could be used to recover metagenome-assembled genomes in a scenario where the reference genome of a species of interest within a metagenome is unknown. We anticipate that genome assembly would be most affected by within-species strain diversity within a sample, as well as localized identity with components of other unrelated species’ genomes. When we measured assemblies containing within-species strain diversity, we saw a greater degradation of assembly metrics in the strain pair with a lower pairwise ANI. However, if the competing strains had an even lower ANI than those tested here, they may be divergent enough for assemblies of each strain to be unaffected by co-assembly.
Plasmids are often found at higher copy numbers than the chromosome in bacterial species’ genomes. To mimic this real world scenario, we opted to not normalize plasmid coverage in our spike-in experiments. Thus, there was an overrepresentation of plasmid reads in Illumina data and an underrepresentation in ONT data for small (<5 kb) plasmids. Additionally, our set of ARGs was limited to those seen in the isolates, and other sets of ARGs may present new challenges to each assembly approach. Alternative tools may potentially offer greater sensitivity for ARG detection, which we did not consider here. k-mer-based methods can identify ARGs directly from SRs when targets are known [41, 42] but provide no information about genomic context. Targeted graph-based methods [43] may be useful for obtaining context surrounding specific ARGs. Future studies comparing the sensitivity of assembly and k-mer- and graph-based approaches would be of interest.
We did not address differences in computational demands in this study, since these have been well documented in previous benchmarking studies [16, 20], and will not differ for the specific use case of targeted genome assembly explored here. Nevertheless, we recognize that this will be an important additional consideration, especially for high throughput analysis pipelines and in resource-limited settings. Furthermore, we did not explicitly evaluate the cost–benefit analysis of different sequence generation approaches, though it is likely cost-prohibitive to generate both SR and LR sequence data routinely for metagenomic samples in most settings. Our results quantify the additional insight provided by LR sequencing, which may justify the additional cost, depending on study-specific goals. Finally, we acknowledge that the ongoing improvement in sequencing technologies and computational algorithms is likely to improve assembly metrics in the coming years. Ongoing benchmarking efforts will be valuable to keep track of these developments and to facilitate optimal choice of data types and tools in the future.
Conclusions
Looking specifically at metagenomic assembly approaches for low-abundance E. coli in human fecal samples, we found that SR assemblies contained the most gene content, with the highest accuracy, but were fragmented; LR assemblies generated longer contigs with sufficient genomic context to link genes to species, but had lower accuracy; and HY assemblies provided a balance of intermediate accuracy and high contiguity. These findings will guide the selection of an assembler approach for metagenomic assembly in research and clinical settings.
Methods
Sequencing data used to construct semi-synthetic metagenomes
For the metagenomic background in our spike-in experiments, we used fecal samples previously sequenced using both ONT and Illumina technologies [12, 19] (SI Table S1).
To use for spiking into these backgrounds, we sequenced three E. coli genomes isolated from feces collected from US international travelers returning from South East Asia in 2018, using both Illumina and Oxford Nanopore technologies (SI Table S2) [45]. Illumina sequencing was as described in Salamzade et al., while ONT sequencing was done using Oxford Nanopore library construction protocol SQK-LSK109 on 600 ng of DNA, following the manufacturer’s recommendations [45]. As stated Salamzade et al., ‘samples were barcoded using the Native Barcoding Expansion 1-12 kit to run in batches of between 1 and 4 samples per flow cell on a GridIon’ [45]. These isolates were chosen due to their varying numbers of plasmids, as well as their clinical relevance, particularly due to their multidrug resistance and membership in sequence types (STs) with global presence. ST38 was among the top 20 extraintestinal global emerging pathogenic E. coli lineages, and ST224 was identified as a high-risk ST with potential to transmit from infected pets to humans [46].
Near-finished assemblies were generated for these three isolates, incorporating both Illumina and Oxford Nanopore data using Unicycler (v0.4.4) [47] with default parameters (SI Table S2). Illumina reads were trimmed using TrimGalore (v0.5.0) (https://github.com/FelixKrueger/TrimGalore), then subsampled to ~100x genome coverage. ONT reads went through the seQc NanoTrim pipeline (https://github.com/broadinstitute/seQuoia/tree/master/seQuoia/tasks) which used Porechop (v0.2.3_seqan2.1.1) (https://github.com/rrwick/Porechop). Of the 15 total plasmids predicted by OPERA-MS across these isolates, only one plasmid, in GTEN_24, was not predicted to be circular. We chose to keep all 15 plasmids in our truth set. Sequencing reads and assemblies were deposited at NCBI (Table S2).
Assembly of semi-synthetic metagenomic datasets
We generated semi-synthetic metagenomic datasets, including reads from a metagenomic background together with different amounts of E. coli isolate reads. Depending on the assembler type, we created semi-synthetic datasets containing either Illumina (SR), ONT (LR) or both (HY), from the fecal sample background and the isolate(s). A random subset of isolate reads was drawn independently for each coverage level using Rasusa [48]. For benchmarking the HY assemblers (metaFlye with Pilon polishing and OPERA-MS), we created semi-synthetic metagenomes with an equal spike-in coverage of Illumina and ONT reads. For example, at 5x spike-in level, the assemblers were given 5x Illumina data and 5x ONT data. Because only one data type was used to construct the original assembly, the same amount of data went into the original assembly, with the additional data used only for polishing. For multi-strain spike-in experiments, we generated semi-synthetic metagenomes containing the reference and competing strains at ratios of 1:1, 1:10 and 10:1.
MEGAHIT (v1.2.9) assemblies were created using default parameters. Illumina SRs used varied from 100 to 150 bp. metaSPAdes (v3.11.1) assemblies were generated using the spades.py script in python (v2.7.1). The metagenomic mode was enabled and one iteration was used for reading error correction. Default assembly parameters were used. metaFlye (v2.9) assemblies were generated using the parameters ‘-meta’, ‘-genome-size 10000000’, and ‘-min-ovlp 1000’. The asm_raw_reads.cfg file from the Flye developers was used for configuration. Pilon (v1.23) polishing with Illumina data was done with default parameters, and a minimum depth of 2 to correct single nucleotide errors and indels. OPERA-MS (v0.9.0) assemblies were generated using default parameters. MEGAHIT v1.0.4 was used as part of OPERA-MS.
Metrics to assess metagenomic assembly
Each metagenomic assembly was assessed by metaQUAST, a tool designed to evaluate noisy metagenomes, using default parameters with the isolate genome as the reference [30]. For E. coli plasmid assembly metrics, each set of plasmids from each isolate assembly was given to metaQUAST as a reference. For multi-strain spike-in experiments, only the target isolate genome was given to metaQUAST as a reference. Percentage of E. coli in background assemblies was calculated using Kraken2 [44].
Identification of resistance genes
RGI (v5.1.0) was used together with the Comprehensive Antibiotic Resistance Database (v3.1.4) to identify the truth set by searching for ARGs within the I1, I2,and I3 reference assemblies, as well as to search for assembled ARGs from this truth set within the metagenomic assemblies [31]. The flags ‘—low-quality’ and ‘—include_loose’ were used. However, only strict and perfect hits were considered. The ARG sequence identified in each isolate by RGI was put in a BLASTn [49] database and used to search for ARGs in each metagenomic assembly and classify each ARG as missing, partial, fragmented, full or perfect. ARGs were counted as missing if they were <25% assembled and/or had <95% identity; partial if 25–99.9% assembled; fragmented if 100% assembled across contigs; full if 100% assembled end-to-end with 95–99.9% identity; and perfect if 100% assembled end-to-end with 100% identity. Perfect genes (genes assembled end-to-end with 100% identity) were defined by BLASTn, which rounds percent identity up to 100% for genes assembled with >99.5% ID. Therefore, our classification of perfect genes may contain mismatches or indels.
RGI first uses Prodigal to identify Open Reading Frames (ORFs), prior to identifying genes from the ORFs. For a small number of genes (<3%) in each spike-in assembly, Prodigal did not correctly identify the ORF corresponding to the ARG, so the ARG went undetected or was truncated.
Determination of species specificity
For the subset of contigs within the metagenomic assemblies that were >1 kb and contained isolate ARGs that were found on the isolate chromosome but not the plasmid, we determined the species specificity. We chose only contigs where chromosome ARGs were assembled over ≥99% in length with ≥95% identity. We searched contigs against the Prokaryotic RefSeq NCBI database, using BLASTn with the flags ‘-task megablast,’ ‘-perc_identity 65,’ ‘max_target_seqs 100,’ ‘-word_size 20,’ ‘-reward 2’ and ‘-penalty −3.’ Contigs where the top hit was to E. coli or any Shigella species with at least 95% identity were considered E. coli-specific contigs.
Visualizations
Circular figures (Figure 3) were generated using Circos [50] using a configuration file made by metaQUAST [30].
Key Points
LR metagenomic assemblers produce long, contiguous assemblies that allow us to consistently recover ARGs that can be traced back to E. coli, even at <1% abundance.
SR metagenomic assemblers recover low-abundance species genomes with the highest accuracy and greatest completeness but in fragmented pieces.
HY metagenomic assemblers provide a balance of accuracy and contiguity.
The presence of a more similar competing strain does not affect the resulting assemblies as much as the presence of a distantly related strain.
Supplementary Material
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
Acknowledgments
Thomas Abeel and the Abeel lab, and Lucas van Dijk provided helpful discussions. Leslie Gaffney assisted with figure generation.
Sosie Yorki is a computational associate in the Bacterial Genomics Group of the Infectious Disease and Microbiome Program at the Broad Institute of MIT and Harvard.
Terrance Shea is a senior computational associate in the Fungal Genomics Group of the Infectious Disease and Microbiome Program at the Broad Institute of MIT and Harvard.
Christina A. Cuomo is the director of the Fungal Genomics Group in the Infectious Disease and Microbiome Program and an associate director in the Genomics Center for Infectious Diseases at the Broad Institute of MIT and Harvard.
Bruce J. Walker is the vice president, technology, at Applied Invention, LLC and a Visiting Scientist at the Broad Institute.
Regina C. LaRocque is a physician investigator at Massachusetts General Hospital, an associate professor of Medicine at Harvard Medical School and an associate physician at the Division of Infectious Disease at Massachusetts General Hospital.
Abigail L. Manson is the senior computational group leader of the Bacterial Genomics Group in the Infectious Disease and Microbiome Program at the Broad Institute of MIT and Harvard.
Ashlee M. Earl is the director of the Bacterial Genomics Group in the Infectious Disease and Microbiome Program at the Broad Institute of MIT and Harvard.
Colin J. Worby is a computational scientist in the Bacterial Genomics Group of the Infectious Disease and Microbiome Program at the Broad Institute of MIT and Harvard.
Contributor Information
Sosie Yorki, Infectious Disease and Microbiome Program, Broad Institute of MIT and Harvard, Cambridge, MA, USA.
Terrance Shea, Infectious Disease and Microbiome Program, Broad Institute of MIT and Harvard, Cambridge, MA, USA.
Christina A Cuomo, Infectious Disease and Microbiome Program, Broad Institute of MIT and Harvard, Cambridge, MA, USA.
Bruce J Walker, Infectious Disease and Microbiome Program, Broad Institute of MIT and Harvard, Cambridge, MA, USA; Applied Invention, LLC, Cambridge, MA, USA.
Regina C LaRocque, Division of Infectious Diseases, Massachusetts General Hospital, Boston, MA, USA.
Abigail L Manson, Infectious Disease and Microbiome Program, Broad Institute of MIT and Harvard, Cambridge, MA, USA.
Ashlee M Earl, Infectious Disease and Microbiome Program, Broad Institute of MIT and Harvard, Cambridge, MA, USA.
Colin J Worby, Infectious Disease and Microbiome Program, Broad Institute of MIT and Harvard, Cambridge, MA, USA.
Data availability
Isolate reads can be found with SRA accessions SRS4897057 (I1), SRS4592135 (I2) and SRS4897068 (I3). Isolate assemblies can be found in Genbank under accession numbers CP113486-CP113492 (I1), CP113493-CP113494 (I3) and CP116480-CP116488 (I2). Human fecal sample metagenomic background reads can be found with SRA accessions SRX10636832 (Illumina; B1), SRX10636834 (ONT; B2), ERR3201913 (Illumina; B2), ERR3201942 (ONT; B2), ERR3201911 (Illumina; B3), and ERR3201949 (ONT; B3).
Funding
This work was supported by the National Institute of Allergy and Infectious Diseases; National Institutes of Health and Department of Health and Human Services [U19AI110818] to the Broad Institute and from the Centers for Disease Control and Prevention (U01CK000633, U01CK000490).
References
- 1. Eloe-Fadrosh EA, Paez-Espino D, Jarett J, et al. Global metagenomic survey reveals a new bacterial candidate phylum in geothermal springs. Nat Commun 2016;7:10476. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 2. Reysenbach A-L, St John E, Meneghin J, et al. Complex subsurface hydrothermal fluid mixing at a submarine arc volcano supports distinct and highly diverse microbial communities. Proc Natl Acad Sci U S A 2020;117:32627–38. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 3. Almeida A, Nayfach S, Boland M, et al. A unified catalog of 204,938 reference genomes from the human gut microbiome. Nat Biotechnol 2021;39:105–14. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 4. Jørgensen TS, Xu Z, Hansen MA, et al. Hundreds of circular novel plasmids and DNA elements identified in a rat cecum metamobilome. PLoS One 2014;9:e87924. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 5. Li A-D, Li L-G, Zhang T. Exploring antibiotic resistance genes and metal resistance genes in plasmid metagenomes from wastewater treatment plants. Front Microbiol 2015;6:1025. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 6. Costa PS, Reis MP, Ávila MP, et al. Metagenome of a microbial community inhabiting a metal-rich tropical stream sediment. PLoS One 2015;10:e0119465. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 7. Zhao R, Yu K, Zhang J, et al. Deciphering the mobility and bacterial hosts of antibiotic resistance genes under antibiotic selection pressure by metagenomic assembly and binning approaches. Water Res 2020;186:116318. [DOI] [PubMed] [Google Scholar]
- 8. Wang S, Yan Z, Wang P, et al. Comparative metagenomics reveals the microbial diversity and metabolic potentials in the sediments and surrounding seawaters of Qinhuangdao mariculture area. PLoS One 2020;15:e0234128. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 9. Lapidus AL, Korobeynikov AI. Metagenomic data assembly – the way of decoding unknown microorganisms. Front Microbiol 2021;12:613791. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 10. Human Microbiome Project Consortium . Structure, function and diversity of the healthy human microbiome. Nature 2012;486:207–14. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 11. Tenaillon O, Skurnik D, Picard B, et al. The population genetics of commensal Escherichia coli. Nat Rev Microbiol 2010;8:207–17. [DOI] [PubMed] [Google Scholar]
- 12. Galata V, Busi SB, Kunath BJ, et al. Functional meta-omics provide critical insights into long- and short-read assemblies. Brief Bioinform 2021;22:2–4, 8. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 13. Brown CL, Keenum IM, Dai D, et al. Critical evaluation of short, long, and hybrid assembly for contextual analysis of antibiotic resistance genes in complex environmental metagenomes. Sci Rep 2021;11:3753. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 14. Ayling M, Clark MD, Leggett RM. New approaches for metagenome assembly with short reads. Brief Bioinform 2020;21:584–94. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 15. Vicedomini R, Quince C, Darling AE, et al. Strainberry: automated strain separation in low-complexity metagenomes using long reads. Nat Commun 2021;12:4485. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 16. Latorre-Pérez A, Villalba-Bermell P, Pascual J, et al. Assembly methods for nanopore-based metagenomic sequencing: a comparative study. Sci Rep 2020;10:13588. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 17. Stoler N, Nekrutenko A. Sequencing error profiles of Illumina sequencing instruments. NAR Genom Bioinform 2021;3:lqab019. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 18. Watson M, Warr A. Errors in long-read assemblies can critically affect protein prediction. Nat Biotechnol 2019;37:124–6. [DOI] [PubMed] [Google Scholar]
- 19. Bertrand D, Shaw J, Kalathiyappan M, et al. Hybrid metagenomic assembly enables high-resolution analysis of resistance determinants and mobile elements in human microbiomes. Nat Biotechnol 2019;37:937–44. [DOI] [PubMed] [Google Scholar]
- 20. Meyer F, Fritz A, Deng Z-L, et al. Critical assessment of metagenome interpretation: the second round of challenges. Nat Methods 2022;19:429–40. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 21. Sczyrba A, Hofmann P, Belmann P, et al. Critical assessment of metagenome interpretation-a benchmark of metagenomics software. Nat Methods 2017;14:1063–71. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 22. Blount ZD. The unexhausted potential of E. coli. Elife 2015;4:4. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 23. Richter TKS, Hazen TH, Lam D, et al. Temporal variability of Escherichia coli diversity in the gastrointestinal tracts of Tanzanian children with and without exposure to antibiotics. mSphere 2018;3(6). [DOI] [PMC free article] [PubMed] [Google Scholar]
- 24. Inouye M, Dashnow H, Raven L-A, et al. SRST2: rapid genomic surveillance for public health and hospital microbiology labs. Genome Med 2014;6:90. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 25. Jain C, Rodriguez-R LM, Phillippy AM, et al. High throughput ANI analysis of 90K prokaryotic genomes reveals clear species boundaries. Nat Commun 2018;9:5114. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 26. Nurk S, Meleshko D, Korobeynikov A, et al. metaSPAdes: a new versatile metagenomic assembler. Genome Res 2017;27:824–34. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 27. Li D, Liu C-M, Luo R, et al. MEGAHIT: an ultra-fast single-node solution for large and complex metagenomics assembly via succinct de Bruijn graph. Bioinformatics 2015;31:1674–6. [DOI] [PubMed] [Google Scholar]
- 28. Kolmogorov M, Bickhart DM, Behsaz B, et al. metaFlye: scalable long-read metagenome assembly using repeat graphs. Nat Methods 2020;17:1103–10. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 29. Walker BJ, Abeel T, Shea T, et al. Pilon: an integrated tool for comprehensive microbial variant detection and genome assembly improvement. PLoS One 2014;9:e112963. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 30. Mikheenko A, Saveliev V, Gurevich A. MetaQUAST: evaluation of metagenome assemblies. Bioinformatics 2016;32:1088–90. [DOI] [PubMed] [Google Scholar]
- 31. Alcock BP, Raphenya AR, Lau TTY, et al. CARD 2020: antibiotic resistome surveillance with the comprehensive antibiotic resistance database. Nucleic Acids Res 2020;48:D517–25. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 32. Johnson M, Zaretskaya I, Raytselis Y, et al. NCBI BLAST: a better web interface. Nucleic Acids Res 2008;36:W5–9. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 33. Pruitt KD, Tatusova T, Maglott DR. NCBI reference sequences (RefSeq): a curated non-redundant sequence database of genomes, transcripts and proteins. Nucleic Acids Res 2007;35:D61–5. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 34. Anyansi C, Straub TJ, Manson AL, et al. Computational methods for strain-level microbial detection in colony and metagenome sequencing data. Front Microbiol 2020;11:1925. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 35. Kunin V, Copeland A, Lapidus A, et al. A bioinformatician’s guide to metagenomics. Microbiol Mol Biol Rev 2008;72:557–78. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 36. Hoffman LR, Pope CE, Hayden HS, et al. Escherichia coli dysbiosis correlates with gastrointestinal dysfunction in children with cystic fibrosis. Clin Infect Dis 2014;58:396–9. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 37. Dicksved J, Ellström P, Engstrand L, et al. Susceptibility to Campylobacter infection is associated with the species composition of the human fecal microbiota. MBio 2014;5:e01212–4. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 38. Dijk LR, Walker BJ, Straub TJ, et al. StrainGE: a toolkit to track and characterize low-abundance strains in complex microbial communities. Genome Biol 2022;23:74. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 39. Kang DD, Li F, Kirton E, et al. MetaBAT 2: an adaptive binning algorithm for robust and efficient genome reconstruction from metagenome assemblies. PeerJ 2019;7:e7359. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 40. Alneberg J, Bjarnason BS, Bruijn I, et al. Binning metagenomic contigs by coverage and composition. Nat Methods 2014;11:1144–6. [DOI] [PubMed] [Google Scholar]
- 41. Titus Brown C, Irber L. Sourmash: a library for MinHash sketching of DNA. J Open Source Softw 2016;1:27. [Google Scholar]
- 42. Shen W, Xiang H, Huang T, et al. KMCP: accurate metagenomic profiling of both prokaryotic and viral populations by pseudo-mapping. Bioinformatics 2023;39: 1–11. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 43. Shafranskaya D, Chori A, Korobeynikov A. Graph-based approaches significantly improve the recovery of antibiotic resistance genes from complex metagenomic datasets. Front Microbiol 2021;12:714836. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 44. Wood DE, Lu J, Langmead B. Improved metagenomic analysis with kraken 2. Genome Biol 2019;20:257. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 45. Salamzade R, Manson AL, Walker BJ, et al. Inter-species geographic signatures for tracing horizontal gene transfer and long-term persistence of carbapenem resistance. Genome Med 2022;14:1–22. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 46. Song J, Oh S-S, Kim J, et al. Extended-spectrum β-lactamase-producing Escherichia coli isolated from raw vegetables in South Korea. Sci Rep 2020;10:19721. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 47. Wick RR, Judd LM, Gorrie CL, et al. Unicycler: resolving bacterial genome assemblies from short and long sequencing reads. PLoS Comput Biol 2017;13:e1005595. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 48. Hall M. Rasusa: randomly subsample sequencing reads to a specified coverage. J Open Source Softw 2022;7:3941. [Google Scholar]
- 49. McGinnis S, Madden TL. BLAST: at the core of a powerful and diverse set of sequence analysis tools. Nucleic Acids Res 2004;32:W20–5. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 50. Krzywinski M, Schein J, Birol I, et al. Circos: an information aesthetic for comparative genomics. Genome Res 2009;19:1639–45. [DOI] [PMC free article] [PubMed] [Google Scholar]
Associated Data
This section collects any data citations, data availability statements, or supplementary materials included in this article.
Supplementary Materials
Data Availability Statement
Isolate reads can be found with SRA accessions SRS4897057 (I1), SRS4592135 (I2) and SRS4897068 (I3). Isolate assemblies can be found in Genbank under accession numbers CP113486-CP113492 (I1), CP113493-CP113494 (I3) and CP116480-CP116488 (I2). Human fecal sample metagenomic background reads can be found with SRA accessions SRX10636832 (Illumina; B1), SRX10636834 (ONT; B2), ERR3201913 (Illumina; B2), ERR3201942 (ONT; B2), ERR3201911 (Illumina; B3), and ERR3201949 (ONT; B3).