

Abstract
The family of NF-κB transcriptional activators controls the expression of many genes, including those involved in cell survival and development. The family consists of homo- and heterodimers constituted by combinations of five subunits. Subunit p50 includes 13 tyrosine residues, but the relationship between specific tyrosine phosphorylations and p50 function is not well understood. Subunits of p50 and p65 prepared in vitro formed a heterodimer, but this NF-κB would not bind to the interleukin-2 (IL-2) promoter DNA. Treatment of p50 with guanosine triphosphate (GTP) and a lysate from activated Jurkat cells, effected rapid p50 phosphorylation, and, in the presence of wild-type subunit p65, was accompanied on the same time scale by IL-2 promoter DNA binding. Modified p50s containing one of seven stoichiometrically phosphorylated tyrosines in NF-κB p50/p65 heterodimers, included three that facilitated binding to the IL-2 DNA promoter region to a greater extent than the wild type. One of these three stoichiometrically phosphorylated p50/p65 heterodimers of NF-κB, containing pTyr60 in the p50 subunit, was treated with a lysate from activated Jurkat cells + GTP and shown to be phosphorylated on the same time scale as wild-type p50. This modified NF-κB also developed IL-2 promoter DNA binding activity on the same time scale as the wild type but exhibited greater binding to the IL-2 DNA promoters than the wild type. The nature of this enhanced binding was studied in greater detail using a metabolically stable pTyr derivative at position 60 of p50 and cellular phosphatases. We suggest that enhanced DNA binding of modified NF-κB containing pTyr60 in the p50 subunit may reflect stoichiometric NF-κB phosphorylation at a site that is not normally fully phosphorylated, or not phosphorylated at all, and is relatively resistant to the effects of Jurkat cell tyrosine phosphatase activity. This conclusion was reinforced by demonstrating that modification of Tyr60 of p50 with a metabolically stable methylenephosphonate moiety further increased the stability of the formed NF-κB p50/p65 heterodimer against the action of activated Jurkat cell phosphatases.
Graphical Abstract
INTRODUCTION
Protein phosphorylation is an important modification linked to the regulation of many cellular networks.1 The most abundant phosphorylation events involve serine, threonine, and tyrosine residues,1–7 and a single protein can be modified at multiple sites post-translationally, typically in a defined order. Confounding the analysis of such systems is the reversible nature of protein phosphorylation, reflecting both phosphorylation by protein kinases, and dephosphorylation by protein phosphatases. Presently, we have focused on key molecular events enabling tyrosine phosphorylation, and on the appearance and persistence of phosphorylated tyrosine (pTyr) in proteins under typical experimental conditions.
Recently, we have described a method for introducing pTyr into specific sites in proteins using a misacylated suppressor tRNA in the presence of modified Escherichia coli ribosomes that had been selected for their ability to incorporate this noncanonical amino acid.8,9 The proteins prepared included modified analogues of NF-κB10 and its inhibitor IκB-α,8 and both were studied in the presence of lysates from activated Jurkat cells. Both types of phosphorylated proteins provided novel insights into their mechanisms of action, suggesting that the stoichiometric introduction of pTyr into cellular proteins normally activated with pTyr in situ might provide a strategy of more general utility.
An issue in extending the use of proteins elaborated with pTyr at specific positions is reported, indicating that some phosphorylated tyrosine residues have very short half-lives. For example, phosphotyrosine residues on epidermal growth factor receptors exhibit very short half-lives and turn over 100–1000 times in an immediate-early response to ligands.11 Luo et al. reported the in cellulo incorporation of phosphotyrosine and its nonhydrolyzable methylene phosphonate analogue (4-phosphomethyl-L-phenylalanine) into position 99 of myoglobin by amber codon suppression.12 Full-length myoglobin was obtained in both cases, but only the myoglobin containing the nonhydrolyzable analogue exhibited the correct mass. The mass spectrum of myoglobin synthesized using phosphorylated tyrosine was consistent with the presence of tyrosine at position 99, an observation attributed to the dephosphorylation of the initial myoglobin product post-translationally by endogenous E. coli phosphatases.
In the present study, we utilized lysates from activated Jurkat cells and modified p50 subunits of NF-κB, each bearing a stoichiometrically phosphorylated tyrosine, to investigate the process by which NF-κB is activated to enable subsequent transcriptional control. Each phosphorylated p50 subunit containing a stoichiometrically phosphorylated tyrosine at a specific site formed a p50/p65 heterodimer with the wild-type p65 subunit, but neither the wild-type nor modified heterodimers were found to be capable of binding to the DNA promoter for interleukin-2 (IL-2) in a native polyacrylamide gel. Brief incubation of p50 with a lysate from activated Jurkat cells in the presence of [γ−32P]-guanosine triphosphate (GTP) afforded phosphorylated p50 and enabled subsequent IL-2 DNA promoter binding in the additional presence of p65. The rapid p50 phosphorylation occurred on a time scale quite similar to the development of DNA promoter binding by the heterodimers, suggesting that phosphorylation in the presence of Jurkat cell lysate was the key event. The wild-type and modified p50/p65 heterodimers all bound to IL-2 DNA promoters, and a subset of them increased DNA promoter binding up to ~3.5-fold, indicating the selectivity of action by the altered NF-κBs.
Also studied was the continued incubation of NF-κB p50/p65 heterodimers in the presence of activated Jurkat cell lysate for a period of time after maximal phosphorylation had been achieved. For both wild-type p50/p65 and analogues in which the p50 subunit was stoichiometrically phosphorylated at position 60, a net loss of IL-2 DNA binding capacity proceeded steadily over a period of 1 h, presumably reflecting phosphatase-mediated loss of phosphate groups essential for IL-2 DNA binding. This possibility was explored further using a modified NF-κB containing a metabolically stable methylenephosphonate derivative of pTyr at position 60 of the p50 subunit.
Though the use of a lysate from activated Jurkat cells was found to be essential for NF-κB phosphorylation and this process seems to suffice to enable IL-2 promoter DNA binding, it is obviously possible that one or more other processes not yet recognized may be required as well. In this context, it is worth noting that in an earlier study,8 endogenous NF-κB from Jurkat cells exhibited behavior in binding exogenous DNA that differed in no obvious way from what is presented here. More recently,10 we found that the lysate was not required for NF-κB subunit assembly, nor did the lysate activate NF-κB in the presence of adenosine triphosphate (ATP).
2. RESULTS AND DISCUSSION
The NF-κB family of transcriptional activators has at least 12 homo- and heterodimers assembled from five subunit proteins having a high degree of sequence homology near or at their N-termini, including RelA (p65), RelB, c-Rel, p50, and p52.13,14 Disorder of the cellular expression of NF-κB is associated with a number of diseases such as autoimmunity, neurodegeneration, and cancer, since it has important roles in cell proliferation, cell death, and the cell cycle, via regulatory control of gene expression.15–17 Because of these central roles, NF-κB represents a plausible target for therapeutic intervention in such diseases. A more detailed understanding of transcriptional regulation by NF-κB may facilitate such efforts.
Numerous studies have explored the phosphorylation of serine, threonine, and tyrosine residues of NF-κB, which regulate its activities and function.10,18–22 In order to be able to analyze the results of the site-specific tyrosine phosphorylation of NF-κB accurately, we prepared two pure subunits, p50 and p65.23,24 The in vitro preparations (Scheme 1) were carried out as described.10
Scheme 1.
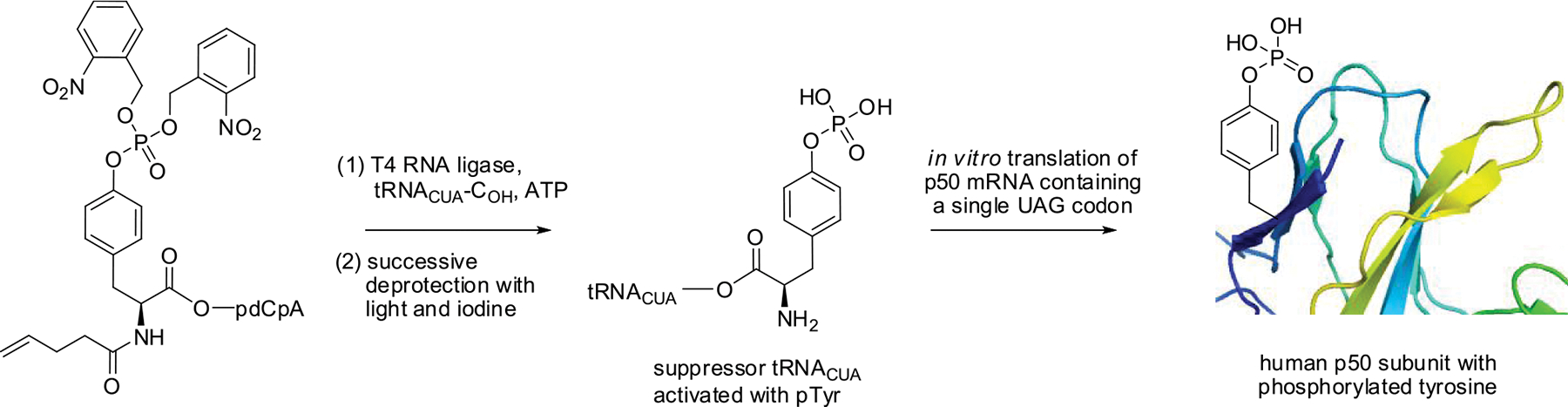
Synthesis of the Human p50 Protein Site Specifically Phosphorylated on Tyrosine
The p50 DNA binding region is present within the N-terminal domain. From the p50/p65-dsDNA complex X-ray crystal structure (PDB 1VKX),25 tyrosines Y44, Y60, Y82, and Y90, located near the p50 N-terminus, were chosen; their nearest distances to the bound DNA were determined. These varied from 4.5 to 33.0 Å, with Tyr60 (4.5 Å) and Tyr82 (13.3 Å) being closest to the DNA, consistent with their possible involvement in DNA binding, and tyrosines Y44 (33.0 Å) and Y90 (31.9 Å) being somewhat further from the DNA. Also studied as controls were the tyrosine residues Y7, Y241, and Y270. To study the way in which phosphorylation of these seven tyrosines impacted the binding of DNA by NF-κB, they were replaced in a systematic fashion by phosphotyrosine, as described.10
In a previous study, we employed a DNA duplex sequence corresponding to the IL-2 gene promoter to verify its binding to cellular NF-κB, which had been activated in a nuclear lysate from Jurkat cells.8 Because this did not permit quantification of the NF-κB derived from the cells, we have employed a new strategy to explore differences in the binding properties of our several singly phosphorylated p50 subunits, each bound to p65 prepared in vitro, to a synthetic 5′−32P-end labeled 20 nt DNA duplex having the sequence of the IL-2 promoter DNA studied previously.8 The formed NF-κB heterodimer, prepared from purified, nonphosphorylated p50 and p65 subunits, did not bind to the radiolabeled IL-2 DNA duplex in a native polyacrylamide gel (Figure 1A, lane 1). The possible formation of a p50/p65 heteroduplex in the absence of incubation with a lysate from Jurkat cells was studied by the preparation of 35S-methionine labeled subunits. We found that their admixture afforded a complex migrating, as expected for p50/p65.10 We also demonstrated that admixture of only a single 35S-radiolabeled subunit (p50) gave a complex with unlabeled p65 that comigrated with p50/65. Also, heterodimer formation was observed between four different p50s that were stoichiometrically phosphorylated and unlabeled wild-type p65.10
Figure 1.
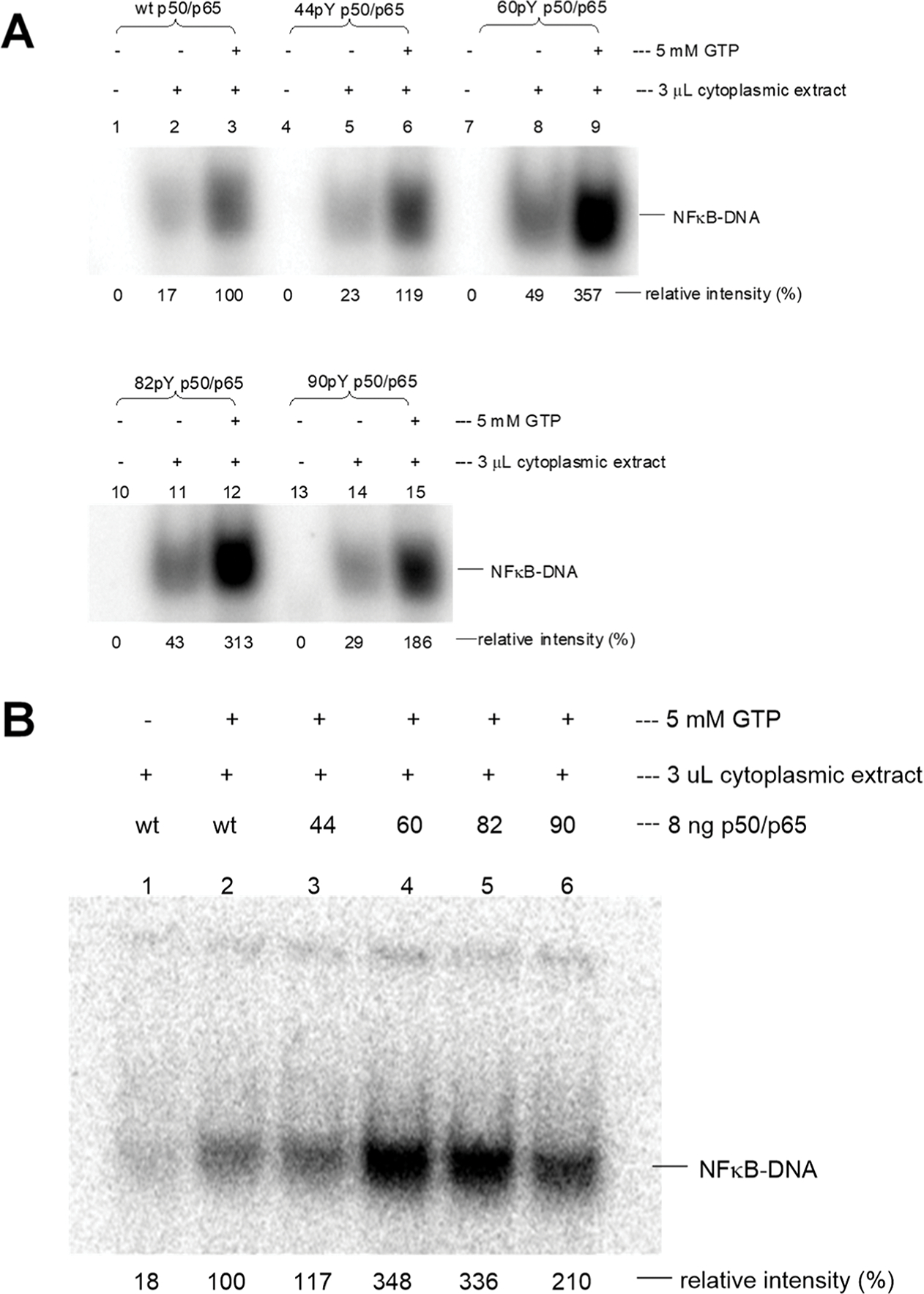
IL-2 promoter DNA binding of modified NF-κBs expressed in a cell-free E. coli protein biosynthesizing system following treatment with a cytoplasmic lysate from activated Jurkat cells in the presence of GTP. The relative intensities were determined using ImageQuant version 5.2 software (Molecular Dynamics) on the basis of the 32P signal. The value of the band for wild-type NF-κB in the presence of GTP was defined as 100%. (A) IL-2 promoter DNA binding with NF-κBs depends on the presence of GTP and cytoplasmic lysate. Lanes 1–3, wild-type NF-κB; lanes 4–15, modified NF-κBs having p50 subunits with pTyr at respective positions 44, 60, 82, or 90. (B) Comparison of IL-2 promoter DNA binding ability to wild-type and modified NF-κBs. Controls included wild-type NF-κB in the absence and presence of GTP (lanes 1 and 2), and NFκBs containing modified p50 subunits with pTyr at positions 44, 60, 82, or 90 (lanes 3, 4, 5, and 6, respectively). The statistical significance was determined using the Student’s t-test: wild-type vs pTyr44-p50, p > 0.05; wild-type vs pTyr60-p50, p < 0.001; wild-type vs pTyr82-p50, p < 0.001; and wild-type vs pTyr90-p50, p < 0.001.
A more detailed experiment was effected using a cytoplasmic lysate from Jurkat cells that had been stimulated with the calcium ionophore A23187 and phorbol 12-myristate 13-acetate (PMA). As shown in Figure 1A, lanes 1 and 2, only a very weak band corresponding to an NF-κB–DNA complex was noted. Significant formation of the NF-κB–DNA complex required GTP (but not ATP, not shown), as illustrated in lane 3. Four modified p50/wild-type p65 complexes exhibited the same behavior qualitatively as wild-type p50/p65 (Figure 1A, lanes 4–15), although the modified p50/wild-type p65 complexes stoichiometrically phosphorylated at positions 60 and 82 exhibited significantly enhanced DNA gel bands compared to wild-type. The weak bands in lanes 2, 5, 8, 11, and 14 (i.e., in the presence of cell lysate but the absence of added GTP) seem likely to be due to adventitious GTP in the activated cell lysate.
In order to judge the reproducibility of the basic experiment in Figure 1A, the experiment was repeated in a completely independent experiment. As shown in Figure 1B, NF-κB composed of wild-type p65 (initially unphosphorylated) and one of four different p50 subunits, those singly phosphorylated at positions 60, 82, or 90, all bound more avidly to the IL-2 promoter DNA duplex than wild-type p50/p65 (lanes 4–6), indicating they had been activated for enhanced promoter DNA binding when incubated with cell lysate from activated Jurkat cells. The extent of DNA binding in Figure 1A,B was quite reproducible for individual phosphorylated p50s, and the figure legend provides a statistical analysis of the binding of NF-κB containing each modified p50 versus wild type (see also Figures S1 and S2, Supporting Information).
The largest increases were observed for the p50s containing pTyr at positions 60 and 82 (348 and 336%, respectively). These tyrosines were closest to the DNA (4.5 and 13.3 Å, respectively) and faced the bases of the DNA strand. In comparison, the formed NF-κB complexes containing stoichiometrically phosphorylated Tyr44 and Tyr90 (closest Tyr–phenolic OH distances from the DNA were 33.0 and 31.9 Å, respectively) exhibited bindings of 117 and 210%, respectively. The presence of pTyr90 thus clearly resulted in enhanced binding relative to the wild type, while the possible binding enhancement due to pTyr44 was less clear. Thus, phosphorylation of the two Tyr residues closest to the DNA produced the greatest enhancement of DNA binding.
Three additional tyrosine residues in p50 were also phosphorylated stoichiometrically and studied as part of heterodimers with p65, including Y7 close to the protein N-terminus, and Y241 and Y270, much farther from the N-terminus. None of the heterodimers formed between these modified p50s and p65 altered the extent of IL-2 DNA binding relative to the wild type (Figures S3 and S4, Supporting Information).
In the absence of added p65, the individual p50 subunits bound to the IL-2 promoter DNA,23 but no enhanced DNA binding relative to wild-type p50 was observed (Figure 2). Thus, the differences observed for the modified p50/p65 heterodimers in Figure 1 required the presence of both NF-κB subunits.
Figure 2.
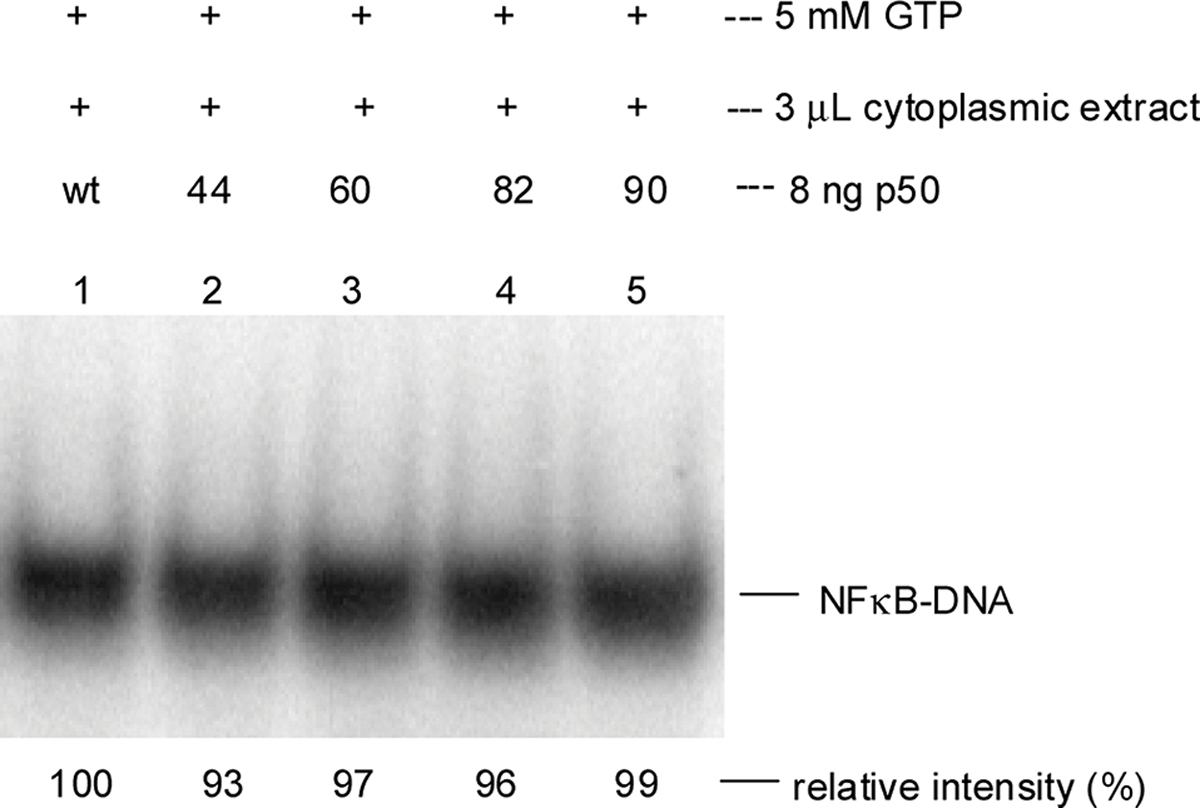
IL-2 promoter DNA binding of modified p50s expressed in a cell-free E. coli protein biosynthesizing system following treatment with a cytoplasmic lysate from activated Jurkat cells in the presence of GTP. Sample analysis was achieved by 5% native polyacrylamide gel electrophoresis (PAGE, 100 V for 1 h). Relative intensities were determined using ImageQuant software (version 5.2, Molecular Dynamics) on the basis of the 32P signal. The band for wild-type NF-κB in the presence of GTP was defined as 100%. Lane 1, wild-type p50; lanes 2–5, altered p50 subunits with pTyr at respective positions 44, 60, 82, or 90. Statistical significance was calculated with the Student’s t-test: p > 0.05 for all proteins.
We next studied the phosphorylation time course of two p50 subunits to understand the extent to which this event might support the observed DNA binding of p50/p65 complexes to the IL-2 DNA promoter construct described above. As shown (Figure S5, Supporting Information), when wild-type p50 (upper panel) and modified p50 phosphorylated stoichiometrically on Tyr60 (middle panel) were incubated solely in the presence of γ−32P-GTP, neither underwent significant phosphorylation (lanes 1). Following the admixture of activated Jurkat cell cytoplasmic lysate, the phosphorylation of wild-type p50 was 76% complete within 30 s and maximal within 30 min. A similar time course was observed for the modified p50 (71% complete in 30 s and complete in 30 min). The time course of phosphorylation is quantified in the lower panel. Also of interest is a comparison of the actual levels of phosphorylation between wild-type and modified p50/p65 complexes. The modified complex, phosphorylated stoichiometrically on Tyr60, was phosphorylated to an extent of 63% of that observed with the wild type.
The time course of IL-2 DNA binding of NF-κB containing the phosphorylated wild-type or modified p50s was studied under the same conditions. As shown (Figure S6, Supporting Information), the time courses of DNA binding were also quite similar for the wild-type and pTyr60 samples. Further, the time courses of p50 phosphorylation and IL-2 DNA binding were also quite similar.
Because the reactions studied in Figures S5 and S6 were largely complete at the first measured time point, it was not possible to use these conditions to judge carefully whether p50 phosphorylation and IL-2 DNA binding by NF-κB occurred on the same time scale. Accordingly, the phosphorylation reactions were carried out in the presence of 1.5 μL (10% as much) of Jurkat cell lysate on ice, rather than at room temperature (Figure 3). The reactions measuring DNA binding were also carried out using 0.15 μL (10% as much) of Jurkat cell lysate on ice, rather than at room temperature (Figure 4). As shown in Figures 3 and 4, the kinetics for both processes were essentially the same, with 19% phosphorylation of the wild type and 21% phosphorylation of the modified p50 after the first 30 s. For DNA binding, the reactions carried out under these modified conditions were 29% complete for wild type and 21% complete for modified p50 after the first 30 s. Thus, the two processes occurred at the same rate, supporting the interpretation of cause and effect.
Figure 3.
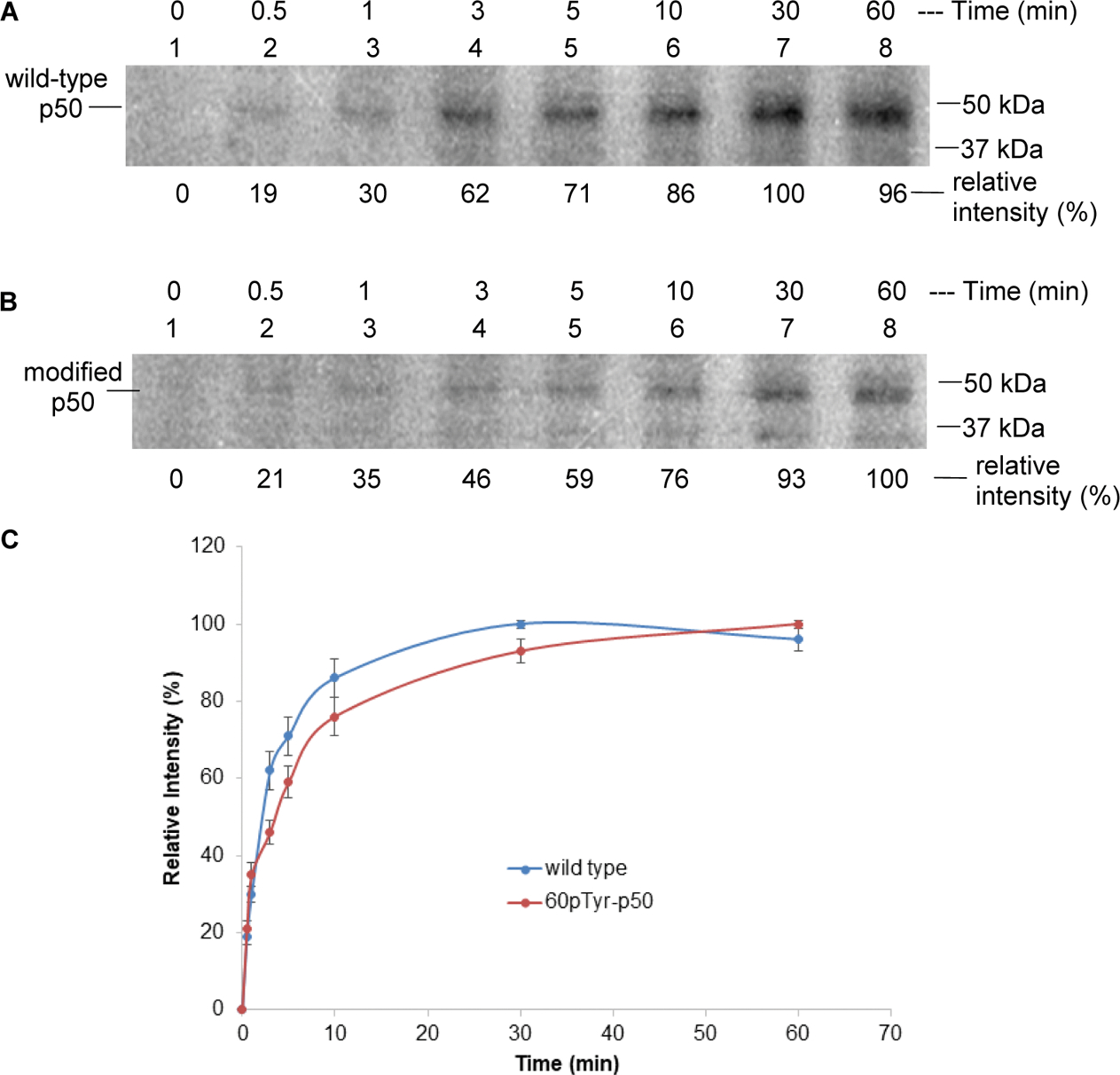
Phosphorylation of wild-type p50 (A) and modified p50 (containing pTyr60) (B) in the presence of Jurkat cell lysate. The results were analyzed by 15% sodium dodecyl sulfate-polyacrylamide gel electrophoresis (SDS-PAGE). The relative intensities were determined using ImageQuant software (version 5.2, Molecular Dynamics) based on the 32P signal. 0 min: no Jurkat cell lysate was added; 0.5–60 min after Jurkat cell lysate addition. (C) Time-dependent phosphorylation of the wild-type and modified p50s in the presence of cell lysate from activated Jurkat cells. The band at 30 or 60 min was defined as 100%.
Figure 4.
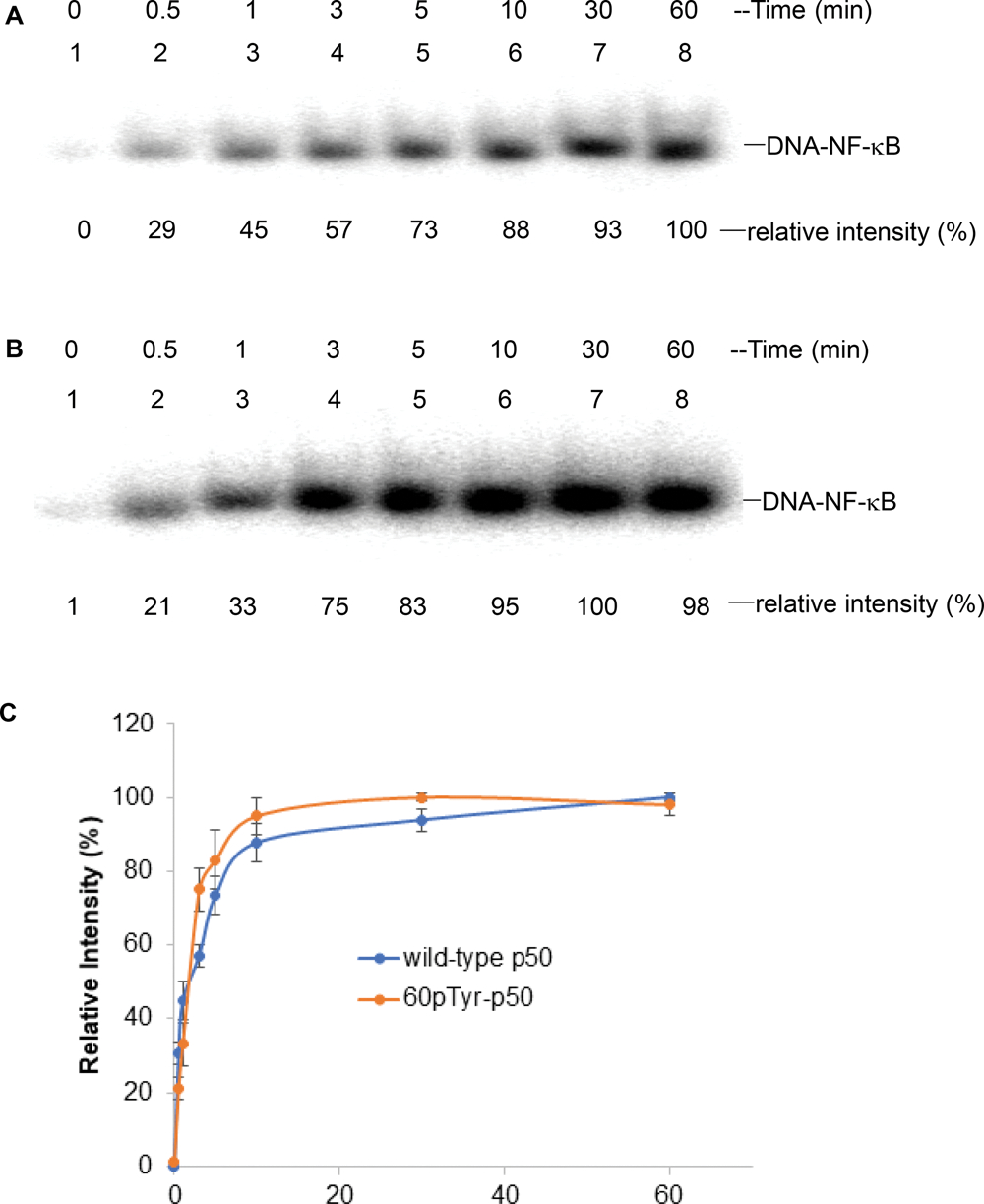
Time-dependent IL-2 promoter DNA binding of phosphorylated wild-type p50 (A) and modified p50 containing pTyr60 (B) in the presence of Jurkat cell lysate. The results were analyzed using 5% native PAGE. 0 min, no activated Jurkat cell lysate was added; 0.5–60 min, following Jurkat cell lysate addition. (C) Time course of binding of wild-type and modified p50s to IL-2 promoter DNA in the presence of cell lysate from activated Jurkat cells. The band having the greatest intensity for each protein was defined as 100%.
These experiments, including the very similar time courses of GTP-mediated 32P-labeling of p50, and newly enabled DNA binding, suggest that NF-κB phosphorylation plays a key role in enabling promoter DNA binding. These experiments also define the persistence and properties of the radiolabeled p50s (Figures 3 and 4). As is clear from Figure S5, which was carried out under ambient conditions using 1.5 μL of Jurkat cell lysate, many Jurkat cell proteins in addition to p50 were phosphorylated under the reaction conditions and persisted throughout the one-hour incubation period, in spite of the phosphatase activities in the cell lysate. Obviously, this likely reflects the competitive effects of kinase-mediated phosphorylation and phosphatase-mediated dephosphorylation.
As is evident from the experiments described above, p50 phosphorylation can be carried out stoichiometrically at single, predetermined sites, and the introduction of phosphate at some positions can alter the affinity of p50, present within an NF-κB p50/p65 heterodimer, for its physiological DNA target. We have shown previously that the altered binding can result in altered transcription in cellulo.10 Though the foregoing experiments illustrate that cellular tyrosine phosphate levels can persist long enough to enable the study of their effects, they do not provide an adequate understanding of the consequences of stoichiometric phosphorylation at a single position, where many sites of phosphorylation may ordinarily involve partial site occupancy. Neither do they provide insight into the consequences of protein modification with metabolically stable tyrosine phosphates. Accordingly, we investigated these issues in a further experiment.
As shown in Figure S7 (Supporting Information), we prepared an analogue of p50 containing the nonhydrolyzable methylene phosphonate analogue (4-phosphomethyl-L-phenylalanine) of pTyr at position 60. In common with the p50 analogue containing pTyr at this position (Figure 1), the formed NF-κB p50/p65 heterodimer bound to IL-2 promoter DNA gave enhanced gel bands as compared with the wild-type NF-κB p50/p65 heterodimer. We then incubated the NF-κB p50/p65 heterodimers containing Tyr60, pTyr60, or pCH2Tyr60 in the presence of activated Jurkat cell lysate under conditions that should permit both phosphorylation and dephosphorylation of p50. The DNA binding properties of the individual NF-κB p50/p65 heterodimers were monitored in a time-dependent fashion (Figure 5). As shown, the NF-κB p50/p65 heterodimer containing Tyr at position 60 of p50, which is known (Figures 1 and S7) to bind least strongly to IL-2 promoter DNA, also lost its DNA binding ability the most quickly and had only 23% of its initial DNA binding ability after 1 h. The effect of incubation on the NF-κB p50/p65 heterodimer initially containing pTyr at position 60 lost its IL-2 DNA binding capacity in a similar time-dependent fashion, and had only 33% of its initial DNA binding ability after 1 h. This would be consistent with the interpretation that the process leading to net dephosphorylation involved the same p50 protein residues. Since NF-κB p50/p65 heterodimers containing pTyr at p50 position 60 bound to the IL-2 DNA 3.5-fold more avidly than those containing Tyr (Figure 1), the data may reflect a lesser rate of dephosphorylation of pTyr60. Interestingly, the NF-κB p50/p65 heterodimer containing nonhydrolyzable pCH2Tyr60 underwent loss of its IL-2 DNA binding capacity least quickly and with an altered time-dependent profile, retaining 56% of its IL-2 DNA binding capacity after 1 h. While these data support the idea that time-dependent maintenance of the NF-κB–promoter DNA complexes reflects the extent of dephosphorylation (e.g., in the relative rates of NF-κB–DNA complex loss by the Tyr60 phosphorylated and methylenephosphonylated NF-κBs), the rate of complex disappearance could also be influenced by protein degradation. Accordingly, the data must be viewed in the context of these two potentially competing pathways.
Figure 5.
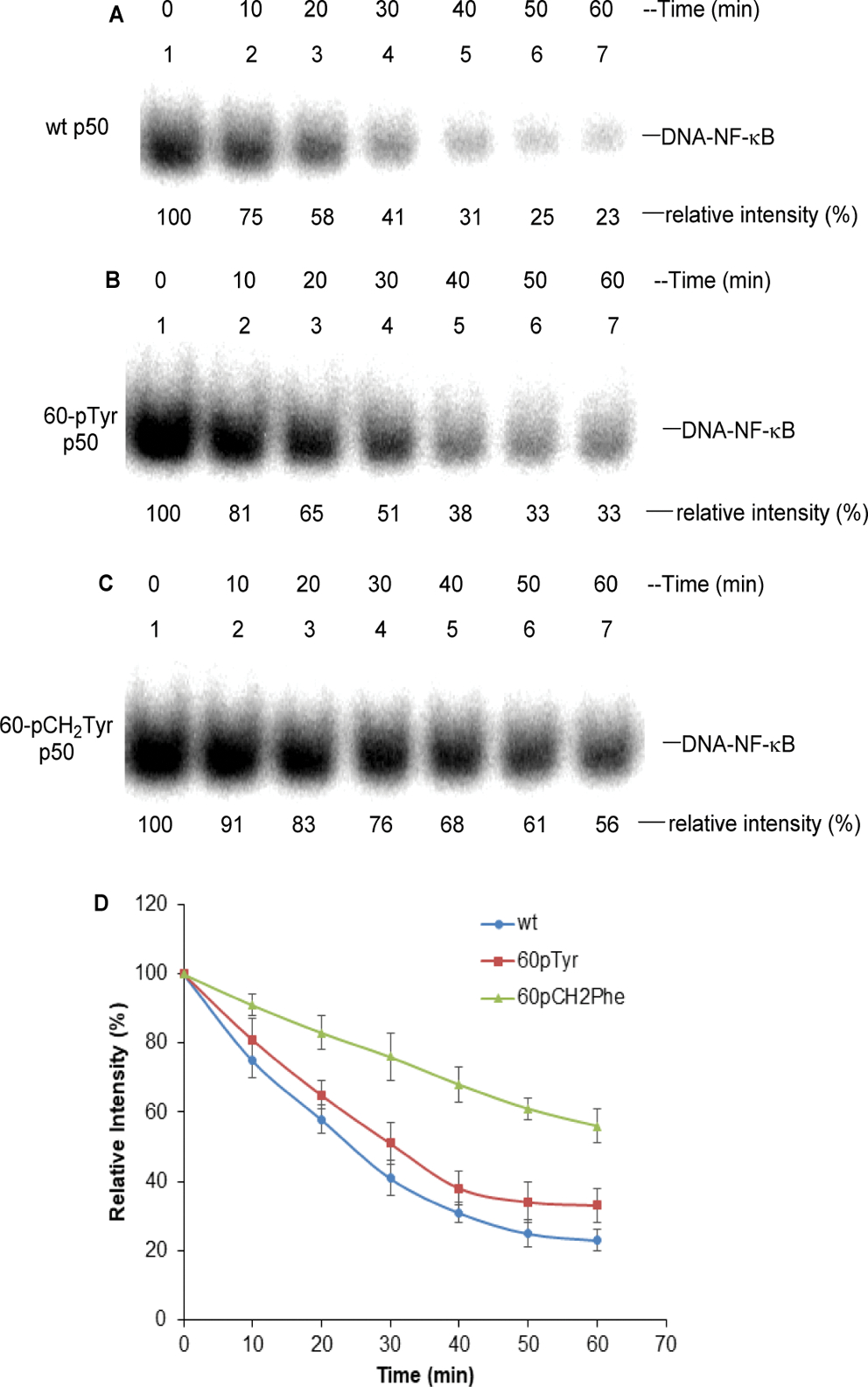
Stability of phosphorylated NF-κB in Jurkat cell lysate at room temperature. (A) IL-2 promoter DNA binding of NF-κB containing wild-type p50 protein in the presence of cell lysate from activated Jurkat cells. The NF-κBs were pretreated with 5 mM GTP at room temperature for 10 min before adding IL-2 promoter DNA. (B) IL-2 DNA binding of NF-κB containing 60p-Tyr p50 in the presence of cell lysate from activated Jurkat cells. (C) IL-2 DNA binding of NF-κB containing 60p-CH2-Tyr p50 in the presence of cell lysate from activated Jurkat cells. (D) Time course of binding of the wild-type and modified phosphorylated NF-κB to IL-2 promoter DNA in the presence of Jurkat cell lysate. The bands at 0 min were defined as 100%.
In order to judge the generality of the observations described above, we employed a Qiagen RT2 Profiler polymerase chain reaction (PCR) Array to determine the possible altered transcription of a set of 96 genes in human NF-κB signaling pathways. These were studied in the presence of both wild-type and six modified NF-κBs. Each of the mutant NF-κBs contained a single stoichiometrically phosphorylated Tyr at a different position. As shown in Figure 6, relative to the wild type, the genes exhibited varying transcription levels under the control of each individual NF-κB. The broad spectrum of altered transcriptional responses can only be due to the presence of the stoichiometrically phosphorylated tyrosines in the NF-κBs, which must have persisted to a significant extent during transcription.
Figure 6.
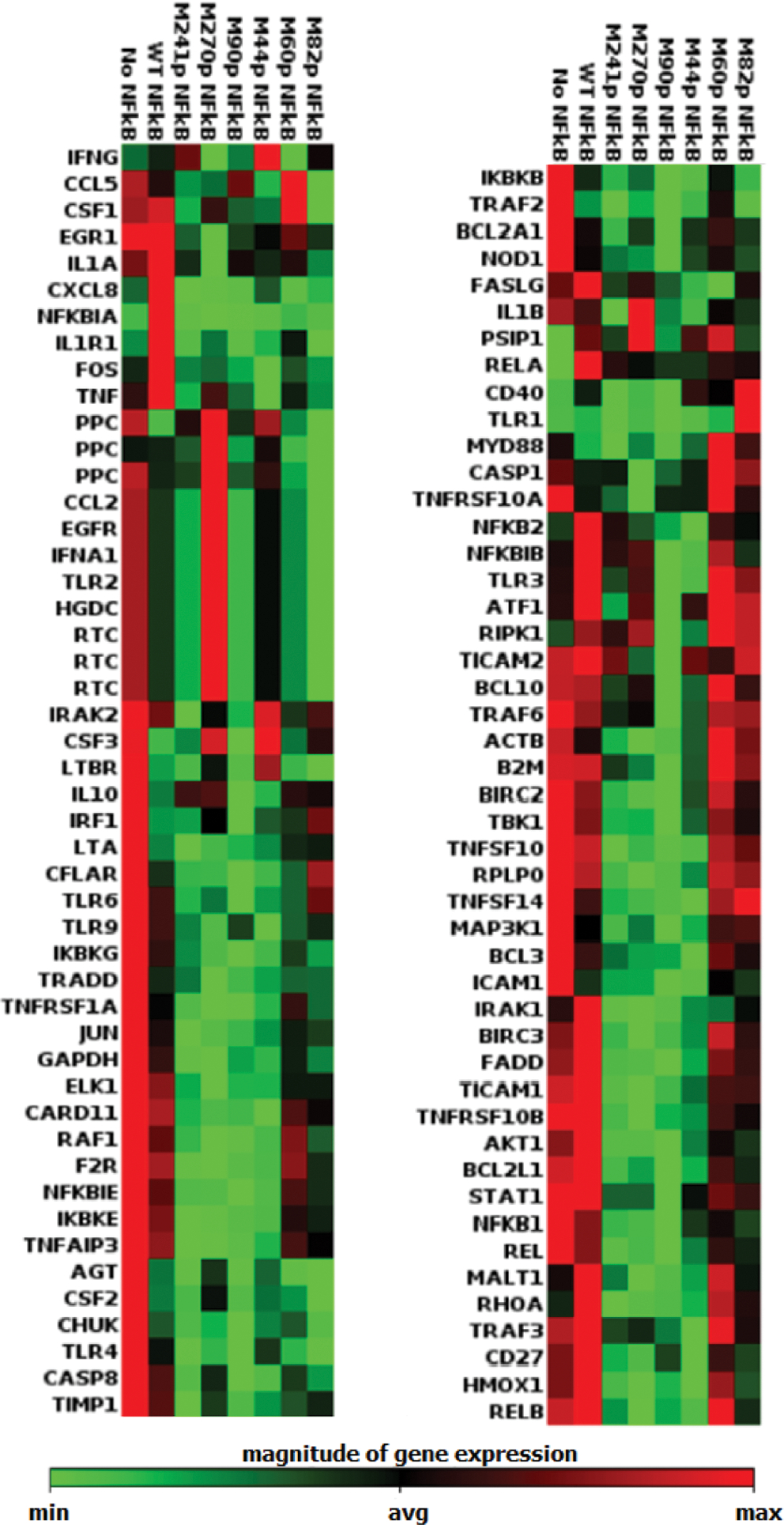
Identification of proteins with potentially altered levels of expression. A Qiagen RT2 Profiler PCR Array was employed to determine the altered transcription of a set of 96 human NF-κB genes involved in signaling pathways. These genes were studied in the presence of wild-type NF-κB and six mutants. Each of the mutants contained a stoichiometrically phosphorylated tyrosine, all differing by their position. The genes exhibited different transcription levels for each individual NF-κB. The letter M preceding the label for each phosphorylated NF-κB protein denotes “mutant”.
In a recently published study, we have shown that NF-κBs having stoichiometrically phosphorylated tyrosines at positions 60 or 82 were bound to three CD40 promoter DNAs more avidly than to NF-κBs phosphorylated at other tyrosine residues.10 Further, the modified NF-κBs supported enhanced transcription and translation of CD40 in cellulo.
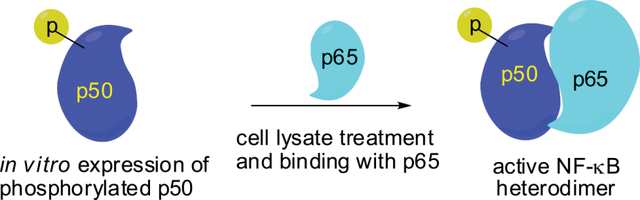
This study further defines the effect of tyrosine phosphorylation in NF-κB activation for selective transcription of a subset of the numerous genes regulated by NF-κB. The enhanced binding of promoter DNAs as a consequence of stoichiometric phosphorylation of certain NF-κB tyrosine residues is consistent with the possibility that selective gene transcription physiologically may involve only a small subset of all NF-κBs that are modified selectively at one or more key positions. Plausibly, this might allow a subset of regulated genes to be expressed by the introduction of DNA plasmids capable of expressing selectively phosphorylated NF-κB subunits in vivo (Scheme 2).
Scheme 2.
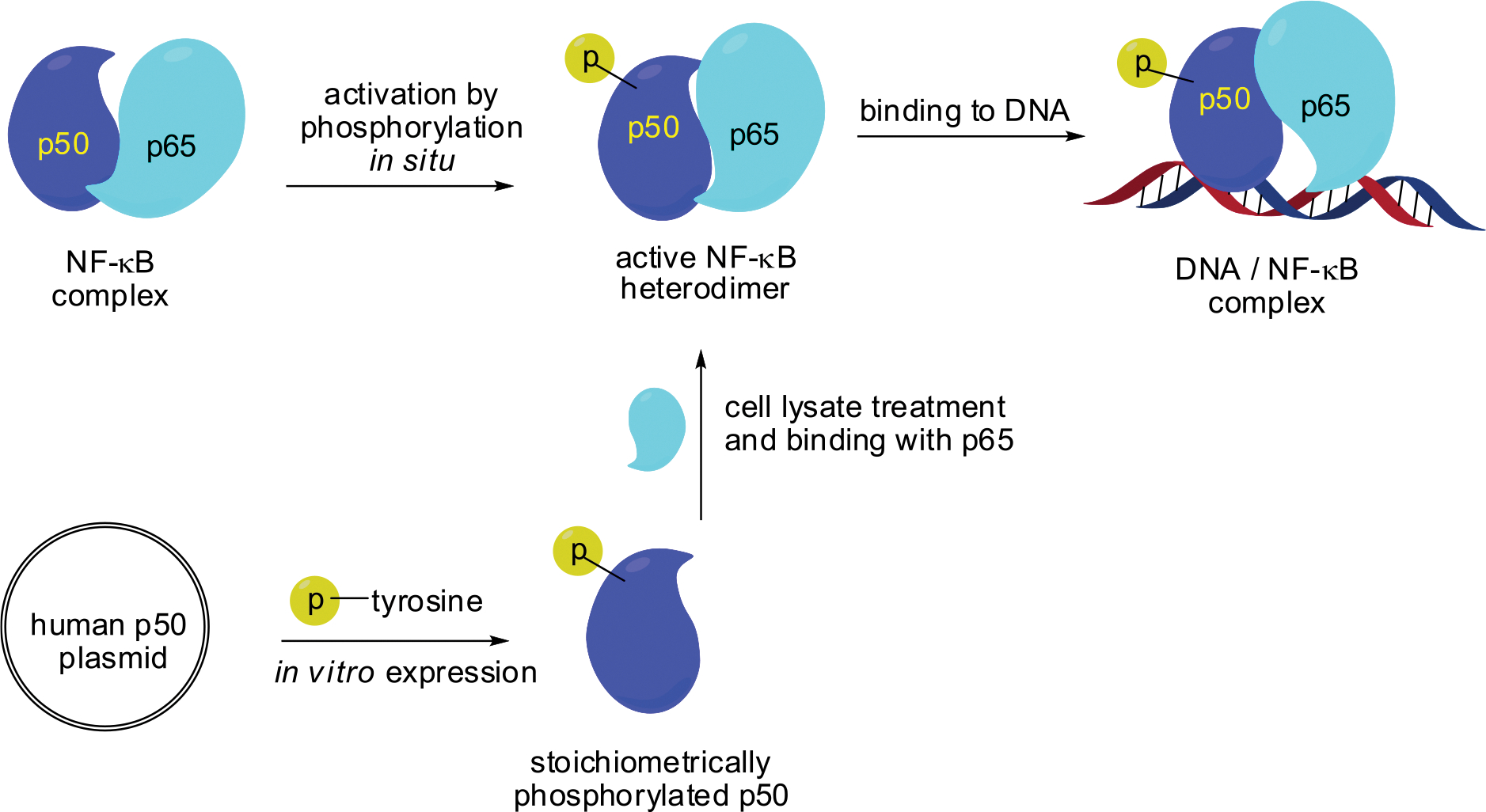
Strategy for the Activation of Human NF-κB Heterodimer Stoichiometrically with Phosphorylated Tyrosine
Also documented in the present study was the ability of stoichiometrically phosphorylated tyrosine residues in the p50 subunit of NF-κB to persist under routine experimental conditions used to study DNA binding and transcription. While physiological tyrosine phosphate levels normally reflect concurrent enzymatic phosphorylation and dephosphorylation, purposeful blockade of either process should enable the study of either process individually and may well occur on a time scale amenable to routine biochemical procedures.
Finally, we note that stoichiometric phosphorylation at some tyrosine residues in p50 resulted in the formation of NF-κB p50/p65 heterodimers exhibiting enhanced IL-2 promoter DNA binding. This enhancement may have resulted in part from the presence of the stoichiometric phosphate group on each NF-κB p50/p65 heterodimer, as compared with presumed partial phosphate occupancy induced by Jurkat cell kinases. Alternatively, it seems possible that cellular phosphatases may dephosphorylate protein phosphates less efficiently at tyrosines that do not normally bear phosphate groups. Consistent with this possibility, there are a number of examples of protein tyrosine phosphatases that operate selectively based on the sequence context of individual Tyr residues.26–28 In either case, the presence of tyrosine phosphates at sites not normally phosphorylated in a protein, but whose phosphorylation nonetheless confers novel or enhanced protein properties, may represent a novel strategy for modulating protein function.
3. CONCLUSIONS
NF-κB regulation involves not only NF-κB–DNA binding, but also many cofactors. The cofactors are essential for the expression of NF-κB regulated genes in a chromatin environment. While still a focus of an ongoing investigation, the mechanism(s) by which gene expression is regulated also include phosphorylation of Rel homology domain protein p65 (Rel A), the phosphorylation of which at Ser276 is essential for proper NF-κB dependent gene expression.29 For this study, we explored the stoichiometric phosphorylation of tyrosine in the NF-κB p50 subunit as a factor in the regulation of the binding of the IL-2 DNA promoter. This represents a new strategy for identifying structural changes in a transcription factor essential for its selectivity of action. While the stoichiometric modification of Tyr residues in NF-κB that leads to an increase in promoter DNA binding does not provide direct evidence that these residues are phosphorylated physiologically, the aggregate data suggest that this may sometimes be the case.
NF-κB regulates the expression of numerous genes, including IL-2. While usually investigated at a cellular level, much of what is still incompletely understood about this long-studied regulatory system involves molecular events, perhaps more logically studied in cell-free systems. In the present study, we elaborated p50 and p65 proteins in vitro and alternatively employed altered bacterial ribosomes to introduce pTyr stoichiometrically at one of several p50 sites usually occupied by tyrosine. The purified p50 and p65 subunits readily formed p50/p65 heterodimers, but unexpectedly, this complex would not bind to the promoter region of IL-2 DNA, an interaction demonstrated previously using NF-κB isolated from Jurkat cells. In the presence of [γ−32P]-GTP and a cytoplasmic lysate from activated Jurkat cells, radiolabeled phosphate was rapidly accumulated into p50, and a p50/p65 complex regained its ability to bind IL-2 promoter DNAs on the same time scale. A subset of the p50s containing pTyr also exhibited up to a 3.5-fold increase in DNA promoter binding. The ability to detect alterations of DNA binding and transcription in vitro and in vivo, as well as coupled translation in vivo,10 as a consequence of certain stoichiometric NF-κB subunit phosphorylations, seems consistent with the possibility that the selectivity of NF-κB-mediated gene activation may normally involve the participation of only a subset of the population of NF-κB heterodimers present in cells and that these have been primed for the expression of specific genes.
4. EXPERIMENTAL SECTION
4.1. Experimental Procedures.
The plasmids containing the p50 and p65 subunit genes of NF-κB were obtained from Synbio Technologies, Inc. Ni-NTA agarose was purchased from Qiagen Inc. DNA oligonucleotides were from IDT (Integrated DNA Technologies). Acrylamide, ammonium persulfate, N,N′-methylene-bis-acrylamide, dithiothreitol, potassium glutamate, acetic acid, ammonium acetate, magnesium acetate, E. coli tRNA, isopropyl β-D-thiogalactopyranoside (IPTG), phospho(enol)pyruvate, CTP, UTP, ATP, GTP, cAMP, amino acids, rifampicin, sodium pyruvate, glutamine, formamide, calcium ionophore A23187, PMA, phenylmethanesulfonyl fluoride (PMSF), and Nonidet P40 were purchased from Sigma-Aldrich, while SDS and Tris were from Bio-Rad Laboratories. [35S]-Met (1000 Ci/mmol, 10 μCi/μL) and [γ−32P]-ATP (6 Ci/mmol, 10 μCi/μL) were obtained from PerkinElmer Inc. T4 polynucleotide kinase and T4 RNA ligase were from New England Biolabs Inc. RPMI 1640 medium, streptomycin, penicillin, and fetal bovine serum (FBS) were purchased from the American Type Culture Collection (ATCC). The human Jurkat leukemia T cell line was from ATCC (TIB-152).
Phosphorimager analysis was carried out with an Amersham Biosciences Storm 820 utilizing ImageQuant software (version 5.2, Molecular Dynamics). UV spectral measurements were determined on a PerkinElmer Lambda 20 UV/vis spectrometer.
4.2. Site-Directed Mutagenesis to Obtain the Mutant p50 Gene of NF-κB Having TAG Codons at Protein Positions 7, 44, 60, 82, 90, 241, and 270.
The wild-type pET16b-Strep-p50 plasmid was purchased from Synbio Technologies. The primers for protein TAG modifications were: 5′-CC ATG GCA GAA GAT GAT CCA TAG TTG GGT CGC CCT GAA CAA ATG-3′ (position 7); 5′-CCA ACA GCA GAT GGC CCA TAG CTT CAA ATT TTA GAG CAA CC-3′ (position 44); 5′-CGC GGT TTT CGT TTC CGT TAG GTA TGT GAA GGC CCA TCT CAT G-3′ (position 60); 5′-CT AGT GAA AAG AAC AAG AAG TCT TAG CCT CAG GTC AAA ATC TGC AAC-3′ (position 82); 5′-CT CAG GTC AAA ATC TGC AAC TAG GTG GGT CCA GCA AAG GTT ATT G-3′ (position 90); 5′-CA GTG GTA TCA GAC GCC ATC TAG GAC AGT AAA GCC CCT AAT GCA TC-3′ (position 241); 5′-GTG ACT GGT GGG GAG GAA ATT TAG CTT CTT TGT GAC AAA GTT CAG AAA GAT G-3′ (position 270).
A reaction mixture (25 μL total volume for each primer) containing 200 pmol of primer, 1 mM ATP, 30 units of T4 polynucleotide kinase, and 2.5 μL of PNK buffer was incubated at 37 °C for 60 min.
The PCR reaction was performed in a 50 μL reaction mixture (200 ng of template plasmid DNA, 16 pmol of the requisite primer, 10 nmol of dNTPs, 5 units of Pfu DNA polymerase, 2.5 μL of Pfu buffer, 40 units of Taq DNA ligase, and 2.5 μL of Taq buffer). An aliquot of 30 μL of mineral oil was added on top of the reaction mixture. Thermal cycle: preincubated at 65 °C for 5 min (allowing the ligase to repair any nicks in the template DNA); denaturation at 95 °C for 2 min; 18 cycles at 95 °C for 1 min, 51 °C for 1 min, 65 °C for 15 min, and post-incubation at 75 °C for 7 min. Restriction enzyme DpnI (4 μL) was added to the reaction mixture which was maintained at 37 °C for 60 min, eliminating methylated and hemimethylated wild-type DNA template. Sample denaturation was then carried out at 95 °C for 30 s, and this was followed by two cycles at 95 °C for 30 s, 51 °C for 1 min, and 70 °C for 7 min. The final sample (10 μL) was then transformed into 100 μL of E. coli competent cells DH5α. The product was placed on an ampicillin plate and incubated at 37 °C overnight.
4.3. Misacylation of Suppressor tRNA-COH with Phosphotyrosyl-pdCpA or Phosphomethyltyrosyl-pdCpA.
The yeast suppressor tRNAPheCUA-COH transcript was isolated as described.30 Suppressor tRNACUA-COH activation was effected in 200 μL (total volume) of 100 mM Hepes, pH 7.5, that also contained 15 mM MgCl2, 2.0 mM ATP, 200 μg of suppressor tRNA-COH, 2.0 A260 units of N-pentenoyl, bis-o-nitrobenzyl protected phosphotyrosyl-pdCpA8 or phosphomethyltyrosyl-pdCpA31 (5–10 fold molar excess, Scheme 1), and 15% DMSO and T4 RNA ligase (400 units). Following incubation (37 °C for 1 h), quenching of the reaction was accomplished by adding 20 μL of 3 M NaOAc, pH 5.2, and then by the addition of 600 μL of ethanol. The incubation mixture was maintained at −20 °C for 30 min and then centrifuged at 15,000g at 4 °C for 30 min. The supernatant was removed by careful decantation, and the pellet containing the tRNA was washed (200 μL of 70% ethanol) and dissolved in 200 μL of (RNase free) H2O. The efficiency of ligation was determined using 8% denaturing PAGE, pH 5.2.32 Deprotection of the pentenoyl-protected aminoacyl-tRNA was effected by treatment with 5 mM aqueous I2 over a period of 10 min.33 The reaction mixture was quenched by treatment with 20 μL of 3 M NaOAc, pH 5.2, and then by the addition of 600 μL of ethanol. The reaction mixture was maintained at −20 °C for 30 min, then the tRNA was recovered by centrifugation at 15,000g at 4 °C for 30 min. Careful decantation of the supernatant afforded the tRNA pellet, which was washed with 200 μL of 70% ethanol and dissolved in 60 μL of H2O (RNase free). After treatment with high-intensity Hg–Xe light, the aminoacylated suppressor tRNA was used in in vitro suppression experiments.
4.4. In Vitro Translation of Mutant NF-κB p50 Subunit Genes (Modified at Protein Positions 7, 44, 60, 82, 90, 241, and 270) and Wild-Type p50 and p65.
The in vitro expression mixture (100 μL total volume) containing 10 μg of plasmid DNA with mutant NF-κB p50 genes (modified at protein positions 7, 44, 60, 82, 90, 241, and 270), 40 μL of a premix (35 mM Tris-acetate, pH 7.0, 30 mM ammonium acetate, 190 mM potassium glutamate, 25 mM magnesium acetate, 2.0 mM dithiothreitol, 20 mM phosphor-(enol)pyruvate, 0.8 mg/mL of E. coli tRNA, 20 mM GTP and ATP, 5 mM UTP and CTP, 4 mM cAMP, and 0.8 mM IPTG), 100 μM of each of the 20 proteinogenic amino acids, 30 μCi of [35S]-L-methionine, 10 μg/μL rifampicin, 30 μL of S-30 lysate from E. coli strain BL21(DE3), and 30–50 μg of phosphotyrosyl-tRNACUA or phosphomethyltyrosyl-tRNACUA (from which the protecting groups had been removed). The reaction mixture was maintained at 30 °C for 45 min to maximize the yields.34 Plasmid DNA containing the wild-type p50 gene was employed as a positive control, and an abbreviated tRNA (tRNA-COH) not activated with any amino acid was used as a negative control. Representative examples of the phosphorylated proteins studied here have been confirmed structurally by mass spectrometry.10
A plasmid DNA containing the wild-type p65 gene was used to prepare the p65 protein. An aliquot containing 2 μL of the incubation mixture was removed and combined with 2 μL of a loading buffer (250 mM Tris-HCl, pH 6.8, containing 10% glycerol, 1% SDS, 80 mM DTT, and 0.01% bromophenol blue). This mixture was heated at 90 °C for 2 min. The sample was then analyzed by means of 15% SDS-PAGE at 100 V for 2 h.
4.5. Purification of the p50 Protein Subunit of NF-κB with Strep-Tactin Sepharose.10
The in vitro expression mixture (500 μL) was treated with 1500 μL of 100 mM Tris-HCl, pH 8.3, and was then applied to a 100 μL column of Strep-Tactin Sepharose. The column was washed with 500 μL of 100 mM Tris-HCl, pH 8.3, and eluted with 100 μL of 100 mM Tris-HCl, pH 8.3, containing 2.5 mM desthiobiotin three times. An aliquot containing 2 μL of each portion was removed; combined with 2 μL of 250 mM Tris-HCl, pH 6.8, containing 10% glycerol, 1% SDS, 0.01% bromophenol blue, and 80 mM DTT; and heated at 90 °C for 2 min. The product was analyzed by 15% SDS-PAGE (100 V, 2 h).
4.6. Purification of the p65 Subunit with Ni-NTA under Native Condition.35,36
The in vitro expression mixture for p65 protein (1000 μL) was treated with 3000 μL of 50 mM Tris-HCl, pH 8.0, and 300 μL of Ni-NTA beads were added. After incubation (4 °C for 5 min), the beads were washed (2000 μL of 50 mM Tris-HCl, pH 8.0, containing 300 mM NaCl and 20 mM imidazole). The p65 protein was eluted three times (300 μL of 50 mM Tris-HCl, pH 8.0, containing 300 mM NaCl and 150 mM imidazole). The p65 protein was concentrated using an Amicon centrifugal filter (cutoff 10 kDa). Aliquots of each fraction were analyzed by 10% SDS-PAGE.
4.7. Preparation of Jurkat Cytosolic Fraction.37
Jurkat cells were cultured (37 °C, 5% CO2 atmosphere) and grown in Gibco RPMI 1640 medium supplemented with 10% FBS that also contained 50 units/mL of penicillin and 50 μg/mL of streptomycin. When grown to 5 × 105 cells/mL density, the cells were stimulated (1.0 μM calcium ionophore A23187 and 0.3 μM PMA) at 37 °C for 1 h. The stimulated Jurkat cells (20 mL) were harvested by centrifugation (500g at room temperature for 5 min) and washed once [5 mL of cold phosphate-buffered saline (PBS)]. The cells were resuspended in 200 μL of 10 mM Hepes, pH 7.9, containing 1.5 mM MgCl2, 0.5 mM DTT, 10 mM KCl, and 0.1% Nonidet P40. They were then incubated at 4 °C for 10 min. Following centrifugation (1000g at room temperature for 2 min), the supernatant containing cytosolic fractions was collected in new tubes and incubated at 37 °C for 6 h. Then this Jurkat cytosolic fraction was stored at −80 °C.
4.8. 5′−32P-Labeling of DNA.8
A reaction mixture (50 μL total volume) containing 400 pmol of IL-2 primer (5′-GATCCAAGGGACTTTCCATG-3′), 6 μL of γ−32P-ATP, T4 polynucleotide kinase (30 units), and 5 μL of PNK buffer was incubated (37 °C for 60 min). The excess γ−32P-ATP was removed using a G-25 column. A complementary DNA strand (400 pmol) was added to the 32P-labeled DNA oligonucleotide. The reaction mixture was heated (90 °C for 5 min) and allowed to cool to room temperature over a period of 1 h to obtain the double-stranded DNA.
4.9. NF-κB and DNA Binding Assay.
The cytoplasmic lysate was incubated at 37 °C for 6 h, then cooled on ice and combined with cold samples of NF-κB and IL-2 promoter DNA. The reaction mixture (10 μL total volume) contained 0–8 ng of purified NF-κB, 8 pmol of double-stranded 5′−32P-labeled DNA probe, 5 mM GTP, and 3 μL of cytoplasmic lysate. The reaction mixture was incubated at 4 °C for 10 min. Analysis of the reactions was carried out by 5% native PAGE at 100 V for 1 h. The relative intensities were determined using ImageQuant software (version 5.2, Molecular Dynamics) based on the 32P signal.
4.10. Phosphorylation of Wild-Type and Mutant p50 Subunits in Jurkat Cell Lysate.
The in vitro expressed p50 protein (80 ng) was added into a 50 μL of reaction mixture containing 15 μL of a Jurkat cell cytoplasmic lysate, 15 μL of Gibco RPMI 1640 medium supplemented with 10% FBS, and 80 nM γ−32P-GTP (6000 Ci/mmol). The reaction mixture was incubated on ice. A 5 μL aliquot of the reaction mixture was treated with loading buffer (5 μL) and heated at 90 °C for 2 min. Analysis of the samples was done by 15% SDS-PAGE at 100 V for 2 h. The gel was quantified using a phosphorimager.
4.11. Time-Dependent DNA Binding with Phosphorylated NF-κB in Jurkat Cell Lysate.
In a 6 μL reaction mixture, the in vitro expressed p50 protein (8 ng; wild-type or containing pTyr60) was combined with 10 ng of wild-type p65 protein, and 1.5 μL of Jurkat cell cytoplasmic lysate was added, as was 1.5 μL of Gibco RPMI 1640 medium supplemented with 10% FBS and 80 nM GTP. At predetermined times (0.5–60 min), 2 pmol of a 32P-labeled oligonucleotide having the IL-2 promoter sequence was added to each reaction mixture. After incubation with the DNA on ice (1 min), the reaction mixtures were treated with loading buffer (4 μL, 50% glycerol in Tris-acetate-EDTA (TAE)) and loaded on a 5% native PAGE gel. The gel was run in TAE (20 mM Tris, 40 mM acetate, containing 1 mM ethylenediaminetetraacetic acid (EDTA), pH 8.0) at 100 V for 1 h. The gel was quantified using a phosphorimager.
4.12. Stability of Phosphorylated NF-κB in Jurkat Cell Lysate.
In vitro expressed wild-type p50 protein, or modified p50s containing pTyr or p-CH2-Tyr at position 60 (80 ng), were mixed with 100 ng of wild-type p65 protein and added to 50 μL reaction mixtures containing 15 μL of lysate from activated Jurkat cells and 15 μL of Gibco RPMI 1640 medium supplemented with 10% FBS in the presence of 5 mM GTP. The reaction mixtures were incubated at room temperature for 10 min. 32P-labeled IL-2 DNA (2 pmol portions) were added to each of the reaction mixtures, and incubation was continued at room temperature for 60 min. Five-μL aliquots were taken at predetermined times and analyzed using 5% native PAGE.
4.13. Transcription in Jurkat Cells of 96 Human NF-κB Signaling Pathway Genes.
Jurkat cells (20 mL) stimulated with 0.3 μM PMA and 1.0 μM calcium ionophore A23187 were collected by centrifugation at 500g (room temperature, 5 min) and washed with 5 mL of cold PBS. The cells were resuspended in 200 μL of 20 mM Hepes, pH 7.9, with 0.5 mM DTT, 50 mM KCl, 0.2 mM EDTA, 0.5 mM PMSF, and 0.17 mM Triton-X100. They were then incubated at 4 °C for 10 min. The pretreated Jurkat cells were stored at −80 °C.
The in vivo expression mixture (60 μL total volume) contained 25 μL of pretreated Jurkat cells, 100 ng of wild-type or modified NF-κB, 5 mM GTP, and 25 μL of RPMI-1640 cell culture medium. The reaction mixture was incubated at 30 °C for 1 h. The total RNAs were extracted with the miRNeasy Mini Kit (Qiagen) following the standard protocol in the handbook (https://www.qiagen.com/us/resources/resourcedetail?id=14e7cf6e-521a-4cf7-8cbc-bf9f6fa33e24&lang=en). After reverse transcription using an RT2 First Strand Synthesis Kit (Qiagen), the RNAs were converted into the corresponding DNAs following a standard protocol (https://www.qiagen.com/us/resources/resourcedetail?id=f4b13eaa-884f-4357-abe6-1a5f9469bc32&lang=en). The generated DNAs were added to a 384-well plate for real-time PCR to identify the overexpression of the target genes. The obtained data were analyzed using the Qiagen online tool “RT2 Profiler PCR Data Analysis” (https://dataanalysis2.qiagen.com/pcr).
Supplementary Material
ACKNOWLEDGMENTS
This study was supported by Research Grants R01GM103861 and R35GM140819 from the National Institute of General Medical Sciences, NIH.
Footnotes
The authors declare no competing financial interest.
ASSOCIATED CONTENT
Supporting Information
The Supporting Information is available free of charge at https://pubs.acs.org/doi/10.1021/acschembio.2c00678.
Expression, phosphorylation, promoter DNA binding, and statistical analysis of modified NF-κBs (PDF)
Contributor Information
Shengxi Chen, Biodesign Center for BioEnergetics, and School of Molecular Sciences, Arizona State University, Tempe, Arizona 85287, United States.
Xun Ji, Biodesign Center for BioEnergetics, and School of Molecular Sciences, Arizona State University, Tempe, Arizona 85287, United States.
Larisa M. Dedkova, Biodesign Center for BioEnergetics, and School of Molecular Sciences, Arizona State University, Tempe, Arizona 85287, United States
Gal Reddy Potuganti, Biodesign Center for BioEnergetics, and School of Molecular Sciences, Arizona State University, Tempe, Arizona 85287, United States.
Sidney M. Hecht, Biodesign Center for BioEnergetics, and School of Molecular Sciences, Arizona State University, Tempe, Arizona 85287, United States
REFERENCES
- (1).Garcia-Garcia T; Poncet S; Derouiche A; Shi L; Mijakovic I; Noirot-Gros M Role of protein phosphorylation in the regulation of cell cycle and DNA-related processes in bacteria. Front. Microbiol. 2016, 7, 184. [DOI] [PMC free article] [PubMed] [Google Scholar]
- (2).Burnett G; Kennedy EP The enzymatic phosphorylation of proteins. J. Biol. Chem. 1954, 211, 969–980. [PubMed] [Google Scholar]
- (3).Eckhart W; Hutchinson MA; Hunter T An activity phosphorylating tyrosine in polyoma T antigen immunoprecipitates. Cell 1979, 18, 925–933. [DOI] [PubMed] [Google Scholar]
- (4).Vincent C; Duclos B; Grangeasse C; Vaganay E; Riberty M; Cozzone AJ; Doublet P Relationship between exopolysaccharide production and protein-tyrosine phosphorylation in gram-negative bacteria. J. Mol. Biol. 2000, 304, 311–321. [DOI] [PubMed] [Google Scholar]
- (5).Pawson T Specificity in signal transduction: from phosphotyrosine-SH2 domain interactions to complex cellular systems. Cell 2004, 116, 191–203. [DOI] [PubMed] [Google Scholar]
- (6).Hunter T; Eckhart W The discovery of tyrosine phosphorylation: It’s all in the buffer! Cell 2004, 116, S35–S39. [DOI] [PubMed] [Google Scholar]
- (7).Grangeasse C; Cozzone A; Deutscher J; Mijakovic I Tyrosine phosphorylation: an emerging regulatory device of bacterial physiology. Trends Biochem. Sci. 2007, 32, 86–94. [DOI] [PubMed] [Google Scholar]
- (8).Chen S; Maini R; Bai X; Nangreave RC; Dedkova LM; Hecht SM Incorporation of phosphorylated tyrosine into proteins: in vitro translation and study of phosphorylated IκB-α and its interaction with NF-κB. J. Am. Chem. Soc. 2017, 139, 14098–14108. [DOI] [PMC free article] [PubMed] [Google Scholar]
- (9).Dedkova LM; Fahmi NE; Paul R; del Rosario M; Zhang L; Chen S; Feder G; Hecht SM β-Puromycin selection of modified ribosomes for in vitro incorporation of β-amino acids. Biochemistry 2012, 51, 401–415. [DOI] [PubMed] [Google Scholar]
- (10).Chen S; Ji X; Dedkova LM; Hecht SM Site-selective incorporation of phosphorylated tyrosine into the p50 subunit of NF-κB and activation of its downstream gene CD40. Chem. Commun. 2021, 57, 12651–12654. [DOI] [PMC free article] [PubMed] [Google Scholar]
- (11).Kleiman LB; Maiwald T; Conzelmann H; Lauffenburger DA; Sorger PK Rapid phosphor-turnover by receptor tyrosine kinases impacts downstream signaling and drug binding. Mol. Cell 2011, 43, 723–737. [DOI] [PMC free article] [PubMed] [Google Scholar]
- (12).Luo X; Fu G; Wang RE; Zhu X; Zambaldo C; Liu R; Liu T; Lyu X; Du J; Xuan W; Yao A; Reed SA; Kang M; Zhang Y; Guo H; Huang C; Yang PY; Wilson IA; Schultz PG; Wang F Genetically encoding phosphotyrosine and its nonhydrolyzable analog in bacteria. Nat. Chem. Biol. 2017, 13, 845–849. [DOI] [PMC free article] [PubMed] [Google Scholar]
- (13).Liou H-C; Baltimore D Regulation of the NF-κB/rel transcription factor and IκB inhibitor system. Curr. Opin. Cell Biol. 1993, 5, 477–487. [DOI] [PubMed] [Google Scholar]
- (14).Grimm S; Baeuerle PA The inducible transcription factor NF-κB: structure–function relationship of its protein subunits. Biochem. J. 1993, 290, 297–308. [DOI] [PMC free article] [PubMed] [Google Scholar]
- (15).Baeuerle P; Henkel T Function and activation of NF-κB in the immune system. Annu. Rev. Immunol. 1994, 12, 141–179. [DOI] [PubMed] [Google Scholar]
- (16).Ghosh S; Hayden MS Celebrating 25 years of NF-κB research. Immunol. Rev. 2012, 246, 5–13. [DOI] [PMC free article] [PubMed] [Google Scholar]
- (17).Hayden MS; Ghosh S NF-κB, the first quarter-century: remarkable progress and outstanding questions. Genes Dev. 2012, 26, 203–234. [DOI] [PMC free article] [PubMed] [Google Scholar]
- (18).Christian F; Smith EL; Carmody RJ The regulation of NF-κB subunits by phosphorylation. Cells 2016, 5, 12. [DOI] [PMC free article] [PubMed] [Google Scholar]
- (19).Hou S; Guan H; Ricciardi RP Phosphorylation of serine 337 of NF-kappaB p50 is critical for DNA binding. J. Biol. Chem. 2003, 278, 45994–45998. [DOI] [PubMed] [Google Scholar]
- (20).Guan H; Hou S; Ricciardi RP DNA binding of repressor nuclear factor-kappaB p50/p50 depends on phosphorylation of Ser337 by the protein kinase A catalytic subunit. J. Biol. Chem. 2005, 280, 9957–9962. [DOI] [PubMed] [Google Scholar]
- (21).Pellegatta F; Bertelli AA; Staels B; Duhem C; Fulgenzi A; Ferrero ME Different short- and long-term effects of resveratrol on nuclear factor-κB phosphorylation and nuclear appearance in human endothelial cells. Am. J. Clin. Nutr. 2003, 77, 1220–1228. [DOI] [PubMed] [Google Scholar]
- (22).Kang JL; Jung HJ; Lee K; Kim HR Src tyrosine kinases mediate crystalline silica-induced NF-κB activation through tyrosine phosphorylation of IκB-α and p65 NF-κB in RAW 264.7 macrophages. Toxicol. Sci. 2006, 90, 470–477. [DOI] [PubMed] [Google Scholar]
- (23).Müller CW; Rey FA; Sodeoka M; Verdine GL; Harrison SC Structure of the NF-kappa B p50 homodimer bound to DNA. Nature 1995, 373, 311–317. [DOI] [PubMed] [Google Scholar]
- (24).Ruben SM; Dillon PJ; Schreck R; Henkel T; Chen CH; Maher M; Baeuerle PA; Rosen CA Isolation of a rel-related human cDNA that potentially encodes the 65-kD subunit of NF-kappa B. Science 1991, 251, 1490–1493. [DOI] [PubMed] [Google Scholar]
- (25).Chen FE; Huang DB; Chen YQ; Ghosh G Crystal structure of p50/p65 heterodimer of transcription factor NF-κB bound to DNA. Nature 1998, 391, 410–413. [DOI] [PubMed] [Google Scholar]
- (26).Sarmiento M; Zhao Y; Gordon SJ; Zhang Z-Y Molecular basis for substrate specificity of protein-tyrosine phosphatase 1B. J. Biol. Chem. 1998, 273, 26368–26374. [DOI] [PubMed] [Google Scholar]
- (27).Zhang Z-Y Protein tyrosine phosphatases: structure and function, substrate specificity, and inhibitor development. Annu. Rev. Pharmacol. Toxicol. 2002, 42, 209–234. [DOI] [PubMed] [Google Scholar]
- (28).Ren L; Chen X; Luechapanichkul R; Selner NG; Meyer TM; Wavreille A-S; Chan R; Iorio C; Zhou X; Neel BG; Pei P Substrate specificity of protein tyrosine phosphatases 1B, RPTPα, SHP-1 and SHP-2. Biochemistry 2011, 50, 2339–2356. [DOI] [PMC free article] [PubMed] [Google Scholar]
- (29).Bhatt D; Ghosh S Regulation of the NF-κB-mediated transcription of inflammatory genes. Front. Immunol. 2014, 5, 71. [DOI] [PMC free article] [PubMed] [Google Scholar]
- (30).Karginov VA; Mamaev SV; An H; Van Cleve MD; Hecht SM; Komatsoulis GA; Abelson JA Probing the role of an active aspartic acid in dihydrofolate reductase. J. Am. Chem. Soc. 1997, 119, 8166–8176. [Google Scholar]
- (31).Arslan T; Mamaev SV; Mamaeva NV; Hecht SM Structurally modified firefly luciferase. Effects of amino acid substitution at position 286. J. Am. Chem. Soc. 1997, 119, 10877–10887. [Google Scholar]
- (32).Chen S; Hecht SM Synthesis of pdCpAs and transfer RNAs activated with derivatives of aspartic acid and cysteine. Bioorg. Med. Chem. 2008, 16, 9023–9031. [DOI] [PubMed] [Google Scholar]
- (33).Lodder M; Golovine S; Hecht SM Chemical deprotection strategy for the elaboration of misacylated transfer RNAs. J. Org. Chem. 1997, 62, 778–779. [Google Scholar]
- (34).Lines JA; Yu Z; Dedkova LM; Chen S Design and expression of a short peptide as an HIV detection probe. Biochem. Biophys. Res. Commun. 2014, 443, 308–312. [DOI] [PubMed] [Google Scholar]
- (35).Yakovleva L; Chen S; Hecht SM; Shuman S Chemical and traditional mutagenesis of Vaccinia DNA topoisomerase provides insights to cleavage site recognition and transesterification chemistry. J. Biol. Chem. 2008, 283, 16093–16103. [DOI] [PMC free article] [PubMed] [Google Scholar]
- (36).Chen S; Zhang Y; Hecht SM p-Thiophenylalanine-induced DNA cleavage and religation activity of a modified vaccinia topoisomerase IB. Biochemistry 2011, 50, 9340–9351. [DOI] [PubMed] [Google Scholar]
- (37).Imbert V; Peyron JF; Farahi Far D; Mari B; Auberger P; Rossi B Induction of tyrosine phosphorylation and T-cell activation by vanadate peroxide, an inhibitor of protein tyrosine phosphatases. Biochem. J. 1994, 297, 163–173. [DOI] [PMC free article] [PubMed] [Google Scholar]
Associated Data
This section collects any data citations, data availability statements, or supplementary materials included in this article.