

Abstract
INTRODUCTION
Lecanemab is a humanized immunoglobulin G1 (IgG1) monoclonal antibody that preferentially targets soluble aggregated Aβ species (protofibrils) with activity at amyloid plaques. Amyloid‐related imaging abnormalities (ARIA) profiles appear to differ for various anti‐amyloid antibodies. Here, we present ARIA data from a large phase 2 lecanemab trial (Study 201) in early Alzheimer's disease.
METHODS
Study 201 trial was double‐blind, placebo‐controlled (core) with an open‐label extension (OLE). Observed ARIA events were summarized and modeled via Kaplan‐Meier graphs. An exposure response model was developed.
RESULTS
In the phase 2 core and OLE, there was a low incidence of ARIA‐E (<10%), with <3% symptomatic cases. ARIA‐E was generally asymptomatic, mild‐to‐moderate in severity, and occurred early (<3 months). ARIA‐E was correlated with maximum lecanemab serum concentration and incidence was higher in apolipoprotein E4 (ApoE4) homozygous carriers. ARIA‐H and ARIA‐E occurred with similar frequency in core and OLE.
DISCUSSION
Lecanemab can be administered without titration with modest incidence of ARIA.
Keywords: Alzheimer's disease, anti‐amyloid, ARIA, exposure response modeling, lecanemab
1. INTRODUCTION
1.1. ARIA in Alzheimer's disease background
Over the past decade, an active area of research has been the development of immunotherapy, with the goal of lowering cerebral amyloid beta burden in patients with Alzheimer's disease (AD). 1 , 2 , 3 , 4 , 5 , 6 , 7 Treatment‐related amyloid‐related imaging abnormalities (ARIA) have been observed on brain imaging after treatment with some anti‐amyloid immunotherapies. The exact pathophysiology underlying ARIA remains uncertain. 8 , 9 , 10 , 11 , 12 ARIA‐E (edema) refers to the magnetic resonance (MR) signal alterations thought to represent vasogenic edema (VE) and related extravasated fluid phenomena, whereas ARIA‐H (hemorrhage) refers to the MR signal alterations attributable to microhemorrhages, macrohemorrhages, and hemosiderosis (superficial siderosis 8 ). ARIA‐E and ARIA‐H are typically detected on magnetic resonance imaging (MRI) sequences. Both appear to relate to the presence of cerebral amyloid angiopathy (CAA), and both occur in the natural history of AD and in the setting of amyloid‐modifying therapeutic approaches. 8 Microhemorrhages naturally occur frequently in AD. Macrohemorrhages, siderosis, and edema occur only rarely in AD without treatments and are thought to be associated with CAA, but are observed at higher rates with antibody treatments. Current practice implements careful clinical and radiographic monitoring for ARIA when treating with an anti‐amyloid therapy. 13
RESEARCH IN CONTEXT
Systematic Review: The authors reviewed the literature utilizing PubMed and recent meeting abstracts. Relevant citations on pathophysiology aspects of amyloid‐related imaging abnormalities (ARIA), as well as clinical reports of ARIA occurring with anti‐amyloid antibodies are cited. ARIA incidence appears to vary among the anti‐amyloid antibodies in development.
Interpretation: Our findings show the incidence of ARIA‐E in the lecanemab (10 mg/kg biweekly, intravenous [IV]) phase 2 Study 201 occurred at a relatively low rate (<10%; symptomatic rate: <3%). ARIA was generally mild‐to‐moderate, occurred early (<3 months), was higher in apolipoprotein E4 (ApoE4) carriers, and was correlated with maximum lecanemab concentrations at steady state. These findings support lecanemab initiation at 10 mg/kg biweekly without titration.
Future Directions: Further evaluation of lecanemab is ongoing in the phase 3 Clarity AD study in early Alzheimer's disease. Research on a lecanemab subcutaneous formulation with a lower Css,max is ongoing, which has the potential for reduced incidence of ARIA‐E versus the IV formulation.
1.2. Lecanemab background
Lecanemab is a humanized immunoglobulin G1 (IgG1) monoclonal antibody that preferentially targets soluble aggregated Aβ species (protofibrils), with activity at insoluble fibrils. 14 , 15 , 16 , 17 , 18 , 19 Lecanemab was evaluated in a large, 18‐month phase 2 proof‐of‐concept double‐blind study (BAN2401‐G000‐201 [Study 201 core]; NCT01767311) using Bayesian design with response adaptive randomization in 856 patients with early AD; mild cognitive impairment (MCI) due to AD or mild AD dementia. 1 , 20 This study also included an open‐label extension (OLE) phase that is ongoing. Although the threshold for the primary Bayesian analysis at 12 months was not met, results from pre‐specified 18‐month frequentist analyses demonstrated that lecanemab treatment produced consistent dose‐dependent reductions in brain amyloid burden assessed by positron emission tomography (PET) scans and were associated with slowing of clinical decline in patients with early AD. 1 Doses were not titrated in this study, and the study identified lecanemab 10 mg/kg IV biweekly as the optimal dose for balancing amyloid clearance, clinical efficacy and safety. This dose is being used in the ongoing phase 3 trial (NCT03887455).
In the double‐blind Study 201 core, lecanemab was generally well‐tolerated, with ARIA‐E and infusion reactions being the most common adverse events observed. 1 ARIA‐E was dose dependent, with an incidence <10% at the highest doses for the overall population and 14.3% for ApoE4 positive subjects, with most events occurring in the first 3 months of treatment and most mild to moderate in severity. Symptomatic ARIA‐E occurred in 3% of participants. There were no symptomatic cases of ARIA‐H reported 1 in the core study. Of note, although increased ARIA occurs with both active immunization as well as many anti‐amyloid antibodies, 1 , 8 , 9 , 21 , 22 , 23 , 24 , 25 the risk of ARIA with lecanemab may be lower due to differences in pharmacological profile and binding affinities of the various anti‐amyloid antibodies.
1.3. Study goals
Given the importance of ARIA as potential adverse events in anti‐amyloid therapies, we provide here an in‐depth update on ARIA from the lecanemab clinical program from Study 201 core and OLE, including exposure response modelling analysis and an evaluation of the relationship of ARIA to ApoE4 genotype. The analysis in this manuscript principally focuses on the highest dose evaluated in the phase 2 study (10 mg/kg biweekly, intravenous [IV]), which is the dose tested in the pivotal phase 3 study. However, since the number of ApoE4 positive subjects in the 10 mg/kg biweekly dose group was limited (30%) by a regulatory authority‐imposed amendment, 1 we also included data where relevant for the second highest dose (10 mg/kg monthly). This 10 mg/kg monthly dosing group has a higher percentage of ApoE4 carriers (90%) and, as we will show in this paper, has a similar maximum concentration (Css,max) of lecanemab to 10 mg/kg biweekly, although total drug exposure (AUC) is lower.
2. METHODS
2.1. Study design and treatments
2.1.1. Study 201 core
The lecanemab Study 201 trial (ClinicalTrials.gov Identifier: NCT01767311) was a multinational, multicenter, double‐blind, placebo‐controlled, parallel‐group study employing response adaptive randomization. Details of the study design have been previously published 1 , 20 and brief description of relevant methods can be found in the supplement.
2.1.2. 201 open‐label extension
The OLE was initiated following analysis of the core phase 2b Study 201 to allow subjects to receive open‐label lecanemab 10 mg/kg biweekly for up to 24 months to assess long‐term safety and tolerability. All subjects who fulfilled OLE inclusion/exclusion criteria and entered the OLE received 10 mg/kg biweekly during the OLE period, regardless of ApoE genotype. Since the OLE was added in an amendment to Study 201 after completion of the core study, there was a gap period between the end of the Study 201 core and OLE baseline when no treatment was provided, which was 9–59 months (mean and median: approximately 24 months). MRI was conducted to monitor for ARIA at 9 weeks, 3 months, 6 months, and 12 months over the first 12 months of 10 mg/kg biweekly lecanemab treatment in the OLE (i.e., identical to the core phase of Study 201 in the first 12 months). After 12 months, MRI was conducted every 6 months. ARIA‐E and ARIA‐H data were summarized according to observed events in the longitudinal OLE period. In contrast to the core study where all cases of ARIA‐E were discontinued, ARIA management in the OLE allowed for uninterrupted dosing in radiographically mild or moderate asymptomatic cases of ARIA‐E, while dosing was interrupted for radiographically severe events, whether symptomatic or asymptomatic, until resolution or stabilization, at which time resumption of treatment was permitted. ARIA‐E was best detected with the 2D‐T2FLAIR sequence used and ARIA‐H was best detected with the T2* sequence. 12 Subjects who developed symptomatic ARIA‐H were also temporarily stopped from study drug administration. Subjects had safety visits (with MRI) at approximately 30 days after event was identified until ARIA resolved, at which time treatment could be resumed.
2.2. Exposure response analysis for safety (ARIA‐E)
ARIA‐E incidence was correlated with lecanemab maximum concentration at steady state (Css,max) using logistic regression analysis data from Study 201 across all dosing regimens. Covariates included age, gender, ApoE4 carrier genotype status, ongoing treatment with AChEIs and/or memantine, and clinical subgroup (MCI due to AD or mild AD dementia). Time‐to‐event (TTE) analysis was performed for time to the first observation of ARIA‐E. Parametric TTE model was constructed by linking an event time and drug exposure through hazard function. ARIA‐E event rate was attenuated over time and log‐hazard model was used as a base model.
3. RESULTS
3.1. Patients
Baseline characteristics were generally similar among the regimens in the core and between the core and OLE (Table 1). The OLE is ongoing and results presented herein are from a data lock of December 31, 2021. In Study 201 core, the proportion of ApoE4 carriers (homozygous and heterozygous) was lower in the 10 mg/kg biweekly group (30.4%) compared to the other treatment groups, including PBO (71.0%). However, 88.9% of subjects in the 10 mg/kg monthly group were ApoE4 carriers. The proportion of ApoE4 carriers was 69.4% in the Study 201 OLE. Lecanemab exposure is summarized in the Supplementary Appendix.
TABLE 1.
Baseline characteristics.
| CORE | OLE | |||||||
|---|---|---|---|---|---|---|---|---|
| Lecanemab | ||||||||
| Category | Placebo (N = 245) | 2.5 mg/kg Biweekly (N = 52) | 5 mg/kg Monthly (N = 51) | 5 mg/kg Biweekly (N = 92) | 10 mg/kg Monthly (N = 253) | 10 mg/kg Biweekly (N = 161) | Lecanemab 10 mg/kg Biweekly (N = 180) | |
| Age (year) | Mean (SD) | 71.1 (8.9) | 70.5 (8.3) | 70.5 (7.3) | 70.6 (7.4) | 71.2 (7.5) | 72.7 (8.7) | 74.0 (7.69) |
| Sex, n (%) | Female | 138 (56.3) | 26 (50.0) | 26 (51.0) | 50 (54.3) | 112 (44.3) | 70 (43.5) | 87 (48.3) |
| Region, n (%) | North America | 201 (82.0) | 47 (90.4) | 43 (84.3) | 73 (79.3) | 222 (87.7) | 142 (88.2) | 139 (77.2) |
| Western Europe | 28 (11.4) | 4 (7.7) | 7 (13.7) | 7 (7.6) | 15 (5.9) | 12 (7.5) | 12 (6.7) | |
| Asia | 16 (6.5) | 1 (1.9) | 1 (2.0) | 12 (13.0) | 16 (6.3) | 7 (4.3) | 29 (16.1) | |
| CDR‐Global, n (%) | 0.5 | 200 (84.0) | 44 (84.6) | 40 (83.3) | 77 (86.5) | 210 (85.4) | 133 (87.5) | 80 (44.4) |
| 1 | 38 (16.0) | 8 (15.4) | 8 (16.7) | 12 (13.5) | 36 (14.6) | 19 (12.5) | 68 (37.8) | |
| ApoE4 status, n (%) | Carrier | 174 (71.0) | 38 (73.1) | 40 (78.4) | 84 (91.3) | 225 (88.9) | 49 (30.4) | 125 (69.4) |
| Heterozygous | 134 (54.7) | 33 (63.5) | 28 (54.9) | 70 (76.1) | 165 (65.2) | 39 (24.2) | 97 (53.9) | |
| Homozygous | 40 (16.3) | 5 (9.6) | 12 (23.5) | 14 (15.2) | 60 (23.7) | 10 (6.2) | 28 (15.6) | |
| Noncarrier | 71 (29.0) | 14 (26.9) | 11 (21.6) | 8 (8.7) | 28 (11.1) | 112 (69.6) | 55 (30.6) | |
| ADCOMS | Mean (SD) | 0.370 (0.1663) | 0.386 (0.1970) | 0.395 (0.1746) | 0.390 (0.1558) | 0.373 (0.1522) | 0.373 (0.1508) | 0.651 (0.3685) |
| CDR‐SB | Mean (SD) | 2.89 (1.454) | 2.98 (1.584) | 2.94 (1.420) | 3.03 (1.314) | 2.91 (1.320) | 2.97 (1.401) | 5.28 (3.514) |
| ADAS‐cog14 | Mean (SD) | 22.56 (7.657) | 22.72 (8.050) | 22.94 (7.735) | 22.75 (6.696) | 21.90 (7.302) | 22.06 (7.667) | 35.105 (13.9699) |
| MMSE | Mean (SD) | 26.01 (2.348) | 25.67 (2.487) | 25.25 (2.622) | 25.60 (2.260) | 25.71 (2.364) | 25.61 (2.351) | 20.7 (6.64) |
Abbreviations: ADAS‐cog, Alzheimer's Disease Assessment Scale‐cognitive subscale; ADCOMS, Alzheimer's Disease Composite Score; CDR, clinical dementia rating; CDR‐SB, Clinical Dementia Rating Scale sum of boxes; MMSE, Mini‐Mental State Examination; OLE, open label extension.
3.2. ARIA in Study 201 core study
3.2.1. ARIA‐E in Study 201 core
ARIA‐E was protocol‐specified as an adverse event of special interest in the lecanemab phase 2 core study and the top‐level ARIA‐E results have been summarized previously 1 (Table 2). A total of 9.9% ARIA‐E on 10 mg/kg biweekly and 9.9% on 10 mg/kg monthly was observed. The incidence of ARIA‐E was dose dependent, occurring more frequently in the highest doses. ARIA‐E was also more frequent in subjects who were ApoE4 carriers (14.3%), with incidence higher in homozygous carriers (5/10; 50.0%) versus heterozygous carriers (2/39; 5.1%) and noncarriers (9/112; 8.0%). Symptomatic ARIA‐E occurred in 3% of subjects on 10 mg/kg biweekly and 0.4% on 10 mg/kg monthly. Four out of seven ApoE4 carriers in lecanemab 10 mg/kg biweekly with ARIA‐E in 201 Core (57%) showed symptomatic ARIA‐E. Symptoms included headaches, visual disturbances, and/or confusion.
TABLE 2.
ARIA‐E events from Study 201 core and OLE.
| CORE | OLE | |||||||
|---|---|---|---|---|---|---|---|---|
| BAN2401 | BAN2401 | |||||||
| Category |
Placebo (N = 245) N (%) |
2.5 mg/kg Biweekly (N = 52) n (%) |
5 mg/kg Monthly (N = 51) n (%) |
5 mg/kg Biweekly (N = 92) n (%) |
10 mg/kg Monthly (N = 253) n (%) |
10 mg/kg Biweekly (N = 161) n (%) |
Newly treated Core Placebo (N = 45) n (%) |
All treated in OLE (N = 180) n (%) |
| ARIA‐E | 2 (0.8) | 1 (1.9) | 1 (2.0) | 3 (3.3) | 25 (9.9) | 16 (9.9) | 4/45 (8.9) | 14/180 (7.8) |
| ApoE4+ | 2/174 (1.1) | 1/38 (2.6) | 1/40 (2.5) | 3/84 (3.6) | 23/225(10.2) | 7/49 (14.3) | 4/31 (12.9) | 13/125 (10.4) |
| Homozygous | 1/40 (2.5) | 0/5 (0) | 1/12 (8.3) | 1/14 (7.1) | 11/60 (18.3) | 5/10 (50.0) | 1/4 (25.0) | 4/28 (14.3) |
| Heterozygous | 1/134 (0.7) | 1/33 (3.0) | 0/28 (0) | 2/70 (2.9) | 12/165 (7.3) | 2/39 (5.1) | 3/27 (11.1) | 9/97 (9.3) |
| ApoE4‐ | 0/71 (0) | 0/14 (0) | 0/11 (0) | 0/8 (0) | 2/28 (7.1) | 9/112 (8.0) | 0/14 (0) | 1/55 (1.8) |
Most ARIA‐E occurred within the first 3 months of treatment (Figure 1) and was mostly (90%) mild to moderate in radiographic severity (71% mild‐to‐moderate in ApoE4 carriers). A table summarizing ARIA by radiographic severity can be found in the supplementary appendix (Table S1). MRI findings resolved within 4–16 weeks for 10 mg/kg biweekly dose. ARIA‐E results in the 10 mg/kg monthly group were similar to the 10 mg/kg biweekly group. One notable exception was the ApoE4 homozygous carrier subgroup in which 18.3% (11/60) of subjects experienced ARIA‐E in the 10 mg/kg monthly group versus 50.0% (5/10) in the 10 mg/kg biweekly group (Table 2). There were no recurrences of ARIA‐E in the core since all subjects with ARIA‐E were discontinued, per protocol.
FIGURE 1.
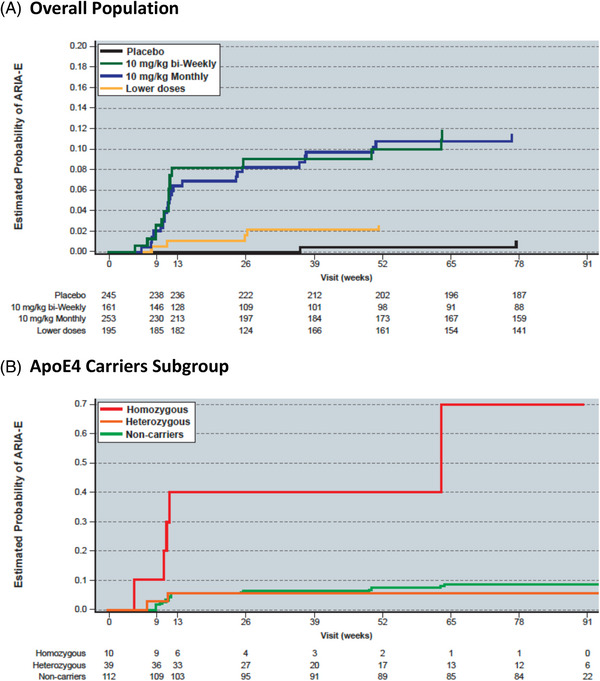
Kaplan‐Meier estimate of ARIA‐E risk in subjects receiving 10 mg/kg biweekly Lecanemab in the Study 201 Core for the (A) overall population and the (B) ApoE4 carriers subgroup.
3.2.2. ARIA‐H in Study 201 core
Overall, ARIA‐H occurred in 6.2% of subjects in the 10 mg/kg biweekly group (Table 3) and events were mostly mild in severity (82%; Table S1). Overall, nine ARIA‐H events were microhemorrhage, one was superficial siderosis, and one was macrohemorrhage. In Study 201 core, there were no dose response trends in incidence of ARIA‐H (PBO 4.9%, lecanemab 2.5 mg/kg biweekly 5.8%, 5 mg/kg monthly 13.7%, 5 mg/kg biweekly 14.1%, 10 mg/kg monthly 9.5%, and 10 mg/kg biweekly 6.2%). There were no symptomatic cases of ARIA‐H in Study 201 core.
TABLE 3.
ARIA‐H events from Study 201 core and OLE.
| Core | OLE | |||||||
|---|---|---|---|---|---|---|---|---|
| Lecanemab | Lecanemab | |||||||
| ARIA term |
Placebo (N = 245) n (%) |
2.5 mg/kg Biweekly (N = 52) n (%) |
5 mg/kg Monthly (N = 51) n (%) |
5 mg/kg Biweekly (N = 92) n (%) |
10 mg/kg Monthly (N = 253) n (%) |
10 mg/kg Biweekly (N = 161) n (%) |
Newly Treated Core Placebo (N = 45) n (%) |
Overall (N = 180) n (%) |
| ARIA‐H | 12 (4.9) | 3 (5.8) | 7 (13.7) | 13 (14.1) | 24 (9.5) | 10 (6.2) | 6 (13.3) | 26 (14.4) |
| ApoE4+ | 9/174 (5.2) | 3/38 (7.9) | 6/40 (15.0) | 13/84 (15.5) | 22/225 (9.8) | 6/49 (12.2) | 5/31 (16.1) | 23/125 (18.4) |
| Homozygous | 1/40 (2.5) | 0/5 (0) | 3/12 (25.0) | 1/14 (7.1) | 9/60 (15.0) | 3/10 (30.0) | 2/4 (50.0) | 11/28 (39.3) |
| Heterozygous | 8/134 (6.0) | 3/33 (9.1) | 3/28 (10.7) | 12/70 (17.1) | 13/165 (7.9) | 3/39 (7.7) | 3/27 (11.1) | 12/97 (12.4) |
| ApoE4‐ | 3/71 (4.2) | 0/14 (0) | 1/11 (9.1) | 0/8 (0) | 2/28 (7.1) | 4/112 (3.6) | 1/14 (7.1) | 3/55 (5.5) |
| ARIA‐E and ARIA‐H | 1 (0.4) | 0 (0) | 1 (2.0) | 0 (0) | 11 (4.3) | 6 (3.7) | 1 (2.2) | 8 (4.4) |
| ApoE4+ | 1/174 (0.6) | 0/38 (0) | 1/40 (2.5) | 0/84 (0) | 10/225 (4.4) | 4/49 (8.2) | 1/31 (3.2) | 7/125 (5.6) |
| Homozygous | 0/40 (0) | 0/5 (0) | 1/12 (8.3) | 0/14 (0) | 5/60 (8.3) | 3/10 (30.0) | ¼ (25.0) | 3/28 (10.7) |
| Heterozygous | 1/134 (0.7) | 0/33 (0) | 0/28 (0) | 0/70 (0) | 5/165 (3.0) | 1/39 (2.6) | 0/27 (0) | 4/97 (4.1) |
| ApoE4‐ | 0/71 (0) | 0/14 (0) | 0/11 (0) | 0/8 (0) | 1/28 (3.6) | 2/112 (1.8) | 0/14 (0) | 1/55 (1.8) |
| Isolated ARIA‐H (no ARIA‐E) | 11 (4.5) | 3 (5.8) | 6 (11.8) | 13 (14.1) | 13 (5.1) | 4 (2.5) | 5 (11.1) | 18 (10.0) |
| ApoE4+ | 8/174 (4.6) | 3/38 (7.9) | 5/40 (12.5) | 13/84 (15.5) | 12/225 (5.3) | 2/49 (4.1) | 4/31 (12.9) | 16/125 (12.8) |
| Homozygous | 1/40 (2.5) | 0/5 (0) | 2/12 (16.7) | 1/14 (7.1) | 4/60 (6.7) | 0/10 (0) | 1/4 (25.0) | 8/28 (28.6) |
| Heterozygous | 7/134 (5.2) | 3/33 (9.1) | 3/28 (10.7) | 12/70 (17.1) | 8/165 (4.8) | 2/39 (5.1) | 3/27 (11.1) | 8/97 (8.2) |
| ApoE4‐ | 3/71 (4.2) | 0/14 (0) | 1/11 (9.1) | 0/8 (0) | 1/28 (3.6) | 2/112 (1.8) | 1/14 (7.1) | 2/55 (3.6) |
3.3. ARIA in OLE
3.3.1. ARIA‐E in OLE
ARIA‐E events in the OLE phase were generally consistent with the rate seen in the lecanemab 10 mg/kg biweekly group in the core. A total of 180 subjects have been dosed in the OLE; all subjects in the OLE were treated with 10 mg/kg biweekly, irrespective of ApoE4 genotype (core allocation for relevant treatment groups: placebo: 45; 10 mg/kg monthly: 60; 10 mg/kg biweekly: 38; Table 2). In the OLE, 14/180 (7.8%) dosed subjects across all core treatment assignments have had ARIA‐E to date (Table 2). Of note, four subjects treated with placebo in the core study had ARIA‐E in the OLE (4 of 45 total; overall incidence of 8.9%). All four of these ARIA‐E cases occurred in ApoE4+ subjects, yielding an incidence of 12.9% (4 of 31 total ApoE4 carriers: 13%) in core placebo‐treated ApoE4 carrier subjects. As in the core, most ARIA‐E occurred within first 3 months of treatment of the OLE (Figure 2) and were mostly mild to moderate in radiographic severity (70%, 10/14; Table S1), with 50% (2/4) mild to moderate in newly treated (all, ApoE4 carriers). ARIA‐E events in the OLE resolved within 4‐16 weeks and six of 14 OLE cases have been dosed through (asymptomatic, mild, or moderate in radiographic severity). Two out of 13 ApoE4 carriers with ARIA‐E in 201 OLE (15%) show symptomatic ARIA‐E. Kaplan‐Meier estimate for ARIA‐E in OLE for core placebo‐treated subjects is approximately 10.0% (Figure 2), consistent with the observed number of ARIA‐E cases for 10 mg/kg biweekly in the core (9.9%) and OLE (8.9%). Three subjects had ARIA‐E recurrence during OLE only, and two subjects had ARIA‐E in both core and OLE. Additional narrative details can be found in the supplement.
FIGURE 2.
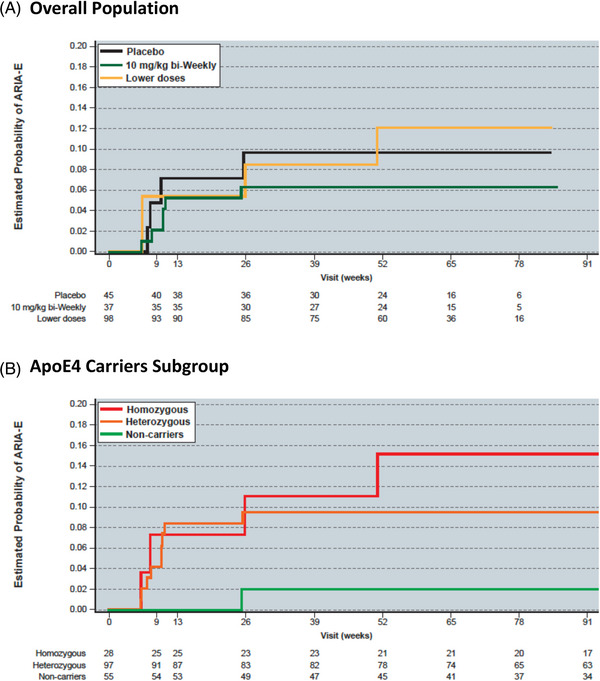
Kaplan‐Meier estimate of ARIA‐E risk in subjects receiving 10 mg/kg biweekly lecanemab in the Study 201 OLE for the (A) overall ole population and the (B) ApoE4 carriers subgroup.
3.3.2. ARIA‐H events in Study 201 OLE
ARIA‐H events in the OLE phase were generally consistent with the rate seen in the lecanemab 10 mg/kg biweekly group in the core (Table 3). ARIA‐H events were mostly mild or moderate in severity (87%; Table S1). Overall, 21 microhemorrhage events, 8 superficial siderosis events, and 1 macrohemorrhage were observed. ARIA‐H occurred in a higher proportion of ApoE4 carriers versus non‐carriers overall in the OLE (13.6% vs. 5.5%), and for newly treated subjects (16.1% vs. 7.1%). Of the 180 subjects who received the study drug, a low proportion of subjects (3.3%) had both ARIA‐E and ARIA‐H events, 4.0% were ApoE4+ and 1.8% who were ApoE4 non‐carriers, consistent with observations from the core study. There was one symptomatic case of macrohemorrhage. This subject had an adverse event of visual field defect (serious and moderate in severity), did not have concurrent with ARIA‐E, and has clinically resolved with residual visual field defect.
Additional information from a pooled analysis of data from Study 201 Core and OLE can be found in the supplement.
3.4. Exposure‐response outcomes for ARIA‐E (phase 2)
The incidence of ARIA‐E as a function of lecanemab exposure was modeled with a logit function. Two measures of exposure, average lecanemab concentration at steady state (Css,av) and maximum lecanemab concentration at steady state (Css,max) were tested as predictors of ARIA‐E using a linear function. Both Css,max and Css,av were statistically significant predictors of ARIA incidence, however, the Akaike information criterion (AIC) for the Css,max model (AIC = 1185.67) was lower than that of the Css,av (AIC = 1207.52), indicating the Css,max model was preferred. Thus Css,max was selected as the lecanemab exposure metric in the model. Subsequently, the significance of ApoE4 genotype was evaluated using the exposure‐response model. It was determined that homozygous ApoE4 carriers were at greater probability of ARIA‐E incidence than noncarriers or heterozygous ApoE4 carriers (p < 0.001). Based on the exposure‐response outcomes ARIA‐E modeling conducted, the observed and model‐predicted proportion of subjects with ARIA‐E increased with an increase in exposure to lecanemab (i.e., observed ARIA‐E is correlated by Css,max quartiles across all doses; Figure 3). Finally, the time‐to‐event profile of the model‐predicted ARIA‐E with 10 mg/kg biweekly dosing regimen confirmed the observed ARIA‐E in OLE (Figure 4). Model‐predicted ARIA‐E incidence was similar to that observed in Study 201 core.
FIGURE 3.
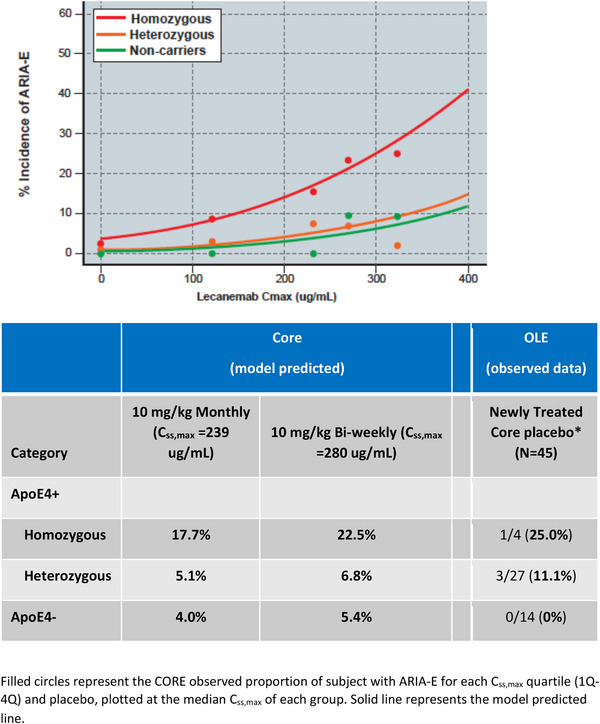
Model‐predicted and observed ARIA‐E versus lecanemab Css,max for ApoE4 subgroups.
FIGURE 4.
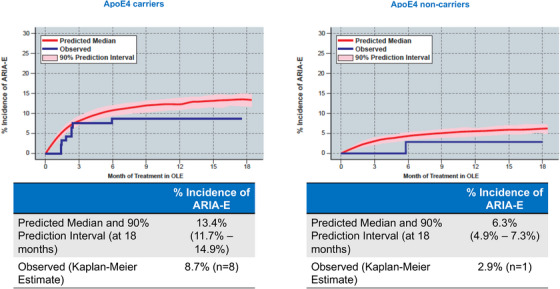
Observed ARIA‐E in OLE versus simulated using PK/PD model in lecanemab‐treated (any dose) subjects from the core study.
4. DISCUSSION
In this report, we present detailed results on the ARIA profile of lecanemab treatment from the phase 2 Study 201 core and OLE phases. The incidence of observed ARIA‐E cases was consistent in the core and OLE for the 10 mg/kg biweekly dose. Observed ARIA in the Study 201 and OLE was largely radiographically mild to moderate and generally occurred within the first 3 months of treatment. Importantly, asymptomatic ARIA‐E can be dosed through in some mild‐to‐moderate cases. The PK/PD model demonstrated that the incidence of ARIA‐E is driven by Css,max and depended on ApoE4 allele number. The model confirmed that ARIA‐E incidence was higher and distinct in ApoE4 homozygous subjects and was similar in ApoE4 heterozygotes and noncarriers.
The risk of ARIA was highest after the first dose of lecanemab (most occurring within 3 months) and was attenuated with time. Of note, an exposure response ARIA‐E model was able to predict the observed incidence rates for ApoE4 carriers in OLE with 10 mg/kg biweekly for those treated with placebo in the core. The predicted ARIA‐E rates in Study 201 and OLE for ApoE4 carriers and noncarriers at Css,max (280 ug/mL) after lecanemab 10 mg/kg biweekly doses were similar with the observed rates in carriers and non‐carriers. The underrepresentation of ApoE4 carriers in the 10 mg/kg biweekly core group make the results difficult to interpret; however, the addition of data from the 10 mg/kg monthly group and the results from the OLE, despite limitations, allows for a fuller analysis. More information is now available with the results of the phase 3 Clarity AD study, which indicated the overall incidence of ARIA‐E was similar to the phase 2 study; however, higher rates of ARIA‐E are noted in the ApoE4 homozygous carriers which has recently completed. 26 ARIA‐H occurred with similar frequency in the core and OLE. There was a relatively low incidence of ARIA‐H in the study and it did not appear to be associated with the dose. As expected, ARIA‐H was more common in ApoE4 carriers; all but one case was asymptomatic (macrohemorrhage; see supplement for additional information). A gene‐dose effect (i.e., heterozygous vs. homozygous ApoE4 carriers) was not apparent for ARIA‐E from the core, but there did appear to be an effect observed for ARIA‐H and for ARIA‐E in the OLE.
The risk of ARIA is a known effect for both active anti‐amyloid immunization and many of the anti‐amyloid antibodies. 1 , 8 , 9 , 10 , 11 , 21 , 22 , 23 , 24 , 25 , 26 , 27 Treatment‐related ARIA‐E has been observed on brain imaging with various emerging anti‐amyloid immunotherapies that target larger aggregated beta‐amyloid species (e.g., bapineuzumab, lecanemab, aducanumab, gantenerumab, donanemab), but not with treatment with antibodies that target monomer and small aggregated beta‐amyloid species (crenezumab, solanezumab). However, the exact pathophysiology underlying ARIA remains uncertain. 6 , 8 , 9 , 10 , 11 , 28 , 29 A recent systematic review on ARIA found that ARIA‐E events occur mostly as asymptomatic events during treatment with amyloid‐modifying therapies. 30 In this analysis, the rate of ARIA‐E among antibody treatments was associated with treatment dose, emerging as a frequent finding in patient groups treated with high‐dose bapineuzumab (26.7% of patients), gantenerumab (28.6%–29.2%), aducanumab (35.9% overall), donanemab (28.8%), and lecanemab (10% overall).
Lecanemab is a monoclonal antibody designed as a potential early AD therapy to selectively target large soluble Aβ protofibrils with activity at the insoluble fibrils that are major components of brain amyloid. 14 , 15 , 16 , 17 Given the association of the occurrence of ARIA with the treatment of anti‐amyloid antibodies in the literature, ARIA‐E and ARIA‐H were pre‐specified as adverse events of special interest and closely monitored in the lecanemab phase 2 proof of concept study. 1 , 3 , 8 , 9 , 10 , 11 For lecanemab (similar for bapineuzumab, donanemab, and aducanumab), the occurrence of ARIA‐E appears to be dose dependent, and increased incidence is associated with the ε4 allele of apolipoprotein E (APOE) gene (APOE ε4). Comparison between trials would suggest that lecanemab has lower ARIA‐E than some of the other published Aβ immunotherapies, including aducanumab, donanemab, and gantenerumab. There are a number of possible reasons why there might be a lower incidence of ARIA with lecanemab at 10 mg/kg biweekly dose including: (a) physicochemical and/or pharmacological profile; (b) degree of brain penetration; (c) specificity of the antibody for vascular amyloid (i.e., the different Aβ target binding site); (d) immune response to the antibody/amyloid complex. ARIA occurs more frequently in ApoE4 carriers relative to non‐carriers and more common in homozygous ApoE4 carriers versus heterozygous ApoE4 carriers. ApoE4 carriers are a high‐risk population for ARIA‐E incidence likely due to the higher presence of severe CAA, 9 even though the major component in CAA is Aβ40 fibrils, whereas neuritic plaques consist of more Aβ42 protofibrils and fibrils. 31 , 32 , 33 Additional preclinical research that may give further insights are ongoing.
4.1. Study limitations and key questions
There was underrepresentation of ApoE carriers in the 10 mg/kg biweekly core group, and particularly ApoE homozygous individuals due to a regulatory authority‐imposed amendment (see supplement), which may have caused underestimated ARIA‐E rates in this group. However, there are several factors that provide reassurance regarding the overall risk of ARIA‐E for lecanemab. First, although ApoE4 carriers were underrepresented in the 10 mg/kg biweekly group in Study 201 core, all participants entering Study 201 OLE (69.4% of whom were ApoE4 carriers) were treated with 10 mg/kg biweekly and ARIA rates in OLE were consistent with those in core. ARIA‐E incidence in ApoE4 carriers in 10 mg/kg biweekly group in core was 14.3% versus 10.4% in OLE (12.9% in newly lecanemab‐treated subjects in the OLE) . Second, subjects that received 10 mg/kg monthly had similar Css,max compared to those receiving the 10 mg/kg biweekly regimen. The PK/PD model demonstrated that the incidence of ARIA‐E is driven primarily by lecanemab Css,max. Similar Css,max levels following 10 mg/kg monthly and biweekly administration are translated in similar ARIA‐E incidence. Thus, the observed 10.2% ARIA‐E incidence in the 10 mg/kg monthly group in core may be viewed as supportive of the expected ARIA‐E incidence for 10 mg/kg biweekly dosing.
4.2. Conclusions
In summary, this analysis provides detailed overview of ARIA occurrences in the lecanemab phase 2 Study 201 core and OLE. There was a modest incidence of ARIA‐E (<10%; symptomatic ARIA rate: <3%) in core and OLE. ARIA was generally radiographically mild to moderate and generally occurred early in the course of treatment (within the first 3 months) for 10 mg/kg monthly and biweekly dosing regimens. The probability of experiencing ARIA‐E was correlated with Css,max and was higher in ApoE4 carriers. As expected, the incidence was higher in homozygous carriers versus heterozygous carriers and noncarriers. For some anti‐amyloid antibodies, the high incidence of ARIA has prompted the utilization of a dosing titration program; however, the lower incidence observed with lecanemab permitted administration without titration, resulting in the initiation of treatment at the dose showing highest efficacy in the phase 2 study. The incidence of observed ARIA‐E cases in the OLE is consistent with the incidence observed at 10 mg/kg biweekly treatment in the core study. ARIA‐H occurred with similar frequency in core and OLE. These findings support initiation of 10 mg/kg biweekly lecanemab at the onset of treatment without titration with a modest incidence of ARIA‐E. Further evaluation of lecanemab is ongoing in the phase 3 Clarity AD study in patients with early AD. In addition, research on a lecanemab subcutaneous formulation with a lower Css,max is ongoing, for which PK/PD modeling suggests the potential for a reduced rate of ARIA‐E relative to the IV formulation. Learnings from the phase 2 study have been incorporated into the phase 3 trial program, including that lecanemab 10 mg/kg biweekly can be used in all subjects regardless of APOE make‐up and that some subjects who are asymptomatic with mild‐moderate ARIA‐E can be dosed through.
CONFLICT OF INTEREST STATEMENT
Lawrence S. Honig; Research Funding from Abbive, Acumen, Alector, Biogen, Eisai, Genentech, Janssen/J&J, Roche, Transposon, UCB, Vaccinex.; Consulting fees from Alector, Biogen, Cortexyme, Eisai, Medscape, Prevail.; Jerome Barakos; Speaker fees from Biogen. Employee of Clario.; Shobha Dhadda, Michio Kanekiyo, Larisa Reyderman, Michael Irizarry, Lynn D. Kramer, and Chad J. Swanson; Authors are employees of Eisai Inc.; Marwan Noel Sabbagh MD; Ownership interest (Stock or stock options): NeuroTau, uMethod Health, Versanum, Athira, TransDermix, Seq BioMarque, NeuroReserve, Cortexyme/Quince Therapeutics, Lighthouse Therapeutics; Consulting: Alzheon, Biogen, Roche‐Genentech, Eisai, KeifeRx, Lilly, Synaptogenix, NeuroTherapia, T3D, Signant Health, Novo Nordisk; Royalties: Humanix; Board of Director:EIP Pharma. Author disclosures are available in the supporting information.
CONSENT STATEMENT
All human subjects provided informed consent.
Supporting information
Supporting Information
Supporting Information
ACKNOWLEDGMENTS
This study was funded by Eisai Inc. We acknowledge with thanks the individuals who enrolled in lecanemab Study 201 as well as their family, care partners, and friends who supported them. We also acknowledge the Site Investigators, Study Coordinators, Raters, and other personnel whose dedication and hard work in collecting data and providing care were essential to the completion of this trial. We acknowledge the manuscript writing, preparation, and editorial efforts of J. David Cox, PhD (Mayville Medical Communications) and Lisa Yarenis (Eisai Inc.).
Honig LS, Barakos J, Dhadda S, et al. ARIA in patients treated with lecanemab (BAN2401) in a phase 2 study in early Alzheimer's disease. Alzheimer's Dement. 2023;9:e12377. 10.1002/trc2.12377
Clinicaltrials.gov identifier: NCT01767311
REFERENCES
- 1. Swanson CJ, Zhang Y, Dhadda S, et al. A randomized, double‐blind, phase 2b proof‐of‐concept clinical trial in early Alzheimer's disease with lecanemab, an anti‐Aβ protofibril antibody. Alzheimers Res Ther. 2021;13:80. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 2. Black RS, Sperling RA, Safirstein B, et al. A single ascending dose study of bapineuzumab in patients with Alzheimer disease. Alzheimer Dis Assoc Disord. 2010;24:198‐203. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 3. Salloway S, Sperling R, Gilman S, et al. A phase 2 multiple ascending dose trial of bapineuzumab in mild to moderate Alzheimer disease. Neurology. 2009;73:2061‐2070. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 4. Rinne JO, Brooks DJ, Rossor MN, et al. 11C‐PiB PET assessment of change in fibrillar amyloid‐beta load in patients with Alzheimer's disease treated with bapineuzumab: a phase 2, double‐blind, placebo‐controlled, ascending‐dose study. Lancet Neurol. 2010;9:363‐372. [DOI] [PubMed] [Google Scholar]
- 5. Ostrowitzki S, Deptula D, Thurfjell L, et al. Mechanism of amyloid removal in patients with Alzheimer disease treated with gantenerumab. Arch Neurol. 2012;69(2):198‐207. [DOI] [PubMed] [Google Scholar]
- 6. Honig LS, Vellas B, Woodward M, et al. Trial of Solanezumab for mild dementia due to Alzheimer's disease. N Engl J Med. 2018;378:321‐330. [DOI] [PubMed] [Google Scholar]
- 7. Sevigny J, Chiao P, Bussière T, et al. The antibody aducanumab reduces Aβ plaques in Alzheimer's disease. Nature. 2016;537:50‐56. [DOI] [PubMed] [Google Scholar]
- 8. Sperling R, Salloway S, Brooks DJ, et al. Amyloid‐related imaging abnormalities in patients with Alzheimer's disease treated with bapineuzumab: a retrospective analysis. Lancet Neurol. 2012;11:241‐249. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 9. Sperling RA, Jack CR Jr., Black SE, et al. Amyloid‐related imaging abnormalities in amyloid‐ modifying therapeutic trials: recommendations from the Alzheimer's Association Research Roundtable Workgroup. Alzheimers Dement. 2011;7:367‐385. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 10. Salloway S, Chalkias S, Barkhof F, et al. Amyloid‐related imaging abnormalities in 2 phase 3 studies evaluating aducanumab in patients with early Alzheimer disease. JAMA Neurol. 2022;79:13‐21. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 11. Lacorte E, Ancidoni A, Zaccaria V, et al. Safety and efficacy of monoclonal antibodies for Alzheimer's disease: a systematic review and meta‐analysis of published and unpublished clinical trials. J Alzheimers Dis. 2022;87:101‐129. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 12. Barakos J, Purcell D, Suhy J, et al. Detection and management of amyloid‐related imaging abnormalities in patients with Alzheimer's disease treated with anti‐amyloid beta therapy. J Prev Alzheimers Dis. 2022;9:211‐220. [DOI] [PubMed] [Google Scholar]
- 13. Cummings J, Aisen P, Apostolova LG, Atri A, Salloway S, Weiner M. Aducanumab: appropriate use recommendations. J Prev Alzheimers Dis. 2021;8(4):398‐410. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 14. Tucker S, Möller C, Tegerstedt K, et al. The murine version of BAN2401 (mAb158) selectively reduces amyloid‐β protofibrils in brain and cerebrospinal fluid of tg‐ArcSwe mice. J Alzheimers Dis. 2015;43:575‐588. [DOI] [PubMed] [Google Scholar]
- 15. Sehlin D, Englund H, Simu B, et al. Large aggregates are the major soluble Aβ species in AD brain fractionated with density gradient ultracentrifugation. PLoS One. 2012;7:e32014. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 16. Sehlin D, Hedlund M, Lord A, et al. Heavy‐chain complementarity‐determining regions determine conformation selectivity of anti‐Aβ antibodies. Neurodegener Dis. 2011;8:117‐123. [DOI] [PubMed] [Google Scholar]
- 17. Lord A, Gumucio A, Englund H, et al. An amyloid‐beta protofibril‐selective antibody prevents amyloid formation in a mouse model of Alzheimer's disease. Neurobiol Dis. 2009;36:425‐434. [DOI] [PubMed] [Google Scholar]
- 18. Magnusson K, Sehlin D, Syvänen S, et al., Specific uptake of an amyloid‐β‐protofibril‐binding antibody‐tracer in AβPP transgenic mouse brain. J Alzheimer's Dis. 2013;37:29‐40. [DOI] [PubMed] [Google Scholar]
- 19. Englund H, Sehlin D, Johansson AS, et al. Sensitive ELISA detection of amyloid‐beta protofibrils in biological samples. J Neurochem. 2007;103:334‐345. [DOI] [PubMed] [Google Scholar]
- 20. Satlin A, Wang J, Logovinsky V, et al. Design of a Bayesian adaptive phase 2 proof‐of‐concept trial for BAN2401, a putative disease‐modifying monoclonal antibody for the treatment of Alzheimer's disease. Alzheimers Dement (N Y). 2016;1:1‐12. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 21. Penninkilampi R, Brothers HM, Eslick GD. Safety and efficacy of anti‐amyloid‐beta immunotherapy in Alzheimer's Disease: a systematic review and meta‐analysis . J Neuroimmune Pharmacol. 2017;12:194‐203. [DOI] [PubMed] [Google Scholar]
- 22. Budd Haeberlein S, Aisen PS, Barkhof F, et al. Two randomized phase 3 studies of aducanumab in early Alzheimer's disease. J Prev Alzheimers Dis. 2022;9:197‐210. [DOI] [PubMed] [Google Scholar]
- 23. Mintun MA, Lo AC, Duggan Evans C, et al. Donanemab in early Alzheimer's disease. N Engl J Med. 2021;384:1691‐1704. [DOI] [PubMed] [Google Scholar]
- 24. Ketter N, Liu E, Di J, et al. A randomized, double‐blind, phase 2 study of the effects of the Vaccine Vanutide Cridificar with QS‐21 adjuvant on immunogenicity, safety and amyloid imaging in patients with mild to moderate Alzheimer's disease. J Prev Alzheimers Dis. 2016;3:192‐201. [DOI] [PubMed] [Google Scholar]
- 25. Farlow MR, Andreasen N, Riviere ME, et al. Long‐term treatment with active Aβ immunotherapy with CAD106 in mild Alzheimer's disease. Alzheimers Res Ther. 2015;7:23. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 26. Cummings J, Aisen P, Apostolova LG, Atri A, Salloway S, Weiner M. Aducanumab: appropriate use recommendations. J Prev Alzheimers Dis. 2021;8(4):398‐410. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 27. Budd Haeberlein S, Gheuens S, Chen T, et al. Aducanumab 36‐month data from PRIME: a randomized, double‐blind, placebo‐controlled Phase 1b study in patients with prodromal or mild Alzheimer's disease (S2.004). Neurology. 2018;90(15):S2.004. [Google Scholar]
- 28. Guthrie H, Honig LS, Lin H, et al. Safety, tolerability, and pharmacokinetics of Crenezumab in Patients with mild‐to‐moderate Alzheimer's disease treated with escalating doses for up to 133 weeks. J Alzheimers Dis. 2020;76:967‐979. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 29. Doody RS, Thomas RG, Farlow M, et al. Phase 3 trials of Solanezumab for mild‐to‐moderate Alzheimer's disease. N Engl J Med. 2014;370:311‐321. [DOI] [PubMed] [Google Scholar]
- 30. Filippi M, Cecchetti G, Spinelli EG, et al. Amyloid‐related imaging abnormalities and β‐amyloid‐targeting antibodies: a systematic review. JAMA Neurol. 2022;79:291‐304. [DOI] [PubMed] [Google Scholar]
- 31. Ringman JM, Sachs MC, Zhou Y, et al. Clinical predictors of severe cerebral amyloid angiopathy and influence of APOE genotype in persons with pathologically verified Alzheimer disease. JAMA Neurol. 2014;71:878‐883. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 32. Blanchard JW, Bula M, Davila‐Velderrain J, et al. Reconstruction of the human blood‐brain barrier in vitro reveals a pathogenic mechanism of APOE4 in pericytes. Nat Med. 2020;26:952‐963. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 33. Weller RO, Massey A, Newman TA, et al. Cerebral amyloid angiopathy: amyloid beta accumulates in putative interstitial fluid drainage pathways in Alzheimer's disease. Am J Pathol. 1998;153:725‐733. [DOI] [PMC free article] [PubMed] [Google Scholar]
Associated Data
This section collects any data citations, data availability statements, or supplementary materials included in this article.
Supplementary Materials
Supporting Information
Supporting Information