

Abstract
Inactivation of the tumor suppressor genes tumor protein p53 (TP53) and cyclin-dependent kinase inhibitor 2A (CDKN2A) occurs early during gastroesophageal junction (GEJ) tumorigenesis. However, because of a paucity of GEJ-specific disease models, cancer-promoting consequences of TP53 and CDKN2A inactivation at the GEJ have not been characterized. Here, we report the development of a wild-type primary human GEJ organoid model and a CRISPR-edited transformed GEJ organoid model. CRISPR-Cas9–mediated TP53 and CDKN2A knockout (TP53/CDKN2AKO) in GEJ organoids induced morphologic dysplasia and proneoplastic features in vitro and tumor formation in vivo. Lipidomic profiling identified several platelet-activating factors (PTAFs) among the most up-regulated lipids in CRISPR-edited organoids. PTAF/PTAF receptor (PTAFR) abrogation by siRNA knockdown or a pharmacologic inhibitor (WEB2086) reduced proliferation and other proneoplastic features of TP53/CDKN2AKO GEJ organoids in vitro and tumor formation in vivo. In addition, murine xenografts of Eso26, an established human esophageal adenocarcinoma cell line, were suppressed by WEB2086. Mechanistically, TP53/CDKN2A dual inactivation disrupted both the transcriptome and the DNA methylome, likely mediated by key transcription factors, particularly forkhead box M1 (FOXM1). FOXM1 activated PTAFR transcription by binding to the PTAFR promoter, further amplifying the PTAF-PTAFR pathway. Together, these studies established a robust model system for investigating early GEJ neoplastic events, identified crucial metabolic and epigenomic changes occurring during GEJ model tumorigenesis, and revealed a potential cancer therapeutic strategy. This work provides insights into proneoplastic mechanisms associated with TP53/CDKN2A inactivation in early GEJ neoplasia, which may facilitate early diagnosis and prevention of GEJ neoplasms.
INTRODUCTION
Gastroesophageal cancers account for more than 1 million deaths annually, representing the second highest cause of cancer death worldwide. One category of these neoplasms, gastroesophageal junction (GEJ) adenocarcinoma, has increased more than 2.5-fold in incidence in the United States and other Western countries in recent decades (1, 2). Compared with other gastroesophageal region cancers, GEJ tumors are particularly aggressive and have a dire prognosis; thus, vigorous investigation of their molecular basis is needed. However, this goal has been difficult to achieve in part due to a lack of biologically relevant GEJ-specific early disease models. Human organoids are robust models that recapitulate and maintain essential genetic, functional, and phenotypic characteristics of their tissues of origin (3). Filling a knowledge gap between transgenic animal models and cell lines, organoids provide a valuable platform for understanding fundamental oncogenic mechanisms and identifying candidate cancer therapeutic targets (4).
Lipid metabolic reprogramming is a known hallmark of cancer (5, 6), and certain lipid profiles have been proposed as either biomarkers for early cancer detection or targets for cancer treatment (7–9). Aberrations in the fate and composition of the lipidome promote tumorigenesis, enhance cancer cell growth, and support cancer cell survival in challenging microenvironments by activating oncogenic pathways and processes (9). By performing lipidomic profiling in gastroesophageal cancers, we recently demonstrated that heightened synthesis of specific lipid species was required for cancer cell viability and proliferation (10, 11). However, metabolic changes in the lipidome of GEJ cancers are still largely unknown.
In the present study, we investigated GEJ tumorigenesis by establishing the first human normal GEJ-derived organoids, modified by tumor protein p53 (TP53) and cyclin-dependent kinase inhibitor 2A (CDKN2A) dual knockout (TP53/CDKN2AKO) using CRISPR-Cas9 genome editing. Using this GEJ-specific disease model, we addressed the phenotypic, metabolic, and epigenomic alterations during the early neoplastic transformation of GEJ cells, with the goal of providing insights into the mechanistic basis of this aggressive and poorly understood malignancy.
RESULTS
Establishment and characterization of human normal GEJ organoids
To address the lack of biologically relevant GEJ-specific disease models, we generated three-dimensional (3D) organoids from human primary endoscopic GEJ biopsies, which were confirmed pathologically to contain neither dysplastic nor neoplastic cells (table S1). Briefly, freshly isolated GEJ crypts were embedded in Matrigel and incubated with conditioned medium containing stem-critical growth factors (Fig. 1A and table S2). Under these culture conditions, we established 3D organoids generated from six normal GEJ biopsies with a 100% success rate. We characterized these organoids using phase-contrast imaging, hematoxylin and eosin (H&E) staining, and viability [water-soluble tetrazolium salt (WST-1)] assays (Fig. 1, B to D). At day 4 after initial seeding, 3D spherical structures were formed and reached a diameter of 25 μm. These structures continued to grow, eventually reaching a size plateau of 106 μm between days 24 and 29 (Fig. 1, B and C). These GEJ organoids consisted of 120 to 250 cells at day 24, indicating that population doubling time in culture was 77.85 ± 5.54 hours. Histologic analyses revealed that normal GEJ organoids formed a 3D structure consisting of single-layered epithelial cells. The cell viability of organoids increased to a peak on day 14 and significantly (P < 0.05) diminished by day 24 (Fig. 1D). We continued to monitor organoids from these six independent normal GEJ biopsies; they propagated continuously for 4 to 6 months ex vivo.
Fig. 1. Establishment and characterization of human normal GEJ organoids.
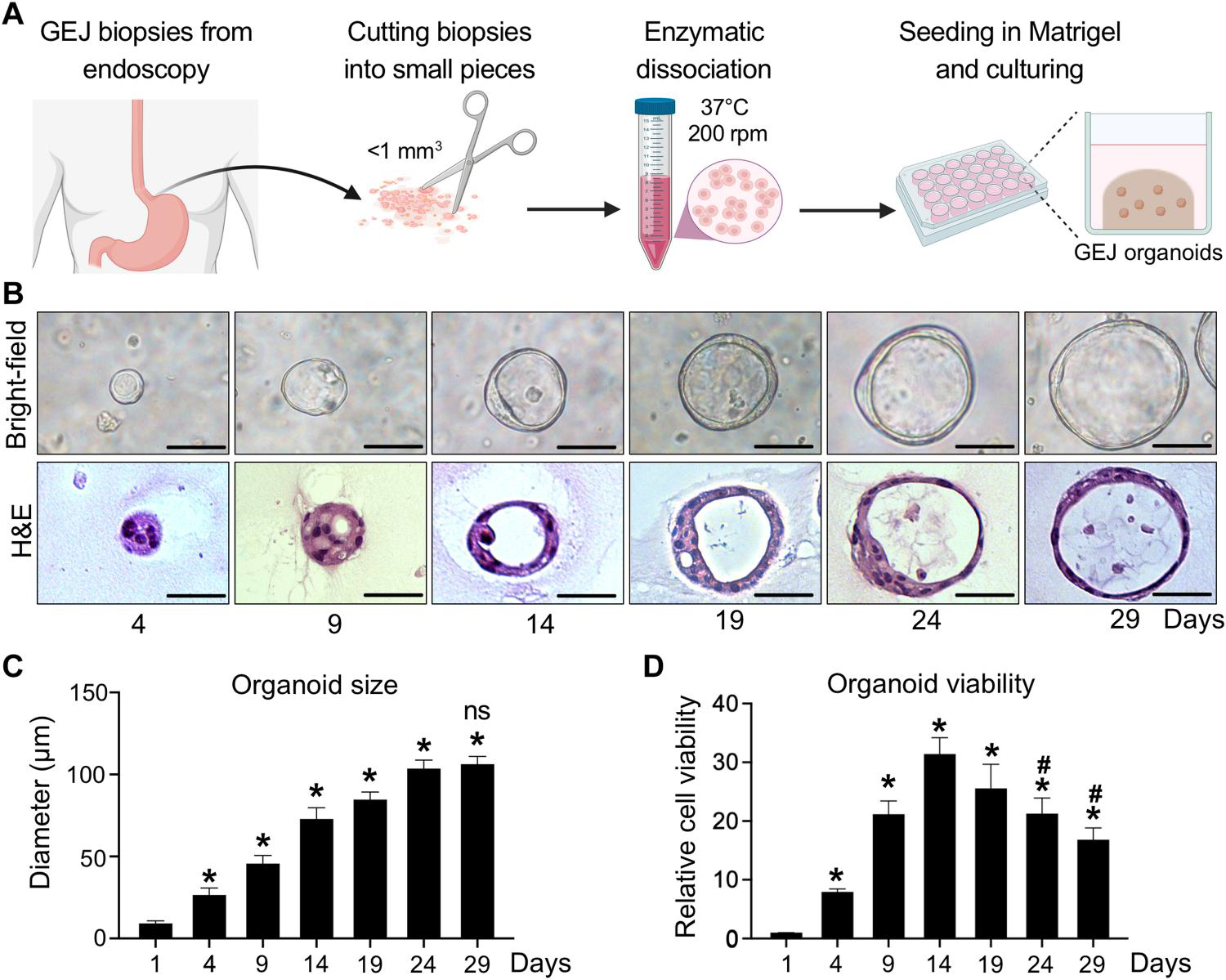
(A) A workflow of organoid generation from human primary endoscopic GEJ biopsies. Biopsies of normal GEJ mucosa were taken by upper endoscopy and then minced and enzymatically dissociated. The cell suspension was mixed with Matrigel to initiate 3D organoid culture in the conditioned medium. (B to D) GEJ organoids were analyzed for structural and growth properties at the indicated time points. 3D organoids were photomicrographed under phase-contrast microscopy (B, top) and collected for H&E staining (B, bottom). Scale bars, 50 μm. Average organoid size (C) and viability (D) were determined at each time point. Data are represented as means ± SD; n = 6 biological replicates. *P < 0.05 versus day 1; #P < 0.05 versus day 14; not significant (ns) versus day 24 by ANOVA.
TP53/CDKN2A loss promotes proliferation, dysplasia, and neoplastic transformation of GEJ organoids
Next, we sought to generate a disease and pathologic model of GEJ carcinoma that could grow more vigorously, survive longer in culture, and better facilitate studies of GEJ neoplastic transformation. Because TP53 and CDKN2A are two of the most frequently inactivated tumor suppressor genes in human GEJ cancers (12–14), we chose to inactivate these two genes in our GEJ organoid model using the CRISPR-Cas9 genome editing system. An all-in-one Cas9:guide RNA (gRNA) ribonucleoprotein (RNP) complex targeting TP53 (exon 4) and CDKN2A (exon 1α) was prepared using a gRNA complex, which combines CRISPR RNA (crRNA) and trans-activating crRNA (tracrRNA), with Cas9 nuclease. We then dissociated human GEJ organoids into small cell clusters of 5 to 15 cells each and delivered the RNP complex by electroporation using an optimized protocol (fig. S1A). For the control organoid group, we electroporated a negative control, nontargeting RNP complex. An electroporation efficiency of 42% was achieved (fig. S1B). Subsequently, we selected for organoids with mutant TP53 using Nutlin-3a, which inhibits mouse double minute 2 (MDM2 proto-oncogene) and hence induces growth arrest of TP53 wild-type cells (fig. S1C) (15). Upon selection, we performed Sanger sequencing to validate specific editing of targeted TP53 and CDKN2A exons. Frameshift mutations, including 1–base pair (bp) insertions or deletions at TP53 and CDKN2A target sites (Fig. 2, A and B), were observed, verifying successful genome editing of GEJ organoids.
Fig. 2. Knockout of TP53/CDKN2A promotes neoplastic transformation in human normal GEJ organoids.
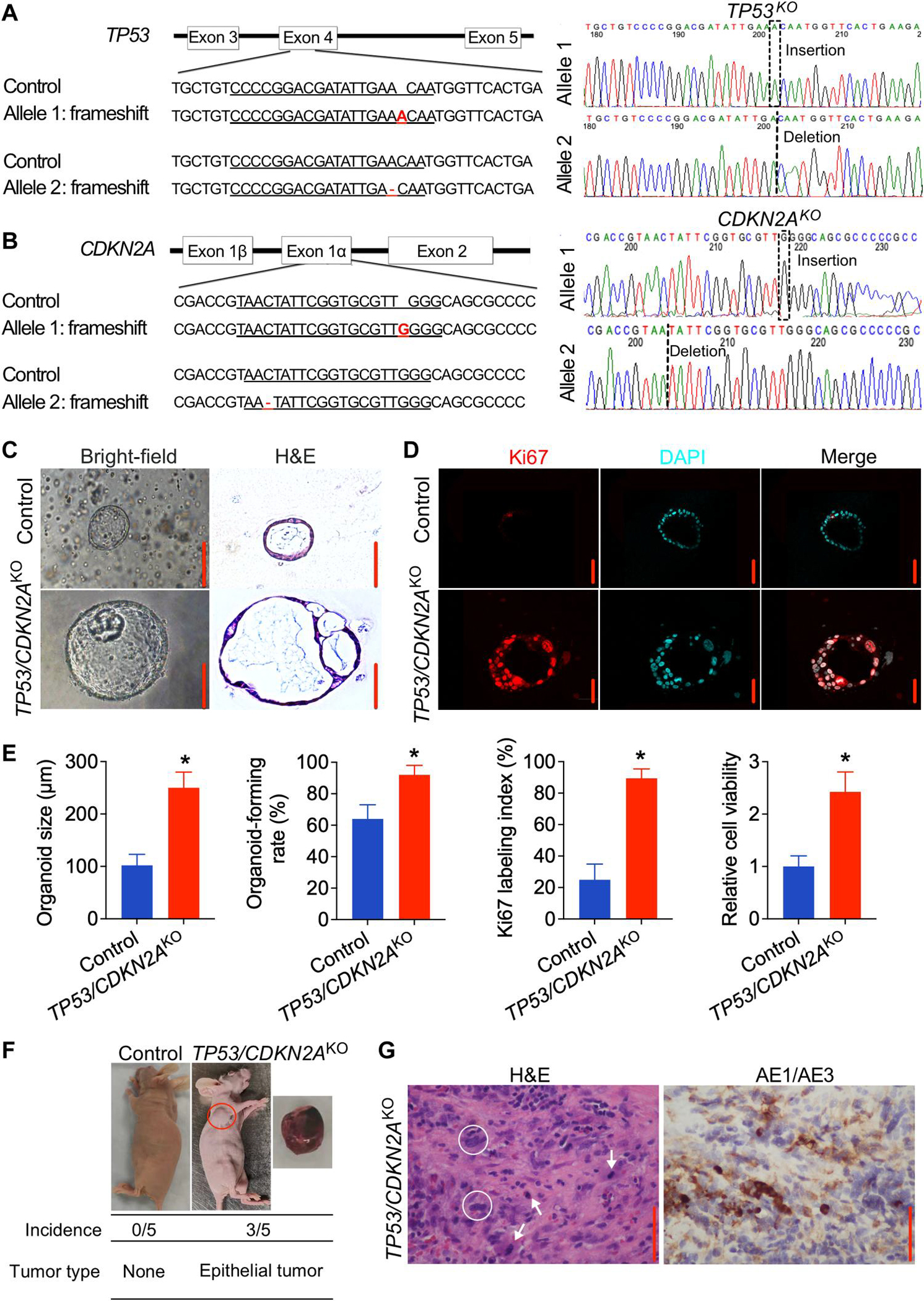
(A and B) Sanger sequencing of TP53/CDKN2AKO GEJ organoids showing 1-bp insertion or deletion in TP53 (A) or CDKN2A (B). Red font indicates corresponding frameshift indels in the genomic DNA. (C and D) On the 10th day after seeding 1 × 105 dissociated organoid cells, organoid cultures were photomicrographed using phase-contrast microscopy and collected for (C) bright-field, H&E, and (D) IF staining for Ki67 (red color). DAPI, 4′,6-diamidino-2-phenylindole. (E) Average organoid size, organoid-forming efficiency, and Ki67 index were determined by measuring >50 organoids. Data are represented as means ± SD; n = 4 biologic replicates. *P < 0.05 by Student’s t test. (F) Representative images of xenografts from mice injected with control or TP53/CDKN2AKO GEJ organoids; the underlying table shows incidence and tumor characteristics. This experiment was repeated once with similar results. (G) Representative H&E and AE1/AE3 pan-keratin IHC staining (brown) in xenografts arising from TP53/CDKN2AKO organoids. White arrows, mitoses; white circles, abnormally large, pleomorphic cells with irregular nuclear envelopes; scale bars, 100 μm.
We next characterized phenotypic changes of GEJ organoids upon loss of TP53 and CDKN2A. Control organoids formed a single layer of epithelial cells with normal nuclei at day 10 after seeding. In contrast, TP53/CDKN2AKO organoids exhibited larger diameters, more complex multicellular structures, increased mitotic activity, and enlarged, atypical nuclei. These changes were consistent with dysplastic morphology (Fig. 2, C and D) (16). In addition, we performed immunofluorescence (IF) staining of trefoil factor 3 (TFF3), a candidate diagnostic marker of intestinal metaplasia, which showed that TFF3 was induced in TP53/CDKN2AKO GEJ organoids (fig. S2). This result suggests that TP53/CDKN2A inactivation promotes intestinal metaplastic change in GEJ columnar cells. Organoid-forming rate also increased significantly in TP53/CDKN2AKO relative to control organoids (92 versus 64%, P < 0.05; Fig. 2E). IF staining showed an elevation of Ki67 labeling index in TP53/CDKN2AKO versus control GEJ organoids (89.4 versus 24.9%; Fig. 2, D and E). Consistently, WST-1 assays revealed a 2.4-fold increase in proliferation of TP53/CDKN2AKO versus control organoids (Fig. 2E). Furthermore, under the same culture conditions, whereas control organoids could only be propagated for up to 6 months, TP53/CDKN2AKO organoids were propagated for more than 19 months, with an estimated population doubling time of 37.86 ± 1.90 hours (fig. S3). These data demonstrate that loss of TP53/CDKN2A enhances proliferation and dysplasia of GEJ organoids in vitro.
To assess the in vivo effect of TP53/CDKN2A inactivation in GEJ organoids, we performed xenotransplantation assays. Control and TP53/CDKN2AKO organoid cells (2 × 106 cells per injection) were subcutaneously injected into the left and right armpit of five nude mice, respectively. Within a 5-month postinjection observation interval, no tumors were formed in five mice injected with control GEJ organoids. In contrast, TP53/CDKN2AKO organoids developed tumors in three of the five injected mice within 8 weeks (Fig. 2F). These tumors appeared morphologically similar to highly differentiated gastroesophageal adenocarcinoma. H&E and immunohistochemistry (IHC) analysis of xenografts arising from TP53/CDKN2AKO organoids showed increased mitosis, abnormally large, pleomorphic cells with irregular nuclear envelopes, and positive expression of anti-cytokeratin 1 antibody (AE1)/AE3 proteins (Fig. 2G). Thus, these in vivo data suggest that inactivation of TP53/CDKN2A directly promotes neoplastic transformation in GEJ organoids.
Lipidomic MALDI-IMS identifies PTAFs as top up-regulated lipids in TP53/CDKN2AKO organoids
Reprogramming and dysregulation of lipid metabolism is a hallmark of cancer (17). However, it is unknown whether and how lipid metabolic processes are altered during early GEJ carcinogenesis. To address this knowledge gap, we applied lipidomic matrix-assisted laser desorption/ionization imaging mass spectrometry (MALDI-IMS) to discover altered lipid species in TP53/CDKN2AKO versus control organoids using 2,5-dihydrobenzoic acid (DHB) as a matrix in positive ion mode to ionize a broad range of phospholipids (18). Within the mass range from mass/charge ratio (m/z) 40 to 2000, we obtained mass spectra of lipid species through direct analysis of organoid sections (fig. S4A), whereas ion images were generated from each peak and displayed as the position in the organoid section and relative intensity (fig. S4B). We applied a cutoff of m/z > 450 to minimize the background matrix signal in imaging experiments and focus on phospholipids that are generally larger than m/z 400. We first performed analysis based on fold changes of mean intensity data for individual peaks, identifying 50 up-regulated peaks and 132 down-regulated peaks in TP53/CDKN2AKO versus control organoids under an average fold change of >1.5 (Fig. 3A and table S3). In parallel, we performed receiver operating characteristic (ROC) analysis, using an area-under-the-curve (AUC) threshold value of >0.75. By this criterion, 16 and 50 peaks were respectively up-regulated and down-regulated in TP53/CDKN2AKO versus control organoids (Fig. 3B and table S4). Concordantly, all of these altered peaks identified by the ROC method were dysregulated in the same directions based on their fold changes (Fig. 3B). We next performed on-organoid tandem mass spectrometry (MS/MS) (fig. S5) and identified that, among the 16 shared up-regulated peaks, the one with the highest AUC (m/z = 467.20) was a specific platelet-activating factor (PTAF) lipid PC-O-14:0 (LMGP01020009). Moreover, four additional lipids were identified as either PTAF lipids [m/z 451.16, PC-O-13:1 (LMGP01020146); m/z 550.03, PC-O-20:0 (LMGP01020094); and m/z 549.06, PC-O-20:1 (LMGP01020146)] or a precursor of a PTAF lipid [m/z 482.19, LPC-O-16:0 (LMGP01060010)]. MALDI imaging data and chemical structures of these lipids are displayed in Fig. 3 (C and D). PTAF lipids are a family of glycerophosphocholines, implicated as bioactive mediators in diverse pathologic processes, including tumor angiogenesis and metastasis (19, 20).
Fig. 3. PTAF lipids are increased in TP53/CDKN2AKO as compared to control GEJ organoids.
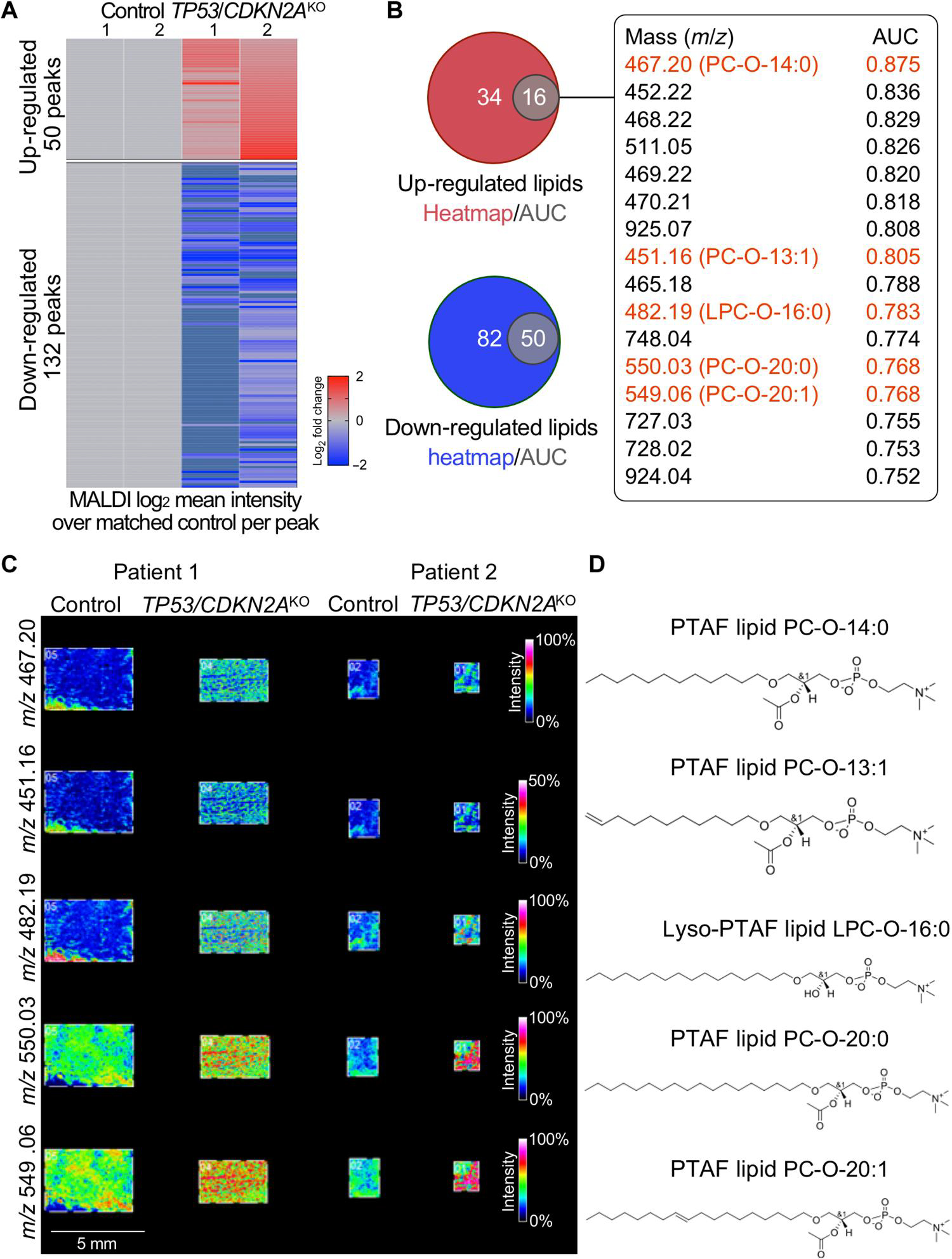
MALDI imaging–based lipidomic analysis was performed on independent paired sets of TP53/CDKN2AKO versus control GEJ organoids derived from two different patients. (A) Heatmap of discriminative lipid peaks (m/z) with a cutoff of m/z >450, a mean absolute fold change value of >1.5, and an individual absolute fold change of >1.2 in each TP53/CDKN2AKO organoids, compared with matched control. (B) Venn diagrams represent the overlap of up-regulated and down-regulated lipids based on AUC and heatmap results. Sixteen overlapping up-regulated lipids and AUC values are listed. (C) MALDI imaging and (D) corresponding chemical structures of representative PTAF lipids in TP53/CDKN2AKO organoids.
Inhibition of PTAF/PTAFR suppresses neoplastic properties of TP53/CDKN2AKO GEJ organoids
After identification of multiple PTAF lipids as increased phospholipids in TP53/CDKN2AKO GEJ organoids, we addressed their potential roles in GEJ neoplasia development. As glycerophosphocholines, PTAFs exert biological effects by binding to their cognate receptor, PTAFR (21). Thus, we first evaluated PTAFR expression in GEJ neoplasia, both in our organoid model and in The Cancer Genome Atlas (TCGA) dataset. Similar to its cognate lipid ligand, PTAFR expression was significantly up-regulated in TP53/CDKN2AKO versus control organoids (P < 0.05; Fig. 4A). Consistent with this finding, TCGA esophageal adenocarcinoma (EAC) samples exhibited higher PTAFR mRNA expression than did nonmalignant TCGA GEJ samples (fig. S6). Encouraged by these observations, we next directly abrogated PTAF/PTAFR function in our early GEJ model system by either small interfering RNA (siRNA) knockdown or pharmacologic inhibition. Silencing PTAFR expression by siRNAs significantly (P < 0.05) decreased the average size, cell viability, and Ki67 index of TP53/CDKN2AKO organoids (Fig. 4, B to E). In parallel, we treated TP53/CDKN2AKO organoids with either vehicle control [0.1% dimethyl sulfoxide (DMSO)] or a specific PTAFR pharmacologic antagonist, WEB2086, at various concentrations. In agreement with the siRNA results, TP53/CDKN2AKO organoids displayed significantly (P < 0.05) reduced size and Ki67 index after WEB2086 treatment (Fig. 4, F to H). WST-1 assays showed that metabolically active cells began to decrease on day 4, and a time- and dose-dependent inhibitory effect was confirmed on days 7 and 10 (Fig. 4H).
Fig. 4. Blockade of PTAF/PTAFR inhibits growth and proliferation of TP53/CDKN2AKO GEJ organoids.
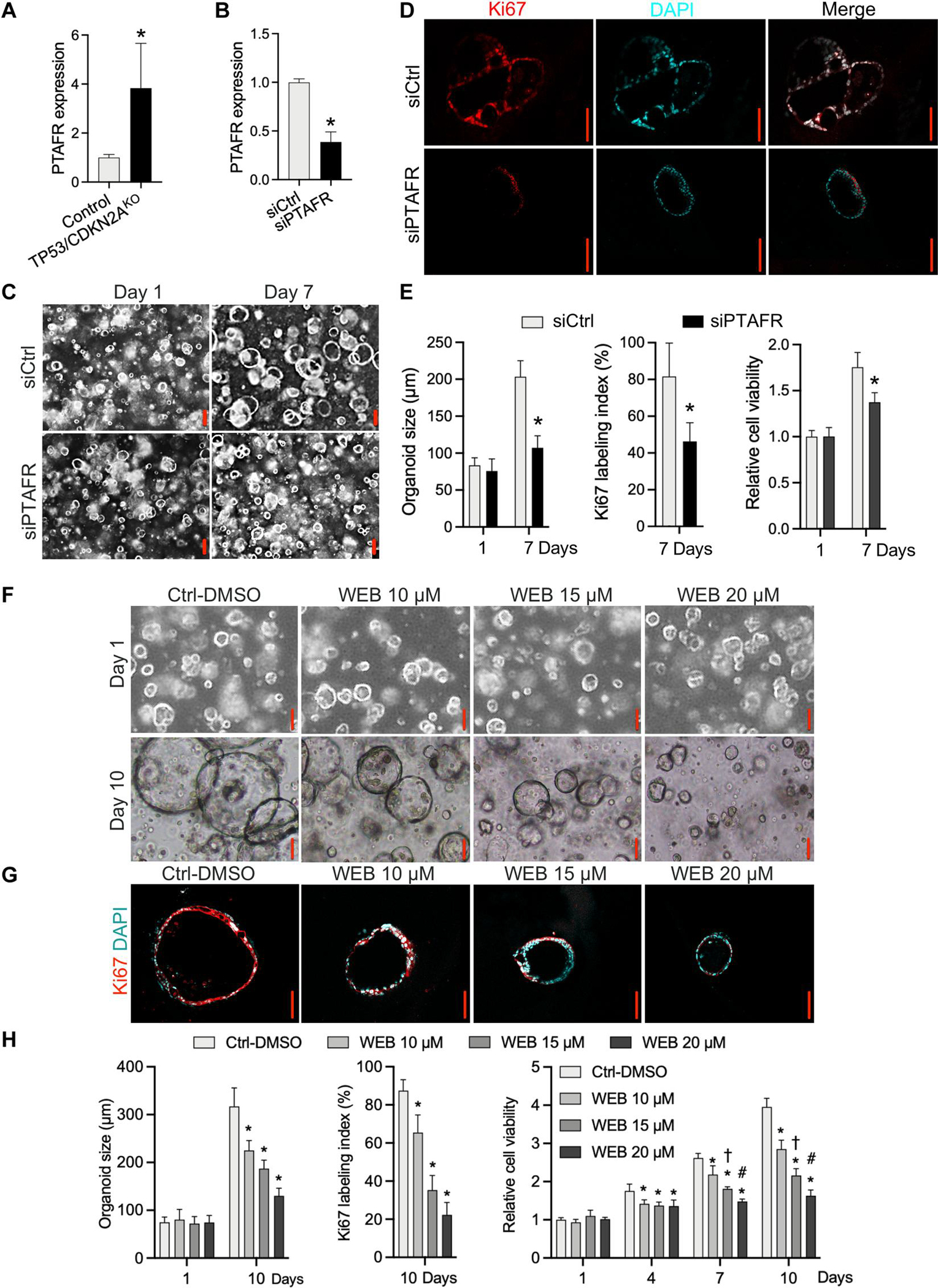
(A) mRNA expression of PTAFR in TP53/CDKN2AKO versus control GEJ organoids. (B) Knockdown of PTAFR mRNA by siRNAs in TP53/CDKN2AKO GEJ organoids. (C) Organoids treated with control silencing RNA (siCtrl) and PTAFR silencing RNA (siPTAFR) were photomicrographed under phase-contrast microscopy and collected for (D) Ki67 IF staining. (E) Average organoid size and Ki67 labeling index were quantified, and cell viability was determined by WST-1 assays at indicated time points. *P < 0.05 versus same-day siCtrl. Scale bars, 100 μm. (F to H) TP53/CDKN2AKO GEJ organoids were treated with vehicle control (0.1% DMSO) or a specific PTAFR pharmacologic antagonist, WEB2086, at various concentrations. Average organoid size in bright-field (F) and Ki67 IF images (G) and cell viability (H) were determined by phase-contrast imaging and WST-1 assays, respectively. (G) Ki67 labeling images and quantification (H) were obtained on day 10. Scale bars, 100 μm. Data are represented as means ± SD; n = 4 biological replicates. *P < 0.05 versus Ctrl-DMSO on the same day; †P < 0.05 versus WEB 10 μM on the same day; #P < 0.05 versus WEB 15 μM on the same day by ANOVA.
Next, we tested the inhibition of PTAFR in vivo. TP53/CDKN2AKO organoid cells (2 × 106 cells per injection) were subcutaneously injected into the armpits of nude mice. After 3 days of inoculation, mice were treated with vehicle control (1.25% DMSO) or WEB2086 (5 mg/kg per day) by intraperitoneal injection every 2 days for 3 weeks. Within a 3-month postinjection observation interval, three of the five injected mice developed tumors in the DMSO control group within 7 weeks (Fig. 5A), similar to what we observed above (Fig. 2F). WEB2086 treatment completely prevented tumor formation in TP53/CDKN2AKO organoids (Fig. 5A). We next assessed the effect of WEB2086 in a xenograft model derived from an established EAC cell line (Eso26). Again, PTAFR inhibition resulted in significant suppression of EAC xenograft growth and proliferation (P < 0.05; Fig. 5, B and C). IHC analysis showed down-regulated expression of Ki67 in WEB2086-treated Eso26 tumor xenografts (Fig. 5D). These results demonstrate that blockade of the PTAF/PTAFR lipid cascade potently inhibits TP53/CDKN2AKO GEJ organoid growth, proliferation, and tumorigenesis in vitro and in vivo, suggesting an important function for PTAF/PTAFR in mediating early neoplastic progression at the GEJ.
Fig. 5. Blocking the PTAF/PTAFR pathway inhibits in vivo tumorigenesis of TP53/CDKN2AKO GEJ organoids and Eso26 cells.
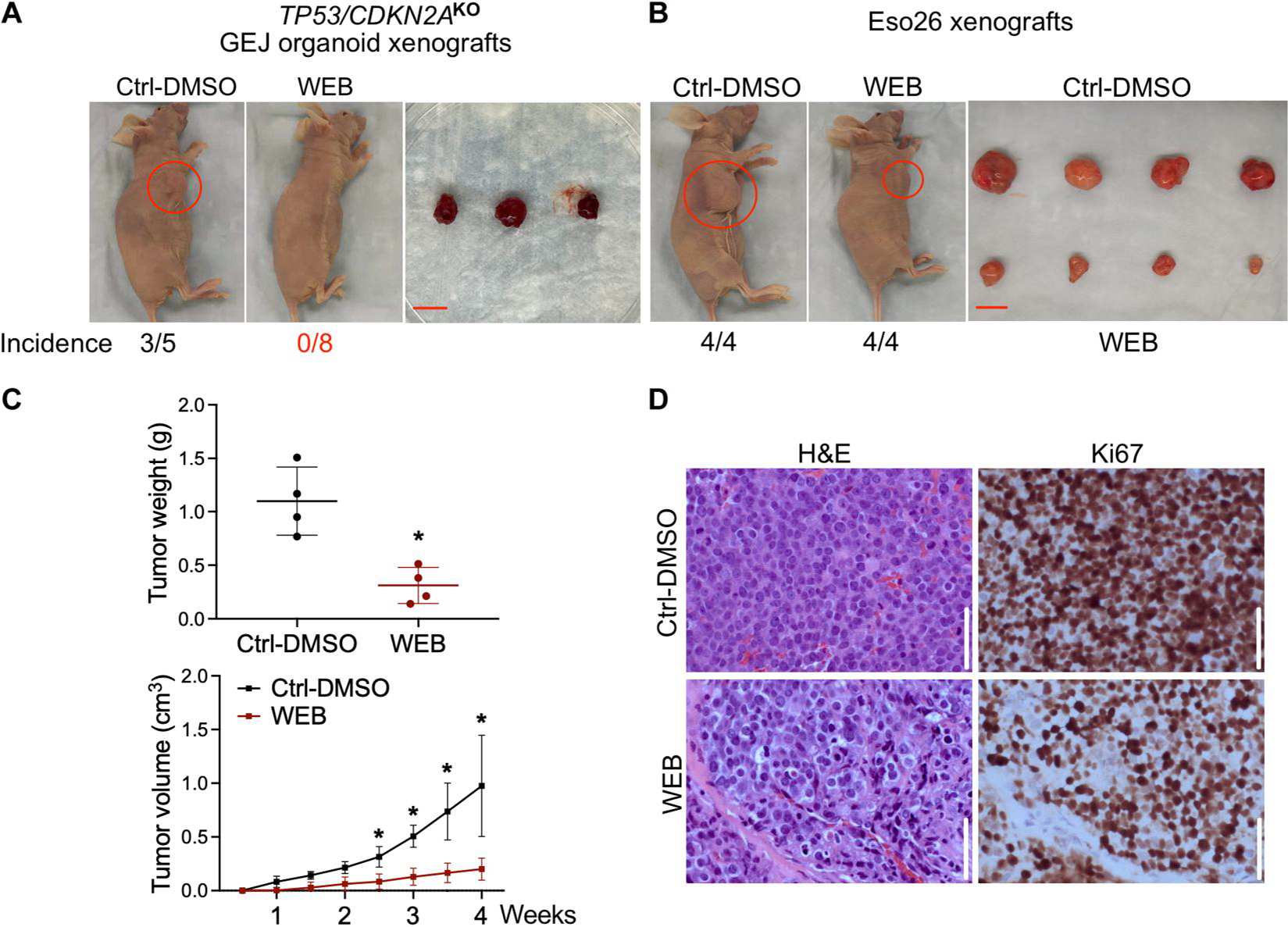
TP53/CDKN2AKO organoid cells or Eso26 cells (2 × 106 cells per injection) were subcutaneously injected into the armpits of nude mice. After 3 days of inoculation, WEB2086 (5 mg/kg per day) or vehicle control (1.25% DMSO in PBS) was administered by intraperitoneal injection every 2 days for 3 weeks. (A) Xenograft images and incidence rates after WEB2086 treatment in TP53/CDKN2AKO GEJ organoids. Scale bar, 1 cm. This experiment was repeated once with similar results. (B) Xenograft images and incidence rates after WEB2086 treatment in Eso26 xenografts. Scale bar, 1 cm. (C) Tumor weight at week 4 and growth of Eso26 xenografts with and without WEB2086 treatment. Data are represented as means ± SD; n = 5. *P < 0.05 versus Ctrl-DMSO on the same day by Student’s t test. (D) H&E staining and Ki67 IHC in Eso26 xenografts. Scale bars, 100 μm.
Inactivation of TP53/CDKN2A alters the DNA methylome and transcriptome of GEJ organoids
To explore differences at the transcriptomic and epigenomic levels between control and TP53/CDKN2AKO organoids, we performed transcriptome sequencing [RNA sequencing (RNA-seq)] and Illumina MethylationEPIC array profiling, respectively. Specifically, paired wild-type and TP53/CDKN2AKO organoids from four patients were subjected to RNA-seq. Compared with the control group, TP53/CDKN2AKO organoids contained 556 significantly (adjusted P < 0.05 and an absolute fold change of >2) differentially expressed genes (DEGs; 312 up-regulated and 244 down-regulated; Fig. 6A and table S5). Gene Ontology (GO) analysis of these genes identified strong enrichment of biological processes and pathways related to mitotic entry and cell cycle progression (Fig. 6B and table S6). This finding is consistent with the above observation of significantly accelerated proliferation and growth of TP53/CDKN2AKO organoids.
Fig. 6. DNA methylome and transcriptome profiling of TP53/CDKN2AKO GEJ organoids.
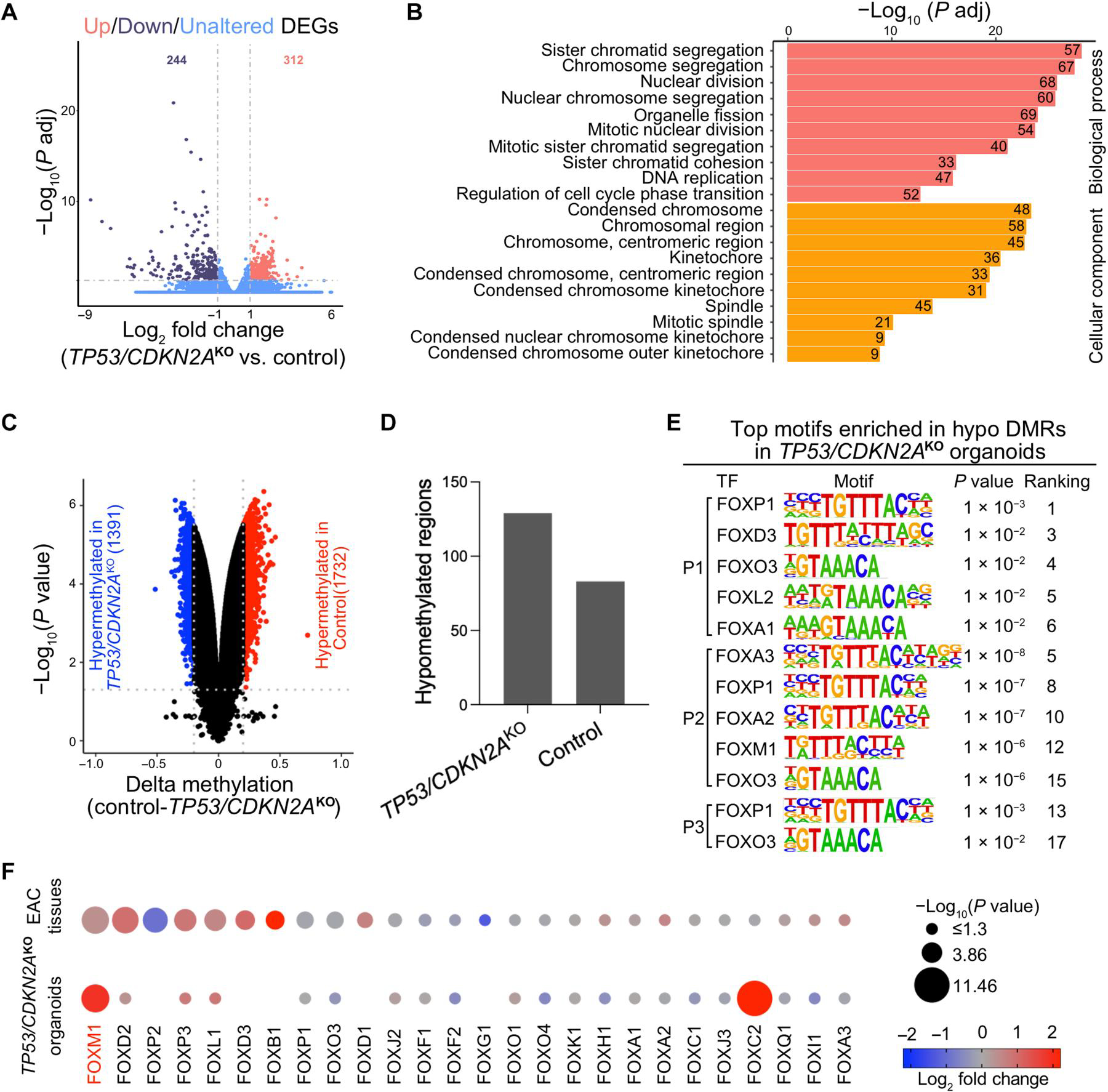
(A) A volcano plot showing differentially expressed genes (DEGs) in TP53/CDKN2AKO organoids versus control organoids. Data represent four biological replicates. (B) Gene ontology (GO) analysis identified top ranked key terms of biological processes and cellular components enriched in DEGs in TP53/CDKN2AKO versus control organoids. (C) A volcano plot showing differentially methylated CpGs (red hypermethylated in control and blue hypermethylated in TP53/CDKN2AKO) and (D) column plots showing DMRs in TP53/CDKN2AKO versus control organoids. (E) Top ranked transcription factor (TF) binding motifs from the forkhead box (FOX) family enriched in hypomethylated differentially methylated regions (DMRs) in TP53/CDKN2AKO versus control organoids. P values were calculated using the HOMER package. P1, patient 1; P2, patient 2; P3, patient 3. (F) Dot plots showing expression of FOX family TFs in TP53/CDKN2AKO versus control organoids (bottom dots, n = 4 biological replicates) and in EAC tumors (n = 88) versus normal GEJ samples (n = 9) from the TCGA (top dots). Genes are ranked on the basis of the −log10 (P value) comparing EAC tumors with normal GEJ samples. The dot size denotes −log10 (P value); color indicates log2 fold change; missing dots correspond to undetectable mRNA expressions.
At the epigenomic level, we identified both differentially methylated CpGs and differentially methylated regions (DMRs) between control and TP53/CDKN2AKO organoids derived from each of the four patients. For example, in organoids derived from patient 1, 1732 and 1391 CpG dinucleotides were significantly hypomethylated (adjusted P < 0.05 and absolute delta methylation change of >0.2) in mutant and control organoids, respectively (Fig. 6C). These CpG sites corresponded to 129 and 83 hypomethylated DMRs in TP53/CDKN2AKO and control organoids, respectively (Fig. 6D and table S7). Results of organoids derived from the other three patients are shown in fig. S7 and table S7.
DNA hypomethylated regions are known to contain regulatory elements associated with the binding of transcription factors (TFs) (22, 23). To identify candidate TFs implicated in our neoplastic GEJ organoid model, we investigated enriched TF recognition motif sequences in hypomethylated DMRs using the Hypergeometric Optimization of Motif EnRichment (HOMER) package (24). Motifs of the forkhead box (FOX) TF family were among the most enriched sequences in hypomethylated DMRs in TP53/CDKN2AKO organoids (Fig. 6E). Because different FOX TF family members recognize similar motif sequences (25), we next sought to identify which factors were involved. Previous studies have shown that expression of a particular TF correlate with amounts of demethylation in regions that it occupies (26, 27). Therefore, we analyzed mRNA expression of the 26 FOX TFs based on RNA-seq data from our organoids as well as on normal GEJ and EAC data from the TCGA. Both FOXM1 and FOXC2 were increased in TP53/CDKN2AKO organoids. FOXM1 was also up-regulated in EAC versus normal GEJ tissues (Fig. 6F), suggesting it as a candidate TF enriched in hypomethylated DMRs with possibly increased activity in TP53/CDKN2AKO organoids. FOXM1 is a known regulator of cell proliferation and cell cycle progression in cancer (28, 29), in line with the proneoplastic phenotypes in our TP53/CDKN2AKO organoids.
PTAFR is a direct transcriptional target of FOXM1
Our above data not only identified PTAF phospholipids as one of the most induced classes of lipid molecules in our lipidomic profiling but also revealed the up-regulation of its cognate receptor, PTAFR, in TP53/CDKN2AKO GEJ organoids (Fig. 4). To further elucidate mechanisms mediating up-regulation of PTAFR expression in TP53/CDKN2AKO organoids, we performed motif enrichment analysis of the PTAFR promoter, which revealed that the FOX motif sequence again was highly enriched (no. 4) (Fig. 7A). This analysis was relevant, given our above data identifying FOXM1 as a potential regulator involved in epigenetic alterations in TP53/CDKN2AKO organoids.
Fig. 7. PTAFR is a direct downstream target of FOXM1.
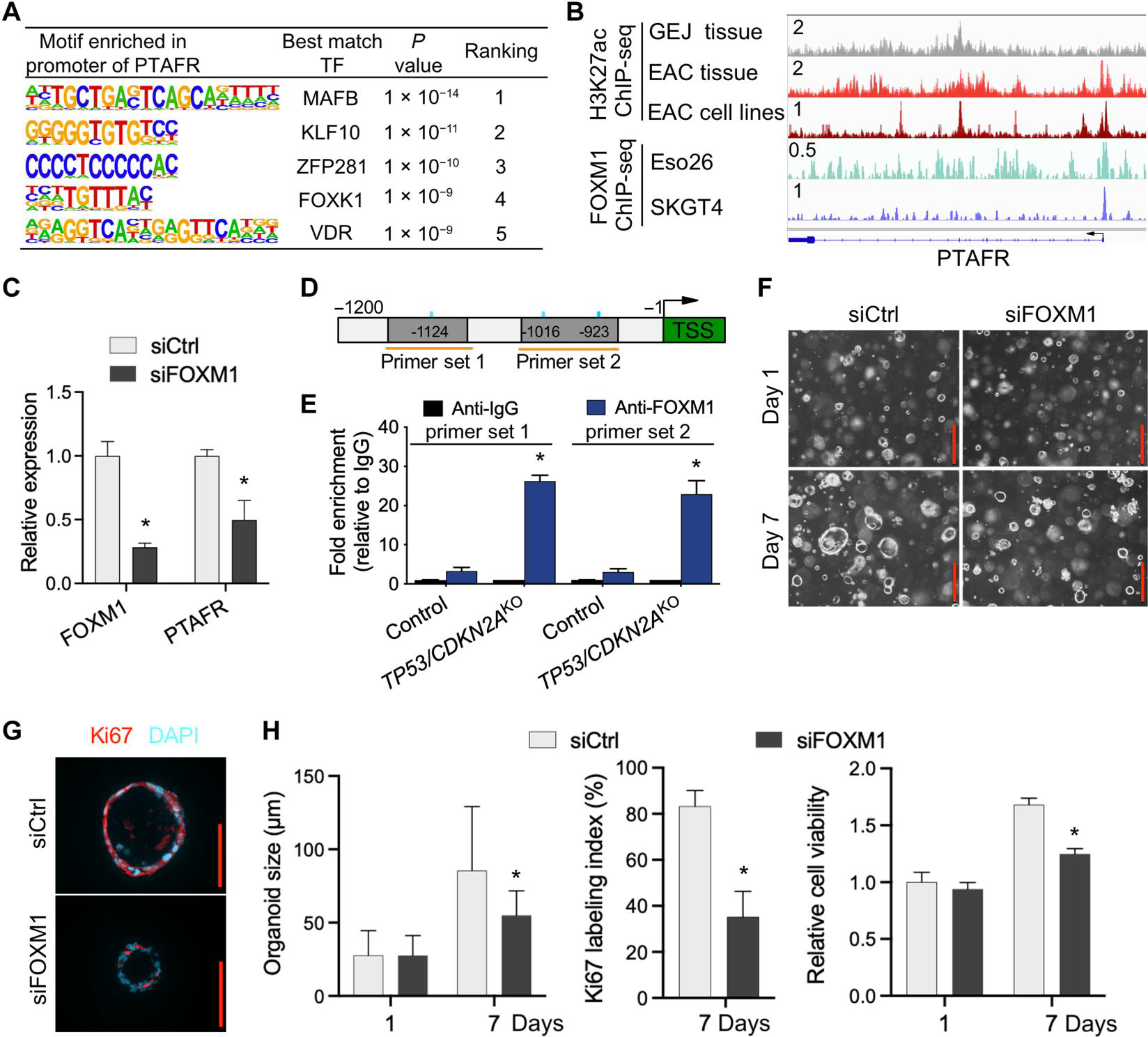
(A) Motif enrichment analysis of PTAFR promoter region in organoids. (B) ChIP-seq profiles for H3K27ac and FOXM1 at the PTAFR locus in indicated tissues and cell lines. All H3K27ac signals are shown at the same scale. (C) Relative expression of PTAFR mRNA after knockdown of FOXM1 by siRNAs in TP53/CDKN2AKO GEJ organoids. (D) Schematic of PTAFR transcriptional start site (TSS) (green) and upstream sequence. PCR primers for ChIP experiments (orange horizontal lines) and FOXM1 binding motifs (blue vertical lines) are indicated. (E) ChIP-qPCR detection of FOXM1 occupancy at the PTAFR promoter region in organoids using primer sets indicated in (D). ChIP was performed with either anti-FOXM1 or anti-IgG antibodies, and fold enrichment relative to anti-IgG is shown. Data are represented as means ± SD; n = 4 biologic replicates. *P < 0.05 versus control by ANOVA. (F) Organoids treated with siCtrl and siFOXM1 were photomicrographed under phase-contrast microscopy and collected for (G) Ki67 IF staining. (H) Average organoid size and Ki67 labeling index were quantified, and cell viability was determined by WST-1 assay. Scale bars, 100 μm. Data are represented as means ± SD; n = 4 biologic replicates. *P < 0.05 versus siCtrl on the same day by ANOVA.
To validate whether FOXM1 regulates the transcription of PTAFR, we first performed chromatin immunoprecipitation sequencing (ChIP-seq) using an anti-FOXM1 antibody. FOXM1 occupied the promoter of PTAFR in two different EAC cell lines (Fig. 7B). Moreover, our recent H3k27ac ChIP-seq data (30) showed that PTAFR harbors strong H3k27ac signals at its promoter and candidate enhancers in both EAC primary tumors and cell lines (Fig. 7B), suggesting robust transcriptional activation. In contrast, in normal GEJ samples, H3k27ac was barely deposited at the PTAFR locus, indicating weak or inactive transcription (Fig. 7B). This pattern of histone modification was consistent with mRNA expression of PTAFR, which was up-regulated in EAC versus normal GEJ samples (fig. S6).
Because the above ChIP-seq data were generated in EAC cell lines and primary tumors, we next performed FOXM1 ChIP–quantitative polymerase chain reaction (qPCR) in GEJ organoids. Endogenous FOXM1 showed prominent occupancy at the PTAFR promoter in TP53/CDKN2AKO, with a more than 7.9-fold increase in enrichment compared to wild-type GEJ organoids (Fig. 7, D and E). To validate the regulation of FOXM1 upon PTAFR transcription, we silenced FOXM1 using siRNA in TP53/CDKN2AKO organoids, which showed that FOXM1 knockdown significantly (P < 0.05) reduced PTAFR expression (Fig. 7C). FOXM1 silencing reduced average organoid size, cell viability, and Ki67 index in TP53/CDKN2AKO organoids (Fig. 7, F to H), phenocopying the effects that we obtained with PTAF/PTAFR blockade. In addition, mRNA expression of PTAFR and FOXM1 were measured in normal control, TP53KO, and TP53/CDKN2AKO organoids (fig. S8). These experiments showed that single knockout of TP53 increased expression of PTAFR and FOXM1 mRNA in GEJ organoids, and this effect was further amplified by double knockout of TP53/CDKN2A. Thus, these data demonstrate that TP53/CDKN2A inactivation enhances direct binding of FOXM1 to the PTAFR promoter, thereby augmenting PTAFR expression and proliferation in GEJ organoids.
DISCUSSION
A major hurdle in understanding the molecular origins and biology of GEJ cancer is a paucity of appropriate biologically relevant models, particularly of early neoplastic events. Primary 3D organoid cultures derived from normal epithelia represent a desirable system for studying critical properties of the original native tissue in vitro, including morphological, histological, and molecular features (3). To our knowledge, we describe here the first human normal GEJ-derived organoid culture model, with a highly reliable protocol ensuring successful organoid culture from endoscopic biopsies. We show that wild-type GEJ organoids can be propagated in vitro for at least 4 months and that TP53/CDKN2AKO organoids can be propagated even longer (for at least 19 months). This platform offers great promise for modeling GEJ-associated diseases, characterizing healthy and diseased GEJ conditions and finding molecular mechanisms underlying the transition from normal to pathological GEJ.
Furthermore, we also demonstrate here that CRISPR-engineered human TP53/CDKN2AKO organoids offer a productive tool for modeling early neoplastic events at the GEJ. TP53 mutations have been detected in patients with benign Barrett’s esophagus (BE) who later develop high-grade dysplasia or EAC (31–33), suggesting their involvement in early neoplasia at the GEJ anatomic region. Likewise, CDKN2A is inactivated in more than 80% of BE and EAC tissues (34–36). Similar molecular abnormalities also occur in gastric cardia neoplasia (37–42), supporting the contribution of TP53 and CDKN2A to diverse types of adenocarcinogenesis involving the GEJ region. In agreement with these static observations, our dynamic model establishes that TP53/CDKN2A inactivation directly causes biologic and molecular features consistent with GEJ neoplastic progression.
TP53 and CDKN2A have been implicated in several lipid metabolic processes (43–46). However, to our knowledge, direct evidence does not yet exist, linking TP53 and CDKN2A to lipid metabolic aberrations occurring during human GEJ carcinogenesis. Here, we demonstrate that TP53/CDKN2A inactivation directly alters lipidomic profiles in GEJ organoids. We identify that the lipids most up-regulated by TP53/CDKN2AKO in GEJ organoids include several PTAFs, a family of phospholipid mediators with known signaling effects in important biological processes, including cancer initiation (21, 47–50). PTAF phospholipid is elevated in a feline model of esophagitis (51); PTAF phospholipid is also more frequently elevated in gastric cancer than in healthy control patients (52). PTAFs function primarily by binding to their cognate receptor, PTAFR (53–55). PTAFR is abundantly expressed in human gastric adenocarcinoma tissues; moreover, PTAFR overexpression is strongly associated with poor prognostic clinical parameters (56–58). Consistent with these known clinical and biological data, we identify here PTAF/PTAFR as an etiologic mediator in neoplastic progression induced by TP53/CDKN2AKO at the GEJ.
In preclinical settings, the PTAF/PTAFR pathway has been targeted using small-molecule inhibitors in different frank cancers in vitro (59, 60). However, the PTAF/PTAFR pathway has so far not been investigated during early neoplastic transformation, likely because modeling early malignant evolution has been difficult in the absence of organoid technology. In our model, the PTAFR antagonist WEB2086 inhibited early neoplastic changes in TP53/CDKN2AKO organoids and also caused analogous growth suppression in Eso26 EAC cells.
Our data also suggest that TP53/CDKN2AKO elicits epigenetic and transcriptional reprogramming that propels the normal GEJ toward a malignant state. Here, our integrative analyses identify enrichment of FOXM1 binding sequences in demethylated regions coupled with the up-regulation of this TF, directly caused by TP53/CDKN2A inactivation. FOXM1 is overexpressed in multiple solid tumors (61–63), and signaling downstream of this TF contributes to cancer development and progression by cross-talk with multiple cell signaling pathways. We also demonstrate a mechanistic link between FOXM1 and PTAFR, where FOXM1 binds to the PTAFR gene promoter, directly activating the transcription of PTAFR. Consistent with results upon PTAF/PTAFR inhibition, FOXM1 knockdown in TP53/CDKN2AKO GEJ organoids leads to severe cell growth inhibition.
Our human primary benign GEJ organoid model and its proneoplastic induction by TP53/CDKN2A knockout now enable molecular deconstruction of early GEJ tumorigenesis and neoplastic progression. However, our study still has some limitations, because in vitro organoid models cannot fully recapitulate the in vivo complexity of the neoplastic process or mirror system-level and immune system interactions. Small sample size is another limitation of our study. Further improvements to the culture system, such as organ-on-a-chip methods and increases in patient sample sizes, and further research to assess the therapeutic prospect of PTAF/PTAFR inhibition in GEJ tumors may reinforce the power of future studies.
In summary, we have established a robust organoid model system for investigating early GEJ neoplastic events; furthermore, our lipidomic, epigenetic, and transcriptional profiling studies yield valuable insights into mechanistic underpinnings of GEJ malignancy. The highly induced phospholipid family, PTAFs, and their receptor, PTAFR, together show strong proneoplastic activity in GEJ evolution and represent potential therapeutic targets against GEJ cancers. Conceivably, our strategies for transforming organoids derived from human primary normal epithelia into more neoplastic entities are generalizable to modeling other cancer-driving events and other types of malignancies, where they may expand our understanding of tumor-specific gene regulatory networks and identify potential therapeutic targets.
MATERIALS AND METHODS
Study design
The objectives of this study were to establish a robust model for investigating early GEJ neoplastic transformation, identify crucial events occurring during GEJ model tumorigenesis, and discover potential cancer therapeutic strategies. The study used patient-derived normal GEJ organoids and engineered these into TP53/CDKN2AKO organoids using the CRISPR-Cas9 genome editing system. After genetic and chemical perturbations, we investigated phenotypic, metabolic, transcriptomic, and epigenomic alterations as well as tumorigenesis in normal control versus TP53/CDKN2AKO organoids. The number of replications is indicated in each figure legend.
Patient samples
In accordance with the approved Institutional Review Board protocols at the Johns Hopkins Hospital, primary human endoscopic biopsy samples were acquired from the GEJs of patients undergoing diagnostic esophagogastroduodenoscopy at the Johns Hopkins Hospital under written informed consent. Tissue samples used to generate organoids were pathologically confirmed as normal cardio-oxyntic mucosa: No goblet cells or specialized epithelium characteristic of Barrett’s mucosa were observed (table S1).
Cell lines and maintenance
L Wnt-3A cells (CRL-2647) were purchased from the American Type Culture Collection and maintained in Dulbecco’s modified Eagle medium (DMEM) with 10% fetal bovine serum (FBS) to produce Wnt-3A–conditioned medium. Cultrex HA-R-Spondin1-Fc 293T cells (3710-001-01) were purchased from Bio-Techne and maintained in DMEM with 10% FBS to generate R-spondin-1–conditioned medium. Eso26 cells were purchased from Jennio Biological Technology and grown in RPMI 1640 with 10% FBS.
GEJ organoid cultures
GEJ organoids were established as described in Fig. 1A. Briefly, fresh endoscopic GEJ biopsies were preserved in ice-cold conditioned phosphate-buffered saline (PBS) [PBS containing 10 μM rho-associated kinase (ROCK) inhibitor Y27632, 2% (v/v) penicillin/streptomycin, and 1× Primocin] until further processing within 24 hours. After washing biopsies with conditioned PBS at least five times, samples were minced into fragments of <1 mm3 using micro-dissecting scissors. Tissue fragments were digested in DMEM containing 2.5% (v/v) FBS, 1% (v/v) penicillin/streptomycin, collagenase type IX (1 mg/ml), and dispase type II (120 μg/ml) at 37°C with 200 rpm shaking for 40 to 90 min. After centrifugation at 400g at 4°C for 3 min, pelleted cell clusters were resuspended in Matrigel. Using 24-well plates, 2000 cells were seeded per well in 50 μl of Matrigel. After incubating at 37°C for 10 min to solidify the Matrigel, 500 μl of growth medium was added to each well. The growth medium for GEJ organoids was Advanced DMEM/F-12 supplemented with 50% (v/v) Wnt-3A–conditioned medium (homemade), 20% (v/v) R-spondin-1–conditioned medium (homemade), 1% (v/v) penicillin/streptomycin, 10 nM prostaglandin E2, human fibroblast growth factor 10 (100 ng/ml), human epidermal growth factor (50 ng/ml), Noggin (100 ng/ml), 1 mM N-acetyl cysteine, 10 mM nicotinamide, 10 nM gastrin I, 500 nM A-83-01, 10 μM SB202190, 10 μM Y27632, 5 μM CHIR99021 (only for the first one to two passages), 1× Primocin, and 1× B-27 supplement. Culture medium was changed once every 3 days until the organoids were ready to passage. For passaging, organoids were washed in PBS and digested with TrypLE containing 10 μM Y27632 for 5 to 7 min at 37°C. After incubation, DMEM/F12 was added to stop digestion. Organoids were mechanically dissociated by pipetting and centrifuged at 500g for three min. After resuspending the pellet in Matrigel, 50 to 100 μl per droplet of the cell Matrigel suspension were plated onto a new culture plate. Reagents used for organoid culture are listed in table S2.
Organoid viability assay (WST-1 assay)
To quantify metabolically active viable cells, organoids were seeded onto 96-well plates and cultured. At indicated time points, 10 μl per well of Cell Proliferation Reagent WST-1 assay kits were added to the 96-well plates and incubated with organoids for 90 min. After incubation, only medium was transferred to the wells of a new 96-well plate, which was read at an absorbance of 450 nm by a Thermo Fisher Scientific microplate reader. All experiments were performed in triplicates.
CRISPR-Cas9 genomic editing of GEJ organoids
Organoids were electroporated using the NEPA21 (Nepa Gene) system and the Alt-R CRISPR-Cas9 System (Integrated DNA Technologies). Cas9:gRNA RNP complex was prepared as follows: To make the 100 μM gRNA complex, 200 μM tracrRNA labeled with ATTO 550 and 200 μM crRNA were mixed in equimolar concentrations, heated at 95°C for 5 min, and then allowed to slowly cool to room temperature; to produce the RNP complex for each electroporation, 6 μl of gRNA complex (100 μM), 8.5 μg of Cas9 nuclease (10 μg/μl), and 10.5 μl of duplex buffer were combined and incubated at room temperature for 10 min. RNP complex was stored for further use at −80°C. Two days before electroporation, organoids were passaged and maintained in organoid culture medium without antibiotics including 5 μM CHIR99021. Organoids were dissociated into clusters of 10 to 15 cells, resuspended in 80 μl of electroporation buffer containing 4 μM electroporation enhancer, and then mixed with 25 μl of RNP complex targeting TP53 and 25 μl of RNP complex targeting CDKN2A. The mixture was transferred into a precooled 2-mm electroporation cuvette. Electroporation parameters were set according to Fujii et al. (64). After electroporation, 400 μl of prewarmed culture medium including 5 μM CHIR99021 was immediately added to the electroporation cuvette. Cells were seeded after incubation for 40 min at 37°C. Two days after electroporation, transfection efficiency was measured by fluorescence microscopy. Three days after electroporation, organoids were treated with 10 μM Nutlin-3a for functional selection of TP53 mutant cells for 2 to 3 weeks, on the basis that Nutlin-3a inhibits the proliferation of TP53 wild-type cells. Organoids electroporated with the negative control RNP complex were used as the control group. To validate targeted mutations, genomic DNA from edited organoids was extracted, followed by PCR amplification, topoisomerase-based cloning, and Sanger sequencing. All reagents and single-guide RNA sequences used in this section are provided in table S2.
MALDI-IMS of the lipidome in GEJ organoids
Organoids were transferred to even molds and immersed in M-1 Embedding Matrix after being isolated from Matrigel using Cell Recovery Solution and washed with cold PBS for three times. Organoid molds were wrapped in aluminum foil and floated on liquid nitrogen for progressive freezing. Frozen organoids were equilibrated to −20°C, cryosectioned at a thickness of 10 μm, and thaw-mounted onto temperature-equilibrated, hexane-and-ethanol–washed indium tin oxide slides (Delta Technologies) on a Leica CM1860 UV cryostat. All organoids were sectioned in a layout that maximizes the number of sections per slide to compare TP53/CDKN2AKO versus control GEJ organoids. Several serial sections were cryosectioned with the same layout for technical repeats. Slides were sprayed with DHB (40 mg/ml) and dissolved in 70% high-performance liquid chromatography (HPLC)–grade methanol/30% HPLC-grade water using an HTX M5 sprayer with the following parameters: nozzle temperature of −75°C, eight passes, a flow rate of 0.1 ml/min, a nozzle velocity of 1200 mm/min, a 3-mm track spacing, a crisscross pattern, a pressure of 10 psi, a gas flow rate of 3 liters/min, and a drying time of 10 s. The final matrix density was 8.89 × 10−3 mg/mm2, and the linear flow rate was 8.33 × 10−5 ml/mm.
MALDI-IMS was acquired on Bruker MALDI–time-of-flight (TOF)/TOF rapifleX instrument (Bruker Daltonics) in the Johns Hopkins Applied Imaging Mass Spectrometry Core in reflectron-positive mode at 20-μm pixel size with a 20-μm raster and 20-μm imaging laser, 200 laser shots per pixel, and a mass range of m/z 40 to 2000. Imaging data were collected using flexImaging version 5.0 and flexControl version 4.2 (Bruker Daltonics), and data were normalized to total ion current (TIC) for all data analysis. MALDI lipid imaging data were imported into SCiLS Lab software (version 2020a, SCiLS GmbH), and alignment was performed with TIC to remove any drift artifacts from imaging. Quantitative spectral, pixel-based, paired comparisons (65–67) were done between TP53/CDKN2AKO and control GEJ organoids. For structural identification, m/z’s of the top lipids were identified by on-tissue MS/MS using collision-induced decay with argon using the single beam laser with a resultant field of 54 μm by 54 μm with 4000 laser shots and an isolation window of ±2 Da. MS/MS spectra were collected from both knockout organoids. For initial identification, the LIPID MAPS Structure Database (68) was used by uploading a peak list, searching [M + H]+ and [M + Na]+ with a mass tolerance of ±0.2 m/z, and all lipid classes are selected. This generated a list of potential hits, which were used to solve MS/MS spectra in ChemDraw Professional version 16.0 (PerkinElmer).
Genome-wide DNA methylation profiling and data analysis
DNA methylation profiles for four paired sets of control and double-knockout GEJ organoids were generated using the Illumina MethylationEPIC array platform, which combines bisulfite conversion of genomic DNA and whole-genome amplification with direct, array-based capture and scoring of CpG loci. Genomic DNA was extracted from organoids using DNeasy Blood & Tissue kits. All DNA samples were quantified by the Qubit double-stranded DNA broad range (BR) Assay, assessed for purity by A260/280 and A260/230 ratio, and examined for integrity by electrophoresis on 0.8% agarose gels. DNA samples were then hybridized to Infinium MethylationEPIC BeadChips, following the Infinium HD Methylation Assay Protocol (69).
The SeSAME package (70) was used to extract the DNA methylation value of each probe using the OpenSesame function. Recommended general masking probes were removed according to the annotation file of Infinium DNA methylation arrays. Differentially methylated probes were identified by the Limma package (version 3.46.0) with adjusted P < 0.05 and an absolute delta methylation change of >0.2. We further identified DMRs based on differentially methylated probes by the DMRcate package, with Fisher’s exact test P of <0.05.
RNA-seq and data analysis
RNA-seq was performed on four paired sets of control and double-knockout GEJ organoids derived from four patients. Total RNA was extracted and treated with deoxyribonuclease I (DNase I) before sequencing. Libraries were constructed using NEBNext Ultra Directional RNA Library Prep kits. Quantified libraries were sequenced on the Illumina NovaSeq 6000 platform, and paired-end reads were generated. An index of the reference genome was built, and paired-end clean reads were aligned to the reference genome using HISAT2 v2.0.5. Read counts for each gene were generated by FeatureCounts v1.5.0-p3. Fragments per kilobase of exon per million mapped fragments of each gene were calculated on the basis of the length of the gene and the read count mapped to this gene. DESeq2 results were used for differential expression analysis, and genes with an adjusted P < 0.05 and an absolute fold change of >2 found by DESeq2 were designated as differentially expressed. GO enrichment analysis of DEGs was implemented using the clusterProfiler R package (71).
Real-time quantitative PCR
Total RNA was extracted using RNeasy kits, and DNA was eliminated using on-column DNase digestion. A total of 500 μg of RNA was reversely transcribed using iScript Select cDNA Synthesis kits (Bio-Rad). qPCR was performed using iTaq Universal SYBR Green Supermix. All results were normalized to β-actin expression. Primers used for quantitative reverse transcription (qRT)–PCR are listed in table S2.
siRNA-directed gene silencing
Two days before electroporation, organoids were passaged and cultured in organoid culture medium without antibiotics including 5 μM CHIR99021. Organoids were dissociated into clusters of 10 to 15 cells, resuspended in 100 μl of electroporation buffer containing 4 μM electroporation enhancer, and then mixed with 10 μl of 50 mM siRNA. The organoids were electroporated using the same procedure mentioned above 24 hours after electroporation. Total DNA from cloned organoids was then extracted and reversely transcribed into cDNA, followed by qRT-PCR to validate knockdown efficiency. siRNA sequences are provided in table S2.
Histology, IF, and IHC
Organoid cultures and tissues were fixed overnight in 10% formalin at room temperature. Paraffin-embedded organoids and tissues were serially sectioned into 10-μm slices. Paraffin sections were deparaffinized and rehydrated, followed by either staining for H&E or antigen retrieval in subboiling 10 mM sodium citrate buffer (pH 6.0) for 10 min. For IF, slides were permeabilized in 0.5% Triton X-100 in PBS and blocked in 1% goat serum in PBS for 30 min at room temperature. After blocking, slides were incubated with anti-Ki67 (1:200, Abcam) or anti-TFF3 (1:100, Abcam) overnight in a humidified chamber at 4°C. Sections were washed by PBS with 0.1% Tween 20 (PBST) (three times for 5 min each) and incubated with Alexa Fluor secondary antibodies (1:500) for 1 hour. After washing with PBST, slides were mounted with Fluoroshield with 4′,6-diamidino-2-phenylindole (Sigma-Aldrich). IHC was performed using the automated BOND-MAX autostainer by Leica for the following antibodies: cytokeratin AE1/AE3 (1:200; Santa Cruz Biotechnology). Images were acquired with a Leica Inverted Confocal SP8 (Johns Hopkins Medicine Ross Imaging Center). Reagents used here are listed in table S2.
Chromatin immunoprecipitation
ChIP experiments were performed using the EZ-Magna ChIP A/G ChIP Kit (Merck Millipore, 17-10086) according to the manufacturer’s procedures. Cells were cross-linked with 1% formaldehyde and lysed with cell lysis buffer containing 1× Protease Inhibitor Cocktail II. Nuclei were isolated with nuclear lysis buffer supplemented with 1× Protease Inhibitor Cocktail II. The chromatin extract was sonicated (for 8 min in total, 30% amplitude, pulse on for 10 s, and pulse off for 20 s) and sheared to a length between 200 and 1000 bp on wet ice. The sheared cross-linked chromatin was immunoprecipitated with antibodies coincubated with magnetic protein A/G beads. Antibodies included anti-FOXM1 (5 μg per ChIP reaction; Invitrogen, 702664) and normal mouse immunoglobulin G (IgG) (1 μg per reaction). Purified DNAs were subjected to qPCR. qPCR primers are listed in table S2.
Xenotransplantation in nude mice
All procedures and experimental protocols involving mice were approved by the Animal Experiment Committee of the Johns Hopkins University School of Medicine. For xenotransplantation, 2 × 106 organoid cells or Eso26 cells resuspended in cold 50% Matrigel were injected into the axillary of nude mice. For the WEB2086 treatment assay, WEB2086 (5 mg/kg per day) or vehicle control (1.25% DMSO in PBS) was administered by intraperitoneal injection every 2 days for 3 weeks. Xenograft size was measured twice a week. The xenograft volume (V) was monitored by measuring the length (L) and width (W) with a caliper based on the formula V = 1/2 × (L × W2). Mice were euthanized at the end of the experiment, and xenografts were excised for further analyses.
Statistical analysis
The data are presented as means ± SD unless indicated otherwise. Statistical analysis was assessed using GraphPad Prism 9.2. For in vitro experiments, we used Student’s t test or one-way analysis of variance (ANOVA) unless otherwise noted in the figure legends. For xenograft experiments, we used unpaired Student’s t test. P < 0.05 was considered statistically significant.
Supplementary Material
Acknowledgments:
We are indebted to the patients who participated in the study. We also thank H. Clevers and J. Drost for key reagents. We thank B. Koo, G. McNamara, Q. Cao (University of Southern California), and K. Waters (Cedars-Sinai Medical Center) for expert technical assistance. We acknowledge the Johns Hopkins Applied Imaging Mass Spectrometry (AIMS) Core Facility at the Johns Hopkins University School of Medicine. Figure 1A was created with BioRender software (https://biorender.com/).
Funding:
The research was supported by National Institutes of Health grants R01-DK118250 (to S.J.M.), UH3-CA211457 (to S.J.M.), and R37-CA237022 (to D.-C.L.); Lynn DeGregorio Family Foundation (to S.J.M.); and Emerson Collective Cancer Research Fund (to S.J.M.). H.Z., B.Z., and D.-C.L. are partially supported by institutional funds from the Herman Ostrow School of Dentistry of USC’s Center for Craniofacial Molecular Biology.
Footnotes
Competing interests: The authors have filed a provisional patent application (JHU Ref. C17281, P17281-01, or CJ Ref. 40856.101) related to the study. The authors declare that they have no other competing interests.
Supplementary Materials
This PDF file includes:
Other Supplementary Material for this manuscript includes the following:
Data files S1 and S2
Data and materials availability:
All data associated with this study are present in the paper or the Supplementary Materials. Reagents, resources, and materials used in this study are detailed in table S2. RNA-seq and methylation array data generated in this study were deposited to Gene Expression Omnibus (GEO) under the series GSE216198 and GSE216814. RNA-seq datasets of TCGA EAC and normal GEJ tissues can be obtained from the XENA data portal (http://xena.ucsc.edu/).
REFERENCES AND NOTES
- 1.Sung H, Ferlay J, Siegel RL, Laversanne M, Soerjomataram I, Jemal A, Bray F, Global Cancer Statistics 2020: GLOBOCAN estimates of incidence and mortality worldwide for 36 cancers in 185 countries. CA Cancer J. Clin. 71, 209–249 (2021). [DOI] [PubMed] [Google Scholar]
- 2.Bartel M, Brahmbhatt B, Bhurwal A, Incidence of gastroesophageal junction cancer continues to rise: Analysis of Surveillance, Epidemiology, and End Results (SEER) database. J. Clin. Orthod. 37, 40 (2019). [Google Scholar]
- 3.Lo YH, Karlsson K, Kuo CJ, Applications of organoids for cancer biology and precision medicine. Nat. Cancer 1, 761–773 (2020). [DOI] [PMC free article] [PubMed] [Google Scholar]
- 4.Kim J, Koo BK, Knoblich JA, Human organoids: Model systems for human biology and medicine. Nat. Rev. Mol. Cell Biol. 21, 571–584 (2020). [DOI] [PMC free article] [PubMed] [Google Scholar]
- 5.Snaebjornsson MT, Janaki-Raman S, Schulze A, Greasing the wheels of the cancer machine: The role of lipid metabolism in cancer. Cell Metab. 31, 62–76 (2020). [DOI] [PubMed] [Google Scholar]
- 6.Beloribi-Djefaflia S, Vasseur S, Guillaumond F, Lipid metabolic reprogramming in cancer cells. Oncogenesis 5, e189 (2016). [DOI] [PMC free article] [PubMed] [Google Scholar]
- 7.Cha J-Y, Lee H-J, Targeting lipid metabolic reprogramming as anticancer therapeutics. J. Cancer Prev. 21, 209–215 (2016). [DOI] [PMC free article] [PubMed] [Google Scholar]
- 8.Srivastava A, Srivastava P, Mathur S, Abbas S, Rai N, Tiwari S, Tiwari M, Sharma L, Lipid metabolism and mitochondria: Cross talk in cancer. Curr. Drug Targets 23, 606–627 (2022). [DOI] [PubMed] [Google Scholar]
- 9.Butler LM, Perone Y, Dehairs J, Lupien LE, de Laat V, Talebi A, Loda M, Kinlaw WB, Swinnen JV, Lipids and cancer: Emerging roles in pathogenesis, diagnosis and therapeutic intervention. Adv. Drug Deliv. Rev. 159, 245–293 (2020). [DOI] [PMC free article] [PubMed] [Google Scholar]
- 10.Ma S, Zhou B, Yang Q, Pan Y, Yang W, Freedland SJ, Ding L-W, Freeman MR, Breunig JJ, Bhowmick NA, Pan J, Koeffler HP, Lin D-C, A transcriptional regulatory loop of master regulator transcription factors, PPARG, and fatty acid synthesis promotes esophageal adenocarcinoma. Cancer Res. 81, 1216–1229 (2021). [DOI] [PMC free article] [PubMed] [Google Scholar]
- 11.Li L-Y, Yang Q, Jiang Y-Y, Yang W, Jiang Y, Li X, Hazawa M, Zhou B, Huang G-W, Xu X-E, Gery S, Zhang Y, Ding L-W, Ho AS, Zumsteg ZS, Wang M-R, Fullwood MJ, Freedland SJ, Meltzer SJ, Xu L-Y, Li E-M, Koeffler HP, Lin D-C, Interplay and cooperation between SREBF1 and master transcription factors regulate lipid metabolism and tumor-promoting pathways in squamous cancer. Nat. Commun. 12, 4362 (2021). [DOI] [PMC free article] [PubMed] [Google Scholar]
- 12.Frankell AM, Jammula S, Li X, Contino G, Killcoyne S, Abbas S, Perner J, Bower L, Devonshire G, Ococks E, Grehan N, Mok J, O’Donovan M, MacRae S, Eldridge MD, Tavaré S; Oesophageal Cancer Clinical and Molecular Stratification (OCCAMS) Consortium, Fitzgerald RC, The landscape of selection in 551 esophageal adenocarcinomas defines genomic biomarkers for the clinic. Nat. Genet. 51, 506–516 (2019). [DOI] [PMC free article] [PubMed] [Google Scholar]
- 13.Lin D-C, Dinh HQ, Xie J-J, Mayakonda A, Silva TC, Jiang Y-Y, Ding L-W, He J-Z, Xu X-E, Hao J-J, Wang M-R, Li C, Xu L-Y, Li E-M, Berman BP, Phillip Koeffler H, Identification of distinct mutational patterns and new driver genes in oesophageal squamous cell carcinomas and adenocarcinomas. Gut 67, 1769–1779 (2018). [DOI] [PMC free article] [PubMed] [Google Scholar]
- 14.Cancer Genome Atlas Research Network; Analysis Working Group: Asan University; BC Cancer Agency; Brigham and Women’s Hospital; Broad Institute; Brown University; Case Western Reserve University; Dana-Farber Cancer Institute; Duke University; Greater Poland Cancer Centre; Harvard Medical School; Institute for Systems Biology; Leuven KU; Mayo Clinic; Memorial Sloan Kettering Cancer Center; National Cancer Institute; Nationwide Children’s Hospital; Stanford University; University of Alabama; University of Michigan; University of North Carolina; University of Pittsburgh; University of Rochester; University of Southern California; University of Texas MD Anderson Cancer Center; University of Washington; Van Andel Research Institute; Vanderbilt University; Washington University; Genome Sequencing Center: Broad Institute; Washington University in St. Louis; Genome Characterization Centers: BC Cancer Agency; Broad Institute, Harvard Medical School; Sidney Kimmel Comprehensive Cancer Center at Johns Hopkins University; University of North Carolina, University of Southern California Epigenome Center; University of Texas MD Anderson Cancer Center; Van Andel Research Institute; Genome Data Analysis Centers: Broad Institute, Brown University; Harvard Medical School, Institute for Systems Biology; Memorial Sloan Kettering Cancer Center; University of California Santa Cruz; University of Texas MD Anderson Cancer Center; Biospecimen Core Resource: International Genomics Consortium; Research Institute at Nationwide Children’s Hospital; Tissue Source Sites: Analytic Biologic Services; Asan Medical Center; Asterand Bioscience; Barretos Cancer Hospital; BioreclamationIVT; Botkin Municipal Clinic; Chonnam National University Medical School; Christiana Care Health System; Cureline; Duke University; Emory University; Erasmus University; Indiana University School of Medicine; Institute of Oncology of Moldova; International Genomics Consortium; Invidumed; Israelitisches Krankenhaus Hamburg; Keimyung University School of Medicine; Memorial Sloan Kettering Cancer Center; National Cancer Center Goyang; Ontario Tumour Bank; Peter MacCallum Cancer Centre; Pusan National University Medical School; Ribeirão Preto Medical School; St. Joseph’s Hospital &Medical Center; St. Petersburg Academic University; Tayside Tissue Bank; University of Dundee; University of Kansas Medical Center; University of Michigan; University of North Carolina at Chapel Hill; University of Pittsburgh School of Medicine; University of Texas MD Anderson Cancer Center; Disease Working Group: Duke University; Memorial Sloan Kettering Cancer Center; National Cancer Institute; University of Texas MD Anderson Cancer Center; Yonsei University College of Medicine; Data Coordination Center: CSRA Inc.; Project Team: National Institutes of Health, Integrated genomic characterization of oesophageal carcinoma. Nature 541, 169–175 (2017). [DOI] [PMC free article] [PubMed] [Google Scholar]
- 15.Vassilev LT, Vu BT, Graves B, Carvajal D, Podlaski F, Filipovic Z, Kong N, Kammlott U, Lukacs C, Klein C, Fotouhi N, Liu EA, In vivo activation of the p53 pathway by small-molecule antagonists of MDM2. Science 303, 844–848 (2004). [DOI] [PubMed] [Google Scholar]
- 16.Ramanathan SP, Krajnc M, Gibson MC, Cell-size pleomorphism drives aberrant clone dispersal in proliferating epithelia. Dev. Cell 51, 49–61.e4 (2019). [DOI] [PMC free article] [PubMed] [Google Scholar]
- 17.Li Y, Zhao J, Chen R, Chen S, Xu Y, Cai W, Integration of clinical and transcriptomics reveals programming of the lipid metabolism in gastric cancer. BMC Cancer 22, 955 (2022). [DOI] [PMC free article] [PubMed] [Google Scholar]
- 18.Leopold J, Popkova Y, Engel KM, Schiller J, Recent developments of useful MALDI matrices for the mass spectrometric characterization of lipids. Biomolecules 8, 173 (2018). [DOI] [PMC free article] [PubMed] [Google Scholar]
- 19.Melnikova V, Bar-Eli M, Inflammation and melanoma growth and metastasis: The role of platelet-activating factor (PAF) and its receptor. Cancer Metastasis Rev. 26, 359–371 (2007). [DOI] [PubMed] [Google Scholar]
- 20.Tsoupras AB, Iatrou C, Frangia C, Demopoulos CA, The implication of platelet activating factor in cancer growth and metastasis: Potent beneficial role of PAF-inhibitors and antioxidants. Infect. Disord. Drug Targets 9, 390–399 (2009). [DOI] [PubMed] [Google Scholar]
- 21.Ishii S, Nagase T, Shimizu T, Platelet-activating factor receptor. Prostaglandins Other Lipid Mediat. 68–69, 599–609 (2002). [DOI] [PubMed] [Google Scholar]
- 22.Héberlé É, Bardet AF, Sensitivity of transcription factors to DNA methylation. Essays Biochem. 63, 727–741 (2019). [DOI] [PMC free article] [PubMed] [Google Scholar]
- 23.Silva TC, Coetzee SG, Gull N, Yao L, Hazelett DJ, Noushmehr H, Lin D-C, Berman BP, ELMER v.2: An R/Bioconductor package to reconstruct gene regulatory networks from DNA methylation and transcriptome profiles. Bioinformatics 35, 1974–1977 (2019). [DOI] [PMC free article] [PubMed] [Google Scholar]
- 24.Heinz S, Benner C, Spann N, Bertolino E, Lin YC, Laslo P, Cheng JX, Murre C, Singh H, Glass CK, Simple combinations of lineage-determining transcription factors prime cis-regulatory elements required for macrophage and B cell identities. Mol. Cell 38, 576–589 (2010). [DOI] [PMC free article] [PubMed] [Google Scholar]
- 25.Obsil T, Obsilova V, Structure/function relationships underlying regulation of FOXO transcription factors. Oncogene 27, 2263–2275 (2008). [DOI] [PubMed] [Google Scholar]
- 26.Blattler A, Farnham PJ, Cross-talk between site-specific transcription factors and DNA methylation states. J. Biol. Chem. 288, 34287–34294 (2013). [DOI] [PMC free article] [PubMed] [Google Scholar]
- 27.Yao L, Shen H, Laird PW, Farnham PJ, Berman BP, Inferring regulatory element landscapes and transcription factor networks from cancer methylomes. Genome Biol. 16, 105 (2015). [DOI] [PMC free article] [PubMed] [Google Scholar]
- 28.Wierstra I, Alves J, FOXM1, a typical proliferation-associated transcription factor. Biol. Chem. 388, 1257–1274 (2007). [DOI] [PubMed] [Google Scholar]
- 29.Costa RH, FoxM1 dances with mitosis. Nat. Cell Biol. 7, 108–110 (2005). [DOI] [PubMed] [Google Scholar]
- 30.Chen L, Huang M, Plummer J, Pan J, Jiang YY, Yang Q, Silva TC, Gull N, Chen S, Ding LW, An O, Yang H, Cheng Y, Said JW, Doan N, Dinjens WN, Waters KM, Tuli R, Gayther SA, Klempner SJ, Berman BP, Meltzer SJ, Lin D-C, Koeffler HP, Master transcription factors form interconnected circuitry and orchestrate transcriptional networks in oesophageal adenocarcinoma. Gut 69, 630–640 (2020). [DOI] [PMC free article] [PubMed] [Google Scholar]
- 31.Stachler MD, Camarda ND, Deitrick C, Kim A, Agoston AT, Odze RD, Hornick JL, Nag A, Thorner AR, Ducar M, Noffsinger A, Lash RH, Redston M, Carter SL, Davison JM, Bass AJ, Detection of mutations in Barrett’s esophagus before progression to high-grade dysplasia or adenocarcinoma. Gastroenterology 155, 156–167 (2018). [DOI] [PMC free article] [PubMed] [Google Scholar]
- 32.Ross-Innes CS, Becq J, Warren A, Cheetham RK, Northen H, O’Donovan M, Malhotra S, di Pietro M, Ivakhno S, He M, Weaver JMJ, Lynch AG, Kingsbury Z, Ross M, Humphray S, Bentley D, Fitzgerald RC, Whole-genome sequencing provides new insights into the clonal architecture of Barrett’s esophagus and esophageal adenocarcinoma. Nat. Genet. 47, 1038–1046 (2015). [DOI] [PMC free article] [PubMed] [Google Scholar]
- 33.Stachler MD, Taylor-Weiner A, Peng S, McKenna A, Agoston AT, Odze RD, Davison JM, Nason KS, Loda M, Leshchiner I, Stewart C, Stojanov P, Seepo S, Lawrence MS, Ferrer-Torres D, Lin J, Chang AC, Gabriel SB, Lander ES, Beer DG, Getz G, Carter SL, Bass AJ, Paired exome analysis of Barrett’s esophagus and adenocarcinoma. Nat. Genet. 47, 1047–1055 (2015). [DOI] [PMC free article] [PubMed] [Google Scholar]
- 34.Wang JS, Guo M, Montgomery EA, Thompson RE, Cosby H, Hicks L, Wang S, Herman JG, Canto MI, DNA promoter hypermethylation of p16 and APC predicts neoplastic progression in Barrett’s esophagus. Am. J. Gastroenterol. 104, 2153–2160 (2009). [DOI] [PMC free article] [PubMed] [Google Scholar]
- 35.Klump B, Hsieh C-J, Holzmann K, Gregor M, Porschen R, Hypermethylation of the CDKN2/p16 promoter during neoplastic progression in Barrett’s esophagus. Gastroenterology 115, 1381–1386 (1998). [DOI] [PubMed] [Google Scholar]
- 36.Bian Y-S, Osterheld M-C, Fontolliet C, Bosman FT, Benhattar J, p16 inactivation by methylation of the CDKN2A promoter occurs early during neoplastic progression in Barrett’s esophagus. Gastroenterology 122, 1113–1121 (2002). [DOI] [PubMed] [Google Scholar]
- 37.Erdem B, Kucukyildirim S, Saglar E, Polat Z, Mergen H, Promoter hypermethylation of p16 and APC in gastrointestinal cancer patients. Turk. J. Gastroenterol. 25, 512–517 (2014). [DOI] [PubMed] [Google Scholar]
- 38.Alves MKS, Ferrasi AC, Lima VP, Ferreira MVP, de Moura Campos Pardini MI, Rabenhorst SHB, Inactivation of COX-2, HMLH1 and CDKN2A gene by promoter methylation in gastric cancer: Relationship with histological subtype, tumor location and Helicobacter pylori genotype. Pathobiology 78, 266–276 (2011). [DOI] [PubMed] [Google Scholar]
- 39.Vo QN, Geradts J, Gulley ML, Boudreau DA, Bravo JC, Schneider BG, Epstein-Barr virus in gastric adenocarcinomas: Association with ethnicity and CDKN2A promoter methylation. J. Clin. Pathol. 55, 669–675 (2002). [DOI] [PMC free article] [PubMed] [Google Scholar]
- 40.Vo QN, Geradts J, Boudreau DA, Bravo JC, Schneider BG, CDKN2A promoter methylation in gastric adenocarcinomas: Clinical variables. Hum. Pathol. 33, 1200–1204 (2002). [DOI] [PubMed] [Google Scholar]
- 41.Xue L, Ouyang Q, Li J, Meng X, Li Y, Xing L, Wang J, Yan X, Zhang X, Different roles for p16INK4a-Rb pathway and INK4a/ARF methylation between adenocarcinomas of gastric cardia and distal stomach. J. Gastroenterol. Hepatol. 29, 1418–1426 (2014). [DOI] [PubMed] [Google Scholar]
- 42.Sarbia M, Geddert H, Klump B, Kiel S, Iskender E, Gabbert HE, Hypermethylation of tumor suppressor genes (p16INK4A, p14ARF and APC) in adenocarcinomas of the upper gastrointestinal tract. Int. J. Cancer 111, 224–228 (2004). [DOI] [PubMed] [Google Scholar]
- 43.Lee SC, Lin K-H, Balogh A, Norman DD, Bavaria M, Kuo B, Yue J, Balázs L, Benyó Z, Tigyi G, Dysregulation of lysophospholipid signaling by p53 in malignant cells and the tumor microenvironment. Cell. Signal. 78, 109850 (2021). [DOI] [PMC free article] [PubMed] [Google Scholar]
- 44.Tew KD, Fisher PB, Sphingolipids in Cancer (Academic Press, 1st ed., 2018). [Google Scholar]
- 45.Lv F, Wu J, Miao D, An W, Wang Y, p16 deficiency promotes nonalcoholic steatohepatitis via regulation of hepatic oxidative stress. Biochem. Biophys. Res. Commun. 486, 264–269 (2017). [DOI] [PubMed] [Google Scholar]
- 46.Zhang Z, Wen H, Peng B, Weng J, Zeng F, CDKN2A deregulation in fatty liver disease and its accelerative role in the process of lipogenesis. FASEB J. 35, e21230 (2021). [DOI] [PubMed] [Google Scholar]
- 47.Kim K-J, Cho K-D, Jang KY, Kim H-A, Kim H-K, Lee H-K, Im S-Y, Platelet-activating factor enhances tumour metastasis via the reactive oxygen species-dependent protein kinase casein kinase 2-mediated nuclear factor-κB activation. Immunology 143, 21–32 (2014). [DOI] [PMC free article] [PubMed] [Google Scholar]
- 48.Mathonnet M, Descottes B, Valleix D, Truffinet V, Labrousse F, Denizot Y, Platelet-activating factor in cirrhotic liver and hepatocellular carcinoma. World J. Gastroenterol. 12, 2773–2778 (2006). [DOI] [PMC free article] [PubMed] [Google Scholar]
- 49.Denizot Y, Descottes B, Truffinet V, Valleix D, Labrousse F, Mathonnet M, Platelet-activating factor and liver metastasis of colorectal cancer. Int. J. Cancer 113, 503–505 (2005). [DOI] [PubMed] [Google Scholar]
- 50.Bussolati B, Biancone L, Cassoni P, Russo S, Rola-Pleszczynski M, Montrucchio G, Camussi G, PAF produced by human breast cancer cells promotes migration and proliferation of tumor cells and neo-angiogenesis. Am. J. Pathol. 157, 1713–1725 (2000). [DOI] [PMC free article] [PubMed] [Google Scholar]
- 51.Cheng L, Cao W, Fiocchi C, Behar J, Biancani P, Harnett KM, HCl-induced inflammatory mediators in cat esophageal mucosa and inflammatory mediators in esophageal circular muscle in an in vitro model of esophagitis. Am. J. Physiol. Gastrointest. Liver Physiol. 290, G1307–G1317 (2006). [DOI] [PubMed] [Google Scholar]
- 52.Konturek PC, Bielański W, Konturek SJ, Hahn EG, Helicobacter pylori associated gastric pathology. J. Physiol. Pharmacol. 50, 695–710 (1999). [PubMed] [Google Scholar]
- 53.Platelet activating factor receptor (PTAFR; PAFR). Science-Business eXchange 3, 1386–1386 (2010). [Google Scholar]
- 54.Seyfried CE, Schweickart VL, Godiska R, Gray PW, The human platelet-activating factor receptor gene (PTAFR) contains no introns and maps to chromosome 1. Genomics 13, 832–834 (1992). [DOI] [PubMed] [Google Scholar]
- 55.Ammit AJ, O’Neill C, Platelet-activating factor (PAF) receptor antagonists inhibit arachidonic acid induced platelet aggregation in rabbit whole blood. Lipids 26, 1189–1192 (1991). [DOI] [PubMed] [Google Scholar]
- 56.Giaginis C, Kourou E, Giagini A, Goutas N, Patsouris E, Kouraklis G, Theocharis S, Platelet-activating factor (PAF) receptor expression is associated with histopathological stage and grade and patients’ survival in gastric adenocarcinoma. Neoplasma 61, 309–317 (2014). [DOI] [PubMed] [Google Scholar]
- 57.Darst M, Al-Hassani M, Li T, Yi Q, Travers JM, Lewis DA, Travers JB, Augmentation of chemotherapy-induced cytokine production by expression of the platelet-activating factor receptor in a human epithelial carcinoma cell line. J. Immunol. 172, 6330–6335 (2004). [DOI] [PubMed] [Google Scholar]
- 58.Chen J, Lan T, Zhang W, Dong L, Kang N, Zhang S, Fu M, Liu B, Liu K, Zhan Q, Feed-forward reciprocal activation of PAFR and STAT3 regulates epithelial-mesenchymal transition in non–small cell lung cancer. Cancer Res. 75, 4198–4210 (2015). [DOI] [PubMed] [Google Scholar]
- 59.Hou T, Lou Y, Li S, Zhao C, Ji Y, Wang D, Tang L, Zhou M, Xu W, Qian M, Wu Z, Zhao J, Wei H, Li Z, Xiao J, Kadsurenone is a useful and promising treatment strategy for breast cancer bone metastases by blocking the PAF/PTAFR signaling pathway. Oncol. Lett. 16, 2255–2262 (2018). [DOI] [PMC free article] [PubMed] [Google Scholar]
- 60.Souza JL, Martins-Cardoso K, Guimarães IS, de Melo AC, Lopes AH, Monteiro RQ, Almeida VH, Interplay Between EGFR and the platelet-activating factor/PAF receptor signaling axis mediates aggressive behavior of cervical cancer. Front. Oncol. 10, 557280 (2020). [DOI] [PMC free article] [PubMed] [Google Scholar]
- 61.Barger CJ, Branick C, Chee L, Karpf AR, Pan-cancer analyses reveal genomic features of FOXM1 overexpression in cancer. Cancer 11, 251 (2019). [DOI] [PMC free article] [PubMed] [Google Scholar]
- 62.Liang S-K, Hsu C-C, Song H-L, Huang Y-C, Kuo C-W, Yao X, Li C-C, Yang H-C, Hung Y-L, Chao S-Y, Wu S-C, Tsai F-R, Chen J-K, Liao W-N, Cheng S-C, Tsou T-C, Wang I-C, FOXM1 is required for small cell lung cancer tumorigenesis and associated with poor clinical prognosis. Oncogene 40, 4847–4858 (2021). [DOI] [PubMed] [Google Scholar]
- 63.Kimura H, Sada R, Takada N, Harada A, Doki Y, Eguchi H, Yamamoto H, Kikuchi A, The Dickkopf1 and FOXM1 positive feedback loop promotes tumor growth in pancreatic and esophageal cancers. Oncogene 40, 4486–4502 (2021). [DOI] [PMC free article] [PubMed] [Google Scholar]
- 64.Fujii M, Matano M, Nanki K, Sato T, Efficient genetic engineering of human intestinal organoids using electroporation. Nat. Protoc. 10, 1474–1485 (2015). [DOI] [PubMed] [Google Scholar]
- 65.Chughtai K, Jiang L, Greenwood TR, Glunde K, Heeren RMA, Mass spectrometry images acylcarnitines, phosphatidylcholines, and sphingomyelin in MDA-MB-231 breast tumor models. J. Lipid Res. 54, 333–344 (2013). [DOI] [PMC free article] [PubMed] [Google Scholar]
- 66.Jiang L, Chughtai K, Purvine SO, Bhujwalla ZM, Raman V, Paša-Tolić L, Heeren RMA, Glunde K, MALDI-mass spectrometric imaging revealing hypoxia-driven lipids and proteins in a breast tumor model. Anal. Chem. 87, 5947–5956 (2015). [DOI] [PMC free article] [PubMed] [Google Scholar]
- 67.Ščupáková K, Balluff B, Tressler C, Adelaja T, Heeren RMA, Glunde K, Ertaylan G, Cellular resolution in clinical MALDI mass spectrometry imaging: The latest advancements and current challenges. Clin. Chem. Lab. Med. 58, 914–929 (2020). [DOI] [PMC free article] [PubMed] [Google Scholar]
- 68.Sud M, Fahy E, Cotter D, Brown A, Dennis EA, Glass CK, Merrill AH Jr., Murphy RC, Raetz CRH, Russell DW, Subramaniam S, LMSD: LIPID MAPS structure database. Nucleic Acids Res. 35, D527–D532 (2007). [DOI] [PMC free article] [PubMed] [Google Scholar]
- 69.Moran S, Arribas C, Esteller M, Validation of a DNA methylation microarray for 850,000 CpG sites of the human genome enriched in enhancer sequences. Epigenomics 8, 389–399 (2016). [DOI] [PMC free article] [PubMed] [Google Scholar]
- 70.Zhou W, Triche TJ Jr., Laird PW, Shen H, SeSAME: Reducing artifactual detection of DNA methylation by Infinium BeadChips in genomic deletions. Nucleic Acids Res. 46, e123 (2018). [DOI] [PMC free article] [PubMed] [Google Scholar]
- 71.Subramanian A, Tamayo P, Mootha VK, Mukherjee S, Ebert BL, Gillette MA, Paulovich A, Pomeroy SL, Golub TR, Lander ES, Mesirov JP, Gene set enrichment analysis: A knowledge-based approach for interpreting genome-wide expression profiles. Proc. Natl. Acad. Sci. U.S.A. 102, 15545–15550 (2005). [DOI] [PMC free article] [PubMed] [Google Scholar]
Associated Data
This section collects any data citations, data availability statements, or supplementary materials included in this article.
Supplementary Materials
Data Availability Statement
All data associated with this study are present in the paper or the Supplementary Materials. Reagents, resources, and materials used in this study are detailed in table S2. RNA-seq and methylation array data generated in this study were deposited to Gene Expression Omnibus (GEO) under the series GSE216198 and GSE216814. RNA-seq datasets of TCGA EAC and normal GEJ tissues can be obtained from the XENA data portal (http://xena.ucsc.edu/).