

Summary
The X chromosome in non-African humans shows less diversity and less Neanderthal introgression than expected from neutral evolution. Analyzing 162 human male X chromosomes worldwide, we identified fourteen chromosomal regions where nearly identical haplotypes spanning several hundred kilobases are found at high frequencies in non-Africans. Genetic drift alone cannot explain the existence of these haplotypes, which must have been associated with strong positive selection in partial selective sweeps. Moreover, the swept haplotypes are entirely devoid of archaic ancestry as opposed to the non-swept haplotypes in the same genomic regions. The ancient Ust'-Ishim male dated at 45,000 before the present (BP) also carries the swept haplotypes, implying that selection on the haplotypes must have occurred between 45,000 and 55,000 years ago. Finally, we find that the chromosomal positions of sweeps overlap previously reported hotspots of selective sweeps in great ape evolution, suggesting a mechanism of selection unique to X chromosomes.
Keywords: X chromosomes, positive selection, evolution, selective sweeps, archaic admixture, meiotic drive
Graphical abstract
Highlights
-
•
Strong reductions in X chromosome diversity among non-Africans
-
•
Positive selection spread long haplotypes across non-African populations
-
•
The selected haplotypes spread from a population without Neanderthal admixture
-
•
An ancient male genome dates these selective sweeps to 45,000–55,000 years BP
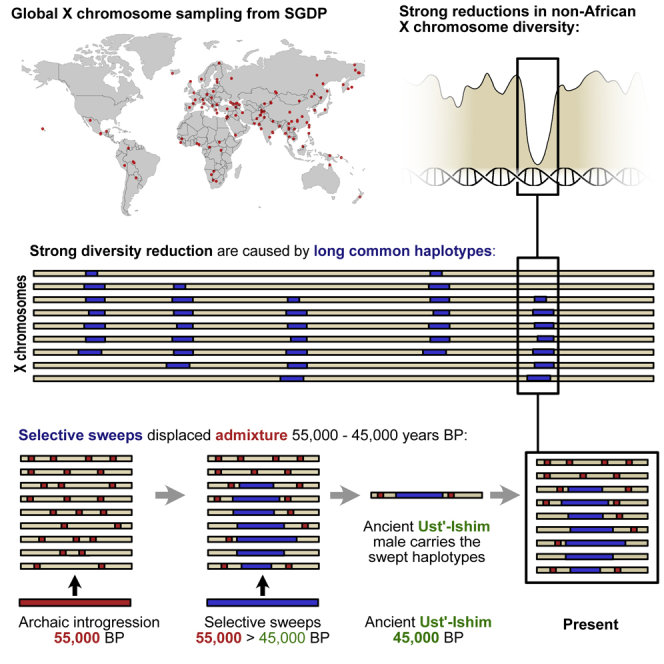
Skov et al. identify fourteen long common haplotypes shared across all non-African populations. These haplotypes are without Neanderthal admixture, and the authors conclude that these spread from an unadmixed population by strong positive selection. Using an ancient human genome, the authors place these events 45,000–55,000 years BP.
Introduction
Mammalian X chromosomes display extraordinary evolution compared with autosomes and a disproportionate effect on male fertility and genetic incompatibilities between species. They are enriched for genes expressed in the testis and undergo specific silencing during male meiosis. In Mus musculus and Bos taurus, there is evidence of an arms race between the X and Y chromosomes.1,2,3 Here, the relative numbers of Y- and X-linked multicopy homologs (ampliconic genes) affect the proportions of viable sperm cells carrying the X or Y chromosome.1,2 This intra-genomic conflict drives a dynamic co-amplification of Y- and X-linked homologs that is expected to accelerate sex-chromosome evolution and the accumulation of incompatibilities between emerging species. Reported evidence shows that the X chromosome was repeatedly subject to strong positive selection in the great apes. This selection is revealed as a loss of diversity in large regions of the X chromosome.4 The regions targeted by selection in each ape species overlap and tend to fall inside larger regions repeatedly targeted by selection in the ancestral species of humans and chimpanzees5 as well as overlapping regions similarly affected in the human-orangutan ancestor (K.M., unpublished data). Together, these findings lead to the hypothesis that meiotic drive plays a prominent role in primate X chromosome evolution.4
The patterns of archaic human introgression on the X and Y chromosomes differ from those observed on the autosomes.6 More than 250,000 years ago, modern humans admixed into Neanderthals, replacing their Y chromosome.7 On this occasion, Neanderthals also received at least as much modern human admixture on their X chromosome as they did on the autosomes.8 In the more recent meeting in the Middle East about 55,000 years ago, the direction of admixture was reversed and qualitatively different. Here, the Neanderthal Y chromosome did not introgress into the out-of-Africa modern humans, and the Neanderthal X chromosome introgressed to a much smaller extent than its autosomes.9 Today, several Mb-long sections of the X chromosome are depleted of Neanderthal introgression. These sections overlap the regions of the X chromosome repeatedly targeted by natural selection in the great apes, as discussed above.
Here, we investigate whether the reduced nucleotide diversity in regions of the human X chromosome is due to positive selection. We find that large regions of the X chromosome, which total more than 17 Mb, contain haplotypes shared by all non-African populations that rapidly rose to high frequency. To our surprise, all these haplotypes are entirely devoid of archaic introgression. We then speculate, based on our findings, which selective mechanism could cause this puzzling combination of observations.
Results
Mb-long high-frequency haplotypes are common in non-African X chromosomes
We analyzed high-coverage genomes from across the world in the Simons Genome Diversity Project (SGDP).10 We first surveyed the nucleotide diversity of each population in 200 kb windows along the X chromosome and compared it with the diversity on a representative autosome with a similar length, chromosome 7. Figures 1A and 1B show the diversity patterns for a representative set of African and non-African populations, located as shown in Figure 1C. Figure 1D shows the distribution of pairwise differences in these 200 kb windows (see Figures S1 and S2 for diversity in all populations). Whereas African populations show a relatively even amount of diversity across the X chromosome, as we observe along a representative autosome, all populations outside Africa display Mb-sized regions with extremely reduced diversity, often in similar parts on the X chromosome. Similar instances of such low diversity are not seen on the representative autosome, indicating that such extreme depressions of diversity are a unique property of the X chromosome.
Figure 1.

Population diversity across chromosomes X and 7
(A) Each panel shows the mean pairwise differences in 200 kb windows (y axis of each panel) across chromosome X (x axis) for a set of representative populations where at least four X chromosomes are sampled. We discarded constituent 100 kb windows with fewer than 50,000 called positions and considered these windows missing data. For a better visual comparison, the y axis is truncated at 0.0015 to remove outliers. Non-African and African populations are colored green and brown, respectively. Light gray regions represent missing data.
(B) As (A) but showing diversity along chromosome 7.
(C) Sampling origin of the selected populations.
(D) Distribution of the mean pairwise differences shown in (A) and (B) divided into African or non-African populations.
These initial observations suggest that high-frequency haplotypes are shared across non-African populations. To identify such haplotypes, we restricted the subsequent analysis to males for which the X chromosome is haploid. Following Lucotte et al.,11 we excluded males with missing data and males not showing the XY karyotype. We further removed African males with any evidence of recent European admixture (STAR Methods). This filtering left us with 162 males, of which 140 were non-Africans. We list these individuals in the Table S3. Sampling locations for the populations analyzed are shown in Figure 2A.
Figure 2.

ECHs identified on each male X chromosome
(A) World map showing sample locations. Colors represent the geographical region of each sample.
(B) Graphical depiction of the criteria used to define an ECH.
(C) ECHs on the X chromosome of each sampled male. Colors represent the geographical region of each sample as in (A), with African samples shown at the top. Colored vertical bar on the right groups African and non-African samples with the colors used in Figure 1.
Next, we screened these X chromosomes for clades of long haplotypes (>500 kb) where the maximum genetic divergence between all member haplotypes is at most 0.005%. This divergence threshold corresponds to an expected common ancestry no more than 60,000 years ago (51,119–67,059, 50% probability mass), i.e., such clades should have a most recent common ancestor, which is more recent than the out-of-Africa event.10 When such haplotype clades contain at least 25% of the individuals in our dataset, we refer to them as extended common haplotypes (ECHs) (Figure 2B). We identified clades of ECHs in sliding windows of 500 kb with a step size of 100 kb using a clique-finding algorithm.12 The ECHs identified in each individual are shown in Figure 2C (see Figures S3 and S4 for the effect of alternative minimum clade sizes). They are almost exclusively found in non-African populations with no apparent geographical differentiation in frequency outside of Africa. The sharing of haplotypes among non-Africans indicates that they rose to high frequency after the main exodus from Africa but before the subsequent diversification of non-African populations. The combined length of all ECHs is 17.3 Mb, or 11% of the entire X chromosome. In five of the nineteen affected chromosomal regions, the ECHs are carried by more than 75% of non-African males and, in fourteen regions, by more than 50% of non-African males. We will refer to this latter set as the fourteen most extreme regions (see Table S1 for hg19 coordinates of all ECH regions). The ECHs are characterized by a higher proportion of high-frequency derived variants, which is expected if each ECH recently arose from a single initial haplotype (Figure S7).
Next, we visualized the haplotype structure of the chromosomal regions where each ECH is found and compared it with regions without ECHs. Figure 3A shows the core 900 kb region of the most extended and most frequent ECHs, with non-reference SNPs marked as black tics and the individual haploid X chromosomes clustered by unweighted pair group method with arithmetic mean (UPGMA). All non-African X chromosomes form a single clade with highly reduced diversity compared with the African X chromosomes, implying that a single ancestral haplotype rose to high frequency in non-Africans. In this example, the ECH spans at least 1.8 Mb. The haplotype structure of the remaining ECHs shows the same pattern, with one haplotype (and in some cases two) shared by a large subset of non-African X chromosomes (Figure S10). For comparison, Figure 3B shows a typical 900 kb region where we find no ECHs. Here, non-African haplotypes do not form a single clade, as more African X chromosome diversity is represented in non-Africans.
Figure 3.

Example of the relationship among haplotypes in ECH regions
(A) The core 900 kb region (where the ECH frequency is at least 90% of its peak value) of the longest and most frequent ECH. The left side of each figure is a UPGMA tree with the x axis representing Jukes-Cantor-corrected sequence distances between male haplotypes. The individual haplotypes are shown as horizontal lines on the right with the x axis in hg19 Mb coordinates. Haplotypes are color coded according to geographical region. The ancient Ust’-Ishim individual is marked with gray. Vertical black bars on each haplotype represents non-reference SNPs.
(B) For comparison, a 900 kb region where no ECHs were identified (see Figure S10 for visualizations of all ECHs).
Each individual carries many ECHs; on average, each non-African male carries an ECH in 9.7 of the 14 regions where the ECH frequency is >50% (2.5th and 97.5th percentiles: 6 and 13). Figures 4 and S6 show the frequency of ECHs among non-Africans across the X chromosome and the core regions of each ECH shared by most individuals.
Figure 4.

Frequencies of ECHs along the X chromosome
Proportion of non-African haplotypes called ECHs in each 100 kb window across the X chromosome (blue). In fourteen cases, more than 50% of individual haplotypes are called ECHs. Vertical blue bands in the bottom part each show the core regions around each peak where the proportion of non-Africans’ called ECHs is at least 90% of its peak value. ECHs in the Ust’-Ishim male, called using the clique finding approach, are shown in red (an additional track below shows the center 500 kb regions of each ECH that share an UPGMA clade with the Ust’-Ishim). Regions depleted of incomplete lineage sorting between humans, chimpanzees, and gorillas are shown in orange.
The rise in ECH frequency predated the Ust’-Ishim male
We included the ancient Ust’-Ishim genome dated at 45,000 before the present (BP; 46,880–43,210 BP)13 to estimate further when these frequency changes occurred. The Ust’-Ishim is equally related to Europeans and Asians, suggesting that its lineage split off before the European/Asian population split and that it did not contribute directly to present-day diversity.13 If haplotypes rose to high frequency soon after the main out-of-Africa event, we would expect the Ust'-Ishim individual to carry ECHs in a way similar to present-day non-Africans. To investigate this, we repeated our ECH-finding procedure, including the Ust’-Ishim male. The Ust’-Ishim male shared six ECHs of the fourteen most frequent ECHs (STAR Methods). Using an alternative approach, robust to false-positive SNP calls in the ancient sequence, we added the Ust’-Ishim male to haplotype plots of 500 kb windows centered at each peak in Figure 4 (STAR Methods). The Ust'-Ishim falls inside a cluster of ECHs in 9 of the 14 most extreme regions, close to the mean of 9.7 among present-day non-Africans. This similar number of ECHs carried by the Ust'-Ishim implies that most of the ECHs had already risen to high frequencies 45,000 years ago.
Estimating the probability of observing the ECHs without natural selection
Our observations suggest that each ECH rose to high frequency from a single haplotype after the out-of-Africa event (60,000–80,000 BP) but before Ust’-Ishim lived (45,000 BP). This rapid change in frequency of such long haplotypes is striking and is characteristic of strong positive selection. However, genetic drift, particularly during extreme population bottlenecks, can also cause the frequency of long haplotypes to increase, and X chromosomes are more affected by bottlenecks than autosomes because of their smaller effective population size (Ne).
First, we performed simple frequency simulations of genetic drift to assess the probability of observing the haplotypes under neutral evolution. We simulated frequency trajectories to compute the probability that one haplotype, across the entire chromosome, rose to a high frequency by genetic drift in the span of time between the 45,000 BP of the Ust’-Ishim and the expected maximum ECH divergence of 60,000 years (STAR Methods). In the simulation, we assume haplotypes of 500 kb, although most ECHs are longer. To cover a range of scenarios, we perform simulations for time spans of 5,000, 10,000, 15,000, 20,000, and 25,000 years. To find an appropriate bottleneck Ne for our simulations, we estimated a population demography, using SMC++, of the non-African samples used in our analysis (STAR Methods). From this, we estimate the lowest autosomal Ne in the bottleneck as 4,125. In our simulations, we conservatively assume an autosomal Ne of 3,000 and include, for comparison, a more unlikely autosomal Ne of 1,500. We perform simulations for two different X/autosome ratios of effective population size: one of 0.65 corresponds to the median ratio among African individuals in our dataset, and a lower one of 0.51 corresponds to the median ratio among non-Africans. The latter estimate may be smaller due to male-driven migration but may also be depressed by the removal of diversity in the ECHs, as discussed above. For an autosomal Ne of 3,000, the two X/autosome ratios correspond to X chromosomal Nes of 1,950 and 975 (as shown in the legend of Figure 5). We further assume a female X chromosome recombination rate of 1.16e−8.14 We perform 500,000 simulations for each combination of parameters.
Figure 5.

Estimated probabilities of neutrally evolving 500 kb ECHs
(A) Evenly spaced probabilities as the fraction of simulations in which a 500 kb haplotype along the chromosome rises to at least the specified frequency in 500,000 simulations. Each panel show simulations for the duration of the bottleneck, which increases from left (5,000 years) to right (25,000 years) in intervals of 5,000 years. Colors represent X chromosome population sizes listed in the legend, with the assumed autosomal population size and X/autosome ratio in parentheses. Crosses represent the upper bounds on the probability in each case where no haplotypes reached the target frequency in the simulation. A dashed horizontal line marks the 0.05 value. Vertical dotted lines represent the frequencies of the ECHs we identify in the SGDP male dataset.
(B) Same as (A) but represents the joint probability of the identified 500 kb ECHs below a given frequency. In each facet, the leftmost point thus represents the probability of observing only the lowest frequency ECH, and the rightmost point shows the joint probability of observing nineteen 500 kb ECHs at the frequency we identify.
Simulations are summarized in Figure 5, where “A” presents the probability that at least one 500 kb haplotype along the chromosome rises to the given frequency inside the given span of time. Assuming realistic parameters, a time span of 5,000 to 15,000 years, and an autosomal bottleneck population size of 3,000, we find that while drift may create a low-frequency ECH, the probabilities of creating each high-frequency ECH are extremely low. Figure 5B quantifies the joint probability of observing all the identified ECHs at the frequencies they each occur. Each point represents the joint probability of the observed ECH at its frequency together with all observed ECHs at lower frequencies. The rightmost points in each facet thus show the joint probability of all observed ECHs. For the parameter range stated above, the observed ECHs are extremely unlikely to have arisen neutrally. Gray and black points show the probabilities for the autosomal population size of 1,500. Even with this unrealistically low autosomal population size, the available time also needs to be unrealistically long (20,000–25,000 years) for genetic drift to explain all observed ECHs. Using additional evidence, we show later in the article why only spans of 5,000 to 15,000 years are realistic. This shows that neutral processes alone are very unlikely to have caused the observed ECHs.
Second, we performed forward simulations of full X chromosomes using a published population demography and a recombination map. Since the SGDP data results from a very extensive population structure and demographics not well suited for simulation, we chose the homogeneous CEU population (Utah residents with Northern and Western European ancestry) from the 1000 Genomes dataset as our model population.15 We repeated our inference procedure on the CEU population and then performed forward simulations to assess the probability that the ECHs called in this population were produced by genetic drift. In our inference of ECHs on the 49 CEU male X chromosomes, we need to consider that a large sample from this single homogeneous population will much more often find recent common ancestry than the highly structured SGDP. To accommodate this contribution to the clade size of such early coalescences, we increase the ECH clade size from 25%, used in our main analysis, to 50%. Inference using this clade size reproduces 15 of the 19 ECH peaks identified among males from the SGDP (STAR Methods). In the CEU population, the ECHs together cover 10% of the entire chromosome, similar to the 11% observed in the SGDP dataset. In the subsequent forward simulations of the past 200,000 generations, performed using SLiM3,16 we use the previously published population size trajectory for CEU that, to our knowledge, estimates the strongest bottleneck.17 The simulations also use a fine-scale pedigree-based recombination map of the X chromosome.14 We perform simulations using the same X/autosome Ne ratios (0.51 and 0.65) used in our frequency simulations. Across 500 simulations of 49 male X chromosomes, we compute the analyzed proportion of the X chromosome where an ECH is called and the analyzed proportion of 100 kb sequence window called ECHs across all simulated chromosomes. Assuming the African X/autosome ratio of 0.65, these two proportions are 0.3% (0%–2%) and 0.2% (0%–1.2%) (interval is 2.5th and 97.5th percentiles across whole-chromosome simulations). Assuming instead a lower non-African X/autosome ratio of 0.51 (which would be lower in part due to the sweeps), the numbers are 1.1% (0%–4%) and 0.7% (0%–2.5%). In our analysis of the 49 CEU samples, we find these two proportions to be a magnitude larger (11.1% and 7.6%). Considering further that the two proportions in the CEU population are also 2.8 and 3 times larger than the 97.5th percentile of proportions obtained from simulations (Figure 6), it is extremely unlikely that the ECHs arose neutrally in the CEU population. It is even less likely that they arose neutrally before the Ust'-Ishim 45,000 years ago, as revealed by the analysis of the SGDP male dataset.
Figure 6.

Proportions of ECHs in realistic forward simulations of CEU X chromosomes
500 forward simulations of 49 male X chromosomes using a CEU population size demography and a parent-offspring recombination map.
(A) The proportion of the X chromosome where an ECH is called. Colors represent the African and non-African X-to-autosome population size ratios used in simulations. The gray arrow shows the observed value of this statistic in the CEU population.
(B) As for (A) but showing the proportion of 100 kb sequence window called ECHs across all simulated sample sequences.
The two separate analyses both indicate that positive selection must have driven the ECHs to high frequencies.
Selective sweeps on the X chromosome may be recurrent
We have previously reported evidence of selective sweeps in the human-chimpanzee ancestor5 (K.M., unpublished data; STAR Methods). We therefore tested if these overlap with the independent observation of ECH regions we report here. We find a strong overlap between the ECHs and the sections of the chromosome swept at least once during the 2–4 million years that separated the human-chimpanzee and human-gorilla speciation events,18,19 shown as orange blocks in Figure 4 (Jaccard stat.: 0.13, p = 1.6e−4).
Selective sweeps displaced archaic introgressed sequence on the X chromosome
The admixture with archaic humans that followed the main out-of-Africa event left a far smaller proportion of introgressed sequence in the X chromosome than in the autosomes. In non-African populations, the Neanderthal component is thus only 0.3% compared with 1.4% for the autosome. Denisovan admixture is virtually absent on the X chromosome, with only Oceanians carrying a small proportion (0.18%).9 To investigate a relationship between archaic admixture and the selective sweeps we detect, we applied hmmix20 to call genomic segments of archaic human ancestry in each non-African male X chromosome (STAR Methods). This approach uses a hidden Markov model to search for clusters of derived SNPs not observed in an unadmixed group of African genomes. Compared with inference in a previous report,9 the method we apply can identify archaic segments not represented in sequenced archaic genomes. We estimate a mean admixture proportion across individuals of 0.8%. However, when we restrict to archaic segments that share derived variants with the high coverage archaic genomes (Denisova, Vindija, and Altai), we identify a proportion of introgressed sequence (0.36%) similar to that previously reported9 (STAR Methods; Table S2).
Restricting the analysis to the 100 kb windows where some individual carries an ECH (11% of the X chromosome), we observe that the archaic proportion here is 0.3% compared with 0.9% in windows where no individuals carry an ECH (t test p = 6e−26). To investigate if this reduction is caused by low levels of admixture in ECHs specifically, we first extracted the subset of chromosomal 100 kb windows where any individuals carry an ECH. In each of these 100 kb windows, we computed the mean archaic admixture proportion of the ECHs and of the haplotypes in the same positions that are not part of the ECH clade. We find that the ECHs are almost completely without inferred archaic admixture. In contrast, the remaining haplotypes in the same genomic windows have a mean admixture proportion of 0.70%, ranging between 0.35% and 1.03%, depending on the geographical region. This admixture proportion is close to the archaic contribution in chromosomal regions not overlapping an ECH. The analysis thus reveals that the absence of admixture in the ECHs themselves entirely explains the reduced archaic admixture in the chromosomal regions where they appear. Within the ECHs, the mean proportion of archaic admixture is 0.0045%, corresponding to a reduction of 99% compared with the non-ECH haplotypes in the same chromosomal windows. This proportion is consistent with a complete absence of archaic admixture since our admixture inference is associated with a small false-positive rate.20 Each geographical region displays this absence of archaic admixture in ECHs (Figure 7A) and so does the core region of each ECH (Figure 7B).
Figure 7.

Admixture proportions in chromosomal regions of partial sweeps
Mean admixture proportions of ECHs (purple) and in remaining haplotypes (black) are computed separately for each 100 kb window. Error bars with wide caps designate the standard 95% confidence intervals obtained from 10,000 bootstrapping iterations. Error bars with narrow caps designate Jeffrey’s binomial confidence interval, which better represents confidence when frequencies are zero or very low (in computing this interval, we assume that the sample size equals the number of 100 kb sequence windows). The dotted line represents zero admixture. The dashed line shows the mean admixture proportion on the X chromosome.
(A) Mean admixture proportions of haplotypes from each geographical region shown as labels on the x axis.
(B) Admixture proportions at each individual ECH. Here, x axis labels represent chromosomal positions where each ECH has the highest frequency (peaks in Figure 4). For legibility, one outlier is shown separately.
Archaic introgression contributed to modern human diversity. Hence, we might falsely conclude that ECHs were admixture free simply because the contributed archaic admixture was not compatible with the low diversity required in our definition of ECHs. However, repeating our ECH inference after masking admixture segments identified in each individual (STAR Methods; Figure S9), we still find ECHs to be without admixture, thus ruling out this potential ascertainment bias.
Archaic displacement provides further support for positive selection
While EHCs displaced archaic admixture, their rise in frequency cannot be a result of purifying selection against archaic contribution. While such negative selection has most likely occurred21 it cannot explain why a single admixture-free haplotype, rather than many, would rapidly rise in frequency. In contrast, this would be expected if a single haplotype was under positive selection for reasons unrelated to archaic admixture. In this context, archaic admixture thus merely serves as a backdrop against which the selection on admixture-free ECHs is visible.
Our finding that ECHs also displaced Neanderthal admixture in the Ust’-Ishim, which lived 45,000 BP,13 further allows us to narrow the period in which the ECHs rose in frequency, as the sweeps must necessarily have happened between the admixture event and the Ust’-Ishim. From the length of Neanderthal admixture segments in the Ust'-Ishim male, the main Neanderthal admixture event has been dated to 9,599 years before the Ust'-Ishim.13 Using four standard errors as the confidence interval on this estimate implies that the sweeps must have occurred in a span of time that is at least 3,724 years and at most 15,341 years. The time intervals of 20,000 and 25,000 years included in our simulations (Figure 5) thus fall outside the range of relevant parameters, further decreasing the probability that ECHs arose from neutral processes.
Discussion
Our analyses suggest that regions of the X chromosome, totaling 11% of its length, carry long high-frequency haplotypes (ECHs) that are shared across all non-African populations. The frequency of these haplotypes rose after the out-of-Africa event and the subsequent archaic admixture events. These dramatic frequency changes appear to have been fully or almost fully completed by the time of the Ust'-Ishim, dated to 45,000 BP. The large size of these haplotypes is consistent with a rapid increase in frequency not compatible with a neutral process of genetic drift. We conclude that they must have increased in frequency by positive selection. Surprisingly, the ECHs are entirely free of archaic admixture, suggesting that whatever variants drove their rise in frequency, these arose on haplotypes without archaic admixture.
Fourteen of the identified ECHs each span between 500 kb and 1.8 Mb in at least 50% of non-Africans (Table S1). The strongest sweep spans 900 kb in 91% of non-Africans and affects 53% of non-Africans across a 1.8 Mb region. In comparison, the strongest selective sweep reported from human diversity data is at the lactase gene and spans 800 kb in 77% of European Americans.22 The selection coefficient on the causal variant has been estimated to be 1.6%–1.8%,23,24 suggesting that the selection coefficients responsible for several ECHs may have been well above 1%.
We have not identified specific genetic elements associated with the ECH rise in frequency. However, the ECHs strongly overlap regions depleted of incomplete lineage sorting in the common ancestor of humans and chimpanzees, suggesting that similar selection affected this ancestral species. This raises the possibility that our observations reflect processes repeatedly affecting the X chromosome across evolutionary timescales.
We have previously suggested a role of ampliconic genes that show post-meiotic expression in mouse testis25,26 and are involved in sex chromosomal meiotic drive processes in mouse1,27 and fruit files.28 However, while human ampliconic regions are significantly proximal to the swept regions (permutation test, p = 0.024), they do not generally overlap. The core regions of each ECH, shared by most individuals, each include several genes, and we do not detect enrichment for any Gene Ontology (STAR Methods). Protein-coding genes also show no enrichment of genes with elevated expression in the testis. One sweep, however, only has a single protein-coding gene, ACTRT1, at its center, which is linked to spermatid formation.29
At the present time, we cannot envision a likely scenario that explains all our observations. We cautiously hypothesize that the selective sweeps may be due to sex-chromosome meiotic drive: if an averagely even transmission of X and Y sperm in meiosis is maintained by a dynamic equilibrium of antagonizing drivers on X and Y, the bottlenecked main out-of-Africa population may have been invaded by sex-chromosome drivers retained in an earlier out-of-Africa population. This hypothesis is indirectly supported by recent evidence suggesting that the rapid expansion of the FT Y chromosome haplotype originated in East/Southeast Asia 50,000–55,000 BP and displaced Y lineages carried by the later main wave out of Africa.30 The spread of a Y driver across Eurasia would be followed by X haplotypes spreading from the same source population to restore an even meiotic transmission in the populations invaded by Y drivers. The date of the Ust’-Ishim male, which carries this FT haplotype, would place these events in the 45,000–55,000 BP window, where we conclude the sweeps occurred. An Asian origin of EHCs and subsequent displacement of the main wave out of Africa is also consistent with our observation that ECHs displaced not only Neanderthal admixture in west Eurasia but also the Denisovan admixture in Asia. If this hypothesis is true, the swept regions represent the only remaining haplotypes from early non-African populations not admixed with Neanderthals. This hypothesis provides testable predictions that will guide our future work.
Whatever the explanation, we believe it must be unique to X chromosomes, possibly in the form of other consequences to fertility or the fidelity of meiosis. We thus suggest that future studies could focus on surveying any male fertility consequences of the ECHs that we report, perhaps in combination with specific Y chromosome haplogroups. Large cohorts with male fertility data and genome-wide sequencing or genotyping will soon be available for such a study.
Limitations of the study
Our inference of ECHs has limited resolution because we require that an ECH spans at least 500 kb. This resolution does not allow fine mapping of any candidate genes responsible for the sweeps and leaves our study unable to identify Gene Ontology enrichments. Another limitation is that the SGDP dataset does not include enough African males to identify ECHs private to African populations with a diversity comparable to non-Africans. Our assessment of the probability that our observations arise neutrally potentially also has limitations. First, we cannot meaningfully estimate an X chromosome demography from X chromosome diversity if the X chromosome is strongly affected by positive selection. We thus resort to constructing an X demography by scaling the autosomal demography by the X/autosome ratio. Second, we base our estimate that sweeps occurred over a period of 5,000–15,000 years on a single high-coverage archaic genome. Access to more high-coverage archaic genomes from this period could further narrow this interval. Finally, we are not able to provide direct evidence to support any speculation on the cause of the many selective sweeps. The hypothesis we tentatively propose will require much further investigation to be scientifically tested.
STAR★Methods
Key resources table
| REAGENT or RESOURCE | SOURCE | IDENTIFIER |
|---|---|---|
| Biological samples | ||
| Human genomes, Simons Genome Diversity Panel (SGDP) B, SGDP-lite | Mallick et al. 201610 | https://reichdata.hms.harvard.edu/pub/datasets/sgdp/ |
| Human genomes, The International Genome Sample Resource (1000 genomes phase 3) | The 1000 Genomes Project Consortium 201515 | https://doi.org/10.1038/nature15393 |
| Software and algorithms | ||
| Hmmix | Skov et al.20 | https://github.com/LauritsSkov/Introgression-detection |
| Repository for code produced | GitHub | https://github.com/kaspermunch/humanXsweeps/tree/cellgenpaper |
| Permanent link and DOI for code produced | Zenodo | https://doi.org/10.5281/zenodo.7528246 |
Resource availability
Lead contact
Further information should be directed to and will be fulfilled by the lead contact, Kasper Munch (kaspermunch@birc.au.dk).
Materials availability
This study did not generate new unique reagents.
Method details
Data processing and filtering
We analyzed male X chromosomes from the fully public subset of genomes of the Simons Genome Diversity Project10 as initially published. We use the SGDP-lite version of the dataset that is run-length compressed sequences representing whether each base differs from the hg19 reference. We exclude African individuals with evidence of gene flow from non-African populations: Mozabite and Saharawi show extensive non-African ancestry and Neanderthal admixture,10 and Masai and Somali show a non-African component in STRUCTURE analysis,10 Luhya and close populations Luo and BantuKenya, show a non-African component in Admixture analysis.15 Following,10 we also exclude five samples (S_Finnish-1, S_Finnish-2, S_Mansi-1, S_Mansi-2, S_Palestinian-2) "based on missing X chromosome data in the initial processing, for themselves or a second sample." We further exclude S_Lezgin-1, for which sex is not assigned, and S_Palestinian-2 and S_Naxi-2, which show patterns of sequencing coverage not congruent with their assigned sex.11 This filtering leaves us with 239 individuals, of which 162 are males. We list these individuals in the Table S3.
Quantification and statistical analysis
Archaic admixture inference
We use a hidden Markov model to infer archaic segments.20 The method finds archaic genomic segments in non-Africans by identifying segments with a strong enrichment of single nucleotide variants that are not seen in an unadmixed outgroup population (in this case, Sub-Saharan populations). Non-African variants not present in the outgroup show a 10-fold enrichment in archaic segments because these variants have been accumulated since the common ancestor of archaic and modern humans. We use an outgroup consisting of individuals from two datasets. We use all Sub-Saharan Africans (populations: YRI, MSL, ESN) from the 1000 Genomes Project that does not have a component shared with the European population (Auton et al. 2015,15 Figure 2A) and all Sub-Saharan African populations from SGDP except Masai, Somali, Sharawi, and Mozabite (Mallick et al.10 see Figure S8), which show signs of out-of-Africa admixture. For all data, we removed sites that fell within repeat-masked regions (downloaded from the UCSC genome browser) as well as sites that were not in the strict callability mask for the 1000 Genomes Project: (ftp://ftp.1000genomes.ebi.ac.uk/vol1/ftp/release/20130502/supporting/accessible_genome_masks/StrictMask/). Since the method is sensitive to variation in mutation rate, we calculate the background mutation rate using the variant density of all variants from populations YRI, LWK, GWD, MSL, and ESN in windows of 100kb divided by the mean variant density of the whole genome.
The hidden Markov model was trained using the whole genome and not only the X chromosome. The reason for this is that there are not enough archaic segments to accurately infer transition and emission parameters just using the X chromosome. To accommodate the smaller effective population size of the X chromosome, we scaled the emission values using the following approach: Since males contribute 3/4 of mutations, but the X chromosome only spends a third of its time in males, then the X chromosome mutation rate is 5/6 = 0.8333 of the autosomal one, i.e., if the autosomal emission of state 2 is 0.38, it should be 0.3166 for X. We show the effect of decoding with haploid X chromosome parameters versus decoding with autosomal parameters in Table S2. This table also lists the proportions of archaic admixture in autosomes. Not relying on the sequence of the admixing archaic human allows this method to predict admixture not represented by the diversity of sequenced archaic genomes. However, when we condition that fragments share derived alleles with the archaic individual (Altai, Vindija, or Denisova), we obtain admixture proportions similar to those of Sankararaman 2016.9 Archaic segments are identified as regions where the posterior probability of the archaic state in the HMM is at least 0.8. Segments interleaved by more than 25Mb of missing data are split in two. 1kb windows with less than 200 bases called are excluded. We then compute the mean proportion of archaic sequence in each non-overlapping 100kb window. Windows, where over 20,000 bases are not called, are treated as missing data. Because several swept regions show a local reduction in Ne, it is important to note that this does not impede our ability to detect admixture. The power of our inference method relies on the fact that the density of SNPs private to non-Africans is low compared to the density of SNPs in archaic segments. For this reason, reduced effective population size in the regions where we identify ECHs, will increase rather than decrease our power to detect admixture.
ECH inference
We initially remove the two pseudoautosomal regions of chromosome X because these recombine with the Y chromosome. We then computed the proportion of pairwise differences along the chromosome for each pair of male individuals in non-overlapping windows of 100kb. We discarded windows with fewer than 50,000 called positions (50%) and considered these missing data. We then computed the pairwise distance between all the male X chromosomes in sliding windows of 500kb with a step of 100kb. For each window, we identify sets of haplotypes where the pairwise distance between all pairs is smaller than 5e-5. These sets must include at least 25% of individuals in the dataset (at least 40 males). We do this by constructing a graph where nodes represent individuals, and edges connect individuals with a distance smaller than 5e-5. We then use a clique-finding algorithm12 to extract fully connected sets of at least 40 individuals (25% of individuals). We refer to each haplotype in such a set as an Extended Common Haplotype (ECH). An ECH may be longer than 500kb if it spans several overlapping 500kb windows. Figures S3 and S4 show the distribution of ECHs called for alternative clique sizes of 32 and 49 (20% and 30% of individuals, respectively). The maximal pairwise distance of 5e-5 corresponds to an expected common ancestry of 59,520 years BP and was chosen to postdate the out-of-Africa event. The date is obtained assuming a mutation rate of 4.2e-10 = 4.3e-10 ∗ 0.8 ∗ 1.221. 4.3e-10 is the autosomal mutation rate,13 0.8 corrects for sex-specific mutation rate and hemizygosity of the X, and 1.221 corrects for a contribution of false-positive SNPs in the SGDP dataset. The latter correction is computed as the ratio between divergence to the human reference of sample S_Eskimo_Sireniki-1 at two filtering levels provided in the SGDP dataset: filtering level 9 (used in this study and recommended for population genetic analysis) and the strictest filtering level 1 (used for estimation of mutation rate).10 As described above, 100kb sequence windows with fewer than 50,000 called positions (50%) are considered missing data. Chromosomal 100kb windows, where this is true for more than 10% of individuals, are masked, leaving 138,5 Mb of analyzed chromosome in the downstream analyses.
Peak proportions of ECHs
In each chromosomal region where we identify ECHs, we use a peak detection algorithm to find the chromosomal position where most individuals carry the ECH and identify from that the consecutive 100kb windows that share this maximal number of individuals. We report the coordinates of these peak regions as "peak start" and "peak end" in Table S1. In addition, we operationally define a region around each peak where the proportion of individuals called ECH is at least 90% of the peak value (Figure S5). Similarly, we define a wider region where the proportion of individuals called ECH rises above 75% of the peak value. Table S1 lists chromosomal coordinates of both 90%-regions and 75%-regions along with the lowest ECH frequency in each region and the frequency of ECH haplotypes that span each region.
Data quality in ECH sequence
We find that ECH windows have fewer missing bases than other windows: The proportion of missing bases in sequence windows called ECH is 23% compared to 31% in windows we do not call as ECH. As described above, we discard 100kb windows of individuals where more than 50% of bases are missing. Pairwise distances to individuals with missing data in a 100kb window are not computed. Chromosomal windows where ECHs are called have fewer missing pairwise distances than the remaining windows: In ECH windows, the proportion of missing pairwise distances is 0.009%, whereas it is 0.1% across the remaining windows.
ECH inference with archaic admixture masking
We observe an almost total depletion of archaic admixture in ECH haplotypes (see main text). However, archaic admixture will increase pairwise differences between the male haplotypes. If this reduces our ability to call ECHs that overlap admixed segments, it could explain why we only identify ECHs without admixture. To rule out this possibility, we repeated our inference procedure after masking all called admixture segments as missing data. The resulting calls of ECHs are thus unaffected by contributions to diversity by admixture. The result of this analysis is almost identical to the original analysis: Only an additional eighteen 100kb windows across chromosomes across samples are identified as ECHs. In comparison, the original analysis finds 13,808 such windows. ECHs are only called when five consecutive 100kb windows each contain more than 50,000 called bases. To allow the calling of ECHs in the same set of windows after admixed segments are masked, we do not remove windows that fall below this cut-off after masking. However, in a small number of cases, admixture segments span such a 100kb window, which means that a distance to other haplotypes cannot be computed after masking. 111 of these 100kb windows overlap haplotypes that are identified as ECH in other individuals. However, this only creates a very small uncertainty in the admixture proportion of ECH haplotypes: Even if we conservatively assume that all the 111 uncalled windows were, in fact, ECH haplotypes and only subsequently admixed, the admixture proportion of ECH haplotypes would only increase by a factor less than 1.01.
Derived allele frequencies
We polarized all variants segregating in our sampled individuals using the chimpanzee (panTro3) variant provided in the Simons Genome Diversity Panel. Our analysis proceeds with X chromosomal SNPs in non-Africans, where we keep only SNPs called in at least 80% of non-Africans. This leaves 482,113 SNPs, each assigned to a 100kb window along the X chromosome. To characterize the relationship between non-African allele frequencies and ECH haplotypes, we compute across 100kb windows the proportion of derived variants. Computing the SFS for windows binned by ECH frequency shows the expected shift toward high-frequency alleles in ECH haplotypes (Figure S7).
ECHs shared with the ancient Ust’-Ishim male
The sequence of the Ust’-Ishim male is available as part of the Simons Genome Diversity dataset. We computed pairwise sequence distances in 100kb windows between the Ust’-Ishim male and all present-day males. Windows, where the pairwise difference is based on fewer than 27,217 positions due to uncalled bases, are considered missing data. The Ust’-Ishim has more uncalled bases than the contemporary samples, and the cutoff is adjusted from 50,000 to 27,217 to ensure the average number of 100kb pairwise comparisons to Ust’-Ishim matches the average number of 100kb pairwise comparisons between all contemporary samples. The distribution of these sequence distances to Ust’-Ishim shows the same characteristic enrichment of short pairwise distances as present-day non-Africans. To take the sampling time of the Ust’-Ishim into account when calling ECHs, we add a distance corresponding to 45,000 years when computing pairwise distances to present-day samples. We repeated our clique-finding procedure to find ECHs, including the Ust’-Ishim male. The ECHs identified in the Ust’-Ishim male total 5.3 Mb. We find that the Ust’-Ishim shares the ECH with the contemporary non-Africans in six of the 14 most extreme regions (where the proportion of non-Africans sharing the ECH is at least 50%). The very small number of sequence differences in ECHs implies that even a few base-calling errors will preclude inference of ECHs. For this reason, we performed a more robust and simpler inference of ECH sharing with Ust’-Ishim. We simply included the Ust’-Ishim in haplotype plots of each ECH. These plots are identical to those shown in Figure 3 but span only the of the 500kb centered at each peak in Figure 4. This identifies additional three of the 14 most extreme regions where the Ust’-Ishim groups with the ECH haplotypes in contemporary non-Africans: 64,550,000–65,050,000; 76,800,000–77,300,000; 129,700,000–130,200,000. The ECH regions shared by the Ust’-Ishim are listed in Table S1.
Estimation of the population bottleneck for SGDP samples
We used SMC++ on chromosome 7 to construct the autosomal population demography for the male samples analyzed. While the extensive population structure in the SGDP dataset will inflate population sizes, this is not the case for the initial out-of-Africa bottlenecked population. The minimum Ne of this shared demography thus provides a good estimation of the bottleneck Ne relevant for the frequency simulations in Figure 5. In the SMC++ analysis, we use one male individual from each non-African region as the “dedicated individuals”: S_Greek-2 (WestEurasia), S_Korean-1 (EastAsia), S_Irula-2 (SouthAsia), S_Yakut-2 (CentralAsiaSiberia), S_Papuan-9 (Oceania), S_Pima-1 (America). The remaining individuals contribute information through the SFS. We use 35 time points and estimate Ne for discrete epochs to better accommodate dramatic changes in population size.
ECH frequency simulations
We use multinomial sampling to produce frequency trajectories of a population of haplotypes of length L, starting at frequency 1/N. Each simulation is run for G generations. We generate n = 500,000 such simulations. For the subset of simulations where any haplotype reaches the target frequency f, we identify the number of generations g until this happens. In each such case, we compute the probability that the haplotype did not recombine onto a different haplotype in the intervening g generations: , where r is the recombination rate, and g is the generation in which frequency f is first reached. If f is not reached before generation G, then . The probability that a haplotype reaches frequency f, without recombining onto the background, is then estimated as the mean across sampled trajectories: . The probability that at least one haplotype rises to frequency f along the entire chromosome is computed as: , where S is the size of chromosome X (excluding the pseudoautosomal regions) and W is the window size of 500kb.
Analysis of CEU population
We use the phase3 version of the 1000 genome dataset and extract the 49 males from the CEU population. We then repeat our ECH inference procedure using a clique size of 50% instead of 25%, which we used in the main analysis of SGDP. The larger clique size accommodates the additional recent ancestry among males from the homogeneous CEU population not found among males sampled from the many separate populations in the SGDP. Using this clique size, the proportion of the X chromosome overlapping an ECH is 10%, similar to the 11% observed in our SGDP analysis. Fifteen of the ECH peaks in the CEU analysis match exactly one of the nineteen peaks in the SGDP analysis (Figure S8). Four SGDP ECHs are not found in the CEU analysis, which in turn finds five ECHs not found in the SGDP analysis.
Forward simulations of CEU X chromosomes
We simulate the ancestral relationship of 49 male samples, mirroring the 49 male samples included in our analysis of ECHs in the CEU population. Among the two most detailed population demographic estimates of the CEU population previously published,17,31 we chose the demography produced by Gravel et al. 2011.17 Although the two are very similar in their representation, Gravel et al. report a marginally stronger bottleneck. We follow the approach by Pool and Nielsen 200732 to scale the autosomal demography so that it reproduces the mean number of pairwise differences between CEU males on chromosome 7 (arbitrarily chosen for its intermediate size). We further apply two different scalings to represent the median X/autosome ratios of 0.51 (non-African samples) and 0.65 (African samples). For a realistic representation of recombination on the X chromosome, we use the sex-averaged deCODE recombination map derived from parent-offspring pairs.14 This map is not confounded by differences in effective population size or selection along the chromosome. We use SLiM316 to simulate fifteen consecutive 10Mb segments of the X chromosome from position 4,000,000 to 154,000,000, which excludes the pseudoautosomal regions. From each simulation, we extract 49 male X chromosomes (matching the number of CEU males) from the population and perform ECH inference as described above. On each simulated chromosome, we mask the same regions as missing data as we do in our analysis of the CEU population. We do this to ensure that the analyzed chromosomal regions from simulations correspond to the analyzed chromosomal regions in the CEU analysis.
Regions with low incomplete lineage sorting
In a previous analysis,19 we estimated the proportion of incomplete lineage sorting (ILS) between humans, chimpanzees, and gorillas along the human genome. There, we analyzed autosomes and the X chromosome but published only estimates for autosomes. Extending this analysis, we computed the mean proportion of ILS in non-overlapping 1Mb windows along the X chromosome. Following Munch et al. 2016,19 windows where the proportion of non-missing data falls below 30%, are not reported. The average proportion of ILS along the X chromosome is 13%. Mega-base windows are called low-ILS windows if the proportion of ILS falls below 5%. These regions, totaling 12% of the chromosome, are shown as orange blocks in Figure 4. Jaccard tests are performed following Favorov et al. 2012.33
Gene ontology enrichment
To identify a list of candidate genes, we first identify the set of 100kb windows where the frequency of each ECH is maximal (Peak start and end in Table S1). We further include the immediately flanking 100kb windows to allow for the possibility that distant enhancers, rather than the transcribed region, is the target of selection on a gene. We tested the 65 protein-coding genes overlapping these regions for gene ontology enrichment using all protein-coding genes on the X chromosome as background. Performing this test against all gene ontology terms GOATOOLS34 yielded no significant enrichments.
Acknowledgments
This work was supported by the Danish Council for Independent Research (grant number 4181-00358) to K.M. and by grant NNF18OC0031004 from the Novo Nordisk Foundation to M.H.S.
Author contributions
K.M. conceived and supervised the study. L.S., M.C.M., E.A.L., M.I.A.C., D.C., and K.M. performed the analyses. M.H.S. provided analytical support. K.M. and M.H.S. wrote the manuscript with input from all co-authors.
Declaration of interests
The authors declare no competing interests.
Published: February 28, 2023
Footnotes
Supplemental information can be found online at https://doi.org/10.1016/j.xgen.2023.100274.
Supplemental information
Data and code availability
This paper analyzes existing, publically available data listed in the key resources table.
All code used to produce results and visualizations are deposited on GitHub. A DOI and permanent link is available via Zenodo: https://doi.org/10.5281/zenodo.7528246.
Any additional information required to reanalyze the data reported in this paper is available from the lead contact upon request.
References
- 1.Larson E.L., Keeble S., Vanderpool D., Dean M.D., Good J.M. The composite regulatory basis of the large X-effect in mouse speciation. Mol. Biol. Evol. 2016;34:282–295. doi: 10.1093/molbev/msw243. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 2.Rathje C.C., Johnson E.E.P., Drage D., Patinioti C., Silvestri G., Affara N.A., Ialy-Radio C., Cocquet J., Skinner B.M., Ellis P.J.I. Differential sperm motility mediates the sex ratio drive shaping mouse sex chromosome evolution. Curr. Biol. 2019;29:3692–3698.e4. doi: 10.1016/j.cub.2019.09.031. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 3.Hughes J.F., Skaletsky H., Pyntikova T., Koutseva N., Raudsepp T., Brown L.G., Bellott D.W., Cho T.-J., Dugan-Rocha S., Khan Z., et al. Sequence analysis in Bos taurus reveals pervasiveness of X–Y arms races in mammalian lineages. Genome Res. 2020;30:1716–1726. doi: 10.1101/gr.269902.120. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 4.Nam K., Munch K., Hobolth A., Dutheil J.Y., Veeramah K.R., Woerner A.E., Hammer M.F., Great Ape Genome Diversity Project. Mailund T., Schierup M.H., et al. Extreme selective sweeps independently targeted the X chromosomes of the great apes. Proc. Natl. Acad. Sci. USA. 2015;112:6413–6418. doi: 10.1073/pnas.1419306112. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 5.Dutheil J.Y., Munch K., Nam K., Mailund T., Schierup M.H. Strong selective sweeps on the X chromosome in the human-chimpanzee ancestor explain its low divergence. PLoS Genet. 2015;11:e1005451. doi: 10.1371/journal.pgen.1005451. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 6.Schierup M.H. The last pieces of a puzzling early meeting. Science. 2020;369:1565–1566. doi: 10.1126/science.abe2766. [DOI] [PubMed] [Google Scholar]
- 7.Petr M., Hajdinjak M., Fu Q., Essel E., Rougier H., Crevecoeur I., Semal P., Golovanova L.V., Doronichev V.B., Lalueza-Fox C., et al. The evolutionary history of Neanderthal and Denisovan Y chromosomes. Science. 2020;369:1653–1656. doi: 10.1126/science.abb6460. [DOI] [PubMed] [Google Scholar]
- 8.Hubisz M.J., Williams A.L., Siepel A. Mapping gene flow between ancient hominins through demography-aware inference of the ancestral recombination graph. PLoS Genet. 2020;16:e1008895. doi: 10.1371/journal.pgen.1008895. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 9.Sankararaman S., Mallick S., Patterson N., Reich D. The combined landscape of denisovan and Neanderthal ancestry in present-day humans. Curr. Biol. 2016;26:1241–1247. doi: 10.1016/j.cub.2016.03.037. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 10.Mallick S., Li H., Lipson M., Mathieson I., Gymrek M., Racimo F., Zhao M., Chennagiri N., Nordenfelt S., Tandon A., et al. The Simons genome diversity Project: 300 genomes from 142 diverse populations. Nature. 2016;538:201–206. doi: 10.1038/nature18964. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 11.Lucotte E.A., Skov L., Jensen J.M., Macià M.C., Munch K., Schierup M.H. Dynamic copy number evolution of X- and Y-linked ampliconic genes in human populations. Genetics. 2018;209:907–920. doi: 10.1534/genetics.118.300826. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 12.Bron C., Kerbosch J. Algorithm 457: finding all cliques of an undirected graph. Commun. ACM. 1973;16:575–577. doi: 10.1145/362342.362367. [DOI] [Google Scholar]
- 13.Fu Q., Li H., Moorjani P., Jay F., Slepchenko S.M., Bondarev A.A., Johnson P.L.F., Aximu-Petri A., Prüfer K., de Filippo C., et al. Genome sequence of a 45,000-year-old modern human from western Siberia. Nature. 2014;514:445–449. doi: 10.1038/nature13810. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 14.Kong A., Thorleifsson G., Gudbjartsson D.F., Masson G., Sigurdsson A., Jonasdottir A., Walters G.B., Jonasdottir A., Gylfason A., Kristinsson K.T., et al. Fine-scale recombination rate differences between sexes, populations and individuals. Nature. 2010;467:1099–1103. doi: 10.1038/nature09525. [DOI] [PubMed] [Google Scholar]
- 15.1000 Genomes Project Consortium. Auton A., Brooks L.D., Durbin R.M., Garrison E.P., Kang H.M., Korbel J.O., Marchini J.L., McCarthy S., McVean G.A., et al. A global reference for human genetic variation. Nature. 2015;526:68–74. doi: 10.1038/nature15393. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 16.Haller B.C., Messer P.W. SLiM 3: forward genetic simulations beyond the Wright–Fisher model. Mol. Biol. Evol. 2019;36:632–637. doi: 10.1093/molbev/msy228. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 17.Gravel S., Henn B.M., Gutenkunst R.N., Indap A.R., Marth G.T., Clark A.G., Yu F., Gibbs R.A., 1000 Genomes Project. Bustamante C.D., et al. Demographic history and rare allele sharing among human populations. Proc. Natl. Acad. Sci. USA. 2011;108:11983–11988. doi: 10.1073/pnas.1019276108. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 18.Scally A., Dutheil J.Y., Hillier L.W., Jordan G.E., Goodhead I., Herrero J., Hobolth A., Lappalainen T., Mailund T., Marques-Bonet T., et al. Insights into hominid evolution from the gorilla genome sequence. Nature. 2012;483:169–175. doi: 10.1038/nature10842. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 19.Munch K., Nam K., Schierup M.H., Mailund T. Selective sweeps across twenty millions years of primate evolution. Mol. Biol. Evol. 2016;33:3065–3074. doi: 10.1093/molbev/msw199. [DOI] [PubMed] [Google Scholar]
- 20.Skov L., Hui R., Shchur V., Hobolth A., Scally A., Schierup M.H., Durbin R. Detecting archaic introgression using an unadmixed outgroup. PLoS Genet. 2018;14:e1007641. doi: 10.1371/journal.pgen.1007641. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 21.Harris K., Nielsen R. The genetic cost of Neanderthal introgression. Genetics. 2016;203:881–891. doi: 10.1534/genetics.116.186890. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 22.Bersaglieri T., Sabeti P.C., Patterson N., Vanderploeg T., Schaffner S.F., Drake J.A., Rhodes M., Reich D.E., Hirschhorn J.N. Genetic signatures of strong recent positive selection at the lactase gene. Am. J. Hum. Genet. 2004;74:1111–1120. doi: 10.1086/421051. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 23.Mathieson S., Mathieson I. FADS1 and the timing of human adaptation to agriculture. Mol. Biol. Evol. 2018;35:2957–2970. doi: 10.1093/molbev/msy180. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 24.Stern A.J., Wilton P.R., Nielsen R. An approximate full-likelihood method for inferring selection and allele frequency trajectories from DNA sequence data. PLoS Genet. 2019;15:e1008384. doi: 10.1371/journal.pgen.1008384. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 25.Mueller J.L., Skaletsky H., Brown L.G., Zaghlul S., Rock S., Graves T., Auger K., Warren W.C., Wilson R.K., Page D.C. Independent specialization of the human and mouse X chromosomes for the male germ line. Nat. Genet. 2013;45:1083–1087. doi: 10.1038/ng.2705. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 26.Mueller J.L., Mahadevaiah S.K., Park P.J., Warburton P.E., Page D.C., Turner J.M.A. The mouse X chromosome is enriched for multicopy testis genes showing postmeiotic expression. Nat. Genet. 2008;40:794–799. doi: 10.1038/ng.126. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 27.Cocquet J., Ellis P.J.I., Mahadevaiah S.K., Affara N.A., Vaiman D., Burgoyne P.S. A genetic basis for a postmeiotic X versus Y chromosome intragenomic conflict in the mouse. PLoS Genet. 2012;8:e1002900. doi: 10.1371/journal.pgen.1002900. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 28.Ellison C., Bachtrog D. Recurrent gene co-amplification on Drosophila X and Y chromosomes. PLoS Genet. 2019;15:e1008251. doi: 10.1371/journal.pgen.1008251. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 29.Heid H., Figge U., Winter S., Kuhn C., Zimbelmann R., Franke W. Novel actin-related proteins arp-T1 and Arp-T2 as components of the cytoskeletal calyx of the mammalian sperm head. Exp. Cell Res. 2002;279:177–187. doi: 10.1006/excr.2002.5603. [DOI] [PubMed] [Google Scholar]
- 30.Hallast P., Agdzhoyan A., Balanovsky O., Xue Y., Tyler-Smith C. A Southeast Asian origin for present-day non-African human Y chromosomes. Hum. Genet. 2020;140:299–307. doi: 10.1007/s00439-020-02204-9. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 31.Terhorst J., Kamm J.A., Song Y.S. Robust and scalable inference of population history from hundreds of unphased whole genomes. Nat. Genet. 2017;49:303–309. doi: 10.1038/ng.3748. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 32.Pool J.E., Nielsen R. Population size changes reshape genomic patterns of diversity. Evolution. 2007;61:3001–3006. doi: 10.1111/j.1558-5646.2007.00238.x. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 33.Favorov A., Mularoni L., Cope L.M., Medvedeva Y., Mironov A.A., Makeev V.J., Wheelan S.J. Exploring massive, genome scale datasets with the GenometriCorr package. PLoS Comput. Biol. 2012;8:e1002529. doi: 10.1371/journal.pcbi.1002529. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 34.Klopfenstein D.V., Zhang L., Pedersen B.S., Ramírez F., Warwick Vesztrocy A., Naldi A., Mungall C.J., Yunes J.M., Botvinnik O., Weigel M., et al. GOATOOLS: a Python library for Gene Ontology analyses. Sci. Rep. 2018;8:10872. doi: 10.1038/s41598-018-28948-z. [DOI] [PMC free article] [PubMed] [Google Scholar]
Associated Data
This section collects any data citations, data availability statements, or supplementary materials included in this article.
Supplementary Materials
Data Availability Statement
This paper analyzes existing, publically available data listed in the key resources table.
All code used to produce results and visualizations are deposited on GitHub. A DOI and permanent link is available via Zenodo: https://doi.org/10.5281/zenodo.7528246.
Any additional information required to reanalyze the data reported in this paper is available from the lead contact upon request.