

ABSTRACT
Background
Progressive supranuclear palsy (PSP)–Richardson's syndrome (RS) presents with a distinctive clinical phenotype of supranuclear ophthalmoplegia, parkinsonism, postural instability with falls, and cognitive impairment. Several rare neurological conditions have been described that mimic PSP, and the co‐occurrence of dual pathologies has also been described.
Cases
In this article, we present 2 cases of patients who presented with a parkinsonian phenotype suggestive of PSP‐RS. In 1 case, a family history and early levodopa‐induced chorea led to testing for Huntington's disease, and a pathogenic HTT mutation was found. In the second case, magnetic resonance imaging findings led to genetic confirmation of a pathogenic FMR1 mutation.
Conclusions
These observations raised the possibility that HD and fragile‐X tremor‐ataxia syndrome may on occasion present with PSP‐RS. Alternatively, and perhaps more likely, is the co‐occurrence of 2 rare neurodegenerative conditions. Neuropathological studies of cases involving complex phenotypes in rare genetic conditions are required to better understand the likely pathologies in cases such as these.
Keywords: neurodegeneration, Huntington's disease, FXTAS, phenotyping, genetics
Introduction
Progressive supranuclear palsy (PSP)–Richardson's syndrome (RS) presents with supranuclear vertical gaze palsy, parkinsonism, postural instability with falls, and cognitive impairment. Recent changes to the recognized phenotypic spectrum in PSP recognize the heterogeneity characteristic of neurodegenerative conditions, which affect neuronal networks in an incompletely selective fashion. A number of rare genetic conditions can mimic PSP, including C9orf72 mutations, Perry syndrome, Kufor–Rakeb, CSF1R mutations, and Niemann‐Pick Type C. Huntington's disease (HD) and fragile‐X tremor‐ataxia syndrome (FXTAS) are triplet repeat disorders that often present in later life. Although most commonly associated with cerebellar dysfunction, parkinsonism in seen in up to 60% of people with FXTAS. 1 Parkinsonism may also develop late in the disease course of HD. However, cases presenting in later life with parkinsonism have been reported with occasional examples mimicking atypical parkinsonian syndromes. We report 2 cases presenting with PSP‐RS phenotypes in which an underlying genetic neurodegenerative condition (HD and FXTAS, respectively) was diagnosed.
Case Series
Case 1
A 58‐year‐old right‐handed man presented to the clinic with oculomotor disturbances and cognitive impairment. His motor function had also declined.
He reported slower movement, difficulty rising from a chair, stiffness of all limbs, and nocturnal hypokinesis. There was stiffness and discomfort in his neck and shoulders. He complained of postural instability and tended to collapse backward into a chair when trying to sit. He had longstanding hyposmia. He had difficulty in reading as he found it difficult to track text on a page or phone.
He reported concerns regarding his memory for 1 year. He had become increasingly reliant on aides‐mémoire. He felt that he had difficulty registering and retaining new information. He had also noticed difficulty with executive function, which manifested as marked difficulty with rapid attention shifting. He became easily frustrated and upset if distracted from a task. His behavioral repertoire had narrowed, and his grooming and hygiene had deteriorated. He had become more passive and had demonstrated emotional lability and tearfulness. There was marked bradyphrenia.
His family history was positive for HD. The patient's mother had developed HD with a classical choreic phenotype in her late 70s and died at the age of 83. The patient's maternal grandparents had died at relatively advanced ages without any neuropsychiatric symptoms. The patient had 6 siblings, including 2 brothers with alcohol dependence syndrome and an older sister with refractory depression requiring hospitalization. He had 3 children who had not suffered any neuropsychiatric symptoms.
On examination, he had a flexed posture and anterocollis (see Video 1). There was hypomimia and frontalis overactivation. There were square wave jerks, hypometric saccades, and slow vertical with normal velocity horizontal saccades. There was prominent axial rigidity as well as appendicular rigidity, more prominent on the right than the left. There was a right upper limb rest tremor. There was bradykinesia, worse on the right than the left, with interruptions and hesitations. Reflexes were brisk, and the left plantar was upgoing. Examination of the gait revealed a good stride length, but the arms were held abducted in a manner suggestive of a “gunslinger” posture, although the typical retrocollis was absent. 2 He had a positive grasp reflex. There were no parietal signs and no chorea. He scored 74/100 on the Addenbrooke's Cognitive Examination with particular deficits in fluency and memory and scored 11/18 on the Frontal Assessment Battery.
Video 1.
Section 1: nuchal rigidity and severe bilateral bradykinesia. Section 2: slowed vertical saccades and “apraxia of eyelid opening.” Section 3: postural instability with positive “pull test.”
Magnetic resonance imaging (MRI) of his brain demonstrated generalized atrophy and mild periventricular white matter hyperintensities. Given his family history, he also underwent testing for HD. This confirmed an HTT allele with a CAG repeat number of 41 ± 1 repeat. Given the unusual features of predominant parkinsonism suggestive of PSP, dopamine active transporter single photon emission computerised tomography imaging was performed, which demonstrated rounded morphology of binding in the caudate and absent putaminal activity.
He was treated with levodopa, which led to a modest improvement in his axial rigidity, gait, and mobility. However, within 5 months he had developed orolingual dyskinesia.
Case 2
A 75‐year‐old man presented with a 4‐year history of gait disturbance. He described a 6‐year history of declining mobility. He recalled a sense of postural instability, particularly with tandem gait. He subsequently developed a right upper limb tremor. He suffered his first fall approximately 2 years after symptoms onset and went on to have repeated backward falls. There were symptoms of orthostatic hypotension, but autonomic function was otherwise unimpaired. There was mild dysphagia for which he had seen a speech and language therapist.
Examination (see Video 2) revealed hypomimia, slow vertical saccades, a reduced upgaze, and subtle “round‐the‐houses” movement on vertical saccades and square wave jerks. There was nuchal rigidity. There was no family history of neurodegenerative disease, autism, or learning disability. There was a mild postural tremor with terminal accentuation.
Video 2.
Section 1: slow vertical saccades. Section 2: bilateral bradykinesia and hypomimia. Section 3: postural instability.
An MRI of his brain demonstrated generalized atrophy with disproportionate atrophy of the cerebellum. There was hyperintensity of the middle cerebellar peduncle on axial T2 imaging and diffusion‐weighted imaging (Fig. 1).
FIG. 1.
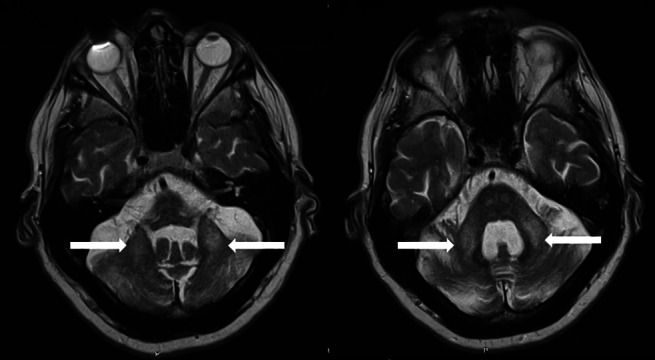
T2 axial imaging demonstrating hyperintensity in the middle cerebellar peduncle bilaterally as indicted by the arrows.
This imaging pattern prompted genetic testing, which confirmed 90 CGG triplet repeats in the FMR1 gene.
Discussion
Both of these cases presented with apparent PSP‐RS with features outside of the clinical presentation (family history in case 1 and MRI appearances in case 2), prompting testing for an alternative disorder. These cases raise the question of whether they represent highly atypical manifestations of HD and FXTAS as “PSP syndromes” or the co‐occurrence of rare neurodegenerative conditions. Perhaps more interesting is the possibility of 1 form of pathology provoking or potentiating the evolution of another. None of these questions are easily answered, and we present these cases to provoke awareness of the potential complexity that can exist behind otherwise apparently simple clinical presentations.
Phenotypic mimicry of a PSP‐RS–like syndrome is a particularly challenging and relevant phenomenon as, in the absence of reliable serological or imaging biomarkers, the PSP‐RS is considered relatively specific for tau‐mediated disease and the Richardson's phenotype is used as an inclusion criterion for research studies and clinical trials, including recent trials of monoclonal antibodies directed against tau, as well as trials currently in progress. Therefore, awareness of potential mimics and vigilance for historical, imaging, and biochemical clues that may help to identify alternative pathologies is vital not only to deliver accurate diagnosis to patients but also to develop study cohorts with a high degree of pathological homogeneity for natural history and interventional studies.
Mimicry in neurological phenotypes may arise as a result of disruption of common pathways by multiple pathologies (eg, corticobasal syndrome as a manifestation of Alzheimer's pathology or corticobasal degeneration). Alternatively, disruption of different anatomical pathways may give rise to a similar phenotype, for example, deficits in vertical gaze imply dysfunction of the supranuclear pathways generating voluntary gaze that in PSP arises as a result of prominent midbrain pathology affecting the medial longitudinal fasciculus and the interstitial nucleus of Cajal, whereas abnormal saccadic and pursuit movements may be associated with lesions of the frontal eye fields and premotor cortices, which may explain the gaze abnormalities seen in HD. PSP‐RS is associated with widespread pathology involving the brainstem, basal ganglia, dentate nucleus, and frontal lobes. It is not surprising, therefore, that many conditions may mimic aspects of PSP. The co‐occurrence of PSP with additional pathologies has been well documented. A study of copathology in patients with PSP demonstrated Lewy bodies and TAR DNA binding protein‐43 deposits in 15% and 14% of cases, respectively, and rates of copathology increased with age. 3
Typical presentations of HD include chorea, neuropsychiatric disease, dementia, and gait abnormalities. Occasionally, cases of HD with parkinsonism may mimic so‐called “atypical parkinsonisms.” HD‐associated levodopa‐responsive parkinsonism combined with autonomic dysfunction mimicking multiple systems atrophy has been reported. 4 PSP‐RS phenotypes in HD are extremely rare. 5 Oculomotor abnormalities are a common component of HD presentations. Interestingly, a study of saccadic abnormalities in HD found that vertical saccades were more commonly impaired than horizontal saccades (9/11 vs. 2/11 cases). 6 Supranuclear gaze palsy is less common but has been described. 5
Case 1 presented with prominent axial rigidity, slowed vertical saccades, apraxia of eyelid opening, reduced amplitude of movements without clear decrement, a “gunslinger” gait, and a positive DaTSCAN, mimicking PSP‐RS. The early onset of dyskinesia, including orofacial dyskinesia, following treatment with levodopa is a recognized phenomenon in presymptomatic HD. Indeed, levodopa administration been used as a provocative test in clinically unaffected offspring the pre‐genetic testing era. However, the presence of dyskinesia in this case probably does not help to resolve the central dilemma. 7 It is unclear whether this case represents a highly atypical HD presentation or whether dual pathology may have modified the clinical manifestation of HD.
Case 2 presented with vertical gaze abnormalities, postural instability, and parkinsonism as well as MRI findings suggestive of FXTAS. Although FXTAS is most commonly associated with signs of cerebellar dysfunction such as ataxia and tremor, parkinsonism is also common and was present in all 5 of the patients reported in the initial cohort. 8 Parkinsonian syndromes indistinguishable from idiopathic Parkinson's disease have been described; however, in general parkinsonism is relatively mild and is rarely a presenting symptom. 9 Cases of PSP co‐occurring with FXTAS have been described, although to our knowledge no pathologically proven case of isolated FXTAS pathology producing a PSP phenotype has been reported. 10
These cases illustrate a dilemma in the assessment of neurodegenerative diseases where broad phenotypes and imperfect clinicopathological correlation are increasingly recognized. The lack of neuropathological data limits our ability to make confident statements about these cases, a circumstance reflected in clinical practice. Further neuropathological studies of patients with overlapping phenotypes would help to inform diagnosis, determine patterns of co‐occurrence, and offer valuable insight into any potential for dynamic interactions where simultaneous pathologies exist.
Author Roles
(1) Research Project: A. Conception, B. Organization, C. Execution; (2) Manuscript: A. Writing of the First Draft, B. Review and Critique.
S.L.: 1A, 1B, 1C, 2A
T.L.: 2B
R.W.: 1A, 2B
S.O.: 1A, 2B
Disclosures
Ethical Compliance Statement: The authors confirm that the approval of an institutional review board was not required for this work. Patient‐informed consent was obtained for this work and for the publication of videos. We confirm that we have read the Journal's position on issues involved in ethical publication and affirm that this work is consistent with those guidelines.
Funding Sources and Conflicts of Interest: There are no conflicts of interest relevant to this work. Shane Lyons was funded by a grant from the Meath Foundation.
Financial Disclosures for the Previous 12 Months: Prof Tim Lynch holds grants from the Health Research Board Ireland, EU‐HRB JPND, Michael J Fox Foundation and the Irish Insitute of Clinical Neuroscience. Dr Sean O'Dowd has served on advisory boards for Roche Pharma, Jannssen, Biogen, and Abbvie Pharmceuticals in the past 12 months and has received support for travel/conferences from Roche Pharma and Novartis.
Relevant disclosures and conflicts of interest are listed at the end of this article.
References
- 1. Niu YQ, Yang JC, Hall DA, et al. Parkinsonism in fragile X‐associated tremor/ataxia syndrome (FXTAS): Revisited. Parkinsonism Relat Disord 2014;20(4):456–459. 10.1016/j.parkreldis.2014.01.006. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 2. Marsili L, Colosimo C. The "gunslinger" sign in progressive supranuclear palsy ‐ Richardson variant. J Neurol Sci 2020;418:117108. 10.1016/j.jns.2020.117108. [DOI] [PubMed] [Google Scholar]
- 3. Robinson JL, Yan N, Caswell C, Xie SX, Suh E, Van Deerlin VM, et al. Primary tau pathology, not Copathology, correlates with clinical symptoms in PSP and CBD. Journal of Neuropathol Exp Neurol 2019;79(3):296–304. 10.1093/jnen/nlz141. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 4. Trosch RMLP. Westphal variant Huntington's disease masquerading as MSA. Mov Disord 1996;13:181. [Google Scholar]
- 5. Bittenbender JB, Quadfasel FA. Rigid and akinetic forms of Huntington's chorea. Arch Neurol 1962;7:275–288. 10.1001/archneur.1962.04210040027003. [DOI] [PubMed] [Google Scholar]
- 6. Bollen E, Reulen JP, Den Heyer JC, Van der Kamp W, Roos RA, Buruma OJ. Horizontal and vertical saccadic eye movement abnormalities in Huntington's chorea. J Neurol Sci 1986;74(1):11–22. 10.1016/0022-510x(86)90187-5. [DOI] [PubMed] [Google Scholar]
- 7. Klawans HL Jr, Paulson GW, Ringel SP, Barbeau A. Use of L‐dopa in the detection of presymptomatic Huntington's chorea. N Engl J Med 1972;286(25):1332–1334. 10.1056/NEJM197206222862503. [DOI] [PubMed] [Google Scholar]
- 8. Hagerman RJ, Leehey M, Heinrichs W, et al. Intention tremor, parkinsonism, and generalized brain atrophy in male carriers of fragile X. Neurology 2001;57(1):127–130. 10.1212/wnl.57.1.127. [DOI] [PubMed] [Google Scholar]
- 9. Juncos JL, Lazarus JT, Graves‐Allen E, et al. New clinical findings in the fragile X‐associated tremor ataxia syndrome (FXTAS). Neurogenetics 2011;12(2):123–135. 10.1007/s10048-010-0270-5. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 10. Paucar M, Nennesmo I, Svenningsson P. Pathological study of a FMR1 Premutation carrier with progressive Supranuclear palsy. Front Genet 2018;9:317. 10.3389/fgene.2018.00317. [DOI] [PMC free article] [PubMed] [Google Scholar]