

Abstract
The aim of this study was to determine the interaction between cerebral amyloid angiopathy (CAA) and Braak staging on cognition in the elderly. The study used a total of 141 subjects consisting of 72 non-cognitively impaired (NCI), 33 mild cognitive impairment (MCI), 36 Alzheimer’s disease (AD) cases displaying Braak stages 0-II and III from the Rush Religious Order Study cohort. The association between Braak stage and CAA status and cognition was evaluated using a series of regression models that adjusted for age at death, sex, education, APOE ε4 status, and Consortium to Establish a Registry for Alzheimer’s Disease (CERAD) neuropathological diagnosis. Individuals with CAA were more likely to be classified as Braak stage III relative to those without CAA [OR = 2.33, 95% CI (1.06, 5.14), p = 0.04]. A significant interaction was found between Braak stage and CAA status on a global cognitive score (β = −0.58, SE = 0.25, p = 0.02). Episodic memory also showed a significant association between Braak stage and CAA (β = −0.75, SE = 0.35, p = 0.03). These data suggest that there is a significant interaction between tau pathology and cerebrovascular lesions on cognition within the AD clinical spectrum.
Keywords: Alzheimer’s disease, Braak stage, cerebral amyloid angiopathy, cognition, cognitive aging, mild cognitive impairment, neurofibrillary tangles
INTRODUCTION
Braak stages I to VI mark the progression of tau-based neurofibrillary tangle (NFT) pathology from least to greatest within the medial temporal lobe memory circuit in Alzheimer’s disease (AD) [1]. Stages I and II display limited NFTs within the transentorhinal and entorhinal cortex and is often associated with normal cognitive and clinical profiles [1]. Braak stage III and IV display NFTs in the hippocampus and neocortex adjoining the transentorhinal cortex refers to the limbic phase when limited cognitive dysfunction is initially observed [2, 3]. Stages V and VI, labeled the neocortical phase, presents with the greatest number and extent of NFTs and is often associated with significant cognitive and functional impairment [2, 3]. While NFT pathology correlates well with cognitive decline and clinical status [3, 4], significant cognitive and clinical heterogeneity exists within the Braak stages [3, 5, 6]. Neuropathological analysis revealed a similar prevalence of Braak stage III in individuals with a clinical dementia rating (CDR) of 0 indicative of no cognitive impairment (55%) and those with a CDR of 0.5 corresponding to mild cognitive impairment (MCI; 56%) [3, 7], suggesting that other factors play a role in the transition from non-cognitively impaired (NCI) to MCI.
Given that many NCI individuals show significant AD pathology at autopsy in the absence of cognitive decline [8], the field has turned to the study of preclinical AD. For example, previous work by our group has shown that NCI individuals exhibit Braak stages ranging from I to V [9] and that an interaction between Braak stage, age, and APOE ε4 carrier status drives cognitive decline [10]. It has been suggested that Braak stage III represents an important transitional stage during the progression of AD [3]. However, the factors that precipitate cognitive decline among this group are unclear. Although inflammation [11], insulin resistance [12], and oxidative stress [13] have been proposed as additive variables contributing to cognitive impairment, the interaction between cerebrovascular factors and NFT pathology upon cognition remains an under-investigated area.
The role that cerebral amyloid angiopathy (CAA) plays in dementia is well established [14–17]. The presence of vascular CAA hastens the onset of AD clinical symptoms [18] and is associated with faster cognitive decline in NCI individuals [16, 18]. Although vascular dysregulation has been linked with amyloid pathology [19–21], there is increasing evidence that cerebrovascular damage is also associated with tau pathology. For example, focal ischemia is related with tau hyperphosphorylation [22, 23] and tau-dependent cerebrovascular repair mechanisms precede the development of CAA, which may contribute to downstream pathological events reported in AD animal models [24]. In human autopsy cases, hyperphosphorylated tau deposits were significantly more likely to be found in cerebral arteries displaying amyloid than those without [25]. The presence of CAA together with NFTs may also differentiate AD from primary age-related tauopathy (PART) [26]. Taken together, these data suggest that cerebrovascular factors moderate NFT pathology [27]. In this regard, positron emission tomography (PET) imaging has found that both the severity of small vessel cerebrovascular disease (SVCD) and amyloid binding (18F-florbetaben uptake) correlated positively with tau binding (18F-flortaucipir uptake) and these associations were independent of each other providing evidence for an interaction between ischemia and tau pathology [28]. Data derived from these studies support the hypothesis that cerebrovascular and NFT pathology interact to effect cognition during early Braak stages. Therefore, we investigated whether CAA plays a critical role in cognitive decline in cases that were neuropathologically categorized as Braak stage 0-II versus III.
MATERIALS AND METHODS
Data was derived from 141 older deceased and autopsied persons who died with a premortem clinical diagnosis of NCI (n = 72), MCI (n = 33), and AD (n = 46) and postmortem were classified as Braak stage 0 to III that were participants in the Rush Religious Orders Study (RROS). The RROS participants had no coexisting clinical or neurological conditions judged to contribute to cognitive impairment at their last clinical evaluation [29, 30], agreed to an annual clinical evaluation, and signed an informed consent and an Anatomic Gift Act donating their brains at time of death. Data from these subjects have been used in numerous clinical pathological studies supported by our ongoing NIA program project grant entitled the “Neurobiology of Mild Cognitive Impairment in the Elderly” (P01AG14449). At the time of these studies, individuals were chosen from all available RROS participants that came to autopsy during a rolling admission (n = 663) [29]. In addition, those taking anticholinesterases or medication for depression were also excluded from this study. The Human Investigation Committee of Rush University Medical Center approved this study.
Clinical evaluation
Each of the participants underwent a uniform, structured, and clinical evaluation performed by a neurologist and a trained neuropsychological test technician [29, 31]. Medications used by the subjects within the previous fourteen days of the examination were reviewed and classified. A neurologist reviewed the medical history, medication use, neurologic examination, results of cognitive performance testing, and the neuropsychologist’s opinion of cognitive impairment and dementia. Each participant was evaluated in their home, emphasizing findings deemed clinically relevant. Clinical diagnostic classification was performed as described previously [31]. Petersen criteria [32] were used to diagnose MCI while NINCDS-ADRDA criteria were used to diagnose AD [33]. Individuals classified as NCI had cognitive test scores within normal limits for age and education and had no significant functional deficits. Among those who progressed to MCI, 12 were classified as amnestic and 21 were classified as non-amnestic. Previous work by our group has shown that plaque and tangle pathology does not differ significantly between amnestic and non-amnestic MCI subjects in this cohort [34].
Tissue preparation and neuropathological diagnosis
Brain accruement and processing was described in previous publications [31, 35, 36]. Briefly, each brain was cut into 1 cm thick coronal slabs using a brain slice apparatus and hemisected. One hemisphere was immersion fixed in 4% paraformaldehyde (24–72 h) and cryoprotected (10% glycerol and 2% dimethyl sulfoxide in phosphate buffer solution) until processing for immunohistochemistry.
Diagnostic blocks (mid-frontal, superior temporal, entorhinal cortex, hippocampus, inferior parietal cortex, basal ganglia, thalamus, and substantia nigra) from the opposite hemisphere were paraffin embedded and sectioned at 6 μm. Examination for cerebral infarctions was conducted as described previously [37]. Bielschowsky silver stain was used to visualize neuritic plaques (NPs), diffuse plaques (DPs), and NFTs. Sections were also immunostained for Aβ using antibody M0872 (1:100; Dako, CA) raised against Aβ1–40 and Aβ1–42. Paired helical filament tau (AT8; 1:800, Covance) immunohistochemistry was also used to label NFTs. Neuropathological diagnoses were determined according to CERAD [38] and Braak staging [1] as recommended by the NIA-Reagan criteria [39]. Exclusion criteria included mixed dementias, Parkinson’s disease, frontotemporal dementia, argyrophilic grain disease, vascular dementia, hippocampal sclerosis, stroke, and Lewy body disease. Cortical and subcortical Lewy body pathology was detected using α-synuclein (αSyn) immunohistochemistry as previously described [40] and scored semi-quantitatively according to the severity and anatomical distribution, separating brainstem predominant, limbic/transitional and diffuse neocortical types, depending on the anatomical distribution of αSyn-positivity [41, 42].
A board-certified neuropathologist or trained technician, blinded to clinical diagnosis, counted number of NPs and DPs revealed by Bielschowsky silver stain and tau immunohistochemistry using the phosphorylated paired helical filament tau AT8 marker for NFTs, respectively, in one square mm area (100x magnification) per cortical region [32, 43]. CAA was assessed using a semiquantitative summary [16, 44] from the angular gyrus, inferior temporal gyrus, midfrontal gyrus, and the calcarine cortices. Paraffin-embedded sections were immunostained for Aβ using 1 of 3 monoclonal anti-human antibodies: 4G8 (1:9000; Covance Labs, Madison, WI), 6F/3D (1:50; Dako North America Inc., Carpinteria, CA), and 10D5 (1:600; Elan Pharmaceuticals, San Francisco, CA). In the group of RROS subjects used in this study, the 6F/3D antibody was used for all cases. For each region, meningeal and parenchymal vessels were assessed for amyloid deposition and scored from 0 to 4, where: 0 = no deposition, 1 = scattered segmental but no circumferential deposition, 2 = circumferential deposition up to 10 vessels, 3 = circumferential deposition up to 75% of the region, 4 = circumferential deposition over 75% of the total region. CAA score for each region was the maximum of the meningeal and parenchymal CAA scores. Scores were averaged across regions and summarized as a continuous measure of CAA pathology. CAA severity was then converted to a semi-quantitative summary and graded on a 0 to 3 scale based on the neuropathologist’s examination (0 = None, 1 = Mild, 2 = Moderate, 3 = Severe) [16].
Cognitive composite scores
Composite scores are based on the results of 17 cognitive tests categorized into five domains of cognition [45, 46]. Mini-Mental State Examination (MMSE) was used to describe the cohort, but was not used in the composite scores. Briefly, episodic memory was evaluated with tests including immediate and delayed recall of Story A from Logical Memory and of the East Boston Story, and Word List Memory, Recall, and Recognition from the Consortium to Establish a Registry for AD (CERAD). Semantic memory was assessed with three tests including a 15-item version of the Boston Naming Test, Verbal Fluency, which involves naming examples of semantic categories (i.e., animals, vegetables) in 1-min trials; and a reading test that involves reading single words aloud and a 10-item reading test. Working memory was assessed using Digit Span Forward and Backward and Digit Ordering. Two tests of perceptual speed included Symbol Digit Modalities Test, and Number Comparison. Finally, two tests of visuospatial ability included a 15-item version of Judgment of Line Orientation and a 9-item version of Ravens Standard Progressive Matrices [46]. For each test, raw scores were converted into z-scores based on the mean and standard deviation of the sample. The z-scores from the individual tests were averaged to create individual domain composite scores. The Global Composite Score (GCS) is an average of the 17 individual test z-scores.
Statistical analysis
Between-group frequency differences for categorical variables were analyzed using the Chi-square test while between-group differences for continuous variables were compared with the independent two-sample t-test procedure. CAA was converted to a dichotomous variable (CAA-Absent = None; CAA-Present = Mild, Moderate, Severe). Braak stage was also dichotomized into the following groups: 0 – II and III. Logistic regression was used to estimate the odds of being in the Braak stage III group given the presence of CAA after adjusting for age at death, sex, years of education, APOE ε4 status, and CERAD diagnosis. Linear regression models were used to test whether the interaction of CAA status and Braak stage was significantly associated with measures of cognition. Separate analyses with the same type of model were carried out for the GCS, Episodic Memory, Semantic Memory, Working Memory, Perceptual Speed, and Visuospatial domains. Each model adjusted for age at death, sex, years of education, APOE ε4 status, and CERAD diagnosis. Group differences for DP and NP counts were analyzed using the Mann-Whitney test due to their highly skewed distributions. Follow-up analyses for the DP and NP group differences used negative binomial regression models that adjusted for age at death, sex, years of education, APOE ε4 status, clinical diagnosis, and CAA status. p value was set at 0.05.
RESULTS
Table 1 shows the demographic and neuropathologic characteristics of the cases evaluated. Braak stage III subjects had significantly older ages at death relative to stages 0-II (p < 0.001), but did not differ on years of education (p = 0.50). The proportion of APOE ε4 carriers was similar between the Braak stage 0-II and stage III groups (p = 0.58). There was no difference in the frequency of clinical diagnoses between groups (p = 0.12), although it was noted that the prevalence of clinical AD in the Braak stage III group was twice that of the Braak stage 0-II group (n = 24 versus n = 12). Postmortem interval (p = 0.18), duration between last clinical assessment and autopsy (p = 0.35), and brain weight at autopsy (p = 0.17) were not significantly different between stage 0-II and III cases. DP load was not a significantly different (p = 0.23), but NP load was significantly higher in the Braak stage III group (p < 0.001). NP difference between groups remained significant even after adjusting for age at death, sex, education, CERAD diagnosis, APOE ε4 status, and CAA (β = 0.77, SE = 0.38, p = 0.04; Fig. 1) while the adjusted group difference for DPs was not statistically significant (β = 0.35, SE = 0.38, p = 0.36). CAA presence was also similar among the three clinical groups (p = 0.31). However, the presence of CAA was significantly higher in Braak stage III (p = 0.01) even after adjusting for CERAD diagnosis, age at death, sex, education, and APOE ε4 status [OR = 2.33, 95% CI (1.06, 5.14), p = 0.04]. A separate analysis using CAA scores from the inferior temporal gyrus alone yielded a similar result [OR = 3.16, 95% CI (1.26, 7.93), p = 0.01].
Table 1.
Demographic and neuropathological characteristics by braak stage
| Braak 0 – II (n = 66) |
Braak III (n = 75) |
p | |
|---|---|---|---|
| Age at death (years) | 82.25 ± 6.56 | 86.43 ± 5.70 | <0.001 |
| Sex (M/F) | 41/25 | 37/38 | 0.13 |
| Education (y) | 18.27 ± 4.03 | 17.83 ± 3.74 | 0.50 |
| APOE ε4 (Carrier/Non-Carrier) | 11/54 | 10/64 | 0.58 |
| Clinical diagnosis (NCI/MCI/AD) | 39/15/12 | 33/18/24 | 0.12 |
| Postmortem interval (hours) | 7.01 ± 4.07 | 6.03 ± 3.77 | 0.18 |
| Duration between last clinical | 0.73 ± 0.64 | 0.64 ± 0.49 | 0.35 |
| assessment and autopsy (y) | |||
| Brain weight at autopsy (g) | 1,256.28 ± 151.55 | 1,222.82 ± 133.93 | 0.17 |
| Cerebral amyloid angiopathy | 32/29 | 50/18 | 0.01 |
| (present/absent) | |||
| Neuritic plaque count | 0 (0–11) | 10 (0–37) | <0.001 |
| Diffuse plaque count | 4 (0–38) | 21.50 (0–60) | 0.23 |
Mean ± standard deviation; median (25th %ile, 75th %ile).
Fig. 1.
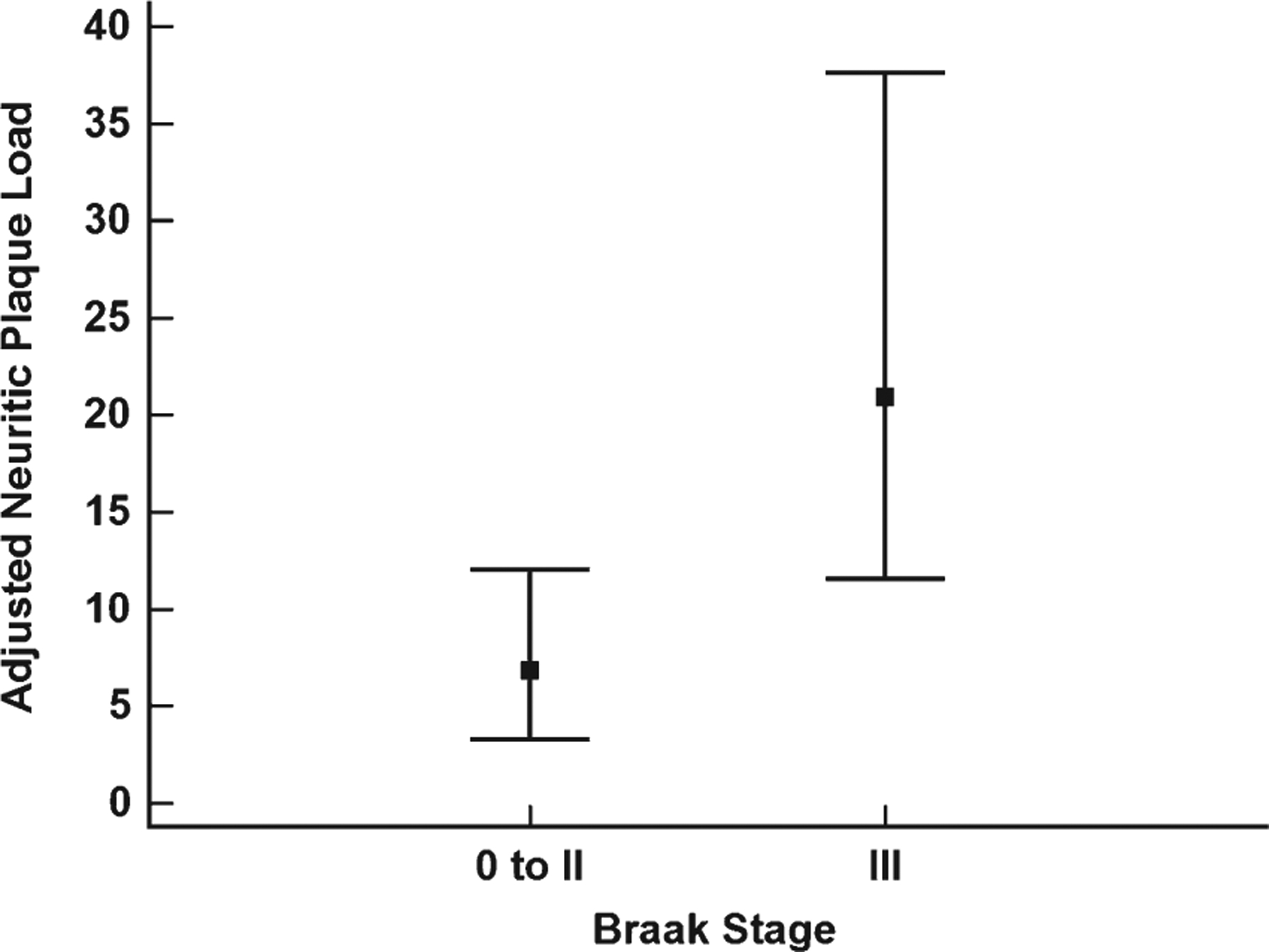
Adjusted neuritic plaque load differences for braak stage 0 to II and III. Braak stage 0-II and Braak stage III group difference for adjusted neuritic plaque load. Boxes represent the median and error bars are the 25th and 75th percentiles. Neuritic plaque load was adjusted age at death, education, sex, APOE ε4 carrier status, clinical diagnosis, and CAA status.
For the cognitive variables, the Braak stage III group had significantly lower GCS (p = 0.02), episodic memory (p = 0.03), semantic memory (p = 0.01), and perceptual speed (p = 0.03) scores relative to the Braak stage 0-II group, while working memory (p = 0.11) and the visuospatial domain (p = 0.81) were not significantly different (Table 2). After adjusting for age at death, sex, education, APOE ε4 status, and CERAD diagnosis, the interaction between Braak stage and CAA status was statistically significant for GCS (β = −0.58, SE = 0.25, p = 0.02). Groupwise comparisons of this interaction found that among those with CAA, the Braak stage III group had significantly lower performance than Braak stage 0-II individuals (p = 0.04; Fig. 2). The episodic memory domain also showed a significant effect for Braak by CAA interaction (β = −0.75, SE = 0.35, p = 0.03). However, the Braak stage and CAA interaction was not significant for semantic memory (p = 0.13), working memory (p = 0.12), visuospatial function (p = 0.13), and perceptual speed (p = 0.33). A separate analysis revealed no significant association of the Braak stage by CAA interaction with GCS and episodic memory using CAA scores from the inferior temporal gyrus (GCS: (β = −0.51, SE = 0.28, p = 0.07); Episodic Memory: (β = −0.51, SE = 0.34, p = 0.13). When the clinical groups were analyzed separately, the CAA by Braak stage interaction was not significantly associated with the GCS (NCI: [β = −0.08, SE = 0.16, p = 0.61]; MCI: [β = 0.11, SE = 0.31, p = 0.73]; AD: [β = −1.05, SE = 0.67, p = 0.13]).
Table 2.
Cognitive score differences by braak stage
| Braak 0 – II (n = 66) |
Braak III (n = 75) |
p | Cohen’s d | |
|---|---|---|---|---|
| MMSE | 26.54 ± 4.65 | 24.78 ± 5.91 | 0.05 | 0.33 |
| Global cognitive score | −0.22 ± 0.76 | −0.53 ± 0.81 | 0.02 | 0.39 |
| Episodic memory | −0.04 ± 1.01 | −0.42 ± 1.09 | 0.03 | 0.36 |
| Semantic memory | −0.05 ± 0.85 | −0.47 ± 0.95 | 0.01 | 0.47 |
| Working memory | −0.32 ± 0.74 | −0.53 ± 0.79 | 0.11 | 0.27 |
| Perceptual speed | −0.65 ± 1.13 | −1.04 ± 0.99 | 0.03 | 0.37 |
| Visuospatial | −0.45 ± 0.81 | −0.48 ± 0.80 | 0.81 | 0.04 |
Mean ± standard deviation; z-scores are reported for all cognitive variables except the MMSE.
Fig. 2.
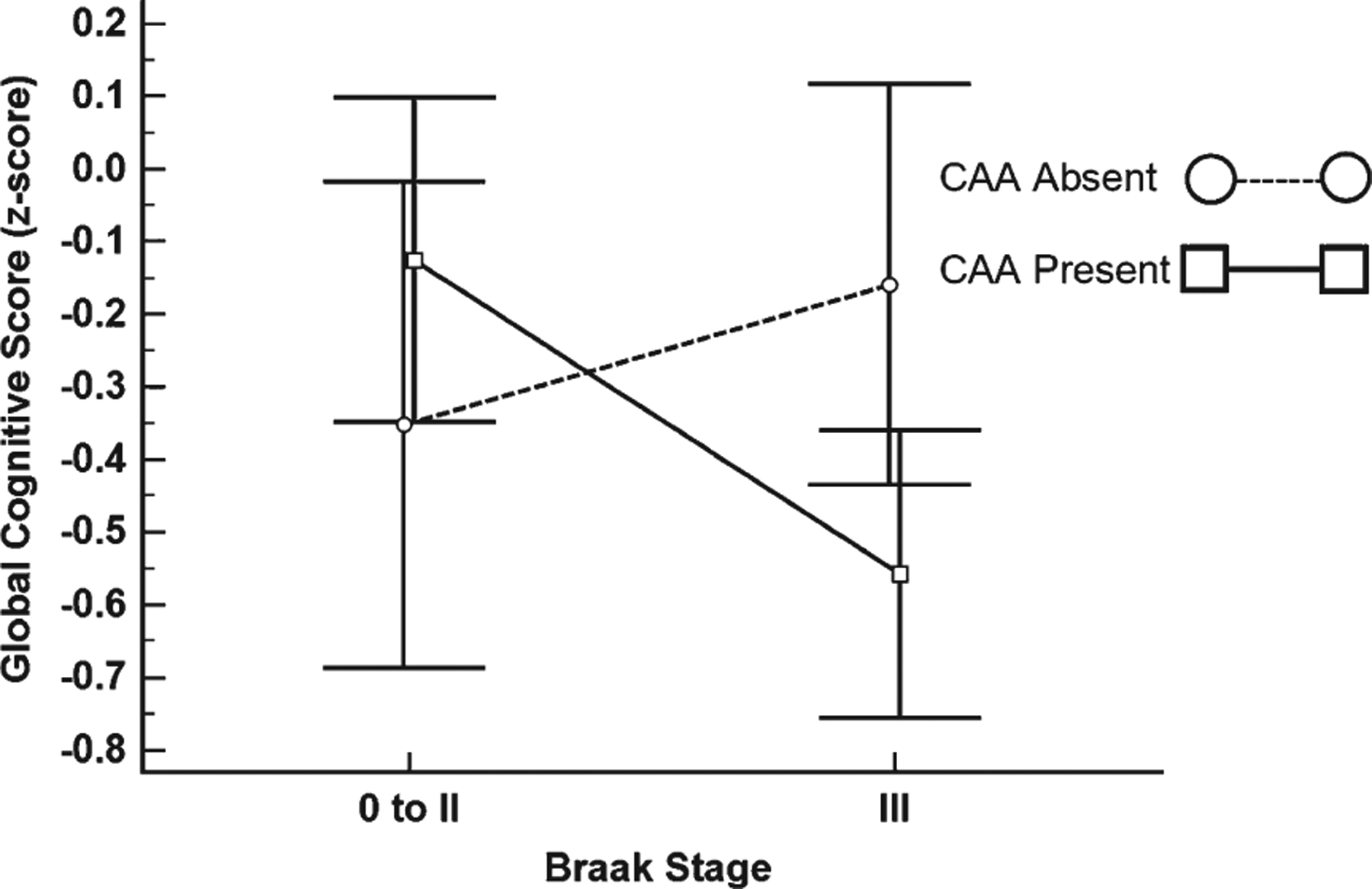
CAA-dependent global cognitive score differences for braak stage 0 to II and III. Global cognitive score stratified by Braak stage and CAA status. Squares and circles represent the mean and error bars are 95% confidence intervals. Dashed line represents the CAA Absent group and the solid line represents the CAA Present group.
DISCUSSION
We found that individuals with CAA were significantly more likely to be classified postmortem as Braak stage III. This was independent of APOE ε4 status, demographic characteristics, and CERAD diagnosis. We also found that Braak stage III individuals with CAA had significantly lower global cognition and episodic memory scores compared to Braak stage 0-II individuals with CAA, which was independent of plaque load, APOE ε4 status, and demographic characteristics. Others have reported similar results showing that the presence of both tau and vascular pathologies are associated with lower delayed recall memory scores among NCI individuals [47]. We also demonstrated that Braak stage III is heterogeneous in terms of clinical diagnosis [3, 5, 6] suggesting that this tau pathological phase is transitional both neuropathologically and cognitively. Although amyloid deposition plays a key role in CAA, it is possible that CAA induces downstream ischemic events that trigger tau hyperphosphorylation resulting in a greater NFT burden in these individuals [22–24].
However, it is possible that other vascular pathologies such as atherosclerosis, superficial siderosis, arteriosclerosis, and white matter rarefaction may also trigger ischemic-related tau hyperphosphorylation. Regardless of which vascular pathology underlies an increase in tau pathology, there is an opportunity to intervene in the pathogenesis of AD that is not focused primarily on amyloid. Although previous studies [48–51], including our own [52, 53], have shown that increases in cortical amyloid are associated with decreases in cognition among NCI individuals, evidence suggesting that brain vascular dysregulation precedes and initiates amyloid deposition [21] highlights the need to include cerebrovascular damage prevention in conjunction with current treatment approaches [54–57]. Although the amyloid cascade hypothesis for AD serves as the rationale for many current therapeutic approaches [58], previous failures of amyloid-based therapies in symptomatic AD has spurred the development of tau-based therapies, since tau pathologies are better correlates of cognitive decline than amyloid lesions [59]. Given the associations between cerebrovascular and tau pathology reported here, tau-directed therapies might be augmented by interventions that ameliorate or prevent cerebral vascular damage.
The present findings offer additional insight into the interaction between the cerebrovascular system and tau pathology in AD. A weakness of most hypotheses of AD pathogenesis is the need to define the pathogenic factors that precede amyloid and tau deposition. Although vascular dysfunction may be just one of several possible mechanisms that trigger or interact with amyloid, here we have shown that cerebrovascular lesions are associated with NFTs, which is supported by experimental evidence showing that CAA-induced tau phosphorylation and misfolding leads to tau-associated neurotoxicity [60]. This raises the possibility that cerebrovascular pathologies influence the progression of NP and NFT pathology during the course of AD. In fact, there is evidence suggesting that increased tau load is the result of a synergistic relationship between vascular factors and amyloid pathology [61]. Previous findings have demonstrated a main effect of CAA on cognitive decline in NCI older adults [16]; however, our findings show that CAA is a moderating factor in the association between Braak stage and cognition. Others have found that vascular factors are not related to amyloid load, but are significantly associated with tau load [62, 63]. Whether CAA interacts with amyloid and/or tau and its effect on cognition remain an under-investigated area in the field of AD.
The role that cognitive reserve (CR) plays in the differences in cognition seen between Braak stage 0-II and stage III groups remains to be determined. In particular, CR likely plays a role in the heterogeneity of clinical diagnoses seen in Braak stage III. Neuroimaging results have shown that CR mitigates the negative effect of white matter hyperintensities on cognition suggesting that CR confers protection against cerebrovascular lesions [61] in the same way that it is thought to mitigate the effects of NP and NFT lesions [64]. In our sample, fifty Braak stage III individuals also had CAA, of which 21 (42%) were NCI suggesting a mitigating effect of CR upon both AD and vascular pathology resulting in the maintenance of normal cognition.
A limitation of this study is its cross-sectional design, which prevents temporal associations from being investigated, particularly as they relate to progression from early to late Braak stages. It is also important to consider that CAA is just one of several vascular lesions that may influence the pathogenesis and cognitive outcomes of people with AD [65]. It is important to consider that age of biological disease onset for AD, which may be earlier for APOE ε4 carriers, may result in greater CAA and NFT burdens for these individuals. Another caveat is the relatively small number of APOE ε4 carriers, particularly homozygous individuals, which may affect the associations reported here. Future studies with a greater balance of APOE ε4 carriers and non-carriers are needed to extend these findings. The cases examined here were from a community-based cohort of highly educated retired clergy who had excellent health care and nutrition and were used in multiple clinical pathological [66, 67] and epidemiological investigations [30, 32, 35]. Individuals who volunteer may introduce bias by decreasing pathology but this is partially mitigated by high follow-up and autopsy rates of the RROS [36]. Strengths include uniform premortem clinical and postmortem pathological evaluation and that final the pathologic classification was performed without knowledge of the clinical evaluation.
The results of this study show that Braak stage III is heterogeneous with respect to clinical diagnosis and represents an important transition stage of AD disease progression. We have also shown that cerebrovascular factors are associated with both the neuropathological and clinical progression of AD and should be given greater consideration in the development of therapeutic strategies.
ACKNOWLEDGMENTS
This study was supported by grants P01AG014449, R01AG043375, P30AG010161, and P30AG042146 from the National Institute on Aging, National Institutes of Health, Barrow Neurological Institute Barrow, and Beyond and the Fine Foundation. We thank the participants of the Rush Religious Orders study. The funders had no role in study design, data collection and analysis, decision to publish, or preparation of the manuscript.
Footnotes
Authors’ disclosures available online (https://www.j-alz.com/manuscript-disclosures/19-1151r1).
REFERENCES
- [1].Braak H, Braak E (1991) Neuropathological stageing of Alzheimer-related changes. Acta Neuropathol 82, 239–259. [DOI] [PubMed] [Google Scholar]
- [2].Gold G, Bouras C, Kövari E, Canuto A, Glaría BG, Malky A, Hof PR, Michel JP, Giannakopoulos P (2000) Clinical validity of Braak neuropathological staging in the oldest-old. Acta Neuropathol 99, 579–582. [DOI] [PubMed] [Google Scholar]
- [3].Carlson JO, Gatz M, Pedersen NL, Graff C, Nennesmo I, Lindström AK, Gerritsen L (2015) Antemortem prediction of Braak stage. J Neuropathol Exp Neurol 74, 1061–1070. [DOI] [PMC free article] [PubMed] [Google Scholar]
- [4].Qian J, Hyman BT, Betensky RA (2017) Neurofibrillary tangle stage and the rate of progression of Alzheimer symptoms: Modeling using an autopsy cohort and application to clinical trial design. JAMA Neurol 74, 540–548. [DOI] [PMC free article] [PubMed] [Google Scholar]
- [5].Abner EL, Kryscio RJ, Schmitt FA, Santacruz KS, Jicha GA, Lin Y, Neltner JM, Smith CD, Van Eldik LJ, Nelson PT (2011) “End-stage” neurofibrillary tangle pathology in pre-clinical Alzheimer’s disease: Fact or fiction? J Alzheimers Dis 25, 445–453. [DOI] [PMC free article] [PubMed] [Google Scholar]
- [6].Riley KP, Snowdon DA, Markesbery WR (2002) Alzheimer’s neurofibrillary pathology and the spectrum of cognitive function: Findings from the Nun Study. Ann Neurol 51, 567–577. [DOI] [PubMed] [Google Scholar]
- [7].Morris JC, Storandt M, Miller JP, McKeel DW, Price JL, Rubin EH, Berg L (2001) Mild cognitive impairment represents early-stage Alzheimer disease. Arch Neurol 58, 397–405. [DOI] [PubMed] [Google Scholar]
- [8].Langbaum JBS, Fleisher AS, Chen K, Ayutyanont N, Lopera F, Quiroz YT, Caselli RJ, Tariot PN, Reiman EM (2013) Ushering in the study and treatment of preclinical Alzheimer disease. Nat Rev Neurol 9, 371–381. [DOI] [PMC free article] [PubMed] [Google Scholar]
- [9].Mufson EJ, Malek-Ahmadi M, Perez SE, Chen K (2016) Braak staging, plaque pathology, and APOE status in elderly persons without cognitive impairment. Neurobiol Aging 37, 147–153. [DOI] [PMC free article] [PubMed] [Google Scholar]
- [10].Mufson EJ, Malek-Ahmadi M, Perez SE, Snyder N, Ausde-more J, Chen K, Perez SE (2016) Braak stage and trajectory of cognitive decline in non-cognitively impaired elders. Neurobiol Aging 43, 101–110. [DOI] [PMC free article] [PubMed] [Google Scholar]
- [11].Martins IJ, Hone E, Foster JK, Sünram-Lea SI, Gnjec A, Fuller SJ, Nolan D, Gandy SE, Martins RN (2006) Apolipoprotein E, cholesterol metabolism, diabetes, and the convergence of risk factors for Alzheimer’s disease and cardiovascular disease. Mol Psychiatry 11, 721–736. [DOI] [PubMed] [Google Scholar]
- [12].Kulstad JJ, Green PS, Cook DG, Watson GS, Reger MA, Baker LD, Plymate SR, Asthana S, Rhoads K, Mehta PD, Craft S (2006) Differential modulation of plasma beta amyloid by insulin in patients with Alzheimer disease. Neurology 66, 1506–1510. [DOI] [PubMed] [Google Scholar]
- [13].Mufson EJ, Ikonomovic MD, Counts SE, Perez SE, Malek-Ahmadi M, Scheff SW, Ginsberg SD (2016) Molecular and cellular pathophysiology of preclinical Alzheimer’s disease. Behav Brain Res 311, 54–69. [DOI] [PMC free article] [PubMed] [Google Scholar]
- [14].Kumar-Sing S (2008) Cerebral amyloid angiopathy: Pathogenetic mechanisms and link to dense amyloid plaques. Genes Brain Behav 7(Suppl 1), 67–82. [DOI] [PubMed] [Google Scholar]
- [15].Brenowitz WD, Nelson PT, Besser LM, Heller KB, Kukull WA (2015) Cerebral amyloid angiopathy and its co-occurrence with Alzheimer’s disease and other cerebrovascular neuropathologic changes. Neurobiol Aging 36, 2702–2708. [DOI] [PMC free article] [PubMed] [Google Scholar]
- [16].Boyle PA, Yu L, Nag S, Leurgans S, Wilson RS, Bennett DA, Schneider JA (2015) Cerebral amyloid angiopathy and cognitive outcomes in community-based older persons. Neurology 85, 1930–1936. [DOI] [PMC free article] [PubMed] [Google Scholar]
- [17].Vidoni ED, Yeh H-W, Morris JK, Newell KL, Alqahtani A, Burns NC, Burns JM, Billinger SA (2016) Cerebral amyloid angiopathy is associated with earlier dementia onset in Alzheimer’s disease. Neurodegener Dis 16, 218–224. [DOI] [PMC free article] [PubMed] [Google Scholar]
- [18].Bos I, Verhey FR, Ramakers IHGB, Jacobs HIL, Soininen H, Freund-Levi Y, Hampel H, Tsolaki M, Wallin ÅK, van Buchem MA, Oleksik A, Verbeek MM, Olde Rikkert M, van der Flier WM, Scheltens P, Aalten P, Visser PJ, Vos SJB (2017) Cerebrovascular and amyloid pathology in predementia stages: The relationship with neurodegeneration and cognitive decline. Alzheimers Res Ther 9, 101. [DOI] [PMC free article] [PubMed] [Google Scholar]
- [19].Iadecola C (2010) The overlap between neurodegenerative and vascular factors in the pathogenesis of dementia. Acta Neuropathol 120, 287–296. [DOI] [PMC free article] [PubMed] [Google Scholar]
- [20].Di Marco LY, Venneri A, Farkas E, Evans PC, Marzo A, Frangi AF (2015) Vascular dysfunction in the pathogenesis of Alzheimer’s disease - A review of endothelium-mediated mechanisms and ensuing vicious circles. Neurobiol Dis 82, 593–606. [DOI] [PubMed] [Google Scholar]
- [21].Iturria-Medina Y, Sotero RC, Toussaint PJ, Mateos-Pérez JM, Evans AC; Alzheimer’s Disease Neuroimaging Initiative (2016) Early role of vascular dysregulation on late-onset Alzheimer’s disease based on multifactorial data-driven analysis. Nat Commun 7, 11934. [DOI] [PMC free article] [PubMed] [Google Scholar]
- [22].Wen Y, Yang S-H, Liu R, Brun-Zinkernagel AM, Koulen P, Simpkins JW (2007) Cdk5 is involved in NFT-like tauopathy induced by transient cerebral ischemia in female rats. Biochim Biophys Acta 1772, 473–483. [DOI] [PubMed] [Google Scholar]
- [23].Basurto-Islas G, Gu JH, Tung YC, Liu F, Iqbal K (2018) Mechanism of tau hyperphosphorylation involving lysosomal enzyme asparagine endopeptidase in a mouse model of brain ischemia. J Alzheimers Dis 63, 821–833. [DOI] [PubMed] [Google Scholar]
- [24].Merlini M, Wanner D, Nitsch RM (2016) Tau pathologydependent remodelling of cerebral arteries precedes Alzheimer’s disease-related microvascular cerebral amyloid angiopathy. Acta Neuropathol 131, 737–752. [DOI] [PMC free article] [PubMed] [Google Scholar]
- [25].Williams S, Chalmers K, Wilcock GK, Love S (2005) Relationship of neurofibrillary pathology to cerebral amyloid angiopathy in Alzheimer’s disease. Neuropathol Appl Neurobiol 31, 414–421. [DOI] [PubMed] [Google Scholar]
- [26].Weller RO, Hawkes CA, Carare RO, Hardy J (2015) Does the difference between PART and Alzheimer’s disease lie in the age-related changes in cerebral arteries that trigger the accumulation of Aβ and propagation of tau? Acta Neuropathol 129, 763–766. [DOI] [PubMed] [Google Scholar]
- [27].Kalaria RN (2000) The role of cerebral ischemia in Alzheimer’s disease. Neurobiol Aging 21, 321–330. [DOI] [PubMed] [Google Scholar]
- [28].Kim HJ, Park S, Cho H, Jang YK, San Lee J, Jang H, Kim Y, Kim KW, Ryu YH, Choi JY, Moon SH, Weiner MW, Jagust WJ, Rabinovici GD, DeCarli C, Lyoo CH, Na DL, Seo SW (2018) Assessment of extent and role of tau in subcortical vascular cognitive impairment using 18F-AV1451 positron emission tomography imaging. JAMA Neurol 75, 999–1007. [DOI] [PMC free article] [PubMed] [Google Scholar]
- [29].Bennett DA, Schneider JA, Arvanitakis Z, Wilson RS (2012) Overview and findings from the Religious Order Study. Curr Alzheimer Res 9, 628–645. [DOI] [PMC free article] [PubMed] [Google Scholar]
- [30].Mufson EJ, Chen EY, Cochran EJ, Beckett LA, Bennett DA, Kordower JH (1999) Entorhinal cortex beta-amyloid load in individuals with mild cognitive impairment. Exp Neurol 158, 469–490. [DOI] [PubMed] [Google Scholar]
- [31].Bennett DA, Wilson RS, Schneider JA, Evans DA, Beckett LA, Aggarwal NT, Barnes LL, Fox JH, Bach J (2002) Natural history of mild cognitive impairment in older persons. Neurology 59, 198–205. [DOI] [PubMed] [Google Scholar]
- [32].Petersen RC, Negash S (2008) Mild cognitive impairment: An overview. CNS Spect 13, 45–53. [DOI] [PubMed] [Google Scholar]
- [33].McKhann G, Drachman D, Folstein M, Katzman R, Price D, Stadlan EM (1984) Clinical diagnosis of Alzheimer’s disease: Report of the NINCDS-ADRDA Work Group under the auspices of Department of Health and Human Services Task Force on Alzheimer’s Disease. Neurology 34, 939–944. [DOI] [PubMed] [Google Scholar]
- [34].Malek-Ahmadi M, Lu S, Chan Y, Perez SE, Chen K, Mufson EJ (2017) Cognitive domain dispersion association with Alzheimer’s pathology. J Alzheimers Dis 58, 575–583. [DOI] [PMC free article] [PubMed] [Google Scholar]
- [35].Bennett DA, Schneider JA, Buchman AS, Mendes de Leon C, Bienias JL, Wilson RS (2005) The Rush Memory and Aging Project: Study design and baseline characteristics of the study cohort. Neuroepidemiology 25, 163–175. [DOI] [PubMed] [Google Scholar]
- [36].Bennett DA, Schneider JA, Wilson RS (2004) Neurofibrillary tangles mediate the association of amyloid load with clinical Alzheimer disease and level of cognitive function. Arch Neurol 61, 378–184. [DOI] [PubMed] [Google Scholar]
- [37].Schneider JA, Aggarwal NT, Barnes L, Boyle P, Bennett DA (2009) The neuropathology of older persons with and without dementia from community versus clinic cohorts. J Alzheimers Dis 18, 691–701. [DOI] [PMC free article] [PubMed] [Google Scholar]
- [38].Mirra SS (1997) The CERAD neuropathology protocol and consensus recommendations for the postmortem diagnosis of Alzheimer’s disease: A commentary. Neurobiol Aging 18, S91–S94. [DOI] [PubMed] [Google Scholar]
- [39].The National Institute on Aging and Reagan Institute Working Group on Diagnostic Criteria for the Neuropathological Assessment of Alzheimer’s Disease (1997) Consensus recommendations for the postmortem diagnosis of Alzheimer’s disease. The National Institute on Aging, and Reagan Institute Working Group on Diagnostic Criteria for the Neuropathological Assessment of Alzheimer’s Disease. Neurobiol Aging 18, S1–S2. [PubMed] [Google Scholar]
- [40].Schneider JA, Arvanitakis Z, Bang W, Bennett DA (2007) Mixed brain pathologies account for most dementia cases in community-dwelling older persons. Neurology 69, 2197–2204. [DOI] [PubMed] [Google Scholar]
- [41].McKeith IG, Galasko D, Kosaka K, Perry EK, Dickson DW, Hansen LA, Salmon DP, Lowe J, Mirra SS, Byrne EJ, Lennox G, Quinn NP, Edwardson JA, Ince PG, Bergeron C, Burns A, Miller BL, Lovestone S, Collerton D, Jansen EN, Ballard C, de Vos RA, Wilcock GK, Jellinger KA, Perry RH (1996) Consensus guidelines for the clinical and pathologic diagnosis of dementia with Lewy bodies (DLB): Report of the consortium on DLB international workshop. Neurology 47, 1113–1124. [DOI] [PubMed] [Google Scholar]
- [42].Jellinger KA (2009) A critical evaluation of current staging of α-synuclein pathology in Lewy body disorders. Biochim Biophys Acta 1792, 730–740. [DOI] [PubMed] [Google Scholar]
- [43].Mitchell TW, Nissanov J, Han LY, Mufson EJ, Schneider JA, Cochran EJ, Bennett DA, Lee VM, Trojanowski JQ, Arnold SE (2000) Novel method to quantify neuropil threads in brains from elders with or without cognitive impairment. J Histochem Cytochem 48, 1627–1638. [DOI] [PubMed] [Google Scholar]
- [44].Love S, Chalmers K, Ince P, Esiri M, Attems J, Jellinger K, Yamada M, McCarron M, Minett T, Matthews F, Greenberg S, Mann D, Kehoe PG (2014) Development, appraisal, validation and implementation of a consensus protocol for the assessment of cerebral amyloid angiopathy in post-mortem brain tissue. Am J Neurodegener Dis 3, 19–32. Erratum in: Am J Neurodegener Dis 4, 49. [PMC free article] [PubMed] [Google Scholar]
- [45].Bennett DA, Schneider JA, Arvanitakis Z, Kelly JF, Aggarwal NT, Shah RC, Wilson RS (2006) Neuropathology of older persons without cognitive impairment from two community-based studies. Neurology 66, 1837–1844. [DOI] [PubMed] [Google Scholar]
- [46].Wilson RS, Leurgans SE, Boyle PA, Bennett DA (2011) Cognitive decline in prodromal Alzheimer disease and mild cognitive impairment. Arch Neurol 68, 351–356. [DOI] [PMC free article] [PubMed] [Google Scholar]
- [47].Wennberg AM, Whitwell JL, Tosakulwong N, Weigand SD, Murray ME, Machulda MM, Petrucelli L, Mielke MM, Jack CR Jr, Knopman DS, Parisi JE, Petersen RC, Dickson DW, Josephs KA (2019) The influence of tau, amyloid, alpha-synuclein, TDP-43, and vascular pathology in clinically normal elderly individuals. Neurobiol Aging 77, 26–36. [DOI] [PMC free article] [PubMed] [Google Scholar]
- [48].Dowling NM, Tomaszewski FS, Reed BR, Reed BR, Sonnen JA, Strauss ME, Schneider JA, Bennett DA, Mungas D (2011) Neuropathological associates of multiple cognitive functions in two community-based cohorts of older adults. J Int Neuropsychol Soc 17, 602–614. [DOI] [PMC free article] [PubMed] [Google Scholar]
- [49].Hedden T, Oh H, Younger AP, Patel TA (2013) Meta-analysis of amyloid-cognition relations in cognitively normal older adults. Neurology 80, 1341–1348. [DOI] [PMC free article] [PubMed] [Google Scholar]
- [50].Bancher C, Jellinger K, Lassmann H, Fischer P, Leblhuber F (1996) Correlations between mental state and quantitative neuropathology in the Vienna Longitudinal Study on Dementia. Eur Arch Psychiatry Clin Neurosci 246, 137–146. [DOI] [PubMed] [Google Scholar]
- [51].Nelson PT, Jicha GA, Schmitt FA, Liu H, Davis DG, Mendiondo MS, Abner EL, Markesbery WR (2007) Clinicopathologic correlations in a large Alzheimer disease center autopsy cohort: Neuritic plaques and neurofibrillary tangles “do count” when staging disease severity. J Neuropathol Exp Neurol 66, 1136–1146. [DOI] [PMC free article] [PubMed] [Google Scholar]
- [52].Malek-Ahmadi M, Chen K, Perez SE, He A, Mufson EJ (2018) Cognitive composite score association with Alzheimer’s disease plaque and tangle pathology. Alzheimers Res Ther 10, 90. [DOI] [PMC free article] [PubMed] [Google Scholar]
- [53].Malek-Ahmadi M, Perez SE, Chen K, Mufson EJ (2016) Neuritic and diffuse plaque associations with memory in non-cognitively impaired elderly. J Alzheimers Dis 53, 1641–1652. [DOI] [PMC free article] [PubMed] [Google Scholar]
- [54].A study of CAD106 and CNP520 versus placebo in participants at risk for the onset of clinical symptoms of Alzheimer’s disease (Generation S1). Accessed electronically on 12/20/18 at https://clinicaltrials.gov/ct2/show/NCT02565511
- [55].A study of CNP520 versus placebo in participants at risk for the onset of clinical symptoms of Alzheimer’s disease (Generation S2). Accessed electronically on 12/20/18 at https://clinicaltrials.gov/ct2/show/NCT03131453
- [56].Clinical trial of solanezumab for older individuals who may be at risk for memory loss (A4). Accessed electronically on 12/20/18 at https://clinicaltrials.gov/ct2/show/NCT02008357
- [57].A study of gantenerumab in participants with prodromal Alzheimer’s disease. Accessed electronically on 12/20/18 at https://clinicaltrials.gov/ct2/show/NCT01224106
- [58].Karran E, De Strooper B (2016) The amyloid cascade hypothesis: Are we poised for success or failure? J Neurochem 139 Suppl 2, 237–252. [DOI] [PubMed] [Google Scholar]
- [59].Boutajangout A, Wisniewski T (2014) Tau-based therapeutic approaches for Alzheimer’s disease - a mini-review. Gerontology 60, 381–385. [DOI] [PMC free article] [PubMed] [Google Scholar]
- [60].You Y, Perkins A, Cisternas P, Muñoz B, Taylor X, You Y, Garringer HJ, Oblak AL, Atwood BK, Vidal R, Lasagna-Reeves CA (2019) Tau as a mediator of neurotoxicity associated to cerebral amyloid angiopathy. Acta Neuropathol Commun 7, 26. [DOI] [PMC free article] [PubMed] [Google Scholar]
- [61].Brickman AM, Siedlecki KL, Muraskin J, Manly JJ, Luchsinger JA, Yeung LK, Brown TR, DeCarli C, Stern Y (2011) White matter hyperintensities and cognition: Testing the reserve hypothesis. Neurobiol Aging 32, 1588–1598. [DOI] [PMC free article] [PubMed] [Google Scholar]
- [62].Rabin JS, Yang HS, Schultz AP, Hanseeuw BJ, Hedden T, Viswanathan A, Gatchel JR, Marshall GA, Kilpatrick E, Klein H, Rao V, Buckley RF, Yau WW, Kirn DR, Rentz DM, Johnson KA, Sperling RA, Chhatwal JP (2019) Vascular risk and β-amyloid are synergistically associated with cortical tau. Ann Neurol 85, 272–279. [DOI] [PMC free article] [PubMed] [Google Scholar]
- [63].Vemuri P, Lesnick TG, Przybelski SA, Knopman DS, Lowe VJ, Graff-Radford J, Roberts RO, Mielke MM, Machulda MM, Petersen RC, Jack CR Jr (2017) Age, vascular health, and Alzheimer disease biomarkers in an elderly sample. Ann Neurol 82, 706–718. [DOI] [PMC free article] [PubMed] [Google Scholar]
- [64].Vemuri P, Lesnick TG, Przybelski SA, Knopman DS, Preboske GM, Kantarci K, Raman MR, Machulda MM, Mielke MM, Lowe VJ, Senjem ML, Gunter JL, Rocca WA, Roberts RO, Petersen RC, Jack CR Jr (2015) Vascular and amyloid pathologies are independent predictors of cognitive decline in normal elderly. Brain 138(Pt 3), 761–771. [DOI] [PMC free article] [PubMed] [Google Scholar]
- [65].Brown WR, Thore CR (2011) Review: Cerebral microvascular pathology in ageing and neurodegeneration. Neuropathol Appl Neurobiol 37, 56–74. [DOI] [PMC free article] [PubMed] [Google Scholar]
- [66].Mufson EJ, Binder L, Counts SE, DeKosky ST, de Toledo-Morrell L, Ginsberg SD, Ikonomovic MD, Perez SE, Scheff SW (2012) Mild cognitive impairment: Pathology and mechanisms. Acta Neuropathol 123, 13–30. [DOI] [PMC free article] [PubMed] [Google Scholar]
- [67].Bennett DA, Wilson RS, Boyle PA, Buchman AS, Schneider JA (2012) Relation of neuropathology to cognition in persons without cognitive impairment. Ann Neurol 72, 599–609. [DOI] [PMC free article] [PubMed] [Google Scholar]