

Abstract
The principal component of the protein homeostasis network is the ubiquitin-proteasome system. Ubiquitination is mediated by an enzymatic cascade involving, i.e. E3 ubiquitin ligases, many of which belong to the cullin-RING ligases family. Genetic defects in the ubiquitin-proteasome system components, including cullin-RING ligases, are known causes of neurodevelopmental disorders. Using exome sequencing to diagnose a pediatric patient with developmental delay, pyramidal signs and limb ataxia, we identified a de novo missense variant c.376G>C; p.(Asp126His) in the FEM1C gene encoding a cullin-RING ligase substrate receptor. This variant alters a conserved amino acid located within a highly constrained coding region and is predicted as pathogenic by most in silico tools. In addition, a de novo FEM1C mutation of the same residue p.(Asp126Val) was associated with an undiagnosed developmental disorder, and the relevant variant (FEM1CAsp126Ala) was found to be functionally compromised in vitro. Our computational analysis showed that FEM1CAsp126His hampers protein substrate binding. To further assess its pathogenicity, we used the nematode Caenorhabditis elegans. We found that the FEM-1Asp133His animals (expressing variant homologous to the FEM1C p.(Asp126His)) had normal muscle architecture yet impaired mobility. Mutant worms were sensitive to the acetylcholinesterase inhibitor aldicarb but not levamisole (acetylcholine receptor agonist), showing that their disabled locomotion is caused by synaptic abnormalities and not muscle dysfunction. In conclusion, we provide the first evidence from an animal model suggesting that a mutation in the evolutionarily conserved FEM1C Asp126 position causes a neurodevelopmental disorder in humans.
Introduction
Neurodevelopmental disorders (NDDs) are a group of neurological and psychiatric conditions that arise from disruption of brain development. The defining clinical features are impairments of motor skills, cognition, communication and/or behavior. NDDs encompass a wide range of disabilities, such as developmental delay (DD), intellectual disability (ID), cerebral palsy (CP), autism spectrum disorder (ASD), attention deficit hyperactivity disorder (ADHD), epilepsy and others. The etiology of NDDs is heterogeneous and includes multifactorial, acquired and monogenic causes, the latter often caused by damaging de novo mutations (1,2). Interestingly, whereas the pathogenesis of brain dysfunction in NDDs is still poorly understood, at least 62 forms of NDDs are caused by mutations in genes encoding components of the ubiquitin-proteasome system (UPS), especially E3 ubiquitin ligases (3).
The UPS orchestrates protein degradation in a process known as ubiquitination, where a small protein ubiquitin is covalently attached to its target (4). Ubiquitination is mediated by an enzymatic cascade involving ubiquitin-activating (E1), ubiquitin-conjugating (E2) and ubiquitin ligase (E3) enzymes (5). The proteasome complex recognizes ubiquitinated proteins and, through proteolysis, degrades them into short peptides that can be further processed (6). Most E3s belong to the RING family, and within it, the largest subfamily is the cullin-RING ligases (CRLs) (7). CRL complexes are large molecular machineries consisting of a cullin protein that acts as a scaffold, a Really Interesting New Gene (RING) box protein coordinating ubiquitination, a substrate receptor that recruits the target protein, and adaptor proteins that link the substrate receptor to the cullin (7). FEM1A, FEM1B and FEM1C are evolutionarily conserved substrate recognition subunits of cullin 2-RING E3, which selectively bind their target proteins via a specific sequence motif called a degron (8) (Fig. 1A). Degron is a destabilizing short linear motif that may be located anywhere in the protein sequence, including its N-/C-terminus, which may contain motifs belonging to the N-/C-degron pathways, respectively (9). Recent studies demonstrated that FEM1C receptors recognize various arginine-terminated motifs (Arg/C-degrons) (10,11), directing them to proteasomal degradation. The known FEM1C substrates include SIL1, a nucleotide exchange factor for the BiP endoplasmic reticulum chaperone associated with Marinesco-Sjögren syndrome—an autosomal recessive cerebellar ataxia (12,13).
Figure 1.
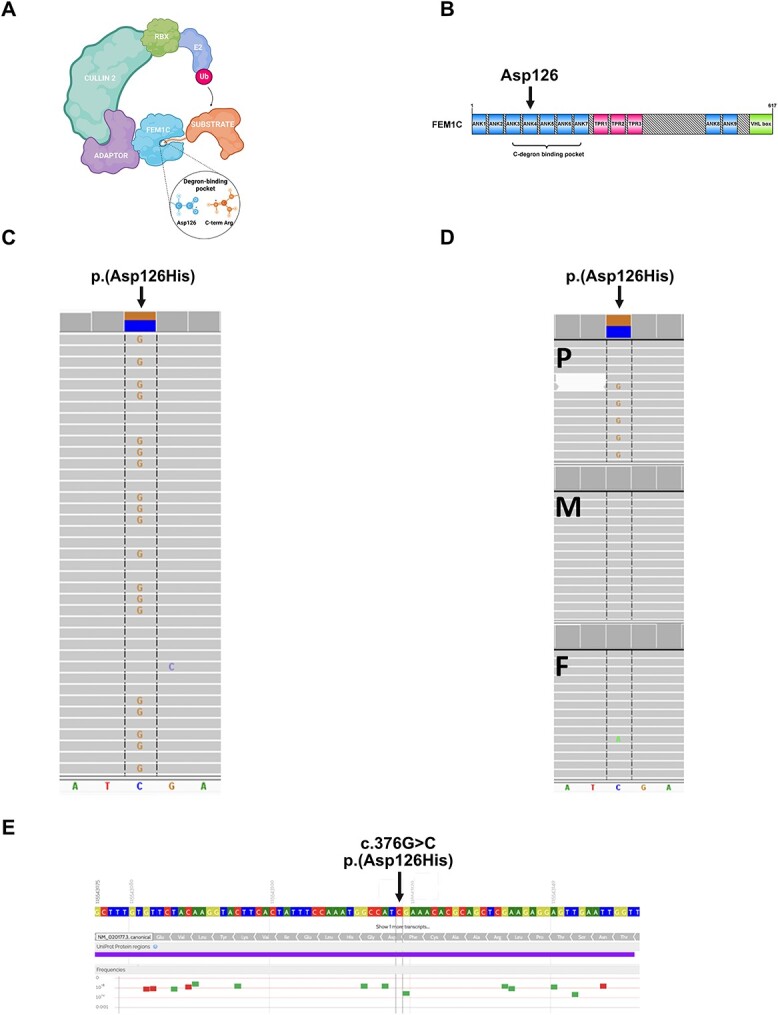
Results of the genetic study. (A) Components of CRL complex on the example of FEM1C. (B) Scheme of the domain architecture of human FEM1C visualized using the IBS software (14). ANK—ankyrin repeat; TPR—tetratricopeptide repeat; VHL box—von Hippel–Lindau box. Modified from (15). (C) Missense p.(Asp126His) variant in FEM1C gene identified in the proband by WES (coverage 119×, reads with alternate allele 57). (D) Amplicon deep sequencing-based family study of p.(Asp126His) (P—proband: coverage 6459×, reads with alternate allele 3097, M—mother: coverage 6171×, reads with alternate allele 0, F—father: coverage 5931, reads with alternate allele 0). (E) Overview of genetic variation in the proximity of p.(Asp126His) in a healthy population (GnomAD and Bravo cohorts, based on https://varsome.com).
We describe a nine-year-old boy with severe DD, lack of speech, pyramidal signs and limb ataxia. Whole exome sequencing (WES) identified a de novo missense variant c.376G>C; p.(Asp126His) in the FEM1C gene. De novo mutation in the same residue, FEM1CAsp126Val, was previously associated with an uncharacterized DD (2) pointing to the importance of this position in FEM1C function in humans. Yet another such variant (FEM1CAsp126Ala) was shown to be functionally relevant by in vitro studies (15,16). Furthermore, a de novo mutation at the corresponding amino acid residue in another member of the FEM1 family, FEM1BArg126Gln (closely related to FEM1C), was associated with syndromic intellectual disability (17). The FEM1C disease-linked variant Asp126His occurs within its degron-binding pocket and impacts a residue directly involved in binding the critical C-terminus arginine of FEM1C substrates (15,16) (Fig. 1A and B).
Here, we show that the pathogenicity of the p.(Asp126His) variant found in the patient is supported by studies of nematode Caenorhabditis elegans with a corresponding mutation in FEM-1 (a worm homolog of human FEM1C). Our results provide the first evidence of an NDD associated with a novel FEM1C mutation that has functional consequences for the nervous system in the C. elegans model.
Results
Exome sequencing and family studies
WES performed in the proband revealed three ultra-rare (0 frequency in gnomAD v3.1.2 https://gnomad.broadinstitute.org and an inhouse database of >5500 Polish exomes) heterozygous missense variants in CSMD2, RALYL and FEM1C genes, which were selected for family studies. Variants in both CSMD2 and RALYL were found to be inherited from a healthy mother and father and thus were considered as not disease-causing. The FEM1C variant (hg38, chr5:115543118-C>G, NM_020177.3:c.376G>C, p.(Asp126His); Fig. 1C) was absent in both parents, indicating a de novo event (Fig. 1D). The FEM1C p.(Asp126His) was predicted as damaging by 13 different pathogenicity predictors implemented in the Varsome web server (https://varsome.com), including BayesDel addAF, BayesDel noAF, EIGEN, EIGEN PC, FATHMM-MKL, FATHMM-XF, LRT, M-CAP, MutPred, MutationTaster, PROVEAN, PrimateAI, SIFT and as ‘Tolerated’ by six other predictors (DEOGEN2, FATHMM, LIST-S2, MVP, Mutation assessor, SIFT4G). The c.376G position is highly conserved (PhastCons100way score 1.0, GERP++ value 5.5), and the p.(Asp126His) variant affects the conserved Asp126 residue located in the ankyrin repeat domain ANK4 (Fig. 1B) (phyloP100way = 7.9). Moreover, FEM1C p.(Asp126His) variant is located in the highly constrained coding region (CCR = 98.6). The scarcity of coding variants in the proximity of p.(Asp126His), characteristic of CCR, is illustrated in Fig. 1E, based on the gnomAD and Bravo datasets.
FEM1CAsp126His disrupts degron binding
Recent structural studies on FEM1C revealed a crucial role for Asp126 in degron recognition and its direct interactions with arginine residue of the Arg/C-degron substrates. Isothermal titration calorimetric performed by Yan and colleagues showed that Asp126Ala mutation in FEM1C lowers its binding affinity towards Arg/C-degron substrates ~2-fold (16). In addition, the global protein stability (GPS) assay conducted by Chen and colleagues revealed that FEM1C double mutant Asp77Ala/Asp126Ala failed to induce the degradation of three different Arg/C-degron substrates (15), further indicating the importance of Asp126 in FEM1C functioning. Therefore, we hypothesized that Asp126His mutation carried by the patient might also abrogate the degron binding by its repulsive interactions with C-terminal arginine and thus underlie the disease by impairing ubiquitination and turnover of FEM1C substrates. To gain insights into the binding modes of FEM1C and its mutated variants—Asp126His, Asp126Ala and Asp126Val (as in another patient with DD), we performed the peptide-protein docking using a novel protocol utilizing the AlphaFold2 modeling (18). To this end, we docked the SIL1 C-terminus peptide to FEM1C and its single-point mutants, and since the experimental structure of FEM1C bound to the Arg/C-degron of its native substrate SIL1 was resolved (PDB ID: 6LBN) (15), we checked whether the obtained AlphaFold2 models managed to recapitulate the interactions present in the experimentally resolved complex. We obtained such similar complexes only for the wild-type FEM1C, where 14/17 (model 1) or 13/17 (model 4′) of native interactions were recapitulated (Fig. 2A), including the interaction of FEM1C Asp126 with the C-terminal arginine of SIL1 (Fig. 2B and C). Remarkably, none of the mutants exhibited the peptide docked to its degron-binding pocket (Fig. 2A, Supplementary Material, Table S1), although the root-mean-square deviation (RMSD), a widely used metric to assess the similarity between structures, indicated that the degron-binding region of all AlphaFold2 FEM1C models was predicted as similarly folded to the experimental structure (RMSD in ranges of 0.322–0.431 Å) (Supplementary Material, Table S1). These results confirm the critical role of FEM1C Asp126 residue in the Arg/C-degron recognition and implicate the pathogenic character of both Asp126His and Asp126Val mutations which likely induce severe changes in the environment of the FEM1C degron-binding pocket.
Figure 2.
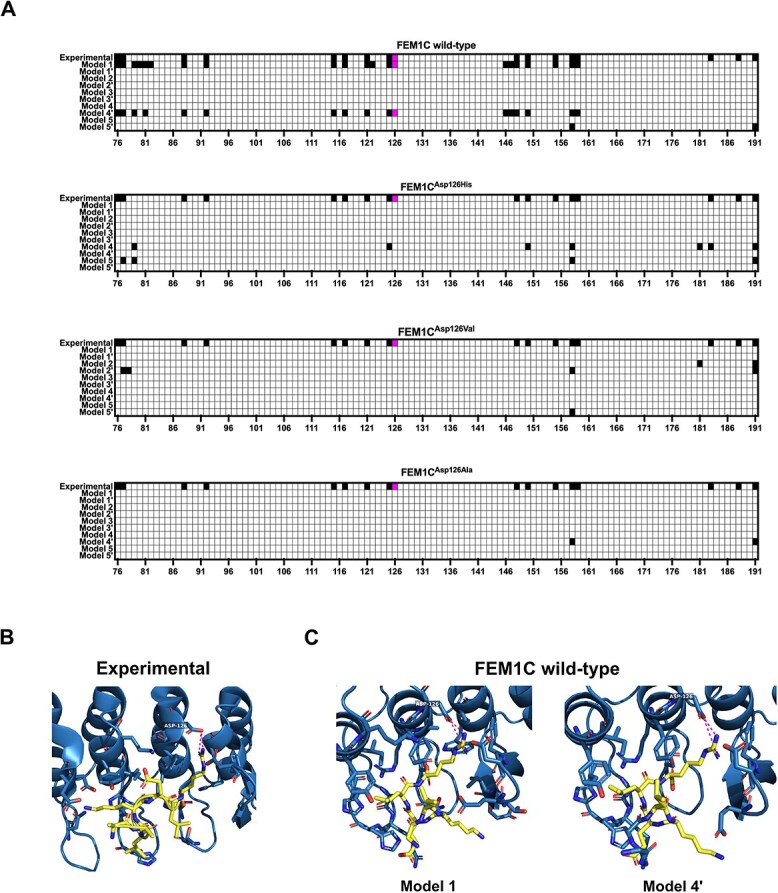
Arg/C-degron from SIL1 docked to human FEM1C and its variants. (A) Contact heatmaps between FEM1C and its variants and the LKELR Arg/C-degron of SIL1 obtained from the AlphaFold2 protein-peptide docking; experimental—interactions present in the resolved structure of the complex (PDB ID: 6LBN); interaction between Asp126 and the C-terminal arginine of SIL1 is colored in magenta. Contacts were defined as not exceeding the 4 Å distance between any heavy atoms. Data visualized in the GraphPad Prism 9 software. (B) Visualization of interactions between Arg/C-degron of SIL1 with FEM1C from the experimentally resolved complex; blue—FEM1C, yellow—LKELR Arg/C-degron of SIL1, magenta—hydrogen bonds between FEM1C Asp126 and C-terminal arginine of SIL1. Visualized in the PyMOL software (19). (C) Visualization of interactions between Arg/C-degron of SIL1 with FEM1C from two AlphaFold2 models (of wild-type FEM1C) where the interaction of the C-terminal arginine of SIL1 with Asp126 of FEM1C was recapitulated; blue—FEM1C, yellow—LKELR Arg/C-degron of SIL1, magenta—hydrogen bonds between FEM1C Asp126 and C-terminal arginine of SIL1. Visualized in the PyMOL software (19).
The nematode C. elegans as a model to study the functional consequences of the FEM1C p.(Asp126His) mutation
The degron-binding pocket of FEM1C is located within its N-terminal ankyrin domain (15,16) (Fig. 1B), a well-conserved motif in all FEM1 proteins and their orthologs in other organisms, including C. elegans nematode. C. elegans is a well-established model organism to study multiple human diseases, including neurodegeneration and ataxias (20,21). Therefore, we used worms to investigate the physiological impact of the FEM1C p.(Asp126His) mutation. The sequence alignment of degron-binding regions of FEM1C and its C. elegans ortholog FEM-1 showed high evolutionary conservation, including the critical Asp126 position (Asp133 in worms) (Fig. 3A, left). We also compared the structures of both proteins, as protein folding directly underlies molecular interactions and may point to the conserved functional role. Superposition of the experimental structure of the FEM1C degron-binding region (residues 1–390) and model of its FEM-1 analog obtained from the AlphaFold database (22,23) showed very high structural similarity, evidenced in particular by a low RMSD value of 1.213 Å.
Figure 3.

Computational and functional analysis of the C. elegans fem-1 mutant. (A) Left: Global alignment of binding regions of human FEM1C (residues 70–220; UniProt ID: Q96JP0) and C. elegans FEM-1 (residues 76–227; UniProt ID: P17221). Sequences were aligned using the EMBOSS Needle web server (24) with default parameters and visualized in the Jalview Desktop software (25) with residues colored by their physicochemical properties; the arrow indicates Asp126/Asp133 position; Right: Comparison of residues involved in SIL1 Arg/C-degron binding in FEM1C (cyan; PDB code: 6LBN) and FEM-1 (magenta; AlphaFold2 model corresponding to P17221). FEM-1 residues that differ from FEM1C are marked in yellow; the arrow indicates the Asp126/Asp133 position. (B) Track lengths, turn counts, a number of reversals, and omega bends in wild-type (n = 146) and FEM-1Asp133His (n = 148) worms; N = 3. n represents the total number of worms; N represents the number of experimental repeats. The stars denote the significance levels per the P-value obtained by the Mann–Whitney test. P-values are shown adjacent to the respective graphs (ns—not significant, *P ≤ 0.05, ***P ≤ 0.001, ****P ≤ 0.0001), dashes represent median of collective data from three independent biological replicates. Data was plotted and analyzed in the GraphPad Prism 9 software. (C) Phalloidin staining of F-actin-containing I-bands of wild-type and FEM-1Asp133His worms. Dashed lines separate two body wall muscle cells. The scale bar is 50 μm. (D) Aldicarb sensitivity assay of wild-type (n = 207) and FEM-1Asp133His (n = 215) worms; N = 3. n represents the total number of worms; N represents the number of experimental repeats. P-value was calculated using the Log-rank (Mantel-Cox) test and is shown adjacent to the graph (***P ≤ 0.001). Data was plotted and analyzed in the GraphPad Prism 9 software. (E) Aldicarb sensitivity assay in neuronal-specific RNAi of fem-1, [fem-1 RNAi (n = 183)] and vector control [control RNAi (n = 177)]; N = 3 in the neurons. n represents the total number of worms; N represents the number of experimental repeats. P-value was calculated using the Log-rank (Mantel-Cox) test and is shown adjacent to the graph (**P ≤ 0.01). Data was plotted and analyzed in the GraphPad Prism 9 software.
Recent structural studies of the FEM1C basis of degron recognition pointed to the critical role of several residues (15,16). Therefore, we compared their evolutionary conservation to assess the similarity of the degron-binding pocket between humans and C. elegans. Among 15 residues of FEM1C indicated as involved in Arg/C-degron binding of its substrate SIL1 (15), 13 were conserved in sequence and structure in FEM-1 (Fig. 3A, right). Notably, the two differing residues showed similar physicochemical properties (phenylalanine/tyrosine and lysine/arginine), further supporting the high conservation of the degron binding pocket and indicating a likely similar substrate pool of FEM1C and FEM-1. Thus, the high similarity of the degron-binding pocket between human FEM1C and its worm ortholog, as well as the conservation of Asp126 residue, are strong premises to employ C. elegans as an FEM1C p.(Asp126His) disease model.
C. elegans FEM-1Asp133His mutants develop locomotor defects due to neuronal dysfunction
Ataxias are a heterogeneous group of disorders in which cerebellar dysfunction underlies neurological symptoms such as impaired muscle coordination (26). As pyramidal signs and limb ataxia were prominent symptoms in our proband, we examined the locomotion behavior of worms expressing the putative disease-causing FEM-1Asp133His mutant protein (hereafter fem-1 mutants) generated using the CRISPR/Cas9 gene-editing tool (Supplementary Material, Fig. S1A). Locomotion is a complex reflection of the functionality of the C. elegans nervous system. It encompasses a range of motor activities, such as omega bends (deep bends usually on the ventral side of the body that change the movement direction) and reversals (transition from crawling forward to crawling backward). We used the WormLab system (MBF Bioscience) to measure animal locomotor parameters in wild-type and fem-1 worms in detail. We found that the fem-1 mutants had impaired mobility as assessed by track length and turn counts (Fig. 3B). In particular, the fem-1 mutants experienced difficulty performing omega bends but showed no defects in reversals (Fig. 3B). This suggests a functional deficit of distinct neurons in fem-1 mutants, as the execution of reversals and omega bends is encoded by separate neurons (27).
Moreover, FEM-1 is essential for sperm production, and its deficiency leads to the sterility of C. elegans hermaphrodites (28). However, expression of FEM-1Asp133His did not affect fertilization, as the number of unhatched eggs (usually accounting for a few percent of all eggs) laid by the mother within 16 h was similar to that in the wild-type strain, indicating that fertilization and embryo viability remained unchanged (Supplementary Material, Fig. S1B). However, fem-1 mutants laid a lower number of eggs over 16 h compared to wild-type worms (Supplementary Material, Fig. S1C), which may suggest defects related to the development of vulva muscles or the neuronal circuit (six cholinergic and two serotonergic neurons) controlling their contraction, and thus, egg expulsion (29).
The organization of sarcomeres and their components in C. elegans body wall muscles is a highly evolutionary conserved feature (30). To determine whether locomotion and egg-laying defects in fem-1 mutants occur due to the loss of muscle fiber structure, we performed phalloidin staining of F-actin filaments containing I bands (31). We did not find myofilament disorganization or reduced muscle cell size in fem-1 mutants (Fig. 3C), suggesting that the observed deficits in locomotion are not the result of compromised body wall muscle organization.
To confirm that the mobility phenotype observed in the fem-1 mutants is due to synaptic defects, we examined the rate of worm paralysis in response to aldicarb and levamisole. Acetylcholine (Ach) is necessary for the transmission of an action potential between neurons, and it exerts its action by binding to nicotinic (nAchR) and muscarinic Ach receptors (32,33). Synaptic acetylcholinesterase (AchE) manages neuromuscular transmission by catabolizing Ach. Aldicarb inhibits AchE and induces spastic muscle paralysis due to the accumulation of Ach in the synaptic gap (34). Similar paralysis is also induced by levamisole, an nAchR agonist (35,36) (Supplementary Material, Fig. S1D). However, in the aldicarb assay, a higher percentage of paralyzed worms at a given time indicates a higher general Ach neurotransmission, whereas the levamisole test selectively evaluates nAchR activity (33,34,37). We found that the fem-1 mutants were significantly (P < 0.001) more sensitive to aldicarb but not to levamisole (P = 0.34) compared to wild-type worms (Fig. 3D and Supplementary Material, Fig. S1E). In addition, neuronal-specific ribonucleic acid (RNA) interference (RNAi) depletion of FEM-1 also sensitized animals to aldicarb treatment (P < 0.01, Fig. 3E) suggesting that FEM-1Asp133His acts through the decreasing function of the protein. Our data suggest that inefficient turnover of neuronal substrates associated with the mutation in FEM-1 may lead to increased Ach release, e.g. via impaired calcium sensing, overactivity of the machinery responsible for synaptic vesicle release, or excessive amounts of neuromodulatory peptides (36–39), resulting in aldicarb sensitivity, and motor and egg-laying defects.
Discussion
We describe a pediatric case of severe DD, lack of speech, pyramidal signs and limb ataxia associated with a novel de novo variant p.(Asp126His) in the FEM1C gene. The variant altered a conserved amino acid position and was predicted to be pathogenic by most in silico tools. Moreover, the variant is located within a highly constrained coding region, i.e. a region with a scarcity of coding variation among healthy people likely to be enriched in pathogenic variants for autosomal dominant disorders (40). Importantly, another de novo variant (Asp126Val) in the same residue of FEM1C was associated with a neurodevelopmental disorder (2,41). Moreover, a de novo variant (Arg > Gln) at homologous substrate-binding residue 126 in another member of the FEM-1 family, FEM1B, was found in a patient with a similar broad phenotype, including global development delay (2,17), pinpointing the significance of the FEM1 family in the nervous system development.
The FEM1C disease-linked mutation Asp126His occurs within its degron-binding pocket and impacts a residue directly interacting with FEM1C substrates. Structural studies revealed specific FEM1C preferences for binding proteins with arginine (R) or lysine (K) at the preantepenultimate or antepenultimate positions, which indicates that the degron motifs preferred by FEM1C are C-terminal R/K-X-X-R or R/K-X-R sequences (X stands for any amino acid) (15,16). Interestingly, multiple somatic mutations in FEM1C were reported to occur in different cancers, and some affect residues responsible for substrate binding: Asp77Tyr (identified in colorectal cancer), Asp77Gly (identified in uterine corpus endometrioid carcinoma) and Glu191Gln (identified in bladder urothelial carcinoma) (42). Yan and colleagues showed that these variants lower FEM1C binding affinity toward its Arg/C-degron substrate by 1.6–2.4-fold (16), further implying the possible physiological consequence of FEM1C functioning due to its improper substrate recognition.
A misregulation of SIL1, which is linked to an autosomal recessive cerebellar ataxia and DD (12,13), might be a possible cause of the diseases associated with FEM1CAsp126His or FEM1CAsp126Val mutations. This is supported by experimental evidence showing that FEM1C recognizes a peptide at the C-terminus of SIL1 and that the FEM1CAsp126Ala mutation significantly reduced this interaction (15,16). However, the aforementioned C-terminal R/K-X-X-R or R/K-X-R motifs preferred by FEM1C occur in 292 human proteins, making them all its putative substrates (Supplementary Material, Table S2). Furthermore, a small amino acid such as glycine (G) or alanine (A) consecutive to the last arginine in the FEM1C substrate may also be tolerated allowing weaker or transient interactions (15,16). This could extend the pool of putative FEM1C substrates, as there are 14 and 19 proteins in the human proteome yielding R/K-X-X-R-G or R/K-X-R-G and R/K-X-X-R-A or R/K-X-R-A C-terminal motifs, respectively. Furthermore, it has been shown that cleavage by proteolytic enzymes produces new protein ends that can contain destabilizing residues and act as degron motifs (43), which implies that under certain circumstances, the number of potential FEM1C substrates could be much higher. Therefore, it would be crucial to perform proteomic studies on the patient’s material to look for proteins with elevated levels and containing relevant degron motifs as likely substrates of FEM1C, whose dysregulation may be responsible for the disease. To search for proteins stabilized by the FEM1CAsp126His mutation, a fluorescent protein timer-based technique, such as GPS (44), might also be used. Briefly, a GPS library expressing each human open reading frame could be introduced into HEK293T cells expressing FEM1CAsp126His (produced using the CRISPR/Cas9 method) to identify proteins whose turnover is impacted by the FEM1C mutation. Further efforts could involve determining if their stability results from defective ubiquitination.
Pathogenicity of the p.(Asp126His) variant found in our patient is supported by studies of transgenic C. elegans, a well-established model organism to study multiple human diseases, including neurodegeneration and ataxias (20,21). We found that the patient’s allele reduced animal mobility by affecting omega bends but not reversals. Since the patient’s allele did not affect muscle organization, the reduced mobility was likely of neuronal origin. As the execution of reversals and omega bends is encoded by separate neurons in the worm, the observed functional deficit is likely discreet (27). Consequently, we analyzed the functionality of the nervous system in fem-1 worms. Using the acetylcholinesterase inhibitor aldicarb, we observed greater sensitivity of mutant animals, as evidenced by a faster rate of muscle paralysis. This finding suggests that locomotor defects induced by a mutation in the fem-1 gene occur from neuronal synaptic deficits and excessive ACh release in the synaptic cleft. Interestingly, levamisole did not have a similar effect, suggesting that excessive ACh does not exert its effect through nicotinic Ach receptors in mutant worms. ACh also has a complex role in controlling C. elegans egg-laying behavior (45). In this regard, fem-1 mutants exhibited reduced egg laying, which may be attributed to cholinergic neuronal abnormalities (neurons capable of producing ACh). The phenotypes we have identified suggest perturbations in the degradation of neuron-specific proteins in fem-1 worms. Proteomic tools based on neuron-selective bio-orthogonal non-canonical amino acid labeling or proximity labeling (46,47) could be used to identify proteins whose levels increase specifically in FEM-1Asp133His expressing neurons.
As neuronal-specific RNAi depletion of FEM-1 also sensitized animals to aldicarb treatment, the FEM-1Asp133His likely acts through decreasing protein function. However, since the complete loss of function (LOF) of FEM-1 causes sterility of C. elegans (28), a feature not observed in our FEM-1Asp133His mutant worms, the decrease due to the variant is likely to be partial. Given that human FEM1C does not appear to be a haploinsufficient gene (3 LOF variants in GnomAD, pLI = 0.63, https://gnomad.broadinstitute.org), we speculate that in the heterozygous patient p.(Asp126His) decreases the protein function by >50%, perhaps acting as a dominant negative hypomorphic allele. Interestingly, a gain of function mechanism was suggested to underlie the disease association of FEM1BArg126Gln (48).
Our results pointing to the functional relevance of FEM1C Asp126His are entirely consistent with previously reported biochemical studies showing that another variant at the same residue—Asp126Ala—abolished or severely abrogated substrate binding by FEM1C (15,16).
In conclusion, we have identified an NDD associated with a novel FEM1C mutation that exerts functional consequences on the nervous system in a C. elegans model. Since optimal modulation of synaptic activity is balanced by the differential activity of multiple pathways, further targeted genetic and pharmacological interventions using our fem-1 worms are needed to explore detailed neuronal mechanisms affected by the FEM1C/FEM-1 variant.
Materials and Methods
The proband family commercially ordered genetic testing for diagnostic purposes. The patient’s legal guardians have written consent to publish genetic and clinical data.
Clinical report
The patient is the first child of non-consanguineous parents. The family history was unremarkable. The pregnancy was uncomplicated, with normal screening ultrasounds. He was delivered at term via C-section, and all mensuration at birth was normal. Developmental concerns became apparent to the family at around six months of age due to gross motor delay. Neurologic evaluation at nine months of age showed axial hypotonia and increased muscle tone in the lower limbs with brisk tendon reflexes. Magnetic Resonance Imaging (MRI) of the brain with spectroscopy performed at the age of 11 months showed symmetrical enlargement of the posterior horns of the lateral ventricles. The boy could not sit independently until 2.5 years of age and crawl until three years. Brain MRI at the age of four showed mildly enlarged lateral ventricles comparable to the previous examination and small areas of incomplete myelination at the posterior parts of the lateral ventricles. A recent neurological examination at the age of nine years was notable for global developmental delay, lack of speech, ataxia, and pyramidal signs. Anthropometric measurements were normal: weight 26 kg (25 cc), height 134 cm (25 cc), head circumference 54 cm (50-75 cc), BMI—14,5. Normal male sexual organs were observed, with testicles in the scrotum and a normal penis. Head MRI performed at nine years was normal. The patient was cheerful, reacted to simple commands, and communicated using assistive devices. He was not able to walk independently. He stood with support and crossed his legs when held by the caregiver. There was massive nystagmus in all directions of gaze, left divergent strabismus, cerebellar signs in the upper limbs (ataxia, intention tremor, dysmetria, dysdiadochokinesia), axial hypotonia, pyramidal signs (spasticity with brisk deep tendon reflexes in the upper and lower limbs), and bilateral pes planovalgus. Cognitive assessment performed at the age of nine using Stanford-Binet 5 test showed moderate intellectual disability. Due to the lack of verbal speech in the boy, the study only included the development of cognitive skills in the non-verbal areas. Results of metabolic evaluation, including urine organic acid analysis, tandem mass spectrometry screening, and neurotransmitters in cerebrospinal fluid, were normal. Chromosomal microarray did not show any disease-causing copy number variants.
Genetic study
Whole exome sequencing (WES) was conducted for the proband only using DNA purified from the whole blood. According to the manufacturer’s instructions, the library was constructed using SureSelect All Human Exon V7 (Agilent Technologies, Cedar Creek, TX, USA). The library was paired-end sequenced (2 × 100 bp) on HiSeq 1500 (Illumina, San Diego, CA, USA) to the mean depth >100× (the min. 10× coverage was 96.7%, and 20×—91.6%). Reads were aligned to the GRCh38 (hg38) reference genome. Data analysis and variants prioritization was performed as previously described (49). Variants considered causative were validated in the proband and studied in his parents by deep amplicon sequencing performed using Nextera XT Kit (Illumina) and paired-end sequencing as described above for WES.
Peptide-protein docking
Peptide-protein docking was performed as described in (18). Briefly, the sequence of the first 390 residues of FEM1C (corresponding to the experimentally resolved region of FEM1C; PDB ID: 6LBN) or its single-point mutants (Asp126His, Asp126Val, Asp126Ala), followed by a 30 glycine linker and the LKELR Arg/C-degron sequence (corresponding to the C-terminus of SIL1, also present in the 6LBN structure) served as an input for the local version of AlphaFold2 run for the monomer model. Ten models were generated for each variant (in two separate runs); multiple sequence alignment was constructed based on all genetic databases; max_template_date was set to July 28, 2022. Root-mean-square deviation (RMSD) was calculated for C-alpha atoms between the degron-binding pocket of the predicted models and the experimental structure (residues 76–191) using the PyMOL software (19).
Worm maintenance and strains
Worms were maintained on nematode growth medium (NGM) plates seeded with OP50 E. coli bacteria at 20°C unless otherwise stated (50). Wild-type nematodes were the N2 (Bristol) strain. PHX5163 fem-1(syb5163) mutant worms were generated at SunyBiotech (https://www.sunybiotech.com) by CRISPR/Cas9 editing using the sgRNAs sg1-CCA TTA AGA GGT GCA TGT TAC GA and sg2-TTA AGA GGT GCA TGT TAC GAT G. The editing was confirmed by sequencing (Supplementary Material, Fig. S1A). Strain PHX5163 was outcrossed 2× to N2 to generate strain WOP497. Neuron-specific silencing was carried out using strain TU3311 [uls60 (unc-119p::YFP + unc-119p::sid-1)] (51).
RNA interference
RNA interference (RNAi) in C. elegans was performed using the standard RNAi feeding method and RNAi clone from the Ahringer C. elegans RNAi feeding library (52). For experiments, we used NGM plates supplemented with 1 mm IPTG and 25 μg/μl carbenicillin seeded with HT115 E. coli bacteria expressing double-stranded RNA from L4440 plasmid against the fem-1 gene. N2 and TU3311 worms were placed on freshly prepared RNAi plates as age-synchronized L1 larvae. Worms fed with E. coli HT115(DE3) containing empty L4440 plasmid were used as control.
Worm mobility assay
Day 1 adult worms were placed onto a single NGM plate, and worm movements were recorded for 2 min using the WormLab system (MBF Bioscience), keeping the frame rate, exposure time, and gain set to 7.5 frames/s. Parameters such as the track length, turn count, number of reversals, and reversal distance travelled pattern of individual worms were recorded and analyzed using the WormLab software (MBF Bioscience). The assay consisted of three independent biological replicates. 75 worms were recorded for one biological replicate.
Phalloidin staining
Sarcomere assembly was monitored by staining F-actin with Phalloidin-Atto 390 (Sigma Aldrich) using the protocol described in (53). Briefly, synchronized day 1 adults were washed from NGM plates seeded with E. coli OP50, and the worm pellet was snap-frozen in liquid nitrogen. The pellet was dried in the SpeecVac concentrator, followed by worm permeabilization using 100% ice-cold acetone for 5 min. After acetone removal, the worm pellet was stained using Phalloidin-Atto 390 for 30 min in the dark. Stained worms were washed twice, mounted onto a 2% (w/v) agarose pad on a glass slide, and imaged on Zeiss LSM800 inverted confocal microscope.
Aldicarb and levamisole sensitivity assay
Aldicarb and levamisole sensitivity assay was performed as described in (20,54). Briefly, 20 adult day 1 worms (also these subjected to RNAi) were placed on NGM plates supplemented with 0.5 mm aldicarb or 0.5 mm levamisole, and worm paralysis was assessed by their inability to move within 5 s in response to touch in the head and tail region. Paralysis was assessed every 20 min until all worms were utterly paralyzed. The assay consisted of three independent biological replicates.
Egg-laying assay
Synchronized N2 and fem-1 mutant worms were grown till day 1 adulthood. The egg-laying assay was carried out by transferring a single day 1 adult animal on NGM plate seeded with E. coli OP50 and kept at 20°C for 16 h. After 16 h, the worm was removed from the plate, and the eggs were counted. At least 25 animals were scored in each independent set, and the experiment was repeated three times as independent biological replicates.
Data availability
Data obtained during this study are available at doi.org/10.18150/9TQUTI
Supplementary Material
{kind=link}
Acknowledgements
We thank Dr. Filip Stefaniak for his advice on molecular docking and sharing computational resources. We acknowledge the Caenorhabditis Genetics Centre (funded by the NIH National Centre for Research Resources, P40 OD010440) for the N2 and TU3311 strains and OP50 E. coli.
Contributor Information
Abhishek Anil Dubey, Laboratory of Protein Metabolism, International Institute of Molecular and Cell Biology in Warsaw, 02-109 Warsaw, Poland.
Magdalena Krygier, Department of Developmental Neurology, Medical University of Gdańsk, 80-952 Gdańsk, Poland.
Natalia A Szulc, Laboratory of Protein Metabolism, International Institute of Molecular and Cell Biology in Warsaw, 02-109 Warsaw, Poland.
Karolina Rutkowska, Department of Medical Genetics, Medical University of Warsaw, 02-106 Warsaw, Poland.
Joanna Kosińska, Department of Medical Genetics, Medical University of Warsaw, 02-106 Warsaw, Poland.
Agnieszka Pollak, Department of Medical Genetics, Medical University of Warsaw, 02-106 Warsaw, Poland.
Małgorzata Rydzanicz, Department of Medical Genetics, Medical University of Warsaw, 02-106 Warsaw, Poland.
Tomasz Kmieć, Department of Neurology and Epileptology, The Children's Memorial Health Institute, 04-730 Warsaw, Poland.
Maria Mazurkiewicz-Bełdzińska, Department of Developmental Neurology, Medical University of Gdańsk, 80-952 Gdańsk, Poland.
Wojciech Pokrzywa, Laboratory of Protein Metabolism, International Institute of Molecular and Cell Biology in Warsaw, 02-109 Warsaw, Poland.
Rafał Płoski, Department of Medical Genetics, Medical University of Warsaw, 02-106 Warsaw, Poland.
Conflict of Interest statement
The authors have declared no competing interests.
Funding
The Norwegian Financial Mechanism 2014–2021 and operated by the Polish National Science Centre, Poland (under the project contract number 2019/34/H/NZ3/00691 to A.A.D., N.A.S., and W.P.); the National Science Centre, Poland (grant PRELUDIUM number 2021/41/N/NZ1/03473 to N.A.S.).
References
- 1. Thapar, A., Cooper, M. and Rutter, M. (2017) Neurodevelopmental disorders. Lancet Psychiat, 4(4), 339–346. [DOI] [PubMed] [Google Scholar]
- 2. Deciphering Developmental Disorders Study (2017) Prevalence and architecture of de novo mutations in developmental disorders. Nature, 542(7642), 433–438. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 3. Ebstein, F., Kury, S., Papendorf, J.J. and Kruger, E. (2021) Neurodevelopmental Disorders (NDD) caused by genomic alterations of the Ubiquitin-Proteasome System (UPS): the possible contribution of immune dysregulation to disease pathogenesis. Front. Mol. Neurosci., 14, 733012. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 4. Kerscher, O., Felberbaum, R. and Hochstrasser, M. (2006) Modification of proteins by ubiquitin and ubiquitin-like proteins. Annu. Rev. Cell Dev. Biol., 22(1), 159–180. [DOI] [PubMed] [Google Scholar]
- 5. Komander, D. (2009) The emerging complexity of protein ubiquitination. Biochem. Soc. Trans., 37(5), 937–953. [DOI] [PubMed] [Google Scholar]
- 6. Hochstrasser, M. (1996) Ubiquitin-dependent protein degradation. Annu. Rev. Genet., 30(1), 405–439. [DOI] [PubMed] [Google Scholar]
- 7. Nguyen, H.C., Wang, W. and Xiong, Y. (2017) Cullin-RING E3 ubiquitin ligases: bridges to destruction. Subcell. Biochem., 83, 323–347. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 8. Dankert, J.F., Pagan, J.K., Starostina, N.G., Kipreos, E.T. and Pagano, M. (2017) FEM1 proteins are ancient regulators of SLBP degradation. Cell Cycle, 16, 556–564. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 9. Varshavsky, A. (2019) N-degron and C-degron pathways of protein degradation. Proc. Natl. Acad. Sci. U. S. A., 116, 358–366. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 10. Koren, I., Timms, R.T., Kula, T., Xu, Q., Li, M.Z. and Elledge, S.J. (2018) The eukaryotic proteome is shaped by E3 ubiquitin ligases targeting C-terminal degrons. Cell, 173, 1622–1635.e1614. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 11. Lin, H.C., Yeh, C.W., Chen, Y.F., Lee, T.T., Hsieh, P.Y., Rusnac, D.V., Lin, S.Y., Elledge, S.J., Zheng, N. and Yen, H.S. (2018) C-terminal end-directed protein elimination by CRL2 ubiquitin ligases. Mol. Cell, 70, 602–613 e603. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 12. Senderek, J., Krieger, M., Stendel, C., Bergmann, C., Moser, M., Breitbach-Faller, N., Rudnik-Schöneborn, S., Blaschek, A., Wolf, N.I., Harting, I. et al. (2005) Mutations in SIL1 cause Marinesco-Sjögren syndrome, a cerebellar ataxia with cataract and myopathy. Nat. Genet., 37, 1312–1314. [DOI] [PubMed] [Google Scholar]
- 13. Anttonen, A.K., Mahjneh, I., Hämäläinen, R.H., Lagier-Tourenne, C., Kopra, O., Waris, L., Anttonen, M., Joensuu, T., Kalimo, H., Paetau, A. et al. (2005) The gene disrupted in Marinesco-Sjögren syndrome encodes SIL1, an HSPA5 cochaperone. Nat. Genet., 37, 1309–1311. [DOI] [PubMed] [Google Scholar]
- 14. Liu, W., Xie, Y., Ma, J., Luo, X., Nie, P., Zuo, Z., Lahrmann, U., Zhao, Q., Zheng, Y., Zhao, Y. et al. (2015) IBS: an illustrator for the presentation and visualization of biological sequences. Bioinformatics, 31, 3359–3361. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 15. Chen, X., Liao, S., Makaros, Y., Guo, Q., Zhu, Z., Krizelman, R., Dahan, K., Tu, X., Yao, X., Koren, I. et al. (2021) Molecular basis for arginine C-terminal degron recognition by Cul2FEM1 E3 ligase. Nat. Chem. Biol., 17, 254–262. [DOI] [PubMed] [Google Scholar]
- 16. Yan, X., Wang, X., Li, Y., Zhou, M., Li, Y., Song, L., Mi, W., Min, J. and Dong, C. (2021) Molecular basis for ubiquitin ligase CRL2FEM1C-mediated recognition of C-degron. Nat. Chem. Biol., 17, 263–271. [DOI] [PubMed] [Google Scholar]
- 17. Lecoquierre, F., Duffourd, Y., Vitobello, A., Bruel, A.L., Urteaga, B., Coubes, C., Garret, P., Nambot, S., Chevarin, M., Jouan, T. et al. (2019) Variant recurrence in neurodevelopmental disorders: the use of publicly available genomic data identifies clinically relevant pathogenic missense variants. Genet. Med., 21, 2504–2511. [DOI] [PubMed] [Google Scholar]
- 18. Tsaban, T., Varga, J.K., Avraham, O., Ben-Aharon, Z., Khramushin, A. and Schueler-Furman, O. (2022) Harnessing protein folding neural networks for peptide-protein docking. Nat. Commun., 13, 176. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 19. Schrödinger, L., and DeLano, W. (2020) PyMOL. Available at: http://www.pymol.org/pymol.
- 20. Sorkaç, A., Alcantara, I.C. and Hart, A.C. (2016) In vivo modelling of ATP1A3 G316S-induced ataxia in C. elegans Using CRISPR/Cas9-Mediated Homologous Recombination Reveals Dominant Loss of Function Defects. PLoS One, 11, e0167963. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 21. Alexander, A.G., Marfil, V. and Li, C. (2014) Use of Caenorhabditis elegans as a model to study Alzheimer’s disease and other neurodegenerative diseases. Front. Genet., 5, 5. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 22. Jumper, J., Evans, R., Pritzel, A., Green, T., Figurnov, M., Ronneberger, O., Tunyasuvunakool, K., Bates, R., Žídek, A., Potapenko, A. et al. (2021) Highly accurate protein structure prediction with AlphaFold. Nature, 596, 583–589. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 23. Varadi, M., Anyango, S., Deshpande, M., Nair, S., Natassia, C., Yordanova, G., Yuan, D., Stroe, O., Wood, G., Laydon, A. et al. (2021) AlphaFold protein structure database: massively expanding the structural coverage of protein-sequence space with high-accuracy models. Nucleic Acids Res., 50, D439–D444. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 24. Madeira, F., Pearce, M., Tivey, A.R.N., Basutkar, P., Lee, J., Edbali, O., Madhusoodanan, N., Kolesnikov, A. and Lopez, R. (2022) Search and sequence analysis tools services from EMBL-EBI in 2022. Nucleic Acids Res., 50, W276–W279. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 25. Waterhouse, A.M., Procter, J.B., Martin, D.M.A., Clamp, M. and Barton, G.J. (2009) Jalview Version 2—a multiple sequence alignment editor and analysis workbench. Bioinformatics, 25, 1189–1191. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 26. Shakkottai, V.G. and Paulson, H.L. (2009) Physiologic alterations in ataxia: channeling changes into novel therapies. Arch. Neurol., 66, 1196–1201. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 27. Gray, J.M., Hill, J.J. and Bargmann, C.I. (2005) A circuit for navigation in Caenorhabditis elegans. Proc. Natl. Acad. Sci. U. S. A., 102, 3184–3191. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 28. Doniach, T. and Hodgkin, J. (1984) A sex-determining gene, fem-1, required for both male and hermaphrodite development in Caenorhabditis elegans. Dev. Biol., 106, 223–235. [DOI] [PubMed] [Google Scholar]
- 29. Jose, A.M., Bany, I.A., Chase, D.L. and Koelle, M.R. (2007) A specific subset of transient receptor potential vanilloid-type channel subunits in Caenorhabditis elegans endocrine cells function as mixed heteromers to promote neurotransmitter release. Genetics, 175, 93–105. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 30. Gieseler, K., Qadota, H. and Benian, G.M. (2017) Development, structure, and maintenance of C. elegans body wall muscle. WormBook, 2017, 1–59. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 31. Shaham, S. (2006) ed., WormBook: Methods in Cell Biology. WormBook, ed. The C. elegans Research Community, WormBook, doi/10.1895/wormbook.1.49.1, http://www.wormbook.org. [Google Scholar]
- 32. Spensley, M., Del Borrello, S., Pajkic, D. and Fraser, A.G. (2018) Acute effects of drugs on Caenorhabditis elegans movement reveal complex responses and plasticity. G3 (Bethesda), 8, 2941–2952. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 33. Sammi, S.R., Jameson, L.E., Conrow, K.D., Leung, M.C.K. and Cannon, J.R. (2022) Caenorhabditis elegans neurotoxicity testing: novel applications in the adverse outcome pathway framework. Front. Toxicol., 4, 826488. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 34. Mahoney, T.R., Luo, S. and Nonet, M.L. (2006) Analysis of synaptic transmission in Caenorhabditis elegans using an aldicarb-sensitivity assay. Nat. Protoc., 1, 1772–1777. [DOI] [PubMed] [Google Scholar]
- 35. Qian, H., Robertson, A.P., Powell-Coffman, J.A. and Martin, R.J. (2008) Levamisole resistance resolved at the single-channel level in Caenorhabditis elegans. FASEB J., 22, 3247–3254. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 36. Treinin, M. and Jin, Y. (2021) Cholinergic transmission in C. elegans: functions, diversity, and maturation of ACh-activated ion channels. J. Neurochem., 158, 1274–1291. [DOI] [PubMed] [Google Scholar]
- 37. Miller, K.G., Alfonso, A., Nguyen, M., Crowell, J.A., Johnson, C.D. and Rand, J.B. (1996) A genetic selection for Caenorhabditis elegans synaptic transmission mutants. Proc. Natl. Acad. Sci. U. S. A., 93, 12593–12598. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 38. Hu, Z., Pym, E.C., Babu, K., Vashlishan Murray, A.B. and Kaplan, J.M. (2011) A neuropeptide-mediated stretch response links muscle contraction to changes in neurotransmitter release. Neuron, 71, 92–102. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 39. McEwen, J.M., Madison, J.M., Dybbs, M. and Kaplan, J.M. (2006) Antagonistic regulation of synaptic vesicle priming by Tomosyn and UNC-13. Neuron, 51, 303–315. [DOI] [PubMed] [Google Scholar]
- 40. Havrilla, J.M., Pedersen, B.S., Layer, R.M. and Quinlan, A.R. (2019) A map of constrained coding regions in the human genome. Nat. Genet., 51, 88–95. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 41. Turner, T.N., Wilfert, A.B., Bakken, T.E., Bernier, R.A., Pepper, M.R., Zhang, Z., Torene, R.I., Retterer, K. and Eichler, E.E. (2019) Sex-based analysis of de novo variants in neurodevelopmental disorders. Am. J. Hum. Genet., 105, 1274–1285. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 42. Tate, J.G., Bamford, S., Jubb, H.C., Sondka, Z., Beare, D.M., Bindal, N., Boutselakis, H., Cole, C.G., Creatore, C., Dawson, E. et al. (2019) COSMIC: the catalogue of somatic mutations in cancer. Nucleic Acids Res., 47, D941–D947. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 43. Varshavsky, A. (2011) The N-end rule pathway and regulation by proteolysis. Protein Sci., 20, 1298–1345. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 44. Yen, H.C., Xu, Q., Chou, D.M., Zhao, Z. and Elledge, S.J. (2008) Global protein stability profiling in mammalian cells. Science, 322, 918–923. [DOI] [PubMed] [Google Scholar]
- 45. Rand, J.B. (2007) Acetylcholine. WormBook, ed. The C. elegans Research Community, WormBook, doi/10.1895/wormbook. 1.131.1, http://www.wormbook.org.
- 46. Aburaya, S., Yamauchi, Y., Hashimoto, T., Minakuchi, H., Aoki, W. and Ueda, M. (2020) Neuronal subclass-selective proteomic analysis in Caenorhabditis elegans. Sci. Rep., 10, 13840. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 47. Artan, M., Barratt, S., Flynn, S.M., Begum, F., Skehel, M., Nicolas, A. and de Bono, M. (2021) Interactome analysis of Caenorhabditis elegans synapses by TurboID-based proximity labeling. J. Biol. Chem., 297, 101094. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 48. Manford, A.G., Mena, E.L., Shih, K.Y., Gee, C.L., McMinimy, R., Martinez-Gonzalez, B., Sherriff, R., Lew, B., Zoltek, M., Rodriguez-Perez, F. et al. (2021) Structural basis and regulation of the reductive stress response. Cell, 184, 5375, e5316–5390. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 49. Rydzanicz, M., Zwolinski, P., Gasperowicz, P., Pollak, A., Kostrzewa, G., Walczak, A., Konarzewska, M. and Ploski, R. (2021) A recurrent de novo variant supports KCNC2 involvement in the pathogenesis of developmental and epileptic encephalopathy. Am. J. Med. Genet. A, 185, 3384–3389. [DOI] [PubMed] [Google Scholar]
- 50. Brenner, S. (1974) The genetics of Caenorhabditis elegans. Genetics, 77, 71–94. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 51. Calixto, A., Chelur, D., Topalidou, I., Chen, X. and Chalfie, M. (2010) Enhanced neuronal RNAi in C. elegans using SID-1. Nat. Methods, 7, 554–559. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 52. Kamath, R.S. and Ahringer, J. (2003) Genome-wide RNAi screening in Caenorhabditis elegans. Methods, 30, 313–321. [DOI] [PubMed] [Google Scholar]
- 53. Romani, M. and Auwerx, J. (2021) Phalloidin staining of actin filaments for visualization of muscle fibers in Caenorhabditis elegans. Bio. Protoc., 11, e4183. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 54. Felton, C.M. and Johnson, C.M. (2014) Dopamine signaling in C. elegans is mediated in part by HLH-17-dependent regulation of extracellular dopamine levels. G3 (Bethesda), 4, 1081–1089. [DOI] [PMC free article] [PubMed] [Google Scholar]
Associated Data
This section collects any data citations, data availability statements, or supplementary materials included in this article.
Supplementary Materials
Data Availability Statement
Data obtained during this study are available at doi.org/10.18150/9TQUTI