

Abstract
Kirsten rat sarcoma viral oncogene homolog mutations are observed in 25% of lung adenocarcinoma and 40% of these are G12C mutations. Historically, no approved targeted agents were available for patients with any KRAS mutation, and response rates to standard-of-care therapies were suboptimal. Newly developed inhibitors directed toward KRASG12C have been successful in clinical trials with overall response rates ranging between 32% and 46%, and two FDA approvals were granted in May 2021 and December 2022 as second-line or later monotherapies. However, rapid tumor resistance complicates their use as a monotherapy. With the rapid development of this novel class of inhibitors, it is important to discern the different types of tumor resistance that may arise and how each can differently contribute to tumor growth and survival. G12C inhibitor resistance is under investigation and combinations of therapies with G12C inhibitors have been proposed. Much of this insight is gleaned from preclinical investigations, as our knowledge of clinical resistance is in its infancy. In this review, we summarize the preclinical development of KRASG12C inhibitors, their clinical evaluations, different types of resistance mechanisms to these compounds, and ways of overcoming them. Finally, we underscore the importance of basic and translational investigations of these molecules in a landscape where their clinical evaluations garner the most attention, and we set the stage for what is to come.
Keywords: clinical trial, drug resistance, G12C, KRAS, preclinical, targeted therapy
Introduction
Over the last several decades, many oncogenes have been identified in non-small-cell lung cancer (NSCLC).1 Research has led to the development and approval of small-molecule inhibitors which specifically target mutant oncogenic proteins.2 However, until recently,3 the Kirsten rat sarcoma viral oncogene homolog (KRAS) was deemed undruggable by small-molecule inhibitors. With the identification of a previously under-appreciated pocket in the GDP-bound form of KRASG12C, compounds capable of trapping KRASG12C in its inactive, GDP-bound state have been generated.4 Many inhibitors of this class are currently under clinical investigation for NSCLC and colorectal cancer (CRC) (Table 1), and objective response rate (ORR) or disease control rates (DCRs) of leading compounds, sotorasib and adagrasib, ranging between 32% and 46% and 88% and 96% for NSCLC, respectively.5,6 To date, both sotorasib and adagrasib are approved as a second-line or later monotherapy in locally advanced or metastatic KRASG12C NSCLC.7,8
Table 1.
List of current clinical trials evaluating GDP-KRASG12C inhibitors for all indications.
| Drug | Trial name | Participants | Indication | Combinations | Trial identifier |
|---|---|---|---|---|---|
| (Phase #) | |||||
| Leading clinical candidates | |||||
| Sotorasib | CodeBreak 100 | 793 (PI/II) | Adv. solid tumors | PD-1i, PD-L1i | NCT03600883 |
| CodeBreak 101 | 1054 (PI/II) | Adv. solid tumors | Chemotherapy (NSCLC, CRC), CDK4/6i, EGFR TKI, MEKi, mTORi, PD-1i, PD-L1i, SHP2i, anti-EGFR Ab, anti-VEGF Ab | NCT04185883 | |
| CodeBreak 105 | 12 (PI) | Adv. solid tumors | – | NCT04380753 | |
| CodeBreak 200 | 345 (PIII) | Adv. NSCLC | Versus docetaxel | NCT04303780 | |
| CodeBreak 201 | 170 (PII) | Adv. NSCLC, as a first line | – | NCT04933695 | |
| CodeBreak 300 | 153 (PIII) | Adv. CRC | Versus chemotherapy (CRC), combination with anti-EGFR Ab | NCT05198934 | |
| – | Expanded access | Adv. NSCLC | – | NCT04667234 | |
| – | 43 (PI/II) | Adv. NSCLC with untreated brain mets | Anti-VEGF Ab | NCT05180422 | |
| – | 59 (PI/II) | Adv. pancreatic cancer | Chemotherapy (pancreatic cancer) | NCT05251038 | |
| LungKG12Ci | 300 (Retrospective) | Adv. NSCLC | – | NCT05273047 | |
| – | 27 (PII) | Neoadjuvant, Stage IIA-IIIB non-squamous NSCLC | Chemotherapy (NSCLC) | NCT05118854 | |
| – | 46 (PII) | Adv. NSCLC | SHP2i | NCT05054725 | |
| Lung-MAP | 116 (PII) | Stage IV/recurrent non-squamous NSCLC | – | NCT04625647 | |
| RAMP203 | 53 (PI/II) | Adv. NSCLC | Raf/MEKi | NCT05074810 | |
| – | 140 (PI) | Adv. NSCLC | AURKAi | NCT05374538 | |
| MERIT-lung | 43 (PII) | Stage III unresectable NSCLC | – | NCT05398094 | |
| – | 25 (PII) | Stage Ib-IIIA NSCLC | – | NCT05400577 | |
| Argonaut | 85 (PI) | Adv. solid tumors | SHP2i | NCT05480865 | |
| – | 30 (PI/II) | All NSCLC | Pan-HER TKI | NCT05313009 | |
| 37 (PII) | Adv. NSCLC | – | NCT05451056 | ||
| SOLUCOM | 100 (PII) | Adv. NSCLC | – | NCT05311709 | |
| HERKULES-2 | 200 (PI/II) | Adv. NSCLC | ERK1/2i, SHP2i, EGFR TKI | NCT04959981 | |
| Adagrasib | KRYSTAL-1 | 740 (PI/II) | Adv. solid tumors | EGFR TKI, PD-1i, anti-EGFR Ab | NCT03785249 |
| KRYSTAL-2 | 86 (PI/II) | Adv. solid tumors | SHP2i | NCT04330664 | |
| KRYSTAL-7 | 250 (PII) | Adv. NSCLC | PD-1i | NCT04613596 | |
| KRYSTAL-10 | 420 (PIII) | Adv. CRC | Versus chemotherapy (CRC), combination with anti-EGFR Ab | NCT04793958 | |
| KRYSTAL-12 | 340 (PIII) | Adv. NSCLC | Versus docetaxel | NCT04685135 | |
| KRYSTAL-14 | 100 (PI) | Adv. NSCLC | SOS1i | NCT04975256 | |
| KRYSTAL-16 | 50 (PI) | Adv. NSCLC | CDK4/6i | NCT05178888 | |
| RAMP204 | 85 (PI/II) | Adv. NSCLC | Raf/MEKi | NCT05375994 | |
| Neo-Kan | 42 (PII) | Stage IB-IIIA NSCLC | PD-1i | NCT05472623 | |
| – | 24 (PI) | Adv. solid tumors | – | NCT05263986 | |
| – | Expanded access | Adv. solid tumors | – | NCT05162443 | |
| – | 133 (PI/II) | Adv. solid cancers | SHP2i, PD-1i | NCT04418661 | |
| JDQ443 | kONTrasT-01 | 425 (PI/II) | Adv. solid tumors | PD-1i, SHP2i | NCT04699188 |
| kONTrasT-02 | 360 (PIII) | Adv. NSCLC | Versus docetaxel | NCT05132075 | |
| kONTrasT-03 | 346 (PI/II) | Adv. solid tumors | MEKi, anti-EGFR-Ab, CDK4/6i | NCT05358249 | |
| – | 120 (PII) | Adv. NSCLC with <1% PD-L1 or ⩾1% and STK11 co-mutation | – | NCT05445843 | |
| GDC-6036 | – | 498 (PI) | Adv. solid tumors | EGFR TKI, PD-L1i, SHP2i, anti-EGFR Ab, anti-VEGF Ab, PI3Kαi | NCT04449874 |
| B-FAST | 1000 (PII/III) | Adv. NSCLC | Versus docetaxel | NCT03178552 | |
| Additional promising clinical compounds | |||||
| BI 1823911 | – | 72 (PI) | Adv. lung, CRC, pancreatic, bile duct cancers | SOS1i | NCT04973163 |
| D-1553 | – | 200 (PI/II) | Adv. solid tumors | NSCLC standard-of-care | NCT04585035 |
| – | 144 (PI/II) | Adv. NSCLC | NSCLC standard-of-care | NCT05492045 | |
| – | 203 (PI/II) | Adv. NSCLC | – | NCT05383898 | |
| LY3537982 | – | 360 (PI) | Adv. solid tumors | AURKAi, CDK4/6i, EGFR TKI, ERK1/2i, PD-1i, Anti-EGFR Ab, SHP2i | NCT04956640 |
| Other KRASG12C inhibitors in development | |||||
| BPI-421286 | – | 80 (PI) | Adv. solid tumors | – | NCT05315180 |
| D3S-001 | 98 (PI) | Adv. solid tumors | – | NCT05410145 | |
| GFH925/IBI351 | – | 128 (PI/II) | Adv. NSCLC | – | NCT05005234 |
| 102 (PI) | Metastatic CRC | Anti-EGFR Ab | NCT05497336 | ||
| 144 (PI) | Adv. NSCLC | Chemotherapy, PD-1i | NCT05504278 | ||
| HBI-2438 | – | 44 (PI) | Adv. solid tumors | – | NCT05485974 |
| HS-10370 | – | 176 (PI/II) | Adv. solid tumors | NCT05367778 | |
| JAB-21822 | – | 144 (PI/II) | Adv. solid tumors | – | NCT05009329 |
| – | 100 (PI/II) | Adv. solid tumors, NSCLC, CRC | Anti-EGFR Ab | NCT05002270 | |
| – | 62 (PI/II) | Adv. CRC, intestinal, appendiceal | Anti-EGFR Ab | NCT05194995 | |
| – | 104 (PI/II) | Adv. NSCLC with STK11 mutation and KEAP1WT | – | NCT05276726 | |
| – | 124 (PI/II) | Adv. solid tumors | SHP2i | NCT05288205 | |
| MK-1084 | – | 185 (PI) | Adv. solid tumors | PD-1i | NCT05067283 |
| YL-15293 | – | 55 (PI/II) | Adv. solid tumors | – | NCT05119933 |
| – | 90 (PI/II) | Adv. solid tumors | – | NCT05173805 | |
CRC, colorectal cancer; EGFR, epidermal growth factor receptor; NSCLC; non-small-cell lung cancer; PD-1i, programmed death-1 inhibitor; PD-L1i, programmed death-ligand 1 inhibitor; TKI, tyrosine kinase inhibitor; VEGF, vascular endothelial growth factor.
Preclinical studies have identified secondary resistance mechanisms to these G12C inhibitors,9–17 and investigators have recently begun a search for potential combination strategies to overcome these resistance mechanisms using cell lines and patient-derived xenograft (PDX) models.9–17 While clinical data for these resistance mechanisms are scarce thus far18–20 likely due to the novelty of these inhibitors, it is expected that more data are forthcoming. Here, we summarize the rapidly progressing study of KRASG12C inhibitors (G12Ci) and the advances in our understanding of intrinsic, adaptive, and acquired resistance to this novel class of inhibitor.
KRAS
KRAS is a 21 kDa GTPase which cycles between its inactive GDP-bound and active GTP-bound forms with the help of guanine exchange factors (GEFs) and GTPase activating proteins (GAPs).21 Three mammalian RAS genes (KRAS, NRAS, and HRAS) encode for four proteins (KRAS4A/4B, NRAS, and HRAS), and all RAS proteins possess highly conserved G-domains but dissimilar hypervariable regions22,23 (Figure 1(a)). The effector lobe (residues 1–86) of the G-domain contains the p-loop (facilitates guanine nucleotide binding), and the switch-I and switch-II regions24,25 (binding interfaces for effector proteins and RAS regulators) (Figure 1(b)). The allosteric lobe (residues 87–166) contains membrane-interacting regions and motifs that allow for guanine nucleotide specificity and binding.26
Figure 1.
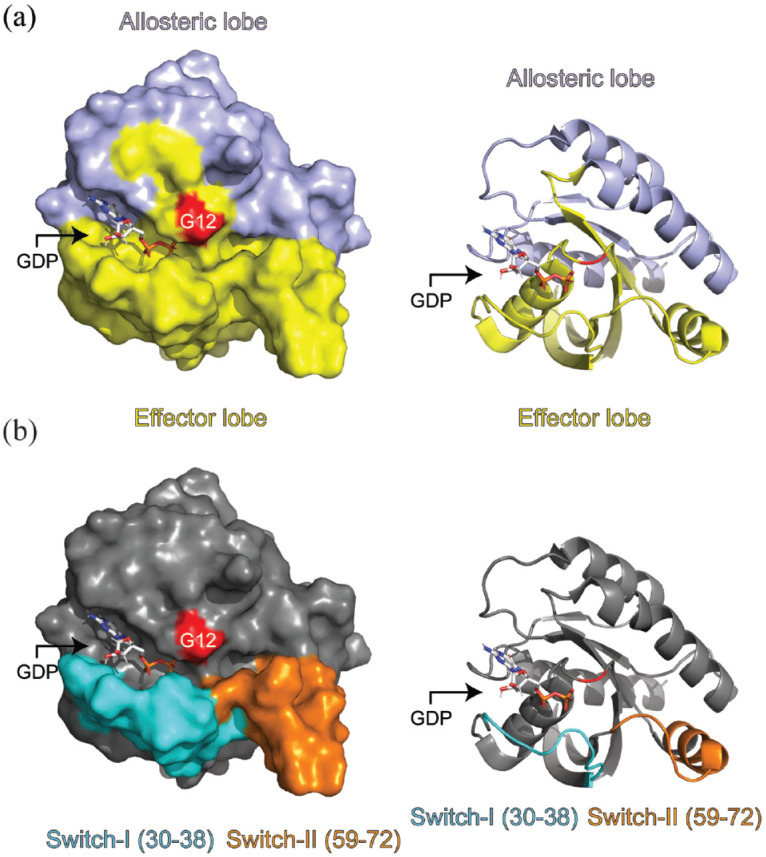
KRASWT complexed with GDP (PDB: 4OBE). (a) Allosteric and effector domains denoted by either purple or yellow colors, respectively. (b) Switch-I and Switch-II regions are colored cyan and orange, respectively. The red highlighted residue is G12. Switch-I is residues 30–38 inclusive while switch-II was considered residues 59–72 inclusive. Images were generated using PyMol version 2.5.2.
Several KRAS downstream pathways exist and the best defined in the NSCLC repertoire are MAPK, PI3K/AKT, and RalGEF, with each playing sometimes overlapping roles in cell proliferation (MAPK), differentiation (MAPK), apoptosis (PI3K/AKT), survival (PI3K/AKT), and migration (RalGEF), among other cellular processes.23
KRAS gain-of-function mutations are observed in ~25% of lung adenocarcinomas (LUAD),27,28 are more prevalent in Caucasians versus Asians, and in current or former smokers versus never smokers.29 KRAS glycine (G)-12 mutations lead to accumulation of active, GTP-bound KRAS30 and account for roughly 90% of all KRAS mutations, where KRASG12C is the most prevalent G12 mutation in LUAD (~40%).31,32 G12 mutations were previously believed to lock KRAS in its active, GTP-bound state.21 However, active intrinsic GTP hydrolysis does occur in mutant-KRAS,30,33 with KRASG12C having the highest rate of the most common KRAS mutants, comparable to wild-type KRAS.30 GTP-bound KRASG12C also uniquely engaged the rasGEF, sons-of-sevenless (SOS), in a similar fashion to wild-type KRAS.34 These data highlight the structural variation between KRAS-mutants and the uniqueness of KRASG12C.
Changes in spatial molecular dynamics also lead to functional differences. Immortalized human bronchial epithelial cells (HBEC) stably expressing KRASG12C and shRNA knockdown of TP53 (HBECsi-TP53) expressed less p-AKT than the KRASG12D HBECs, and this was validated in KRASG12C NSCLC lines, which expressed less p-AKT than other mutants and KRASWT.35 As well, KRASG12C-expressing HBECsi-TP53 expressed the RalA and RalB effectors to a greater extent than other mutants.35 These data suggest the need for unique therapeutic regimens depending on the specific KRAS mutation identified. Extensive efforts have been made to target mutant-KRAS, and the first clinical success has come with inhibitors directed toward KRASG12C.
KRASG12C direct inhibitors
In 1997, Ganguly et al. first targeted the switch-II pocket of GDP-KRASG12C with the non-covalent inhibitors SCH 54292 and SCH5434.36 Ostrem et al.4 expanded on this work and in 2013 led an exploration of cysteine reactive small molecules that bound to this underappreciated pocket, and the lead compound 12 was identified as the first GDP-KRASG12C direct-targeting irreversible inhibitor specific for KRASG12C. Upon binding to KRAS, two mechanisms impaired KRAS signaling: (1) nucleotide affinity was shifted to prefer GDP versus GTP, leading to the accumulation of inactive KRAS and (2) interactions with effectors and regulatory proteins were hindered, further reducing KRAS signaling. Unfortunately, this compound could not be studied in vivo due to poor pharmacologic properties.
Patricelli et al. subsequently utilized structure-based drug design to identify ARS-853, which engaged KRASG12C to a 600-fold greater extent than compound 12 in the NCI-H358 cell line.4,37 The group later optimized the scaffold of ARS-853 using a quinazoline core38 to make ARS-1620, which had a 10-fold improvement in covalently modifying KRASG12C over ARS-853 and possessed pharmacokinetic and pharmacodynamic properties suitable for in vivo use.39 ARS-1620 reduced the pool of active KRAS, and a resultant inhibition in MAPK and PI3K/AKT signaling was observed in all NSCLC cell lines tested.11,13,37,40 Cell-derived xenografts also had varying responses to ARS-1620 monotherapy, between complete regression to no impact whatsoever.10,11,39,41 Overall, while these data were promising, it was understood that improvements to ARS-1620 were necessary before this class of compound could enter clinical trials. For this review, we will summarize G12Ci that are being investigated clinically for KRASG12C-mutant tumors but not mRNA vaccines42,43 (NCT03948763 and NCT04853017) or pan-KRAS inhibitors.44–46
Leading clinical candidates
Among several G12Ci under clinical evaluation, sotorasib (AMG 510), adagrasib (MRTX849), JDQ443, and GDC-6036 are the furthest along in their scrutinization (Table 1).
Sotorasib
Sotorasib/LUMAKRAS®/AMG510 (Amgen, Thousand Oaks, CA, USA) was conceived by a KRAS H95 groove-binding molecule optimization.47 The isopropyl-methylpyridine component of sotorasib that occupies the KRAS H95 groove engages in 25 ligand–protein van der Waals interactions, leading to improved binding over ARS-1620, despite sharing significant molecular structure. This improved binding translated to a 10-fold improved potency over ARS-1620 in a nucleotide exchange assay, and a 40-fold increase in reducing NCI-H358 LUAD and MIA PaCa-2 pancreatic ductal adenocarcinoma (PDAC) cell line viabilities versus ARS-1620.47 In vivo, sotorasib led to maximal inhibition of p-ERK 2–4 h after treatment which was sustained for 48 h, and cell-derived xenografts regressed over 3 weeks of treatment.
Sotorasib was the first KRASG12C inhibitor to enter phase I/II clinical trials targeting CRC48 and LUAD49 (CodeBreak 100; NCT03600883) (Table 1). At the highest dose (960 mg daily) of sotorasib, this rate reached 37.1% in 126 NSCLC phase II patients.50 Of these, 125 patients had adverse events, the most common being diarrhea (50.8%), nausea (31%), and increases in aspartate/alanine aminotransferase levels (21.4%; 20.6%).50 An updated ORR from combining phases I and II NSCLC patients (n = 174) was 40.7% [95% confidence interval (CI): 33.2–48.4] and median progression-free survival (PFS) and overall survival were 6.3 months (95% CI: 5.3–8.2) and 12.5 months (95% CI: 10.5–17.8), respectively.51
After the success of CodeBreak 100 for NSCLC, the phase I/II CodeBreak 101 was initiated to study sotorasib combinations in all advanced-stage tumors with chemotherapies, targeted agents, or various antibodies (NCT04185883). These combinations are based on preclinical studies by Amgen,47 and they align with combinations of targeted agents in KRAS-mutant tumors proposed by studies predating G12Ci.52–56 With positive data from CodeBreak 100 and CodeBreak 101, the FDA granted approval as a second-line or later therapy in locally advanced or metastatic KRASG12C NSCLC in May 2021.57 The cell-free DNA (cfDNA) Guardant360® CDx liquid biopsy test was also approved by the FDA as a companion diagnostic tool for sotorasib at the same time.58
CodeBreak 200 is a phase III trial to compare sotorasib to the standard-of-care chemotherapy, docetaxel, in advanced-stage NSCLC as a second-line therapy (NCT04303780). An ORR of 28.1% was observed for sotorasib versus 13.2% for docetaxel (p < 0.001), while PFS was 24.8% for sotorasib versus 10.1% with docetaxel (hazard ratio: 0.66, 95% CI: 0.51–0.86; p = 0.002).59 Also, a phase II trial was initiated to assess sotorasib as a first-line therapy in Stage IV NSCLC patients (NCT04933695) (Table 1). Other trials are ongoing to study sotorasib in combination with an anti-vascular endothelial growth factor (VEGF) antibody in Stage IV NSCLC with untreated brain metastases (NCT05180422), and in combination with a dual RAF/MEK inhibitor (NCT05074810) or with a SHP2 inhibitor (NCT05054725)60 in advanced-stage NSCLC.
Adagrasib
Mirati Therapeutics (San Diego, CA, USA) developed their own KRASG12C inhibitor called compound 4.61 This molecule shared similarities with ARS-1620 and sotorasib, despite being identified from an independent screen62 (Figure 2). Improvements to compound 4 led to compounds 18 and 19, which reduced cellular IC50 to 1nM.63 Slight electrophilic substitution led to compound 20 (adagrasib/KRAZATI™/MRTX849).63 Adagrasib has a 3-h cellular IC50 of 14 nM (NCI-H358) and 5 nM (MIA PaCa-2). These IC50 values were comparable to those observed with sotorasib, where NCI-H358 and MIA PaCa-2 cells had IC50 values of 6 nM and 9 nM, respectively.12,47
Figure 2.

Binding of different inhibitors on KRASG12C. (a) Molecular structure of sotorasib, adagrasib, and JDQ443. (b) Two different angles of aligned GDP with aligned sotorasib, adagrasib, and JDQ443 on KRASG12C. (c) Illustration of how different KRAS-G12Ci binding impacts KRAS protein conformation. (d) Two different angles of sotorasib, adagrasib, and JDQ443 binding on GDP-KRASG12C with several key KRAS residues highlighted. AMG 510 (PDB: 6OIM); MRTX849 (PDB: 6UT0); JDQ443 (PDB: 7R0M). Images were generated using PyMol version 2.5.2.
Adagrasib was the second GDP-KRASG12C inhibitor to enter clinical trials. In the KRYSTAL-1 trial, 42.9% (48/112) of NSCLC patients had an objective response (NCT03785249).64 Adagrasib is also being evaluated in combination with the programmed death-1 (PD-1) inhibitor, pembrolizumab, the anti-epidermal growth factor receptor (EGFR) antibody, cetuximab, or the EGFR/Her2 inhibitor, afatinib, in the phase Ib portion of the trial. On 12 December 2022, based on data from the KRYSTAL-1 trial, adagrasib was granted accelerated approval as a second-line therapy for KRASG12C locally advanced or metastatic NSCLC.8 In addition, the QIAGEN therascreen KRAS RGQ PCR kit (tissue) and the Agilent Resolution ctDx FIRST Assay (plasma) were both approved by the FDA as companion diagnostic tools for adagrasib.8 Finally, adagrasib received breakthrough designation in combination with cetuximab in KRASG12C advanced CRC after chemotherapy and anti-VEGF therapy.65 This designation came the same day as a KRYSTAL-1 report highlighting a 23% (10/43) ORR in advanced CRC patients with adagrasib alone, whereas the ORR with an adagrasib and cetuximab combination jumped to 46% (13/28).66
With a modest ORR of 42.9% in NSCLC, several phase I–III trials in advanced-stage NSCLC have been initiated to investigate adagrasib in combination with SHP2 inhibitors (NCT04330664), PD-1 antibodies (NCT04613596), SOS1 inhibitors (NCT04975256), or CDK4/6 inhibitors (NCT05178888) as second-line therapies (Table 1). Adagrasib is also being compared to docetaxel as a second-line therapy in advanced-stage NSCLC (NCT04685135).
Adagrasib can also penetrate the blood–brain barrier. At a dose of 100 mg/kg BID in mice, adagrasib led to near complete tumor regression of intracranially implanted LU99-Luc and NCI-H23-Luc KRASG12C NSCLC cells with reductions in p-ERK and proliferation (KI-67).67 In two stage IIIA and IV, NSCLC patients from the KRYSTAL-16,68 (NCT03785249) trial, adagrasib monotherapy led to the disappearance of or reduced the size of three brain lesions relative to baseline, respectively, supporting the use of adagrasib to treat brain metastases.67 The data from KRYSTAL-16,68 are forthcoming and future developments should be watched closely.
JDQ443
Novartis Pharmaceuticals (Basel, Switzerland) first reported their KRASG12C covalent and irreversible inhibitor, JDQ443 in October 2021 (Figure 2).69 This compound binds to the KRAS switch-II pocket with unique interactions not found in sotorasib- and adagrasib- KRASG12C binding.69 Antitumor activity in mouse tumor xenografts was comparable to the leading clinical compounds, and in mouse, rat, and dog, JDQ443 was well tolerated and orally bioavailable. Combinations with SHP2 inhibitors enhanced KRASG12C occupancy in vivo and improved antitumor activity over combinations with MEK or CDK4/6 inhibitors.70
The phase I/II KontRASt-01 trial studies JDQ443 in advanced-stage solid tumor patients. The experimental arms include combinations of JDQ443 with a SHP2 inhibitor, JDQ443 with the anti-PD1 antibody tislelizumab, or JDQ443 triple combination (NCT04699188). Two patients in this trial were administered 200 mg JDQ443 BID (21-day cycle) or 200 mg JDQ443 BID (21-day cycle) with 20 mg QD 2 weeks on/1 week off TNO155 (SHP2i); each reduced the target lesion sizes by ~30.4% and ~44.2%, respectively, versus baseline.70 At the time of cutoff, Grade 3 treatment-related adverse events (TRAEs) occurred in 10.3% of 39 patients with no Grade 4 or 5 TRAE observed, and the ORR across all dose levels in 20 NSCLC patients was 30% (6/20), and 43% (3/7) at the recommended dose of 200 mg BID.71
The phase III KontRASt-02 trial was initiated in March 2022 comparing JDQ443 with docetaxel in an estimated 360 treated advanced-stage NSCLC patients previously treated with one platinum-based chemotherapy and one immune checkpoint inhibitor (NCT05132075).
GDC-6036
Genentech (South San Francisco, CA, USA) has also developed their own KRAS direct inhibitor, GDC-6036. GDC-6036 has greater potency and selectivity versus sotorasib and adagrasib in vitro and in vivo, with median IC50 values in the sub-nanomolar range.72 A phase I trial for advanced-stage KRASG12C-positive solid tumors has been initiated. This trial includes 342 participants and combinations of GDC-6036 with atezolizumab, cetuximab, bevacizumab, erlotinib, inavolisib, and their own SHP2 inhibitor, GDC-1971 (NCT04449874). As a monotherapy, confirmed ORR reached 46% (26/57) patients.73 The international phase II/III B-FAST study also includes an arm evaluating GDC-6036 versus docetaxel (NCT03178552) with forthcoming data.
Additional promising clinical compounds
Several other G12Ci exist, but the clinical assessments of many of them are not as mature as those for sotorasib, adagrasib, JDQ443, and GDC-6036. After these, the next three compounds with the most data are listed below in alphabetical order.
BI 1823911
The method of development for BI 1823911 (Boehringer Ingelheim, Ingelheim am Rhein, Germany) is undisclosed, but it was reported to possess comparative antiproliferative activity to sotorasib and adagrasib in a panel of 2 CRC, 12 NSCLC, and 1 PDAC cell lines with KRASG12C mutations.74 MIA PaCa-2 and CRC cell-derived xenografts and PDX were used to demonstrate similarity in tumor response between BI 1823911, sotorasib, and adagrasib. Of note, the NSCLC SW1573 cell-derived xenograft was resistant to BI 1823911(60 mg/kg),74 similarly to ARS-1620 (200 mg/kg),10 sotorasib (in vitro IC50 > 7.5 µM)12,47 and adagrasib (100 mg/kg).75 Combinations with the SOS1:KRAS inhibitor, BI 1701963, led to tumor regression in 9/9 NSCLC NCI-H2122 cell-derived xenografts, whereas monotherapy led to regression in either 1/8 (BI 1823911) or 0/8 (BI 1701963) mice.
A phase I trial of BI 1823911 was initiated in August 2021 to investigate its combination with SOS1:KRAS inhibitors (NCT04973163) in prior-treated advanced-stage solid tumors.
D-1553
InventisBio (Shanghai, China) reported the discovery of D-1553.76 Activity was observed in NSCLC and PDAC cell-derived xenografts, and combinations with a MEKi, SHP2i, or other cytotoxic agents led to tumor regression.
InventisBio is collaborating with Merck Sharp & Dohme (Kenilworth, NJ, USA) on a phase I/II trial to study D-1553 as a monotherapy or with standard-of-care agents in previously treated solid tumors (NCT04585035). In this China-based trial, there was no dose-limiting toxicity and an ORR of 40.4% (21/52) and DCR 90.4% (47/52) in NSCLC KRASG12C patients.77 In another phase I/II study based in China, ORR reached 37.8% (28/74) and DCR 91.9% (68/74) in a 100% Asian and 11.4% (9/79) female population.78
LY3537982
Eli Lilly (Indianapolis, IN, USA) reported their covalent KRASG12C inhibitor, LY3537982.79 IC50 values of KRAS-GTP loading and inhibiting p-ERK in NCI-H358 cells were 3.35 nM and 0.65 nM, respectively, which were lower than values for sotorasib and adagrasib,12,47 and LY3537982 was predicted to have a >90% clinical target occupancy. A phase I trial is ongoing for patients with KRASG12C solid tumors at any stage (NCT04956640). LY3537982 is being investigated as a monotherapy or in combination with CDK4/6, AURKA, EGFR, ERK inhibitors, or PD-1 or anti-EGFR antibodies. This comes after the failure of their earlier G12Ci, LY3499446 (NCT04165031).
Other KRASG12C inhibitors in development
There are several other G12C inhibitors under clinical investigation, but these have little data available and are subject to scrutinization in the future. All G12Ci under clinical investigation are summarized in Table 1.
Resistance to KRAS G12Ci
Studies investigating oncogene driver-mutant tumors and their respective treatments have highlighted the need to understand mechanisms of therapeutic resistance, as this invariably occurs during treatment. KRASG12C-mutant cancers are no exception to this rule.9–20 Sharma et al. broadly defined resistance to immunotherapy into three separate but interrelated phenomena: primary, adaptive, and acquired.80 For this review, we refer to primary resistance as intrinsic resistance, and define acquired resistance mechanisms as more permanent tumor changes like mutations or cell transformation. Here, for the sake of conceptualization, we consider intrinsic, adaptive, and acquired resistance as three separate phenomena that are fundamentally different, although there is likely overlap between them.80
Intrinsic resistance
To translate to the clinical perspective, we define intrinsic resistance as resulting in either progressive disease (PD) or stable disease (SD) according to the response evaluation criteria in solid tumors (RECIST) V1.1.81 Here, we relate intrinsic resistance to the tumor cell independence from the driver oncogene which may have initiated the tumor. Intrinsic resistance has been observed in KRAS-mutant cell lines since at least 1997, when knockdown of KRAS via transfection of a plasmid containing an antisense gene fragment in cancer cell lines resulted in non-uniform growth suppression.82 KRAS dependence was analyzed more systematically in 2009 using KRAS-directed shRNA.83 Here, after shRNA treatment, there was a wide variety of KRAS dependence in KRAS-mutant cancer cell lines irrespective of KRAS zygosity, and lines were classified as KRAS-dependent or independent based on the expression of the apoptotic marker cleaved caspase-3. Cell lines that were more mesenchymal in nature or cells undergoing epithelial-to-mesenchymal transition (EMT) were more likely to be KRAS independent.83 Indeed, the initiation of EMT in KRAS-dependent NCI-H358 cells induced by TGFβ1 led to characteristic traits of KRAS-independency (E-cadherin loss and gain of vimentin expression).83
Shao and colleagues demonstrated that upon shRNA knockdown of KRAS in CRC and PDAC cell lines that were survival dependent on KRAS, Yes-associated protein 1 (YAP1) signaling led to independence from PI3K/AKT and MAPK pathway activity for survival, and induction of EMT.84 In addition, withdrawing doxycycline in engineered mice containing a doxycycline inducible KRASG12D transgene and conditional TP53 null alleles led to tumor relapse where KRASG12D was absent and YAP1 was amplified.85 Thus, both YAP1 amplification and signaling lead to KRAS-independent tumor growth, possibly through EMT.
KRAS protein expression is correlated with KRAS dependency,83 and both KRAS protein expression and GTP activity are associated with sotorasib sensitivity.12 However, although NCI-H358 and NCI-H23 LUAD cell lines are both G12Ci sensitive,9–13 they were categorized as KRAS dependent and KRAS independent, respectively.83 Importantly, cell lines resistant to sotorasib had GSEA enrichments of EMT gene sets.12 While evidence points to EMT, the relationship of EMT and G12Ci intrinsic resistance has not been conclusively demonstrated in vivo.
Adaptive resistance
We consider adaptive resistance as short-term changes in gene and protein expression. These adaptive mechanisms have been widely reported for KRAS-directed therapies (prior to G12Ci)53,84,54,86 and to KRASG12C inhibitors themselves.9–14 Like intrinsic resistance, we define adaptive resistance as resulting in either PD or SD according to RECIST 1.1.81 However, the mechanisms to reach these tumor response criteria may be different (Figure 3). For that reason, while intrinsic resistance is primary resistance, we consider adaptive (and acquired) resistance as secondary resistance mechanisms.
Figure 3.

Detailed schematic of adaptive and acquired GDP-KRASG12C inhibitor resistance. The MAPK and PI3K/AKT pathways are shown here. Any protein in green indicates mRNA or protein upregulation after GDP-KRASG12C inhibitor (G12Ci) treatment, while any protein in red indicates any secondary mutation or copy number amplification that occurred after G12Ci treatment. Clinical trials investigating G12Ci combination therapies with targeted molecules against specific proteins are listed here. This schematic fails to address the resistance mechanisms including but not limited to histologic transformation, genetic rearrangements, immunomodulation, or global proteome changes in metabolic programming.
Epithelial-to-mesenchymal transition
The role of EMT in adaptive G12Ci resistance is better understood than in intrinsic resistance.83,87 Historical KRAS-dependent signatures53,83,86 and those developed post-G12Ci treatment using RNA-seq12 and protemics13 both have EMT genes significantly represented. Artificial means of inducing EMT via TGFβ treatment confirmed these findings in NCI-H35812,13 and LU6512 cell lines. Upon EMT induction and the acquisition of mesenchymal features, both lines became significantly less sensitive to ARS-1620 and sotorasib, with little effect on MAPK signaling. KRAS oncogene addiction via EMT is facilitated through YAP1/Tead2 signaling,84,85 which can regulate FGFR1.88 Indeed, in a recent autopsy case of a LUAD patient rapidly resisting sotorasib, transcriptome analysis cited one of the seven gene sets upregulated in five of six post-treatment (relative to two pre-treatment) lymph node metastases as EMT.20 In these post-treatment samples, significant upregulation of signaling pathways involving YAP1 was observed.
Epithelial cells seemingly have separate adaptive resistance programs versus mesenchymal cells. In epithelial-like cancer cells, ErbB2/3 is upregulated after ARS-1620 treatment,11,40 and reactivates the MAPK and PI3K/AKT pathways after MEK inhibition53 and ARS-1620 treatment.13 Relating to EMT, gene expression patterns in 59 KRASG12C LUAD tumors identified a significant negative correlation of a TGFβ-EMT gene signature and ErbB2/3 gene expression.13 While ErbB2/3 are suggested receptor tyrosine kinases (RTKs) to target in combination with G12Ci, they are not the only RTKs responsible for adaptive resistance.
RTK/RTK ligand upregulation and SHP2
Treatment with KRASG12C inhibitors can alter phospho-RTK and RTK/RTK ligand expression profiles in NSCLC, CRC, and PDAC cell lines.9,11,40,89 Some RTKs and their signaling intermediates have been assessed in detail. EGFR, FGFR, and AXL were identified in NCI-H358 and MIA PaCa-2 cells as dependencies in genome-wide knockout screens after ARS-1620 treatment.89 Unfortunately, RTK/RTK ligand expression profiles are different in each cell line, obscuring any attempt to derive a universal adaptive resistance strategy used by cancer cell lines.40,75 Instead, groups decided to focus heavily on a more common node in KRAS signaling.
Extracellular signals feed into RTKs which are then relayed through adaptor proteins that can activate the phosphatase SHP2.90 This phosphatase can recruit the GRB2:SOS1 complex to the plasma membrane to facilitate RAS activation90–92 and promotes RAS activation itself by dephosphorylating the Src phosphorylation sites of Y32 and Y64 on KRAS.93 Knockout of SHP2 significantly delayed development of pancreatic intraepithelial neoplasias and the course of atypical adenomatous hyperplasia–adenoma–adenocarcinoma in KRASG12D mice, suggesting an indispensable role for SHP2 in tumor initiation and progression.92 ShRNA targeting SHP2 impacted NCI-H2122 cell fitness in vitro and in vivo,75 and relevant to KRASG12C inhibitors, a genome-wide CRISPR/Cas9 screen identified SHP2 as a dependency in KRASG12C cancer cells treated with ARS-1620.89 Finally, SHP2 inhibition in LUAD NCI-H1944 cell-derived xenografts stopped in vivo tumor growth with a total loss of p-ERK.94 Thus, SHP2 is a key common node in the KRAS signaling axis and may be better suited as a target in combination with a G12C inhibitor compared to individual RTKs.
RAS activation and upregulation
KRASG12C inhibitors very potently reduce pools of KRAS-GTP.4,37 In CRC and NSCLC cell lines, NRAS-GTP and HRAS-GTP have been shown to be upregulated (possibly by RTKs) by fivefold within 48 h of ARS-1620 or sotorasib treatment leading to MAPK signaling rebound11 and G12Ci adaptive resistance.95 This illuminates a potential RTK-driven mechanism of MAPK pathway reactivation that is KRASG12C independent, although it is not widely reported.11,40,95 In this scenario, inhibiting SHP2 is sufficient to dampen NRAS-GTP increases and resultant MAPK signaling after G12Ci treatment, supporting its feasibility in overcoming several adaptive resistance programs.
Protein upregulation of KRAS is observed in cell lines 48–72 h after G12Ci treatment.9–12,14,40,75 Cell lines treated with G12Ci and subjected to single-cell RNA-seq and trajectory inference analyses have informed us of at least two distinct cell fates including cells with inhibited KRAS signaling or cells that have dampened and subsequent rapid reactivation of KRAS signaling.9 Translation of new, drug-free KRASG12C through EGFR and Aurora Kinase A signaling was noted as a major contributor to adaptive G12Ci resistance in these models.9 In this scenario, KRAS-dependent cells continue to activate KRAS signaling after G12Ci treatment through these mechanisms. More broadly, the determinant of the adaptive resistance measures induced in response to targeted agents may be linked the oncogenic dependence of cells, and this may be heterogeneous within a tumor, complicating efforts to identify viable G12C combinations.9
Proteomic adaptation
A focus on the expression patterns of individual genes and proteins helped choreograph the identification of targets that may be leveraged to sensitize tumors to G12C inhibitors.13 Quantitative global and phospho-proteomic isobaric-tag-based mass spectrometry analyses have also been utilized to understand broad changes after G12Ci administration.14 In the PDAC MIAPaCa-2 and the KRAS-dependent83 LUAD NCI-H358 cell lines, increased amino acid, fatty acid, and lipid metabolism and TCA cycle were noted in cell lines at 24 h and 7 days relative to control, while decreases in cell cycle regulation, transcription, translation, and mRNA processing occurred in the same time-frame.14 The contribution of these broad adaptive changes in the context of G12Ci resistance requires further exploration.
Acquired resistance
Tumors undergo partial or complete responses to therapies81 and sometime later may relapse or regrow leading to disease progression. In this scenario, we postulate that the tumor develops G12Ci resistance by acquiring permanent alterations, for example, mutational changes or histologic transformation that supersede KRAS as the driver mechanism for tumor cell growth. In the context of lung cancer, EGFR-mutant tumors may overcome EGFR-tyrosine kinase inhibitors (TKI), erlotinib and gefitinib, by developing T790M mutations.96 Similarly, ALK-rearranged NSCLC have been shown to develop secondary L1152R mutations after treatment with the ALK inhibitor, crizotinib.97 Before the discovery of the switch-II pocket in GDP-KRASG12C, it was not known whether KRAS-mutant tumors also developed secondary mutations leading to resistance, whereas KRAS copy number changes were better understood, at least for pancreatic cancer.
In a cohort of 38 mouse KRASG12D PDAC tumors, chromothripsis-induced changes led to the identification of KRAS as the most amplified locus globally.87 Here, the copy number of KRAS was referred to as gene dosage, with four distinct groupings of tumors: (i) focal gain, (ii) arm-level gain, (iii) loss of KRASWT (copy number neutral), and (iv) no change. Tumors with increased KRAS gene dosage (groups i–iii) had increased metastatic potential, while tumors with no change (group iv) were largely non-metastatic. Indeed, a LUAD case study including pre- and post-sotorasib patient metastases identified a decrease in KRASG12C mutant allele frequency (MAF) in post-treatment samples as well as an inverse correlation of KRASG12C MAF with G12Ci resistance pathway activation while loss of G2/M cycle checkpoints positively correlated with KRASG12C MAF.20 This is plausible considering how KRASWT impairs mutant-KRAS upon heterodimerization in LUAD by sacrificing MAPK pathway output for increased resistance to MEK inhibition, whereas mutant-KRAS homodimerization increases MAPK pathway output while also increasing sensitivity to MEK inhibitors.98 Thus, while KRAS MAF and zygosity fail to predict G12Ci antitumor activity,10,75 they may be important determinants in predicting likelihood of acquired resistance.
A major roadblock in understanding resistance to G12Ci is the heterogeneity of tumors. Even within cell lines sensitive to G12Ci, resistant subclones after G12Ci treatment have been observed,9 and outgrowth of clones with G12Ci resistance mutations have also been noted.15–19 The earliest work exploring resistance to G12Ci via KRAS secondary mutations came in 2016 from Lito and colleagues,99 even before sotorasib and adagrasib were known. KRASA59G mutations displaced the Q61 residue essential for GTP hydrolysis, and KRASG12C/A59G HEK293 or NCI-H358 double mutant cells were resistant to ARS-853. Other G12C co-mutations in residues impacting GTP hydrolysis, GTPase activity, and nucleotide exchange including Y40A, Q61L, Y64A, N116H, and A146V also led to ARS-853 resistance.99 More recently, Koga and colleagues used N-ethyl-N-nitrosourea mutagenesis to identify secondary mutations in sotorasib- or adagrasib-resistant KRASG12C-transduced Ba/F3 cells.15 While several mutations including KRAS V8E, G13D, A59S/T, R68M/S, M72I, Q61L, Q99L, and Y96D/S were observed, only A59S and Y96D were shared between both sotorasib- and adagrasib-resistant cells. Sotorasib or adagrasib treatment suppressed P-ERK and p-S6 in Ba/F3 cells with various secondary KRAS mutations, but not with Y96D/S. IC50 values of NCI-H358 cells with retrovirally introduced Y96D/S were 30 times higher to both G12Ci than parental NCI-H358 cells, while cells with A59T were much more sensitive.15 Several of the mutations listed above have been found through independent saturation mutagenesis screening100 and in patient tumors Table 2.18,19
Table 2.
Observed alterations leading to GDP-KRASG12C inhibitor resistance for all indications.
| Gene | Mutation | Context of the alteration | Observing studies (PMID) |
|---|---|---|---|
| KRAS | A146V | Preclinical | 26841430 |
| A59G/S/T | Preclinical | 26841430; 33971321; 35471904 | |
| C12F | Preclinical | 35471904 | |
| D69P | Preclinical | 35471904 | |
| D92R | Preclinical | 35471904 | |
| F156L | Preclinical | 35471904 | |
| G12C AMP** | Clinical | 34161704 | |
| G12R/V/W | Clinical | 34161704; 33824136 | |
| G13D/E | G13D: preclinical and clinical; G13E: preclinical | 33824136; 34161704; 33971321; 35471904 | |
| H95D/N/R/Q/V | H95D/R: preclinical and clinical; H95Q: clinical; H95N/V: Preclinical | 35471904; 34161704 | |
| M72I | Preclinical | 33971321; 35471904 | |
| N116H | Preclinical | 26841430 | |
| Q61H/L | Q61L: preclinical; Q61H: clinical | 26841430; 33971321; 34161704 | |
| Q99F/L/W | Preclinical | 35471904; 33971321 | |
| R68L/M/S | R68L/M: preclinical; R68S: Preclinical and clinical | 33971321; 35471904; 34161704 | |
| S65W | Preclinical | 35471904 | |
| T58I | Preclinical | 35471904 | |
| V8A/E/L | Preclinical | 33971321; 35471904 | |
| V9Y | Preclinical | 35471904 | |
| Y40A | Preclinical | 26841430 | |
| Y64A | Preclinical | 26841430 | |
| Y96C/D/H/S | Y96C: preclinical and clinical; Y96D: clinical; Y96H/S: preclinical | 35471904; 34161704; 33824136; 33971321 | |
| AKAP9-BRAF | Fusion | Clinical | 34161704 |
| BRAF | V600E | Clinical | 34161704; 33824136 |
| CCDC6-RET | Fusion | Clinical | 34161704 |
| EGFR | A289V | Clinical | 34161704 |
| EML4-ALK | Fusion | Clinical | 34161704 |
| ERBB2 | Gain | Clinical | 34715459 |
| FGFR3-TACC3 | Fusion | Clinical | 34161704 |
| MAP2K1 | E102_I103del | Clinical | 34161704; 33824136 |
| I99_K104del | Clinical | 34161704 | |
| K57N/T | Clinical | 34161704; 33824136 | |
| Q56P | Clinical | 33824136 | |
| MET | AMP** | Preclinical and clinical | 34161704; 34365406 |
| NF1 | R2637* | Clinical | 34161704 |
| NRAS | Q61K/L/R | Clinical | 34161704; 33824136 |
| NRF1-BRAF | Fusion | Clinical | 34161704 |
| PIK3CA | H1047R | Clinical | 34161704 |
| PIK3R1 | S361fs | Clinical | 34161704 |
| PTEN | G209V | Clinical | 34161704 |
| N48K | Clinical | 34161704 | |
| RAF1-CCDC176 | Fusion | Clinical | 34161704 |
| RAF1-TRAK1 | Fusion | Clinical | 34161704 |
| RET | M918T | Clinical | 34161704 |
| RIT1 | P128L | Clinical | 34161704 |
| TP53 | F338fs | Clinical | 33824136 |
Nonsense mutation.
Amplification (AMP).
In 38 KRASG12C patients (27 NSCLC, 10 CRC, and 1 appendiceal cancer) treated with adagrasib, circulating tumor DNA (ctDNA) analysis detected 17 (10 NSCLC, 6 CRC, and 1 appendiceal cancer) cases of acquired secondary KRAS mutations including G12D/V/R/W, G13D, Q61H, R68S, H95D/R, and Y96C, and multiple mutations could be found in individual patients.19 Additional alterations along the MAPK axis were found in NRAS, MEK1, EGFR, and several gene fusions were confirmed. There is also at least one instance of a patient who acquired an ERBB2 amplification as a means of sotorasib resistance, identified by fluorescence in situ hybridization.17 MET amplifications have also been identified as an acquired G12Ci resistance mechanism.16 A combination of SHP2i + G12Ci were able to overcome both MET amplification in sotorasib-resistant NCI-H23 cell lines and xenografts16 and ERBB2 overexpression in NCI-H358 or Calu-1 cell lines and xenografts,17 suggesting the feasibility of this combination.
cfDNA from a metastatic KRASG12C NSCLC patient treated with adagrasib identified secondary KRAS, NRAS, BRAF, and MEK mutations,18 many of which overlapped with the other studies.15,19,99 In this patient, secondary mutations within KRAS including Y96D arose in trans with KRASG12C, although wild-type KRAS was retained.18 Until these studies, KRASY96D mutations were unknown. It is now understood that Y96 interacts with sotorasib and adagrasib through hydrogen bonds that are abolished with a Y96D mutation.101 Y96D-mutants sustained MAPK pathway activity and IC50 of sotorasib and adagrasib in NCI-H358 were increased by >100-fold.18 While KRASG12C co-mutation with KRAS G13D, A59S, R68S, H95D/Q/R, and Q99L was resistant to either sotorasib or adagrasib, Y96-mutations were the only ones resistant to both.15,18,19 Indeed, mutations in residues that impact GTP hydrolysis and nucleotide binding like G13D, A59S, K117N, and A146P were less resistant to G12Ci compared to mutations which prevent drug binding like G12R or Y96C.19,99 While Tsai and colleagues did not observe any secondary KRAS or MAPK pathway resistance mutations to G12Ci by whole-exome sequencing (WES) in their case report,20 secondary mutations were identified elsewhere in patients using cfDNA18 and ctDNA.19 It is likely the case that the 200× WES coverage used by Tsai and colleagues20 was not deep enough to capture the low-frequency variants,102 and cfDNA and ctDNA detection methods are superior for this purpose.
With such a dramatic shift in G12Ci sensitivity, it is intriguing how Y96 mutations have only been identified in a few patients thus far. As multiple resistance mutations were identified within individual patients, there may be coordination within the tumor to determine which mutation is most resistant to G12Ci and promotes tumor fitness the best. This is in line with the notion of treatment-induced clonal evolution contrary to the presence of pre-existing, drug-resistant clones within a tumor.103 Thus, the acquisition of G12Ci-resisting KRAS mutations may be dictated by their ability to prevent drug binding, and more clinical data should be compiled to evaluate the prevalence of these mutations in the G12Ci-treated patient population.
Finally, akin to EGFR-mutant LUAD transforming to small-cell carcinoma on EGFR-TKIs,104,105 two LUAD patient tumors treated with adagrasib transformed to squamous cell carcinoma without any genomic alterations as a means of acquired resistance.19 Thus, secondary mutations or KRAS copy number gains may not be universal G12Ci-acquired resistance mechanisms.
Therapeutic combinations to overcome KRASG12C inhibitor resistance
Years of research and development have culminated into a growing list of covalent KRASG12C inhibitors. Despite intense validation of protein binding and antitumor efficacy, adaptive resistance can develop in as little as a few hours. As such, potential G12Ci combination partners have been proposed to re-sensitize tumors to G12Ci and overcome resistance, and many are being investigated in clinical trials (Table 1).
Receptor TKIs
Activation of RTKs after G12Ci as a resistance mechanism to maintain MAPK signaling is well established.9,11,40,89 Recent works have identified an increasing number of RTKs as combination partners, including EGFR, ErbB2/3, IGF1R, FGFR, and AXL in different cell lines and contexts.10,13 EGFR, FGFR, and AXL are suggested to be dependencies in cells specifically in a G12Ci-induced state,89 although these are cell-line dependent. Nonetheless, several clinical trials are evaluating EGFR TKI or EGFR antibodies with G12Ci (Table 1, Figure 3). Once clinical G12Ci resistance is better characterized, G12Ci + specific TKI responses may be better predicted.
SHP2 inhibitors
It is hinted that the SHP2i + G12Ci combination works well in cancer cells with an epithelial subtype, whereas RTKi + G12Ci is more effective in mesenchymal-like cancer cells.13 However, overwhelming evidence of reductions in cell viability and KRAS signaling across several mesenchymal-like and epithelial-like pancreatic, colon, and lung cancer cell lines suggests broad activity of the SHP2i + G12Ci combination.9,11,12,75,89 In addition, SHP2 inhibition promotes an antitumor immune program that can be leveraged with immunotherapy.40
PI3K inhibition is suggested to be a lethal combination with G12Ci10,13,89 and leads to SW1573 cell-derived xenograft tumor regression,10 despite SW1573 NSCLC cells being intrinsically resistant to G12Ci monotherapy.13 However, SW1573 possess a PIK3CAK111E gain-of-function allele, possibly rendering them KRAS independent.40 While PI3Ki + G12Ci fails to account for the MAPK pathway reactivation that comes with G12Ci adaptive resistance, SHP2i + PI3Ki + G12Ci can,12 and this triple combination reduced viability in this cell line.13 Unfortunately, the regimen is unlikely to be evaluated in the clinic due to the high toxicity of PI3Ki combinations observed clinically.106,107
SHP2 inhibitor combinations with G12Ci are well documented to be effective in KRASG12C cell lines, but the same cannot be said about cancer cell lines expressing KRASG13D or KRASQ61H/R/X, which were resistant to SHP2i with continued expression of p-ERK.90,94,108 As secondary KRAS mutations can be acquired after G12Ci,15,18,19 it is possible that these specific KRAS mutations (G13D, Q61H/R/X) may occur after SHP2i + G12Ci combination treatment to escape SHP2i antitumor activity. As there are several active clinical investigations looking at this combination (Table 1), an investigation to answer this question may be warranted. Nonetheless, this combination has seen preclinical success. The combination with the SHP2 inhibitor, RMC-4630, with sotorasib was safe and tolerable in NSCLC patients, and confirmed partial responses were observed in 3/11 (27%) and 3/6 (50% – with two highest doses of RMC-4630) of pre-treated and treatment-naive patients, respectively.60
SOS1 inhibitors
SOS1 is a RasGEF activated by SHP2 and it facilitates RAS activation by promoting GTP binding. Downregulation of SOS1 phenocopies SHP2 inhibition,40 suggesting an alternative to SHP2i. Indeed, SOS1 + MEK inhibition reduced MAPK pathway output and led to tumor regression in cell-derived xenografts.109 SOS1i + sotorasib reduced phosphorylation of ERK stronger than sotorasib monotherapy in NCI-H358 cells,109 although signal rebounded within this and other cell lines within 48–72 h.13 Especially relevant to G12Ci-acquired resistance, sotorasib- and adagrasib-resistant KRASG12C/Y96D NCI-H358 cells were sensitive to a combination of SOS1 and MEK inhibitors,15 and this combination is being assessed clinically for advanced-stage KRASG12C solid tumors (NCT04111458).
SOS1 been implicated as a negative feedback regulator of KRAS signaling, and the GEF can activate wild-type N/HRAS via oncogenic KRAS.110 SOS1 interacts with Ribosomal S6 kinase 1 (RSK1) to achieve negative regulator status, but RSK1 also complexes with the RAS GAP neurofibromin 1 (NF1) to negatively regulate wildtype KRAS, at least in pancreatic cancer.111 It is therefore reasonable that as 7 of 14 solid cancer cell lines with aberrations in NF1 were sensitive to SOS1 inhibitors regardless of KRAS mutation status,109 a SOS1i + G12Ci combination may be useful in patients with NF1 alterations. The SOS1i + G12Ci is under investigation in three clinical trials (NCT04585035, NCT04973163, and NCT04975256), and should shed light on whether SOS1 or SHP2 inhibitors are the superior G12Ci combination partner, or if there is no difference between the two.
Immunotherapy
KRAS-mutant LUAD have complex tumor immune microenvironments, and immune modulation by G12Ci has been investigated. Single-agent adagrasib or sotorasib increased CD3/4/8+ T-cell tumor infiltration and decreased myeloid suppressor cell populations in vivo, which ultimately sensitized tumors to PD-1/PD-L1 blockade.20,40,47,112 Resistance to sotorasib in patient lymph node metastases was linked to immunological dampening, and reductions in adaptive immune cell populations and neoantigens.20 Combination of immune checkpoint inhibition (ICI) and G12Ci led to improved antitumor activity over single-agent G12Ci and abrogation of G12Ci resistance.47,112 Several clinical trials are investigating PD-1/PD-L1 blockade in combination with G12C inhibitors (Figure 3, Table 1). In CodeBreak 100/101, 58 G12Ci-naïve NSCLC patients were treated with sotorasib and atezolizumab or pembrolizumab with an ORR of 29% (17/58) in all cohorts, although major hepatoxicity (Grade ⩾3 = 79% in all cohorts; any grade = 89.5%) was observed.113 In contrast, 75 treatment-naïve KRASG12C advanced-stage NSCLC patients better tolerated the combination of adagrasib and pembrolizumab with grade 3–4 TRAEs occurring in 44% of patients, while increased lipase (11%) and increased alanine transaminase/aspartate transaminase (8%/9%) were all grade 3 (NCT03785249 and NCT04613596).114 ORR was 49% (26/53) for the 53 patients with at least one on-study scan and DCR was 89%.114 Thus, while adagrasib may be the superior combination partner for immunotherapy versus sotorasib in terms of tolerability, further investigations to fully explain this discrepancy are warranted.
KRAS itself can direct immunosuppression,115 but a classification of tumors into immune ‘hot’ or ‘cold’ categories by KRAS co-mutations with TP53 or STK11, respectively, has also been proposed.116 Indeed, STK11 alterations are a key contributor of intrinsic resistance to PD-1 inhibitors in KRAS-mutant NSCLC.117,118 KRASG12C/STK11 co-mutant LUAD patients had shorter time to next treatment, time to discontinuation, and overall survival after ICI treatment than patients without STK11 co-mutations.119 Meanwhile, TP53 co-mutations increase PD-L1 expression and tumor mutation burden, which both contribute to ICI response and improved patient outcomes.120,121 Adagrasib antitumor activity was not predicted by STK11 or TP53 co-mutations in preclinical studies.75 However, patients with STK11 co-mutations that were previously treated with ICI and/or chemotherapy had a 64%122 (9/14 patients) ORR to adagrasib monotherapy, versus the 42.9% (48/112) ORR observed in all NSCLC patients.6,64 Thus, there may be an added benefit of treating KRASG12C/STK11 co-mutated patients with adagrasib after or with ICI. More work must be done to understand the role of STK11 in G12Ci immunomodulation.
Other small-molecule combinations
mTOR signaling is activated upon ARS-1620 treatment,13 although its downstream signaling targets p70-S6 kinase and p-S6 were partially inhibited after exposure to adagrasib.75 The ATP-competitive mTOR inhibitor, vistusertib, in combination with adagrasib inhibited several components of the PI3K/mTOR signaling pathway and led to tumor regression in LUAD NCI-H2030 xenografts, suggesting the applicability of this combination.75 A triple combination of mTORi + IGF1Ri + G12Ci has also been proposed.41
Cell cycle kinases have been supported as targets for KRAS-mutant tumors for several years. Large decreases in cell cycle proteomes were noted after 24 h of a single G12Ci treatment, and CDK4/6i + G12Ci reduced growth in 2D- and 3D-HCC44 cells.14 The combination of the CDK4/6i, palbociclib, and adagrasib had profound antitumor efficacy in NCI-H2122 and in the G12Ci-resistant SW1573 cell line and xenograft tumors.75 A combination of CDK4/6i + MEKi is being assessed in KRAS-mutant NSCLC (NCT02022982).
A unique tricomplex inhibitor of GTP-KRASG12C by Revolution Medicines called RM-018 was unveiled in late 2019.123 This inhibitor creates a ternary complex with the immunophilin cyclophilin A and KRASG12C, forming non-covalent interactions within the switch-I and switch-II regions of KRASG12C, irreversibly inhibiting KRASG12C and preventing RAF binding. RM-018 monotherapy in NCI-H358 and MIA PaCa-2 cells suppressed proliferation longer than with G12Ci alone, and in vivo administration led to dose-dependent tumor regression.123 Moreover, sotorasib- and adagrasib-resistant KRASG12C/Y96D LUAD NCI-H358, PDAC MIA PaCa-2, HEK293T, and NSCLC MGH1138-1 cells were sensitive to RM-018 alone, with reductions in p-ERK and p-RSK levels and cell viability comparable to the effects of sotorasib and adagrasib in these lines with only KRASG12C mutations.18 Finally, RM-018 IC50 levels in KRASG12C/Y96D NCI-H358, MIA PaCa-2, and Ba/F3 cells were 7.3 nM, 3.4 nM, and 2.8 nM, respectively, whereas these values were all >2 µM for ARS-1620, sotorasib, and adagrasib.18 While limited experimental data on RM-018 have been released, it may be a key contender in the fight against KRASG12C.
Other novel strategies
The latest information on other types of therapies being investigated for KRASG12C including peptide/dendritic cells/mRNA cancer vaccines42,43 (NCT03948763 and NCT04853017), adoptive T-cell therapy, PROTACs, and CRISPR/Cas9, is nicely summarized elsewhere.124,125 While switch-II pocket inhibitors are superior to nucleotide pocket inhibitors,126 mathematical modeling suggests the inverse to be true if secondary KRAS mutations that result in faster nucleotide dissociation are acquired in KRASG12C cells.127 Thus, while small-molecule inhibitor combinations with G12Ci are being heavily investigated due to their widespread availability and understanding, esoteric therapies may be better suited for overcoming G12Ci resistance. At least one clinical trial evaluating a KRAS-targeted vaccine with nivolumab and ipilimumab for advanced-stage KRAS-mutant NSCLC patients has been initiated (NCT05254184).
Finally, as opposed to overcoming resistance, one strategy may be to prevent resistance from occurring in the first place. Multiple low dosing has been studied in EGFR-mutant NSCLC.128 A combination of IC20 doses of EGFR TKIs + RAFi + MEKi + ERKi completely inhibited MAPK signaling and significantly reduced cell viability and proliferation in parental PC9 cells, TKI-resistant PC9 cells, and patient-derived organoids and xenografts.128 In addition, Hayashi et al. evaluated alternating monotherapy with the third-generation EGFR TKI osimertinib and second-generation EGFR TKI afatinib in 46 treatment-naïve advanced EGFR-mutant NSCLC patients.129 Although the trial failed to meet its primary endpoint of 77% 12-month PFS (70.2%; 95% CI, 54.2–81.5%), the median PFS in the trial was 21.3 months (95% CI, 16.3 months–not reached), hinting at a utility for alternating therapies in NSCLC. Similarly, Koga and colleagues suggested that resistance via acquisition of certain secondary KRAS mutations during sotorasib (G13D, A59S/T, R68M) and adagrasib (Q99L) treatment can be overcome by treating with the opposite G12Ci.15 Therefore, an alternating dosing regimen of sotorasib and adagrasib may be worth investigating.
Conclusions
Targeting KRAS has been a longstanding goal for several decades. While G12Ci poised for clinical use have only been studied since 20193,7,8,47,57,75 and our clinical understanding of KRASG12C inhibitor resistance is in its infancy,17–20 tremendous strides have been made in treating KRASG12C NSCLC patients and understanding resistance preclinically. Diverse mechanisms of adaptive and acquired resistance have been explained and numerous clinical trials investigating a wide range of different combination therapies supported by preclinical data are already underway (Table 1). Little information on G12Ci intrinsic resistance is available. As such, specific therapeutic combinations may be more effective in cases of intrinsic resistance versus adaptive/acquired resistance, but there is not enough information at this time to answer this question.
With the success of G12Ci pipelines, researchers are beginning to investigate inhibitors specific to other KRAS mutations, such as G12D130–133 and G12V.130,134 To date, we have seen the tip of the proverbial iceberg. It will only be a matter of time before we amass an arsenal of diverse inhibitors directed toward the once ‘undruggable’ KRAS protein.
Acknowledgments
We thank Dr. Nhu-An Pham for her review of a draft of this manuscript. We thank Drs. Chris Marshall and Geneviève Seabrook for their constructive critiques of the Pymol-generated figures.
Footnotes
ORCID iD: Ming-Sound Tsao  https://orcid.org/0000-0002-9160-5405
https://orcid.org/0000-0002-9160-5405
Contributor Information
Joshua C. Rosen, Princess Margaret Hospital Cancer Centre, University Health Network, Toronto, ON, Canada Department of Laboratory Medicine and Pathobiology, Temerty Faculty of Medicine, University of Toronto, Toronto, ON, Canada.
Adrian Sacher, Princess Margaret Hospital Cancer Centre, University Health Network, Toronto, ON, Canada; Division of Medical Oncology, Department of Medicine, Princess Margaret Cancer Centre, Temerty Faculty of Medicine, University of Toronto, Toronto, ON, Canada; Department of Immunology, Temerty Faculty of Medicine, University of Toronto, Toronto, ON, Canada.
Ming-Sound Tsao, Princess Margaret Hospital Cancer Centre, University Health Network, 101 College Street, Toronto, ON M5G1L7, Canada; Department of Laboratory Medicine and Pathobiology, Temerty Faculty of Medicine, University of Toronto, Toronto, ON, Canada; Department of Medical Biophysics, Temerty Faculty of Medicine, University of Toronto, Toronto, ON, Canada.
Declarations
Ethics approval and consent to participate: Not applicable.
Consent for publication: Not applicable.
Author contribution(s): Joshua C. Rosen: Conceptualization; Formal analysis; Visualization; Writing – original draft; Writing – review & editing.
Adrian Sacher: Conceptualization; Supervision; Writing – original draft; Writing – review & editing.
Ming-Sound Tsao: Conceptualization; Funding acquisition; Supervision; Writing – original draft; Writing – review & editing.
Funding: The authors disclosed receipt of the following financial support for the research, authorship, and/or publication of this article: This work is partially supported by the Canadian Institutes of Health Research Foundation Stream Grant (FDN-148395).
Adrian Sacher – Research funding or support: AstraZeneca, Genentech, BMS, Guardant. Institutional Sponsored Research Funding: AstraZeneca, Genentech, Amgen, Merck, GSK, Spectrum, Pfizer, Lilly, RevMed, Iovance, CRISPR Therapeutics
Ming-Sound Tsao – Consultancy honoraria: Abbvie, Amgen, AstraZeneca, Bayer, BMS, Daiichi-Sankyo, Lilly, Regeneron, Sanofi, Nucleix. Research grant: Bayer, AstraZeneca, Sanofi
Availability of data and materials: Not applicable.
References
- 1. Gower A, Wang Y, Giaccone G. (2014) Oncogenic drivers, targeted therapies, and acquired resistance in non-small-cell lung cancer. J Mol Med 2014; 92: 697–707. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 2. Zhong L, Li Y, Xiong L, et al. Small molecules in targeted cancer therapy: advances, challenges, and future perspectives. Signal Transd Target Therap 2021; 6: 201. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 3. Amgen. LUMAKRAS® (SOTORASIB) Receives approval in japan for patients with Kras G12c-mutated advanced non-small cell lung cancer, https://www.amgen.com/newsroom/press-releases/2022/01/lumakras-sotorasib-receives-approval-in-japan-for-patients-with-kras-g12cmutated-advanced-nonsmall-cell-lung-cancer (2022, accessed 20 January 2022).
- 4. Ostrem JM, Peters U, Sos ML, et al. K-Ras(G12C) inhibitors allosterically control GTP affinity and effector interactions. Nature 2013; 503: 548–551. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 5. Li B, Skoulidis F, Falchook G, et al. PS01.07 registrational phase 2 trial of sotorasib in KRAS p.G12C mutant NSCLC: first disclosure of the codebreak 100 primary analysis. J Thorac Oncol 2021; 16: S61. [Google Scholar]
- 6. Jänne PA, Rybkin II, Spira AI, et al. (2020). KRYSTAL-1: activity and safety of adagrasib (MRTX849) in advanced/ metastatic non-small-cell lung cancer (NSCLC) harboring KRAS G12C mutation. Eur J Cancer 2020; 138: S1–S2. [Google Scholar]
- 7. FDA. (2021, May 28). FDA grants accelerated approval to sotorasib for KRAS G12C mutated NSCLC. https://www.fda.gov/drugs/resources-information-approved-drugs/fda-grants-accelerated-approval-sotorasib-kras-g12c-mutated-nsclc
- 8. FDA. (2022, December 12). FDA grants accelerated approval to adagrasib for KRAS G12C-mutated NSCLC. https://www.fda.gov/drugs/resources-information-approved-drugs/fda-grants-accelerated-approval-adagrasib-kras-g12c-mutated-nsclc
- 9. Xue JY, Zhao Y, Aronowitz J, et al. Rapid non-uniform adaptation to conformation-specific KRAS(G12C) inhibition. Nature 2020; 577: 421–425. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 10. Misale S, Fatherree JP, Cortez E, et al. KRAS G12C NSCLC models are sensitive to direct targeting of KRAS in combination with PI3K inhibition. Clin Cancer Res 2019; 25: 796. [DOI] [PubMed] [Google Scholar]
- 11. Ryan MB, Fece de la Cruz F, Phat S, et al. Vertical pathway inhibition overcomes adaptive feedback resistance to KRASG12C inhibition. Clin Cancer Res 2020; 26: 1633. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 12. Adachi Y, Ito K, Hayashi Y, et al. Epithelial-to-mesenchymal transition is a cause of both intrinsic and acquired resistance to KRAS G12C inhibitor in KRAS G12C–mutant non–small cell lung cancer. Clin Cancer Res 2020; 26: 5962. [DOI] [PubMed] [Google Scholar]
- 13. Solanki HS, Welsh EA, Fang B, et al. Cell type–specific adaptive signaling responses to KRASG12C inhibition. Clin Cancer Res 2021; 27: 2533. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 14. Santana-Codina N, Chandhoke AS, Yu Q, et al. Defining and targeting adaptations to oncogenic KRASG12C inhibition using quantitative temporal proteomics. Cell Rep 2020; 30: 4584.e4–4599.e4. [DOI] [PubMed] [Google Scholar]
- 15. Koga T, Suda K, Fujino T, et al. KRAS Secondary mutations that confer acquired resistance to KRAS G12C inhibitors, sotorasib and adagrasib, and overcoming strategies: Insights from in vitro experiments. J Thorac Oncol 2021; 16: 1321–1332. [DOI] [PubMed] [Google Scholar]
- 16. Suzuki S, Yonesaka K, Teramura T, et al. KRAS inhibitor-resistance in MET-amplified KRASG12C non-small cell lung cancer induced by RAS- And non-RAS-mediated cell signaling mechanisms. Clin Cancer Res 2021; 27: 5697–5707. [DOI] [PubMed] [Google Scholar]
- 17. Ho CSL, Tüns AI, Schildhaus HU, et al. HER2 mediates clinical resistance to the KRASG12C inhibitor sotorasib, which is overcome by co-targeting SHP2. Eur J Cancer 2021; 159: 16–23. [DOI] [PubMed] [Google Scholar]
- 18. Tanaka N, Lin JJ, Li C, et al. Clinical acquired resistance to KRASG12C inhibition through a novel KRAS switch-II pocket mutation and polyclonal alterations converging on RAS–MAPK reactivation. Cancer Discov 2021; 11: 1913–1922. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 19. Awad MM, Liu S, Rybkin II, et al. Acquired resistance to KRASG12C inhibition in cancer. N Engl J Med 2021; 384: 2382–2393. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 20. Tsai YS, Woodcock MG, Azam SH, et al. Rapid idiosyncratic mechanisms of clinical resistance to KRAS G12C inhibition. J Clin Investig 2022; 132: e155523. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 21. Cox AD, Der CJ. Ras history: the saga continues. Small GTPases 2010; 1: 2–27. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 22. Hancock JF, Parton RG. Ras plasma membrane signalling platforms. Biochem J 2005; 389: 1–11. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 23. Aviel-Ronen S, Blackhall FH, Shepherd FA, et al. K-ras mutations in non-small-cell lung carcinoma: a review. Clin Lung Cancer 2006; 8: 30–38. [DOI] [PubMed] [Google Scholar]
- 24. Marcus K, Mattos C. Direct attack on RAS: intramolecular communication and mutation-specific effects. Clin Cancer Res 2015; 21: 1810–1818. [DOI] [PubMed] [Google Scholar]
- 25. Pantsar T. The current understanding of KRAS protein structure and dynamics. Comput Struct Biotechnol J 2020; 18: 189. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 26. Johnson CW, Reid D, Parker JA, et al. The small GTPases K-Ras, N-Ras, and H-Ras have distinct biochemical properties determined by allosteric effects. J Biol Chem 2017; 292: 12981. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 27. Villalobos P, Wistuba II. Lung Cancer biomarkers. Hematol/Oncol Clin N Am 2017; 31: 13. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 28. Dearden S, Stevens J, Wu YL, et al. Mutation incidence and coincidence in non-small-cell lung cancer: meta-analyses by ethnicity and histology (mutMap). Ann Oncol 2013; 24: 2371. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 29. Dogan S, Shen R, Ang DC, et al. Molecular epidemiology of EGFR and KRAS mutations in 3026 lung adenocarcinomas: higher susceptibility of women to smoking-related KRAS-mutant cancers. Clin Cancer Res: Off J Am Assoc Cancer Res 2012; 18: 6169. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 30. Hunter JC, Manandhar A, Carrasco MA, et al. Biochemical and structural analysis of common cancer-associated KRAS mutations. Mol Cancer Res 2015; 13: 1325–1335. [DOI] [PubMed] [Google Scholar]
- 31. Prior IA, Lewis PD, Mattos C. A comprehensive survey of ras mutations in cancer. Cancer Res 2012; 72: 2457–2467. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 32. Yu HA, Sima CS, Shen R, et al. Prognostic impact of KRAS mutation subtypes in 677 patients with metastatic lung adenocarcinomas. J Thorac Oncol: Off Public Int Assoc Stud Lung Cancer 2015; 10: 431–437. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 33. Addeo A, Banna GL, Friedlaender A. KRAS G12C mutations in NSCLC: from target to resistance. Cancers 2021; 13: 2541. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 34. Moghadamchargari Z, Shirzadeh M, Liu C, et al. Molecular assemblies of the catalytic domain of SOS with KRas and oncogenic mutants. Proc Natl Acad Sci USA 2021; 118: e2022403118. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 35. Ihle NT, Byers LA, Kim ES, et al. Effect of KRAS oncogene substitutions on protein behavior: implications for signaling and clinical outcome. J Natl Cancer Inst 2012; 104: 228. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 36. Ganguly AK, Pramanik BN, Huang EC, et al. Detection and structural characterization of ras oncoprotein-inhibitors complexes by electrospray mass spectrometry. Bioorg Med Chem 1997; 5: 817–820. [DOI] [PubMed] [Google Scholar]
- 37. Patricelli MP, Janes MR, Li LS, et al. Selective inhibition of oncogenic KRAS output with small molecules targeting the inactive state. Cancer Discov 2016; 6: 316–329. [DOI] [PubMed] [Google Scholar]
- 38. Zeng M, Lu J, Li L, et al. Potent and selective covalent quinazoline inhibitors of KRAS G12C. Cell Chem Biol 2017; 24: 1005.e3–1016.e3. [DOI] [PubMed] [Google Scholar]
- 39. Janes MR, Zhang J, Li L-S, et al. Targeting KRAS mutant cancers with a covalent G12C-specific inhibitor. Cell 2018; 172: 578.e17–589.e17. [DOI] [PubMed] [Google Scholar]
- 40. Fedele C, Li S, Teng KW, et al. SHP2 inhibition diminishes KRASG12C cycling and promotes tumor microenvironment remodeling. J Exp Med 2021; 218: e20201414. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 41. Molina-Arcas M, Moore C, Rana S, et al. Development of combination therapies to maximize the impact of KRAS-G12C inhibitors in lung cancer. Sci Transl Med 2019; 11: eaaw7999. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 42. ClinicalTrials.gov. (n.d.). A Study of mRNA-5671/V941 as Monotherapy and in Combination With Pembrolizumab (V941-001). https://clinicaltrials.gov/ct2/show/NCT03948763 (2022, accessed 8 Febraury 2022).
- 43. ClinicalTrials.gov. (n.d.). A Study of ELI-002 in Subjects With KRAS Mutated Pancreatic Ductal Adenocarcinoma (PDAC) and Other Solid Tumors. https://clinicaltrials.gov/ct2/show/NCT04853017 (2022, accessed 8 Febraury 2022).
- 44. Singh S, Murillo G, Singh A, et al. Effect of KRAS and P-STAT3 inhibition by SBT-100 on gemcitabine and pancreatic cancer growth in vivo. J Clin Oncol 2017; 35: e15727. [Google Scholar]
- 45. Pipeline. (n.d.). BridgeBio. https://bridgebio.com/pipeline (2022, accessed 8 February 2022).
- 46. Pipeline Schrödinger. (n.d.). https://www.schrodinger.com/pipeline (2022, accessed 8 February 2022).
- 47. Canon J, Rex K, Saiki AY, et al. The clinical KRAS(G12C) inhibitor AMG 510 drives anti-tumour immunity. Nature 2019; 575: 7781. [DOI] [PubMed] [Google Scholar]
- 48. Fakih MG, Kopetz S, Kuboki Y, et al. Sotorasib for previously treated colorectal cancers with KRAS G12C mutation (CodeBreaK100): a prespecified analysis of a single-arm, phase 2 trial. Lancet Oncol 2022; 23: 115–124. [DOI] [PubMed] [Google Scholar]
- 49. Hong DS, Fakih MG, Strickler JH, et al. KRASG12C inhibition with sotorasib in advanced solid tumors. N Engl J Med 2020; 383: 1207. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 50. Skoulidis F, Li BT, Dy GK, et al. Sotorasib for lung cancers with KRAS p.G12C mutation. N Engl J Med 2021; 384: 2371–2381. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 51. Dy GK, Govindan R, Velcheti V, et al. Abstract CT008: long-term outcomes with sotorasib in pretreated KRASp.G12C-mutated NSCLC: 2-year analysis of CodeBreaK100. Cancer Res 2022; 82: CT008. [Google Scholar]
- 52. Manchado E, Weissmueller S, Morris JP, et al. A combinatorial strategy for treating KRAS mutant lung cancer. Nature 2016; 534: 647. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 53. Kitai H, Ebi H, Tomida S, et al. Epithelial-to-mesenchymal transition defines feedback activation of receptor tyrosine kinase signaling induced by MEK inhibition in KRAS mutant lung cancer. Cancer Discov 2016; 6: 754. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 54. Sun C, Hobor S, Bertotti A, et al. Intrinsic resistance to MEK inhibition in KRAS mutant lung and colon cancer through transcriptional induction of ERBB3. Cell Rep 2014; 7: 86–93. [DOI] [PubMed] [Google Scholar]
- 55. Lee JW, Zhang Y, Eoh KJ, et al. The combination of MEK inhibitor with Immunomodulatory antibodies targeting PD-1 and PD-L1 results in prolonged survival in Kras/p53-driven lung cancer. J Thorac Oncol: Off Public Int Assoc Stud Lung Cancer 2019; 14: 1046. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 56. Engelman JA, Chen L, Tan X, et al. Effective use of PI3K and MEK inhibitors to treat mutant Kras G12D and PIK3CA H1047R murine lung cancers. Nat Med 2008; 14: 1351–1356. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 57. Blair HA. Sotorasib: first approval. Drugs 2021; 81: 1573. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 58. Bauml JM, Li BT, Velcheti V, et al. Clinical validation of guardant360 CDx as a blood-based companion diagnostic for sotorasib. Lung Cancer 2022; 166: 270–278. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 59. Johnson ML, de Langen AJ, Waterhouse DM, et al. LBA10 Sotorasib versus docetaxel for previously treated non-small cell lung cancer with KRAS G12C mutation: CodeBreaK 200 phase III study. Ann Oncol 2022; 33: S1417–S1418. [Google Scholar]
- 60. Falchook G, Li BT, Marrone KA, et al. OA03.03 Sotorasib in combination with RMC-4630, a SHP2 inhibitor, in KRAS p.G12C-mutated NSCLC and other solid tumors. J Thorac Oncol 2022; 17: S8. [Google Scholar]
- 61. Fell JB, Fischer JP, Baer BR, et al. Discovery of tetrahydropyridopyrimidines as irreversible covalent inhibitors of KRAS-G12C with in vivo activity. ACS Med Chem Lett 2018; 9: 1230–1234. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 62. Gabizon R, London N. Hitting KRAS when it’s down. J Med Chem 2020; 63: 6677. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 63. Fell JB, Fischer JP, Baer BR, et al. Identification of the clinical development candidate MRTX849, a covalent KRASG12CInhibitor for the treatment of cancer. J Med Chem 2020; 63: 6679–6693. [DOI] [PubMed] [Google Scholar]
- 64. Jänne PA, Riely GJ, Gadgeel SM, et al. Adagrasib in non–small-cell lung cancer harboring a KRAS G12C mutation. N Engl J Med 2022; 387: 120–131. [DOI] [PubMed] [Google Scholar]
- 65. Mirati Therapeutics, Inc. Mirati announces Adagrasib (KRAZATI™) Receives Breakthrough Therapy Designation from FDA for Patients with Advanced, KRAS-Mutated Colorectal Cancer and NEJM Publishes Phase 1b/2 Data from Adagrasib With or Without Cetuximab in Colorectal Cancer. PRNewswire. https://www.prnewswire.com/news-releases/mirati-announces-adagrasib-krazati-receives-breakthrough-therapy-designation-from-fda-for-patients-with-advanced-kras-mutated-colorectal-cancer-and-nejm-publishes-phase-1b2-data-from-adagrasib-with-or-without-cetuximab-in-col-301708551.html (2022, accessed 25 December 2022).
- 66. Yaeger R, Weiss J, Pelster MS, et al. Adagrasib with or without cetuximab in colorectal cancer with mutated KRAS G12C. N Engl J Med 2022; 388: 44–54. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 67. Sabari JK, Velcheti V, Shimizu K, et al. Activity of Adagrasib (MRTX849) in brain metastases: preclinical models and clinical data from patients with KRASG12C-mutant non-small cell lung cancer preliminary activity of adagrasib in brain metastases. Clin Cancer Res 2022; 28: 3318–3328. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 68. Bekaii-Saab TS, Spira AI, Yaeger R, et al. KRYSTAL-1: Updated activity and safety of adagrasib (MRTX849) in patients (Pts) with unresectable or metastatic pancreatic cancer (PDAC) and other gastrointestinal (GI) tumors harboring a KRASG12C mutation. JCO 2022; 40: 519. [Google Scholar]
- 69. Brachmann SM, Weiss A, Guthy DA, et al. Abstract P124: JDQ443, a covalent irreversible inhibitor of KRAS G12C, exhibits a novel binding mode and demonstrates potent anti-tumor activity and favorable pharmacokinetic properties in preclinical models. Mol Cancer Therap 2021; 20: P124. [Google Scholar]
- 70. Weiss A, Lorthiois E, Barys L, et al. Discovery, preclinical characterization, and early clinical activity of JDQ443, a structurally novel, potent and selective, covalent oral inhibitor of KRASG12CDiscovery and characterization of JDQ443. Cancer Discov 2022; 12: 1500–1517. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 71. Tan DS, Shimizu T, Solomon B, et al. Abstract CT033: KontRASt-01: A phase Ib/II, dose-escalation study of JDQ443 in patients (pts) with advanced, KRAS G12C-mutated solid tumors. Cancer Res 2022; 82: CT033. [Google Scholar]
- 72. Purkey H. Abstract ND11: discovery of GDC-6036, a clinical stage treatment for KRAS G12C-positive cancers. Cancer Res 2022; 82: ND11. [Google Scholar]
- 73. Sacher A, Patel MR, Miller WH, et al. OA03.04 phase I a study to evaluate GDC-6036 monotherapy in patients with non-small cell lung cancer (NSCLC) with KRAS G12C mutation. J Thorac Oncol 2022; 17: S8–S9. [Google Scholar]
- 74. Savarese F, Gollner A, Rudolph D, et al. Abstract 1271: in vitro and in vivo characterization of BI 1823911-a novel KRASG12C selective small molecule inhibitor. Cancer Res 2021; 81: 1271. [Google Scholar]
- 75. Hallin J, Engstrom LD, Hargis L, et al. The KRAS(G12C) inhibitor MRTX849 provides insight toward therapeutic susceptibility of KRAS-mutant cancers in mouse models and patients. Cancer Discov 2020; 10: 54–71. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 76. Shi Z, Weng J, Fan X, et al. Abstract 932: discovery of D-1553, a novel and selective KRas-G12C inhibitor with potent anti-tumor activity in a broad spectrum of tumor cell lines and xenograft models. Cancer Res 2021; 81: 932. [Google Scholar]
- 77. InventisBio Co., Ltd. InventisBio Reported Promising Phase I Study Results of a Novel KRAS G12C Inhibitor D-1553 in Cancer Patients. https://www.prnewswire.com/news-releases/inventisbio-reported-promising-phase-i-study-results-of-a-novel-kras-g12c-inhibitor-d-1553-in-cancer-patients-301522575.html (2022, accessed 11 April 2022).
- 78. Lu S, Jian H, Zhang Y, et al. OA03.07 safety and efficacy of D-1553 in patients with KRAS G12C mutated non-small cell lung cancer: a phase 1 trial. J Thorac Oncol 2022; 17: S11. [Google Scholar]
- 79. Peng S-B, Si C, Zhang Y, et al. Abstract 1259: preclinical characterization of LY3537982, a novel, highly selective and potent KRAS-G12C inhibitor. Cancer Res 2021; 81: 1259. [Google Scholar]
- 80. Sharma P, Hu-Lieskovan S, Wargo JA, et al. Primary, adaptive, and acquired resistance to cancer immunotherapy. Cell 2017; 168: 707–723. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 81. Eisenhauer EA, Therasse P, Bogaerts J, et al. New response evaluation criteria in solid tumours: revised RECIST guideline (version 1.1). Eur J Cancer 2009; 45: 228–247. [DOI] [PubMed] [Google Scholar]
- 82. Aoki K, Yoshida T, Matsumoto N, et al. Suppression of Ki-ras p21 levels leading to growth inhibition of pancreatic cancer cell lines with Ki-ras mutation but not those without Ki-ras mutation - PubMed. Mol Carcinog 1997; 20: 251–258. [PubMed] [Google Scholar]
- 83. Singh A, Greninger P, Rhodes D, et al. A gene expression signature associated with “K-Ras addiction” reveals regulators of EMT and tumor cell survival. Cancer Cell 2009; 15: 489. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 84. Shao DD, Xue W, Krall EB, et al. KRAS and YAP1 converge to regulate EMT and tumor survival. Cell 2014; 158: 171–184. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 85. Kapoor A, Yao W, Ying H, et al. Yap1 activation enables bypass of oncogenic kras addiction in pancreatic cancer. Cell 2014; 158: 185–197. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 86. Loboda A, Nebozhyn M, Klinghoffer R, et al. A gene expression signature of RAS pathway dependence predicts response to PI3K and RAS pathway inhibitors and expands the population of RAS pathway activated tumors. BMC Med Genom 2010; 3: 1–11. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 87. Mueller S, Engleitner T, Maresch R, et al. Evolutionary routes and KRAS dosage define pancreatic cancer phenotypes. Nature 2018; 554: 7690. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 88. Lu T, Li Z, Yang Y, et al. The Hippo/YAP1 pathway interacts with FGFR1 signaling to maintain stemness in lung cancer. Cancer Lett 2018; 423: 36–46. [DOI] [PubMed] [Google Scholar]
- 89. Lou K, Steri V, Ge AY, et al. KRASG12C inhibition produces a driver-limited state revealing collateral dependencies. Sci Signal 2019; 12: eaaw9450. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 90. Gebregiworgis T, Kano Y, St-Germain J, et al. The Q61H mutation decouples KRAS from upstream regulation and renders cancer cells resistant to SHP2 inhibitors. Nat Commun 2021; 12: 1–15. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 91. Bunda S, Burrell K, Heir P, et al. Inhibition of SHP2-mediated dephosphorylation of Ras suppresses oncogenesis. Nat Commun 2015; 6: 8859. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 92. Ruess DA, Heynen GJ, Ciecielski KJ, et al. Mutant KRAS-driven cancers depend on PTPN11/SHP2 phosphatase. Nat Med 2018; 24: 954–960. [DOI] [PubMed] [Google Scholar]
- 93. Kano Y, Gebregiworgis T, Marshall CB, et al. Tyrosyl phosphorylation of KRAS stalls GTPase cycle via alteration of switch I and II conformation. Nat Commun 2019; 10: 1–14. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 94. Mainardi S, Mulero-Sánchez A, Prahallad A, et al. SHP2 is required for growth of KRAS-mutant non-small-cell lung cancer in vivo. Nat Med 2018; 24: 961–967. [DOI] [PubMed] [Google Scholar]
- 95. Ryan MB, Coker O, Sorokin A, et al. KRASG12C-independent feedback activation of wild-type RAS constrains KRASG12C inhibitor efficacy. Cell Rep 2022; 39: 110993. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 96. Suda K, Onozato R, Yatabe Y, et al. EGFR T790M mutation: a double role in lung cancer cell survival? J Thorac Oncol 2009; 4: 1–4. [DOI] [PubMed] [Google Scholar]
- 97. Sasaki T, Koivunen J, Ogino A, et al. A novel ALK secondary mutation and EGFR signaling cause resistance to ALK kinase inhibitors. Cancer Res 2011; 71: 6051–6060. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 98. Ambrogio CA, Köhler J, Zhou Z-W, et al. KRAS dimerization impacts MEK inhibitor sensitivity and oncogenic activity of mutant KRAS the tumor-suppressive function of wild-type KRAS depends on its dimerization capacity with mutant KRAS. Cell 2018; 172: 857.e15–868.e15. [DOI] [PubMed] [Google Scholar]
- 99. Lito P, Solomon M, Li L-S, et al. Allele-specific inhibitors inactivate mutant KRAS G12C by a trapping mechanism. Science 2016; 351: 604–608. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 100. Feng S, Callow MG, Fortin JP, et al. A saturation mutagenesis screen uncovers resistant and sensitizing secondary KRAS mutations to clinical KRASG12C inhibitors. Proc Natl Acad Sci USA 2022; 119: e2120512119. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 101. Zhuang H, Fan J, Li M, et al. Mechanistic insights into the clinical Y96D mutation with acquired resistance to AMG510 in the KRASG12C. Front Oncol 2022; 12: 915512. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 102. Yan YH, Chen SX, Cheng LY, et al. Confirming putative variants at ⩽ 5% allele frequency using allele enrichment and Sanger sequencing. Sci Rep 2021; 11: 1–9. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 103. Mikubo M, Inoue Y, Liu G, et al. Mechanism of drug tolerant persister cancer cells: the landscape and clinical implication for therapy. J Thorac Oncol 2021; 16: 1798–1809. [DOI] [PubMed] [Google Scholar]
- 104. Schmid S, Stewart EL, Martins-Filho SN, et al. Early detection of multiple resistance mechanisms by ctDNA profiling in a patient with EGFR-mutant lung adenocarcinoma treated with osimertinib. Clin Lung Cancer 2020; 21: e488–e492. [DOI] [PubMed] [Google Scholar]
- 105. Marcoux N, Gettinger SN, O’Kane G, et al. EGFR-mutant adenocarcinomas that transform to small-cell lung cancer and other neuroendocrine carcinomas: clinical outcomes. J Clin Oncol 2019; 37: 278. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 106. Bedard PL, Tabernero J, Janku F, et al. A phase Ib dose-escalation study of the oral pan-PI3K inhibitor buparlisib (BKM120) in combination with the oral MEK1/2 inhibitor trametinib (GSK1120212) in patients with selected advanced solid tumors. Clin Cancer Res 2015; 21: 730–738. [DOI] [PubMed] [Google Scholar]
- 107. Grilley-Olson JE, Bedard PL, Fasolo A, et al. A phase Ib dose-escalation study of the MEK inhibitor trametinib in combination with the PI3K/mTOR inhibitor GSK2126458 in patients with advanced solid tumors. Investig N Drugs 2016; 34: 740–749. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 108. Ahmed TA, Adamopoulos C, Karoulia Z, et al. SHP2 drives adaptive resistance to ERK signaling inhibition in molecularly defined subsets of ERK-dependent tumors. Cell Rep 2019; 26: 65.e5–78.e5. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 109. Hofmann MH, Gmachl M, Ramharter J, et al. Bi-3406, a potent and selective sos1–kras interaction inhibitor, is effective in kras-driven cancers through combined mek inhibition. Cancer Discov 2021; 11: 142–157. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 110. Rozakis-Adcock M, van der Geer P, Mbamalu G, et al. MAP kinase phosphorylation of mSos1 promotes dissociation of mSos1-Shc and mSos1-EGF receptor complexes. Oncogene 1995; 11: 1417–1426. [PubMed] [Google Scholar]
- 111. Cheng DK, Oni TE, Thalappillil JS, et al. Oncogenic KRAS engages an RSK1/NF1 pathway to inhibit wild-type RAS signaling in pancreatic cancer. Proc Natl Acad Sci USA 2021; 118: e2016904118. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 112. Briere DM, Li S, Calinisan A, et al. The KRASG12C inhibitor MRTX849 reconditions the tumor immune microenvironment and sensitizes tumors to checkpoint inhibitor therapy. Mol Cancer Therap 2021; 20: 975–985. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 113. Li BT, Falchook GS, Durm GA, et al. OA03.06 CodeBreaK 100/101: First report of safety/efficacy of sotorasib in combination with pembrolizumab or atezolizumab in advanced KRAS p.G12C NSCLC. J Thorac Oncol 2022; 17: S10–S11. [Google Scholar]
- 114. Jänne PA, Smit EF, de Marinis F, et al. LBA4 - preliminary safety and efficacy of adagrasib with pembrolizumab in treatment-naïve patients with advanced non-small cell lung cancer (NSCLC) harboring a KRASG12C mutation. Ann Oncol 2022; 16: 100104. [Google Scholar]
- 115. Kortlever RM, Sodir NM, Wilson CH, et al. Myc cooperates with Ras by programming inflammation and immune suppression. Cell 2017; 171: 1301.e14–1315.e14. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 116. Skoulidis F, Byers LA, Diao L, et al. Co-occurring genomic alterations define major subsets of KRAS - mutant lung adenocarcinoma with distinct biology, immune profiles, and therapeutic vulnerabilities. Cancer Discov 2015; 5: 860. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 117. Skoulidis F, Goldberg ME, Greenawalt DM, et al. STK11/LKB1 mutations and PD-1 inhibitor resistance in KRAS-mutant lung adenocarcinoma. Cancer Discov 2018; 8: 822. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 118. di Federico A, de Giglio A, Parisi C, et al. STK11/LKB1 and KEAP1 mutations in non-small cell lung cancer: prognostic rather than predictive? Eur J Cancer 2021; 157: 108–113. [DOI] [PubMed] [Google Scholar]
- 119. Heist RS, Yu J, Donderici EY, et al. Impact of STK11 mutation on first-line immune checkpoint inhibitor (ICI) outcomes in a real-world KRAS G12C mutant lung adenocarcinoma cohort. J Clin Oncol 2021; 39: 9106. [Google Scholar]
- 120. Dong ZY, Zhong WZ, Zhang XC, et al. Potential predictive value of TP53 and KRAS mutation status for response to PD-1 blockade immunotherapy in lung adenocarcinoma. Clin Cancer Res 2017; 23: 3012–3024. [DOI] [PubMed] [Google Scholar]
- 121. Gandara DR, Paul SM, Kowanetz M, et al. Blood-based tumor mutational burden as a predictor of clinical benefit in non-small-cell lung cancer patients treated with atezolizumab. Nat Med 2018; 24: 1441–1448. [DOI] [PubMed] [Google Scholar]
- 122. Helwick C. KRAS inhibitor adagrasib shows activity in non–small cell lung cancer - the ASCO Post. The ASCO Post. https://ascopost.com/issues/november-25-2020/kras-inhibitor-adagrasib-shows-activity-in-non-small-cell-lung-cancer/, 2020.
- 123. Schulze CJ, Bermingham A, Choy TJ, et al. Abstract PR10: tri-complex inhibitors of the oncogenic, GTP-bound form of KRASG12C overcome RTK-mediated escape mechanisms and drive tumor regressions in vivo. Mol Cancer Therap 2019; 18: PR10. [Google Scholar]
- 124. Nagasaka M, Potugari B, Nguyen A, et al. KRAS inhibitors– yes but what next? direct targeting of KRAS– vaccines, adoptive T cell therapy and beyond. Cancer Treat Rev 2021; 101: 102309. [DOI] [PubMed] [Google Scholar]
- 125. Asimgil H, Ertetik U, Çevik NC, et al. Targeting the undruggable oncogenic KRAS: the dawn of hope. JCI Insight 2022; 7: e153688. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 126. Lim SM, Westover KD, Ficarro SB, et al. Therapeutic targeting of oncogenic K-Ras by a covalent catalytic site inhibitor. Angewandte Chemie 2014; 53: 199. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 127. Stites EC, Shaw AS. Quantitative systems pharmacology analysis of KRAS G12C covalent inhibitors. CPT: Pharmacometr Syst Pharmacol 2018; 7: 342. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 128. Fernandes Neto JM, Nadal E, Bosdriesz E, et al. Multiple low dose therapy as an effective strategy to treat EGFR inhibitor-resistant NSCLC tumours. Nat Commun 2020; 11: 1–9. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 129. Hayashi H, Yonesaka K, Nakamura A, et al. Alternating therapy with osimertinib and afatinib for treatment-naive patients with EGFR-mutated advanced non-small cell lung cancer: a single-group, open-label phase 2 trial (WJOG10818L). Lung Cancer 2022; 168: 38–45. [DOI] [PubMed] [Google Scholar]
- 130. Vasta JD, Peacock DM, Zheng Q, et al. KRAS is vulnerable to reversible switch-II pocket engagement in cells. Nat Chem Biol 2022; 18: 596–604. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 131. Wang X, Allen S, Blake JF, et al. Identification of MRTX1133, a noncovalent, potent, and selective KRAS G12D inhibitor. J Med Chem 2022; 65: 3123–3133. [DOI] [PubMed] [Google Scholar]
- 132. Kulkarni AM, Kumar V, Parate S, et al. Identification of New KRAS G12D inhibitors through computer-aided drug discovery methods. Int J Mol Sci 2022; 23: 1309. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 133. Kemp SB, Cheng N, Markosyan N, et al. Efficacy of a small molecule inhibitor of KrasG12D in immunocompetent models of pancreatic cancer. Cancer Discov 2022; 13: 298–311. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 134. Koltun E, Cregg J, Rice MA, et al. Abstract 1260: first-in-class, orally bioavailable KRASG12V(ON) tri-complex inhibitors, as single agents and in combinations, drive profound anti-tumor activity in preclinical models of KRASG12V mutant cancers. Cancer Res 2021; 81: 1260. [Google Scholar]