

Abstract
Endothelial cells are located at the critical interface between circulating blood and semi-solid tissues and have many important roles in maintaining systemic physiological function. The vascular endothelium is particularly susceptible to pathogenic stimuli that activate tumour suppressor pathways leading to cellular senescence. In the past few years, the literature on cellular senescence has expanded and we now understand that senescent endothelial cells are highly active, secretory and pro-inflammatory, and exhibit an aberrant morphological phenotype. Moreover, endothelial senescence has been identified as an important contributor to various cardiovascular and metabolic diseases. In this Review, we discuss the consequences of endothelial cell exposure to damaging stimuli (haemodynamic forces, circulating and endothelial derived factors) and the cellular and molecular mechanisms that induce endothelial cell senescence. We also discuss how endothelial cell senescence causes arterial dysfunction and contributes to clinical cardiovascular diseases and metabolic disorders. Finally, we summarize the latest evidence on the impact of eliminating senescent endothelial cells (senolysis), and identify important remaining questions that can be addressed in future studies.
Introduction
The endothelium is a highly dynamic monolayer of cells that lines the vascular network. Endothelial cells initiate and dictate the formation of blood vessels via vasculogenesis. The endothelium is also vital for maintaining blood in a fluid state and controls vascular tone through the production of vasoactive compounds, which is important for maintaining blood pressure and preventing orthostasis. Moreover, the endothelium is selectively permeable and mediates trafficking of macromolecules and immune cells out of the blood.
Endothelial cells are continuously exposed to many circulating factors and pathogenic stimuli that predispose them to damage, leading to impaired tissue and organ function via dysregulation of blood flow and barrier function. Furthermore, endothelial-cell-derived pro-inflammatory cytokines and chemokines transmit damaging signals to other vascular cells and non-vascular tissues and organs. A consequence of endothelial damage is cellular senescence, a state of permanent cell cycle arrest that occurs in response to a variety of stressors and acts to prevent uncontrolled proliferation of damaged cells and tumorigenesis.1 However, senescence can also be deleterious, and a growing body of literature suggests that endothelial cell damage could lead to senescence and the development of a host of diseases.
Several lines of evidence provide the impetus for investigating endothelial cell senescence. Characteristics of vascular architecture, as well as the rate of endothelial turnover, are likely to predispose these cells to senescence. Tissues with a high proportion of endothelial cells have been identified as having the greatest burden of senescent cells,2 and endothelial cells are known to be one of the first cell types to become senescent with advancing age.3 In vivo, endothelial senescence occurs in multiple vascular beds including the kidney,4 the retina,5 the liver,3 the brain6 and the aorta,7,8 suggesting that endothelial senescence contributes to a variety of pathological processes associated with vascular dysfunction. The development of models that allow for selective ablation of senescent cells, as well as drugs that selectively kill senescent cells (senolytics), has provided additional evidence that senescent endothelial cells represent a viable therapeutic target.9 Endothelial cellular senescence has, therefore, become a topic of intense investigation.
Evidence demonstrates that endothelial senescence impairs endothelium-dependent dilation, angiogenesis and barrier function. Furthermore, numerous studies have identified a role for endothelial senescence in atherosclerosis, heart failure, pulmonary hypertension and metabolic dysfunction. Thus, understanding the role of endothelial senescence in these settings could lead to the development of novel therapeutics for cardiometabolic diseases.
In this Review, we provide a detailed discussion of advances in the field of endothelial cell senescence, including the phenotype of senescent endothelial cells and the molecular mechanisms that contribute to or protect from senescence. We also discuss the consequences of an increased abundance of senescent endothelial cells and their contribution to arterial dysfunction and cardiometabolic diseases. We also summarize the latest evidence on approaches to senolysis; although several senolytic drugs have entered clinical trials, these studies have been reviewed in detail elsewhere9,10,14–17 and are beyond the scope of this article.
Phenotype of senescent endothelial cells
Two main tumor suppressor pathways are upregulated in response to a variety of senescence-inducing stressors. These pathways include the p53/p21 and pRB/p16 pathways.1 p21 and cyclin-dependent kinase inhibitor 2A (CDKN2A; also known as p16) enforce the senescent state and, therefore, are commonly used as markers of senescence. p21 and p16 are differentially activated depending on the cell type and the stressor.11 However, in some cell types, convergence is evident between p21-positive and p16-positive senescent cells, whereby damage leads to an early rise in p21 that decreases with time, followed by a later rise in p16.12,13 Although the specific kinetics of p21 and p16 in vivo remain elusive, evidence from mouse models suggests that cells with high levels of p21 or p16 represent distinct populations.14,15 The severity, duration and type of damage determines the molecular response and cell fate. For example, minor damage leads to p53 stabilization, allowing transcription of p21 until the damage is repaired, after which cell cycle re-entry is permitted.16–18 However, delayed activation of p21, or more severe or sustained damage leads to senescence, which is often associated with elevated p16 expression.16–18 In addition to senescence, p53 induces apoptosis, which occurs in response to even greater stress than is required for senescence.16,17 In healthy individuals, both senescence and apoptosis lead to clearance of damaged cells.17 However, in pathological states and advanced age, the balance between senescence and clearance is disrupted due to excessive damage or immune dysfunction (immunosenescence), leading to an accumulation of senescent cells.17
In addition to p21 and p16, senescent cells can also be identified by elevated activity of lysosomal senescence associated β-galactosidase.1,19 Moreover, additional markers of senesce have been identified in the past 2 years (for example, lysosomal and proliferative features, expression of senescence-associated genes), and a consensus has been reached that whenever possible a combination of two or more markers should be used to identify senescent cells with high fidelity.20,21
Senescent cells are highly active and characterized by the senescence-associated secretory phenotype (SASP), which is heterogeneous and determined by the cell type and mechanisms of senescence.5–8 Canonical SASP factors are a collection of inflammatory chemokines, cytokines, growth factors and, in some cases, reactive oxygen species (ROS) secreted by senescent cells.5 In endothelial cells, senescence produces a distinct morphological and secretory phenotype — senescent endothelial cells are flat, enlarged and are refractory to phenotypic changes and alignment in response to laminar shear stress22. Mechanistically, senescent endothelial cells increase adhesion to basement membranes preventing the correct alignment to blood flow22 (FIG. 2). The functional consequences of increased adhesion are that cells resist denudation and can persist for long periods of time. Additionally, the inability of these cells to align to blood flow might contribute to the reductions in NO and increased inflammatory signalling seen in senescent endothelial cells.23
Fig. 2 |. Exposure of endothelial cells to circulating and endogenous stimuli.
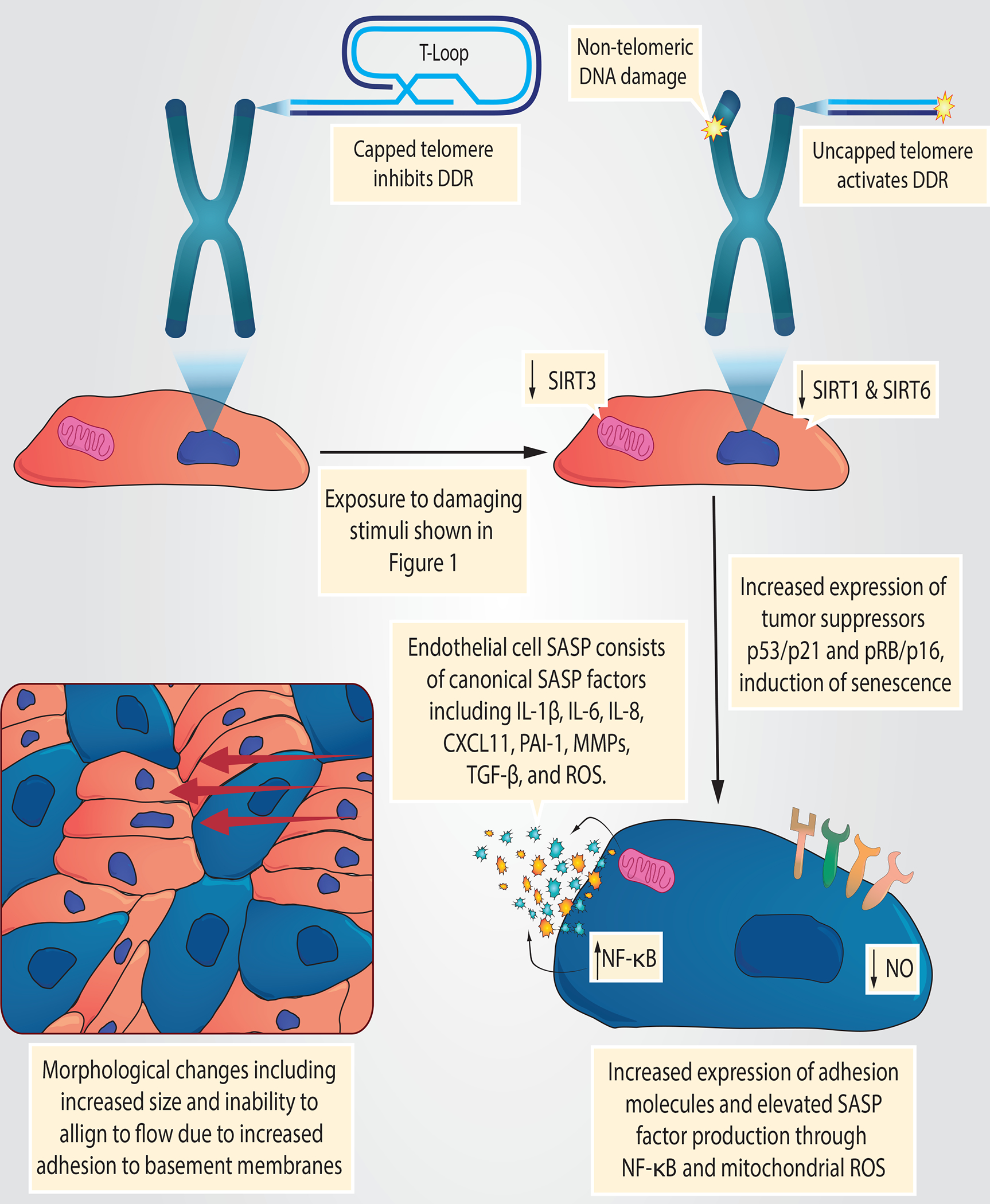
Owing to their anatomical position, endothelial cells are continually exposed to circulating biochemical factors and haemodynamic forces. These factors include oxygen, nutrients (e.g. amino acids, glucose, hormones (e.g. insulin, angiotensin II, endothelin 1 and lipids) and cytotoxic drugs (e.g. chemotherapeutics). Haemodynamic forces include low and oscillatory shear stress at bifurcations and curvature points, as well as high and pulsatile pressure in other regions. Endothelial cells are also exposed to inflammatory cytokines and reactive oxygen species (ROS) derived from circulating immune cells and directly from every organ and tissue. This combination of unique damaging stimuli renders endothelial cells particularly susceptible to cellular senescence. Moreover, these interactions are prolonged due to the long-lived nature of endothelial cells, particularly in large arteries where they are rarely replaced by progenitors.
Interestingly, the SASP of endothelial cells mirrors age-related changes that are important mediators of arterial dysfunction11, but were not originally attributed to senescence. Evidence suggests that endothelial senescence results in elevated expression of inflammatory cytokines and ROS akin to the canonical SASP. Thus, an increase in the abundance of senescent endothelial cells could cause inflammation and oxidative stress that impair endothelial function and stiffen large arteries through secretion of SASP factors. The source of ROS in senescent endothelial cells has not been fully elucidated. However, evidence suggests mitochondrial dysfunction as a potential source. For example, senescent cells have impaired mitochondrial respiration and increased mitochondrial superoxide production compared with non-senescent cells.24 Conversely, targeting mitochondrial ROS reduces senescence induced by hydrogen peroxide.25 The pro-inflammatory transcription factor nuclear factor NF-kappa-B (NF-κB) is associated with impaired endothelial function11,26 and seems to be involved in the endothelial cell SASP. For example, chronic exposure to the inflammatory cytokine tumour necrosis factor (TNF) results in premature senescence that is characterized by elevated NF-κB activity and ROS production.27 In this setting, inhibiting NF-κB or treating cells with antioxidants delays senescence and prevents induction of the SASP.27 Senescent endothelial cells also produce numerous inflammatory chemokines and cytokines including IL-1β,28 IL-6,27,29 IL-8,27,30 C-X-C motif chemokine 1131 and plasminogen activator inhibitor 1 (PAI-1),32,33 and demonstrate reduced expression of the anti-inflammatory cytokine IL-10,34 as well as increased expression of a variety of cellular adhesion molecules.27,35–38 Despite observations that the endothelial SASP is pro-inflammatory, several studies have demonstrated an opposite, anti-inflammatory effect of senescence in endothelial cells,39–41 highlighting the need for more research on this phenomenon both in vivo and ex vivo.
Remodelling that reduces the elastic properties of arteries, resulting in stiffening, contributes to the development of cardiovascular diseases (CVD). Senescent endothelial cells upregulate pathways involved in arterial remodelling42, such as the transforming growth factor-β (TGF-β)33,36 and matrix metalloproteinase (MMP) pathways,25 which could contribute to arterial stiffening. Indeed, in young mice, a genetic DNA repair defect in endothelial cells increases endothelial cell expression of cyclin-dependent kinase inhibitor 1A (CDKN1A; also known as p21) and aortic remodelling and stiffening, thereby phenocopying advanced age.43
Evidence from endothelial cells44 and other cell types45 suggests that secreted SASP factors can induce senescence in neighbouring cells. Even if endothelial SASP production is insufficient to drive senescence in neighbouring cells, such as vascular smooth muscle cells (VSMCs), nitric oxide (NO) is reduced in pathways associated with arterial remodelling, and elevated production of inflammatory SASP factors and ROS from senescent endothelial cells could have widespread deleterious effects on other cells in arteries and peripheral tissues.
Origins of endothelial senescence
Owing to their anatomical location, endothelial cells are continuously exposed to flowing blood containing many substances that can contribute to senescence, including oxygen, glucose, lipids, amino acid metabolites, immune cells, ROS and inflammatory cytokines46 (FIG. 1). The haemodynamic forces of blood flow, such as shear stress and high or pulsatile pressure, can also induce senescence. Endothelial cell turnover is high in areas exposed to disturbed blood flow; therefore, these cells experience damaging haemodynamic and replicative stress. In regions that do not experience disturbed blood flow, endothelial cells are long lived,47 with some cells estimated to survive for up to 80% of the average human lifespan. 48 However, long-lived cells accumulate damage through prolonged exposure to harmful substances in the blood. Therefore, both short-lived and long-lived endothelial cells can become senescent through a variety of stimuli. The various environmental challenges that contribute to senescence are discussed in the following sections.
Fig. 1 |. Morphological changes in senescent endothelial cells.
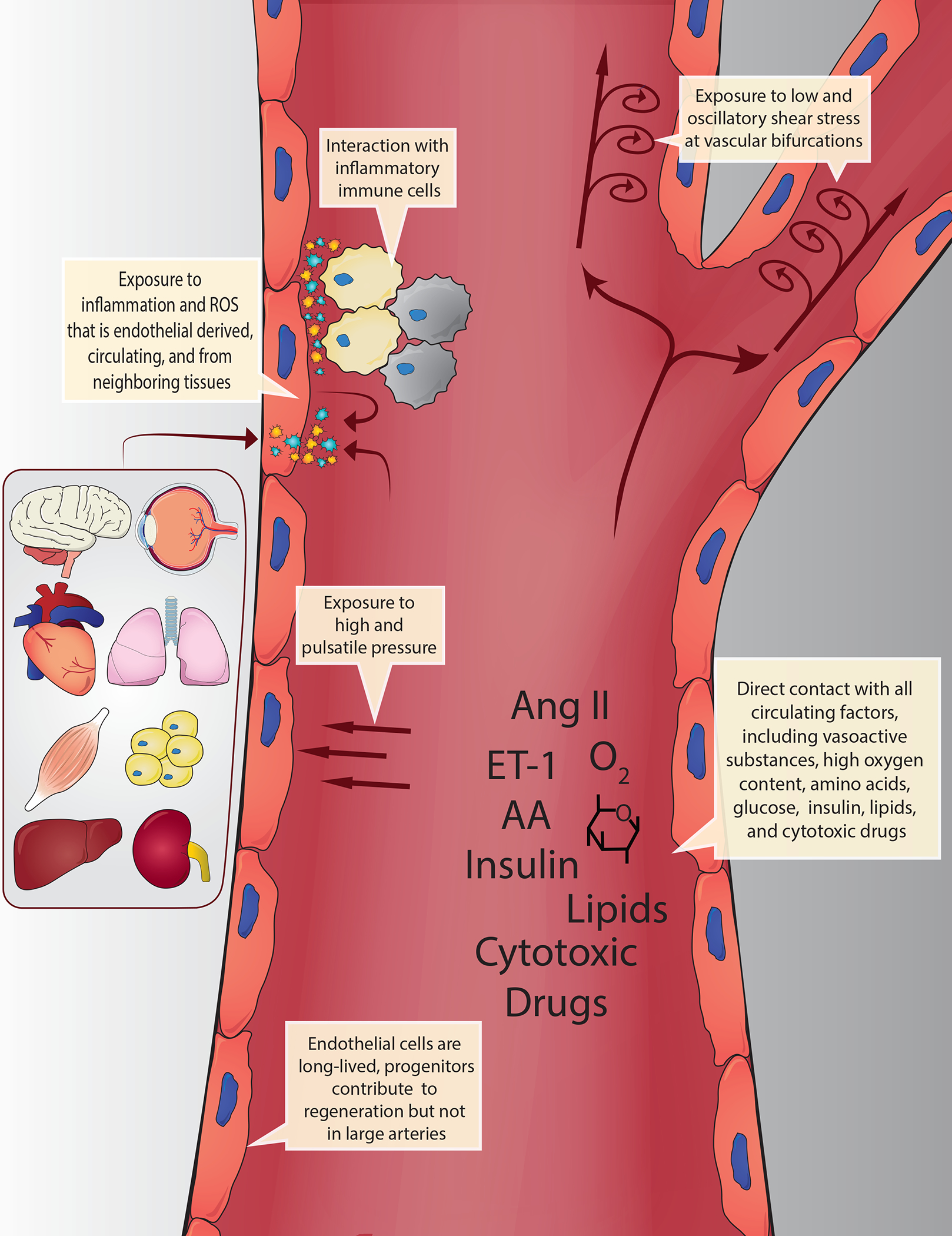
Senescent cells (shown in blue) become enlarged and adhesion to basement membranes is increased, thereby hindering their alignment to laminar shear stress.
Haemodynamic environment
The location of endothelial cells within the vascular tree dictates the haemodynamic environment. Within large conduit arteries, endothelial cells are exposed to high blood pressure and pulsatile circumferential stresses, both of which can induce damage. Endothelial cells at the sites of branches and curvatures, such as the carotid sinus and aortic arch, experience low and oscillatory shear stress, which is deleterious and leads to a phenotype akin to the SASP49,50. By contrast, endothelial cells in the distal internal carotid artery or descending thoracic aorta are exposed to high laminar shear stress, which exerts beneficial effects.
The importance of the haemodynamic environment is highlighted by studies of cell cultures and mouse arteries, which show that endothelial cells exposed to disturbed flow display high cellular turnover, leading to replicative senescence.50,51 Furthermore, evidence from human tissue samples shows that cell turnover and replicative senescence are greater in endothelial cells proximal to arterial bifurcations than in endothelial cells from arteries that experience less haemodynamic stress, venous endothelial cells and VSMCs.52 The effects of exposure to damaging haemodynamic environments could explain the increased burden of atherosclerosis at sites of low and oscillatory shear stress. This notion is supported by the higher abundance of senescent endothelial cells in human atherosclerotic plaques compared with healthy vasculature.38
Oxygen and metabolites
At rest and normoxia, the partial pressure of oxygen in the blood is approximately 100 mmHg in arteries and 40 mmHg in veins.53,54 By contrast, the interstitial partial pressure of oxygen is approximately 66 mmHg in the spleen53, 34 mmHg in skeletal muscle54 and 25 mmHg in the heart, liver and kidney53. The gradient between partial pressure of oxygen in arterial blood and peripheral tissue is greatly exaggerated during times of increased metabolic demand, such as exercise, when intracellular oxygen in peripheral tissues can be as low as 3 mmHg.54 Cellular exposure to high partial pressure of oxygen is well known to hasten the onset of senescence.55–59 Therefore, endothelial cells within the arterial tree continuously face a senescence-inducing stimulus not experienced by other cells within the body, which might explain their early onset of senescence. As such, it is important to consider that most cell culture experiments occur in supraphysiological oxygen conditions, and that mouse cells are more susceptible than human cells to oxidative DNA damage-induced senescence.57,60
Endothelial cells are continuously exposed to nutrients, metabolites and hormones (such as glucose, lipids, amino acids and insulin) and the concentration of these solutes increases postprandially.61,62 Likewise, pathological states such as obesity and diabetes mellitus, as well as advancing age, result in elevated circulating levels of insulin, glucose, triglycerides, cholesterol and amino acids61–64, which could contribute to endothelial cell senescence.8,65–72 Indeed, culturing endothelial cells with high levels of insulin results in premature senescence and reduced proliferation.73 Mechanistically, insulin activates the RAC-alpha serine/threonine-protein kinase (AKT1, also known as Akt) signalling pathway that inhibits forkhead box protein O3a resulting in reduced manganese superoxide dismutase activity and a subsequent increase in ROS.73 Several studies have demonstrated that glucose promotes endothelial cell senescence and SASP production both in vivo and in vitro.8,65,68–70 Culturing endothelial cells with high levels of glucose suppresses the expression of sirtuins and histone acetylases, as well as the activity of forkhead box protein O1, resulting in oxidative stress.69,70 Elevated ROS attenuates NO bioavailability, induces DNA damage and activates the cellular tumour antigen p53 (p53)–p21 signalling pathway, inducing proliferative arrest and cell senescence.73 Evidence also exists that high glucose levels reduce telomerase activity, telomere length and phosphorylation of endothelial NO synthase (eNOS) thereby reducing NO production, all of which foster endothelial cell senescence.8,66,74
Amino acid metabolites and lipids have also been shown to induce endothelial senescence in culture75–78 and in nonhuman primates.79 Homocysteine, which is produced during metabolism of the amino acid methionine, hastens the onset of senescence.78,80–82 In cultured human umbilical vein endothelial cells, chronic homocysteine treatment upregulates markers of senescence, due to demethylation of telomerase reverse transcriptase resulting in telomere shortening78 (discussed later in this Review). In mice, a high methionine diet increases serum homocysteine levels and promotes several features of vascular aging, including arterial senescence.83 Interestingly, reducing methionine is one of few interventions that has been shown to extend the lifespan of an organism in numerous models,84 which could be partly explained by a delay in the onset of endothelial cell senescence. The interaction between circulating lipids and the endothelium is also vital to our understanding of how senescence promotes disease. Atherosclerosis is initiated by endothelial dysfunction, leading to deposition of lipids underneath the endothelial barrier.85 Endothelial cell senescence could contribute to this lipid deposition, due to reduced expression of junctional proteins and impaired barrier function.86,87 Together, these findings suggest that the elevated levels of insulin, glucose, amino acids and lipids caused by physiological dysfunction and pathological states lead to endothelial cell senescence that, in turn, contributes to disease progression.
Vasodilators and vasoconstrictors
Endothelial cells produce numerous vasodilators and vasoconstrictors, and have receptors for circulating vasoactive substances of non-endothelial origin. Notably, endothelial cells produce the potent vasodilatory molecule NO, which diffuses to VSMCs to induce vasodilation, reduces endothelial expression of adhesion molecules, improves barrier function, prevents coagulation and is generally vasoprotective. Furthermore, evidence suggests that NO could be the key vasodilatory molecule in the prevention of cell senescence.67,74 Bradykinin is a vasoactive peptide that induces vasodilation via increased production of NO and other endothelial-derived relaxing factors. Importantly, pharmacological inhibition of NO production impairs the senescence-blocking effects of bradykinin88. This finding is bolstered by evidence from cell culture studies that inhibition of eNOS increases senescence.67,89 Importantly, NO production is impaired in senescent endothelial cells90; therefore, a lack of NO results in endothelial senescence that further reduces NO bioavailability — a hallmark of arterial aging that contributes to the progression of numerous diseases74.
Other vasoconstrictors with senescence-inducing effects in endothelial cells include endothelin-1, which also impairs endothelium-dependent dilation (EDD)91,92, and angiotensin-235,93–95. The angiotensin-receptor blocker valsartan reduces angiotensin-2-induced senescence35, indicating that the effects of this vasoconstrictor are mediated by type 1 angiotensin-2 receptors. Interestingly, repeated passaging of endothelial cells (a cell culture model of aging) increases their susceptibility to angiotensin-2-induced senescence.35 Together, these findings suggest a feedforward mechanism by which vasodilators and vasoconstrictors modulate endothelial cell senescence, leading to changes in bioavailability of NO and physiological dysfunction.
Inflammation and oxidative stress
Inflammation and oxidative stress derived from many sources, including immune cells, endothelial cells and circulating factors produced by other tissues and exogenous sources, lead to endothelial dysfunction, which could be partly caused by senescence. In support of this concept, evidence suggests that exposure of endothelial cells to TNF induces premature replicative senescence27,96 and increases endothelial cell expression of cytokines, chemokines96,97 and adhesion molecules, such as intercellular adhesion molecule 1 (ICAM-1) and vascular cell adhesion protein 1 (VCAM-1).96 TNF-induced premature senescence and the consequential inflammatory milieu are suppressed by the administration of antioxidants or inhibition of NF-κB.27 Likewise, treating endothelial cells with antioxidants in combination with co-factors necessary for enzymatic production of NO, partially prevents glucose-induced senescence.67 Mechanistically, ROS and inflammatory factors are likely to drive endothelial senescence through DNA damage and activation of DNA damage-response pathways.96 Senescent endothelial cells act as a source of inflammatory cytokines and ROS, which could induce senescence in healthy neighbouring cells in a feedforward fashion, thereby propagating and perpetuating the inflammatory microenvironment.44 Collectively, this evidence suggests a complex interplay among inflammation, oxidative stress and endothelial cell senescence.
Iatrogenic senescence
Medical treatment that leads to cell senescence is known as iatrogenic senescence. Notably, endothelial dysfunction98,99 and CVD100 resulting from cancer therapy could, in part, be caused by endothelial cell senescence. This notion is supported by the fact that several chemotherapeutic drugs induce endothelial senescence in culture29,31,32,101 and are administered to patients intravenously, thus interacting directly with endothelial cells (FIG. 1). As would be expected with induction of endothelial cell senescence, treating mice with the chemotherapeutic drug, doxorubicin, impairs EDD due to increased mitochondrial ROS102 and leads to elevated expression of arterial SASP factors IL-1β and IL-6.103 Furthermore, genetic ablation of senescent cells following doxorubicin treatment improves EDD, suggesting a direct connection between chemotherapeutics and endothelial cell senescence.104 Another chemotherapeutic drug — the vascular endothelial growth factor inhibitor, axitinib — has also been shown to induce endothelial cell senescence via ROS, in human umbilical vein endothelial cells.101,105
The clinical consequences of chemotherapy on the cardiovascular system often do not present until over a decade after treatment,106 which supports the idea that accumulation of senescent endothelial cells contributes to CVD. Another consequence of chemotherapy-induced senescence in endothelial cells is the production of SASP factors that can promote tumour growth.31 Thus, targeting endothelial cell senescence using senolytic drugs could be an effective strategy to prevent cancer progression acutely and reduce the risk of chemotherapy-related CVD. Senolytics are currently being evaluated for use in an ongoing clinical trial in survivors of childhood cancer.107Although the primary outcomes of this trial are unrelated to endothelial function or CVD development, if senolytics prove to be safe in this setting, future studies examining endothelial function prior to and after administration of senolytics, as well as long-term studies on the future incidence of CVD, would be of great interest and importance.9
Mechanisms of endothelial senescence
The damaging stimuli described above induce endothelial senescence through a variety of cellular and molecular mechanisms. These include telomere dysfunction, non-telomeric DNA damage, and alterations to energy-sensitive pathways. Consequently, cells activate tumour-suppressor pathways that permanently inhibit cell cycle progression, undergo morphological changes and upregulate production of SASP factors (FIG. 2).
Telomeres and DNA damage
In large arteries, endothelial cell turnover occurs primarily via mitotic cell division rather than replacement by progenitor cells.108 A consequence of increased cell division without progenitor replacement is telomere attrition, which increases the likelihood of replicative senescence.52 Telomeres at the ends of chromosomes are comprised of repeat sequences (TTAGGG) that shorten with each cell division due to the nature of the DNA replication machinery.109 When telomeres become critically short, they lose the ability to maintain the telomere-loop (T-loop) structure that caps chromosome ends.110 Telomere uncapping initiates a DNA damage response that activates the p53–p21 tumor suppressor pathway leading to cellular senescence.110
The contribution of telomere shortening to endothelial senescence is supported by evidence that disturbed blood flow shortens endothelial cell telomeres in culture.50 A similar phenotype is seen in humans, whereby telomere attrition is greater in vascular regions exposed to disturbed flow than in regions that experience laminar flow.52 These studies did not directly demonstrate that telomere attrition was sufficient to reach replicative senescence. However, as would be expected with critically short telomeres, disturbed flow increases endothelial cell senescence in mice and cell culture via the p53–p21 pathway51, which is prevented by exogenous activation of telomerase (the enzyme that elongates telomeres).38 Attrition due to replication might only be partially responsible for critical shortening of telomeres at these sites, however, because telomere attrition is also accelerated by oxidative stress111,112, which is elevated in regions of high endothelial cell turnover.49
In regions of laminar blood flow, endothelial cells are thought to be long lived and experience prolonged exposure to damaging agents. Telomeres are prone to oxidative DNA damage because their sequence contains an abundance of guanine triplets (TTAGGG), which are oxidation targets.113,114 Telomere oxidation can lead to telomere uncapping and cellular senescence, independent of telomere length115–117. This concept is supported by the finding that older individuals and patients with hypertension have higher levels of telomere uncapping associated with p53–p21-induced senescence than younger and normotensive control individuals, irrespective of telomere length.117,118 Although the precise mechanism leading to telomere uncapping remains unknown, evidence from mouse models supports the idea that uncapping due to replication and oxidative stress both lead to endothelial dysfunction. For example, genetic induction of telomere shortening or telomere uncapping results in an aged arterial phenotype in young mice.119,120 The role of telomere dysfunction in vascular aging has been reviewed in more-depth elsewhere.116,121,122 In addition to telomere dysfunction, non-telomeric DNA damage induces endothelial senescence both in vitro37,123,124 and in vivo.43 Akin to the consequences of telomere uncapping, studies have shown that impairments in the DNA damage repair response lead to an accelerated arterial aging phenotype in young mice.43
Sirtuins
Sirtuins (SIRTs) are NAD+-dependent deacetylases and ADP-ribosyl transferases.125 In response to elevated cellular NAD+, SIRT1 and SIRT6 promote cellular repair and stress resistance by mediating tumour-suppressor and DNA damage-repair pathways.125 SIRTs have, therefore, emerged as important mediators of endothelial cell senescence.
Exposure to oxidative or replicative stress reduces endothelial expression of both SIRT1 and SIRT6.37,126–128 Additionally, inhibition of SIRT1 and SIRT6 increases endothelial cell senescence in culture,37,127–129 suggesting the presence of a positive-feedback loop that predisposes cells to senescence over time through replication. Inhibition of SIRT1 in endothelial cells increases p53 activation,129 which might explain how SIRT1 mediates endothelial cell senescence. Conversely, the mechanisms by which a reduction in SIRT6 leads to senescence seem to be upstream of p53–p21 activation, and are associated with the role of SIRT6 in telomere protection and DNA repair. For example, inhibition of SIRT6 in endothelial cells results in increased telomere uncapping127,130 as well as in total nuclear DNA damage.127 In concordance with the canonical role of SIRT6 as a mediator of heterochromatin and DNA, SIRT6 buffers against the induction of senescence by ROS,126 which are known to induce DNA damage123 and telomere dysfunction.111 The totality of evidence suggests that SIRT1 and SIRT6 prevent endothelial senescence by protecting DNA and reducing activation of the p53–p21 senescence pathway.
SIRT3 also has a role in preventing endothelial cell senescence. This molecule is localized to the mitochondria and protects cells from oxidative stress.125 Elevated SIRT3 expression reduces endothelial cell senescence in culture.83,131 Conversely, levels of SIRT3 are reduced in senescent endothelial cells.131 Whole-body overexpression of SIRT3 protects mice from endothelial dysfunction induced by angiotensin-2 and reduces aortic senescence.132 These findings allude to an important role for SIRT3 and mitochondrial function in endothelial cell senescence. Mitochondrial dysfunction has been implicated as an inducer and a consequence of senescence in many cell types.133,134 However, the direct contribution of mitochondrial function to senescence within endothelial cells remains understudied. Endothelial cells are highly glycolytic, and do not rely heavily on oxidative phosphorylation for ATP production.135 Nonetheless, alterations in mitochondrial function seem to precede the induction of senescence, as replicative aging of human brain microvascular endothelial increases reliance on oxidative phosphorylation for energy production.136 Furthermore, in endothelial cells, radiation-induced senescence has been shown to increase mitochondrial ROS production, whereas protecting cells with antioxidants reduces the burden of senescence24 by maintaining telomere length and reducing both lipid peroxidation and total nuclear DNA damage,137 thereby extending replicative capacity.138 Together, these findings suggest that molecular mediators of mitochondrial function, such as SIRT3, have a critical role in preventing endothelial senescence.
Consequences of endothelial senescence
The role of endothelial cell senescence on vascular function and in cardiometabolic diseases is summarized below and in FIG. 3.
Fig. 3 |. Mechanisms of endothelial cell senescence induced by damaging stimuli.
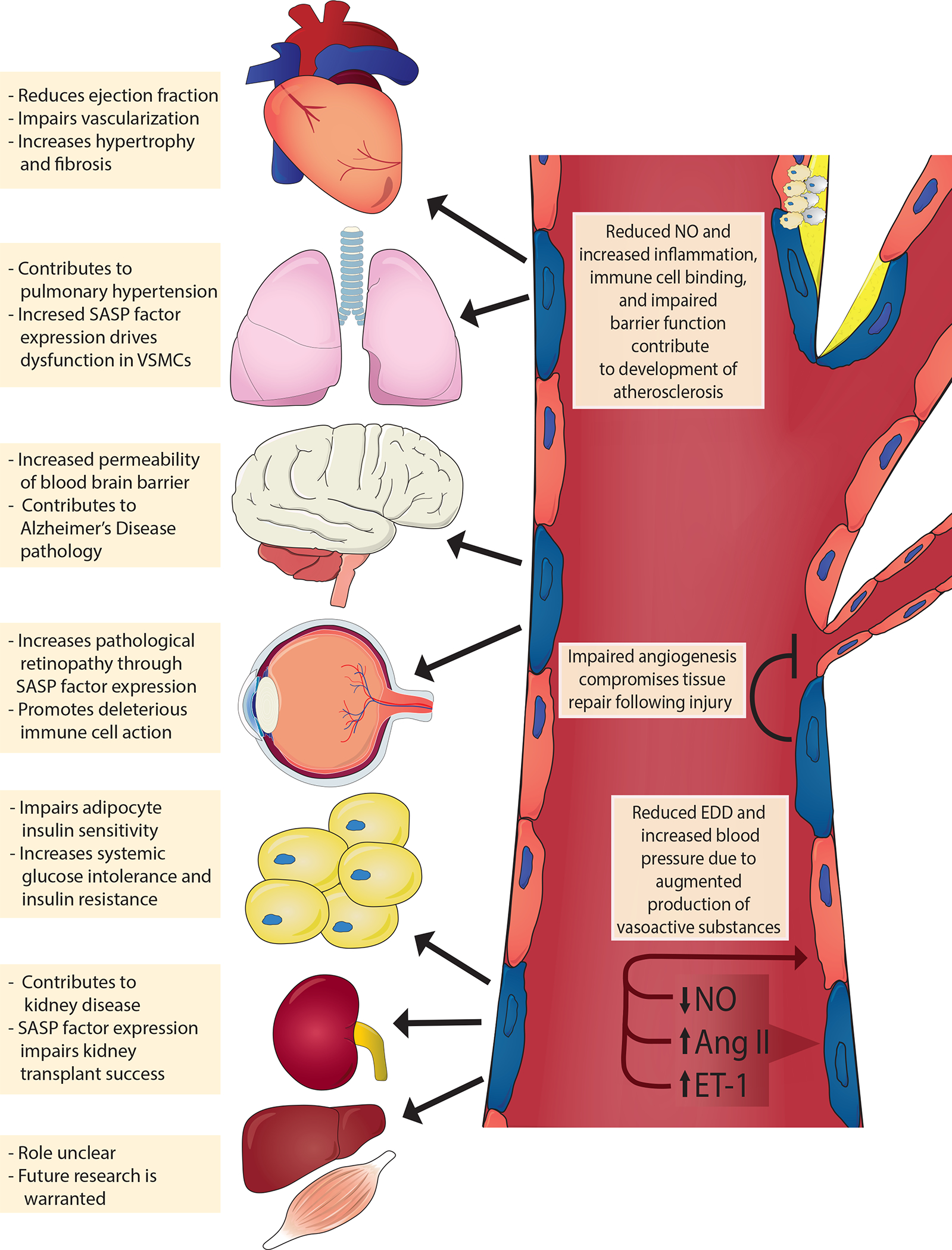
Circulating and endogenous stimuli result in telomeric and non-telomeric DNA damage and alter energy sensor pathways, such as those involving the sirtuins (SIRT1, SIRT3 and SIRT6). Consequently, endothelial cells activate tumour suppressor pathways (tumour antigen p53–cyclin-dependent kinase inhibitor 1A (also known as p21) and retinoblastoma protein (pRb)–cyclin-dependent kinase inhibitor 2A (also known as p16), which reprogrammes endothelial gene expression resulting in a host of cellular and molecular changes. For example, endothelial cells lose their proliferative potential and become senescent (blue cells); activate the senescence-associated secretory phenotype (SASP), which includes inflammatory pathways such as nuclear factor NF-kappa B (NF-κB) and secretion of inflammatory cytokines and reactive oxygen species (ROS); and attenuate the production of critical vasoactive molecules (e.g. nitric oxide (NO)). Collectively, the cellular and molecular changes promote arterial dysfunction resulting in cardiometabolic diseases. CXCL11, C-X-C motif chemokine 11; DDR, DNA damage response; MMPs, matrix metalloproteinases; PAI-1, plasminogen activator inhibitor 1; TGF-β, transforming growth factor-β.
Endothelium-dependent dilation
EDD is impaired with advancing age,134 and is associated with CVD, morbidity and mortality.134 Elevated oxidative stress and inflammation are implicated as causal factors in diminished EDD.139,140 An increase in resident senescent cells could plausibly represent the origin of such oxidative stress and inflammation. Furthermore, senescent endothelial cells have reduced eNOS expression, which contributes to impaired EDD.72,90,141,142 Although, no studies have yet demonstrated unequivocally that endothelial cell senescence alone is sufficient to disrupt EDD, many reports allude to this being the case. For example, in aged mice, treatment with the senolytic drugs dasatinib and quercetin improves carotid artery EDD and reduces the number of uncapped telomeres in aortic endothelial cells, with no change in the smooth muscle vasodilatory response to NO.143 This study provides compelling evidence that reducing the burden of senescent cells in advanced age is particularly beneficial for endothelial function with little effect on smooth muscle. These data are supported by a study of vascular endothelial cells from healthy, sedentary humans, in which increased senescence was associated with impaired EDD.144
Telomere uncapping has also been associated with EDD. In mice, whole-body induction of telomere uncapping induces arterial senescence and reduces EDD and NO bioavailability due to elevated ROS production, despite having no effects on smooth muscle vasodilation in the presence of NO.119 Furthermore, mice bred to have critically short telomeres accumulate senescent endothelial cells, exhibit oxidative damage in large arteries and reduced EDD due to oxidative stress, with a preserved smooth muscle vasodilatory response to NO.120 In humans, arterial telomere uncapping is associated with senescence and expression of inflammatory cytokines known to impair EDD.117 These studies support the hypothesis that the endothelium is particularly susceptible to senescence-inducing stimuli, resulting in impaired EDD. However, the effect of senescence in other cell types that interact with the endothelium, or changes in circulating factors, cannot be discounted. Therefore, investigations that directly induce or prevent senescence selectively in the endothelium are warranted.
Angiogenesis
The process of forming new blood vessels, known as angiogenesis, is an essential physiological process. For example, adaptation to exercise requires angiogenesis to meet the elevated demand for oxygen and nutrients; therefore, impaired angiogenesis contributes to exercise intolerance seen in elderly adults.145 Likewise, repairing cardiac or other tissues after injury requires angiogenesis, and disruption to this process delays and negatively impacts recovery.145 Importantly, aging results in reduced angiogenic capacity of the visceral adipose tissue vasculature.146 Endothelial cells are the main cell type that initiates and sustains the angiogenic process, primarily via vascular endothelial growth factor, fibroblast growth factor and hypoxia-inducible factor 1-α. Therefore, understanding age-related changes in endothelial cells that affect these signalling pathways and impair angiogenesis could reveal novel therapeutic targets.
Cellular senescence could be a fundamental mechanism underlying age-associated and disease-associated changes in angiogenesis. Experimental evidence demonstrates that disruption of telomeric repeat-binding factor 2 (TRF2) in endothelial cell senescence impairs angiogenic capacity.147 Likewise, genetic deletion or inhibition of telomerase impairs angiogenesis in rodents, compromising tissue repair in a model of ischaemia–reperfusion injury.148,149 Evidence also exists that ionizing radiation induces senescence and reduces angiogenic capacity in cerebrovascular endothelial cells.150 Furthermore, genetic deletion of the gene for endothelial p53 (Trp53) ameliorates vascular density in a murine model of heart failure, implicating a role of endothelial senescence and angiogenesis in cardiac injury.151 Mechanistically, endothelial cell senescence lowers VEGF gene expression, which could contribute to attenuated angiogenic capacity.152
Several lines of evidence support targeting cellular senescence to restore and maintain angiogenesis. For example, deletion of the genes for p21 (Cdkn1a)153 or p53 (Trp53)154,155 in mice attenuates endothelial cell senescence, and improves proliferation and angiogenesis.156 Deletion of endothelial Trp53 also results in improvement in angiogenesis, prevention of pressure overload-induced cardiac fibrosis and diabetic cardiomyopathy in mice.154,155 Although these studies provide convincing evidence that endothelial senescence inhibits angiogenesis, a report published in 2021 paradoxically demonstrated that, in a mouse model of retinopathy, endothelial senescence induces pathological angiogenesis and expression of SASP factors that promote abnormal neovascularization and fibrosis157. Treatment with senolytics attenuates disease severity by reducing angiogenesis and allowing regrowth of functional blood vessels157. Taken together, the literature supports the assertion that endothelial cell senescence strongly influences angiogenesis and can be targeted therapeutically; however, future studies on this topic are warranted.
Barrier function
As the inner lining of blood vessels, the endothelium forms a selectively permeable barrier that moderates the passage of molecules between blood and peripheral cells and tissues.158 Permeability of the endothelium is tissue specific and is mediated by junctional proteins.158 Maintenance of barrier function is critical, as barrier breakdown is involved in numerous diseases.158 Interestingly, culturing endothelial cells until they become senescent leads to disorganization of the junctional proteins cadherin-5 (also known as vascular endothelial cadherin) and tight junction protein ZO-1 and reductions in junctional proteins occludin and claudin-5.87 Furthermore, a bystander effect is apparent, whereby non-senescent cells display dysfunctional tight junctions when co-cultured with senescent endothelial cells,87 which could be explained by the SASP, as inflammation is known to increase permeability of the endothelial monolayer.158 Importantly, induction of endothelial senescence impairs blood brain barrier function in vitro86,87,159 and in vivo86. The consequences of endothelial cell senescence, and the subsequent impairments in barrier function, are exemplified in the disease states discussed below.
Hypertension
Hypertension is one of the greatest contributors to morbidity and mortality worldwide, and is associated with elevated oxidative stress and inflammation160. Endothelial dysfunction is implicated in the connection between oxidative stress, inflammation and the development of hypertension.160 These processes negatively impact the ratio between vasoprotective dilatory molecules, such as NO, and vasoconstrictor molecules, such as angiotensin-2 and endothelin-1, thus contributing to increases in systemic vascular resistance and systolic blood pressure. Therefore, the molecular mechanisms and subsequent phenotype that characterizes senescent endothelial cells strongly indicate that endothelial cell senescence contributes to the development of hypertension. Although evidence of this link exists161–163, few studies have demonstrated a direct cause-and-effect relationship between endothelial cell senescence and hypertension.
Perhaps the most convincing area of research pointing to senescence as a driver of hypertension is the study of telomere dysfunction. For example, mice that are genetically bred to have critically short telomeres are hypertensive due to elevated endothelin-1 levels.164 Moreover, genetic induction of telomere uncapping, independent of telomere length, results in arterial senescence and elevated systolic blood pressure in mice.119 Human patients with hypertension were found to have significant increases in telomere uncapping, which was associated with cellular senescence, when compared with normotensive individuals.118 This finding is supported by evidence that, of all the risk factors for atherosclerosis, hypertension is the best predictor of endothelial senescence.161
As well as having a causative role in hypertension, endothelial senescence could also be a consequence of increased blood pressure because pro-hypertensive stimuli, including endothelin-1 and angiotensin-2, induce endothelial cell senescence.66,35,93–95 Moreover, elevated pulse pressure exposes endothelial cells to mechanical stress that can drive senescence, as demonstrated by murine studies of transverse aortic constriction, which results in acute increases in pulse pressure leading to p53–p21-mediated senescence in the brain microvasculature.165 These data suggest a positive feedback loop, whereby senescence reduces NO and increases endothelin-1 and angiotensin-2, which induces more senescence that further elevates blood pressure and leads to even greater senescence. The role of endothelial cell senescence as a stand-alone mechanism in hypertension requires further investigation. To this end, mouse models of endothelial cell-specific induction of senescence, such as telomere uncapping, or manipulation of p16, p21 or p53, could be instrumental. Likewise, genetic or pharmacological ablation of senescent endothelial cells in models of hypertension (for example, high-salt high-fat diet, or eNOS inhibition) could shed light on the basic and translational value of targeting endothelial cell senescence in hypertension.
Atherosclerosis
Atherosclerosis is an inflammatory disease initially characterized by development of a fatty streak in the lumen of arteries that progresses to the development of advanced plaques containing oxidized lipids, a necrotic core and neovessels.85,166 The severity of disease is determined by the size of the plaque and its composition, which determines stability.167 Unstable plaques with thin fibrous caps are prone to rupture and, in the face of altered haemodynamic forces, become dislodged and occlude capillaries resulting in clinical cardiovascular events, such as myocardial ischaemia and infarction, stroke and peripheral artery diseases.85,167 Endothelial cells have a critical role in the formation and progression of atherosclerotic plaque, and in plaque stability and rupture.85 A healthy endothelial monolayer forms tight junctions preventing blood-borne factors, such as LDL-cholesterol (LDL-C) and immune cells, from entering the subintimal space.166,167 Likewise, endothelial cells prevent immune cell adhesion and rolling as well as platelet aggregation via secretion of NO.167 However, in atheroprone areas of the arterial tree, endothelial dysfunction is the first detectable change that occurs in response to damaging stimuli.85 Endothelial dysfunction attenuates NO production and impairs barrier function, fostering LDL-C and immune cell infiltration.167 Moreover, activation of endothelial inflammatory signals and ROS promotes disease progression and contributes to plaque instability.167 Endothelial cells within atherosclerotic plaques form leaky blood vessels, known as vasa vasorum, facilitating nutrient delivery, immune cell infiltration and haemorrhage, which accentuate plaque growth and instability.167
Many features of endothelial cell dysfunction in atherosclerosis are akin to the senescent phenotype. The first evidence on the role of endothelial cell senescence in atherosclerosis came from an observational study published in 2002, which demonstrated that endothelial senescence is upregulated in human atherosclerotic plaques.38 Over the past 20 years, more studies have been performed elucidating the causal role, mechanisms and the therapeutic potential of endothelial cell senescence in atherosclerosis. For example, together with macrophage foam cells and VSMCs, endothelial cells have been identified as vital contributors to atherosclerotic plaque formation and stability.168 In support of a causal role of endothelial senescence in atherosclerosis, induction of endothelial cell-specific senescence by overexpressing a dominant negative TRF2 promotes atherosclerosis, without affecting plasma triglyceride levels.169 Mechanistically, senescent endothelial cells contribute to atherosclerotic disease progression by impairing barrier function,87 resulting in infiltration of LDL-C and immune cells.87,170 Likewise, endothelial senescence attenuates NO production and increases NADPH oxidase and prostaglandin G/H synthase 2 (also known as cyclooxygenase-2) production, leading to platelet activation, adhesion and aggregation.85,171,172 Attenuated NO production promotes leukocyte rolling, adhesion and infiltration90,170, increases VSMC contractility and proliferation85,173 and reduces red blood cell oxygen delivery,85 all of which foster atherosclerotic diseases progression.
As discussed earlier, senescent endothelial cells secrete a host of SASP factors including inflammatory cytokines, matrix proteins and proteases and growth factors, all of which are known to contribute to atherosclerosis. Notably, compared with healthy cells, senescent endothelial cells produce higher levels of VCAM-1 in response to TNF and lipopolysaccharide in an NF-κB-dependent manner.169 Endothelial cells from human and murine atherosclerotic plaques also upregulate neurogenic locus notch homolog protein 1 signalling, which promotes ICAM-1, IL-6, IL-8 and IL-1α expression and transendothelial migration of monocytes.174 In addition to promoting plaque growth, compelling evidence suggests that senescent cells also drive plaque instability via the production of MMPs.168 However, these findings are from a study of MMP production in a mixed population of senescent cells from atherosclerotic plaques. Therefore, the individual role of endothelial cell senescence in plaque instability requires future investigation. Assuming that inducing apoptosis in senescent endothelial cells does not adversely affect plaque stability, clearing senescent endothelial cells is a potential therapeutic target for treating atherosclerosis. Studies have shown that elimination or inhibition of senescent endothelial cells using genetic ablation180 or senolytics76,168or via inhibition of angiopoietin-related protein 2 (also known as angiopoietin-like 2)175 and cyclin-dependent kinase 5176, or activation of peroxisome proliferator-activated receptor-α177 can suppress atherosclerotic disease progression. Collectively, these studies suggest that endothelial cell senescence not only contributes to, but also represents a viable therapeutic target for atherosclerosis
Heart failure
Heart failure is characterized by reductions in cardiac muscle function that impairs filling of the heart chambers (diastolic dysfunction, known as heart failure with preserved ejection fraction: HFpEF) or the ability to properly eject blood (systolic dysfunction, known as heart failure with reduced ejection fraction: HFrEF).178
In HFpEF, endothelial dysfunction akin to the senescent phenotype has been proposed to contribute to disease progression.179 Comorbidities, such as diabetes, and risk factors, such as advanced age (both of which induce endothelial senescence), promote endothelial-derived inflammation and oxidative stress within the coronary circulation, which impairs EDD due to reductions in NO and contributes to pathological remodelling.179 Although direct evidence is sparse, several studies support a role for endothelial senescence in HFpEF. For example, in a mouse model of accelerated senescence, a high-fat high-salt western diet exacerbated endothelial senescence, inflammation and impaired NO-mediated vasodilation, ultimately resulting cardiac hypertrophy, fibrosis and reduced diastolic function.180 Conversely, systemic inhibition of p53 in a mouse model of diabetes ameliorates cardiac inflammation, oxidative stress and subsequent remodelling155, which could result partly from the prevention of endothelial cell senescence.151,154 Additional evidence that endothelial senescence is a potential therapeutic target in HFpEF comes from studies of aged mice, in which genetic or pharmacological clearance of senescent cells reduces cardiac hypertrophy and fibrosis.115 Although both methods of senescent cell ablation in this study targeted multiple cell types, the senolytic (navitoclax) is known to selectively kill senescent, but not non-senescent endothelial cells.181 Effective pharmacotherapies for HFpEF are lacking. Targeting cells other than the cardiomyocytes could be a promising, novel approach, as HFpEF is the result of dysfunction within multiple organ systems, including the vasculature.182 Furthermore, concerns about a potential lack of cell replacement after clearance of senescent cardiomyocytes (although post-mitotic, cardiomyocytes display many features of senescence)183 means that senolytics targeting endothelial cells could be advantageous.
Evidence in the literature also supports the role of endothelial senescence in HFrEF. For example, in a transverse aortic constriction mouse model of cardiac hypertrophy and HFrEF,184 the p53–p21 pathway is upregulated in endothelial cells.151 Conversely, blunting the induction of senescence via genetic deletion of Trp53 specifically from endothelial cells reduces expression of the SASP factors PAI-1, TNF, C-C motif chemokine 2 (also known as monocyte chemotactic protein 1) and the adhesion molecule ICAM-1, thereby decreasing cardiac fibrosis and cardiomyocyte hypertrophy, resulting in improved systolic function and survival.151,154 These findings are supported by a mouse model of ischaemia–reperfusion injury that impairs systolic function and recapitulates the increased risk of heart failure after myocardial infarction.185 Treatment with the senolytic navitoclax attenuated impaired systolic function and infarct size by increasing proliferative capacity of endothelial cells and, thus, stimulating angiogenesis.185 Collectively, these studies indicate that endothelial cell senescence contributes to both HFpEF and HFrEF and senolytics that ablate senescent endothelial cells are beneficial for cardiac function. Despite these encouraging findings, the use of senolytics in this setting could affect other cell types that lack regenerative potential183,186 and so additional research in this area is warranted.
Pulmonary hypertension
Pulmonary hypertension has a broad range of aetiologies, from rare and idiopathic, to common and occurring as a complication of cardiovascular or respiratory conditions.187 Numerous cell types are involved in the pathophysiology of pulmonary hypertension. However, studies have identified endothelial cell senescence as an important contributor to the development of this disease.187 Senescent endothelial cells have been identified in rodent models of pulmonary hypertension, cell cultures from patients with pulmonary hypertension and single-cell RNA sequencing data from lung tissue,.188,189 Increased susceptibility to damage is a likely cause of endothelial cell senescence in pulmonary hypertension. For example, cultured lung endothelial cells from patients with pulmonary hypertension are more susceptible to disturbed blood flow and radiation-induced senescence compared with controls.188 Another study has provided further mechanistic insight into endothelial cell senescence in pulmonary hypertension. The researchers found that deficiency in the mitochondrial iron–sulfur cluster biogenesis protein, frataxin, leads to endothelial cell senescence, which increases inflammation and vascular remodelling and contributes to pulmonary hypertension.189
In pulmonary hypertension, endothelial cell senescence seems to drive dysfunctional phenotypes through upregulation of several canonical SASP factors, such as IL-6, TNF and PAI-1, leading to dysfunction in other vascular cells.188,189 For example, conditioned media derived from the senescent endothelial cells of patients with pulmonary hypertension increased inflammatory markers and proliferation in non-senescent smooth muscle cells.188,189 Importantly, the consequences of endothelial senescence in pulmonary hypertension are attenuated by treatment with senolytics, which reduce the burden of senescence and expression of SASP factors and improve clinical manifestations of pulmonary hypertension, such as reducing medial hypertrophy and pulmonary blood pressure.188,189 Further studies are warranted to examine the impact of eliminating senescent cells in pre-clinical models of pulmonary hypertension and in clinical trials of patients with this condition.
Metabolic function
Systemic metabolic disorders, such as hyperglycaemia and dyslipidaemia, drive vascular dysfunction.190 However, emerging evidence demonstrates that endothelial cell senescence and dysfunction precedes and, in many instances, has a causal role in these conditions. For example, endothelial cell-specific inactivation of NF-κB prevents metabolic dysfunction induced by a high fat diet by suppressing vascular senescence and the SASP.191 Evidence also exists that endothelial upregulation of p53 and induction of senescence attenuates phosphorylation of AKT1, indicating an impairment in insulin signalling.72 Another study revealed that conditioned media from senescent endothelial cells increases SASP factors and oxidative stress in adipocytes.44 Furthermore, this treatment attenuated protein expression of insulin receptor substrate 1, leading to reduced insulin stimulated phosphorylation of AKT1 in adipocytes, consistent with impaired insulin signaling.44 In mice, endothelial cell-specific Trp53 deletion reduces obesity, systemic glucose intolerance and insulin resistance induced by a high-fat diet72. Conversely, endothelial cell-specific upregulation of p53 exacerbated these adverse metabolic phenotypes, demonstrating the critical role of endothelial senescence in systemic metabolic function.72 In another study, endothelial cell-specific inhibition of the telomere capping protein TRF2 resulted in systemic insulin resistance, partly via circulating SASP factors.44 Taken together, these findings indicate that endothelial cell senescence leads to inflammatory cytokine secretion that adversely effects neighbouring cells in a paracrine and endocrine manner, drives systemic metabolic dysfunction and can be targeted to improve metabolic function. The mechanistic link between endothelial cell senescence and systemic metabolic impairments is likely to be due to alterations in immune cell infiltration, blood flow, vasodilation, capillary density and capillary recruitment in metabolically active tissues such as adipose tissues and skeletal muscle. Further research into these processes is warranted.
Adverse effects of senolysis
Although eliminating senescent cells is generally thought to be beneficial, killing certain senescent cells with senolytics can be detrimental192,183. The deleterious effects of senolysis stem from systemic drug administration leading to the unintentional death of post-mitotic cells that express senescence markers, but that cannot be replaced. Evidence from a mouse model of heart failure suggests that clearance of senescent cells, using genetic (p16-positive cells) or pharmacological (navitoclax) approaches, attenuates hypertrophy of cardiomyocytes and fibrosis115,183. However, these interventions also result in multinucleated cardiomyocytes, a potential cause of future cardiomyocyte hypertrophy and maladaptive remodelling.115,183 The method of killing senescent cells seems to be an important determinant of adverse effects, as genetic and pharmacologic senolysis have been shown to have differential effects on atherosclerosis.193 In this study, navitoclax, suppressed atherosclerosis and attenuated inflammation, but had no effect on senescence markers in vivo, suggesting a potential limitation of commonly used markers.193 The systemic consequences of attenuating senescence are supported by evidence from mice, whereby whole-body deletion of p53 (Trp53−/−) or p16 (Cdkn2a−/−) exacerbates cardiac fibrosis in models of heart failure induced by transverse aortic constriction or left carotid artery ligation.194,195 Moreover, genetic removal of p16-positive cells induces hepatic and perivascular fibrosis, as well as blood vessel permeability.3 Collectively, the evidence suggests that effective elimination of senescent cells in a clinical setting requires the target cell type to be determined, and senolytics selective to that particular cell type and disease state to be used. Interestingly, quercetin and fisetin, two of the senolytic drugs currently being evaluated for use in clinical trials, were first identified based on their ability to selectively kill senescent, but not non-senescent, endothelial cells.196,197
Conclusions
Cellular senescence is a critical biological phenomenon whereby cells undergo permanent proliferative arrest, remain metabolically active, and become secretory in nature. Senescent cells have important roles in development and wound healing and are, therefore, essential for maintaining physiological homeostasis. In normal physiological conditions, senescent cells are cleared by immune cells preventing their accumulation over time. However, with advancing age and in pathological states, senescent cells accumulate in tissues and organs where they contribute to disease progression and physiological dysfunction. The development of genetically modified mouse models, which enable researchers to track and ablate senescent cells, as well as induce senescence, has greatly advanced our understanding of the role of cellular senescence in disease. Furthermore, the development of senolytic drugs demonstrated that cellular senescence is a viable therapeutic target, leading to numerous ongoing clinical trials.
In this Review, we have explored the idea that endothelial cells are particularly susceptible to cellular senescence due to their anatomical location and exposure to unique mechanical and biochemical stimuli. We have also described the cellular and molecular mechanisms and consequences of endothelial cell senescence, as well as the physiological effects in the vasculature and in diseases that encompass multiple tissue types and organ systems — specifically in cardiometabolic diseases.
General limitations of studies in the field leave some questions still to be addressed in future research. For example, many models are not cell-type specific — owing to interactions between endothelial cells, circulating factors and other cell types, studies using whole-body manipulations in mice, or clinical samples from humans, cannot rule out affects from other cell types. Although technically challenging, endothelial cell-specific manipulation will be required to improve our understanding of endothelial senescence in aging and disease. Investigations into how, and in what setting, senescence-inducing damage occurs would provide foundational knowledge about how endothelial cells become senescent. Genetically modified mouse models and technologies, such as lineage tracing, live-cell imaging, single-cell RNA-sequencing and other omics approaches, will be instrumental in furthering our understanding of how senescence is induced. This information is important because the method of senescence induction effects the SASP, which is largely responsible for the deleterious effects of increased senescence burden. Furthermore, understanding how senescence occurs could provide novel therapeutic opportunities to prevent senescence-inducing damage or kill senescent endothelial cells. From a translational perspective, ongoing clinical trials of senolytics will shed light on the role of endothelial cell senescence in human aging and disease.
Key points.
Forming the inner lining of blood vessels, endothelial cells are exposed to a unique milleu of damaging stimuli, including haemodynamic forces as well as circulating and endothelial-derived factors.
Exposure to damaging stimuli results in telomeric and non-telomeric DNA-damage, mitochondrial dysfunction and alterarations in energy sensor pathways in endothelial cells.
Changes induced by damaging stimuli lead to the activation of tumour suppressor pathways, such as p53–p21 and pRb–p16, resulting in proliferative arrest and senescence.
Senescent endothalial cells are enlarged, flat and refractory to changes in response to shear stress; they are also metabolically active and secrete a host of inflammatory molecules.
Senescent endothelial cells and their secreted factors are major contributors to arterial dysfunction and the pathophysiology of various cardiometabolic diseases.
Emerging evidence suggests that targeting senescent endothalial cells can be an effective strategy to supresses cardiometabolic diseases.
Acknowledgements
The authors are supported by funding from National Institute of Health Awards R01 AG060395 (A.J.D.), R01 AG050238 (A.J.D.), R01 AG048366 (L.A.L.), F31AG076312 (S.I.B.) and Veteran’s Affairs Merit Review Award I01 BX004492 (L.A.L.) from the United States Department of Veterans Affairs Biomedical Laboratory Research and Development Service. The contents do not represent the views of the United States Department of Veterans Affairs, the National Institutes of Health or the United States Government.
Footnotes
Competing interests
A.J.D. is a scientific advisor and stockholder in Recursion Pharmaceuticals. None of the work done with Recursion is outlined or discussed in this Review. The other authors declare no competing interests.
Peer review information
Nature Reviews Cardiology thanks Jorge Erusalimsky and the other, anonymous, reviewer(s) for their contribution to the peer review of this work.
References
- 1.Campisi J & d’Adda di Fagagna F Cellular senescence: when bad things happen to good cells. Nat Rev Mol Cell Biol 8, 729–740 (2007). 10.1038/nrm2233 [DOI] [PubMed] [Google Scholar]
- 2.Yousefzadeh MJ et al. Tissue specificity of senescent cell accumulation during physiologic and accelerated aging of mice. Aging Cell 19, e13094 (2020). 10.1111/acel.13094 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 3.Grosse L et al. Defined p16(High) Senescent Cell Types Are Indispensable for Mouse Healthspan. Cell Metab 32, 87–99.e86 (2020). 10.1016/j.cmet.2020.05.002 [DOI] [PubMed] [Google Scholar]
- 4.Cohen C et al. Glomerular endothelial cell senescence drives age-related kidney disease through PAI-1. EMBO Mol Med 13, e14146 (2021). 10.15252/emmm.202114146 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 5.Shosha E et al. Mechanisms of Diabetes-Induced Endothelial Cell Senescence: Role of Arginase 1. Int J Mol Sci 19 (2018). 10.3390/ijms19041215 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 6.Kiss T et al. Single-cell RNA sequencing identifies senescent cerebromicrovascular endothelial cells in the aged mouse brain. Geroscience 42, 429–444 (2020). 10.1007/s11357-020-00177-1 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 7.Yokoi T et al. Apoptosis signal-regulating kinase 1 mediates cellular senescence induced by high glucose in endothelial cells. Diabetes 55, 1660–1665 (2006). 10.2337/db05-1607 [DOI] [PubMed] [Google Scholar]
- 8.Hayashi T et al. Endothelial cellular senescence is inhibited by liver X receptor activation with an additional mechanism for its atheroprotection in diabetes. Proc Natl Acad Sci U S A 111, 1168–1173 (2014). 10.1073/pnas.1322153111 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 9.Gasek NS, Kuchel GA, Kirkland JL & Xu M Strategies for Targeting Senescent Cells in Human Disease. Nat Aging 1, 870–879 (2021). 10.1038/s43587-021-00121-8 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 10.Dolgin E in Nat Biotechnol Vol. 38 1371–1377 (2020). [DOI] [PubMed] [Google Scholar]
- 11.Donato AJ, Morgan RG, Walker AE & Lesniewski LA Cellular and molecular biology of aging endothelial cells. J Mol Cell Cardiol 89, 122–135 (2015). 10.1016/j.yjmcc.2015.01.021 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 12.De Cecco M et al. L1 drives IFN in senescent cells and promotes age-associated inflammation. Nature 566, 73–78 (2019). 10.1038/s41586-018-0784-9 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 13.Uryga AK et al. Telomere damage promotes vascular smooth muscle cell senescence and immune cell recruitment after vessel injury. Commun Biol 4, 611 (2021). 10.1038/s42003-021-02123-z [DOI] [PMC free article] [PubMed] [Google Scholar]
- 14.Wang B et al. An inducible p21-Cre mouse model to monitor and manipulate p21-highlyexpressing senescent cells in vivo. Nat Aging 1, 962–973 (2021). 10.1038/s43587-021-00107-6 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 15.Wang L et al. Targeting p21(Cip1) highly expressing cells in adipose tissue alleviates insulin resistance in obesity. Cell Metab 34, 75–89.e78 (2022). 10.1016/j.cmet.2021.11.002 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 16.d’Adda di Fagagna F Living on a break: cellular senescence as a DNA-damage response. Nat Rev Cancer 8, 512–522 (2008). 10.1038/nrc2440 [DOI] [PubMed] [Google Scholar]
- 17.Childs BG, Baker DJ, Kirkland JL, Campisi J & van Deursen JM Senescence and apoptosis: dueling or complementary cell fates? EMBO Rep 15, 1139–1153 (2014). 10.15252/embr.201439245 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 18.Hsu CH, Altschuler SJ & Wu LF Patterns of Early p21 Dynamics Determine Proliferation-Senescence Cell Fate after Chemotherapy. Cell 178, 361–373.e312 (2019). 10.1016/j.cell.2019.05.041 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 19.Lee BY et al. Senescence-associated beta-galactosidase is lysosomal beta-galactosidase. Aging Cell 5, 187–195 (2006). 10.1111/j.1474-9726.2006.00199.x [DOI] [PubMed] [Google Scholar]
- 20.Kohli J et al. Algorithmic assessment of cellular senescence in experimental and clinical specimens. Nat Protoc 16, 2471–2498 (2021). 10.1038/s41596-021-00505-5 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 21.González-Gualda E, Baker AG, Fruk L & Muñoz-Espín D A guide to assessing cellular senescence in vitro and in vivo. Febs j 288, 56–80 (2021). 10.1111/febs.15570 [DOI] [PubMed] [Google Scholar]
- 22.Chala N et al. Mechanical Fingerprint of Senescence in Endothelial Cells. Nano Lett 21, 4911–4920 (2021). 10.1021/acs.nanolett.1c00064 [DOI] [PubMed] [Google Scholar]
- 23.Wang C, Baker BM, Chen CS & Schwartz MA Endothelial cell sensing of flow direction. Arterioscler Thromb Vasc Biol 33, 2130–2136 (2013). 10.1161/atvbaha.113.301826 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 24.Lafargue A et al. Ionizing radiation induces long-term senescence in endothelial cells through mitochondrial respiratory complex II dysfunction and superoxide generation. Free Radic Biol Med 108, 750–759 (2017). 10.1016/j.freeradbiomed.2017.04.019 [DOI] [PubMed] [Google Scholar]
- 25.Huo J et al. Coenzyme Q10 Prevents Senescence and Dysfunction Caused by Oxidative Stress in Vascular Endothelial Cells. Oxid Med Cell Longev 2018, 3181759 (2018). 10.1155/2018/3181759 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 26.Donato AJ et al. Direct evidence of endothelial oxidative stress with aging in humans: relation to impaired endothelium-dependent dilation and upregulation of nuclear factor-kappaB. Circ Res 100, 1659–1666 (2007). 10.1161/01.RES.0000269183.13937.e8 [DOI] [PubMed] [Google Scholar]
- 27.Khan SY et al. Premature senescence of endothelial cells upon chronic exposure to TNFα can be prevented by N-acetyl cysteine and plumericin. Sci Rep 7, 39501 (2017). 10.1038/srep39501 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 28.Yin Y et al. Vascular endothelial cells senescence is associated with NOD-like receptor family pyrin domain-containing 3 (NLRP3) inflammasome activation via reactive oxygen species (ROS)/thioredoxin-interacting protein (TXNIP) pathway. Int J Biochem Cell Biol 84, 22–34 (2017). 10.1016/j.biocel.2017.01.001 [DOI] [PubMed] [Google Scholar]
- 29.Bent EH, Gilbert LA & Hemann MT A senescence secretory switch mediated by PI3K/AKT/mTOR activation controls chemoprotective endothelial secretory responses. Genes Dev 30, 1811–1821 (2016). 10.1101/gad.284851.116 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 30.Hampel B et al. Increased expression of extracellular proteins as a hallmark of human endothelial cell in vitro senescence. Exp Gerontol 41, 474–481 (2006). 10.1016/j.exger.2006.03.001 [DOI] [PubMed] [Google Scholar]
- 31.Hwang HJ et al. Endothelial cells under therapy-induced senescence secrete CXCL11, which increases aggressiveness of breast cancer cells. Cancer Lett 490, 100–110 (2020). 10.1016/j.canlet.2020.06.019 [DOI] [PubMed] [Google Scholar]
- 32.Ghosh AK et al. A small molecule inhibitor of PAI-1 protects against doxorubicin-induced cellular senescence. Oncotarget 7, 72443–72457 (2016). 10.18632/oncotarget.12494 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 33.Grillari J, Hohenwarter O, Grabherr RM & Katinger H Subtractive hybridization of mRNA from early passage and senescent endothelial cells. Exp Gerontol 35, 187–197 (2000). 10.1016/s0531-5565(00)00080-2 [DOI] [PubMed] [Google Scholar]
- 34.Chen L, Holder R, Porter C & Shah Z Vitamin D3 attenuates doxorubicin-induced senescence of human aortic endothelial cells by upregulation of IL-10 via the pAMPKα/Sirt1/Foxo3a signaling pathway. PLoS One 16, e0252816 (2021). 10.1371/journal.pone.0252816 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 35.Li R et al. Long-term stimulation of angiotensin II induced endothelial senescence and dysfunction. Exp Gerontol 119, 212–220 (2019). 10.1016/j.exger.2019.02.012 [DOI] [PubMed] [Google Scholar]
- 36.Shelton DN, Chang E, Whittier PS, Choi D & Funk WD Microarray analysis of replicative senescence. Curr Biol 9, 939–945 (1999). 10.1016/s0960-9822(99)80420-5 [DOI] [PubMed] [Google Scholar]
- 37.Lee OH et al. Sirtuin 6 deficiency induces endothelial cell senescence via downregulation of forkhead box M1 expression. Aging (Albany NY) 12, 20946–20967 (2020). 10.18632/aging.202176 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 38.Minamino T et al. Endothelial cell senescence in human atherosclerosis: role of telomere in endothelial dysfunction. Circulation 105, 1541–1544 (2002). 10.1161/01.cir.0000013836.85741.17 [DOI] [PubMed] [Google Scholar]
- 39.Coleman PR et al. Stress-induced premature senescence mediated by a novel gene, SENEX, results in an anti-inflammatory phenotype in endothelial cells. Blood 116, 4016–4024 (2010). 10.1182/blood-2009-11-252700 [DOI] [PubMed] [Google Scholar]
- 40.Coleman PR et al. Age-associated stresses induce an anti-inflammatory senescent phenotype in endothelial cells. Aging (Albany NY) 5, 913–924 (2013). 10.18632/aging.100622 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 41.Powter EE et al. Caveolae control the anti-inflammatory phenotype of senescent endothelial cells. Aging Cell 14, 102–111 (2015). 10.1111/acel.12270 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 42.Kamino H et al. Searching for genes involved in arteriosclerosis: proteomic analysis of cultured human umbilical vein endothelial cells undergoing replicative senescence. Cell Struct Funct 28, 495–503 (2003). 10.1247/csf.28.495 [DOI] [PubMed] [Google Scholar]
- 43.Bautista-Niño PK et al. Local endothelial DNA repair deficiency causes aging-resembling endothelial-specific dysfunction. Clin Sci (Lond) 134, 727–746 (2020). 10.1042/cs20190124 [DOI] [PubMed] [Google Scholar]
- 44.Barinda AJ et al. Endothelial progeria induces adipose tissue senescence and impairs insulin sensitivity through senescence associated secretory phenotype. Nat Commun 11, 481 (2020). 10.1038/s41467-020-14387-w [DOI] [PMC free article] [PubMed] [Google Scholar]
- 45.Nelson G et al. A senescent cell bystander effect: senescence-induced senescence. Aging Cell 11, 345–349 (2012). 10.1111/j.1474-9726.2012.00795.x [DOI] [PMC free article] [PubMed] [Google Scholar]
- 46.Erusalimsky JD Vascular endothelial senescence: from mechanisms to pathophysiology. J Appl Physiol (1985) 106, 326–332 (2009). 10.1152/japplphysiol.91353.2008 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 47.Woywodt A, Bahlmann FH, De Groot K, Haller H & Haubitz M Circulating endothelial cells: life, death, detachment and repair of the endothelial cell layer. Nephrol Dial Transplant 17, 1728–1730 (2002). 10.1093/ndt/17.10.1728 [DOI] [PubMed] [Google Scholar]
- 48.Hobson B & Denekamp J Endothelial proliferation in tumours and normal tissues: continuous labelling studies. Br J Cancer 49, 405–413 (1984). 10.1038/bjc.1984.66 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 49.Chiu JJ & Chien S Effects of disturbed flow on vascular endothelium: pathophysiological basis and clinical perspectives. Physiol Rev 91, 327–387 (2011). 10.1152/physrev.00047.2009 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 50.Kotla S et al. Endothelial senescence is induced by phosphorylation and nuclear export of telomeric repeat binding factor 2-interacting protein. JCI Insight 4 (2019). 10.1172/jci.insight.124867 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 51.Warboys CM et al. Disturbed flow promotes endothelial senescence via a p53-dependent pathway. Arterioscler Thromb Vasc Biol 34, 985–995 (2014). 10.1161/atvbaha.114.303415 [DOI] [PubMed] [Google Scholar]
- 52.Chang E & Harley CB Telomere length and replicative aging in human vascular tissues. Proc Natl Acad Sci U S A 92, 11190–11194 (1995). 10.1073/pnas.92.24.11190 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 53.Vaupel P, Kallinowski F & Okunieff P Blood flow, oxygen and nutrient supply, and metabolic microenvironment of human tumors: a review. Cancer Res 49, 6449–6465 (1989). [PubMed] [Google Scholar]
- 54.Richardson RS et al. Human skeletal muscle intracellular oxygenation: the impact of ambient oxygen availability. J Physiol 571, 415–424 (2006). 10.1113/jphysiol.2005.102327 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 55.Chen Q, Fischer A, Reagan JD, Yan LJ & Ames BN Oxidative DNA damage and senescence of human diploid fibroblast cells. Proc Natl Acad Sci U S A 92, 4337–4341 (1995). 10.1073/pnas.92.10.4337 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 56.von Zglinicki T, Saretzki G, Döcke W & Lotze C Mild hyperoxia shortens telomeres and inhibits proliferation of fibroblasts: a model for senescence? Exp Cell Res 220, 186–193 (1995). 10.1006/excr.1995.1305 [DOI] [PubMed] [Google Scholar]
- 57.Parrinello S et al. Oxygen sensitivity severely limits the replicative lifespan of murine fibroblasts. Nat Cell Biol 5, 741–747 (2003). 10.1038/ncb1024 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 58.Lee AC et al. Ras proteins induce senescence by altering the intracellular levels of reactive oxygen species. J Biol Chem 274, 7936–7940 (1999). 10.1074/jbc.274.12.7936 [DOI] [PubMed] [Google Scholar]
- 59.Packer L & Fuehr K Low oxygen concentration extends the lifespan of cultured human diploid cells. Nature 267, 423–425 (1977). 10.1038/267423a0 [DOI] [PubMed] [Google Scholar]
- 60.Busuttil RA, Rubio M, Dollé ME, Campisi J & Vijg J Oxygen accelerates the accumulation of mutations during the senescence and immortalization of murine cells in culture. Aging Cell 2, 287–294 (2003). 10.1046/j.1474-9728.2003.00066.x [DOI] [PubMed] [Google Scholar]
- 61.Basu R et al. Effects of age and sex on postprandial glucose metabolism: differences in glucose turnover, insulin secretion, insulin action, and hepatic insulin extraction. Diabetes 55, 2001–2014 (2006). 10.2337/db05-1692 [DOI] [PubMed] [Google Scholar]
- 62.Issa JS, Diament J & Forti N [Postprandial lipemia: influence of aging]. Arq Bras Cardiol 85, 15–19 (2005). [DOI] [PubMed] [Google Scholar]
- 63.Lindberg O, Tilvis RS & Strandberg TE Does fasting plasma insulin increase by age in the general elderly population? Aging (Milano) 9, 277–280 (1997). 10.1007/bf03341830 [DOI] [PubMed] [Google Scholar]
- 64.Trott DW et al. T lymphocyte depletion ameliorates age-related metabolic impairments in mice. Geroscience 43, 1331–1347 (2021). 10.1007/s11357-021-00368-4 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 65.Prattichizzo F et al. Short-term sustained hyperglycaemia fosters an archetypal senescence-associated secretory phenotype in endothelial cells and macrophages. Redox Biol 15, 170–181 (2018). 10.1016/j.redox.2017.12.001 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 66.Zhong W, Zou G, Gu J & Zhang J L-arginine attenuates high glucose-accelerated senescence in human umbilical vein endothelial cells. Diabetes Res Clin Pract 89, 38–45 (2010). 10.1016/j.diabres.2010.03.013 [DOI] [PubMed] [Google Scholar]
- 67.Hayashi T et al. Endothelial cellular senescence is inhibited by nitric oxide: implications in atherosclerosis associated with menopause and diabetes. Proc Natl Acad Sci U S A 103, 17018–17023 (2006). 10.1073/pnas.0607873103 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 68.Maeda M, Hayashi T, Mizuno N, Hattori Y & Kuzuya M Intermittent high glucose implements stress-induced senescence in human vascular endothelial cells: role of superoxide production by NADPH oxidase. PLoS One 10, e0123169 (2015). 10.1371/journal.pone.0123169 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 69.Mortuza R, Chen S, Feng B, Sen S & Chakrabarti S High glucose induced alteration of SIRTs in endothelial cells causes rapid aging in a p300 and FOXO regulated pathway. PLoS One 8, e54514 (2013). 10.1371/journal.pone.0054514 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 70.Liu J et al. Glucose-induced oxidative stress and accelerated aging in endothelial cells are mediated by the depletion of mitochondrial SIRTs. Physiol Rep 8, e14331 (2020). 10.14814/phy2.14331 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 71.Brodsky SV et al. Prevention and reversal of premature endothelial cell senescence and vasculopathy in obesity-induced diabetes by ebselen. Circ Res 94, 377–384 (2004). 10.1161/01.res.0000111802.09964.ef [DOI] [PubMed] [Google Scholar]
- 72.Yokoyama M et al. Inhibition of endothelial p53 improves metabolic abnormalities related to dietary obesity. Cell Rep 7, 1691–1703 (2014). 10.1016/j.celrep.2014.04.046 [DOI] [PubMed] [Google Scholar]
- 73.Miyauchi H et al. Akt negatively regulates the in vitro lifespan of human endothelial cells via a p53/p21-dependent pathway. Embo j 23, 212–220 (2004). 10.1038/sj.emboj.7600045 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 74.Hayashi T et al. Nitric oxide and endothelial cellular senescence. Pharmacol Ther 120, 333–339 (2008). 10.1016/j.pharmthera.2008.09.002 [DOI] [PubMed] [Google Scholar]
- 75.Oh ST, Park H, Yoon HJ & Yang SY Long-Term Treatment of Native LDL Induces Senescence of Cultured Human Endothelial Cells. Oxid Med Cell Longev 2017, 6487825 (2017). 10.1155/2017/6487825 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 76.Jiang YH, Jiang LY, Wang YC, Ma DF & Li X Quercetin Attenuates Atherosclerosis via Modulating Oxidized LDL-Induced Endothelial Cellular Senescence. Front Pharmacol 11, 512 (2020). 10.3389/fphar.2020.00512 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 77.Liu R, Cheng F, Zeng K, Li W & Lan J GPR120 Agonist GW9508 Ameliorated Cellular Senescence Induced by ox-LDL. ACS Omega 5, 32195–32202 (2020). 10.1021/acsomega.0c03581 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 78.Zhang D et al. Homocysteine accelerates senescence of endothelial cells via DNA hypomethylation of human telomerase reverse transcriptase. Arterioscler Thromb Vasc Biol 35, 71–78 (2015). 10.1161/atvbaha.114.303899 [DOI] [PubMed] [Google Scholar]
- 79.Shi Q et al. Endothelial senescence after high-cholesterol, high-fat diet challenge in baboons. Am J Physiol Heart Circ Physiol 292, H2913–2920 (2007). 10.1152/ajpheart.01405.2006 [DOI] [PubMed] [Google Scholar]
- 80.Albertini E, Kozieł R, Dürr A, Neuhaus M & Jansen-Dürr P Cystathionine beta synthase modulates senescence of human endothelial cells. Aging (Albany NY) 4, 664–673 (2012). 10.18632/aging.100491 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 81.Scalera F et al. Effect of L-arginine on asymmetric dimethylarginine (ADMA) or homocysteine-accelerated endothelial cell aging. Biochem Biophys Res Commun 345, 1075–1082 (2006). 10.1016/j.bbrc.2006.05.015 [DOI] [PubMed] [Google Scholar]
- 82.Xu D, Neville R & Finkel T Homocysteine accelerates endothelial cell senescence. FEBS Lett 470, 20–24 (2000). 10.1016/s0014-5793(00)01278-3 [DOI] [PubMed] [Google Scholar]
- 83.Xing SS et al. Salidroside attenuates endothelial cellular senescence via decreasing the expression of inflammatory cytokines and increasing the expression of SIRT3. Mech Ageing Dev 175, 1–6 (2018). 10.1016/j.mad.2017.12.005 [DOI] [PubMed] [Google Scholar]
- 84.Parkhitko AA, Jouandin P, Mohr SE & Perrimon N Methionine metabolism and methyltransferases in the regulation of aging and lifespan extension across species. Aging Cell 18, e13034 (2019). 10.1111/acel.13034 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 85.Gimbrone MA Jr. & García-Cardeña G Endothelial Cell Dysfunction and the Pathobiology of Atherosclerosis. Circ Res 118, 620–636 (2016). 10.1161/circresaha.115.306301 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 86.Yamazaki Y et al. Vascular Cell Senescence Contributes to Blood-Brain Barrier Breakdown. Stroke 47, 1068–1077 (2016). 10.1161/strokeaha.115.010835 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 87.Krouwer VJ, Hekking LH, Langelaar-Makkinje M, Regan-Klapisz E & Post JA Endothelial cell senescence is associated with disrupted cell-cell junctions and increased monolayer permeability. Vasc Cell 4, 12 (2012). 10.1186/2045-824x-4-12 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 88.Oeseburg H et al. Bradykinin protects against oxidative stress-induced endothelial cell senescence. Hypertension 53, 417–422 (2009). 10.1161/hypertensionaha.108.123729 [DOI] [PubMed] [Google Scholar]
- 89.Vasa M, Breitschopf K, Zeiher AM & Dimmeler S Nitric oxide activates telomerase and delays endothelial cell senescence. Circ Res 87, 540–542 (2000). 10.1161/01.res.87.7.540 [DOI] [PubMed] [Google Scholar]
- 90.Matsushita H et al. eNOS activity is reduced in senescent human endothelial cells: Preservation by hTERT immortalization. Circ Res 89, 793–798 (2001). 10.1161/hh2101.098443 [DOI] [PubMed] [Google Scholar]
- 91.Olmos G et al. Hyperphosphatemia induces senescence in human endothelial cells by increasing endothelin-1 production. Aging Cell 16, 1300–1312 (2017). 10.1111/acel.12664 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 92.Donato AJ et al. Vascular endothelial dysfunction with aging: endothelin-1 and endothelial nitric oxide synthase. Am J Physiol Heart Circ Physiol 297, H425–432 (2009). 10.1152/ajpheart.00689.2008 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 93.Shan H, Bai X & Chen X Angiotensin II induces endothelial cell senescence via the activation of mitogen-activated protein kinases. Cell Biochem Funct 26, 459–466 (2008). 10.1002/cbf.1467 [DOI] [PubMed] [Google Scholar]
- 94.Khan I et al. Low Dose Chronic Angiotensin II Induces Selective Senescence of Kidney Endothelial Cells. Front Cell Dev Biol 9, 782841 (2021). 10.3389/fcell.2021.782841 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 95.Kim MY et al. The PPARδ-mediated inhibition of angiotensin II-induced premature senescence in human endothelial cells is SIRT1-dependent. Biochem Pharmacol 84, 1627–1634 (2012). 10.1016/j.bcp.2012.09.008 [DOI] [PubMed] [Google Scholar]
- 96.Kandhaya-Pillai R et al. TNFα-senescence initiates a STAT-dependent positive feedback loop, leading to a sustained interferon signature, DNA damage, and cytokine secretion. Aging (Albany NY) 9, 2411–2435 (2017). 10.18632/aging.101328 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 97.Yamagata K, Suzuki S & Tagami M Docosahexaenoic acid prevented tumor necrosis factor alpha-induced endothelial dysfunction and senescence. Prostaglandins Leukot Essent Fatty Acids 104, 11–18 (2016). 10.1016/j.plefa.2015.10.006 [DOI] [PubMed] [Google Scholar]
- 98.Luu AZ et al. Role of Endothelium in Doxorubicin-Induced Cardiomyopathy. JACC Basic Transl Sci 3, 861–870 (2018). 10.1016/j.jacbts.2018.06.005 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 99.Terwoord JD, Beyer AM & Gutterman DD Endothelial dysfunction as a complication of anti-cancer therapy. Pharmacol Ther, 108116 (2022). 10.1016/j.pharmthera.2022.108116 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 100.Yeh ET et al. Cardiovascular complications of cancer therapy: diagnosis, pathogenesis, and management. Circulation 109, 3122–3131 (2004). 10.1161/01.cir.0000133187.74800.b9 [DOI] [PubMed] [Google Scholar]
- 101.Mongiardi MP et al. Axitinib exposure triggers endothelial cells senescence through ROS accumulation and ATM activation. Oncogene 38, 5413–5424 (2019). 10.1038/s41388-019-0798-2 [DOI] [PubMed] [Google Scholar]
- 102.Clayton ZS et al. Doxorubicin-Induced Oxidative Stress and Endothelial Dysfunction in Conduit Arteries Is Prevented by Mitochondrial-Specific Antioxidant Treatment. JACC CardioOncol 2, 475–488 (2020). 10.1016/j.jaccao.2020.06.010 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 103.Clayton ZS et al. Tumor Necrosis Factor Alpha-Mediated Inflammation and Remodeling of the Extracellular Matrix Underlies Aortic Stiffening Induced by the Common Chemotherapeutic Agent Doxorubicin. Hypertension 77, 1581–1590 (2021). 10.1161/hypertensionaha.120.16759 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 104.Hutton D et al. Cellular senescence mediates doxorubicin-induced arterial dysfunction via activation of mitochondrial oxidative stress and the mammalian target of rapamycin. The FASEB Journal 35 (2021). 10.1096/fasebj.2021.35.S1.00283 [DOI] [Google Scholar]
- 105.Merolle M, Mongiardi MP, Piras M, Levi A & Falchetti ML Glioblastoma Cells Do Not Affect Axitinib-Dependent Senescence of HUVECs in a Transwell Coculture Model. Int J Mol Sci 21 (2020). 10.3390/ijms21041490 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 106.Wang Y, Boerma M & Zhou D Ionizing Radiation-Induced Endothelial Cell Senescence and Cardiovascular Diseases. Radiat Res 186, 153–161 (2016). 10.1667/rr14445.1 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 107.Medicine, U. N. L. o. ClinicalTrials.gov, <https://www.clinicaltrials.gov/ct2/show/NCT04733534> (2022).
- 108.McDonald AI et al. Endothelial Regeneration of Large Vessels Is a Biphasic Process Driven by Local Cells with Distinct Proliferative Capacities. Cell Stem Cell 23, 210–225.e216 (2018). 10.1016/j.stem.2018.07.011 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 109.Hastings R, Qureshi M, Verma R, Lacy PS & Williams B Telomere attrition and accumulation of senescent cells in cultured human endothelial cells. Cell Prolif 37, 317–324 (2004). 10.1111/j.1365-2184.2004.00315.x [DOI] [PMC free article] [PubMed] [Google Scholar]
- 110.Roake CM & Artandi SE Control of Cellular Aging, Tissue Function, and Cancer by p53 Downstream of Telomeres. Cold Spring Harb Perspect Med 7 (2017). 10.1101/cshperspect.a026088 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 111.Kurz DJ et al. Chronic oxidative stress compromises telomere integrity and accelerates the onset of senescence in human endothelial cells. J Cell Sci 117, 2417–2426 (2004). 10.1242/jcs.01097 [DOI] [PubMed] [Google Scholar]
- 112.von Zglinicki T Oxidative stress shortens telomeres. Trends Biochem Sci 27, 339–344 (2002). 10.1016/s0968-0004(02)02110-2 [DOI] [PubMed] [Google Scholar]
- 113.Hewitt G et al. Telomeres are favoured targets of a persistent DNA damage response in ageing and stress-induced senescence. Nat Commun 3, 708 (2012). 10.1038/ncomms1708 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 114.Oikawa S, Tada-Oikawa S & Kawanishi S Site-specific DNA damage at the GGG sequence by UVA involves acceleration of telomere shortening. Biochemistry 40, 4763–4768 (2001). 10.1021/bi002721g [DOI] [PubMed] [Google Scholar]
- 115.Anderson R et al. Length-independent telomere damage drives post-mitotic cardiomyocyte senescence. Embo j 38 (2019). 10.15252/embj.2018100492 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 116.Morgan RG, Donato AJ & Walker AE Telomere uncapping and vascular aging. Am J Physiol Heart Circ Physiol 315, H1–h5 (2018). 10.1152/ajpheart.00008.2018 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 117.Morgan RG et al. Age-related telomere uncapping is associated with cellular senescence and inflammation independent of telomere shortening in human arteries. Am J Physiol Heart Circ Physiol 305, H251–258 (2013). 10.1152/ajpheart.00197.2013 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 118.Morgan RG et al. Role of arterial telomere dysfunction in hypertension: relative contributions of telomere shortening and telomere uncapping. J Hypertens 32, 1293–1299 (2014). 10.1097/hjh.0000000000000157 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 119.Morgan RG et al. Induced Trf2 deletion leads to aging vascular phenotype in mice associated with arterial telomere uncapping, senescence signaling, and oxidative stress. J Mol Cell Cardiol 127, 74–82 (2019). 10.1016/j.yjmcc.2018.11.014 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 120.Bhayadia R, Schmidt BM, Melk A & Hömme M Senescence-Induced Oxidative Stress Causes Endothelial Dysfunction. J Gerontol A Biol Sci Med Sci 71, 161–169 (2016). 10.1093/gerona/glv008 [DOI] [PubMed] [Google Scholar]
- 121.Dominic A, Banerjee P, Hamilton DJ, Le NT & Abe JI Time-dependent replicative senescence vs. disturbed flow-induced pre-mature aging in atherosclerosis. Redox Biol 37, 101614 (2020). 10.1016/j.redox.2020.101614 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 122.Liu Y, Bloom SI & Donato AJ The role of senescence, telomere dysfunction and shelterin in vascular aging. Microcirculation 26, e12487 (2019). 10.1111/micc.12487 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 123.Zhan H, Suzuki T, Aizawa K, Miyagawa K & Nagai R Ataxia telangiectasia mutated (ATM)-mediated DNA damage response in oxidative stress-induced vascular endothelial cell senescence. J Biol Chem 285, 29662–29670 (2010). 10.1074/jbc.M110.125138 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 124.Kim KS, Kim JE, Choi KJ, Bae S & Kim DH Characterization of DNA damage-induced cellular senescence by ionizing radiation in endothelial cells. Int J Radiat Biol 90, 71–80 (2014). 10.3109/09553002.2014.859763 [DOI] [PubMed] [Google Scholar]
- 125.Houtkooper RH, Pirinen E & Auwerx J Sirtuins as regulators of metabolism and healthspan. Nat Rev Mol Cell Biol 13, 225–238 (2012). 10.1038/nrm3293 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 126.Liu R, Liu H, Ha Y, Tilton RG & Zhang W Oxidative stress induces endothelial cell senescence via downregulation of Sirt6. Biomed Res Int 2014, 902842 (2014). 10.1155/2014/902842 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 127.Cardus A, Uryga AK, Walters G & Erusalimsky JD SIRT6 protects human endothelial cells from DNA damage, telomere dysfunction, and senescence. Cardiovasc Res 97, 571–579 (2013). 10.1093/cvr/cvs352 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 128.Zu Y et al. SIRT1 promotes proliferation and prevents senescence through targeting LKB1 in primary porcine aortic endothelial cells. Circ Res 106, 1384–1393 (2010). 10.1161/circresaha.109.215483 [DOI] [PubMed] [Google Scholar]
- 129.Ota H et al. Sirt1 modulates premature senescence-like phenotype in human endothelial cells. J Mol Cell Cardiol 43, 571–579 (2007). 10.1016/j.yjmcc.2007.08.008 [DOI] [PubMed] [Google Scholar]
- 130.Yuen LH et al. A Focused DNA-Encoded Chemical Library for the Discovery of Inhibitors of NAD(+)-Dependent Enzymes. J Am Chem Soc 141, 5169–5181 (2019). 10.1021/jacs.8b08039 [DOI] [PubMed] [Google Scholar]
- 131.Chen T et al. SIRT3 protects endothelial cells from high glucose-induced senescence and dysfunction via the p53 pathway. Life Sci 264, 118724 (2021). 10.1016/j.lfs.2020.118724 [DOI] [PubMed] [Google Scholar]
- 132.Dikalova AE et al. Mitochondrial Deacetylase Sirt3 Reduces Vascular Dysfunction and Hypertension While Sirt3 Depletion in Essential Hypertension Is Linked to Vascular Inflammation and Oxidative Stress. Circ Res 126, 439–452 (2020). 10.1161/circresaha.119.315767 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 133.Correia-Melo C & Passos JF Mitochondria: Are they causal players in cellular senescence? Biochim Biophys Acta 1847, 1373–1379 (2015). 10.1016/j.bbabio.2015.05.017 [DOI] [PubMed] [Google Scholar]
- 134.Wiley CD et al. Mitochondrial Dysfunction Induces Senescence with a Distinct Secretory Phenotype. Cell Metab 23, 303–314 (2016). 10.1016/j.cmet.2015.11.011 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 135.Eelen G et al. Endothelial Cell Metabolism. Physiol Rev 98, 3–58 (2018). 10.1152/physrev.00001.2017 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 136.Sakamuri S et al. Glycolytic and Oxidative Phosphorylation Defects Precede the Development of Senescence in Primary Human Brain Microvascular Endothelial Cells. Geroscience (2022). 10.1007/s11357-022-00550-2 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 137.Voghel G et al. Chronic treatment with N-acetyl-cystein delays cellular senescence in endothelial cells isolated from a subgroup of atherosclerotic patients. Mech Ageing Dev 129, 261–270 (2008). 10.1016/j.mad.2008.01.004 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 138.Lener B et al. The NADPH oxidase Nox4 restricts the replicative lifespan of human endothelial cells. Biochem J 423, 363–374 (2009). 10.1042/bj20090666 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 139.Donato AJ, Machin DR & Lesniewski LA Mechanisms of Dysfunction in the Aging Vasculature and Role in Age-Related Disease. Circ Res 123, 825–848 (2018). 10.1161/circresaha.118.312563 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 140.Csiszar A, Wang M, Lakatta EG & Ungvari Z Inflammation and endothelial dysfunction during aging: role of NF-kappaB. J Appl Physiol (1985) 105, 1333–1341 (2008). 10.1152/japplphysiol.90470.2008 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 141.Rippe C et al. MicroRNA changes in human arterial endothelial cells with senescence: relation to apoptosis, eNOS and inflammation. Exp Gerontol 47, 45–51 (2012). 10.1016/j.exger.2011.10.004 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 142.Tarantini S et al. Treatment with the BCL-2/BCL-xL inhibitor senolytic drug ABT263/Navitoclax improves functional hyperemia in aged mice. Geroscience 43, 2427–2440 (2021). 10.1007/s11357-021-00440-z [DOI] [PMC free article] [PubMed] [Google Scholar]
- 143.Roos CM et al. Chronic senolytic treatment alleviates established vasomotor dysfunction in aged or atherosclerotic mice. Aging Cell 15, 973–977 (2016). 10.1111/acel.12458 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 144.Rossman MJ et al. Endothelial cell senescence with aging in healthy humans: prevention by habitual exercise and relation to vascular endothelial function. Am J Physiol Heart Circ Physiol 313, H890–h895 (2017). 10.1152/ajpheart.00416.2017 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 145.Lähteenvuo J & Rosenzweig A Effects of aging on angiogenesis. Circ Res 110, 1252–1264 (2012). 10.1161/circresaha.111.246116 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 146.Islam MT et al. Aging differentially impacts vasodilation and angiogenesis in arteries from the white and brown adipose tissues. Exp Gerontol 142, 111126 (2020). 10.1016/j.exger.2020.111126 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 147.El Maï M et al. The Telomeric Protein TRF2 Regulates Angiogenesis by Binding and Activating the PDGFRβ Promoter. Cell Rep 9, 1047–1060 (2014). 10.1016/j.celrep.2014.09.038 [DOI] [PubMed] [Google Scholar]
- 148.Franco S, Segura I, Riese HH & Blasco MA Decreased B16F10 melanoma growth and impaired vascularization in telomerase-deficient mice with critically short telomeres. Cancer Res 62, 552–559 (2002). [PubMed] [Google Scholar]
- 149.Zaccagnini G et al. Telomerase mediates vascular endothelial growth factor-dependent responsiveness in a rat model of hind limb ischemia. J Biol Chem 280, 14790–14798 (2005). 10.1074/jbc.M414644200 [DOI] [PubMed] [Google Scholar]
- 150.Ungvari Z et al. Ionizing radiation promotes the acquisition of a senescence-associated secretory phenotype and impairs angiogenic capacity in cerebromicrovascular endothelial cells: role of increased DNA damage and decreased DNA repair capacity in microvascular radiosensitivity. J Gerontol A Biol Sci Med Sci 68, 1443–1457 (2013). 10.1093/gerona/glt057 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 151.Yoshida Y et al. p53-Induced inflammation exacerbates cardiac dysfunction during pressure overload. J Mol Cell Cardiol 85, 183–198 (2015). 10.1016/j.yjmcc.2015.06.001 [DOI] [PubMed] [Google Scholar]
- 152.Chang H et al. Telomerase- and angiogenesis-related gene responses to irradiation in human umbilical vein endothelial cells. Int J Mol Med 31, 1202–1208 (2013). 10.3892/ijmm.2013.1300 [DOI] [PubMed] [Google Scholar]
- 153.Brühl T et al. p21Cip1 levels differentially regulate turnover of mature endothelial cells, endothelial progenitor cells, and in vivo neovascularization. Circ Res 94, 686–692 (2004). 10.1161/01.res.0000119922.71855.56 [DOI] [PubMed] [Google Scholar]
- 154.Gogiraju R et al. Endothelial p53 deletion improves angiogenesis and prevents cardiac fibrosis and heart failure induced by pressure overload in mice. J Am Heart Assoc 4 (2015). 10.1161/jaha.115.001770 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 155.Gu J et al. Inhibition of p53 prevents diabetic cardiomyopathy by preventing early-stage apoptosis and cell senescence, reduced glycolysis, and impaired angiogenesis. Cell Death Dis 9, 82 (2018). 10.1038/s41419-017-0093-5 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 156.Akimoto S, Mitsumata M, Sasaguri T & Yoshida Y Laminar shear stress inhibits vascular endothelial cell proliferation by inducing cyclin-dependent kinase inhibitor p21(Sdi1/Cip1/Waf1). Circ Res 86, 185–190 (2000). 10.1161/01.res.86.2.185 [DOI] [PubMed] [Google Scholar]
- 157.Crespo-Garcia S et al. Pathological angiogenesis in retinopathy engages cellular senescence and is amenable to therapeutic elimination via BCL-xL inhibition. Cell Metab 33, 818–832.e817 (2021). 10.1016/j.cmet.2021.01.011 [DOI] [PubMed] [Google Scholar]
- 158.Claesson-Welsh L, Dejana E & McDonald DM Permeability of the Endothelial Barrier: Identifying and Reconciling Controversies. Trends Mol Med 27, 314–331 (2021). 10.1016/j.molmed.2020.11.006 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 159.Salvador E et al. Senescence and associated blood-brain barrier alterations in vitro. Histochem Cell Biol 156, 283–292 (2021). 10.1007/s00418-021-01992-z [DOI] [PMC free article] [PubMed] [Google Scholar]
- 160.Buford TW Hypertension and aging. Ageing Res Rev 26, 96–111 (2016). 10.1016/j.arr.2016.01.007 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 161.Voghel G et al. Cellular senescence in endothelial cells from atherosclerotic patients is accelerated by oxidative stress associated with cardiovascular risk factors. Mech Ageing Dev 128, 662–671 (2007). 10.1016/j.mad.2007.09.006 [DOI] [PubMed] [Google Scholar]
- 162.Westhoff JH et al. Hypertension induces somatic cellular senescence in rats and humans by induction of cell cycle inhibitor p16INK4a. Hypertension 52, 123–129 (2008). 10.1161/hypertensionaha.107.099432 [DOI] [PubMed] [Google Scholar]
- 163.McCarthy CG, Wenceslau CF, Webb RC & Joe B Novel Contributors and Mechanisms of Cellular Senescence in Hypertension-Associated Premature Vascular Aging. Am J Hypertens 32, 709–719 (2019). 10.1093/ajh/hpz052 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 164.Pérez-Rivero G et al. Mice deficient in telomerase activity develop hypertension because of an excess of endothelin production. Circulation 114, 309–317 (2006). 10.1161/circulationaha.105.611111 [DOI] [PubMed] [Google Scholar]
- 165.de Montgolfier O et al. High Systolic Blood Pressure Induces Cerebral Microvascular Endothelial Dysfunction, Neurovascular Unit Damage, and Cognitive Decline in Mice. Hypertension 73, 217–228 (2019). 10.1161/hypertensionaha.118.12048 [DOI] [PubMed] [Google Scholar]
- 166.Islam T Impact of statins on vascular smooth muscle cells and relevance to atherosclerosis. J Physiol 598, 2295–2296 (2020). 10.1113/jp279774 [DOI] [PubMed] [Google Scholar]
- 167.Bentzon JF, Otsuka F, Virmani R & Falk E Mechanisms of plaque formation and rupture. Circ Res 114, 1852–1866 (2014). 10.1161/circresaha.114.302721 [DOI] [PubMed] [Google Scholar]
- 168.Childs BG et al. Senescent intimal foam cells are deleterious at all stages of atherosclerosis. Science 354, 472–477 (2016). 10.1126/science.aaf6659 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 169.Honda S et al. Cellular senescence promotes endothelial activation through epigenetic alteration, and consequently accelerates atherosclerosis. Sci Rep 11, 14608 (2021). 10.1038/s41598-021-94097-5 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 170.Yanaka M et al. Increased monocytic adhesion by senescence in human umbilical vein endothelial cells. Biosci Biotechnol Biochem 75, 1098–1103 (2011). 10.1271/bbb.100909 [DOI] [PubMed] [Google Scholar]
- 171.Silva GC et al. Replicative senescence promotes prothrombotic responses in endothelial cells: Role of NADPH oxidase- and cyclooxygenase-derived oxidative stress. Exp Gerontol 93, 7–15 (2017). 10.1016/j.exger.2017.04.006 [DOI] [PubMed] [Google Scholar]
- 172.Bochenek ML, Schütz E & Schäfer K Endothelial cell senescence and thrombosis: Ageing clots. Thromb Res 147, 36–45 (2016). 10.1016/j.thromres.2016.09.019 [DOI] [PubMed] [Google Scholar]
- 173.Tsihlis ND et al. Nitric oxide inhibits vascular smooth muscle cell proliferation and neointimal hyperplasia by increasing the ubiquitination and degradation of UbcH10. Cell Biochem Biophys 60, 89–97 (2011). 10.1007/s12013-011-9179-3 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 174.Liu ZJ et al. Notch activation induces endothelial cell senescence and pro-inflammatory response: implication of Notch signaling in atherosclerosis. Atherosclerosis 225, 296–303 (2012). 10.1016/j.atherosclerosis.2012.04.010 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 175.Caland L et al. Knockdown of angiopoietin-like 2 induces clearance of vascular endothelial senescent cells by apoptosis, promotes endothelial repair and slows atherogenesis in mice. Aging (Albany NY) 11, 3832–3850 (2019). 10.18632/aging.102020 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 176.Bai B et al. Cyclin-dependent kinase 5-mediated hyperphosphorylation of sirtuin-1 contributes to the development of endothelial senescence and atherosclerosis. Circulation 126, 729–740 (2012). 10.1161/circulationaha.112.118778 [DOI] [PubMed] [Google Scholar]
- 177.Dou F et al. PPARα Targeting GDF11 Inhibits Vascular Endothelial Cell Senescence in an Atherosclerosis Model. Oxid Med Cell Longev 2021, 2045259 (2021). 10.1155/2021/2045259 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 178.Borlaug BA & Redfield MM Diastolic and systolic heart failure are distinct phenotypes within the heart failure spectrum. Circulation 123, 2006–2013; discussion 2014 (2011). 10.1161/circulationaha.110.954388 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 179.Paulus WJ & Tschöpe C A novel paradigm for heart failure with preserved ejection fraction: comorbidities drive myocardial dysfunction and remodeling through coronary microvascular endothelial inflammation. J Am Coll Cardiol 62, 263–271 (2013). 10.1016/j.jacc.2013.02.092 [DOI] [PubMed] [Google Scholar]
- 180.Gevaert AB et al. Endothelial Senescence Contributes to Heart Failure With Preserved Ejection Fraction in an Aging Mouse Model. Circ Heart Fail 10 (2017). 10.1161/circheartfailure.116.003806 [DOI] [PubMed] [Google Scholar]
- 181.Zhu Y et al. Identification of a novel senolytic agent, navitoclax, targeting the Bcl-2 family of anti-apoptotic factors. Aging Cell 15, 428–435 (2016). 10.1111/acel.12445 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 182.Shah SJ et al. Phenotype-Specific Treatment of Heart Failure With Preserved Ejection Fraction: A Multiorgan Roadmap. Circulation 134, 73–90 (2016). 10.1161/circulationaha.116.021884 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 183.Owens WA, Walaszczyk A, Spyridopoulos I, Dookun E & Richardson GD Senescence and senolytics in cardiovascular disease: Promise and potential pitfalls. Mech Ageing Dev 198, 111540 (2021). 10.1016/j.mad.2021.111540 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 184.Riehle C & Bauersachs J Small animal models of heart failure. Cardiovasc Res 115, 1838–1849 (2019). 10.1093/cvr/cvz161 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 185.Dookun E et al. Clearance of senescent cells during cardiac ischemia-reperfusion injury improves recovery. Aging Cell 19, e13249 (2020). 10.1111/acel.13249 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 186.Childs BG, Li H & van Deursen JM Senescent cells: a therapeutic target for cardiovascular disease. J Clin Invest 128, 1217–1228 (2018). 10.1172/jci95146 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 187.Lawrie A & Francis SE Frataxin and endothelial cell senescence in pulmonary hypertension. J Clin Invest 131 (2021). 10.1172/jci149721 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 188.van der Feen DE et al. Cellular senescence impairs the reversibility of pulmonary arterial hypertension. Sci Transl Med 12 (2020). 10.1126/scitranslmed.aaw4974 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 189.Culley MK et al. Frataxin deficiency promotes endothelial senescence in pulmonary hypertension. J Clin Invest 131 (2021). 10.1172/jci136459 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 190.Graupera M & Claret M Endothelial Cells: New Players in Obesity and Related Metabolic Disorders. Trends Endocrinol Metab 29, 781–794 (2018). 10.1016/j.tem.2018.09.003 [DOI] [PubMed] [Google Scholar]
- 191.Hasegawa Y et al. Blockade of the nuclear factor-κB pathway in the endothelium prevents insulin resistance and prolongs life spans. Circulation 125, 1122–1133 (2012). 10.1161/circulationaha.111.054346 [DOI] [PubMed] [Google Scholar]
- 192.Jurk D & Passos JF Senolytic drugs: Beyond the promise and the hype. Mech Ageing Dev 202, 111631 (2022). 10.1016/j.mad.2022.111631 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 193.Garrido AM et al. Efficacy and limitations of senolysis in atherosclerosis. Cardiovasc Res (2021). 10.1093/cvr/cvab208 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 194.Zhu F et al. Senescent cardiac fibroblast is critical for cardiac fibrosis after myocardial infarction. PLoS One 8, e74535 (2013). 10.1371/journal.pone.0074535 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 195.Meyer K, Hodwin B, Ramanujam D, Engelhardt S & Sarikas A Essential Role for Premature Senescence of Myofibroblasts in Myocardial Fibrosis. J Am Coll Cardiol 67, 2018–2028 (2016). 10.1016/j.jacc.2016.02.047 [DOI] [PubMed] [Google Scholar]
- 196.Zhu Y et al. The Achilles’ heel of senescent cells: from transcriptome to senolytic drugs. Aging Cell 14, 644–658 (2015). 10.1111/acel.12344 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 197.Zhu Y et al. New agents that target senescent cells: the flavone, fisetin, and the BCL-X(L) inhibitors, A1331852 and A1155463. Aging (Albany NY) 9, 955–963 (2017). 10.18632/aging.101202 [DOI] [PMC free article] [PubMed] [Google Scholar]