

Abstract
Arboviruses are medically important arthropod-borne viruses that cause a range of diseases in humans from febrile illness to arthritis, encephalitis and hemorrhagic fever. Given their transmission cycles, these viruses face the challenge of replicating in evolutionarily divergent organisms that can include ticks, flies, mosquitoes, birds, rodents, reptiles and primates. Furthermore, their cell attachment receptor utilization may be affected by the opposing needs for generating high and sustained serum viremia in vertebrates such that virus particles are efficiently collected during a hematophagous arthropod blood meal but they must also bind sufficiently to cellular structures on divergent organisms such that productive infection can be initiated and viremia generated. Sulfated polysaccharides of the glycosaminoglycan (GAG) groups, primarily heparan sulfate (HS), have been identified as cell attachment moieties for many arboviruses. Original identification of GAG binding as a phenotype of arboviruses appeared to involve this attribute arising solely as a consequence of adaptation of virus isolates to growth in cell culture. However, more recently, naturally circulating strains of at least one arbovirus, eastern equine encephalitis, have been shown to bind HS efficiently and the GAG binding phenotype continues to be associated with arbovirus infection in published studies. If GAGs are attachment receptors for many naturally circulating arboviruses, this could lead to development of broad-spectrum antiviral therapies through blocking of the virus–GAG interaction. This review summarizes the available data for GAG/HS binding as a phenotype of naturally circulating arbovirus strains emphasizing the importance of avoiding tissue culture amplification and artifactual phenotypes during their isolation.
Keywords: gycosaminoglycan, heparan sulfate, cell culture adaptation, arbovirus, alphavirus, flavivirus
Introduction
Arbovirus attachment receptor utilization
Arboviruses are viruses that infect and replicate in the cells of hematophagous arthropod vectors (e.g. ticks, flies, mosquitoes) and are spread to vertebrates during blood meals. Clinically important members of these viruses are found in the Togaviridae, Flaviviridae, Bunyaviridae, Arenaviridae and Reoviridae families of RNA viruses and the Asfarviridae family of DNA viruses (classical swine fever virus only). A subset of vertebrate species serves as reservoirs for arboviruses that include reptilian, amphibian, avian and mammalian members. These species can become infected by virus present in the arthropod vector’s saliva and generate sufficient serum viremia for acquisition of the virus by another blood-feeding arthropod, thus perpetuating the transmission cycle [1]. In addition to arthropod vector feeding preferences, the balance between high and sustained viremia levels and severity of viral illness is considered critical for the functioning of a particular species as an efficient reservoir host [1, 2]. Therefore, the ability of an arbovirus to generate sufficient viremia to achieve vector infection is an important aspect of its sylvatic transmission cycle. Given the evolutionary diversity of hosts in which these viruses replicate, two predominant theories have been promoted regarding the nature of cell attachment structures to which they bind to initiate infection: (i) arboviruses bind to highly evolutionarily conserved structures in multiple hosts; or (ii) arboviruses utilize different attachment receptors in different hosts and possibly, on different cell types within a single host. Interestingly, the identification of arbovirus attachment receptors has generally lagged behind those for non-arboviruses, even for closely related viruses within the same family [1]. Although, more recently, advanced techniques for genetic manipulation of protein expression in cells have greatly accelerated the discovery of arbovirus cell attachment/entry factors [3–13].
As might be expected, the results of these studies have indicated that both of the above theories are partially correct: receptor utilization by arboviruses may simultaneously be highly divergent in different hosts and/or tissues but also involves evolutionarily conserved structures. For example, flaviviruses, alphaviruses and bunyaviruses can bind to C-type lectin receptors on specialized cells through lectin binding to conserved carbohydrate modifications of the viral glycoproteins [3, 4, 14, 15]. Studies also have been published providing evidence for infection dependence or binding of alphaviruses [Sindbis virus (SINV) [16] and Venezuelan equine encephalitis virus (VEEV) [17]], flaviviruses [Dengue virus (DENV), Japanese encephalitis virus (JEV), yellow fever virus (YFV), West Nile virus (WNV) and tick-borne encephalitis virus (TBEV) [18–23]] and classical swine fever virus (CSFV) [24] through the high-affinity laminin receptor (HALR), which is a highly conserved ribosomal and cell surface protein [25, 26].
Since the 1990s, many publications have implicated host-expressed and highly conserved sulfated glycosaminoglycans (GAGs) as universal attachment factors for viruses from each of the arbovirus families (Table 1) (earliest reports for each family [27–32]). Clearly, identification of a common cell surface molecule that is conserved between reservoir, arthropod and dead-end hosts and utilized by many arboviruses could lead to development of broad-spectrum antiviral therapeutics that interrupt the infection process. Such drugs could significantly improve treatment of these infections, which plague millions of humans each year. However, in the case of GAG binding, careful attention must be paid to the problem of acquisition or enhancement of this phenotype as an artifact of conventional in vitro passage of virus isolates and the relationship of laboratory arbovirus strains to those circulating in nature. The subject of this review is a consideration of existing literature on the interactions of arbovirus strains with GAG molecules as attachment receptors, focusing initially on differences and similarities between GAG interaction studies in vitro with different arbovirus types and subsequently summarizing the evidence for these molecules functioning as receptors in the natural arbovirus transmission cycle.
Table 1.
Arboviruses examined for heparan sulfate binding and attachment protein residues implicated in the phenotype
|
Arbovirus family (genus) |
Type |
Arbovirus strains tested for GAG interactions* (validated wild type residues involved in GAG interactions)† |
Cell amplification mutations that increase GAG interactions‡ |
Reference |
|---|---|---|---|---|
|
Togaviridae (Alphavirus) |
|
|
||
|
|
SINV |
AR339 |
– |
[31] |
|
|
|
TR339 |
– |
[52] |
|
|
|
Toto1101 |
E2-E70K |
[52] |
|
|
|
AR339 cell passaged |
E2-S1R E2-E70K E2-S114R |
[52] |
|
|
SFV |
pSFV-4 |
– |
[53] |
|
|
RRV |
RRV64 |
– |
[57] |
|
|
|
RRV64 cell passaged |
E2-N218R |
[57] |
|
|
CHIKV |
L Reunion |
– |
[75] |
|
|
|
SL15629 37997 |
– |
[61] |
|
|
|
181/25 Vaccine |
E2-G82R |
[60] |
|
|
|
La Reunion cell passaged |
E2-G55R E2-E79K E2-S159R E2-E166Δ E2-E166K E2-E168K |
[75] |
|
|
VEEV |
Trinidad Donkey |
– |
[54] |
|
|
|
TC83 Vaccine |
E2-E120R |
[54] |
|
|
|
Trinidad Donkey cell passaged |
E2-E3K E2-E4K E2-E76K E2-E116K E2-E209K |
[54] |
|
|
EEEV |
FL93-939 (E2-K71) (E2-K74) (E2-K77) (E2-R84) (E2-R119) (E2-K156) (E2-R157) |
– |
|
|
|
EEEV |
Various isolates§ (E2-K71Q) (E2-K71T) (E2-T72K) |
– |
[49] |
|
|
WEEV |
McMillan |
– |
Gardner unpublished observations |
|
|
|
CBA87 |
– |
[62] |
|
|
MAYV |
Ohio |
– |
Klimstra unpublished observations |
|
Flaviviridae (Flavivirus) |
|
|
||
|
|
YFV |
Asibi |
– |
[55] |
|
|
|
17D Vaccine |
E-T380R |
[55] |
|
|
|
YF5.2iv generated from 17D |
E-T380R |
[93] |
|
|
DENV |
DEN2 New Guinea C Cell passaged |
E-E126K |
|
|
|
|
DEN 2 PUO-218 |
– |
[67] |
|
|
|
Cell passaged DENV3 MG-20 DANV3 PV_BR |
E-N124D/E202K E-T120K E-E126K E- E202K – – |
[67] [130] [130] |
|
|
DENV|| |
DEN 1 45AZ5 PDK-27 |
– |
[66] |
|
|
DENV|| |
DEN 1 TH-Sman |
– |
[66] |
|
|
WNV |
NY99 |
– |
[73] |
|
|
JEV |
RP-9 cell passaged |
– |
[131] |
|
|
|
RP-2ms cell passaged |
– |
[131] |
|
|
JEV |
Nakayama |
– |
[72] |
|
|
|
Nakayama variants isolated from mice |
E- E306K |
[72] |
|
|
JEV JEV JEV |
SA14-14-2 FU cell passaged SA14/CDC cell passaged |
E- E138K E-E128K E-E49K E-E138K E-E306K E-D389G |
[94] [73] [73] |
|
|
MVEV |
– |
[59] |
|
|
|
|
MVE-1–51 |
E-D390V E-D390L E-D390G E-D390H E-D390A |
[59] |
|
|
TBEV |
NeuDoerfl |
– |
|
|
|
|
NeuDoerfl |
E-E201K E-E122G E-S158R/G159R |
|
|
|
|
Ya10/89 clone 125 |
E-E122G |
[95] |
|
|
|
80 k |
E-D67N E-T68A |
[95] |
|
|
ZIKV |
Expressed E glycoprotein |
– |
[70] |
|
|
ZIKV¶ |
MR766 |
– |
[90] |
|
|
ZIKV |
MRS OPY |
[92] |
|
|
|
ALFV |
ALFV3929 |
E-L327K |
[96] |
|
Bunyaviridae |
|
– |
|
|
|
|
RVFV |
RVFVns |
– |
[74] |
|
|
|
MP-12 |
– |
[88] |
|
|
|
ZH-501 |
– |
[88] |
|
|
|
Kenya 9 800 523 |
– |
[88] |
|
|
|
Kenya 2007002444 |
– |
[88] |
|
|
TOSCV |
ISS.Phi.32 |
– |
[132] |
|
|
AKAV |
JaGAr-39 TS-C2 OBE-1 |
– – – |
[29] [29] [133] |
|
|
|
Iriki |
– |
[133] |
|
|
SBV |
SBV |
– |
[133] |
|
Arenaviridae |
|
|
||
|
|
JUNV§ |
? |
– |
[30] |
|
|
TCRV§ |
? |
– |
[30] |
|
|
LUJV# |
VSV-LUJV pseudotype |
– |
[97] |
|
Reoviridae (Orbivirus) |
|
|
||
|
|
BTV |
BTV-10 |
– |
[28] |
|
Asfarviridae |
|
|
||
|
|
CSFV |
Brescia |
– |
[27] |
|
|
|
C |
– |
[27] |
|
|
|
Brescia cell passaged |
ERNS-S476R |
[27] |
*Different strains with virus types exhibit a wide range of GAG/HS interaction efficiencies. HS dependence is proposed on the basis of the results of in vitro infection inhibition assays using soluble heparin or other GAG mimetics or more specific assays.
†Residues in strains validated to reflect the sequence of naturally circulating unpassaged viruses. With EEEV, these comprise three separate HS binding domains.
‡Known mutations in low-passage strains adapted by cell and/or mouse brain passage that bind GAGs with higher efficiency than progenitor strains.
§Alterations in one HS binding domain of EEEV found in different isolates that alter HS interactions and virulence in animals.
||Recombinant subviral particles of high passage clinical samples were shown bind heparin, and heparin and heparan sulfate inhibited binding to cells.
¶ZIKV infectivity was enhanced by heparin interaction.
#Reduction of infection on HAPI cells following heparinase treatment.
SINV, Sindbis virus; SFV, Semliki Forest virus; RRV, Ross River virus; CHIKV, chikungunya virus; VEEV, Venezuelan equine encephalitis virus; EEEV, eastern equine encephalitis virus; WEEV, western equine encephalitis virus; MAYV, Mayaro virus; YFV, yellow fever virus; DENV, dengue virus; WNV, West Nile virus; JEV, Japanese encephalitis virus; MVEV, Murray Valley encephalitis virus; TBEV, tick-borne encephalitis virus; ZIKV, Zika virus; RVFV, Rift Valley Fever virus; TOSCV, Toscana virus; AKAV, Akabane virus; SBV, Schmallenberg virus; JUNV, Junin virus; TCRV, Tacaribe virus; LUJV, Lujo virus; BTV, bluetongue virus; CSFV, classical swine fever virus.
Glycosaminoglycan structure and binding by microbial pathogens
Many reports have been published that demonstrate a role for GAGs, particularly heparan sulfate (HS) for in vitro cellular attachment of both DNA and RNA viruses (reviewed in [33–35]). GAGs are long chain polymers of hexuronic acid (d-glucuronic acid or l-iduronic acid) and d-glucosamine or d-galactosamine heterodimers with the specific heterodimer distinguishing the different GAG groups, of which there are four [36] (Fig. 1). As a post-translational modification, these chains are variously sulfated at the 3-O and 6-O positions leading to a net negative charge on the molecule (reviewed in [37]). GAG chains are displayed as post-translational modifications of cell membrane-bound (integral membrane or glycophosphatidyl inositol-linked) or secreted core proteins resulting in their abundant expression on most cell-surfaces and in the extracellular matrix as proteoglycans [38–40]. Although specific patterns of GAG sulfation can vary between tissues and hosts, GAG primary structures are highly conserved between diverse organisms, being found in invertebrate and vertebrate animals and plants [37].
Fig. 1.
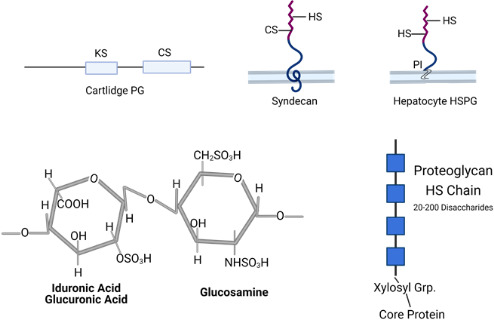
Top panels: Schematic showing general arrangement GAGs on proteoglycans, including cartlidge PG, syndecan and hepatocyte HSPG. GAGs in proteoglycans can consist of long chains of keratan sulfates (KS), dermatan sulfate (DS), chondroitin sulfates (CS), or heparan sulfates (HS). PI, phosphatidyl inositol linkage to cell membrane in hepatocyte PG. Bottom panels: The core disaccharide unit of HS (sulfonated uronic acid and glucosamines) is shown on the left. On the right is depicted a canonical proteoglycan HS modification showing the disaccharide chains linked with a xylosyl group to a core protein. Sulfonation of HS chains is not uniform, and the blue boxes represent highly sulfated regions of HS chains that may interact strongly with protein ligands. Figure generated using BioRender (www.biorender.com).
The interaction of protein ligands with GAGs plays a role in cell adhesion, tissue structure, anticoagulation, sequestration and concentration of cell signaling factors, regulation of transcription factor activity, and, particularly with HS proteoglycans, in vitro infection of cells by a number of bacterial, protozoan and viral pathogens including representatives of essentially all arboviruses (reviewed in [33, 36, 41–43]). In most instances, it is unclear whether or not the proteoglycan molecule can serve as a stand-alone entry receptor or if it acts as a pathogen concentration factor, handing organisms off to other structures that directly promote infection (Fig. 2). This latter model was proposed originally by Spear and colleagues in their pioneering work on HS utilization by herpesviruses (reviewed in [44]).
Fig. 2.
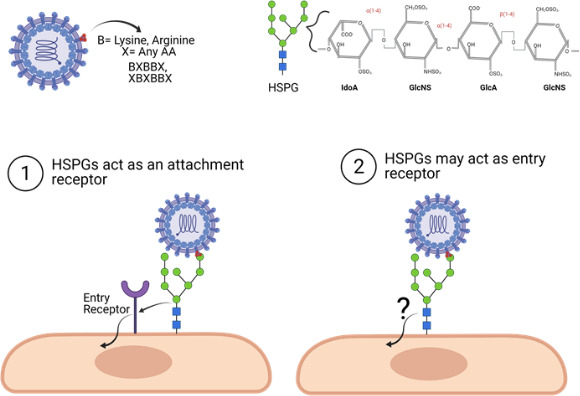
Positively charged amino acids on the virus surface will interact with the highly sulfated uronic acid and glucosamines on cellular HS proteoglycans (HSPGs), probably via HS binding motifs (indicated in the top panel). These interactions can lead to virus entry by (1) the HSPG acting as an attachment receptor only, potentially by increasing the amount of virus particles interacting with the cellular surface, thus increasing the probability of virus interaction with a proteinaceous entry receptor or leading to recruitment of entry receptors to a site of virus binding. (2) HSPGs may also act as both attachment and entry receptors, promoting entry without the recruitment or interactions of any other receptor. Figure generated using BioRender (www.biorender.com).
Positively-charged amino acids (especially Lys and Arg) either in linear HS binding consensus sequences [45–47] or in conformationally-determined three-dimensional binding sites [45, 48] on protein ligands are presumed to interact with sulfated and carboxylated components of HS chains (reviewed in [49]). While originally thought to be relatively non-specific ionic interactions, more recent work has revealed a great deal of specificity in the interaction of specific protein ligands with particular arrays of carboxyl and sulfate moieties on GAGs (reviewed in [37, 50]), although, this aspect of the arbovirus–GAG interaction remains relatively unexplored. With arboviruses, the capability to bind to HS or GAG receptors is associated with positively charged amino acids in the virus receptor attachment proteins (E2 with alphaviruses; E with flaviviruses, G with rhabdoviruses, GN/GC with bunyaviruses and E2 or envelope Erns with CSFV) [19, 49, 51–57], Due to the fact that alphaviruses and flaviviruses were the first arboviruses shown to interact with HS/GAGs [31, 52, 58, 59] and they are the most studied with respect to this phenotype, they will be the subject of the majority of this review. Occasionally, references to non-arboviruses are included when they support conclusions drawn from studies of arboviruses.
Alphaviruses are mosquito-borne arboviruses that cause two major types of human disease. The so-called Old World alphaviruses [e.g. Sindbis (SINV), Chikungunya (CHIKV), and Ross River viruses (RRV)] cause arthritis and arthralgia while the New World alphaviruses [e.g. eastern equine encephalitis virus (EEEV), western equine encephalitis virus (WEEV) and Venezuelan equine encephalitis virus (VEEV)] cause encephalitis [1]. Laboratory strains within each group of alphaviruses have been shown to interact with GAG/HS receptors on cells in vitro [52, 53, 57, 60, 61]. However, with the exception of EEEV, for which the attachment protein sequences of cDNA cloned viruses have been compared with those of unpassaged field samples, these phenotypes have not been directly confirmed as reflective of circulating viruses [62]. Many studies identifying GAGs as attachment receptors have also been published with flaviviruses [e.g. dengue virus (DENV) [32, 63–66], yellow fever virus (YFV) [55, 63], West Nile virus (WNV) [67], Murray Valley encephalitis virus (MVEV)] [67], tick-borne encephalitis virus (TBEV) [68], Japanese encephalitis virus (JEV) [65, 69] and Zika virus (ZIKV) [70]] which, using the nomenclature of the field, also can be broadly segregated into encephalitis-causing (e.g. MVEV, JEV, TBEV, WNV) and viscerotropic (e.g. DENV, YFV, ZIKV) groups [2, 71].
As with most RNA viruses, arboviruses possess an error-prone polymerase and highly adaptable genomes, making use of HS as a receptor controversial, as this phenotype can be rapidly acquired during amplification of virus isolates in vitro [48, 52, 53, 72]. Early studies with the alphavirus, SINV, demonstrated that within as few as three high multiplicity passages in cultured fibroblast cells, a positive charge mutation in the E2 protein became predominant in the population that conferred efficient (used as a measure of affinity and/or avidity) HS attachment to a virus that initially did not exhibit significant interaction with HS and, in fact, bound to multiple cell types very poorly (e.g. less than 5 % of added radiolabeled virus bound to an excess of cells [52]). An analogous strain has not been available for other arboviruses, although, one study utilized observed HS binding among cloned DENV human clinical isolates which were minimally passaged in cell culture [66]. Other studies of flaviviruses have compared very low passage viruses (e.g. fewer than four) to highly cell-adapted viruses [73] as has been done with CHIKV [61] leading to the conclusion that HS binding can be a phenotype of low-passage strains. Perhaps more definitively, one study with EEEV alphavirus and one with Rift Valley fever bunyavirus have compared the attachment protein sequences found in unpassaged field samples with those of laboratory strains shown to interact with HS during infection [62, 74]. In both cases, the sequence in the unpassaged primary samples was either identical to [62] or did not contain additional positively charged residues [74] with respect to the laboratory strain.
Yet, in cases where it has been examined, efficient binding to HS has been identified as an attenuating factor in live attenuated arbovirus vaccines (LAVs) including YFV 17D, VEEV TC83, CHIKV 181/25, and JEV SA14-14-2 [54, 55, 75–78], all of which were created by blind passage of virulent parental virus stocks in cultured cells. While the passage history and GAG binding phenotype of parental strains from which the LAVs were derived is frequently unclear, the vaccine strains show an increased efficiency of attachment to and dependency of infection on GAGs versus parental strains. Since nearly all studies assessing HS dependence of infection have utilized cell-adapted laboratory strains, and efficient HS binding is rapidly acquired during cell passage, rigorous assessment of the effects of cell passage on virus phenotypes should be applied to both in vitro and in vivo studies of HS dependence as is discussed below.
Studies of the arbovirus GAG binding phenotype in vitro
Traditional techniques for assessment of GAG interactions
Typically, in vitro HS dependence is evaluated by examining the effects upon cell infectivity of treatment with high salt (typically NaCl) concentration, which disrupts ionic interactions, direct competition of binding and/or infectivity with soluble heparin, a highly sulfated HS analogue, or other GAG molecules, heparin lyase (heparinase) digestion of cell surface HS structures or lyases specific to other GAG molecules, and comparing relative infectivity for wild type and HS- or GAG-deficient cells (e.g. CHOK1 mutants created by Jeffrey Esko and colleagues [79]) (e.g. [32, 48, 52–54, 57, 58]). In general, a virus for which HS is a cell attachment factor, will exhibit decreased infectivity for cells with all of the treatments with the exception of lyases specific to other GAGs. However, it has been shown with a number of viruses that other GAGs such as chondroitin sulfate or dermatan sulfate may be involved in cell attachment to some degree, with the phenotype varying even between different isolates of a given virus [60, 61, 75, 80]. Furthermore, we have observed significant differences in the susceptibility of genetic variants of a single alphavirus type to the three different heparinases [52, 81], which have specificity for different HS sulfation patterns and structures [82], indicating that there can be divergence in the particular HS structure bound even by very closely related viruses. With different HS binding alphaviruses and flaviviruses, twofold to 100-fold decreases in infection efficiency are typically observed when using these treatments [52, 54, 72, 81], which may reflect underlying efficiency of binding or dependence upon a particular GAG structure for infectivity. The infectivity of most viruses can be affected by one or more of the above experimental treatments, for example SINV strain TR339, derived from a cDNA clone generated from an original 1952 SINV isolate with no history of in vitro passage [52] (R. Swanopoel and R.E. Johnston, personal communication), shows infectivity reduction of approximately 10–30 % with heparin competition and also significant infectivity reduction with increased ionic strength [52]. However, the virus binds poorly to cells and infectivity is not appreciably reduced when infecting GAG-deficient CHOK1 cells [52]. Therefore, it is possible that other ionic interactions may be affected by some experimental conditions used to identify HS binding. Ultimately the degree to which infection is inhibited by treatments designed to disrupt ionic interactions or specific GAG binding is probably related to the efficiency of GAG binding by the virus and may be a critical factor in assessing the influence of the phenotype on virus transmission and disease processes (e.g, is binding sufficiently strong to significantly alter virus spread, tissue infectivity or other aspects of pathogenesis in animals or replication/transmission efficiency with arthropod vectors?).
Cell binding versus infectivity
It is also highly relevant to in vitro analysis of GAG dependence to determine attachment efficiency of virus particles to cells. In general, low-passage alphaviruses bind poorly to many types of cultured cells [52, 81], while cell-adapted viruses can attach with much greater efficiency. Generally, the reductions in cell binding after the above treatments are consistent with reductions in infectivity [52], indicating that greater attachment of arboviruses mediated by HS promotes greater infection. As stated above, whether or not this is direct promotion of entry or concentration of virus particles on the cell surface is unclear and whether or not the phenotype reflects inefficient interaction with cell surfaces versus low receptor abundance is also unclear.
In vitro data generated with some flaviviruses indicate that the interaction with HS may be less efficient than that for alphaviruses (e.g. reductions in infectivity of flaviviruses may be twofold to threefold after ablation of HS structures or with heparin competition) [52, 59, 64, 72, 83], although, it is difficult to determine due to the various methodologies used to examine the phenotype. Binding efficiency differences may be due to the fact that binding sites for HS have been mapped to the disordered and flexible E3 loop region as well as the E1/E2 domain interface or E2 domain of the flavivirus E protein (Table 1, Fig. 3b), whereas, alphavirus HS binding domains are mostly present in highly structured, surface-exposed areas of the analogous E2 glycoprotein (Table 1, Fig. 3a). It is possible that the repeating structure of the alphavirus trimeric spike within which stable GAG interaction sites are positioned (resulting in 80 copies of stable E2 glycoprotein in each particle [84]), may provide a three-dimensional, multivalent and highly avid binding site. However, specific HS or GAG structures bound by individual alphaviruses or flaviviruses have yet to be identified and direct comparisons of binding efficiency are lacking. It is remarkable, however, that E2 glycoprotein sites associated with use of HS receptors by naturally circulating viruses such as EEEV can essentially be identically positioned to those acquired as a result of cell culture passage [52, 62] (Fig. 3a). Furthermore, the difference between weak to undetectable interaction with HS and efficient binding can be the result of acquisition of a single positively charged amino acid mutation in the alphavirus or flavivirus surface glycoproteins [52, 72, 73, 85].
Fig. 3.
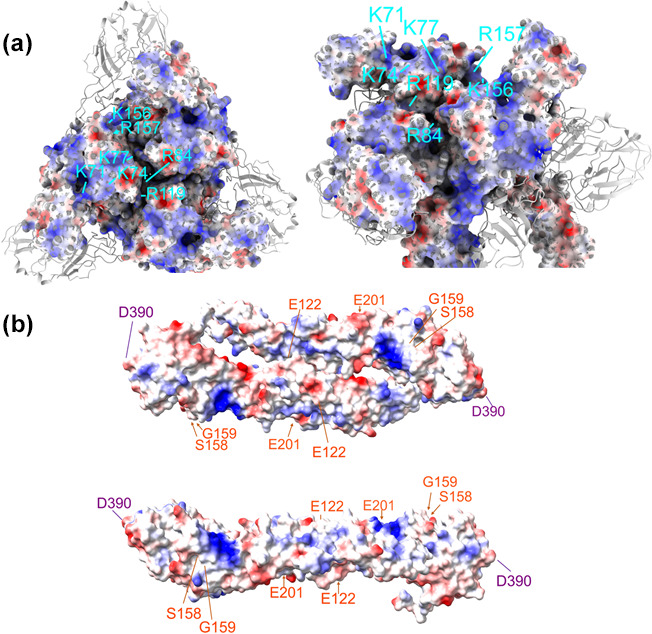
(a) Top and rotated and cropped three-dimensional model of the mature trimeric spike complex of EEEV E1 and E2 heterodimers (PDB: 6M×4). E1 is shown as a ribbon diagram and E2 as a residue electrostatic map (red=negative charge and blue=positive charge). E2 residues K71/K74/K77, R84/R119, K156/R157 (identified with lines in cyan) have been shown to be three separate interaction sites involved in HS binding by naturally circulating strains. (b) Top and side view of an electrostatic three-dimensional model of the JEV E dimer ectodomain (PDB: 5MV1, dimer was reconstructed using Swiss model programming). Corresponding residues on both E proteins where mutations increase HS interactions for TBEV (E122, S158, G159 and E201) are highlighted in orange, and for MVEV (D390) is highlighted in purple. The figures were made using UCSF ChimeraX software.
We and others have speculated that weak cell attachment, which could be mediated by either a low-abundance receptor or inefficient binding, may be a ubiquitous feature of arbovirus particles that promotes generation of high titer sustained viremia in vertebrate hosts [4, 14, 52, 73, 85]. However, this raises a conundrum with respect to cell binding by arboviruses. In our hands, with common fibroblastic cell lines, nearly all of the measurable cell binding by laboratory strains of SINV strain AR339, for example, is dependent upon the HS interaction and is greatly reduced by manipulating the availability of HS receptors [52, 54, 81]. Therefore, at least on these cell types, it is likely that interaction of virus particles with receptors other than GAGs, at least at the virus population level measured in conventional binding assays, is of low efficiency, again either through weak interaction with a receptor or low receptor abundance. These results indicate that interactions with other identified alphavirus fibroblast receptors [e.g. high affinity laminin receptor (HALR) [16, 17], natural resistance-associated macrophage protein (NRAMP) [5], matrix remodeling associated 8 (MxrA8) [7] or low-density lipoprotein receptor class A domain-containing 3 (LDLRAD3) [6]] may either be linked to HS binding, are relatively inefficient, or receptor abundance is low on multiple permissive cell types. Notably, it has been proposed that the prion protein interacts with the HALR via binding of each protein to HS in a ‘sandwich’ structure [86] and this may account for its identification as an arbovirus receptor. It is also worth considering whether or not inhibitors used to block virus interactions with other attachment receptors possess properties that may non-specifically disrupt ionic interactions as this may also block GAG binding leading to reductions in cell attachment, or that HS binding and protein receptor interaction domains overlap.
In contrast, with specialized cell types such as DC- or l-SIGN expressing myeloid cells, alphaviruses that contain high-mannose carbohydrate modifications of the surface glycoproteins but do not bind HS well, can bind cells efficiently and to similar levels to HS binding viruses [4]. Alternatively, HS-independent binding of cells by different arboviruses could vary greatly between virus types. Regardless, a comparison of direct GAG and/or cell binding efficiency either between virus isolates or between representatives of different families would be of assistance in placing the GAG binding phenotype of individual viruses in context, for example, in attempts to understand the relationship of cell binding efficiency to virus phenotypes in animal models. Finally, it should be noted that HS/GAGs are generally not found to be the sole attachment/entry mediator for arboviruses on common fibroblast-derived cell lines such that a significant base-line infectivity for these cell lines cells is retained in the complete absence of GAG molecules. This confirms that other attachment/entry receptors are present on these cell types. Indeed, LDLRAD3 overexpression has been shown recently to increase binding and infectivity of VEEV in the absence of cell surface heparan sulfate [6].
HS receptor identification through global host factor knockdown
Recent attempts to identify host factors associated with arbovirus entry, studies using gene trap mutagenesis, RNAi or CRISPR/Cas9 genome knockdown have demonstrated that GAG synthesis pathways are involved in infectivity of laboratory virus strains including CHIKV or Rift Valley Fever virus in HAP1 human myeloid cells [87, 88], SINV in CHO cells [89] and DENV and ZIKV in MAGI human cervical epithelial cells [90]. These screens relied on differential survival of cells with putative essential entry mediators ablated, therefore, the infection efficiency of viruses for surviving cells was by requirement, very low to none. These results are surprising since significant infectivity of all tested alphaviruses is maintained for HS/GAG-negative CHOK1 cells. As several of these studies utilized non-fibroblast cell types, this indicates that there may be a cell type dependence of virus infection upon GAG molecules. While it is unlikely, yet possible, that these cells express a cell surface form of GAG not expressed by fibroblasts and to which arboviruses can bind, it is also possible that other pathways of cell entry are greatly reduced in comparison with fibroblast cell types, such that a GAG-dependent pathway is essential for substantial entry. However, it is also possible that GAG biosynthetic pathways are required once the cell has been entered for virus trafficking and fusion within endocytic vesicles. For instance, results from two studies have indicated that HS is not vital for ZIKV entry but is important for replication in vitro [91, 92]. Similarly, chondroitin sulfate enhanced CHIKV replication after infection [87]. In this case, comparative cell binding assays, as opposed to infectivity assays, are critical to distinguish between these different possibilities.
Informed passage experiments
Multiple studies have also formally examined differences in GAG interaction between low-passage arbovirus strains and those subjected to greater numbers of in vitro growth cycles [52–55, 57, 72, 73, 75]. These types of studies have demonstrated empirically that significant increases in GAG binding can be acquired by an arbovirus through a single amino acid substitution and that after a small number of passages at high MOI, such mutations can be predominant in a virus population and give the impression that efficient GAG binding is the phenotype of the unpassaged virus. It is sobering to consider the number of commonly used arbovirus biological stocks and the practice still common today to amplify viruses in cultured cells prior to plaque purification and/or generation of cDNA clones.
The first arbovirus study to demonstrate this effect was with SINV strain TR339 showing that particle infectivity and cell binding and competition with HS analogs increased steadily up to three passages where a plateau was observed [52]. Furthermore, sequencing of the thrice-amplified stock revealed the presence of a Glu–Lys substitution at position 70 of the E2 glycoprotein responsible for the receptor utilization change (Table 1). Other studies revealed that the Lys 70 residue was present in multiple commonly used SINV laboratory strains [51]. This phenomenon has been reproduced starting with low cell passage versions of RRV, Semliki Forest virus (SFV) and CHIKV alphaviruses and YFV, MVEV, DENV, WNV, JEV, and TBEV flaviviruses yielding a large panel of known loci associated with enhancement of GAG interaction after in vitro passage (Fig. 3b) (Table 1) [53, 57, 60, 66, 72, 73, 75, 85, 88, 93–97]. These findings have utility in live attenuated vaccine design and also may allow determination of differences in naturally acquired and cell-adaptive GAG binding characteristics. Recent studies with SARS CoV-2 propagation in vitro have reinforced the capacity of RNA viruses to mutate during the first few cell amplification steps [98, 99]. Ultimately, in vitro studies have yet to distinguish mechanistically between natural and cell passage-acquired HS binding and its role in cell infectivity.
Effects of HS binding upon arboviral pathogenesis in animal models
Mouse models of arboviral disease
Early studies examining the phenotypes of HS binding SINV at later stages after infection of mice or encephalomyocarditis virus in horses indicated that viruses adapted to in vivo replication by reducing the affinity of HS binding or losing the HS binding phenotype altogether [48, 100]. Furthermore, viruses with known cell passage-acquired HS binding mutations were attenuated in comparison with their weakly HS binding ‘wild type’ progenitors [48, 58, 101]. Thus, as stated above, original assessments of the GAG binding of RNA viruses were that the phenotype was an artifact of cell culture passage of virus isolates [48, 52, 53, 57]. Indeed, in virtually all circumstances where GAG binding can definitely be shown to be an artifact of cell culture amplification or the deliberate increasing of positive charge in viral attachment proteins, the phenotype is attenuating in vivo when the natural arbovirus infection route [e.g. subcutaneous (sc) or intradermal inoculation] is mimicked.
The mechanism of attenuation of viruses that bind HS efficiently appears to be related to spread of virions as several studies have demonstrated that the in vivo dissemination of HS binding arboviruses is restricted. In comparisons of the blood retention time or viremic spread of HS-binding versus non-binding alphavirus, flavivirus and other RNA virus strains, efficient HS-binding viruses are generally cleared more rapidly from mouse blood and show diminished spread [48, 54, 56, 72, 102]. Similarly, viruses isolated from mouse blood later during infection with HS-binding SINV had frequently lost the HS binding phenotype, indicating that it was an inhibitor of virus spread and serum retention [100]. In general, the inhibition of spread in vivo for a particular virus genotype correlates with the degree of infection dependence upon or binding efficiency to HS as measured in in vitro assays [52, 54, 81]. This indicates that cell culture adaptive mutations may confer GAG interaction that exceeds the optimal efficiency of GAG binding naturally present in virus glycoproteins, thereby, attenuating disease in vivo. However, there are exceptions, for example with SINV, the E2 position 70 lysine mutation acquired after fibroblast passage, can slightly, but significantly, increase neurovirulence of the TR339 strain after intracranial infection of weanling mice although it is attenuating in suckling and adult mice [81]. In addition, this mutation is essential in combination with an in vivo-selected non-charged amino acid mutation for neurovirulence of an adult neurovirulent SINV mutant ([81, 103] described in more detail below). Interestingly, deletion of certain post-translational carbohydrate addition sites from the SINV E2 glycoprotein also increased HS binding and mouse virulence after intracranial infection [104]
EEEV is unique among alphaviruses examined to date in that virus access to peripheral lymphoid tissues and infection of myeloid cells is restricted by efficient binding to HS [49, 62] as well as restriction of myeloid cell replication by virus genome binding to a hematopoietic cell-specific microRNA [105]. In these studies, sequences of the E2 attachment protein of the cDNA cloned FL93-939 strain were compared with those from primary unpassaged field isolates and found to be identical in the majority of samples tested [62]. A combination of biological [62] and cryo-EM structure imaging analyses [106] has identified three domains on the native virus E2 protein capable of interaction with HS (Fig. 3a). These include lysine and arginine residues at positions 71, 74 and 77 (71 and 74 being most critical) [49, 62], of the E2 attachment protein and binding sites comprised of positively charged residues at positions 84/119 and 156/157, which were predicted to bind heparin hexamers in cryo-EM reconstruction experiments [106]. All three domains have been examined by charged-to-alanine mutagenesis and each independently affects EEEV binding to and infectivity for cells in a HS/GAG-dependent manner [62, 106]. In a mouse model, wild-type (WT) EEEV virus exhibited significantly less replication and numbers of infected cells in the draining lymph node (DLN) after sc inoculation than an E2 71–77 charged-to-alanine mutant that no longer bound HS efficiently [62]. Elimination of the HS binding domain increased levels of serum interferon-α/β and survival times due to decreased virus replication and dissemination within the central nervous system (CNS) [49, 62]. This is similar to reports with both Theiler’s murine encephalomyelitis virus [107] and neuroadapted SINV [81]. In contrast, mutation to reduce the efficiency of HS binding decreased replication and spread within the brain [62, 81]. The 84/119 and 156/157 mutations have not been examined for effects upon virulence in vivo (Fig. 3a). Together, these data indicate that the HS binding domain(s) of WT EEEV suppress virus access to permissive cells in DLNs while simultaneously increasing virus infection of neurons and spread in the CNS, contributing to the severe manifestations of EEEV. Furthermore, natural alteration of the HS binding site may be linked to variation in virulence of individual virus isolates [49]. Several other studies have found a positive association of HS binding with arbovirus neurovirulence specifically, especially when the requirement for viremic spread was obviated, and it has been speculated that HS binding may be a neurovirulence factor due to the close association of cells within the brain, which may promote efficient infection of neighboring cells by HS binding viruses [81, 108–110] (reviewed in [111]).
The role of GAGs in arthropod vector infection
Infection of the hematophagous arthropod vector gut epithelium from a blood meal and subsequent spread to and infection of cells in the salivary gland is a requirement for sylvatic transmission of arboviruses (reviewed in [112, 113]). Very few studies have examined the role of HS or GAG binding in this process, and it is unclear if the spread of arboviruses in arthropods is analogous to viremic spread in vertebrates. A recent study has implicated rice arbovirus binding to sperm HS in vertical transmission [114]. In vitro, we have passaged SINV TR339 on C6/36 mosquito cells and obtained clustered mutations in the E2 protein (Ser–Arg at amino acid 60 and Ala–Thr at amino acid 61) that, together, increase HS dependence of infection, indicating that HS proteoglycans can function as attachment receptors on mosquito cells. It has been demonstrated that glycosaminoglycans capable of binding pathogens are present in the mosquito gut and salivary gland [115–118] (reviewed in [119]), although, the diversity of core proteins in proteoglycans is limited in comparison with vertebrates [120]. One recent study has evaluated HS/GAG structures in different mosquito species, finding some differences in predominant type and structure [121].
In preliminary studies, we have determined that with EEEV, HS binding increases virus infectivity for the mosquito gut epithelium after a blood meal, indicating that, at least with a virus that expresses this phenotype in sylvatic transmission cycles, HS binding promotes mosquito infection. In addition, our data supports the notion that with EEEV, HS binding also promotes transit to and infection of the salivary gland (SC Weaver and WB Klimstra unpublished data). Similar results were obtained in a study using SINV strains with different efficiencies of GAG binding [115]. Whether or not HS binding is promoting to or inhibitory of release from the arthropod salivary gland remains to be determined.
HS binding may also play a role in mosquito–vertebrate transmission efficiency. Results from one study indicated that mosquito saliva proteases that were delivered alongside virus particles and that degraded the extracellular matrix, improved DENV engagement of HS receptors on cells [122] Amplification of CHIKV on mosquito cells reduced virion binding to HS, presumably through blockade of HS binding sites on virions by the high-mannose, glycoprotein carbohydrate modifications obtained during invertebrate replication [123]. However, in contrast, growth of a SINV cell-adapted strain in C7/10 mosquito cells increased the efficiency of virion binding to heparin bead columns [104]. While more research is a critical need, these findings indicate that HS binding of mosquito-delivered alphaviruses may be influenced by characteristics of the mosquito vector. This phenomenon may have parallels with differences in mammal- versus arthropod-derived alphavirus and flavivirus glycoprotein interactions with C-type lectins [4, 14].
The relationship between natural and laboratory acquired GAG binding in animal models
It is likely that promiscuous and efficient interaction of HS binding domains with ubiquitously expressed HS results in sequestration of virus particles to non-productive structures in vivo. This may lead to limitations of virus spread from initial sites of inoculation (e.g. footpad to DLN and serum in murine models) and, therefore, in the context of natural infection, may represent an adaptation of some viruses to limit immune responses by suppressing myeloid cell replication. Similar hypotheses regarding suppression of spread in vivo have been proposed in studies of other viruses which exhibit increased GAG binding over other strains [55, 56, 73, 85]. Furthermore, efficiency of infection of cells within isolated organs such as brain may be increased by efficient GAG binding. Therefore, the role of HS binding in suppressing virus spread in vivo could be important in the pathogenesis of a number of viruses.
In contrast, most studies conclude that HS or GAG binding acquired as an artifact of cell adaptation in vitro has an attenuating effect on virus replication and disease in vivo and it is clear that HS binding is an important attenuating characteristic of multiple live attenuated vaccine strains. For unknown reasons, some viruses may have adapted to achieve vertebrate–invertebrate transmission while incorporating the high efficiency HS binding phenotype. With EEEV, this may imply that other features in its attachment protein (or other viral proteins) allow the efficient binding of HS in the face of the inhibitory effects of this phenotype to spread in vivo. As mentioned earlier, neurovirulent Sindbis virus strain, NSV, developed by Griffin and colleagues [81, 103] which is more virulent from the natural sc route of infection in adult mice, was derived by serial passage of cell-adapted HS binding strain in sucking and weanling mice. The adult neurovirulence phenotype is mediated by the E2 E70K HS binding mutation, probably an artifact of cell culture passage, but only in combination with an additional E2 N55H mutation [81]. As mentioned above, increased GAG binding has also been associated with enhanced neurovirulence in some flaviviruses in the absence of a requirement for viremic spread [67, 72, 95]. Therefore, replication in vertebrates can result in adaptation to retain or even enhance virulence in the presence of efficient HS binding. Considering results ablating individual HS binding sites in EEEV [62] and adding HS binding sites to other alphaviruses and flaviviruses [53, 54, 57, 73, 85], it may be that disruption of adapted homeostasis, either increasing or decreasing HS binding versus that of natural viruses, may be the primary attenuating factor in vivo. This can function through suppression of virus spread in vivo (e.g. acquisition/enhancement of HS binding) or increases in lymphotropism/innate immune responses and reduction in neurovirulence (reduction in HS binding). In addition to these points, the effects of cell type-specific expression of particular proteoglycans and/or GAG structural variants and their specific binding by viral attachment proteins must be considered when evaluating effects of GAG binding on tropism and spread in vivo. As mentioned above, it must be noted that these conclusions are almost exclusively derived from arbovirus disease models and do not adequately consider effects upon transmission between reservoir species and arthropod vectors.
Concluding remarks
Arboviruses infect evolutionarily divergent hosts during their transmission cycle. This places constraints on many aspects of their replication including attachment receptor utilization. Research to date has indicated that both cell/host-specific and highly conserved receptors can be utilized for attachment, sometimes in combination as with the interaction of conserved virus carbohydrate modifications with vertebrate C-type lectin receptors. GAG molecules expressed as post-translational modifications of cell surface proteoglycans are also a class of highly conserved receptor structures to which many arbovirus strains attach, however, unlike other identified receptors, the lesser specificity of the ionic GAG–protein interaction appears to allow adaptation to efficient binding of these receptors rapidly and with as little as a single nucleotide change in viral attachment protein genes. Controversy exists because efficient GAG interactions can often be a consequence of minimal in vitro cell adaptation and have little relevance to naturally circulating viruses but can also be observed with truly ‘wild type’ strains.
The efficiency of interactions with GAG molecules during infection can exhibit a very large range, as measured by binding and GAG infection dependence assays, and increasing strength is generally associated with increasing infectivity of viruses for common fibroblastic cell lines leading to the conclusion that the primary selective advantage of in vitro-acquired GAGs interactions is to increase binding of arboviruses to cells and increase infectivity [52]. From this review of the literature, it seems clear that lower numbers of cell passages are most often associated with weaker interactions with GAGs and vice-versa (with EEEV as a potential exception). Therefore, the study of GAG binding by arboviruses has, to date, very probably been biased by the characteristics of HS interaction engaged in by cell-adapted virus strains. In particular, conclusions regarding the potential for use of HS analogs as anti-arboviral therapeutics may have been affected.
From our experience and this review of the literature, we propose that the following procedures be followed during isolation of strains for analysis of GAG interaction phenotypes to avoid acquisition of cell-adaptive GAG binding mutations. (1) attachment proteins should be sequenced directly from clinical material and the predominant sequences directly recapitulated through synthesis of viral genes when creating cDNA clones (2) cDNA cloned viruses should be passaged less than three times in cultured cells prior to evaluation of GAG interaction phenotypes (preferably, stocks would be harvested after a single electroporation of viral genomic RNA into cells), and (3) deep sequencing should be utilized to confirm that the predominant sequence of attachment proteins in virus stocks is reflective of the cDNA clone. These procedures would ensure that cell culture adaptive mutations are not incorporated into ‘wild type’ stocks. While these methods for generation of virus stocks from cDNA clones may constrain quasispecies diversity in the initial stages of virus production, it should be rapidly regenerated during virus production and after infection of experimental animals. Furthermore, this is presumably no less artificial than shifts in quasispecies composition imposed by repeated amplification of virus strains in cultured cells. It is well established that passage of arboviruses in single cell types alters the adaptive balance achieved through the alternating vertebrate–invertebrate replication cycle [124].
Making the assumption that lower passage viruses are genetically closer to viruses circulating in nature, a critical goal of additional research in this area is to determine if the weak interactions with GAGs achieved by low passage and/or authentic natural isolates are relevant to replication and disease in vivo. Furthermore, it will be important to determine if the disruption of HS interactions by HS analogs used therapeutically has a beneficial effect on disease caused by naturally GAG-binding viruses. Given that some evidence exists that closely related viruses may bind different GAG structures (e.g. differential heparinase sensitivity [49, 52, 81] and the fact that different mutations that alter HS interactions can be selected in virus glycoproteins with passage on different cell types [75]), cell type-specific aspects of virus–GAG interactions and proteoglycan core protein identity should also be explored. GAG- and HS-deficient CHOK1 cells have become the standard for assessment of virus–GAG interactions and this approach should be expanded to tissues and cell types relevant to pathogenesis in vivo (recently a few studies have used this approach with arboviruses [6, 7, 121, 125]). In particular, the structural and functional relationship of GAGs to other entry receptors may be cell type-dependent. In a possible analogy with arboviruses, a diversity in function of different HS structures has been observed with herpes simplex virus (reviewed in [44, 126]) where unmodified HS, such as that expressed by CHOK1 cells, can bind viruses but only specifically modified forms of HS, such as those modified by the 3–O-transferases can facilitate entry [127]. A CHIKV laboratory strain, but not JEV or YFV flaviviruses, was shown to rely on N-sulfation of HS for infection of HAP1 cells indicating some similarities [87].
It will be also important to determine if proteoglycans can serve as single attachment and entry receptors or if they merely serve to concentrate viruses on the cell surface. In particular, the relationship of GAG binding and the effect of GAG deficiency upon virus utilization of other receptor structures should be investigated during the identification stage of these receptors. Highly relevant to this idea, recent cryo-EM structures of CHIKV and VEEV bound to proteinaceous receptors, Mxra8 and LDLRAD3 [128, 129], have revealed common E2 contact sites for both receptors and some of these are either immediately adjacent to or overlapping with positively charged clusters involved in HS binding [52, 62, 75, 106]. This raises the possibility that cell culture adaptation to efficient HS binding might alter interactions with other receptors.
Finally, the ease with which arboviruses can transition between GAG binding phenotypes indicates that they could seamlessly respond to an adaptive pressure for increased cell binding and infectivity during the complexities of the arthropod–vertebrate–arthropod transmission cycle or even within individual tissues of a single host. Viruses such as EEEV may express an efficient GAG binding phenotype during the majority of their transmission and disease cycles but other viruses may not. Therefore, the adaptive potential of arboviruses to GAG binding should be considered at each point in the arboviral transmission cycle (reservoir, arthropod vector and dead-end host) as well as within individual tissues within each host. Such considerations may have significant influence on the potential or context for use of GAG analogs as antiviral therapeutics. Separate from these issues, rational manipulation of the native GAG binding phenotypes of arboviruses should become an important factor in the informed design of live attenuated arbovirus vaccines.
Funding information
This work was supported by grants from the National Institutes of Health (R01 AI095436 and R01 AI141646) and (HDTRA1-15-1-0047) from the Defence Threat Reduction Agency to W.B.K. and M.D.H.A was supported by a National Institutes of Health grant [T32 AI049820].
Acknowledgements
We apologize for not being able to cite all our colleagues’ work due to space constraints.
Conflicts of interest
The authors declare that there are no conflicts of interest.
Footnotes
Abbreviations: CHIKV, Chikungunya virus; CSFV, classical swine fever virus; DENV, dengue virus; DLN, draining lymph node; EEEV, eastern equine encephalitis virus; GAG, glycosaminoglycan; HS, heparan sulfate; JEV, Japanese encephalitis virus; MVEV, Murray Valley encephalitis virus; RRV, Ross River virus; sc, subcutaneous; SINV, Sindbis virus; TBEV, tick-borne encephalitis virus; VEEV, Venezuelan equine encephalitis virus; WEEV, western equine encephalitis virus; WNV, West Nile virus; YFV, yellow fever virus; ZIKV, Zika virus.
References
- 1.Griffin DE, Weaver SC. In: Fields Virology: Emerging Viruses. 7th edn. Howley PM, Knipe DM, Whelan S, editors. Philadelphia, PA: Wolters Kluwer; 2020. Alphaviruses; pp. 194–245. [Google Scholar]
- 2.Pierson TC, Lazear HM, Diamond MS. In: Fields Virology: Emerging Viruses. 7th edn. Howley PM, Knipe DM, Whelan S, editors. Philadelphia, PA: Wolters Kluwer; 2020. Flaviviruses: Dengue, Zika, West Nile, Yellow Fever and Other Flaviviruses; p. 346. p. [Google Scholar]
- 3.Tassaneetrithep B, Burgess TH, Granelli-Piperno A, Trumpfheller C, Finke J, et al. DC-SIGN (CD209) mediates dengue virus infection of human dendritic cells. J Exp Med. 2003;197:823–829. doi: 10.1084/jem.20021840. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 4.Klimstra WB, Nangle EM, Smith MS, Yurochko AD, Ryman KD. DC-SIGN and L-SIGN can act as attachment receptors for alphaviruses and distinguish between mosquito cell- and mammalian cell-derived viruses. J Virol. 2003;77:12022–12032. doi: 10.1128/jvi.77.22.12022-12032.2003. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 5.Rose PP, Hanna SL, Spiridigliozzi A, Wannissorn N, Beiting DP, et al. Natural resistance-associated macrophage protein is a cellular receptor for sindbis virus in both insect and mammalian hosts. Cell Host Microbe. 2011;10:97–104. doi: 10.1016/j.chom.2011.06.009. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 6.Ma H, Kim AS, Kafai NM, Earnest JT, Shah AP, et al. LDLRAD3 is a receptor for Venezuelan equine encephalitis virus. Nature. 2020;588:308–314. doi: 10.1038/s41586-020-2915-3. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 7.Zhang R, Kim AS, Fox JM, Nair S, Basore K, et al. Mxra8 is a receptor for multiple arthritogenic alphaviruses. Nature. 2018;557:570–574. doi: 10.1038/s41586-018-0121-3. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 8.Meertens L, Carnec X, Lecoin MP, Ramdasi R, Guivel-Benhassine F, et al. The TIM and TAM families of phosphatidylserine receptors mediate dengue virus entry. Cell Host Microbe. 2012;12:544–557. doi: 10.1016/j.chom.2012.08.009. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 9.Morizono K, Chen ISY. Role of phosphatidylserine receptors in enveloped virus infection. J Virol. 2014;88:4275–4290. doi: 10.1128/JVI.03287-13. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 10.Perera-Lecoin M, Meertens L, Carnec X, Amara A. Flavivirus entry receptors: an update. Viruses. 2013;6:69–88. doi: 10.3390/v6010069. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 11.Jemielity S, Wang JJ, Chan YK, Ahmed AA, Li W, et al. TIM-family proteins promote infection of multiple enveloped viruses through virion-associated phosphatidylserine. PLoS Pathog. 2013;9:e1003232. doi: 10.1371/journal.ppat.1003232. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 12.Richard AS, Zhang A, Park SJ, Farzan M, Zong M, et al. Virion-associated phosphatidylethanolamine promotes TIM1-mediated infection by Ebola, dengue, and West Nile viruses. Proc Natl Acad Sci U S A. 2015;112:14682–14687. doi: 10.1073/pnas.1508095112. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 13.Hamel R, Dejarnac O, Wichit S, Ekchariyawat P, Neyret A, et al. Biology of Zika virus infection in human skin cells. J Virol. 2015;89:8880–8896. doi: 10.1128/JVI.00354-15. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 14.Davis CW, Nguyen H-Y, Hanna SL, Sánchez MD, Doms RW, et al. West Nile virus discriminates between DC-SIGN and DC-SIGNR for cellular attachment and infection. J Virol. 2006;80:1290–1301. doi: 10.1128/JVI.80.3.1290-1301.2006. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 15.Léger P, Tetard M, Youness B, Cordes N, Rouxel RN, et al. Differential use of the C-type lectins L-SIGN and DC-SIGN for phlebovirus endocytosis. Traffic. 2016;17:639–656. doi: 10.1111/tra.12393. [DOI] [PubMed] [Google Scholar]
- 16.Wang KS, Kuhn RJ, Strauss EG, Ou S, Strauss JH. High-affinity laminin receptor is a receptor for Sindbis virus in mammalian cells. J Virol. 1992;66:4992–5001. doi: 10.1128/JVI.66.8.4992-5001.1992. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 17.Ludwig GV, Kondig JP, Smith JF. A putative receptor for Venezuelan equine encephalitis virus from mosquito cells. J Virol. 1996;70:5592–5599. doi: 10.1128/JVI.70.8.5592-5599.1996. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 18.Zidane N, Ould-Abeih MB, Petit-Topin I, Bedouelle H. The folded and disordered domains of human ribosomal protein SA have both idiosyncratic and shared functions as membrane receptors. Biosci Rep. 2012;33:113–124. doi: 10.1042/BSR20120103. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 19.Malygin AA, Bondarenko EI, Ivanisenko VA, Protopopova EV, Karpova GG, et al. C-terminal fragment of human laminin-binding protein contains a receptor domain for Venezuelan equine encephalitis and tick-borne encephalitis viruses. Biochemistry. 2009;74:1328–1336. doi: 10.1134/s0006297909120050. [DOI] [PubMed] [Google Scholar]
- 20.Sakoonwatanyoo P, Boonsanay V, Smith DR. Growth and production of the dengue virus in C6/36 cells and identification of a laminin-binding protein as a candidate serotype 3 and 4 receptor protein. Intervirology. 2006;49:161–172. doi: 10.1159/000089377. [DOI] [PubMed] [Google Scholar]
- 21.Boonsanay V, Smith DR. Entry into and production of the Japanese encephalitis virus from C6/36 cells. Intervirology. 2007;50:85–92. doi: 10.1159/000097394. [DOI] [PubMed] [Google Scholar]
- 22.Tio PH, Jong WW, Cardosa MJ. Two dimensional VOPBA reveals laminin receptor (LAMR1) interaction with dengue virus serotypes 1, 2 and 3. Virol J. 2005;2:25. doi: 10.1186/1743-422X-2-25. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 23.Thepparit C, Smith DR. Serotype-specific entry of dengue virus into liver cells: identification of the 37-kilodalton/67-kilodalton high-affinity laminin receptor as a dengue virus serotype 1 receptor. J Virol. 2004;78:12647–12656. doi: 10.1128/JVI.78.22.12647-12656.2004. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 24.Chen J, He W-R, Shen L, Dong H, Yu J, et al. The laminin receptor is a cellular attachment receptor for classical swine fever virus. J Virol. 2015;89:4894–4906. doi: 10.1128/JVI.00019-15. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 25.Ardini E, Pesole G, Tagliabue E, Magnifico A, Castronovo V, et al. The 67-kDa laminin receptor originated from a ribosomal protein that acquired a dual function during evolution. Mol Biol Evol. 1998;15:1017–1025. doi: 10.1093/oxfordjournals.molbev.a026000. [DOI] [PubMed] [Google Scholar]
- 26.Rea VE, Rossi FW, De Paulis A, Ragno P, Selleri C, et al. 67 kda laminin receptor: structure, function and role in cancer and infection. Le infezioni in medicina: rivista periodica di eziologia, epidemiologia, diagnostica, clinica e terapia delle patologie infettive. 2012;20:8–12. [PubMed] [Google Scholar]
- 27.Hulst MM, van Gennip HG, Moormann RJ. Passage of classical swine fever virus in cultured swine kidney cells selects virus variants that bind to heparan sulfate due to a single amino acid change in envelope protein Erns . J Virol. 2000;74:9553–9561. doi: 10.1128/jvi.74.20.9553-9561.2000. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 28.Mecham JO, McHolland LE. Measurement of bluetongue virus binding to a mammalian cell surface receptor by an in situ immune fluorescent staining technique. J Virol Methods. 2010;165:112–115. doi: 10.1016/j.jviromet.2009.12.011. [DOI] [PubMed] [Google Scholar]
- 29.Nakamura S, Ishihara Y, Ishikawa K, Shimazaki T, Inoue Y, et al. Inhibitory effect of heparin on Rhabdovirus and Bunyavirus isolated from cattle. Aust Vet J. 1993;70:264–265. doi: 10.1111/j.1751-0813.1993.tb08046.x. [DOI] [PubMed] [Google Scholar]
- 30.Andrei G, De Clercq E. Inhibitory effect of selected antiviral compounds on arenavirus replication in vitro. Antiviral Res. 1990;14:287–299. doi: 10.1016/0166-3542(90)90009-v. [DOI] [PubMed] [Google Scholar]
- 31.Mastromarino P, Conti C, Petruzziello R, Lapadula R, Orsi N. Effect of polyions on the early events of Sindbis virus infection of Vero cells. Arch Virol. 1991;121:19–27. doi: 10.1007/BF01316741. [DOI] [PubMed] [Google Scholar]
- 32.Chen Y, Maguire T, Hileman RE, Fromm JR, Esko JD, et al. Dengue virus infectivity depends on envelope protein binding to target cell heparan sulfate. Nat Med. 1997;3:866–871. doi: 10.1038/nm0897-866. [DOI] [PubMed] [Google Scholar]
- 33.Rostand KS, Esko JD. Microbial adherence to and invasion through proteoglycans. Infect Immun. 1997;65:1–8. doi: 10.1128/iai.65.1.1-8.1997. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 34.Cagno V, Tseligka ED, Jones ST, Tapparel C. Heparan sulfate proteoglycans and viral attachment: true receptors or adaptation bias? Viruses. 2019;11:596. doi: 10.3390/v11070596. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 35.Chen Y, Götte M, Liu J, Park PW. Microbial subversion of heparan sulfate proteoglycans. Mol Cells. 2008;26:415–426. [PubMed] [Google Scholar]
- 36.Jackson RL, Busch SJ, Cardin AD. Glycosaminoglycans: molecular properties, protein interactions, and role in physiological processes. Physiol Rev. 1991;71:481–539. doi: 10.1152/physrev.1991.71.2.481. [DOI] [PubMed] [Google Scholar]
- 37.Sarrazin S, Lamanna WC, Esko JD. Heparan sulfate proteoglycans. Cold Spring Harb Perspect Biol. 2011;3:a004952. doi: 10.1101/cshperspect.a004952. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 38.Lander AD, Selleck SB. The elusive functions of proteoglycans: in vivo veritas. J Cell Biol. 2000;148:227–232. doi: 10.1083/jcb.148.2.227. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 39.Bernfield M, Kokenyesi R, Kato M, Hinkes MT, Spring J, et al. Biology of the syndecans: a family of transmembrane heparan sulfate proteoglycans. Annu Rev Cell Biol. 1992;8:365–393. doi: 10.1146/annurev.cb.08.110192.002053. [DOI] [PubMed] [Google Scholar]
- 40.Cole GJ, Halfter W. Agrin: an extracellular matrix heparan sulfate proteoglycan involved in cell interactions and synaptogenesis. Perspect Dev Neurobiol. 1996;3:359–371. [PubMed] [Google Scholar]
- 41.Templeton DM. Proteoglycans in cell regulation. Crit Rev Clin Lab Sci. 1992;29:141–184. doi: 10.3109/10408369209114599. [DOI] [PubMed] [Google Scholar]
- 42.Park PW, Reizes O, Bernfield M. Cell surface heparan sulfate proteoglycans: selective regulators of ligand-receptor encounters. J Biol Chem. 2000;275:29923–29926. doi: 10.1074/jbc.R000008200. [DOI] [PubMed] [Google Scholar]
- 43.Kim SY, Li B, Linhardt RJ. Pathogenesis and inhibition of flaviviruses from a carbohydrate perspective. Pharmaceuticals. 2017;10:E44. doi: 10.3390/ph10020044. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 44.Spear PG. Herpes simplex virus: receptors and ligands for cell entry. Cell Microbiol. 2004;6:401–410. doi: 10.1111/j.1462-5822.2004.00389.x. [DOI] [PubMed] [Google Scholar]
- 45.Hileman RE, Fromm JR, Weiler JM, Linhardt RJ. Glycosaminoglycan–protein interactions: definition of consensus sites in glycosaminoglycan binding proteins. Bioessays. 1998;20:156–167. doi: 10.1002/(SICI)1521-1878(199802)20:2<156::AID-BIES8>3.0.CO;2-R. [DOI] [PubMed] [Google Scholar]
- 46.Cardin AD, Weintraub HJ. Molecular modeling of protein–glycosaminoglycan interactions. Arteriosclerosis. 1989;9:21–32. doi: 10.1161/01.atv.9.1.21. [DOI] [PubMed] [Google Scholar]
- 47.Margalit H, Fischer N, Ben-Sasson SA. Comparative analysis of structurally defined heparin binding sequences reveals a distinct spatial distribution of basic residues. J Biol Chem. 1993;268:19228–19231. doi: 10.1016/S0021-9258(19)36503-2. [DOI] [PubMed] [Google Scholar]
- 48.Neff S, Sá-Carvalho D, Rieder E, Mason PW, Blystone SD, et al. Foot-and-mouth disease virus virulent for cattle utilizes the integrin alphavbeta3 as its receptor. J Virol. 1998;72:3587–3594. doi: 10.1128/JVI.72.5.3587-3594.1998. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 49.Gardner CL, Choi-Nurvitadhi J, Sun C, Bayer A, Hritz J, et al. Natural variation in the heparan sulfate binding domain of the eastern equine encephalitis virus E2 glycoprotein alters interactions with cell surfaces and virulence in mice. J Virol. 2013;87:8582–8590. doi: 10.1128/JVI.00937-13. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 50.Kjellén L, Lindahl U. Specificity of glycosaminoglycan–protein interactions. Curr Opin Struct Biol. 2018;50:101–108. doi: 10.1016/j.sbi.2017.12.011. [DOI] [PubMed] [Google Scholar]
- 51.McKnight KL, Simpson DA, Lin SC, Knott TA, Polo JM, et al. Deduced consensus sequence of Sindbis virus strain AR339: mutations contained in laboratory strains which affect cell culture and in vivo phenotypes. J Virol. 1996;70:1981–1989. doi: 10.1128/JVI.70.3.1981-1989.1996. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 52.Klimstra WB, Ryman KD, Johnston RE. Adaptation of Sindbis virus to BHK cells selects for use of heparan sulfate as an attachment receptor. J Virol. 1998;72:7357–7366. doi: 10.1128/JVI.72.9.7357-7366.1998. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 53.Smit JM, Waarts BL, Kimata K, Klimstra WB, Bittman R, et al. Adaptation of alphaviruses to heparan sulfate: interaction of Sindbis and Semliki forest viruses with liposomes containing lipid-conjugated heparin. J Virol. 2002;76:10128–10137. doi: 10.1128/jvi.76.20.10128-10137.2002. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 54.Bernard KA, Klimstra WB, Johnston RE. Mutations in the E2 glycoprotein of Venezuelan equine encephalitis virus confer heparan sulfate interaction, low morbidity, and rapid clearance from blood of mice. Virology. 2000;276:93–103. doi: 10.1006/viro.2000.0546. [DOI] [PubMed] [Google Scholar]
- 55.Lee E, Lobigs M. E protein domain III determinants of yellow fever virus 17D vaccine strain enhance binding to glycosaminoglycans, impede virus spread, and attenuate virulence. J Virol. 2008;82:6024–6033. doi: 10.1128/JVI.02509-07. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 56.Prestwood TR, Prigozhin DM, Sharar KL, Zellweger RM, Shresta S. A mouse-passaged dengue virus strain with reduced affinity for heparan sulfate causes severe disease in mice by establishing increased systemic viral loads. J Virol. 2008;82:8411–8421. doi: 10.1128/JVI.00611-08. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 57.Heil ML, Albee A, Strauss JH, Kuhn RJ. An amino acid substitution in the coding region of the E2 glycoprotein adapts Ross River virus to utilize heparan sulfate as an attachment moiety. J Virol. 2001;75:6303–6309. doi: 10.1128/JVI.75.14.6303-6309.2001. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 58.Byrnes AP, Griffin DE. Binding of Sindbis virus to cell surface heparan sulfate. J Virol. 1998;72:7349–7356. doi: 10.1128/JVI.72.9.7349-7356.1998. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 59.Lee E, Lobigs M. Substitutions at the putative receptor-binding site of an encephalitic flavivirus alter virulence and host cell tropism and reveal a role for glycosaminoglycans in entry. J Virol. 2000;74:8867–8875. doi: 10.1128/jvi.74.19.8867-8875.2000. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 60.Silva LA, Khomandiak S, Ashbrook AW, Weller R, Heise MT, et al. A single-amino-acid polymorphism in Chikungunya virus E2 glycoprotein influences glycosaminoglycan utilization. J Virol. 2014;88:2385–2397. doi: 10.1128/JVI.03116-13. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 61.McAllister N, Liu Y, Silva LM, Lentscher AJ, Chai W, et al. Chikungunya virus strains from each genetic clade bind sulfated glycosaminoglycans as attachment factors. J Virol. 2020;94:24. doi: 10.1128/JVI.01500-20. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 62.Gardner CL, Ebel GD, Ryman KD, Klimstra WB. Heparan sulfate binding by natural eastern equine encephalitis viruses promotes neurovirulence. Proc Natl Acad Sci U S A. 2011;108:16026–16031. doi: 10.1073/pnas.1110617108. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 63.Germi R, Crance J-M, Garin D, Guimet J, Lortat-Jacob H, et al. Heparan sulfate-mediated binding of infectious dengue virus type 2 and yellow fever virus. Virology. 2002;292:162–168. doi: 10.1006/viro.2001.1232. [DOI] [PubMed] [Google Scholar]
- 64.Hilgard P, Stockert R. Heparan sulfate proteoglycans initiate dengue virus infection of hepatocytes. Hepatology. 2000;32:1069–1077. doi: 10.1053/jhep.2000.18713. [DOI] [PubMed] [Google Scholar]
- 65.Lee E, Pavy M, Young N, Freeman C, Lobigs M. Antiviral effect of the heparan sulfate mimetic, PI-88, against dengue and encephalitic flaviviruses. Antiviral Res. 2006;69:31–38. doi: 10.1016/j.antiviral.2005.08.006. [DOI] [PubMed] [Google Scholar]
- 66.Artpradit C, Robinson LN, Gavrilov BK, Rurak TT, Ruchirawat M, et al. Recognition of heparan sulfate by clinical strains of dengue virus serotype 1 using recombinant subviral particles. Virus Res. 2013;176:69–77. doi: 10.1016/j.virusres.2013.04.017. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 67.Lee E, Wright PJ, Davidson A, Lobigs M. Virulence attenuation of dengue virus due to augmented glycosaminoglycan-binding affinity and restriction in extraneural dissemination. J Gen Virol. 2006;87:2791–2801. doi: 10.1099/vir.0.82164-0. [DOI] [PubMed] [Google Scholar]
- 68.Kroschewski H, Allison SL, Heinz FX, Mandl CW. Role of heparan sulfate for attachment and entry of tick-borne encephalitis virus. Virology. 2003;308:92–100. doi: 10.1016/s0042-6822(02)00097-1. [DOI] [PubMed] [Google Scholar]
- 69.Chen H-L, Her S-Y, Huang K-C, Cheng H-T, Wu C-W, et al. Identification of a heparin binding peptide from the Japanese encephalitis virus envelope protein. Biopolymers. 2010;94:331–338. doi: 10.1002/bip.21371. [DOI] [PubMed] [Google Scholar]
- 70.Kim SY, Zhao J, Liu X, Fraser K, Lin L, et al. Interaction of zika virus envelope protein with glycosaminoglycans. Biochemistry. 2017;56:1151–1162. doi: 10.1021/acs.biochem.6b01056. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 71.Lindenbach B, Randall G, Bartenschlager R. In: Fields Virology Emerging Viruses. 7 ed. Howley P, Knipe DM, editors. Philadelphia, PA: Wolters Kluwer; 2020. CM Rice Flaviviridae: the viruses and their replication; p. 246. edn. p. [Google Scholar]
- 72.Lee E, Lobigs M. Mechanism of virulence attenuation of glycosaminoglycan-binding variants of Japanese encephalitis virus and Murray Valley encephalitis virus. J Virol. 2002;76:4901–4911. doi: 10.1128/jvi.76.10.4901-4911.2002. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 73.Lee E, Hall RA, Lobigs M. Common E protein determinants for attenuation of glycosaminoglycan-binding variants of Japanese encephalitis and West Nile viruses. J Virol. 2004;78:8271–8280. doi: 10.1128/JVI.78.15.8271-8280.2004. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 74.de Boer SM, Kortekaas J, de Haan CAM, Rottier PJM, Moormann RJM, et al. Heparan sulfate facilitates Rift Valley fever virus entry into the cell. J Virol. 2012;86:13767–13771. doi: 10.1128/JVI.01364-12. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 75.Gardner CL, Hritz J, Sun C, Vanlandingham DL, Song TY, et al. Deliberate attenuation of chikungunya virus by adaptation to heparan sulfate-dependent infectivity: a model for rational arboviral vaccine design. PLoS Negl Trop Dis. 2014;8:e2719. doi: 10.1371/journal.pntd.0002719. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 76.Gardner CL, Burke CW, Higgs ST, Klimstra WB, Ryman KD. Interferon-alpha/beta deficiency greatly exacerbates arthritogenic disease in mice infected with wild-type chikungunya virus but not with the cell culture-adapted live-attenuated 181/25 vaccine candidate. Virology. 2012;425:103–112. doi: 10.1016/j.virol.2011.12.020. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 77.Gorchakov R, Wang E, Leal G, Forrester NL, Plante K, et al. Attenuation of Chikungunya virus vaccine strain 181/clone 25 is determined by two amino acid substitutions in the E2 envelope glycoprotein. J Virol. 2012;86:6084–6096. doi: 10.1128/JVI.06449-11. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 78.Liu H, Chiou SS, Chen WJ. Differential binding efficiency between the envelope protein of Japanese encephalitis virus variants and heparan sulfate on the cell surface. J Med Virol. 2004;72:618–624. doi: 10.1002/jmv.20025. [DOI] [PubMed] [Google Scholar]
- 79.Esko JD, Stanley P, et al. In: Essentials of Glycobiology. 2nd ed. Varki A, Cummings RD, Esko JD, Freeze HH, Stanley P, editors. Cold Spring Harbor (NY); 2009. Glycosylation Mutants of Cultured Cells. [PubMed] [Google Scholar]
- 80.Banfield BW, Leduc Y, Esford L, Visalli RJ, Brandt CR, et al. Evidence for an interaction of herpes simplex virus with chondroitin sulfate proteoglycans during infection. Virology. 1995;208:531–539. doi: 10.1006/viro.1995.1184. [DOI] [PubMed] [Google Scholar]
- 81.Ryman KD, Gardner CL, Burke CW, Meier KC, Thompson JM, et al. Heparan sulfate binding can contribute to the neurovirulence of neuroadapted and nonneuroadapted Sindbis viruses. J Virol. 2007;81:3563–3573. doi: 10.1128/JVI.02494-06. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 82.Desai UR, Wang HM, Linhardt RJ. Specificity studies on the heparin lyases from Flavobacterium heparinum . Biochemistry. 1993;32:8140–8145. doi: 10.1021/bi00083a012. [DOI] [PubMed] [Google Scholar]
- 83.Zhao Z, Date T, Li Y, Kato T, Miyamoto M, et al. Characterization of the E-138 (Glu/Lys) mutation in Japanese encephalitis virus by using a stable, full-length, infectious cDNA clone. J Gen Virol. 2005;86:2209–2220. doi: 10.1099/vir.0.80638-0. [DOI] [PubMed] [Google Scholar]
- 84.Kuhn RJ. In: Fields Virology: Emerging Viruses. 7th ed. Howley PM, Knipe DM, Whelan S, editors. Philadelphia, PA: Wolters Kluwer; 2020. Togaviridae: The Viruses and Their Replication; p. 194. p. [Google Scholar]
- 85.Mandl CW, Kroschewski H, Allison SL, Kofler R, Holzmann H, et al. Adaptation of tick-borne encephalitis virus to BHK-21 cells results in the formation of multiple heparan sulfate binding sites in the envelope protein and attenuation in vivo. J Virol. 2001;75:5627–5637. doi: 10.1128/JVI.75.12.5627-5637.2001. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 86.Gauczynski S, Peyrin JM, Haïk S, Leucht C, Hundt C, et al. The 37-kDa/67-kDa laminin receptor acts as the cell-surface receptor for the cellular prion protein. EMBO J. 2001;20:5863–5875. doi: 10.1093/emboj/20.21.5863. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 87.Tanaka A, Tumkosit U, Nakamura S, Motooka D, Kishishita N, et al. Genome-wide screening uncovers the significance of N-sulfation of heparan sulfate as a host cell factor for chikungunya virus Infection. J Virol. 2017;91:13. doi: 10.1128/JVI.00432-17. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 88.Riblett AM, Blomen VA, Jae LT, Altamura LA, Doms RW, et al. A haploid genetic screen identifies heparan sulfate proteoglycans supporting Rift Valley fever virus infection. J Virol. 2016;90:1414–1423. doi: 10.1128/JVI.02055-15. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 89.Jan JT, Byrnes AP, Griffin DE. Characterization of a Chinese hamster ovary cell line developed by retroviral insertional mutagenesis that is resistant to Sindbis virus infection. J Virol. 1999;73:4919–4924. doi: 10.1128/JVI.73.6.4919-4924.1999. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 90.Savidis G, McDougall WM, Meraner P, Perreira JM, Portmann JM, et al. Identification of Zika virus and dengue virus dependency factors using functional genomics. Cell Rep. 2016;16:232–246. doi: 10.1016/j.celrep.2016.06.028. [DOI] [PubMed] [Google Scholar]
- 91.Gao H, Lin Y, He J, Zhou S, Liang M, et al. Role of heparan sulfate in the Zika virus entry, replication, and cell death. Virology. 2019;529:91–100. doi: 10.1016/j.virol.2019.01.019. [DOI] [PubMed] [Google Scholar]
- 92.Tan CW, Sam IC, Chong WL, Lee VS, Chan YF. Polysulfonate suramin inhibits Zika virus infection. Antiviral Res. 2017;143:186–194. doi: 10.1016/j.antiviral.2017.04.017. [DOI] [PubMed] [Google Scholar]
- 93.Nickells J, Cannella M, Droll DA, Liang Y, Wold WSM, et al. Neuroadapted yellow fever virus strain 17D: a charged locus in domain III of the E protein governs heparin binding activity and neuroinvasiveness in the SCID mouse model. J Virol. 2008;82:12510–12519. doi: 10.1128/JVI.00458-08. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 94.Wang X, Li S-H, Zhu L, Nian Q-G, Yuan S, et al. Near-atomic structure of Japanese encephalitis virus reveals critical determinants of virulence and stability. Nat Commun. 2017;8:14. doi: 10.1038/s41467-017-00024-6. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 95.Kozlovskaya LI, Osolodkin DI, Shevtsova AS, Romanova LI, Rogova YV, et al. GAG-binding variants of tick-borne encephalitis virus. Virology. 2010;398:262–272. doi: 10.1016/j.virol.2009.12.012. [DOI] [PubMed] [Google Scholar]
- 96.Westlake D, Bielefeldt-Ohmann H, Prow NAA, Hall RAA. Novel flavivirus attenuation markers identified in the envelope protein of Alfuy virus. Viruses. 2021;13:147. doi: 10.3390/v13020147. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 97.Raaben M, Jae LT, Herbert AS, Kuehne AI, Stubbs SH, et al. NRP2 and CD63 are host factors for Lujo virus cell entry. Cell Host Microbe. 2017;22:688–696. doi: 10.1016/j.chom.2017.10.002. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 98.Lamers MM, Mykytyn AZ, Breugem TI, Wang Y, Wu DC, et al. Human airway cells prevent SARS-CoV-2 multibasic cleavage site cell culture adaptation. Elife. 2021;10:e66815. doi: 10.7554/eLife.66815. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 99.Klimstra WB, Tilston-Lunel NL, Nambulli S, Boslett J, McMillen CM, et al. SARS-CoV-2 growth, furin-cleavage-site adaptation and neutralization using serum from acutely infected hospitalized COVID-19 patients. J Gen Virol. 2020;101:1156–1169. doi: 10.1099/jgv.0.001481. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 100.Byrnes AP, Griffin DE. Large-plaque mutants of Sindbis virus show reduced binding to heparan sulfate, heightened viremia, and slower clearance from the circulation. J Virol. 2000;74:644–651. doi: 10.1128/jvi.74.2.644-651.2000. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 101.Klimstra WB, Ryman KD, Bernard KA, Nguyen KB, Biron CA, et al. Infection of neonatal mice with sindbis virus results in a systemic inflammatory response syndrome. J Virol. 1999;73:10387–10398. doi: 10.1128/JVI.73.12.10387-10398.1999. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 102.Kobayashi K, Sudaka Y, Takashino A, Imura A, Fujii K, et al. Amino acid variation at VP1-145 of enterovirus 71 determines attachment receptor usage and neurovirulence in human scavenger receptor B2 transgenic mice. J Virol. 2018;92:15. doi: 10.1128/JVI.00681-18. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 103.Griffin DE, Levine B, Tyor WR, Tucker PC, Hardwick JM. Age-dependent susceptibility to fatal encephalitis: alphavirus infection of neurons. Arch Virol Suppl. 1994;9:31–39. doi: 10.1007/978-3-7091-9326-6_4. [DOI] [PubMed] [Google Scholar]
- 104.Knight RL, Schultz KLW, Kent RJ, Venkatesan M, Griffin DE. Role of N-linked glycosylation for sindbis virus infection and replication in vertebrate and invertebrate systems. J Virol. 2009;83:5640–5647. doi: 10.1128/JVI.02427-08. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 105.Trobaugh DW, Gardner CL, Sun C, Haddow AD, Wang E, et al. RNA viruses can hijack vertebrate microRNAs to suppress innate immunity. Nature. 2014;506:245–248. doi: 10.1038/nature12869. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 106.Chen C-L, Hasan SS, Klose T, Sun Y, Buda G, et al. Cryo-EM structure of eastern equine encephalitis virus in complex with heparan sulfate analogues. Proc Natl Acad Sci U S A. 2020;117:8890–8899. doi: 10.1073/pnas.1910670117. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 107.Reddi HV, Kumar ASM, Kung AY, Kallio PD, Schlitt BP, et al. Heparan sulfate-independent infection attenuates high-neurovirulence GDVII virus-induced encephalitis. J Virol. 2004;78:8909–8916. doi: 10.1128/JVI.78.16.8909-8916.2004. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 108.Hulst MM, van Gennip HG, Vlot AC, Schooten E, de Smit AJ, et al. Interaction of classical swine fever virus with membrane-associated heparan sulfate: role for virus replication in vivo and virulence. J Virol. 2001;75:9585–9595. doi: 10.1128/JVI.75.20.9585-9595.2001. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 109.Lipton HL, Kumar ASM, Hertzler S, Reddi HV. Differential usage of carbohydrate co-receptors influences cellular tropism of Theiler’s murine encephalomyelitis virus infection of the central nervous system. Glycoconj J. 2006;23:39–49. doi: 10.1007/s10719-006-5436-x. [DOI] [PubMed] [Google Scholar]
- 110.Ferguson MC, Saul S, Fragkoudis R, Weisheit S, Cox J, et al. Ability of the encephalitic arbovirus Semliki Forest virus to cross the blood–brain barrier is determined by the charge of the E2 glycoprotein. J Virol. 2015;89:7536–7549. doi: 10.1128/JVI.03645-14. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 111.Zhu W, Li J, Liang G. How does cellular heparan sulfate function in viral pathogenicity? Biomed Environ Sci. 2011;24:81–87. doi: 10.3967/0895-3988.2011.01.011. [DOI] [PubMed] [Google Scholar]
- 112.Weaver SC, Forrester NL, Liu J, Vasilakis N. Population bottlenecks and founder effects: implications for mosquito-borne arboviral emergence. Nat Rev Microbiol. 2021;19:184–195. doi: 10.1038/s41579-020-00482-8. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 113.Franz AWE, Kantor AM, Passarelli AL, Clem RJ. Tissue barriers to arbovirus infection in mosquitoes. Viruses. 2015;7:3741–3767. doi: 10.3390/v7072795. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 114.Mao Q, Wu W, Liao Z, Li J, Jia D, et al. Viral pathogens hitchhike with insect sperm for paternal transmission. Nat Commun. 2019;10:955. doi: 10.1038/s41467-019-08860-4. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 115.Ciano KA, Saredy JJ, Bowers DF. Heparan sulfate proteoglycan: an arbovirus attachment factor integral to mosquito salivary gland ducts. Viruses. 2014;6:5182–5197. doi: 10.3390/v6125182. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 116.Armistead JS, Wilson IBH, van Kuppevelt TH, Dinglasan RR. A role for heparan sulfate proteoglycans in Plasmodium falciparum sporozoite invasion of anopheline mosquito salivary glands. Biochem J. 2011;438:475–483. doi: 10.1042/BJ20110694. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 117.Dinglasan RR, Alaganan A, Ghosh AK, Saito A, van Kuppevelt TH, et al. Plasmodium falciparum ookinetes require mosquito midgut chondroitin sulfate proteoglycans for cell invasion. Proc Natl Acad Sci U S A. 2007;104:15882–15887. doi: 10.1073/pnas.0706340104. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 118.Sinnis P, Coppi A, Toida T, Toyoda H, Kinoshita-Toyoda A, et al. Mosquito heparan sulfate and its potential role in malaria infection and transmission. J Biol Chem. 2007;282:25376–25384. doi: 10.1074/jbc.M704698200. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 119.Shao L, Devenport M, Jacobs-Lorena M. The peritrophic matrix of hematophagous insects. Arch Insect Biochem Physiol. 2001;47:119–125. doi: 10.1002/arch.1042. [DOI] [PubMed] [Google Scholar]
- 120.Spring J, Paine-Saunders SE, Hynes RO, Bernfield M. Drosophila syndecan: conservation of a cell-surface heparan sulfate proteoglycan. Proc Natl Acad Sci U S A. 1994;91:3334–3338. doi: 10.1073/pnas.91.8.3334. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 121.Kim SY, Koetzner CA, Payne AF, Nierode GJ, Yu Y, et al. Glycosaminoglycan compositional analysis of relevant tissues in Zika virus pathogenesis and in vitro evaluation of heparin as an antiviral against Zika virus infection. Biochemistry. 2019;58:1155–1166. doi: 10.1021/acs.biochem.8b01267. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 122.Conway MJ, Watson AM, Colpitts TM, Dragovic SM, Li Z, et al. Mosquito saliva serine protease enhances dissemination of dengue virus into the mammalian host. J Virol. 2014;88:164–175. doi: 10.1128/JVI.02235-13. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 123.Acharya D, Paul AM, Anderson JF, Huang F, Bai F. Loss of glycosaminoglycan receptor binding after mosquito cell passage reduces Chikungunya virus infectivity. PLoS Negl Trop Dis. 2015;9:e0004139. doi: 10.1371/journal.pntd.0004139. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 124.Coffey LL, Vasilakis N, Brault AC, Powers AM, Tripet F, et al. Arbovirus evolution in vivo is constrained by host alternation. Proc Natl Acad Sci U S A. 2008;105:6970–6975. doi: 10.1073/pnas.0712130105. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 125.Lawrence R, Yabe T, Hajmohammadi S, Rhodes J, McNeely M, et al. The principal neuronal gD-type 3-O-sulfotransferases and their products in central and peripheral nervous system tissues. Matrix Biol. 2007;26:442–455. doi: 10.1016/j.matbio.2007.03.002. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 126.Tiwari V, Tarbutton MS, Shukla D. Diversity of heparan sulfate and HSV entry: basic understanding and treatment strategies. Molecules. 2015;20:2707–2727. doi: 10.3390/molecules20022707. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 127.Shukla D, Liu J, Blaiklock P, Shworak NW, Bai X, et al. A novel role for 3-O-sulfated heparan sulfate in herpes simplex virus 1 entry. Cell. 1999;99:13–22. doi: 10.1016/s0092-8674(00)80058-6. [DOI] [PubMed] [Google Scholar]
- 128.Basore K, Kim AS, Nelson CA, Zhang R, Smith BK, et al. Cryo-EM structure of chikungunya virus in complex with the Mxra8 receptor. Cell. 2019;177:1725–1737. doi: 10.1016/j.cell.2019.04.006. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 129.Basore K, Ma H, Kafai NM, Mackin S, Kim AS, et al. Structure of Venezuelan equine encephalitis virus in complex with the LDLRAD3 receptor. Nature. 2021;598:672–676. doi: 10.1038/s41586-021-03963-9. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 130.Andrade AS, Ferreira RS, Guedes MIMC, Dias J, Pinheiro MA, et al. Dengue virus 3 genotype I shows natural changes in heparan sulphate binding sites, cell interactions, and neurovirulence in a mouse model. J Gen Virol. 2021;102 doi: 10.1099/jgv.0.001630. [DOI] [PubMed] [Google Scholar]
- 131.Su CM, Liao CL, Lee YL, Lin YL. Highly sulfated forms of heparin sulfate are involved in Japanese encephalitis virus infection. Virology. 2001;286:206–215. doi: 10.1006/viro.2001.0986. [DOI] [PubMed] [Google Scholar]
- 132.Pietrantoni A, Fortuna C, Remoli ME, Ciufolini MG, Superti F. Bovine lactoferrin inhibits Toscana virus infection by binding to heparan sulphate. Viruses. 2015;7:480–495. doi: 10.3390/v7020480. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 133.Murakami S, Takenaka-Uema A, Kobayashi T, Kato K, Shimojima M, et al. Heparan sulfate proteoglycan Is an important attachment factor for cell entry of Akabane and Schmallenberg viruses. J Virol. 2017;91:15. doi: 10.1128/JVI.00503-17. [DOI] [PMC free article] [PubMed] [Google Scholar]