

Summary
Estrogen and progesterone have been extensively studied in the mammary gland, but the molecular effects of androgen remain largely unexplored. Transgender men are recorded as female at birth but identify as male and may undergo gender-affirming androgen therapy to align their physical characteristics and gender identity. Here we perform single-cell-resolution transcriptome, chromatin, and spatial profiling of breast tissues from transgender men following androgen therapy. We find canonical androgen receptor gene targets are upregulated in cells expressing the androgen receptor and that paracrine signaling likely drives sex-relevant androgenic effects in other cell types. We also observe involution of the epithelium and a spatial reconfiguration of immune, fibroblast, and vascular cells, and identify a gene regulatory network associated with androgen-induced fat loss. This work elucidates the molecular consequences of androgen activity in the human breast at single-cell resolution.
Keywords: androgen regulation of human breast homeostasis, hormone receptor, transcriptional control, chromatin regulation, single-cell RNA sequencing, single-cell ATAC sequencing, multiplexed immunohistochemistry by co-detection by indexing, cellular metabolism, lactation, breast microenvironment
Graphical abstract
Highlights
-
•
A multi-omic atlas of breast tissues from individuals treated with androgen therapy
-
•
Hormone receptor-expressing (HR+) cells undergo gender-biased reprogramming
-
•
Paracrine signaling relays androgen effects from HR+ cells to other cell types
-
•
Androgen shapes breast morphology, immune composition, and metabolism
Raths et al. analyzed breast tissue from individuals receiving gender-affirming androgen therapy. Androgen shrunk ductal structures, induced gender-biased transcriptional changes, and reshaped the stromal compartment of the breast. Changes were induced directly through the androgen receptor or indirectly through paracrine signaling to cells lacking hormone receptors.
Introduction
Hormones regulate mammary gland development, menstrual cycles, pregnancy, lactation, and malignant transformation. The most critical hormones for tissue homeostasis are estrogen, progesterone, prolactin, and oxytocin, which signal via receptors in luminal and myoepithelial (basal) cells constituting the mammary epithelium. Oxytocin receptors (OTR) are expressed by myoepithelial cells, while the intracellular nuclear receptors for estrogen and progesterone (ER and PR, respectively) and the transmembrane receptor for prolactin (PRLR) are expressed by luminal epithelial cells.1,2,3 These hormone receptor-expressing (HR+) cells communicate hormone signals to proximal cells via paracrine signaling cascades that stimulate growth and secretory activity depending on reproductive status.4
Androgen acting via the androgen receptor (AR) is another important sex hormone in women that modulates breast development and function. AR is more ubiquitously expressed than ER or PR and is detected in multiple breast cell subtypes, including epithelial cells, fibroblasts, and adipocytes.5,6 It is generally accepted that androgens counteract estrogen and can inhibit thelarche (pubertal breast development) in boys and in girls with pathologically elevated androgen levels.7,8 AR is expressed in 60%–90% of all breast cancers, and recent data show AR is a tumor suppressor in ER+ disease.5,9 This opens possibilities for context-specific AR-targeted breast cancer treatments and demands a deeper understanding of androgen action in the breast.9,10
One opportunity to study androgen signaling in the human breast lies in tissues from transgender men receiving gender-affirming treatment. Transgender describes a discordance between the gender identity of a person and their sex recorded at birth.11 Transgender men are recorded female at birth, but identify as male, while cisgender women are recorded female at birth and identify as such. Many transgender men undergo gender-affirming androgen therapy, which is often complemented by gender-affirming mastectomy.12,13,14,15
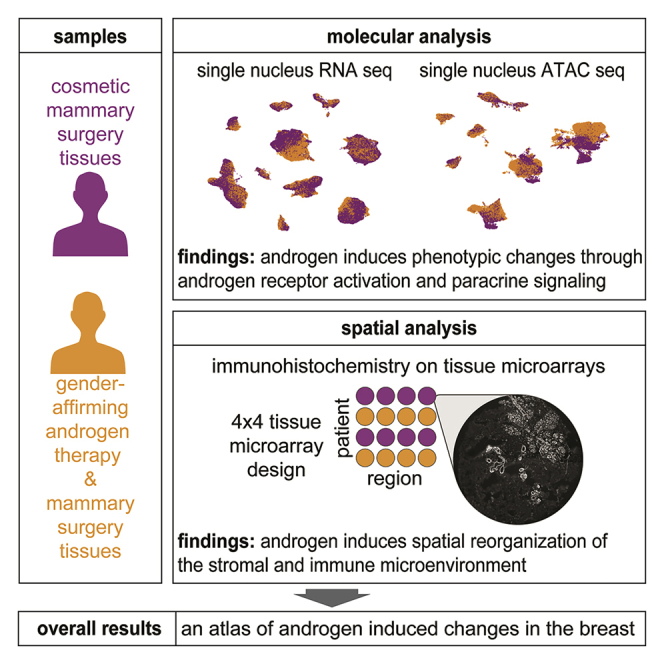
This study elucidates changes induced in the breasts following exposure to androgen. We performed single nucleus RNA and assay for transposase-accessible chromatin (ATAC) sequencing (small nuclear RNA sequencing [snRNA-seq] and small nuclear ATAC sequencing [snATAC-seq]) on mastectomy specimens from transgender men undergoing gender-affirming androgen therapy and compared them to samples from cosmetic mammary surgeries in cisgender women. We also used co-detection by indexing (CODEX) multiplex immunohistochemistry (IHC) staining to study structural changes and how androgen influences cell-cell interactions. This multi-modal single-cell atlas represents a comprehensive resource to study the molecular consequences of androgen activity in the human breast.
Results
Breast cells are silenced by androgen exposure
We analyzed nuclei and tissues from nine transgender men undergoing gender-affirming androgen therapy and subcutaneous mastectomy. For comparison, we studied breast tissue from nine cisgender women who had cosmetic mammary surgery (Figures 1A and S1A–S1C; Table S1; STAR Methods). We refer to samples from cisgender men, transgender men, and cisgender women as cis-male, trans-male, and cis-female respectively. We analyzed 38,762 trans-male and 66,926 cis-female nuclei with snRNA-seq, as well as 30,927 trans-male and 27,459 cis-female nuclei with snATAC-seq. Cells were classified in the transcriptomic data using curated markers, whose promoter accessibilities were used to annotate corresponding snATAC-seq populations (Figures 1B, S1D, and S1E; Table S2). We also created eight tissue microarrays (TMAs) from the left and right breasts of most individuals that we analyzed with a CODEX antibody panel designed based on the nuclei data, providing spatial information for 161,241 trans-male and 156,842 cis-female cells (Figures 1C and S1C; Table S3; STAR Methods).
Figure 1.

Multi-modal single-nuclei sequencing and spatial proteomics identify molecular distinctions between the breasts of transgender men and cisgender women
(A) Color code for breast samples analyzed, as well as the age, menopausal status, and length of androgen therapy for their corresponding patient.
(B) Uniform manifold approximation and projection (UMAP) plots annotated by cell type (left) and gender ID (right), with snRNA-seq on top and snATAC-seq data at the bottom. Luminal-HR+, hormone receptor-expressing luminal cells; luminal-HR–, hormone receptor-negative luminal cells; basal, basal/myoepithelial cells; blood EC, blood endothelial cells; lymph. EC, lymphatic endothelial cells; vasc. acc., vascular accessory cells.
(C) Fluorescence microscopy images of breast tissues, with each marker identifying a major cell class or structure: KRT8, pan-luminal; TP63, basal; AR, luminal-HR+; ACTA2, smooth muscle structures; CD31, endothelial; LYVE1, lymphatic vessels; PDGFRB, fibroblasts; CD45, immune; PLIN1, adipocyte.
(D) Unique molecule identifiers (UMIs) detected in each cell of the snRNA-seq data, split by cell types and gender ID. Horizontal axis limits were set to 350–30,000, excluding 43 outliers to improve interpretability (p values, Wilcoxon: ∗∗∗p ≤ 5.29 × 10−203, ∗∗p ≤ 1.63 × 10−97, ∗p ≤ 2.66 × 10−71).
(E) Mean fraction of spliced transcripts detected in the snRNA-seq data based on velocyto (La Manno et al., 2018).16 Horizontal axis limits were set to 0–50, excluding 49 outliers to improve interpretability of the plot (p values, Wilcoxon: ∗∗∗p ≤ 2.32 × 10−277, ∗∗p ≤ 6.33 × 10−93, ∗p ≤ 5.04 × 10−6).
(F) Ratios of nucleosome bound to nucleosome-free genomic fragments in each cell of the snATAC-seq data, split by cell types and gender ID. Horizontal axis limits were set to 0.3–1.5, excluding 90 outliers to improve interpretability of the plots (p values, Wilcoxon: ∗∗∗p ≤ 3.76 × 10−137, ∗∗p ≤ 4.67 × 10−45, ∗p ≤ 7.64 × 10−5).
Within the snRNA-seq data, the epithelium contained hormone receptor-positive and -negative luminal cells (luminal-HR+ and luminal-HR−, respectively), as well as myoepithelial/basal cells (Figures 1B and S1D). The vasculature contained blood and lymphatic endothelial cells (blood EC and lymph. EC) as well as pericytes and vascular smooth muscle cells, which clustered into a group we termed vascular accessory cells (vasc. acc.). The stroma contained adipocytes and fibroblasts, while the immune compartment harbored myeloid and lymphoid clusters (Figures 1B and S1D). snRNA- and snATAC-seq cell proportions were correlated, except vascular accessory cells were not detected in the chromatin samples, where epithelial cells constituted greater proportions, likely due to cell-type-specific vulnerabilities to the different assay conditions (Figures 1B, S1F, and S1G). Most TMA cells could be correspondingly annotated based on staining patterns, but some stromal cells were negative for all markers (designated stroma-other) and a group of cells that exclusively stained for the actin regulatory protein ENAH could not be identified (Figures S1H and S1I).
Our cohort contained pre- and post-menopausal cis-female samples, enabling analysis of how menopausal status affected comparisons (Figure S1J; Table S1). Relative to androgen-induced changes, there were few differences between cells from pre- and post-menopausal cis-female samples or between cells from different types of cosmetic surgeries (Figures S2A and S2B). Furthermore, similar differential gene-expression patterns were observed when trans-male cells were compared with pre- and post-menopausal cis-female cells (Figure S2C). Based on these observations, pre- and post-menopausal cis-female samples were consolidated for subsequent analyses.
There were fewer unique molecular identifiers and genes captured in trans-male nuclei despite higher corresponding library sequencing saturation rates (Figures 1D and S3A). Analysis of the Genotype-Tissue Expression (GTEx) database also found fewer genes expressed in cis-male breast tissues compared with cis-female samples (Figures S3B and S3C). Pathway analysis showed no indication of cell stress, apoptosis, or degradation, while instead revealing upregulation of splicing factors in the trans-male samples, consistent with increased rates of exon mapping read counts (Figures 1E, S3A, and S3D; Table S4). Genes related to protein translation were also downregulated, coinciding with a higher degree of nuclear condensation, although DAPI staining indicated nuclei were not smaller in trans-male samples, with some cell types showing increased staining areas (Figures 1F, S3D, and S3E; Table S4).
Hormone-responsive luminal cells activate male gene programs during androgen therapy
Luminal-HR+ cells, the only cell type to express RNA for AR, ESR1, and PGR, showed the largest treatment-associated chromatin changes (Figures S3F and S3G). This luminal subclass formed six subclusters that showed strong trans-male and cis-female bias (Figures 2A, S4A, and S4B). RNA-velocity analysis also suggested trans-male and cis-female cells follow diverging trajectories to terminate at two distinct states (Figures 2A and 2B). The luminal-HR+ subcluster containing most trans-male cells (lup 2) was associated with fatty acid metabolism and calcium signaling, while the major cis-female subclusters (lup 1 and lup 3) were associated with growth factors and estrogen signaling as well as mammary gland development (Figure S4B; Table S5). There was also one minor subcluster, making up only 3% of lumina-HR+ cells, which was enriched for ribosomal genes (labeled “ribo”) and another subgroup modestly associated with trans-male cells that expressed cycling genes (labeled “cycling”), but it only constituted 0.4% of the population and cycling genes were unchanged overall (Figures 2A and S4B; Tables S5 and S6). The most evenly shared subcluster (lup 4) associated with steroid metabolism and androgen response genes and had low ESR1 expression (Figures 2A, 2C, and S4B; Table S5). Coincident with luminal progenitors being ER− and lipid metabolism being a regulator of progenitor cell maintenance, various trajectory analyses implied lup 4 might represent a less differentiated state (Figures 2A, 2B, S4C, and S4D).2,17
Figure 2.

Hormone-responsive epithelial cells of the trans-male breast are altered for genes that show sex bias in other tissues
(A) UMAP of luminal-HR+ snRNA-seq data showing detected subclusters with RNA-velocity streams overlayed (left) and gender identity (right).
(B) RNA-velocity pseudotime ordering of trans-male and cis-female luminal-HR+ cells. Time 0 (T0) in the center and respective endpoints of cis-female and trans-male lineages (T1) at the outer maxima. Annotation bars show gender identity and subcluster assignment of each cell. Rows are annotated with highly differentially expressed genes or subcluster markers.
(C) Left panel overlays hormone receptor RNA expression on UMAP from (A). Right panel shows boxplots of hormone receptor staining intensities averaged across luminal-HR+ cells in the CODEX data (p value, Wilcoxon: PGR = 0.00041).
(D) Left panel shows per nucleus ChromVAR motif enrichment z scores for AR (Catalog of Inferred Sequence Binding Preferences [CisBP] M03389_2.00) in luminal-HR+ snATAC-seq data (p value, Wilcoxon: <2.2 × 10−16). Right panel shows average nuclear to cytoplasmic staining ratios for AR in luminal-HR+ cells from each TMA region (p value, Wilcoxon: 0.00021).
(E) RYR2 chromatin accessibility (top) for cis-female (purple) and trans-male (orange) luminal-HR+ cells, with highlighted motif binding sites of AR, FOXA1, and CCCTC-binding factor (CTCF). The RYR2 gene body (light green) is shown with promoter (arrow) and exon boundaries (dark green). Also shown (center) is chromatin accessibility data for the genomic region in tissues with varying RYR2 expression and Hi-C data (bottom) comparing three-dimensional chromatin structure of the same region in PANC-1 (pancreas) and MCF-10A (breast) cell lines.
(F) AR-motif binding sites (red markers) across open chromatin regions of the CUX2 locus in luminal-HR+ cells from cis-females and trans-males. CUX2 gene body (light green), exon boundaries (dark green), and promoter (arrow) are shown below.
(G) Average RNA (left, adjusted p value, model-based analysis of single-cell transcriptomics [MAST]: <2.2 × 10−16) and per-region average staining intensity (right, p value, Wilcoxon: 0.027) of CUX2 in cis-female and trans-male tissues.
(H) Effect sizes of CUX2 sex bias in GTEx tissues, as a function of median AR expression (vertical axis and dot size). Positive and negative values indicate female and male bias, respectively.
(I) Chromatin accessibility (top) around the PGR locus in trans-male and cis-female luminal-HR+ and luminal-HR– cells. The PGR gene body (light green) is shown with promoter (arrow) and exon boundaries (dark green). The one significantly altered chromatin peak is indicated by a gray shaded area (Wilcoxon false discovery rate [FDR] <0.05), magnified on the bottom left. Bottom right shows importance levels of transcription factors inferred through random forest analyses to co-bind with AR and determine the directionality of the transcriptional change, with the corresponding DNA binding sites at the PGR locus shown above the top panel.
(J) Top left panel shows AR, JUN, and ESR1 chromatin footprints in cis-female and trans-male luminal-HR+ cells. Bottom left panel shows average log2FC of chromatin accessibility in peaks containing no ESR1 motif, only ESR1 motifs, or both ESR1 and JUN motifs. Bottom right panel shows the fraction of chromatin peaks overlapping only ESR1 motifs or both ESR1 and JUN motifs among all, cis-female-specific, and trans-male-specific luminal-HR+ peaks. Top right panel shows the fraction of peaks overlapping both ESR1 and JUN motifs that had in vitro ChIP-seq evidence for both JUN and ESR1 binding.
Androgen did not markedly affect AR or ESR1 levels in luminal-HR+ cells, but the canonical ER target PGR was reduced, in accordance with other studies of AR action in the breast (Figures 2C and S4A).9,18 However, AR nuclear to cytoplasmic AR staining ratios were higher in trans-male luminal-HR+ cells and AR-motif accessibility increased, indicating enhanced canonical AR activity (Figures 2D, S4E, and S4F). In addition to androgen response elements (AREs), trans-male cells showed increased binding motif accessibility for other steroid receptors and several members of the forkhead family of transcription factors, consistent with known features of AR signaling (Figure S4G).19,20 This is exemplified by differential upregulation of Ryanodine receptor 2 (RYR2) at both the RNA and protein level (Figures 2E, S2C, and S4H; Table S6).21 RYR2 regulates heart and blood vessel calcium signaling events and calcium-dependent insulin secretion in the pancreas.22,23,24 In these organs, RYR2 loci show accessibility at AR and FOXA1 DNA binding motifs that are natively inaccessible in breast tissue (Figure 2E).23,25
AR activation in luminal-HR+ cells also induced altered chromatin accessibility around the promoter of the most upregulated transcription factor, CUX2 (Figures 2F, 2G, and S2C; Table S6). CUX2 normally shows the highest expression in the prostate, and CUX2 is also higher in cis-male liver and breast samples compared with cis-female tissues (Figures 2H and S5A). Indeed, the top up- and downregulated genes in luminal-HR+ cells showed strong cis-male and -female sex bias, respectively, in breasts and other organs (Figures S5B–S5E).
Most trans-male patients, including all studied here, experience cessation of menses within 6 months of treatment. Hence, we thoroughly examined PR activity, which is critical for ovulation and was the most downregulated transcription factor in androgen-treated luminal-HR+ cells (Figure S2C; Tables S1 and S6).26,27,28 Consistent with published AR chromatin immunoprecipitation sequencing (ChIP-seq) data, our snATAC-seq data showed multiple areas of open chromatin overlapping AREs near the PGR locus, but only one small open chromatin peak emerged after androgen therapy (Figure 2I).9 Notably, that peak and others contain binding motifs for transcription factors identified in a broader analysis as likely co-repressors at accessible ARE-associated downregulated genes, with NFIC and FOXA1 also showing co-regulatory function in the prostate (Figures 2I and S6A; STAR Methods).29,30 PGR motif accessibility gains after androgen therapy were only superseded by those for AR and NR3C1, which is likely an artifact of AR and PGR motif sequence similarity based on reduced levels of PGR RNA and protein expression (Figure S4G).31,32 Indeed, PGR binding sites were enriched near AREs, and linear regression showed PGR motif accessibility could not be predicted from corresponding gene expression like for other nuclear receptors (Figures S6B and S6C).
ER binding motif accessibility was largely unaltered by androgen therapy, but estrogen signaling pathways were downregulated (Figures 2C, 2J, S3F, and S4B; Table S6). AR can sequester essential ER co-activators, and one related mechanism for estrogen signaling regulation in the normal breast may involve AP-1 transcription factors such as BATF, which facilitate ER signaling (Figures S2C, S4B, S4G, and S6D; Table S6).9,33,34 The archetypical AP-1 factor JUN, which physically interacts with both ER and AR, was also decreased in expression and motif accessibility, and ER binding sites co-located with JUN motifs were more likely to decrease in accessibility after androgen therapy (Figures 2J and S2C; Table S6).35,36,37,38
Among genes potentially affected by lowered ER and AP-1 factor binding, we found the growth factors amphiregulin (AREG) and epiregulin (EREG), which are estrogen responsive in breast cancer (Figures S2C, S5E, and S6E–S6G; Table S6).39,40,41,42,43 However, at the protein level, AREG increased and became more concentrated in luminal-HR+ cells (Figure S6G). AREG secretion depends on membrane-bound pro-AREG cleavage via matrix metalloproteases such as ADAM17, which is downregulated in luminal-HR− and basal cells, potentially causing un-cleaved pro-AREG accumulation in luminal-HR+ cells (Figure S6H).44,45 This may also explain why the AREG receptor EGFR was downregulated in neighboring basal cells but upregulated in luminal-HR+ cells (Figure S6I).
Altered fibroblast signaling to epithelial cells through reduced laminin production
Fibroblasts are AR+ cells that influence the breast epithelium through the extracellular matrix (ECM; Figure S3F).46,47 Based on RNA, breast fibroblasts consist of two matrix subtypes, a lipo-fibroblast group expressing PPARG and vascular-like cells that express endothelial genes such as NRP1 (Figures 3A and 3B; Table S5).48,49 Chondrocytes were also detected. After androgen therapy, fibroblasts show increased AR-motif accessibility, centrality of AR-motif occurrence with respect to peak summit, and increased AR nuclear to cytoplasmic staining (Figures 3C–3E). Immunostaining also showed higher proportions of AR+ fibroblasts (fibr-main) and an AR+ epithelial associated (fibr-epi) fibroblast subgroup staining for keratin 8 and 23 due to epithelial cell proximity (Figures 3F and 3G).
Figure 3.

AR activity drives fibroblast’s response to hormone-replacement therapy
(A) UMAP depicting fibroblast subclusters (top) in snRNA-seq data (matrix 1 and 2, matrix-type fibroblasts; lipo-f, lipo-fibroblasts; vasc-f, vascular-like fibroblasts) and the distribution of patient samples in each gender identity (bottom).
(B) Heatmap shows scaled average expression of snRNA-seq markers identified for each of the fibroblast subclusters.
(C) Motif footprints for AR (AR-CisBP M03389_2.00) among trans-male and cis-female fibroblast cells. Top panel shows the transposase bias-corrected signal, and the bottom panel shows the transposase bias.
(D) Right panel shows the enrichment of motifs among unique accessible chromatin peaks of fibroblast cells from cis-female and trans-male samples. Left panel shows the fraction of the peaks of the corresponding cells that overlap with the motif.
(E) Average ratio of AR staining intensity in fibroblast nucleus compared with the cytoplasm in tissue regions of each sample type (p value, Wilcoxon; 1.2 × 10−8).
(F) Left panel boxplots show the fraction of cis-female (purple) and trans-male (orange) cells corresponding to five different classes of fibroblasts detected in tissue regions of CODEX microarray data (p values, Wilcoxon: fibr-main = 0.00011, fibr-epi = 0.0046). Right panel shows the scaled staining intensities of various markers that distinguish the five subtypes of fibroblasts.
(G) Boxplots show per-region average distance for each of the five subtypes of fibroblasts to the most proximal epithelial cell.
(H) Violin plots show the RNA expression of laminins LAMA2 (top) and LAMB1 (bottom) in fibroblast subclusters, split by sample type (adjusted p values, MAST: LAMA2 in lipo-f = 2.60 × 10−63, matrix 1 = 5.11 × 10−175, matrix 2 = 1.03 × 10−69, vasc-f = 1.99 × 10−10; LAMB1 in lipo-f = 3.71 × 10−35, matrix 1 = 1.46 × 10−55, matrix 2 = 3.08 × 10−87, vasc-f = 2.58 × 10−13).
(I) AR binding sites (red markers) across genomic regions of LAMA2 and LAMB1. Gene bodies are shown (light green) with the promoter (arrow) and exon boundaries (dark green). Genomic window shows chromatin accessibility in cis-female (purple) and trans-male (orange) fibroblasts.
(J) Boxplots show per-region average LAMA2 (left) and LAMB1 (right) staining intensities in the LAMB1+ fibroblast subtype on CODEX microarray data (p value, Wilcoxon: LAMA2 = 0.64, LAMB1 = 0.0079).
(K) Boxplots show the average RNA expression of ITGB1 among luminal-HR+, luminal-HR–, and basal epithelial cells (adjusted p value, MAST in basal <2.22 × 10−16).
The ECM constituent laminins LAMB1 and LAMA2 were downregulated after androgen therapy and were also lower in cis-male breast samples (Figures 3H, S2C, and S5E; Table S6). Chromatin data suggest ARE accessibility and a potential for direct AR-mediated repression at these laminins, but no differentially accessible peaks emerged after androgen therapy (Figure 3I). LAMB1 immunostaining was also reduced in trans-male cells from the LAMB1+ subcluster (fibr-LAMB1+), but LAMA2 protein changes were not detected (Figures 3F and 3J). When examining possible responses to altered laminin composition, we found the ITGB1 integrin receptor for LAMB1 was downregulated in ECM interfacing basal cells (Figure 3K).50,51
The breast epithelium shows reduced myoepithelial coverage and becomes infiltrated by stromal cells after androgen therapy
Androgen response was also studied in the basal and luminal-HR− cells that constitute the rest of the epithelium (Figures 1B and S7A). Acini and ductal structures had reduced size and proximal ACTA2 staining coverage after androgen therapy (Figures 4A, 4B, S7B, and S7C). Basal cell proportions were unchanged in the RNA data, but androgen therapy transitioned these cells away from two subclusters (bas 1 and bas 3) toward one other (bas 2) with reduced expression of the lactation-associated genes ACTA2 and OXTR as well as TP63, suggesting contractile impairment, but only TP63 showed corresponding protein changes (Figures 4C, 4D, and S7D; Table S5).52,53,54 OXTR gene expression was also lower in cis-male breast samples, and RNA-velocity analysis indicated androgen-treated basal cells follow a unique trajectory and converge at a terminal state where smooth muscle contraction, focal adhesion, and cell junction organization are reduced (Figures 4C, 4E, S5E, and S7E). For the latter pathway, this likely results from reduced BACH2 expression, whose DNA binding motif is enriched at corresponding genes and whose expression is also lower in cis-male breasts (Figures 4F, S5E, S7F, and S7G; Table S7).
Figure 4.

Epithelial cells without hormone responsiveness lose contractile functions upon androgen therapy
(A) Images from CODEX data showing mammary acini structures from cis-female (top) and trans-male (bottom) tissues marked by expression of ACTA2 (basal cells, purple), TP63 (basal cell nuclei, blue), and KRT8 (luminal cells, green).
(B) Average area of acinar structures (left panel) and average area of acini border that was filled with ACTA2 signal (see Figure S7C and STAR Methods) among cis-female and trans-male tissues (p values, Wilcoxon: area = 0.026, ACTA2 coverage = 0.012).
(C) UMAP of basal cell subclusters in snRNA-seq data (top) and the distribution of trans-male and cis-female cells across them (bottom).
(D) RNA expression of ACTA2, OXTR (lactation markers), and TP63 in basal cells of trans-male and cis-female samples (adjusted p values, MAST: ACTA2 = 8.86 × 10−296, OXTR = 9.59 × 10−262, TP63 = 1.16 × 10−96).
(E) Module scores of enriched pathways overlaid on the basal cell UMAP (REAC, Reactome; KEGG, Kyoto Encyclopedia of Genes and Genomes).
(F) Right panel shows the enrichment of motifs among unique accessible chromatin peaks from trans-male and cis-female basal cells. Left panel shows the fraction of peaks from the corresponding cells that overlap with the motif.
(G) Kernel density estimation of module scores for selected altered structural pathways in luminal-HR– cells (p values, Wilcoxon: KEGG, adherens junction = 4.13 × 10−285; KEGG, focal adhesion = 1.42 × 10−255; KEGG, regulation of actin cytoskeleton <1.42 × 10−255).
(H) Average RNA expression (top) of integrin receptors from the “KEGG: regulation of actin cytoskeleton” pathway in luminal-HR– cells (adjusted p values, MAST: ITGA2 = 4.89 × 10−201, ITGB8 = 6.40 × 10−267) and average expression of the ITGA2 and ITGB8 ligand FN1 in fibroblast subclusters and lymphatic endothelial cells (bottom) from trans-male and cis-female samples (adjusted p values, MAST: matrix 1 = 1.66 × 10−54, matrix 2 = non-significant [n.s.], lipo-f = 1.32 × 10−16, vasc-f = n.s., lymph. EC = 3.13 × 10−99).
(I) Fisher exact test odds ratio (x axis) and –log10 p value (y axis) corresponding to enrichment of each motif among the chromatin accessibility peaks for the genes of the "WikiPathways: focal adhesion pathway.” Colors indicate log2 fold change in gene expression of transcription factors corresponding to each motif. Gray motifs represent transcription factors without differential gene expression among luminal-HR– cells. Right panel shows the fraction of genes (left) and genes annotated within the focal adhesion pathway (right) that contain a chromatin peak with an ESRRG sequence motif (cisBP ESRRG_697).
(J) Spatial distribution of epithelial, stromal, immune, and endothelial cells in an example breast tissue region from cis-female (top) and trans-male (bottom) samples.
(K) Ratios of stromal to epithelial cells in the epithelial neighborhood (see Figure S8C) among regions of cis-female and transgender male tissue in CODEX microarray data (p value, Wilcoxon: 0.0052).
Luminal-HR− cells showed no subcluster redistribution following androgen therapy, but structural pathways of focal adhesion, adherens junctions, and actin cytoskeleton regulation were downregulated (Figures 1B, 4G, S7H, and S7I; Tables S6 and S7). Significant loss of ITGA2 and ITGB8 in androgen-treated luminal-HR− cells and in epithelial enriched cis-male breast samples were observed (Figures 4H and S5C). Among matching integrin ligands, there was decreased expression of the ECM component and critical cell adhesion and morphology regulator FN1 in fibroblasts and lymphatic endothelial cells (Figure 4H).55,56,57 Overall staining intensity of FN1 was not decreased in these cell types after androgen therapy, but fewer FN1+ fibroblasts were identified near the epithelium (Figures 3F, S8A, and S8B). Furthermore, DNA binding motifs for the androgen-responsive gene estrogen-related receptor gamma (ESRRG) are over-represented in chromatin peaks proximal to focal adhesion genes in these cells (Figures 4G and 4I).58,59
Androgen therapy decreases capillary vasculature near the breast epithelium
IHC analysis showed breast acini and ducts have a higher proportion of non-epithelial cells in their proximity after androgen therapy (Figures 4J and 4K). With fibroblasts being the largest group found between individual acini, this might explain previously reported fibrosis in transgender male breast tissue (Figure S8C).60,61 One other infiltrating population was endothelial cells (Figures 5A and S8C). In the snRNA-seq data, blood endothelial cells (blood EC) consisting of arterial, capillary, and venous cells, were called according to established signatures (Figures 5B and S9A–S9C).62 Lymphatic endothelial cells expressing PDPN, LYVE1, and FLT4 formed two subclusters (lymph. EC and lymph. EC2), while pericytes and two subclusters of vascular smooth muscle cells (vasc. SM1 and vasc. SM2) formed what we termed vascular accessory cells. IHC found CD31+ cells separated into an ACTA2+ contractile subgroup (endo-SMA), a CD45+ immune-associated subgroup (endo-immu.), and another group expressing LNX1 and CD36, resembling capillaries (endo-LNX1+; Figures S9D and S9E). Lymphatic ECs, expressing LYVE1 and PDPN, were represented in the epithelial neighborhood and were increased after androgen therapy based on all data modalities (Figures 5C and S9D–S9H). RNA and staining data also indicated capillaries were reduced, particularly near ductal structures (Figures 5C, S9F, and S9H).
Figure 5.

Androgen therapy reduces epithelial vascularization through PPARG activity
(A) Microscopic images show vascularization of two ductal structures in a cis-female (left) and a trans-male (right) breast tissue in CODEX microarray data. KRT8 (green) marks luminal cells, ACTA2 (purple) marks green cells, and CD31 (red) marks endothelial cells. Arrows point out (1) larger vessels with smooth muscle layer and (2) smaller vessels without smooth muscle layer.
(B) UMAP shows vasculature subclusters detected in the snRNA-seq dataset. (left, blood endothelial cells; right, lymphatic endothelial cells; upper-mid, vascular accessory cells).
(C) Boxplots show the proportions of vascular subclusters in each sample of the snRNA-seq data, split by gender ID (general linearized model [GLM] fitting a Poisson, p values: vein = 6.72 × 10−45, capillary = 5.31 × 10−77, artery = 3.58 × 10−5, lymph. EC = 1.33 × 10−22, and lymph. EC 2 = 0.0071).
(D) UMAP (left) shows blood endothelial cells overlaid with scVelo stream plots. The scatterplot shows the ratio of spliced (horizontal axis) and unspliced RNA molecules (vertical axis) of PPARG among vein (blue), capillary (orange), and artery (green) blood endothelial cells. Dashed diagonal indicates the steady-state ratio. Top and bottom arcs indicate the estimated kinetic parameters of PPARG induction and repression, respectively.
(E) PPARG GRN module score overlaid on UMAP plot among cis-female (left) and trans-male (right) blood endothelial cells. Barplot shows GRN importance scores of the top five genes coexpressed with PPARG.
(F) Volcano plot shows the average log2 fold change and –log10 adjusted p value for differential expression of genes within the PPARG module among the trans-male and cis-female blood endothelial cells. Purple data points indicate genes with a chromatin accessibility peak overlapping the PPARG transcription factor sequence motif (CisBP PPARG_676) match. Barplots show the fraction of all genes (left) or genes within PPARG module (right) that contain a chromatin accessibility peak overlapping the PPARG transcription factor sequence motif (purple).
(G) Boxplot shows average expression of PPARG in blood endothelial cells of cis-female (purple) and trans-male (orange) samples in snRNA-seq data.
(H) Heatmap shows the log2 fold change in expression of ligand (left)-receptor (right) pairs among cell types and vascular subclusters in the trans-male and cis-female samples. Colors indicate log2 fold change in expression, and diameter of the circle shows the percent of cis-female cells expressing the gene.
RNA-velocity analysis found capillaries have a distinct terminal state of differentiation that appears driven by PPARG (Figure 5D). This was corroborated by the specific expression of a PPARG gene regulatory network (GRN) in capillaries, which contained PPARG-associated lipid-metabolism markers FABP4 and CD36 and was enriched for angiogenesis and blood vessel development pathways also likely to be downstream of PPARG (Figures 5E, 5F, and S9I; Table S4).63,64 This implies androgen-driven PPARG and PPARG GRN loss drives reduced capillary proportions (Figures 5E and 5G). Additionally, we found that, after androgen therapy, AR+ adipocytes and matrix 2-type fibroblasts upregulate the anti-angiogenic CD36 ligand Thrombospondin 1 (THBS1) also found higher in adipose and fibroblast enriched cis-male breast samples (Figures 5H and S5E).65,66 The vascular growth factor VEGFA is also upregulated in adipocytes after androgen therapy, and lymphatic endothelial cells are the only cell type that increases expression of both the corresponding receptor KDR (VEGFR2) and FLT4, which dimerize to promote angiogenesis (Figure 5H).67,68 This may underlie increased lymphatic endothelial cell numbers after androgen therapy, and pathway analyses find VEGFR2-mediated cell proliferation is also higher at this time (Figure S9J).
Androgen therapy reshapes the breast immune microenvironment
Sub-clustering of immune populations, which are also well represented near the breast epithelium, revealed CD8+ and CD4+ T cells, T effector cells, natural killer (NK) cells, and two classes of B cells, while the myeloid compartment consisted of monocytes, macrophages, monocyte-derived dendritic cells (labeled mono.DC), and dendritic cells (labeled DC) (Figures 6A, S8C, and S10A). A small cluster of hematopoietic stem cells (HSCs) was also detected.
Figure 6.

Androgen therapy dominates helper T lymphocytes and reduces the presence of innate immunity
(A) UMAP showing subclusters of all myeloid (left) and lymphoid (right) cells detected in the snRNA-seq data (CD8. CD8+ T cells; CD4, CD4+ T cells; T-effector, effector T cells; NK, natural killer cells; mono.DC, monocyte-derived dendritic cells; DC, dendritic cells).
(B) Boxplots show the fraction of main immune cell subtypes within entire immune compartment in each sample (GLM p values, generalized linear model fitting a Poisson: CD4 = 0.00035, CD8 = 4.035 × 10−13, T effector = 0.045, NK = 0.00035, mono.DC = 0.017, macrophage = 0.52, monocyte = 0.055, DC = 0.0001).
(C) Boxplot shows the proportion of macrophages within the periphery of epithelial cells in cis-female (purple) and trans-male (orange) tissue regions of the CODEX microarray data (p value, Wilcoxon: 0.003).
(D) Kernel density estimates and boxplots show the module scores of immune-relevant Reactome pathways in macrophages of trans-male (orange) and cis-female (purple) samples (p values, Wilcoxon, class I major histocompatibility complex [MHC]-mediated antigen processing/presentation = 8.32 × 10−17, clathrin-mediated endocytosis = 3.64 × 10−21, toll-like receptor TLR1 TLR2 cascade = 3.89 × 10−16).
(E) Boxplots show the average RNA expression of PROS1 in basal cells of cis-female (purple) and trans-male (orange) samples (adjusted p value, MAST: 3.17 × 10−192).
(F) Volcano plot shows the average log2 fold change and –log10 adjusted p value assessing the differential expression of genes in trans-male macrophages compared with cis-female macrophages. Purple data points indicate scavenger receptors.
(G) UMAP shows four immune cell staining sub-classes (macrophage, red; immune endo., green; immune main, blue; immune-epi., orange) according to the staining pattern in CODEX microarray data. Size of the data points indicates the distance to the most proximal epithelial cell. Boxplot (below) summarizes the average distance of each group of immune cells to their most proximal epithelial cell.
(H) Microscopic images show staining of luminal (KRT8, green), basal (ACTA2, purple), and immune cells (CD45, red) within a trans-male (top) and a cis-female (bottom) breast tissue in CODEX microarray data.
(I) Microscopic image shows IHC staining of luminal (KRT8, red), immune (CD45, green), and T-lymphocyte (CD3, purple) cells within a trans-male (top) and a cis-female (bottom) breast tissue. White cells are double-positive for CD45 and CD3.
(J) Boxplot shows the ratio of immune cells (CD45+) expressing CD3 to those not expressing CD3 (T lymphocytes versus other immune cells) within the epithelial neighborhood of cis-female (purple) and trans-male (orange) breast tissues of IHC scan regions.
There were fewer macrophages after androgen therapy, specifically near the epithelium, and differential pathway analysis revealed these cells had reduced expression of endocytosis and antigen presentation pathways (Figures 6B–6D, S10B, and S10C).69,70 When examining relevant ligand-receptor pairs, we found PROS1 was reduced in basal cells, which stimulates efferocytosis in macrophages (Figure 6E).71 The PROS1 receptors MERTK and AXL were also downregulated in macrophages, alongside other scavenger receptors, and PROS1 was downregulated in cis-male breast samples (Figures 6F and S5E).72,73,74
In the staining data, CD45+ cells formed distinct subsets based on their adjacency to other cell types (Figure 6G). An epithelium-associated immune population was increased after androgen therapy, which further analysis revealed was constituted by T cells (Figures 6G–6J and S10D–S10F). Based on RNA data, CD4+ T cell proportions also increased after androgen therapy, potentially driven by the T cell differentiation factor TCF7 (Figures 6B and S10G).75,76,77 When searching for chemoattractants, we found matrix-type fibroblasts upregulate IL16, which triggers CD4+ T cell migration (Figure S10G).78,79 Pathway enrichment further indicated androgen-treated CD4+ T cells are activated and proliferative after androgen therapy (Figure S10H; Table S4).
Androgen therapy elicits AZGP1 secretion and targets adipogenesis pathways in adipocytes
The human breast contains significant adipose tissue (7%–56%) in large, distinctly organized, homogeneous structures (Figure S11A).80 Adipose tissue was broadly removed during sample collection and hydrophobicity resulted in further exclusion during nuclei preparations, but adipocytes are of interest considering they are AR+ and the impact androgens have on fat distribution and metabolism (Figures S3F and S11B).15,81,82,83,84 Indeed, ligand-receptor analyses found activating ligands and receptors upstream of the phosphoinositide 3-kinase (PI3K) pathway were the most altered signaling components in the snRNA-seq dataset (Figure S11C; Table S4). Across PI3K pathway activating receptors, the insulin receptor (INSR) showed the broadest upregulation after androgen therapy, with adipocytes showing the strongest change and the highest baseline expression, consistent with previous reports (Figures 7A, S11D, and S11E).15,83,85
Figure 7.

Testosterone induces PI3K pathway alterations with adipocytes showing distinct metabolic adaptations
(A) Heatmap showing the log2 fold change in RNA expression of PI3K activating receptors (taken from "KEGG: PI3K-Akt signaling pathway") among 10 breast cell types identified. The circle diameter indicates the fraction of cis-female cells of the cell type expressing the receptor. INS, circulating insulin secreted in the pancreas.
(B) Heatmap showing log2 fold changes in RNA expression of KEGG: PI3K-Akt signaling pathway downstream transcription factors within 10 breast cell types identified.
(C) Pathway enrichment of tissue wide upregulated genes (>7 cell types) that have an NR4A1 motif in an enhancer (>4 cell types). Horizontal axis shows the odds ratio (one-sided Fisher’s exact test) comparing frequency of selected genes in the pathway versus background and vertical axis shows –log10 p value of Fisher’s exact test (FDR < 0.05 = red, p value < 0.05 = yellow, n.s. = gray (PID, Pathway Interaction Database; WP, WikiPathways).
(D) Module scores for the WikiPathways "WP: insulin signaling pathway" in all cell types, split by cis-female and trans-male origin (p value in adipocytes, Wilcoxon: 7.59 × 10−4).
(E) Representative images of computational segmentation of lipid vacuoles (left, see STAR Methods), and resulting average area of adipocyte vacuoles per IHC scan region (p value Wilcoxon: 0.00059).
(F) Sample averages of AZGP1 RNA expression in each cell type in trans-male and cis-female samples (adjusted p values, MAST: adipocyte = 5.95 × 10−12, basal = 2.42 × 10−70, blood EC = 8.67 × 10−83, fibroblast = n.s., luminal-HR− = 4.54 × 10−302, luminal-HR+ = 0.00, lymph. EC = 2.83 × 10−15, lymphoid cells = 5.46 × 10−12, myeloid cells = 9.23 × 10−8).
(G) Microscopic CODEX image of a duct stained against AR (red), AZGP1 (green), and ACTA2 (purple) in a trans-male (top) and cis-female (bottom) breast tissue region of the tissue microarray.
(H) GPAM co-expression module (GRNboost2, 95th percentile, p value, Wilcoxon: <2.22 × 10−16) score in cis-female and trans-male adipocytes.
(I) TCF7L2 expression in trans-male and cis-female adipocytes (adjusted p value, MAST: 3.48 × 10−105).
(J) Differential expression of transcription factors in trans-male compared with cis-female adipocytes. Horizontal axis shows log2 fold change in expression and the vertical axis shows –log10 adjusted p value. Purple data points indicate transcription factors with accessible chromatin matching the AR sequence motif (CisBP AR_689).
(K) Microscopic IHC image showing staining against nuclei (DAPI, blue), adipocytes (PLIN1, green), and TCF7L2 (purple). Boxplot shows the median staining intensity of TCF7L2 among IHC scan regions of cis-female and trans-male adipocytes (p value, Wilcoxon: 0.0069).
Multiple PI3K-related transcription factors showed increased expression in multiple cell types (Figure 7B). In particular, the nuclear receptor NR4A1 and its binding partner RXRA showed consistent upregulation, while the homeostatic regulator and target for inactivation by AKT, FOXO3 and its nuclear chaperone YWHAZ were downregulated.86,87,88,89,90 FOXO3 also showed lowered expression in cis-male breast samples (Figure S5E). Adipocytes showed the highest baseline insulin signaling score and were the only cell type to increase insulin signaling, as illustrated by upregulation of PTEN, PIK3R1, and AKT3 (Figures 7C and S11F). NR4A1 showed a strong increase in adipocytes based on RNA and protein levels and was also upregulated in adipose-enriched cis-male breast samples (Figures 7B, S5E, and S11G). NR4A1 is a target in multiple metabolic and cardiovascular processes, and corresponding binding motifs were enriched at genes associated with insulin signaling, supporting its status as an effector of glucose and lipid metabolism (Figure 7D).91,92,93
Androgen therapy led to smaller lipid vacuoles, indicating metabolic dysfunction and insulin resistance (Figure 7E).94,95,96 Considering this, lipolysis and lipogenesis genes were analyzed, but were not found differentially expressed (not shown). However, the lipolysis regulator zinc-alpha glycoprotein 1 (AZGP1) was upregulated (Figures 7F and 7G). AZGP1 is an androgen-responsive secreted factor that is higher in the cis-male breast and is known for potently inducing lipid degradation and fat loss in smokers and cancer patients.97,98,99,100 Androgen therapy particularly increased AZGP1 expression in luminal cells, where the HR+ subset showed strong cytoplasmic staining (Figures 7G, S5E, and S11H).
Androgen therapy also reduced expression of a GRN module linked to the metabolic marker GPAM, which pathway analysis indicated was associated with adipogenesis and adipocyte differentiation (Figures 7H and S11I; Table S4). In searching for GPAM module regulators, we found accessible binding elements of the upregulated Wnt/β-catenin effector TCF7L2 were enriched at associated gene loci (Figures 7I and S11J). Besides being a repressor of adipogenesis, TCF7L2 is crucial for glucose tolerance and insulin sensitivity.101,102,103 In trans-male adipocytes, TCF7L2 appears to be a direct AR target and is one of the most upregulated transcription factors alongside NR4A1 (Figures 7J and 7K).
Discussion
Our multi-modal single-cell resolution analyses showed that gender-affirming androgen therapy induces both broad and cell-type-specific changes in the human breast that can be mapped directly and indirectly to AR activation. In line with previous reports, the ER target gene PGR was downregulated in luminal-HR+ cells, and evidence for ARE accessibility was prominent at the PGR locus, along with AR co-regulators FOXA1 and NFIC.9,29,30 Another consequence of AR action in luminal-HR+ cells was altered production of ligands and an upregulation of the calcium channel member RYR2, which may facilitate secretion of these factors to communicate hormonal changes to HR− cell types. Related to this, myoepithelial cells lost expression of OXTR and ACTA2, which are essential for ductal contraction during lactation, corresponding to lowered breastfeeding rates in women with polycystic ovary syndrome, where androgen is in excess.52,104,105,106 Many of the gene-expression changes induced by androgen therapy were also consistent with male-specific sex biases uncovered when comparing breast tissues and other organs from cis-gender males and females.
Although androgen therapy caused reduced estrogen signaling, ESR1 expression and overall motif accessibility were not altered, consistent with high doses of selective androgen modulators reducing the mammary epithelium but increasing ER expression in murine mammary fat pad luminal cells.6 AR inhibits ER signaling in ER+ breast cancer cells by sequestering essential co-activators.9 Herein, we inferred that androgen-induced estrogen signaling changes were specific to genes co-regulated by ER and AP-1 factors, indicating AR may sequester these transcription factors as well. Indeed, AP-1 factor blockade appears to overcome endocrine therapy resistance in ER+ breast cancers.107
Since breast cancer rates are lower in transgender men, we also examined how androgen affects cancer-related pathways.9,108 Coincident with BRCA1 being a co-activator of AR, we found androgen suppresses genes that are upregulated in luminal-HR+ cells of BRCA-mutant breast samples (Figures S12A and S12B).109,110 We also found normal luminal cell expression patterns correlated most strongly with PAM50 classifications of breast cancer, while basal cells were not associated with any molecular subtype (Figure S12C). Luminal-HR− cells believed to give rise to basal-like breast tumors were correlated with the corresponding signature, but this was reduced after androgen therapy (Figures S12C and S12D).111,112 However, correlations between luminal-HR+ cells and luminal A- or B-type breast cancers were most decreased, corroborating previous findings.9 In the latter study, androgen inhibited proliferation of estrogen-dependent breast cancer cells, and genes used to make this inference, including PGR, the oncogene BCL2, and the tumor suppressor SEC14L2, were also altered by androgen in normal luminal-HR+ cells (Figure S12E).113,114,115 Androgen also downregulates the epithelial growth factors AREG and EREG in luminal-HR+ cells and their receptor EGFR, which are overexpressed and functionally relevant in ER+ breast cancers.39,40,41,42,43 We hypothesize that the observed reduction of angiogenic activity by capillary cells and increased surveillance by CD4+ T cells could also contribute to breast cancer protection. Overall, this indicates AR activation may be a valid preventive or therapeutic strategy for ER+ breast cancer, and, in accordance, clinical trials testing AR activation in this subtype have been implemented.116,117
Global transcriptional, translational, and chromatin silencing by androgen therapy may also have an oncoprotective effect. However, this observation seems to misalign with increased nucleus sizes observed in luminal epithelial cells after androgen therapy. While chromatin condensation and nuclear size are correlated during development and cell differentiation, a causal relationship is, to our knowledge, not known. Nonetheless, various laminins, which were altered in multiple cell types after androgen therapy, are critical regulators of nuclear morphology and may have contributed to these differences (Table S6).118
We found breast metabolic activity is affected by androgen, in line with other studies describing metabolic changes induced by sex hormones.81,119,120 Adipocytes showed an upregulation of the insulin receptor and altered downstream effectors of the PI3K pathway.120 NR4A1, an orphan nuclear receptor and key regulator of glucose and lipid metabolism, was also broadly upregulated, supporting research on NR4A1, indicating it aids in insulin signaling and is a target for diabetes and metabolic disorders, particularly in transgender patients.90,91,92,93,121 Androgen also caused AZGP1 upregulation, and activation of the anti-adipogenic transcription factor TCF7L2, potentially representing an unrecognized mechanism through which transgender men shed weight.100,101,102
Limitations of the study
Bentz et al. previously assessed pre- and post-treatment tissues from transgender men using gene-expression microarray technology.122 Some of our key results, including the global downregulation of genes, decreased translation, and increased activity of NR4A1, were also found there. However, the single-cell resolution and the multi-modal nature of our approach allowed us to study cell-type-specific chromatin and expression changes, and our spatial analyses elucidated how breast morphology and local microenvironment are altered by androgen therapy.
Other important limitations of this study include low patient numbers and a lack of relevant information for some samples. This study also lacked corresponding AR, ER, and JUN ChIP-seq data that would have allowed better examination of estrogen signaling regulation in luminal-HR+ cells following androgen therapy. Despite these limitations, this study represents a rich resource to study hormonal control of human breast tissue homeostasis.
STAR★Methods
Key resources table
| REAGENT or RESOURCE | SOURCE | IDENTIFIER |
|---|---|---|
| Antibodies | ||
| Rat anti-KRT8 (clone TROMA-1) | Sigma-Aldrich | Cat#MABT329; RRID:AB_2891089 |
| Rabbit anti-PLIN1 (polyclonal) | Thermo-Fisher Scientific | Cat#PA5-81240; RRID:AB_2788467 |
| Mouse anti-TCF4 (clone 6H5-3) | Sigma-Aldrich | Cat#05-511; RRID:AB_309772 |
| Mouse anti-Nur77 AF488 (clone 12.14) | Thermo-Fisher Scientific | Cat#53-5965-82; RRID:AB_2574429 |
| Mouse anti-INSR (clone MM0414-2A12) | Abcam | Cat#ab90940; RRID:AB_2127106 |
| Rabbit anti-CD45 (clone EP322Y) | Abcam | Cat#ab40763; RRID:AB_726545 |
| Mouse anti-CD68 (clone KP1) | Thermo-Fisher Scientific | Cat#MA5-13324; RRID:AB_10987212 |
| Rabbit anti-CD3e AF488 (clone EP449E) | Abcam | Cat#ab271850 |
| Goat anti-rabbit IgG AF488 | Thermo-Fisher Scientific | Cat#A11008; RRID:AB_143165 |
| Goat anti-rabbit IgG AF568 | Thermo-Fisher Scientific | Cat#A11011; RRID:AB_143157 |
| Donkey anti-mouse IgG AF568 | Thermo-Fisher Scientific | Cat#A10037; RRID:AB_2534013 |
| Goat anti-rat IgG Cy5 | Thermo-Fisher Scientific | Cat#A10525; RRID:AB_2534034 |
| See Table S3 for details on antibodies used for CODEX | N/A | N/A |
| Biological samples | ||
| Healthy breast tissue from female-to-male gender-affirmation surgeries and cis-gender cosmetic mammary surgeries | Cedars-Sinai Biobank and Research Pathology Resource | www.cedars-sinai.edu/research/cores/biobank-research-pathology.html |
| Chemicals, peptides, and recombinant proteins | ||
| Nuclei EZ Lysis Buffer | Sigma-Aldrich | Cat#NUC101-1KT |
| RiboLock RNase Inhibitor (40 U/μL) | Thermo-Fisher Scientific | Cat#EO0381 |
| Nonidet P40 Substitute | Sigma-Aldrich | Cat#11754599001 |
| Digitonin (5%) | Thermo-Fisher Scientific | Cat#BN2006 |
| BSA, Molecular Biology Grade | New England Biolabs | Cat#B9200 |
| Critical commercial assays | ||
| Chromium Single Cell 3ʹ GEM, Library & Gel Bead Kit v3 | 10X Genomics | PN-1000075 |
| Chromium Single Cell B Chip Kit | 10X Genomics | PN-1000073 |
| Chromium i7 Multiplex Kit | 10X Genomics | PN-120262 |
| Chromium Next GEM Single Cell ATAC Library & Gel Bead Kit v1.1 | 10X Genomics | PN-1000175 |
| Chromium Next GEM Chip H Single Cell Kit | 10X Genomics | PN-1000161 |
| Single Index Kit N, Set A | 10X Genomics | PN-1000212 |
| 10X Buffer for CODEX | Akoya | Cat#7000001 |
| Assay Reagent for CODEX | Akoya | Cat#7000002 |
| Staining Kit for CODEX | Akoya | Cat#7000008 |
| Antibody Conjugation Kit | Akoya | Cat#7000009 |
| Nuclear Stain for CODEX | Akoya | Cat#7000003 |
| Deposited data | ||
| Raw and analyzed scRNA-seq and scATAC-seq data | This paper | GSE168838 |
| Processed imaging (CODEX) data | This paper | zenodo.6569916 |
| MSigDB; v7.2 | Subramanian & Tamayo et al.,123 Liberzon et al.,124 Liberzon et al.125 | www.gsea-msigdb.org/gsea/msigdb |
| Geno-type-Tissue Expression Project (GTEx); v8 | GTEx Consortium126 | gtexportal.org/home |
| Cis-BP: Catalog of Inferred Sequence Binding Preferences | Weirauch et al.127 | cisbp.ccbr.utoronto.ca |
| Software and algorithms | ||
| Cell Ranger; v6.0.1 | 10x Genomics | support.10xgenomics.com/single-cell-gene-expression/software |
| CellRanger ATAC; v1.2.0 | 10X Genomics | support.10xgenomics.com/single-cell-atac/software |
| Seurat; v4.1.0 | Hao & Hao et al.128 | satijalab.org/seurat |
| scVelo; v0.2.3 | Bergen et al.129 | scvelo.readthedocs.io |
| CellRank; v1.3 | Lange et al.130 | cellrank.readthedocs.io/en/stable |
| scDblFinder; v1.6.0 | Germain et al.131 | github.com/plger/scDblFinder |
| Harmony; v0.1.0 | Korsunsky et al.132 | github.com/immunogenomics/harmony |
| UMAP; v0.5 | McInnes et at.133 | github.com/lmcinnes/umap |
| Augur; v1.0.3 | Skinnider & Squair et al.134 | github.com/neurorestore/Augur |
| MAST; 1.21.3 | Finak et al.135 | rglab.github.io/MAST |
| SCANPY; v1.4.5.1 | Wolf et al.136 | scanpy.readthedocs.io/en/stable |
| g:Profiler; v0.2.1 | Raudvere et al.137 | biit.cs.ut.ee/gprofiler/gost |
| GRNboost2 | Moerman et al.138 | github.com/aertslab/GRNBoost |
| pySCENIC; v0.11.2 | Aibar et al.139 | scenic.aertslab.org |
| DESeq2; v1.32.0 | Love et al.140 | github.com/mikelove/DESeq2 |
| velocyto; v0.17.17 | La Manno et al.16 | http://velocyto.org |
| destiny; v3.0.1 | Angerer et al.141 | github.com/theislab/destiny |
| AICSImageIO; v3.3.7 | AICSImageIO Contributors142 | github.com/AllenCellModeling/aicsimageio |
| StarDist; v0.8.1 | Schmidt et al.143 and Weigert et al.144 | github.com/stardist/stardist |
| Rdist; v0.0.5 | The Comprehensive R Archive Network (CRAN) | github.com/blasern/rdist |
| APCluster; v1.4.9 | Frey & Dueck145 | github.com/UBod/apcluster |
| scikit-image; v0.19.1 | Van der Walt et al.146 | github.com/scikit-image/scikit-image |
| ArchR; v1.0.0 | Granja et al.147 | archrproject.com |
| chromVAR; v1.16.0 | Schep et al.148 | github.com/GreenleafLab/chromVAR |
| MACS2; v2.2.7.1 | Zhang et al.149 | github.com/macs3-project/MACS |
| randomForest; v4.6-14 | The Comprehensive R Archive Network (CRAN) | stat.berkeley.edu/∼breiman/RandomForests |
| Other | ||
| KIMBLE Dounce tissue grinder set | Sigma-Aldrich | Cat#D9063 |
| MACS SmartStrainers (70 μm) | Miltenyi | Cat#130-110-916 |
| Pre-Separation Filters (20 μm) | Miltenyi | Cat#130-101-812 |
Resource availability
Lead contact
The lead contact for this paper is Simon R. V. Knott (simon.knott@cshs.org).
Materials availability
This study did not generate new unique reagents.
Experimental model and subject details
Fresh human breast tissue specimens from cosmetic mammary surgeries in nine cisgender women and subcutaneous mastectomies in nine transgender men receiving intramuscular testosterone-cypionate were collected at the time of surgery. The age of the transgender men ranged from 18 to 36 (Mean = 27.67 and SD = 8.26), while the age of the control cisgender women ranged from 28 to 62 (Mean = 45.44 and SD = 13.78). Five of the cisgender women were pre-menopausal and four were post-menopausal. Seven of the cisgender women received breast reduction surgery, while two received implant exchange capsulectomy. Transgender men received from 6 to 36 months of hormone-replacement therapy before their surgery (Mean = 17.1 and SD = 10.38). We investigated the impact of menopause state and age on our results (Figure S2A). Table S1 contains all recorded clinical information of our cohort. This study was approved by the Institutional Review Board (IRB) of Cedars-Sinai Medical Center and informed consent was obtained from all participants.
Method details
Sample collection
Fresh human breast tissue specimens from cosmetic mammary surgeries in cisgender women and subcutaneous mastectomies in transgender men receiving intramuscular testosterone-cypionate were collected at time of surgery (Table S1). For mammary reductions, samples were taken from the central breast, deep to the nipple-areola complex and slightly inferior (1-2 cm at most), in order to not interfere with nipple perfusion, and ensure that the second and third intercostal artery branches, are not damaged. In this same area, transgender patients on androgen therapy also have glandular tissue. While different operations may be performed through different incisions or involve different ways of modifying the breast envelope and contents, tissue was always extracted from this same location for these subjects. Tissue from cis-gender patients (CF-318-813 and CF-428-112) was collected during breast implant exchanges (capsulectomies). During these procedures, healthy breast tissue from the same area, superficial to the implant site was collected. An analysis of surgery type as a factor driving phenotypic differences found that this was not a discriminating variable for any cell type (Figure S2A). All collected samples were placed in Dulbecco’s Modified Eagle’s Medium (DMEM, Corning) at surgery and processed shortly after. Tissues were washed 3x with DMEM, and any large pieces of fat were grossly removed. Tissue was then cut into 2-3 mm3 pieces before being directly stored at −80°C. Where available, the Cedars-Sinai Biobank collected pieces of tissue from the left and right breast and fixed them with 10% formalin for subsequent paraffin embedding (FFPE). This study was approved by the Institutional Review Board (IRB) of Cedars-Sinai Medical Center and informed consent was obtained from all participants.
Preparation of RNA expression libraries
All RNA-expression libraries were prepared with Chromium Single Cell 3’ (v3) Reagent Kits by 10X Genomics. To assure minimal handling time while reducing batch effects, samples were processed in batches of 3-4 (with each batch containing trans-male and cis-female samples). All steps until library generation were carried out on ice and in pre-cooled instruments. Lysis was done with Nuclei EZ lysis buffer (Sigma) and subsequent steps were carried out in a custom wash buffer (10 mM Tris, 146 mM NaCl, 1 mM CaCl2, 21 mM MgCl2) that was freshly supplemented with 40 U/mL of RNAse inhibitor (Thermo Fisher) and 2% of Molecular Biology Grade BSA (New England Biolabs). About 250 mg of cryopreserved tissue was placed on wet ice and cut with a scalpel into rice grain sized pieces while still frozen. The tissue was then transferred into a chilled 7 mL dounce tissue grinder (Sigma) containing 3 mL of 20% Nuclei EZ lysis buffer (diluted 1:5 with wash buffer). Tissue was soaked on ice for 3 min with occasional pipetting with a wide bore tip. Lysate was homogenized, first with pestle marked “A” (coarse) and then “B” (fine), for 10 complete strokes and for no longer than 5 min. Nuclei suspension was then filtered through a 70 μm filter (Miltenyi) and the lysis buffer was quenched with 9 mL of wash buffer. Filtrate was then spun down in a swing bucket rotor for 8 min at 850 g, and pellet was resuspended in 1.2 mL of wash buffer. Suspension was filtered through a 20 μm filter (Miltenyi), spun down again and final pellet was resuspended in 400 μL of wash buffer supplemented with 2.5 μg/mL DAPI. Solution was immediately loaded onto a FACSAria III Cell Sorter (BD Biosciences) equipped with a 70 μm nozzle (liquid output for this setup was previously measured to be 1.9 nL per event), and a gate around single nuclei was determined using DAPI and side-scatter signals. 22,000 events (∼41.8 μL) were sorted directly into a 96-well round bottom plate harboring RT-buffer (10X Genomics), which was prepared without adding RT-Enzyme (total of 25.1 μL). After sorting was completed, RT-Enzyme (8.3 μL) was added, and nuclei suspension was immediately loaded onto a 10X Chromium controller. Following library preparation was performed according to the Chromium Single Cell 3′ Reagent Kits User Guide (v3 Chemistry) with an assumed input of 10,000 cells. The cDNA amplification was carried out with two extra PCR cycles (13 total); all other steps were kept unaltered. Quality, amount, and size distribution of the final libraries was assessed on a BioAnalyzer (Agilent).
Preparation of single nuclei ATAC libraries
All snATAC libraries were prepared with Chromium Single Cell ATAC Reagent Kits (v1.1) by 10X Genomics. Nuclei extraction method was identical to RNA-expression workflow (see above) with the following buffers (prepared according to 10X Genomics demonstrated protocol: CG000212, Rev.B). Lysis Buffer (10 mM Tris-HCl pH 7.4, 10 mM NaCl, 3 mM MgCl2, 0.01% Tween 20, 0.01% Nonidet P40 Substitute, 0.001% Digitonin, 1% BSA), Wash Buffer (10 mM Tris-HCl pH 7.4, 10 mM NaCl, 3 mM MgCl2, 0.1% Tween 20, 1% BSA). After washing and 20 μm filtration (see RNA-expression workflow above), nuclei were spun down and the final pellet was resuspended in 400 μL of wash buffer, supplemented with 0.5 μg/mL 7-AAD (BioLegend). Solution was immediately loaded onto a FACSAria III Cell Sorter (BD Biosciences) equipped with a 70 μm nozzle and a gate around single nuclei was determined using 7-AAD and side-scatter signals. All available nuclei (on average ∼350.000 events) were sorted directly into a 1.5 mL protein lo-bind tube (Eppendorf) containing 100 μL diluted nuclei buffer (10X Genomics Single Cell ATAC Reagent Kit). The sort volume was calculated, and the sorted nuclei were supplemented with 20x diluted nuclei buffer to a final concentration of 1x. Nuclei were spun down at 850 g for 8 min, supernatant was carefully removed as much as possible and the final pellet was resuspended in 10 μL diluted nuclei buffer. 5 μL of nuclei suspension was mixed with 5 μL of 5 μg/mL DAPI and counted on a hemocytometer to determine loading concentration. If necessary, the remaining nuclei solution was diluted to 3000 nuclei/μL and used immediately in the transposition reaction. Following library preparation was performed according to the Chromium Single Cell ATAC Reagent Kits User Guide (v1.1 Chemistry). Quality, amount, and size distribution of the final libraries was assessed on a BioAnalyzer (Agilent).
Sequencing
Single nuclei RNA expression libraries were sequenced according to 10X Genomics recommended read lengths (read1 = 28bp, read2 = 91bp) on a NovaSeq6000 (Illumina) to an average of ∼40.000 reads per nucleus, resulting in an average sequencing saturation of ∼75% (as reported by 10X Genomics CellRanger v6.0.1). Single nuclei ATAC libraries were sequenced according to 10X Genomics recommended read lengths (read1 = 50bp, read2 = 50bp) on a NovaSeq6000 (Illumina), to an average of ∼32.000 reads per nucleus, resulting in an average of ∼7000 fragments per cell (as reported by 10X Genomics CellRanger ATAC v1.2.0).
Preprocessing of single nuclei RNA expression data
Fastq files were processed using 10X Genomics CellRanger v6.0.1 and aligned to the human reference genome “refdata-gex-GRCh38-2020-A″ provided by 10X Genomics. In order to account for increased amounts of pre-mRNA captured in single nuclei RNA sequencing, CellRanger was run using the option —include-introns. Resulting count matrices were further processed in Seurat (v4.1.0).128 Thresholds for maximum fraction of mitochondrial genes and number of UMIs for each nucleus were set to 2.5% and 20.000 respectively. Barcodes that likely contained doublets were detected and removed with scDblFinder (v1.6.0).131 Further doublet-enriched and low UMI (median UMI count <1000) clusters that emerged while sub-clustering each individual cell type were also removed.
Immunohistochemistry
Immunohistochemistry was performed on sections taken from FFPE blocks that were collected by the Cedars Sinai Biobank at time of surgery. Briefly, fixed sections were incubated in 60°C for 25 min to remove excess paraffin and then immediately deparaffinized and rehydrated. Antigen retrieval was performed using an "Instant Pot Duo" pressure cooker and 1x Universal HIER buffer (Abcam, cat: ab208572). Background fluorescence was quenched by photobleaching for 1.5h in bleaching solution according to Du et al. 2019.150 Sections were then blocked in protein blocking buffer (Abcam, cat: ab64226) for 1 h at room temperature, washed and then incubated with primary antibodies at 4 °C overnight. The primary antibodies used were as follows (all dilutions were performed with protein blocking buffer): KRT8 (Sigma-Aldrich, cat: MABT329, clone TROMA-1, 1:100), PLIN1 (Thermo-Fisher Scientific, cat: PA5-81240, rabbit polyclonal, 1:500), TCF4 (Sigma-Aldrich, cat: 05-511, clone 6H5-3, 1:100), Nur77-AF488 preconjugated (Thermo-Fisher Scientific, cat: 53-5965-82, clone 12.14, 1:500), INSR (Abcam, cat: ab90940, clone MM0414-2A12, 1:100), CD45 (Abcam, cat: 40,763, clone EP322Y, 1:200), CD68 (Thermo-Fisher Scientific, cat: MA5-13324, clone KP1, 1:100), and CD3e-AF488 custom conjugated (Abcam, cat: 271,850, clone EP449E, 1:200).
Sections were then washed and incubated with the appropriate fluorophore-conjugated secondary antibodies at room temperature for 1 h. Secondary antibodies used were as follows (all dilutions were performed with protein blocking buffer): Goat anti-rabbit IgG AF488 (Thermo-Fisher Scientific, cat: A11008, 1:500), Goat anti-rabbit IgG AF568 (Thermo-Fisher Scientific, cat: A11011, 1:500), Donkey anti-mouse IgG AF568 (Thermo-Fisher Scientific, cat: A10037, 1:500), and Goat anti-rat IgG Cy5 (Thermo-Fisher Scientific, cat: A10525, 1:500).
Sections were finally Washed 3 times with 1X PBST (1X PBS with 0.1% Tween 20) for 3-5 min at room temperature, mounted with Vectashield containing DAPI (Vector Laboratories, cat: H-1200), and imaged using a Zeiss Axio Scan.Z1. Automated imaging was set up in Zeiss ZEN pro v3.1. Due to large tissue sizes, the slides were scanned without z stack and divided into ∼12-25 regions with individual focus maps. CZI format images were read with the AICSImageIO package (v3.3.7).142 Nuclei from the DAPI channel of each image were segmented with StarDist, then the average staining intensity per nucleus was tabulated.143,144
Co-detection by indexing (CODEX) of tissue microarrays
Tissue microarrays (TMA) were prepared from 2 mm punches taken from left and right breast representative FFPE blocks where available. To ensure the capture of diverse tissue sections that include epithelial structures, H&E stains were used to pre-annotate regions of interest, which were then transferred onto TMA paraffin blocks. Each (4x4) block contained four regions from four patients (see Figure S1C) and each patient was represented with eight regions across two separate TMA-blocks, resulting in eight TMAs total.
Sections from each of these eight TMAs were then collected onto poly-L-lysine-coated coverslips, which were prepared according to the Akoya Biosciences CODEX protocol. Similar to the IHC protocol (see above), sections were incubated in 60°C for 25 min, then deparaffinized and rehydrated. Following antigen retrieval, sections were then quenched for autofluorescence using a protocol adapted from.150 Subsequently, sections were stained and imaged according to the Akoya Biosciences CODEX protocol. Details regarding primary antibodies and imaging conditions can be found in Table S3. Imaging was performed using a Leica DMi8 equipped with a 20× objective, Lumencor SOLA SE U-nIR LED, and Hamamatsu Orca Flash 4.0 v3.
Primary antibodies were initially screened by performing standard IHC (as above) on FFPE tissue sections to verify positive staining. Primary antibodies were then conjugated to their corresponding barcodes according to the Akoya Biosciences CODEX antibody conjugation protocol. Conjugated antibodies were then titrated by performing CODEX staining on a TMA section using the full panel diluted at either 50x, 100x, or 200x. The dilution that resulted in the optimal signal-to-noise ratio was determined for each antibody individually. The final dilutions obtained from this titration can be found in Table S3.
Quantification and statistical analysis
Dimension reduction, cell type and subcluster identification, and variable discriminatory analysis in snRNA-seq data
All samples were integrated into a single dataset using the standard Seurat workflow (variable features = 5000, principal components = 50, louvain resolution = 0.05), batch-corrected with Harmony v0.1.0, and projected into two dimensional space using uniform manifold approximation and projection (UMAP).133,132 Identification of the main cell types was done by using canonical marker genes (Figure S1D) and further confirmed through existing gene modules in case of the epithelial cells.151 Each individual cell type was then extracted into a separate dataset for further classification of subclusters. Lymphoid and myeloid subtypes were determined by using canonical immune markers (Figure S10A). Blood endothelial subclusters were determined via conserved marker modules from Table S7 (Figure S9B) and pericytes were distinguished from vascular smooth muscle cells through expression of ACTA2 and PDGFRB (Figure S9C).62 Fibroblast subclusters were labeled to reflect the function of their top marker genes. AUC scores measuring phenotypic similarities between pre- and post-menopausal cis-female and trans-male samples as well as between cells from different surgery types were calculated using the Auger algorithm using 100 cell subsamples and a 3-fold cross validation.134
Differential gene expression, pathway enrichment, co-expression module generation, gene module scoring, and receptor ligand interactions in snRNA-seq data
Differential gene expression analysis on snRNA-seq data was done on log-transformed counts using MAST, and filtered for FDR < 0.05.135 Curated gene sets were obtained from MSigDB (v7.2) from the "h:hallmark" and "c2:curated" gene set collections and filtered for Reactome, PID, WikiPathways, Biocarta, KEGG and Hallmark as providers, as well as a minimum set size of 10 genes.125 Nuclei were scored for gene modules according to scanpy’s score_genes tool, using default settings (v1.4.5.1) and full results of these analyses can be found in Table S7.136 Pathway enrichment of gene modules was carried out via the R client for g:Profiler (v0.2.1) and full results for the relevant plots can be found in Table S4.137 Gene regulatory networks were generated using GRNboost2 with a list of 1839 transcription factors and pathway anchors (hs_hgnc_tfs) provided in the resources of pySCENIC.138,139 From the resulting TF-target association table, highly correlated target genes (>95th percentile of importance) of a transcription factor were selected to form a co-expression module. A curated list of ligand-receptor interaction pairs was taken from Cabello-Aguilar et al., 2020, and filtered for pairs with a PMID ref.152.
GTEx breast tissue and sex bias analysis
Gene RNA-expression data in other tissues was acquired through the Geno-type-Tissue Expression Project (version 8) (GTEx) which was supported by the Common Fund of the Office of the Director of the National Institutes of Health, and by NCI, NHGRI, NHLBI, NIDA, NIMH, and NINDS.126 Clustering and dimensionality reduction of GTEx breast samples was done with selected tools of the Seurat preprocessing workflow (variable features = 2000, principal components = 50, louvain resolution = 0.2) (Figures S5C and S5D). The resulting dataset was scored for the top 100 marker genes of the epithelial and vasculature groups, as well as the top 100 marker genes of the adipocyte and fibroblast cell types taken from our snRNA-seq data. The resulting module scores were used to classify the breast samples into epithelial, vasculature and adipose enriched subsets. Since the fibroblast score did not highlight a distinctive cluster, we chose the 50th percentile of the fibroblast score for this subset. We used the GTEx table of gene counts (GTEx Analysis 2017-06-05 v8 RNASeQC v1.1.9) to identify the differentially expressed genes among cis-male and cis-female breast samples using DESeq2 (v1.32.0).140 We then compared these genes to the corresponding significant differentially expressed genes in snRNA-seq data.
For the CUX2 and sex bias analysis, median transcript TPM values were calculated using the aggregated tissue classifier “SMTS” of the GTEx "Sample Attributes" metadata and sex bias effect sizes were used as provided by GTEx “GTEx_Analysis_v8_sbgenes.tar.gz”. In summary: sex-biased gene expression statistics for GTEx v8 tissues present in both sexes are derived from across-tissue meta-analysis with MASH, based on per-tissue sex effect size and corresponding SE values calculated with voom-limma.153,154
Inferring trajectory and diffusion maps from snRNA-seq data
To prepare input for RNA-velocity analysis, the BAM files generated by the CellRanger snRNA-seq workflow were processed into loom files containing spliced and unspliced abundances using velocyto (v0.17.17).16 Here, the same 10X Genomics genome annotation file used in CellRanger “refdata-gex-GRCh38-2020-A” was utilized alongside the GRCh38 repeat mask file from UCSC genome browser.155 The loom files were combined and cells that passed QC in previous gene expression analysis were extracted from the output. RNA-velocity analysis was done on each cell type separately using the standard workflow of the scVelo package (v0.2.3) and CellRank (v1.3).129,130 Moments were calculated using the batch-corrected first 30 harmony principal components, and velocity was estimated using the top 2,000 most variant genes and the dynamical model. For CellRank analysis, we determined the terminal states using the prior information of having two terminal states within each cell type (corresponding to cis-female and trans-male populations), weight connectivity of 0.2, and the Monte Carlo average of randomly sampled velocity vectors. We used the default parameters to identify the initial state.
To extract the diffusion maps, we used the Bioconductor package destiny (v3.0.1).141 We used snRNA-seq data of each cell type with the default parameters of the DiffusionMap function.
Preprocessing and integration of single nuclei ATAC data
We used 10X Genomics CellRanger ATAC (v1.2.0) to align the fastq files to the reference genome “refdata-cellranger-atac-GRCh38-1.2.0” and obtained the fragment and barcode annotation file for each sample. The resulting 500 bp resolution data was further processed using ArchR (v1.0.0) to remove doublets and correct for batch effects using Harmony.147 Reduced representation of the batch-effect–corrected tile matrix was calculated with iterative latent semantic indexing and visualized with UMAP. We then used ArchR to integrate snATAC-seq data with snRNA-seq data by correlating gene activity scores of each snATAC-seq cluster with the transcriptome of each snRNA-seq cluster (Figure S1D).
Footprint analysis, enhancer mapping, and differentially accessible peak identification
All transcription factor binding motifs were provided by the Catalog of Inferred Sequence Binding Preferences (CisBP).127 We used the motif footprint feature of ArchR to investigate the enrichment of each motif among the peaks of each cell type or gender identity while adjusting for the Tn5 bias by dividing the signal to Tn5 bias. ArchR investigates the correlation of each peak with the expression levels of a gene, where we used a Pearson correlation cutoff of 0.2 to identify peak-gene relationships. We used ArchR’s implemented Wilcoxon test to identify differentially accessible peaks adjusting for transcription start site enrichment (TSS) and number of fragments in log10.
Motif enrichment in cell types and single nuclei
To calculate motif enrichment for each cell type on basis of gender identity, we compared the enrichment of each motif in the foreground (trans-male nuclei) versus the background (cis-female nuclei). We performed a motif enrichment analysis through ArchR and using the CisBP motif database. The motif enrichment analysis investigates the peaks accessible within the group that contains the sequence motif of interest. It compares the fraction of peaks containing the motif in the foreground versus the background and calculates a one-sided Fisher exact test. For the top motifs passing the cutoff adjusted p value < 0.05, we visualized the odds ratio of the enrichment for each cell type. Motif enrichment on a single nuclei basis was done using ChromVAR, which calculates z-scores that indicate the enrichment of each sequence motif in each nucleus relative to other nuclei.148
HiC data analysis
We downloaded two publicly available HiC datasets for MCF-10A (Accession: GSE98552) and PANC-1 (ENCODE Accession: ENCLB951HSJ).156,157 MCF-10A data had already been processed using HiC-Pro (v2.8.1), GRCh38 genome assembly, and 40,000 bp genomic bins.158 We used the same software and pipeline to process PANC-1 datasets. We divided the ICE-normalized genomic bin contact values to the maximum value of the chromosome to adjust for the potential variations introduced by library size and experimental protocol. We visualized contact of genomic bins overlapping the RYR2 gene or its enhancers that passed the 75th percentile after pooling data of MCF-10A and PANC-1 around RYR2.
High-resolution peak calling and motif filtering
In addition to the peak set generated through ArchR, we also generated pseudo-bulk BAM files by extracting the reads corresponding to each cell type and sample type (total of 20 BAM files). For each pseudo-bulk BAM file, we used MACS2 peak calling with parameters -f BAMPE -g 2.7 × 109 --nomodel --SPMR --bdg.149 We then used the generated NarrowPeak files for identifying motifs that are within an absolute distance of 200 bp from the summit of the peaks.
Predicting dysregulation direction of AR-regulated genes with random forest
We identified 603 genes upregulated in trans-male luminal-HR+ cells (adjusted p value < 0.01 and log2 FC > 0.25) and 652 genes downregulated (adjusted p value < 0.01 and logFC < −0.25). To consider a gene an AR-regulated gene, we required an ARE containing peak whose accessibility correlates with the gene’s expression (Pearson r > 0.2) as identified by ArchR (see above). AR upregulated 309/603 genes and downregulated 244/652 genes. ArchR identified 1,080,500 bp peaks which correlate with the expression of these 553 genes. Using pseudo-bulk BAM files, we identified 1,427 narrow peaks corresponding to these 500 bp genomic regions. These narrow peaks ranged from 141 bp to 3,011 bp with a median of 792 bp. We annotated each peak according to the chromatin accessibility signal at the exact motif position in the peak in luminal-HR+ trans-male cells for motifs of 189 expressed transcription factors. We labeled these peaks according to their mapping to downregulated genes or upregulated genes. We randomly selected 80% of these peaks as the training set and the remaining 20% as our validation set. We trained a random forest with 500 trees with each tree sampling 100 of the 189 motifs to predict if the gene was downregulated or upregulated. We used the R package ‘randomForest’ (v4.6-14). We allowed each classification tree to reach maximum purity. The model was able to predict downregulated genes with an area under the receiver-operating characteristic curve (auROC) of 0.825 and area under the precision-recall curve (auPR) of 0.741. We used mean decrease in Gini index as the measure of variable importance.
Enrichment of transcription factors in pathways
To investigate if a transcription factor regulates a particular pathway, we identified the number of genes of that pathway which have a chromatin accessibility peak containing the motif of that transcription factor. We also required the chromatin accessibility peak to correlate positively with the expression of the gene (Pearson r > 0.2). We compared the fraction of pathway’s genes containing the motif in their peaks to a background of all the genes using a one-sided Fisher exact test.
Identifying active transcription factors
Sequence motifs corresponding to the same family of transcription factors often show similar binding sites. To identify the active transcription factors, we used ArchR’s correlation of a motif’s Z score versus the expression of its corresponding transcription factor. For the specific case of nuclear receptors in luminal-HR+ cells, we built all of the possible simple linear models which predict a nuclear receptor’s motif’s Z score given the expression of all other nuclear receptors (one at a time) or a background transcription factor (i.e. PPARG). To be able to compare the effect sizes, we standardized both the motif’s Z score as well as the transcription factor’s expression. We compared the linear model effect size, p value, and R2.
CODEX data preprocessing
Raw images of TMA-regions from CODEX experiments were pre-processed with a custom workflow where five preprocessing operations were applied in this order: extended depth of field (EDOF), shading correction, cycle alignment, background subtraction and tile stitching, described briefly here.
1: An EDOF image was produced from the z stack for each tile where each position is taken from the z-plane most in focus. 2: The BaSiC method of optical shading correction was applied to each channel of each imaging cycle.159 3: An image registration transformation was estimated between the first cycle DAPI channel and the DAPI of each subsequent cycle. For each cycle, the registration parameters were saved and applied to all other channels from the same cycle. 4: Blank cycles were used to subtract background from each channel. 5: Finally, neighboring tiles were stitched by applying a registration between the overlapping areas between two tiles. First, the two tiles with the best naive overlap were stitched by applying the appropriate registration shift to one of the tiles. Stitching then proceeded with the next two most nearly aligned tiles, until all tiles were merged. Since each cycle was previously aligned to the first cycle’s DAPI channel, the registrations used for tile stitching were estimated once on the first DAPI and reused for subsequent channels and cycles.
The assembled DAPI images for each TMA region were then visually inspected and any remaining grossly out-of-focus areas due to low tissue adherence were removed from further analysis.
To obtain nuclear segmentations, we applied a pre-trained StarDist model to the first cycle DAPI image.143,144 The model weights of the 2D 2018 Data Science Bowl model released by the original StarDist authors were fine-tuned using a training set of nuclei imaged on our CODEX platform. A whole-cell or "cytoplasmic" segmentation was obtained by expanding the nuclear segmentation area by morphological dilation, without introducing overlaps in adjacent nuclei. The average intensities under each nuclear mask and cytoplasmic mask were extracted for each cell to be used for cell type assignment.
CODEX data analysis
Nuclei and cytoplasmic staining intensities of all data points passing segmentation were merged into a single unfiltered dataset, followed by batch correction, dimensional reduction and clustering with harmony and Seurat. The dataset was then cleaned from over-segmentation (i.e. blood clots, very dim regions, damaged tissue) by visually inspecting sub clusters and removing data points that were not represented by a clearly visible nuclear DAPI signal. Cell classes were defined according to their marker staining and the resulting filtered dataset was then scaled according to the first and third quartiles using RobustScaler across each region, producing the intensity values shown in log scale.160 When calculating nuclear vs. cytoplasmic staining, raw signal values were used. Where region averages of these values are shown, only regions with a minimum of 10 cells of the discussed cell type are shown, unless otherwise stated.
Epithelial neighborhood and cell-cell distance analysis
For all analyses including cell-cell adjacencies (CODEX and IHC), we calculated pairwise Euclidean distances based on the nuclei centroids using the package rdist (v0.0.5). To assess the composition of cell types that are closely associated with the epithelium we first detected spatial clusters of epithelial cells (cells in cluster >35) using affinity propagation from the APCluster (v1.4.9) package.145 To define the epithelial neighborhood populations, we retained all cells within 100px (∼32.5μm) distance from these epithelial cells.
Segmentation of acini and assessment of epithelial smooth muscle layer
To study the staining patterns around epithelial cells in detail, we applied morphological operations to specifically isolate individual acini structures within the CODEX tissue microarray images (Figure S7C). First, masks of the KRT8 and KRT23 stains per TMA region were obtained by multi otsu thresholding (scikit-image) and imposing a minimum intensity of 100 in order to avoid false-positive epithelial segmentations.146 An ACTA2 mask was obtained similarly by multi otsu thresholding, but without the minimum intensity requirement. Morphological dilation was applied to each mask. Then, the KRT8 and KRT23 masks were added together to form a pan-luminal mask, and the ACTA2 mask was subtracted from this pan-luminal mask to divide acini into individual instances. Individual putative acini were labeled by connected components, dilated to fill extraneous holes, filtered for a minimum area of 200, then individually analyzed. Segmented nuclei underneath each acini mask were gathered to tally the cell type composition, and the 15-pixel wide border surrounding the acini was considered the border area. The percentage of the acini border that was positive for ACTA2 above the predetermined threshold was tallied.
Image analysis of adipocyte vacuole area
To study the morphology of adipocytes we used IHC staining of PLIN1 to segment individual adipocyte vacuoles and profile their size. To avoid abundant small-scale staining artifacts, PLIN1 IHC images were analyzed at 1/5th the original resolution. First, the PLIN1 signal was capped at the 90th percentile intensity value and thresholded using Ostu’s method (scikit-image v0.19.1). To join small gaps in the PLIN1 mask we applied morphological dilation (disk structuring element radius 2) and closing (disk structuring element radius 5). A label image was created from the inverse of the PLIN1 mask, where segmented regions represented regions of PLIN1- area that were completely enclosed by PLIN1+ staining. We next filtered these putative vacuoles to exclude artifacts including small regions, and regions corresponding to multiple vacuoles erroneously connected because of e.g. broken membrane. To filter putative vacuoles, we calculated the area, and convex area (area of a convex hull fit around the actual label) and took their ratio. We reasoned that true positive vacuole segmentations would have an area-to-convex-area ratio (Rac) near one and applied a threshold of Rac >0.85. Similarly, we reasoned that erroneously connected vacuoles, or over-segmentations, would be outliers on the distribution of area, and applied a lower threshold determined by Otsu’s method of all vacuole areas exceeding a predetermined absolute minimum of 101.25 pixels, and a fixed upper threshold of ∼103.7 pixels. Following these filters, we noted that the PLIN1-INSR experiment had consistently more coverage of adipose vacuoles than others, and restricted further analysis to these images. Each image scene containing more than 1,000 adipocyte nuclei (determined by DAPI segmentation and PLIN1 threshold) was summarized by the median area of observed vacuoles.
Acknowledgments
M.K. is supported by a Canadian Institutes of Health Research post-doctoral fellowship. W.D.T. and T.E.H. are supported by National Health and Medical Research Council of Australia (ID 1084416 and 1130077), Movember and National Breast Cancer Foundation Collaboration Initiative (MNBCF-17-012), and Hospital Research Foundation (ID 2018-06-Strategic-R) grants. T.E.H. is also supported by an NBCF fellowship (IIRS-19-009). X.C. is supported by NIH (2R01CA151610) and Department of Defense (DOD; W811XWH-18-1-0067) grants. H.G. is a DOD Breast Cancer Research Program (BCRP) Era of Hope Scholar and is supported by National Institute of Health (NIH, R01CA244634 and R01CA240984) grants. A.E.G. is supported by The Fashion Footwear Charitable Foundation of New York Inc. and the Margie and Robert E. Peterson Foundation. S.R.V.K. is supported by the DOD BCRP grant W81XWH-21-2-0035.
We thank Dr. Benjamin Hopkins and Dr. Camilla Dos Santos for helpful discussions as well as the Cedars-Sinai Medical Center Biobank and Translational Genomics Core for their support.
Author contributions
F.R., E.C.R., X.C., and S.R.V.K. conceived of the study. E.C.R. performed the gender-affirming surgeries. A.E.G. performed the cisgender women’s surgeries, with both contributing to the tissue collection process along with Y.Q., T.Y.L., and X.C. F.R., Q.Y., A.M., and T.Y.L. performed the nuclei isolation. F.R. prepared all sequencing libraries. F.R., M.K., N.I., and B.M. performed all computational analysis with B.W., H.G., and S.R.V.K. supervising. F.R., M.K., N.I., B.M., S.D., W.D.T., T.E.H., B.W., H.G., E.C.R., X.C., and S.R.V.K. interpreted the data and wrote the manuscript.
Declaration of interests
S.R.V.K. is a founder and consultant at Faeth Therapeutics. W.D.T. is an advisor at Havah Therapeutics and holds provisional patent 10/05/2007 Australia Application Number 2007905468, Method and Composition for Modulating Androgen Activity.
Published: March 8, 2023
Footnotes
Supplemental information can be found online at https://doi.org/10.1016/j.xgen.2023.100272.
Contributor Information
Hani Goodarzi, Email: hani.goodarzi@ucsf.edu.
Edward C. Ray, Email: edward.ray@cshs.org.
Xiaojiang Cui, Email: xiaojiang.cui@cshs.org.
Simon R.V. Knott, Email: simon.knott@cshs.org.
Supplemental information
Data and code availability
-
•
We deposited all raw snRNA-seq and snATAC-seq data to Gene Expression Omnibus repository under GSE168838. We deposited all processed snRNA-seq, snATAC-seq, and CODEX datasets to Zenodo data repository under https://doi.org/10.5281/zenodo.6569916.
-
•
This paper does not report original code. We provided scripts describing preprocessing of our data under https://doi.org/10.5281/zenodo.6569916.
-
•
Any additional information required to reanalyze the data reported in this paper is available from the lead contact upon request.
References
- 1.Brisken C., O’Malley B. Hormone action in the mammary gland. Cold Spring Harb. Perspect. Biol. 2010;2:a003178. doi: 10.1101/CSHPERSPECT.A003178. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 2.Tornillo G., Smalley M.J. ERrrr…where are the progenitors? Hormone receptors and mammary cell heterogeneity. J. Mammary Gland Biol. Neoplasia. 2015;20:63–73. doi: 10.1007/s10911-015-9336-1. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 3.Liu H., Gruber C.W., Alewood P.F., Möller A., Muttenthaler M. The oxytocin receptor signalling system and breast cancer: a critical review. Oncogene. 2020;39:5917–5932. doi: 10.1038/s41388-020-01415-8. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 4.Oh H., Eliassen A.H., Wang M., Smith-Warner S.A., Beck A.H., Schnitt S.J., Collins L.C., Connolly J.L., Montaser-Kouhsari L., Polyak K., Tamimi R.M. Expression of estrogen receptor, progesterone receptor, and Ki67 in normal breast tissue in relation to subsequent risk of breast cancer. NPJ Breast Cancer. 2016;2:16032-. doi: 10.1038/npjbcancer.2016.32. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 5.Hickey T.E., Robinson J.L.L., Carroll J.S., Tilley W.D. Minireview: the androgen receptor in breast tissues: growth inhibitor, tumor suppressor, oncogene? Mol. Endocrinol. 2012;26:1252–1267. doi: 10.1210/me.2012-1107. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 6.Tarulli G.A., Laven-Law G., Shakya R., Tilley W.D., Hickey T.E. Hormone-sensing mammary epithelial progenitors: emerging identity and hormonal regulation. J. Mammary Gland Biol. Neoplasia. 2015;20:75–91. doi: 10.1007/S10911-015-9344-1. [DOI] [PubMed] [Google Scholar]
- 7.Kadlubar F.F., Berkowitz G.S., Delongchamp R.R., Wang C., Green B.L., Tang G., Lamba J., Schuetz E., Wolff M.S. The CYP3A4∗1B variant is related to the onset of puberty, a known risk factor for the development of breast cancer. Cancer Epidemiol. Biomarkers Prev. 2003;12:327–331. [PubMed] [Google Scholar]
- 8.Dimitrakakis C., Bondy C. Androgens and the breast. Breast Cancer Res. 2009;11:212. doi: 10.1186/bcr2413. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 9.Hickey T.E., Selth L.A., Chia K.M., Laven-Law G., Milioli H.H., Roden D., Jindal S., Hui M., Finlay-Schultz J., Ebrahimie E., et al. The androgen receptor is a tumor suppressor in estrogen receptor–positive breast cancer. Nat. Med. 2021;27:310–320. doi: 10.1038/s41591-020-01168-7. [DOI] [PubMed] [Google Scholar]
- 10.Kensler K.H., Beca F., Baker G.M., Heng Y.J., Beck A.H., Schnitt S.J., Hazra A., Rosner B.A., Eliassen A.H., Hankinson S.E., et al. Androgen receptor expression in normal breast tissue and subsequent breast cancer risk. NPJ Breast Cancer. 2018;4:33. doi: 10.1038/s41523-018-0085-3. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 11.Patel H., Arruarana V., Yao L., Cui X., Ray E. Effects of hormones and hormone therapy on breast tissue in transgender patients: a concise review. Endocrine. 2020;68:6–15. doi: 10.1007/s12020-020-02197-5. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 12.Coleman E., Bockting W., Botzer M., Cohen-Kettenis P., DeCuypere G., Feldman J., Fraser L., Green J., Knudson G., Meyer W.J., et al. Standards of care for the health of transsexual, transgender, and gender-nonconforming people, version 7. Int. J. Transgenderism. 2012;13:165–232. doi: 10.1080/15532739.2011.700873. [DOI] [Google Scholar]
- 13.Irwig M.S. Testosterone therapy for transgender men. Lancet Diabetes Endocrinol. 2017;5:301–311. doi: 10.1016/S2213-8587(16)00036-X. [DOI] [PubMed] [Google Scholar]
- 14.Deutsch M.B., Bhakri V., Kubicek K. Effects of cross-sex hormone treatment on transgender women and men. Obstet. Gynecol. 2015;125:605–610. doi: 10.1097/AOG.0000000000000692. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 15.Irwig M.S. Cardiovascular health in transgender people. Rev. Endocr. Metab. Disord. 2018;19:243–251. doi: 10.1007/s11154-018-9454-3. [DOI] [PubMed] [Google Scholar]
- 16.la Manno G., Soldatov R., Zeisel A., Braun E., Hochgerner H., Petukhov V., Lidschreiber K., Kastriti M.E., Lönnerberg P., Furlan A., et al. RNA velocity of single cells. Nature. 2018;560:494–498. doi: 10.1038/s41586-018-0414-6. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 17.Ramosaj M., Madsen S., Maillard V., Scandella V., Sudria-Lopez D., Yuizumi N., Telley L., Knobloch M. Lipid droplet availability affects neural stem/progenitor cell metabolism and proliferation. Nat. Commun. 2021;12:7362. doi: 10.1038/s41467-021-27365-7. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 18.D’Amato N.C., Gordon M.A., Babbs B., Spoelstra N.S., Carson Butterfield K.T., Torkko K.C., Phan V.T., Barton V.N., Rogers T.J., Sartorius C.A., et al. Cooperative dynamics of AR and ER activity in breast cancer. Mol. Cancer Res. 2016;14:1054–1067. doi: 10.1158/1541-7786.MCR-16-0167. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 19.Zhao Y., Tindall D.J., Huang H. Modulation of androgen receptor by FOXA1 and FOXO1 factors in prostate cancer. Int. J. Biol. Sci. 2014;10:614–619. doi: 10.7150/IJBS.8389. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 20.Jin H.J., Zhao J.C., Wu L., Kim J., Yu J. Cooperativity and equilibrium with FOXA1 define the androgen receptor transcriptional program. Nat. Commun. 2014;5:3972. doi: 10.1038/ncomms4972. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 21.Teng M., Zhou S., Cai C., Lupien M., He H.H. Pioneer of prostate cancer: past, present and the future of FOXA1. Protein Cell. 2021;12:29–38. doi: 10.1007/S13238-020-00786-8/FIGURES/3. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 22.Taur Y., Frishman W.H. The cardiac ryanodine receptor (RyR2) and its role in heart disease. Cardiol. Rev. 2005;13:142–146. doi: 10.1097/01.CRD.0000128709.84812.86. [DOI] [PubMed] [Google Scholar]
- 23.Santulli G., Pagano G., Sardu C., Xie W., Reiken S., D’Ascia S.L., Cannone M., Marziliano N., Trimarco B., Guise T.A., et al. Calcium release channel RyR2 regulates insulin release and glucose homeostasis. J. Clin. Invest. 2015;125:1968–1978. doi: 10.1172/JCI79273. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 24.Kaßmann M., Szijártó I.A., García-Prieto C.F., Fan G., Schleifenbaum J., Anistan Y.M., Tabeling C., Shi Y., le Noble F., Witzenrath M., et al. Role of ryanodine type 2 receptors in elementary Ca2+ signaling in arteries and vascular adaptive responses. J. Am. Heart Assoc. 2019;8:e010090. doi: 10.1161/JAHA.118.010090. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 25.Kushnir A., Marks A.R. The ryanodine receptor in cardiac physiology and disease. Adv. Pharmacol. 2010;59:1–30. doi: 10.1016/S1054-3589(10)59001-X. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 26.Robker R.L., Russell D.L., Espey L.L., Lydon J.P., O’Malley B.W., Richards J.S. Progesterone-regulated genes in the ovulation process: ADAMTS-1 and cathepsin L proteases. Proc. Natl. Acad. Sci. USA. 2000;97:4689–4694. doi: 10.1073/pnas.080073497. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 27.Ahmad S., Leinung M. The response of the menstrual cycle to initiation of hormonal therapy in transgender men. Transgend. Health. 2017;2:176–179. doi: 10.1089/trgh.2017.0023. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 28.Su H.-W., Yi Y.-C., Wei T.-Y., Chang T.-C., Cheng C.-M. Detection of ovulation, a review of currently available methods. Bioeng. Transl. Med. 2017;2:238–246. doi: 10.1002/btm2.10058. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 29.Grabowska M.M., Elliott A.D., DeGraff D.J., Anderson P.D., Anumanthan G., Yamashita H., Sun Q., Friedman D.B., Hachey D.L., Yu X., et al. NFI transcription factors interact with FOXA1 to regulate prostate-specific gene expression. Mol. Endocrinol. 2014;28:949–964. doi: 10.1210/me.2013-1213. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 30.Wang L., Xu M., Kao C.Y., Tsai S.Y., Tsai M.J. Small molecule JQ1 promotes prostate cancer invasion via BET-independent inactivation of FOXA1. J. Clin. Invest. 2020;130:1782–1792. doi: 10.1172/JCI126327. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 31.Verrijdt G., Haelens A., Schoenmakers E., Rombauts W., Claessens F. Comparative analysis of the influence of the high-mobility group box 1 protein on DNA binding and transcriptional activation by the androgen, glucocorticoid, progesterone and mineralocorticoid receptors. Biochem. J. 2002;361:97–103. doi: 10.1042/0264-6021:3610097. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 32.Tan M.H.E., Li J., Xu H.E., Melcher K., Yong E.L. Androgen receptor: structure, role in prostate cancer and drug discovery. Acta Pharmacol. Sin. 2015;36:3–23. doi: 10.1038/aps.2014.18. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 33.Björnström L., Sjöberg M. Mechanisms of estrogen receptor signaling: convergence of genomic and nongenomic actions on target genes. Mol. Endocrinol. 2005;19:833–842. doi: 10.1210/me.2004-0486. [DOI] [PubMed] [Google Scholar]
- 34.Cicatiello L., Mutarelli M., Grober O.M.V., Paris O., Ferraro L., Ravo M., Tarallo R., Luo S., Schroth G.P., Seifert M., et al. Estrogen receptor α controls a gene network in luminal-like breast cancer cells comprising multiple transcription factors and microRNAs. Am. J. Pathol. 2010;176:2113–2130. doi: 10.2353/ajpath.2010.090837. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 35.Bubulya A., Chen S.Y., Fisher C.J., Zheng Z., Shen X.Q., Shemshedini L. c-Jun potentiates the functional interaction between the amino and carboxyl termini of the androgen receptor. J. Biol. Chem. 2001;276:44704–44711. doi: 10.1074/jbc.M107346200. [DOI] [PubMed] [Google Scholar]
- 36.Teyssier C., Belguise K., Galtier F., Chalbos D. Characterization of the physical interaction between estrogen receptor α and JUN proteins. J. Biol. Chem. 2001;276:36361–36369. doi: 10.1074/jbc.M101806200. [DOI] [PubMed] [Google Scholar]
- 37.Zanconato F., Forcato M., Battilana G., Azzolin L., Quaranta E., Bodega B., Rosato A., Bicciato S., Cordenonsi M., Piccolo S. Genome-wide association between YAP/TAZ/TEAD and AP-1 at enhancers drives oncogenic growth. Nat. Cell Biol. 2015;17:1218–1227. doi: 10.1038/NCB3216. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 38.Korkmaz G., Lopes R., Ugalde A.P., Nevedomskaya E., Han R., Myacheva K., Zwart W., Elkon R., Agami R. Functional genetic screens for enhancer elements in the human genome using CRISPR-Cas9. Nat. Biotechnol. 2016;34:192–198. doi: 10.1038/NBT.3450. [DOI] [PubMed] [Google Scholar]
- 39.McBryan J., Howlin J., Napoletano S., Martin F. Amphiregulin: role in mammary gland development and breast cancer. J. Mammary Gland Biol. Neoplasia. 2008;13:159–169. doi: 10.1007/s10911-008-9075-7. [DOI] [PubMed] [Google Scholar]
- 40.Baillo A., Giroux C., Ethier S.P. Knock-down of amphiregulin inhibits cellular invasion in inflammatory breast cancer. J. Cell. Physiol. 2011;226:2691–2701. doi: 10.1002/jcp.22620. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 41.Aupperlee M.D., Leipprandt J.R., Bennett J.M., Schwartz R.C., Haslam S.Z. Amphiregulin mediates progesterone-induced mammary ductal development during puberty. Breast Cancer Res. 2013;15:R44. doi: 10.1186/bcr3431. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 42.Farooqui M., Bohrer L.R., Brady N.J., Chuntova P., Kemp S.E., Wardwell C.T., Nelson A.C., Schwertfeger K.L. Epiregulin contributes to breast tumorigenesis through regulating matrix metalloproteinase 1 and promoting cell survival. Mol. Cancer. 2015;14:138. doi: 10.1186/s12943-015-0408-z. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 43.Peterson E.A., Jenkins E.C., Lofgren K.A., Chandiramani N., Liu H., Aranda E., Barnett M., Kenny P.A. Amphiregulin is a critical downstream effector of estrogen signaling in ERα-positive breast cancer. Cancer Res. 2015;75:4830–4838. doi: 10.1158/0008-5472.CAN-15-0709. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 44.Sternlicht M.D., Sunnarborg S.W. The ADAM17-amphiregulin-EGFR axis in mammary development and cancer. J. Mammary Gland Biol. Neoplasia. 2008;13:181–194. doi: 10.1007/s10911-008-9084-6. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 45.Hosur V., Farley M.L., Burzenski L.M., Shultz L.D., Wiles M.v. ADAM17 is essential for ectodomain shedding of the EGF-receptor ligand amphiregulin. FEBS Open Bio. 2018;8:702–710. doi: 10.1002/2211-5463.12407. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 46.Wang B., Xi C., Liu M., Sun H., Liu S., Song L., Kang H. Breast fibroblasts in both cancer and normal tissues induce phenotypic transformation of breast cancer stem cells: a preliminary study. PeerJ. 2018;6:e4805. doi: 10.7717/peerj.4805. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 47.Morsing M., Kim J., Villadsen R., Goldhammer N., Jafari A., Kassem M., Petersen O.W., Rønnov-Jessen L. Fibroblasts direct differentiation of human breast epithelial progenitors. Breast Cancer Res. 2020;22:102. doi: 10.1186/s13058-020-01344-0. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 48.Soker S., Takashima S., Miao H.Q., Neufeld G., Klagsbrun M. Neuropilin-1 is expressed by endothelial and tumor cells as an isoform- specific receptor for vascular endothelial growth factor. Cell. 1998;92:735–745. doi: 10.1016/S0092-8674(00)81402-6. [DOI] [PubMed] [Google Scholar]
- 49.McGowan S.E. The lipofibroblast: more than a lipid-storage depot. Am. J. Physiol. Lung Cell Mol. Physiol. 2019;316:L869–L871. doi: 10.1152/ajplung.00109.2019. [DOI] [PubMed] [Google Scholar]
- 50.Colognato H., Yurchenco P.D. Form and function: the laminin family of heterotrimers. Dev. Dynam. 2000;218:213–234. doi: 10.1002/(SICI)1097-0177(200006)218:2<213::AID-DVDY1>3.0.CO;2-R. [DOI] [PubMed] [Google Scholar]
- 51.Peng Y., Wu D., Li F., Zhang P., Feng Y., He A. Identification of key biomarkers associated with cell adhesion in multiple myeloma by integrated bioinformatics analysis. Cancer Cell Int. 2020;20:262. doi: 10.1186/s12935-020-01355-z. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 52.Weymouth N., Shi Z., Rockey D.C. Smooth muscle α actin is specifically required for the maintenance of lactation. Dev. Biol. 2012;363:1–14. doi: 10.1016/J.YDBIO.2011.11.002. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 53.Yallowitz A.R., Alexandrova E.M., Talos F., Xu S., Marchenko N.D., Moll U.M. p63 is a prosurvival factor in the adult mammary gland during post-lactational involution, affecting PI-MECs and ErbB2 tumorigenesis. Cell Death Differ. 2014;21:645–654. doi: 10.1038/cdd.2013.199. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 54.Li D., Ji Y., Zhao C., Yao Y., Yang A., Jin H., Chen Y., San M., Zhang J., Zhang M., et al. OXTR overexpression leads to abnormal mammary gland development in mice. J. Endocrinol. 2018;239:121–136. doi: 10.1530/JOE-18-0356. [DOI] [PubMed] [Google Scholar]
- 55.Pankov R., Yamada K.M. Fibronectin at a glance. J. Cell Sci. 2002;115:3861–3863. doi: 10.1242/jcs.00059. [DOI] [PubMed] [Google Scholar]
- 56.Ramos G.d.O., Bernardi L., Lauxen I., Sant'Ana Filho M., Horwitz A.R., Lamers M.L. Fibronectin modulates cell adhesion and signaling to promote single cell migration of highly invasive oral squamous cell carcinoma. PLoS One. 2016;11:e0151338. doi: 10.1371/journal.pone.0151338. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 57.Hsiao C.T., Cheng H.W., Huang C.M., Li H.R., Ou M.H., Huang J.R., Khoo K.H., Yu H.W., Chen Y.Q., Wang Y.K., et al. Fibronectin in cell adhesion and migration via N-glycosylation. Oncotarget. 2017;8:70653–70668. doi: 10.18632/oncotarget.19969. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 58.Yu S., Wang X., Ng C.F., Chen S., Chan F.L. ERRγ suppresses cell proliferation and tumor growth of androgen-sensitive and androgen-insensitive prostate cancer cells and its implication as a therapeutic target for prostate cancer. Cancer Res. 2007;67:4904–4914. doi: 10.1158/0008-5472.CAN-06-3855. [DOI] [PubMed] [Google Scholar]
- 59.Audet-Walsh É., Yee T., McGuirk S., Vernier M., Ouellet C., St-Pierre J., Giguère V. Androgen-dependent repression of ERRγ reprograms metabolism in prostate cancer. Cancer Res. 2017;77:378–389. doi: 10.1158/0008-5472.CAN-16-1204. [DOI] [PubMed] [Google Scholar]
- 60.Burgess H.E., Shousha S. An immunohistochemical study of the long-term effects of androgen administration on female-to-male transsexual breast: a comparison with normal female breast and male breast showing gynaecomastia. J. Pathol. 1993;170:37–43. doi: 10.1002/path.1711700107. [DOI] [PubMed] [Google Scholar]
- 61.Slagter M.H., Gooren L.J.G., Scorilas A., Petraki C.D., Diamandis E.P. Effects of long-term androgen administration on breast tissue of female-to-male transsexuals. J. Histochem. Cytochem. 2006;54:905–910. doi: 10.1369/jhc.6A6928.2006. [DOI] [PubMed] [Google Scholar]
- 62.Kalucka J., de Rooij L.P.M.H., Goveia J., Rohlenova K., Dumas S.J., Meta E., Conchinha N.v., Taverna F., Teuwen L.A., Veys K., et al. Single-cell transcriptome atlas of murine endothelial cells. Cell. 2020;180:764–779.e20. doi: 10.1016/j.cell.2020.01.015. [DOI] [PubMed] [Google Scholar]
- 63.Garin-Shkolnik T., Rudich A., Hotamisligil G.S., Rubinstein M. FABP4 attenuates PPARγ and adipogenesis and is inversely correlated with PPARγ in adipose tissues. Diabetes. 2014;63:900–911. doi: 10.2337/db13-0436. [DOI] [PubMed] [Google Scholar]
- 64.Maréchal L., Laviolette M., Rodrigue-Way A., Sow B., Brochu M., Caron V., Tremblay A. The CD36-PPARγ pathway in metabolic disorders. Int. J. Mol. Sci. 2018;19:1529. doi: 10.3390/ijms19051529. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 65.Dawson D.W., Pearce S.F., Zhong R., Silverstein R.L., Frazier W.A., Bouck N.P. CD36 mediates the in vitro inhibitory effects of thrombospondin-1 on endothelial cells. J. Cell Biol. 1997;138:707–717. doi: 10.1083/jcb.138.3.707. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 66.Lawler J. The functions of thrombospondin-1 and -2. Curr. Opin. Cell Biol. 2000;12:634–640. doi: 10.1016/S0955-0674(00)00143-5. [DOI] [PubMed] [Google Scholar]
- 67.Dixelius J., Mäkinen T., Wirzenius M., Karkkainen M.J., Wernstedt C., Alitalo K., Claesson-Welsh L. Ligand-induced vascular endothelial growth factor receptor-3 (VEGFR-3) heterodimerization with VEGFR-2 in primary lymphatic endothelial cells regulates tyrosine phosphorylation sites. J. Biol. Chem. 2003;278:40973–40979. doi: 10.1074/jbc.M304499200. [DOI] [PubMed] [Google Scholar]
- 68.Reuter M.S., Jobling R., Chaturvedi R.R., Manshaei R., Costain G., Heung T., Curtis M., Hosseini S.M., Liston E., Lowther C., et al. Haploinsufficiency of vascular endothelial growth factor related signaling genes is associated with tetralogy of Fallot. Genet. Med. 2019;21:1001–1007. doi: 10.1038/s41436-018-0260-9. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 69.Gyorki D.E., Asselin-Labat M.L., van Rooijen N., Lindeman G.J., Visvader J.E. Resident macrophages influence stem cell activity in the mammary gland. Breast Cancer Res. 2009;11:R62. doi: 10.1186/bcr2353. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 70.Goff S.L., Danforth D.N. The role of immune cells in breast tissue and immunotherapy for the treatment of breast cancer. Clin. Breast Cancer. 2021;21:e63–e73. doi: 10.1016/j.clbc.2020.06.011. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 71.Lumbroso D., Soboh S., Maimon A., Schif-Zuck S., Ariel A., Burstyn-Cohen T. Macrophage-derived protein S facilitates apoptotic polymorphonuclear cell clearance by resolution phase macrophages and supports their reprogramming. Front. Immunol. 2018;9:358. doi: 10.3389/fimmu.2018.00358. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 72.Sadahiro H., Kang K.D., Gibson J.T., Minata M., Yu H., Shi J., Chhipa R., Chen Z., Lu S., Simoni Y., et al. Activation of the receptor tyrosine kinase AXL regulates the immune microenvironment in glioblastoma. Cancer Res. 2018;78:3002–3013. doi: 10.1158/0008-5472.CAN-17-2433. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 73.Ubil E., Caskey L., Holtzhausen A., Hunter D., Story C., Earp H.S. Tumor-secreted Pros1 inhibits macrophage M1 polarization to reduce antitumor immune response. J. Clin. Invest. 2018;128:2356–2369. doi: 10.1172/JCI97354. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 74.Giroud P., Renaudineau S., Gudefin L., Calcei A., Menguy T., Rozan C., Mizrahi J., Caux C., Duong V., Valladeau-Guilemond J. Expression of TAM-R in human immune cells and unique regulatory function of MerTK in IL-10 production by tolerogenic DC. Front. Immunol. 2020;11:564133. doi: 10.3389/fimmu.2020.564133. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 75.Nish S.A., Zens K.D., Kratchmarov R., Lin W.H.W., Adams W.C., Chen Y.H., Yen B., Rothman N.J., Bhandoola A., Xue H.H., et al. CD4+ T cell effector commitment coupled to self-renewal by asymmetric cell divisions. J. Exp. Med. 2017;214:39–47. doi: 10.1084/jem.20161046. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 76.Escobar G., Mangani D., Anderson A.C. T cell factor 1: a master regulator of the T cell response in disease. Sci. Immunol. 2020;5:eabb9726. doi: 10.1126/SCIIMMUNOL.ABB9726. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 77.Kim C., Jin J., Weyand C.M., Goronzy J.J. The transcription factor tcf1 in t cell differentiation and aging. Int. J. Mol. Sci. 2020;21:6497. doi: 10.3390/ijms21186497. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 78.Kopec A., Michaud Z., Maniero G. The Role of IL-16 as a lymphocyte attractant appears to be conserved through phylogeny: preliminary evidence that recombinant human IL-16 preferentially attracts regulatory lymphocytes in the amphibian, Xenopus laevis. Arch Autoimmune Dis. 2021;1:44–48. doi: 10.46439/AUTOIMMUNE.1.008. [DOI] [Google Scholar]
- 79.Cruikshank W.W., Kornfeld H., Center D.M. Interleukin-16. J. Leukoc. Biol. 2000;67:757–766. doi: 10.1002/JLB.67.6.757. [DOI] [PubMed] [Google Scholar]
- 80.Vandeweyer E., Hertens D. Quantification of glands and fat in breast tissue: an experimental determination. Ann. Anat. 2002;184:181–184. doi: 10.1016/S0940-9602(02)80016-4. [DOI] [PubMed] [Google Scholar]
- 81.Fernandez J.D., Tannock L.R. Metabolic effects of hormone therapy in transgender patients. Endocr. Pract. 2016;22:383–388. doi: 10.4158/EP15950.OR. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 82.Klaver M., de Blok C.J.M., Wiepjes C.M., Nota N.M., Dekker M.J.H.J., de Mutsert R., Schreiner T., Fisher A.D., T’Sjoen G., den Heijer M. Changes in regional body fat, lean body mass and body shape in trans persons using cross-sex hormonal therapy: results from a multicenter prospective study. Eur. J. Endocrinol. 2018;178:163–171. doi: 10.1530/EJE-17-0496. [DOI] [PubMed] [Google Scholar]
- 83.Spanos C., Bretherton I., Zajac J.D., Cheung A.S. Effects of gender-affirming hormone therapy on insulin resistance and body composition in transgender individuals: a systematic review. World J. Diabetes. 2020;11:66–77. doi: 10.4239/wjd.v11.i3.66. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 84.Suppakitjanusant P., Ji Y., Stevenson M.O., Chantrapanichkul P., Sineath R.C., Goodman M., Alvarez J.A., Tangpricha V. Effects of gender affirming hormone therapy on body mass index in transgender individuals: a longitudinal cohort study. J. Clin. Transl. Endocrinol. 2020;21:100230. doi: 10.1016/j.jcte.2020.100230. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 85.Bretherton I., Spanos C., Leemaqz S.Y., Premaratne G., Grossmann M., Zajac J.D., Cheung A.S. Insulin resistance in transgender individuals correlates with android fat mass. Ther. Adv. Endocrinol. Metab. 2021;12 doi: 10.1177/2042018820985681. 2042018820985681. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 86.Brunet A., Bonni A., Zigmond M.J., Lin M.Z., Juo P., Hu L.S., Anderson M.J., Arden K.C., Blenis J., Greenberg M.E. Akt promotes cell survival by phosphorylating and inhibiting a forkhead transcription factor. Cell. 1999;96:857–868. doi: 10.1016/S0092-8674(00)80595-4. [DOI] [PubMed] [Google Scholar]
- 87.Maira M., Martens C., Philips A., Drouin J. Heterodimerization between members of the Nur subfamily of orphan nuclear receptors as a novel mechanism for gene activation. Mol. Cell Biol. 1999;19:7549–7557. doi: 10.1128/mcb.19.11.7549. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 88.Arimoto-Ishida E., Ohmichi M., Mabuchi S., Takahashi T., Ohshima C., Hayakawa J., Kimura A., Takahashi K., Nishio Y., Sakata M., et al. Inhibition of phosphorylation of a Forkhead transcription factor sensitizes human ovarian cancer cells to cisplatin. Endocrinology. 2004;145:2014–2022. doi: 10.1210/en.2003-1199. [DOI] [PubMed] [Google Scholar]
- 89.Santo E.E., Stroeken P., Sluis P.v., Koster J., Versteeg R., Westerhout E.M. FOXO3a is a major target of inactivation by PI3K/AKT signaling in aggressive neuroblastoma. Cancer Res. 2013;73:2189–2198. doi: 10.1158/0008-5472.CAN-12-3767. [DOI] [PubMed] [Google Scholar]
- 90.Lee S.O., Jin U.H., Kang J.H., Kim S.B., Guthrie A.S., Sreevalsan S., Lee J.S., Safe S. The orphan nuclear receptor NR4A1 (Nur77) regulates oxidative and endoplasmic reticulum stress in pancreatic cancer cells. Mol. Cancer Res. 2014;12:527–538. doi: 10.1158/1541-7786.MCR-13-0567. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 91.Fu Y., Luo L., Luo N., Zhu X., Garvey W.T. NR4A orphan nuclear receptors modulate insulin action and the glucose transport system: potential role in insulin resistance. J. Biol. Chem. 2007;282:31525–31533. doi: 10.1074/jbc.M701132200. [DOI] [PubMed] [Google Scholar]
- 92.Smith C.J., Kener K.B., Tessem J.S. Nr4a1 and Nr4a3 knock out mice have impaired glucose clearance and beta-cell function under high-fat feeding. Diabetes. 2018;67:2040-P. doi: 10.2337/db18-2040-. [DOI] [Google Scholar]
- 93.Crean D., Murphy E.P. Targeting NR4A nuclear receptors to control stromal cell inflammation, metabolism, angiogenesis, and tumorigenesis. Front. Cell Dev. Biol. 2021;9:589770. doi: 10.3389/fcell.2021.589770. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 94.Salans L.B., Knittle J.L., Hirsch J. The role of adipose cell size and adipose tissue insulin sensitivity in the carbohydrate intolerance of human obesity. J. Clin. Invest. 1968;47:153–165. doi: 10.1172/jci105705. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 95.Hammarstedt A., Graham T.E., Kahn B.B. Adipose tissue dysregulation and reduced insulin sensitivity in non-obese individuals with enlarged abdominal adipose cells. Diabetol. Metab. Syndr. 2012;4:42. doi: 10.1186/1758-5996-4-42. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 96.Yang J., Eliasson B., Smith U., Cushman S.W., Sherman A.S. The size of large adipose cells is a predictor of insulin resistance in first-degree relatives of type 2 diabetic patients. Obesity. 2012;20:932–938. doi: 10.1038/oby.2011.371. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 97.Russell S.T., Zimmerman T.P., Domin B.A., Tisdale M.J. Induction of lipolysis in vitro and loss of body fat in vivo by zinc-α2-glycoprotein. Biochim. Biophys. Acta. 2004;1636:59–68. doi: 10.1016/j.bbalip.2003.12.004. [DOI] [PubMed] [Google Scholar]
- 98.Vanni H., Kazeros A., Wang R., Harvey B.G., Ferris B., De B.P., Carolan B.J., Hübner R.H., O’Connor T.P., Crystal R.G. Cigarette smoking induces overexpression of a fat-depleting gene AZGP1 in the human. Chest. 2009;135:1197–1208. doi: 10.1378/chest.08-1024. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 99.Mracek T., Stephens N.A., Gao D., Bao Y., Ross J.A., Rydén M., Arner P., Trayhurn P., Fearon K.C.H., Bing C. Enhanced ZAG production by subcutaneous adipose tissue is linked to weight loss in gastrointestinal cancer patients. Br. J. Cancer. 2011;104:441–447. doi: 10.1038/sj.bjc.6606083. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 100.Cao R., Ke M., Wu Q., Tian Q., Liu L., Dai Z., Lu S., Liu P. AZGP1 is androgen responsive and involved in AR-induced prostate cancer cell proliferation and metastasis. J. Cell. Physiol. 2019;234:17444–17458. doi: 10.1002/jcp.28366. [DOI] [PubMed] [Google Scholar]
- 101.Chen X., Ayala I., Shannon C., Fourcaudot M., Acharya N.K., Jenkinson C.P., Heikkinen S., Norton L. The diabetes gene and wnt pathway effector TCF7L2 regulates adipocyte development and function. Diabetes. 2018;67:554–568. doi: 10.2337/db17-0318. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 102.Geoghegan G., Simcox J., Seldin M.M., Parnell T.J., Stubben C., Just S., Begaye L., Lusis A.J., Villanueva C.J. Targeted deletion of Tcf7l2 in adipocytes promotes adipocyte hypertrophy and impaired glucose metabolism. Mol. Metab. 2019;24:44–63. doi: 10.1016/j.molmet.2019.03.003. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 103.Nguyen-Tu M.S., Martinez-Sanchez A., Leclerc I., Rutter G.A., da Silva Xavier G. Adipocyte-specific deletion of Tcf7l2 induces dysregulated lipid metabolism and impairs glucose tolerance in mice. Diabetologia. 2021;64:129–141. doi: 10.1007/s00125-020-05292-4. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 104.Biggs J.S., Hacker N., Andrews E., Munro C. Bromocriptine, methyl testosterone and placebo for inhibition of physiological lactation. A controlled study. Med. J. Aust. 1978;2:23–25. doi: 10.5694/j.1326-5377.1978.tb77383.x. [DOI] [PubMed] [Google Scholar]
- 105.de Gezelle H., Dhont M., Thiery M., Parewyck W. Puerperal lactation suppression and prolactin. Acta Obstet. Gynecol. Scand. 1979;58:469–472. doi: 10.3109/00016347909154069. [DOI] [PubMed] [Google Scholar]
- 106.Vanky E., Isaksen H., Moen M.H., Carlsen S.M. Breastfeeding in polycystic ovary syndrome. Acta Obstet. Gynecol. Scand. 2008;87:531–535. doi: 10.1080/00016340802007676. [DOI] [PubMed] [Google Scholar]
- 107.Malorni L., Giuliano M., Migliaccio I., Wang T., Creighton C.J., Lupien M., Fu X., Hilsenbeck S.G., Healy N., de Angelis C., et al. Blockade of AP-1 potentiates endocrine therapy and overcomes resistance. Mol. Cancer Res. 2016;14:470–481. doi: 10.1158/1541-7786.MCR-15-0423. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 108.de Blok C.J.M., Wiepjes C.M., Nota N.M., van Engelen K., Adank M.A., Dreijerink K.M.A., Barbé E., Konings I.R.H.M., den Heijer M. Breast cancer risk in transgender people receiving hormone treatment: nationwide cohort study in The Netherlands. BMJ. 2019;365:l1652. doi: 10.1136/bmj.l1652. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 109.Pal B., Chen Y., Vaillant F., Capaldo B.D., Joyce R., Song X., Bryant V.L., Penington J.S., Di Stefano L., Tubau Ribera N., et al. A single-cell RNA expression atlas of normal, preneoplastic and tumorigenic states in the human breast. EMBO J. 2021;40:e107333. doi: 10.15252/EMBJ.2020107333. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 110.Park J.J., Irvine R.A., Buchanan G., Koh S.S., Park J.M., Tilley W.D., Stallcup M.R., Press M.F., Coetzee G.A. Breast cancer susceptibility gene 1 (BRCAI) is a coactivator of the androgen receptor. Cancer Res. 2000;60:5946–5949. [PubMed] [Google Scholar]
- 111.Molyneux G., Geyer F.C., Magnay F.A., McCarthy A., Kendrick H., Natrajan R., MacKay A., Grigoriadis A., Tutt A., Ashworth A., et al. BRCA1 basal-like breast cancers originate from luminal epithelial progenitors and not from basal stem cells. Cell Stem Cell. 2010;7:403–417. doi: 10.1016/J.STEM.2010.07.010. [DOI] [PubMed] [Google Scholar]
- 112.Chiche A., Di-Cicco A., Sesma-Sanz L., Bresson L., de La Grange P., Glukhova M.A., Faraldo M.M., Deugnier M.A. P53 controls the plasticity of mammary luminal progenitor cells downstream of Met signaling. Breast Cancer Res. 2019;21:13. doi: 10.1186/s13058-019-1101-8. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 113.Czabotar P.E., Lessene G., Strasser A., Adams J.M. Control of apoptosis by the BCL-2 protein family: implications for physiology and therapy. Nat. Rev. Mol. Cell Biol. 2014;15:49–63. doi: 10.1038/nrm3722. [DOI] [PubMed] [Google Scholar]
- 114.Xiao S., Caglar E., Maldonado P., Das D., Nadeem Z., Chi A., Trinité B., Li X., Saxena A. Induced expression of nucleolin phosphorylation-deficient mutant confers dominant-negative effect on cell proliferation. PLoS One. 2014;9:e109858. doi: 10.1371/journal.pone.0109858. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 115.Li Z., Lou Y., Tian G., Wu J., Lu A., Chen J., Xu B., Shi J., Yang J. Discovering master regulators in hepatocellular carcinoma: one novel MR, SEC14L2 inhibits cancer cells. Aging. 2019;11:12375–12411. doi: 10.18632/aging.102579. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 116.Anestis A., Zoi I., Papavassiliou A.G., Karamouzis M.v. Androgen receptor in breast cancer—clinical and preclinical research insights. Molecules. 2020;25:358. doi: 10.3390/MOLECULES25020358. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 117.Narayanan R., Coss C.C., Dalton J.T. Development of selective androgen receptor modulators (SARMs) Mol. Cell. Endocrinol. 2018;465:134–142. doi: 10.1016/J.MCE.2017.06.013. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 118.Jevtić P., Edens L.J., Vuković L.D., Levy D.L. Sizing and shaping the nucleus: mechanisms and significance. Curr. Opin. Cell Biol. 2014;28:16–27. doi: 10.1016/J.CEB.2014.01.003. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 119.Yanase T., Tanabe M., Nomiyama T. Sex hormones and metabolic function. Nihon Rinsho. 2015;73:571–575. [PubMed] [Google Scholar]
- 120.Shadid S., Abosi-Appeadu K., de Maertelaere A.S., Defreyne J., Veldeman L., Holst J.J., Lapauw B., Vilsbøll T., T’Sjoen G. Effects of gender-affirming hormone therapy on insulin sensitivity and incretin responses in transgender people. Diabetes Care. 2020;43:411–417. doi: 10.2337/dc19-1061. [DOI] [PubMed] [Google Scholar]
- 121.Zhang L., Wang Q., Liu W., Liu F., Ji A., Li Y. The orphan nuclear receptor 4A1: a potential new therapeutic target for metabolic diseases. J Diabetes Res. J. Diabetes Res. 2018;2018:9363461. doi: 10.1155/2018/9363461. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 122.Bentz E.K., Pils D., Bilban M., Kaufmann U., Hefler L.A., Reinthaller A., Singer C.F., Huber J.C., Horvat R., Tempfer C.B. Gene expression signatures of breast tissue before and after cross-sex hormone therapy in female-to-male transsexuals. Fertil. Steril. 2010;94:2688–2696. doi: 10.1016/j.fertnstert.2010.04.024. [DOI] [PubMed] [Google Scholar]
- 123.Subramanian A., Tamayo P., Mootha V.K., Mukherjee S., Ebert B.L., Gillette M.A., Paulovich A., Pomeroy S.L., Golub T.R., Lander E.S., Mesirov J.P. Gene set enrichment analysis: a knowledge-based approach for interpreting genome-wide expression profiles. Proc. Natl. Acad. Sci. USA. 2005;102:15545–15550. doi: 10.1073/pnas.0506580102. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 124.Liberzon A., Subramanian A., Pinchback R., Thorvaldsdóttir H., Tamayo P., Mesirov J.P. Molecular signatures database (MSigDB) 3.0. Bioinformatics. 2011;27:1739–1740. doi: 10.1093/BIOINFORMATICS/BTR260. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 125.Liberzon A., Birger C., Thorvaldsdóttir H., Ghandi M., Mesirov J.P., Tamayo P. The molecular signatures database hallmark gene set collection. Cell Syst. 2015;1:417–425. doi: 10.1016/j.cels.2015.12.004. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 126.GTEx Consortium. Laboratory Data Analysis &Coordinating Center LDACC—Analysis Working Group. Statistical Methods groups—Analysis Working Group. Enhancing GTEx eGTEx groups. NIH Common Fund. Jo B., Mohammadi P., Park Y.S., Parsana P., et al. Biospecimen Collection Source Site—NDRI Genetic effects on gene expression across human tissues. Nature. 2017;550:204–213. doi: 10.1038/nature24277. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 127.Weirauch M.T., Yang A., Albu M., Cote A.G., Montenegro-Montero A., Drewe P., Najafabadi H.S., Lambert S.A., Mann I., Cook K., et al. Determination and inference of eukaryotic transcription factor sequence specificity. Cell. 2014;158:1431–1443. doi: 10.1016/j.cell.2014.08.009. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 128.Hao Y., Hao S., Andersen-Nissen E., Mauck W.M., Zheng S., Butler A., Lee M.J., Wilk A.J., Darby C., Zager M., et al. Integrated analysis of multimodal single-cell data. Cell. 2021;184:3573–3587.e29. doi: 10.1016/j.cell.2021.04.048. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 129.Bergen V., Lange M., Peidli S., Wolf F.A., Theis F.J. Generalizing RNA velocity to transient cell states through dynamical modeling. Nat. Biotechnol. 2020;38:1408–1414. doi: 10.1038/s41587-020-0591-3. [DOI] [PubMed] [Google Scholar]
- 130.Lange M., Bergen V., Klein M., Setty M., Reuter B., Bakhti M., Lickert H., Ansari M., Schniering J., Schiller H.B., et al. CellRank for directed single-cell fate mapping. Nat. Methods. 2022;19:159–170. doi: 10.1038/s41592-021-01346-6. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 131.Germain P.-L., Lun A., Garcia Meixide C., Macnair W., Robinson M.D. Doublet identification in single-cell sequencing data using scDblFinder. F1000Res. 2021;10:979. doi: 10.12688/f1000research.73600.1. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 132.Korsunsky I., Millard N., Fan J., Slowikowski K., Zhang F., Wei K., Baglaenko Y., Brenner M., Loh P.R., Raychaudhuri S. Fast, sensitive and accurate integration of single-cell data with Harmony. Nat. Methods. 2019;16:1289–1296. doi: 10.1038/s41592-019-0619-0. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 133.McInnes L., Healy J., Melville J. UMAP: uniform manifold approximation and projection for dimension reduction. arXiv. 2018 doi: 10.48550/arXiv.1802.03426. Preprint at. [DOI] [Google Scholar]
- 134.Skinnider M.A., Squair J.W., Kathe C., Anderson M.A., Gautier M., Matson K.J.E., Milano M., Hutson T.H., Barraud Q., Phillips A.A., et al. Cell type prioritization in single-cell data. Nat. Biotechnol. 2020;39:30–34. doi: 10.1038/s41587-020-0605-1. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 135.Finak G., McDavid A., Yajima M., Deng J., Gersuk V., Shalek A.K., Slichter C.K., Miller H.W., McElrath M.J., Prlic M., et al. MAST: a flexible statistical framework for assessing transcriptional changes and characterizing heterogeneity in single-cell RNA sequencing data. Genome Biol. 2015;16:278. doi: 10.1186/s13059-015-0844-5. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 136.Wolf F.A., Angerer P., Theis F.J. SCANPY: large-scale single-cell gene expression data analysis. Genome Biol. 2018;19:15. doi: 10.1186/s13059-017-1382-0. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 137.Raudvere U., Kolberg L., Kuzmin I., Arak T., Adler P., Peterson H., Vilo J. G:Profiler: a web server for functional enrichment analysis and conversions of gene lists (2019 update) Nucleic Acids Res. 2019;47:W191–W198. doi: 10.1093/nar/gkz369. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 138.Moerman T., Aibar Santos S., Bravo González-Blas C., Simm J., Moreau Y., Aerts J., Aerts S. GRNBoost2 and Arboreto: efficient and scalable inference of gene regulatory networks. Bioinformatics. 2019;35:2159–2161. doi: 10.1093/bioinformatics/bty916. [DOI] [PubMed] [Google Scholar]
- 139.Aibar S., González-Blas C.B., Moerman T., Huynh-Thu V.A., Imrichova H., Hulselmans G., Rambow F., Marine J.C., Geurts P., Aerts J., et al. SCENIC: single-cell regulatory network inference and clustering. Nat. Methods. 2017;14:1083–1086. doi: 10.1038/nmeth.4463. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 140.Love M.I., Huber W., Anders S. Moderated estimation of fold change and dispersion for RNA-seq data with DESeq2. Genome Biol. 2014;15:550. doi: 10.1186/s13059-014-0550-8. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 141.Angerer P., Haghverdi L., Büttner M., Theis F.J., Marr C., Buettner F. Destiny: diffusion maps for large-scale single-cell data in R. Bioinformatics. 2016;32:1241–1243. doi: 10.1093/bioinformatics/btv715. [DOI] [PubMed] [Google Scholar]
- 142.Contributors Aicsi. 2021. AICSImageIO: Image Reading, Metadata Conversion, and Image Writing for Microscopy Images in Pure Python. [Google Scholar]
- 143.Schmidt U., Weigert M., Broaddus C., Myers G. Lecture Notes in Computer Science (including subseries Lecture Notes in Artificial Intelligence and Lecture Notes in Bioinformatics) 2018. Cell detection with star-convex polygons. [DOI] [Google Scholar]
- 144.Weigert M., Schmidt U., Haase R., Sugawara K., Myers G. Proceedings - 2020 IEEE Winter Conference on Applications of Computer Vision, WACV 2020. 2020. Star-convex polyhedra for 3D object detection and segmentation in microscopy. [DOI] [Google Scholar]
- 145.Frey B.J., Dueck D. Clustering by passing messages between data points. Science. 2007;315:972–976. doi: 10.1126/science.1136800. [DOI] [PubMed] [Google Scholar]
- 146.van der Walt S., Schönberger J.L., Nunez-Iglesias J., Boulogne F., Warner J.D., Yager N., Gouillart E., Yu T., scikit-image contributors Scikit-image: image processing in python. PeerJ. 2014;2:e453. doi: 10.7717/peerj.453. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 147.Granja J.M., Corces M.R., Pierce S.E., Bagdatli S.T., Choudhry H., Chang H.Y., Greenleaf W.J. ArchR is a scalable software package for integrative single-cell chromatin accessibility analysis. Nat. Genet. 2021;53:403–411. doi: 10.1038/s41588-021-00790-6. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 148.Schep A.N., Wu B., Buenrostro J.D., Greenleaf W.J. ChromVAR: inferring transcription-factor-associated accessibility from single-cell epigenomic data. Nat. Methods. 2017;14:975–978. doi: 10.1038/nmeth.4401. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 149.Zhang Y., Liu T., Meyer C.A., Eeckhoute J., Johnson D.S., Bernstein B.E., Nusbaum C., Myers R.M., Brown M., Li W., Liu X.S. Model-based analysis of ChIP-seq (MACS) Genome Biol. 2008;9:R137. doi: 10.1186/gb-2008-9-9-r137. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 150.Du Z., Lin J.R., Rashid R., Maliga Z., Wang S., Aster J.C., Izar B., Sorger P.K., Santagata S. Qualifying antibodies for image-based immune profiling and multiplexed tissue imaging. Nat. Protoc. 2019;14:2900–2930. doi: 10.1038/s41596-019-0206-y. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 151.Lim E., Vaillant F., Wu D., Forrest N.C., Pal B., Hart A.H., Asselin-Labat M.L., Gyorki D.E., Ward T., Partanen A., et al. Aberrant luminal progenitors as the candidate target population for basal tumor development in BRCA1 mutation carriers. Nat. Med. 2009;15:907–913. doi: 10.1038/nm.2000. [DOI] [PubMed] [Google Scholar]
- 152.Cabello-Aguilar S., Alame M., Kon-Sun-Tack F., Fau C., Lacroix M., Colinge J. SingleCellSignalR: inference of intercellular networks from single-cell transcriptomics. Nucleic Acids Res. 2020;48:e55. doi: 10.1093/nar/gkaa183. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 153.Urbut S.M., Wang G., Carbonetto P., Stephens M. Flexible statistical methods for estimating and testing effects in genomic studies with multiple conditions. Nat. Genet. 2019;51:187–195. doi: 10.1038/s41588-018-0268-8. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 154.Law C.W., Chen Y., Shi W., Smyth G.K. Voom: precision weights unlock linear model analysis tools for RNA-seq read counts. Genome Biol. 2014;15:R29. doi: 10.1186/gb-2014-15-2-r29. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 155.Kent W.J., Sugnet C.W., Furey T.S., Roskin K.M., Pringle T.H., Zahler A.M., Haussler D. The human genome browser at UCSC. Genome Res. 2002;12:996–1006. doi: 10.1101/gr.229102. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 156.ENCODE Project Consortium. Kundaje A., Aldred S.F., Collins P.J., Davis C.A., Doyle F., Epstein C.B., Frietze S., Harrow J., Kaul R., et al. An integrated encyclopedia of DNA elements in the human genome. Nature. 2012;489:57–74. doi: 10.1038/nature11247. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 157.Fritz A.J., Ghule P.N., Boyd J.R., Tye C.E., Page N.A., Hong D., Shirley D.J., Weinheimer A.S., Barutcu A.R., Gerrard D.L., et al. Intranuclear and higher-order chromatin organization of the major histone gene cluster in breast cancer. J. Cell. Physiol. 2018;233:1278–1290. doi: 10.1002/jcp.25996. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 158.Servant N., Varoquaux N., Lajoie B.R., Viara E., Chen C.J., Vert J.P., Heard E., Dekker J., Barillot E. HiC-Pro: an optimized and flexible pipeline for Hi-C data processing. Genome Biol. 2015;16:259. doi: 10.1186/s13059-015-0831-x. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 159.Peng T., Thorn K., Schroeder T., Wang L., Theis F.J., Marr C., Navab N. A BaSiC tool for background and shading correction of optical microscopy images. Nat. Commun. 2017;8:14836. doi: 10.1038/ncomms14836. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 160.Pedregosa F., Varoquaux G., Gramfort A., Michel V., Thirion B., Grisel O., Blondel M., Prettenhofer P., Weiss R., Dubourg V., et al. Scikit-learn: machine learning in Python. J. Mach. Learn. Res. 2011;12:2825–2830. [Google Scholar]
Associated Data
This section collects any data citations, data availability statements, or supplementary materials included in this article.
Supplementary Materials
Data Availability Statement
-
•
We deposited all raw snRNA-seq and snATAC-seq data to Gene Expression Omnibus repository under GSE168838. We deposited all processed snRNA-seq, snATAC-seq, and CODEX datasets to Zenodo data repository under https://doi.org/10.5281/zenodo.6569916.
-
•
This paper does not report original code. We provided scripts describing preprocessing of our data under https://doi.org/10.5281/zenodo.6569916.
-
•
Any additional information required to reanalyze the data reported in this paper is available from the lead contact upon request.