

Abstract
The Golgi-associated retrograde protein (GARP) complex is proposed to tether endosome-derived transport vesicles, but the exact function and mechanism of GARP action are not completely understood. To uncover the GARP function in human cells, we employ CRISPR/Cas9 strategy and knock out (KO) the unique VPS54 subunit of the GARP complex. In this chapter, we describe the detailed method of generating CRISPR/Cas9-mediated VPS54-KO in hTERT-RPE1 cells, rescue of resulting KO cells with stable near-endogenous expression of myc-tagged VPS54, and validation of KO and rescued (KO-R) cells using Western blot and immunofluorescence approaches. This approach is helpful in uncovering new functions of the GARP and other vesicle tethering complexes.
Keywords: Golgi complex, GARP, VPS54, Knockout, Knockout rescue, Vesicle tethering, CRISPR/Cas9, Cathepsin D
1. Introduction
Vesicle membrane tethers play a crucial role in intracellular vesicular trafficking [1]. These proteins are classified as either coiled-coil or multisubunit tethers [2, 3]. GARP complex is a multisubunit vesicle tethering complex localized in the trans-Golgi network (TGN) [4]. It is believed to tether vesicles arriving from endosomes to the TGN and takes part in Golgi retrieval of mannose-6-phosphate receptors (MPRs) and the TGN-resident protein TGN46 [2, 5]. GARP is also involved in anterograde transport of GPI-anchored, transmembrane proteins, and sphingolipid homeostasis [1, 3, 4], but the exact function and mechanism of GARP action are not completely understood. It is composed of four different subunits: VPS51, VPS52, VPS53, and VPS54; three subunits (VPS51, VPS52, and VPS53) are shared with endosome-associated recycling protein (EARP) complex, and VPS54 is the only unique subunit of GARP complex [6]. Mutations in the VPS54 subunit of the GARP complex showed progressive death of the motor neurons in wobbler mouse, which is an animal model for amyotrophic lateral sclerosis [7]. Here, we detail the methods employed to study the function of VPS54 in telomerase-immortalized retinal pigment epithelial (hTERT- RPE1) cells [8]. hTERT-RPE1 cells are chosen for study because they can divide indefinitely, have superior microscopic characteristics, and are non-tumorigenic [9].
CRISPR/Cas9 (clustered regularly interspaced short palindromic repeats/CRISPR-associated protein 9) strategies provide some of the simplest, most versatile, and precise methods of genetic manipulation [10–13]. This system mainly consists of two key components: Guide RNA (gRNA) and Cas9 enzyme. The gRNA is approximately 20 base pairs sequence that is complementary to the target sequence of the genome [13]. Cas9 is the endonuclease that can cleave the DNA. The gRNA and Cas9 are combined into a ribonucleoprotein complex when they are used in CRISPR experiments. Once the CRISPR/Cas9 system is introduced into the cell, the gRNA identifies the target sequence of DNA to be edited. The Cas9 now acts as a molecular scissor and cuts across two strands of the target DNA in the genome, resulting in the double-strand break. Once the DNA is cut, the cells begin to repair the breaks in the DNA by non-homologous end joining (NHEJ). This process of repairing the double-strand break of DNA is error-prone as nucleotides can be inserted or deleted by mistake (also known as indels) [14, 15]. As a consequence, no functional protein is made, which will help us to study the role of a particular gene of interest.
To determine the specificity of KO and validate the absence of off-target effects, integration of genes of interest to rescue the KO should be performed. One of the useful ways to re-introduce the gene of interest is by using a lentiviral transduction system [16]. This transduction system has several advantages over other approaches for protein expression. For instance, lentiviruses can be used to stably integrate foreign DNA into the genome of the host, infect both dividing and non-dividing cells, do not generate immunogenic proteins, deliver transgene fragments as large as 8 kb, and are used in broad spectrum of cells and tissues [17–20]. In our study, we have used a third-generation lentivirus packaging system as it is significantly safer than first and second generations. For viral transduction, HEK293FT cells were transfected with destination plasmid encoding mVPS54-13myc under control of the COG4 promoter for expression near to the endogenous level [9] and viral packaging plasmids pMD2.G, pRSV-Rev, and pMDLg/pRRE.
Collected lentivirus was used to transduce the hTERT RPE1 VPS54-KO cells to rescue GARP-KO phenotype.
Here, we describe the methods for making hTERT-RPE1 VPS54-KO cells, validating the absence of VPS54 protein (Fig. 1) and rescuing VPS54-KO cells with re-expression of mVPS54-13myc (Figs. 2 and 3). This approach uncovered novel functions of GARP that were previously unknown. We have shown that hTERT-RPE1, HEK293T, and HeLa cells depleted for either VPS54 or VPS53 proteins, using the methodology described in this chapter, are deficient for the recycling/retention of several Golgi glycosyl-transferases resulting in severe glycosylation defects. Importantly, the re-introduction of VPS54 and VPS53 stabilized the Golgi enzymes and restored normal Golgi glycosylation [9].
Fig. 1.
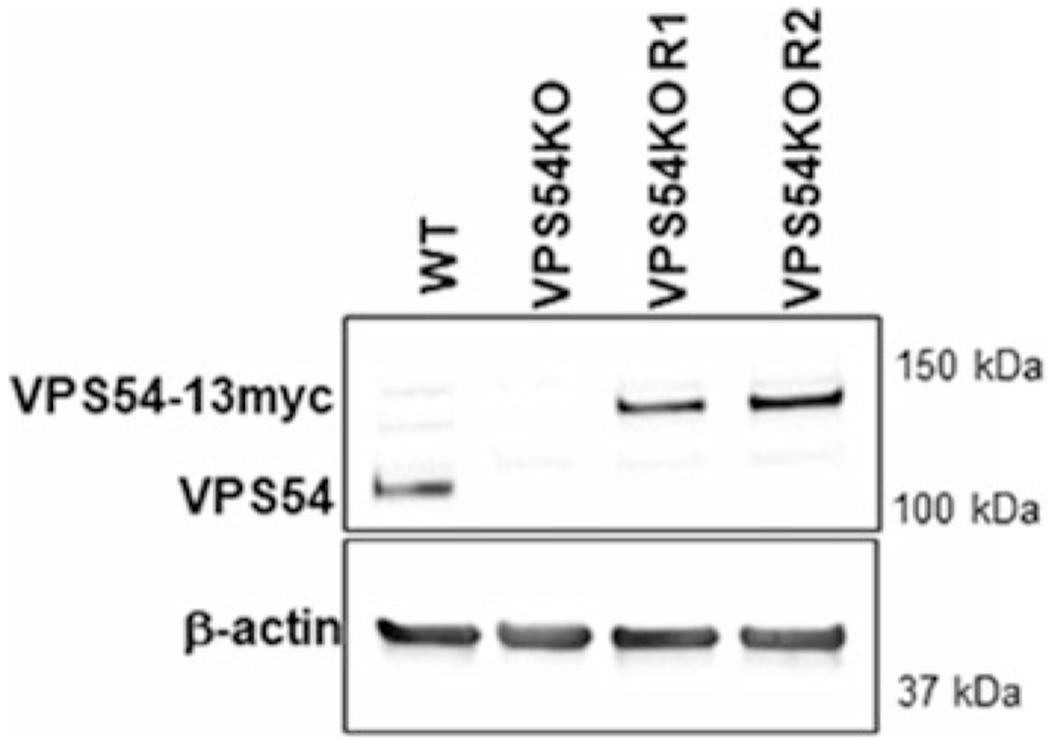
Creation of VPS54-KO and rescue of the VPS54-KO by mVPS54-13myc. WB of hTERT RPE1 cell lysates from wild type (WT), VPS54-KO, and two VPS54-KO clones rescued with mVPS54-13myc (R1 and R2) probed with anti-VPS54 antibodies. β-actin was used as a loading control. VPS54-13myc was expressed under the COG4 promoter and therefore the protein level of the expressed VPS54-13myc was similar to the endogenous VPS54 level
Fig. 2.
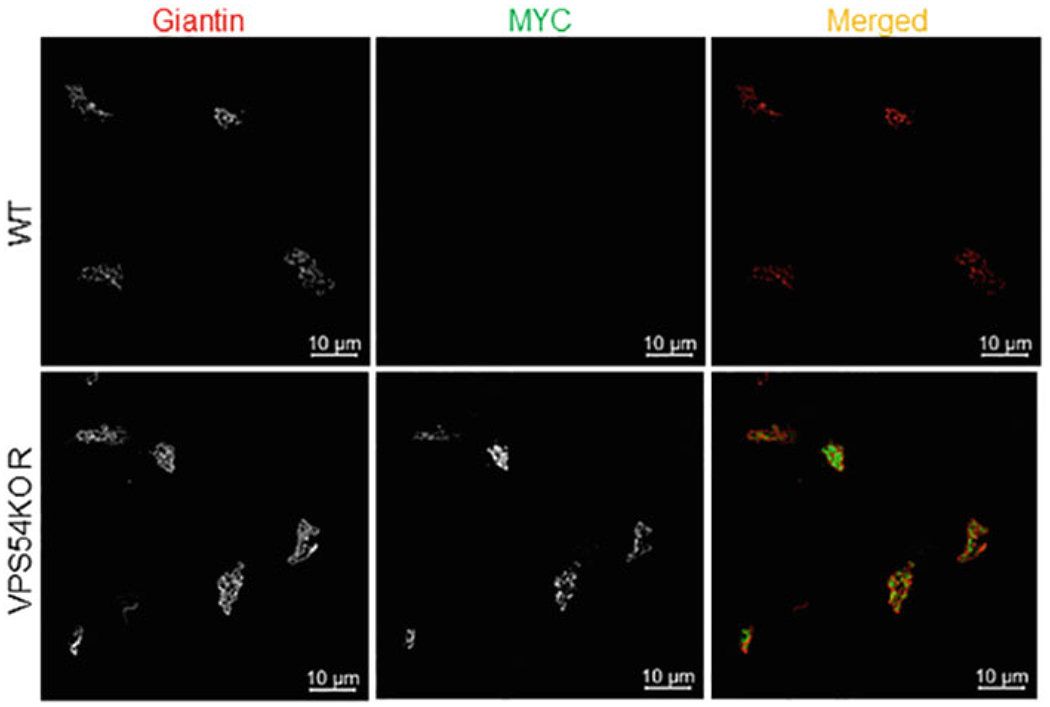
Validation of VPS54-KO and rescue cells by IF. hTERT RPE1 WT and VPS54-KO-R cells stably expressing mVPS54-13myc were plated in glass coverslips for 16 h. Expression and localization of mVPS54-13myc were confirmed by staining the cells with antibodies against myc (green) and the Golgi marker giantin (red). Images were taken using superresolution Airyscan microscopy
Fig. 3.
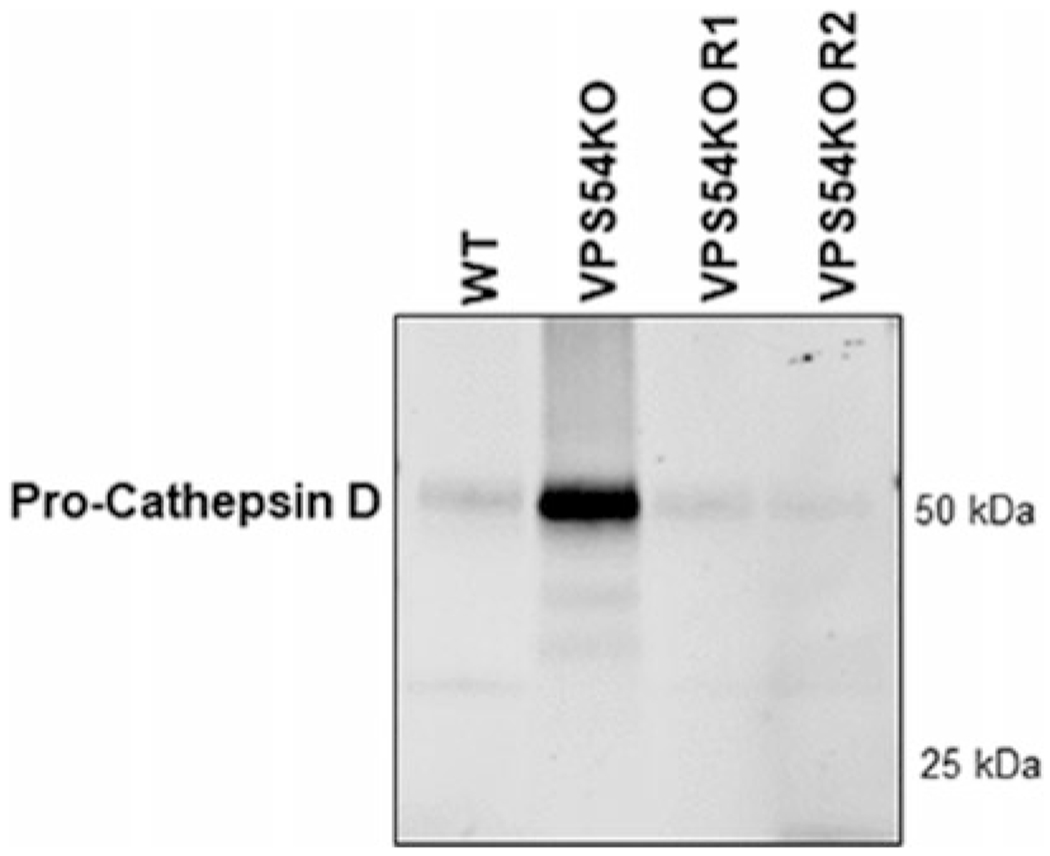
GARP-KO alters sorting of lysosomal protein cathepsin D. hTERT RPE1 WT, VPS54-KO, and VPS54-KO rescued (R1 and R2) cells were grown on three 6-cm dishes in DMEM/F12-FBS for 16h. Next, cells werer insed with PBS and placed in chemically defined medium for 36 h. Following the incubation for 36 h, the medium was collected, concentrated using 10 k concentrator, and samples were prepared for SDS-PAGE. WB analysis was done by incubating blots with anti-cathepsin D antibody
2. Materials
2.1. Cells and Media (See Note 1)
hTERT-RPE1-Cas9 stable cells [9].
HEK293FT cells (Thermo Fisher).
DMEM/F12-FBS: Dulbecco’s Modified Eagle Medium (DMEM) with F-12 50/50 (Corning) supplemented with 10% fetal bovine serum (FBS). Filtered using 0.22 μm polyethersulfone (PES) membrane filter.
Phosphate buffer saline (PBS), pH 7.4, sterile.
0.25% trypsin in PBS, sterile
Cell culture dishes: 6- and 12-well tissue culture plates and 6-cm and 10-cm dishes.
Automatic micropipette sets (P10, P20, P200, and P1000) with sterile Pipette tips.
Disposable sterile pipettes, 10 mL.
Tissue culture CO2 incubator.
Inverted fluorescence microscope with 5× objective.
Centrifuge with the rotor for 15 mL conical tubes (Centrifuge 5430/5430 R).
1.5 mL microcentrifuge tubes.
2.2. Neon Transfection System (Thermo Fisher, Catalog No. MPK10096) to Transfect hTERT-RPE1 Cells
Neon electronic pipette.
Neon electronic 10 μL pipette tips.
Neon electronic pipette tubes.
Neon pipette station.
Electrolytic (E) buffer.
Resuspension (R) buffer.
-
Guide RNA.
transEDIT-dual CRISPR for VPS54 (Transomic, TEDHG1001).
gRNA-a: CAAAAGATAATTCACTGGACACAGAGGTGG.
gRNA-b: CATTCTACCTCCCACAGATCAGCAAGGAAC
Cell sorting medium: PBS, 25 mM K+ HEPES, pH 7.0, 2% FBS (heat inactivated), 1 mM EDTA.
Fluorescence illumination system (EXFO X-Cite™ 120).
2.3. Lentivirus Production and Cell Transduction
pCl-neo-VPS54-13myc [6],
pLenti pCOG4 Neo DEST (705-1) [9],
pLenti VPS54-13myc [9],
pMD2.G (VSV-G envelope expressing plasmid) (a gift from Didier Trono (Addgene plasmid # 12259; http://n2t.net/addgene:12259; RRID: Addgene_12,259)) [21],
pRSV-Rev (Addgene #12253) [21],
pMDLg/pRRE (Addgene #12251) [21],
Reduced serum Opti-MEM.
Reduced serum Opti-MEM containing GlutaMAX (1×).
Reduced serum Opti-MEM containing GlutaMAX (1×) and chloroquine (25 mM).
Sodium butyrate:1 mM sodium butyrate in PBS.
Lipofectamine transfection reagent 3000; P3000.
G418 (Geneticin) stock solution: sterile-filtered 5% G418 sulfate in water.
Polybrene:10 μg/mL in PBS.
0.45 μm polyethersulfone (PES) membrane filter.
Lenti-X concentrator (PT4421-2).
Lenti-X p24 Rapid Titer Kit (Takara, # Cat. No. 632200).
2.4. Validation of VPS54-KO and VPS54-KO-R Cells
2.4.1. Western Blot
SDS: 2% sodium dodecyl sulfate in water.
6× SB (sample buffer): 30% glycerol, 12% SDS, 0.060% bromophenol blue, 4.8% β-mercaptoethanol, 30.3% Tris-HCl pH 6.8
Ponceau S stain: 0.1% Ponceau S in 5% acetic acid.
PBST: PBS with 0.05% Tween 20.
Primary antibody: anti-VPS54 (rabbit) (St John’s Lab, STJ115181).
Secondary antibody: Alexa Fluor 647 AffiniPure donkey anti-rabbit IgG (Jackson Immuno Research/711-605-152).
Intercept (PBS) blocking buffer (LI-COR).
SDS-PAGE Running buffer: 10X Tris/Glycine Buffer (BIO-RAD #1610771).
Cell scrapers.
Sonicator.
Bicinchoninic acid assay kit.
CLARIOstar Plus Microplate Reader.
70 °C heat block
Electrophoresis chamber (SDS-PAGE).
Bio-Rad gradient gel (4–15%).
Filter paper.
Nitrocellulose membrane 0.2 μm.
Pierce G2 Fast Blotter.
Membrane incubation box.
Plastic forceps.
Odyssey imaging system (Licor).
2.4.2. Immunofluorescence (IF) Microscopy
Fixative: 4% paraformaldehyde (PFA) in PBS (see Note 2).
Triton X-100 solution: 0.1% Triton X-100 in PBS.
50 mM ammonium chloride
IF Blocking buffer: 1% bovine serum albumin (BSA), 0.1% saponin in PBS.
IF Antibody buffer: 1% cold fish gelatin, 0.1% saponin in PBS.
Primary antibody: anti-Giantin (rabbit polyclonal; Covance PRB-114C), anti-Myc (mouse monoclonal antibody; Cell signaling #2276).
Secondary antibody: Donkey Anti-Rabbit Cy3 (Jackson Immuno Research/711-165-152) Alexa Fluor 555 and Donkey Anti-Mouse Alexa Fluor 488 (Jackson Immuno Research/711-605-152).
Hoechst 33342 (10 mg/mL in deionized water).
Prolong Gold Antifade reagent (Life Technologies) (Cat no. P36934).
Clear nail polish.
Fine-end metal forceps.
Round glass coverslips (12 mm diameter).
Glass slides.
LSM880 Zeiss Laser inverted microscope with Airyscan and 63× oil 1.4 NA objective using ZEN software.
2.4.3. Cathepsin D Secretion Assay
Serum-free chemically defined medium (BioWhittaker Pro293a-CDM, Lonza).
1× GlutaMAX (100× stock, Life Technologies)
10 k concentrator (Amicon Ultra 10 k, Millipore).
3. Methods
3.1. Knocking out VPS54 in hTERT-RPE-1 Cells
Plate 1 × 106 hTERT-RPE1-Cas9 cells on a 10-cm cell culture dish in DMEM/F12-FBS (without any antibiotic or antimycotics) (see Note 3).
After 2 days the cells will reach 100% confluency. Then, wash the cells twice with PBS.
Quickly after removing the PBS, add 700 μL of 0.25% trypsin and incubate for 3 min in a 37° C incubator (see Note 4).
Add 5 mL of DMEM/F12-FBS to neutralize the trypsin, resuspend cells with 10 mL pipette and transfer to 15 mL conical tube.
Count the cells using Beckman Coulter Vi-Cell XR cell counter to determine the number of cells in 1 mL of cell suspension (see Note 5).
Transfer 1 × 106 cells to a new 15 mL conical tube, pellet at 400 × g for 10 min and wash with PBS.
For a 6-well cell culture plate, resuspend the cells in 50 μL of resuspension (R) buffer.
Prepare high-quality TransEDIT-dual CRISPR plasmid (described in Subheading 2.2, item 7) and dilute it to 1 μg/μL in deionized water.
Preparation of transfection reaction: Add the resuspended cells to the plasmid DNA in the 1.5 mL microcentrifuge tube (3 μg plasmid DNA used per well of 6-well plate) and gently mix (see Note 6).
Turn on neon transfection system and set the transfection parameters for hTERT-RPE1 cell: 1050 V, 30 ms width, 2 pulses (see Note 7).
Add 3 mL of electrolytic (E) buffer in the neon tube. Insert the neon tube in the neon pipette station. There should be a click sound.
In the database, select the correct protocol (1050 V, width 30 ms, 2 pulses) and press “Start.” Use 10 μL neon pipette to pipette up the cells and DNA and gently insert it to the neon pipette station. Once the electroporation is completed, the touch screen will display “Complete.” You are now ready to take out the neon pipette from the neon pipette station. Immediately transfer the samples from the neon tip to a 6-well plate containing 2 mL of DMEM/F12-FBS (see Note 8).
Repeat step 12 of Subheading 3.1 for the remaining samples. Gently shake the 6-well plate up and down to mix up the cells in a 6-well plate and incubate the 6-well cell culture plate in a 37 °C degree cell incubator with 5% CO2 and 90% humidity.
The next day following neon transfection, examine the transfection efficiency using an inverted fluorescence microscope with 5× objective. TransEDIT-dual CRISPR plasmid for VPS54 expresses green fluorescent protein (GFP).
48 h post-transfection, trypsinize cells (see Subheading 3.1, steps 3 and 4), spin them down at 400 × g, 10 min and resuspend in filtered cell sorting medium.
Sort single cells in a 96-well plate containing DMEM/F12-FBS using BD FACS Aria III cell sorter. TransEDIT-dual CRISPR plasmid for VPS54 expresses green fluorescent protein (GFP). Therefore, sort the transfected cells based on high-GFP-Alexa Fluor 488 fluorescence (see Note 9).
Regularly examine the wells with clones and after 10–14 days of sorting expand them to 12-well plates, 6-well plates, and then to 10-cm dishes, respectively (see Note 10).
Analyze for the absence of the targeted protein by Western blotting (WB). Prepare the cell lysate from the single clones, run them on SDS-PAGE (sodium dodecyl sulfate–polyacrylamide gel electrophoresis), incubate with anti-VPS54 primary antibody prepared in Intercept (PBS) blocking buffer with the dilution of 1:1000, and Alexa Fluor tagged secondary antibody prepared in Intercept (PBS) blocking buffer with the dilution of 1:4000 and analyze for the absence of the VPS54 protein band in KO cells.
3.2. Making hTERT RPE1 VPS54-KO-R Cells by Lentiviral Transduction
Plate 1 × 106 HEK293FT cells on a 10-cm cell culture dish with 10 mL of DMEM/F12-FBS. The reason for using HEK293FT cells for lentivirus production is their ability to generate high-titer lentivirus (see Note 11).
To generate the lentivirus encoding VPS54 subunit, pCl-neo-VPS54-13myc was initially subcloned into the pENTR1A no ccDB (w48-1) entry vector and then recombined with pLenti pCOG4 Neo DEST destination vector using Gateway LR Clonase II Enzyme Mix (Thermo Fisher) according to the manufacturer’s instructions. The pLenti pCOG4 Neo DEST mVPS54-13myc plasmid is now ready to transfect the HEK293FT cells.
Once the HEK293FT cells become 90% confluent in 10-cm cell culture dish, transfect the cells with pLenti pCOG4 Neo DEST mVPS54-13myc and viral packaging plasmids [(pMD2.G), (pRSV-Rev), (pMDLg/pRRE)] in 1:1:1:1 ratio using lipofectamine 3000 transfection reagent (see Notes 12 and 13).
Incubate the transfected cells with serum reduced Opti-MEM containing GlutaMAX (1×) and chloroquine (25 μM) for 16 h.
Add sodium butyrate (1 mM final concentration) 5 h post-transfection (see Note 14).
The next day, change the medium with serum reduced Opti-MEM and GlutaMAX (1×) and incubate the cells at 37 °C, 5% CO2, and 90% humidity for 48 h.
Collect the supernatants in two 15 mL sterile conical tubes and centrifuge at 400 × g for 10 min.
Filter the centrifuged supernatants with 0.45 μm PES filter (see Note 15).
Concentrate the virus ten-fold using Lenti-X concentrator.
Quantify the concentration of mVPS54-13myc viral particles using Lenti-X p24 Rapid Titer Kit.
Transduce the hTERT-RPE1 VPS54-KO cells in a 6-well cell culture plate with 100 μL of mVPS54-13myc lentivirus (100 viral particles/cell) and polybrene (10 μg/mL). Polybrene enhances the efficiency of transduction. Plate one control well of untransduced cells (see Notes 16 and 17).
48 h post-transduction, change the medium in all wells to DMEM/F12-FBS with G418 (500 μg/mL final concentration) and incubate for 1 week or longer. Change the medium to a fresh DMEM/F12-FBS with G418 every 3 days. You will observe survival of the transduced cells while untransduced cells should die. Prepare surviving cells for a single-cell sorting as soon as all cells in the control well are dead (see Note 18).
Sort the single cells (see step 16 in Subheading 3.1) into 96-well plates to obtain clonal populations.
Examine the clones in the wells, and 10–14 days after single-cell sorting, expand the cells from 96-well to 12-well plate to 6-well plate to 10-cm dish eventually. For the maintenance of the transduced cells, use maintenance dose of G418 (200 μg/mL).
Perform tests for the expression of the mVPS54-13myc by WB and immunofluorescence microscopy (IF). For WB analysis, prepare the cell lysate from the single clones growing on 6-well plate, run them on SDS-PAGE, incubate with anti-VPS54 primary antibody, Alexa Fluor tagged secondary antibody and analyze samples for the presence of the VPS54-13myc band in VPS54-KO-Rcells (see Subheading 3.3). For IF, grow cells on glass coverslips in 12-well plates, fix and stain them with antimyc antibody, and analyze samples using fluorescent microscopy (see Subheading 3.4).
3.3. Validation of VPS54-KO and VPS54-KO-R Cells by WB
Plate 6 × 105 hTERT-RPE1 WT, VPS54-KO, and VPS54-KO-R cells on 6-well plate with DMEM/F12-FBS (see Note 18).
Once the cells reach 100% confluence, wash the cells twice with 2 mL of PBS.
Transfer SDS solution to the 1.5 mL Eppendorf tube and heat for 5 min to 70 °C using heat block.
Completely remove PBS from cells.
Add 150 μL of hot 2% SDS to the each well of 6-well plate for cell lysis.
Scrap the cells and collect them in 1.5 mL Eppendorf tubes.
Sonicate the cells with a sonicator equipped with a mini tip at 24 kHz for approximately 5 s.
Quantify the protein concentration in each of the samples using Bicinchoninic acid assay and measure the sample absorbance using CLARIOstar plate reader (see Note 19).
Prepare the samples by adding 6× SDS sample buffer.
Denature the protein samples for 10 min at 70 °C using a heat block.
Load samples (10 μg total protein) on a precast 4–15% Bio-Rad gradient gel. Perform electrophoresis at 160 V for 40 min or until the bromophenol blue band reaches the bottom of the gel (see Note 20).
Perform protein transfer from SDS-PAGE to the nitrocellulose membrane using a semi-dry or wet blotting apparatus.
Using plastic forceps, transfer nitrocellulose membrane to a clean membrane incubation box.
Stain the nitrocellulose membrane (blot) with Ponceau S solution for 5 min to determine transfer efficiency (see Note 21).
Wash the blot twice with PBS and block in 4 mL of the Odyssey blocking buffer for 20 min to prevent non-specific binding of the antibodies to the nitrocellulose membrane (see Note 22).
Remove blocking solution and add the primary anti-VPS54 (rabbit) (St John’s Lab, STJ115181) antibody diluted (1:1000) in the Odyssey blocking buffer and incubate overnight in a 4 °C cold room in the shaker (see Note 23).
Next day wash the blot four times (4 min each) with PBST.
Then, add a secondary antibody donkey anti-rabbit labeled with Alexa Fluor 647 diluted 1:4000 in the Odyssey blocking buffer for 1 h at room temperature in the shaker.
Wash the blot four times (4 min each) with PBST.
Wash once with PBS.
Scan the blots using the Odyssey imaging system.
3.4. Validation of VPS54-KO and VPS54-KO-R Cells Rescue by IF
Plate 6 × 105 RPE1 WT and VPS54-KO-R cells on 6-well plate with sterile round glass coverslips in DMEM/F12-FBS (see Notes 24 and 25).
Once the cells reach 80% confluency, place the coverslips on a flat surface with a parafilm on it.
Wash the cells on the coverslips gently with PBS (see Notes 26 and 27).
Fix the cells in coverslip with 200 μL of freshly prepared fixative (4% PFA in PBS) for 15 min.
Permeabilize cells by incubation with 200 μL of 0.1% Triton X-100 for 1 min.
Neutralize PFA with 200 μL of 50 mM NH4Cl in PBS for 5 min.
Rinse the coverslips twice with PBS.
Incubate the coverslips 2 times (10 min each) with IF blocking buffer (filtered) to minimize the non-specific binding of the primary antibody.
Incubate the coverslips for 40 min with primary antibodies anti-giantin (rabbit; Covance PRB-114C) and anti-myc (mouse monoclonal antibody; Cell signaling #2276) diluted in IF antibody buffer (see Note 28).
Wash the coverslips 4 times (2 min each) with PBS.
Incubate with secondary antibody donkey anti-rabbit 555 and donkey anti-mouse 488 diluted 1:500 in IF antibody buffer for 30 min in the dark (see Note 29).
Wash the coverslip with PBS for 2 min.
Rinse the coverslips once with Hoechst (1:2000 dilutions made in PBS) for 2 min.
Wash 4 times with 2 min each with PBS.
Pick-up coverslip using fine-end forceps.
Wash coverslip by immersing in PBS 10 times.
Wash coverslip by immersing in Milli-Q water 10 times.
Put a small drop of Prolong Gold Antifade reagent on the glass slide and mount coverslip with cells upside-down.
Cover the coverslips overnight in the dark.
The next day, apply the clear nail polish on the edges of glass coverslip to prevent it from moving while imaging.
Analyze the coverslips under 63× oil 1.4 NA objective of a LSM880 Zeiss Laser inverted microscope with Airyscan using ZEN software.
3.5. Validation of VPS54-KO and Rescue by Cathepsin D Secretion Assay
GARP complex is required for mannose 6-phosphate-receptor-dependent sorting of Cathepsin D to lysosomes. Dysfunction of GARP complex impairs the recycling of the mannose 6-phosphate-receptor which results in missorting and increasing secretion of immature cathepsin D [2]. Here, we perform the secretion assay to validate the missorting of cathepsin D in the absence of GARP subunit VPS54, and correction of cathepsin D missorting in VPS54-KO-R cells.
Plate 6 × 105 RPE WT, VPS54-KO, and VPS54-KO-R cells (R1 and R2 clones) in three 6-cm dishes and grow them to 100% confluency (see Note 30).
Rinse the cells three times with PBS and add 4 mL serum-free chemically defined medium with 1× GlutaMAX.
After 36 h, collect the medium into 15 mL conical tubes and spin down at 400 × g for 10 min to remove floating cells.
Concentrate the supernatant 20-fold using a 10 k concentrator.
Prepare the protein samples in 2% SDS, add 6× SDS sample buffer containing β-mercaptoethanol and denature samples by incubation at 70 °C for 10 min.
Load 10 μL of samples onto the Bio-Rad (4–15%) gradient gel and perform WB analysis.
Incubate the blot with anti-cathepsin D (mouse; Sigma, C0715) and probe with a donkey anti-mouse with Alexa Fluor 555 secondary antibodies.
Scan the blot using the Odyssey imaging system.
Acknowledgements
This work was supported by the National Institutes of Health (R01GM083144) and UAMS Easy Win Early Victory grant program.
4 Notes
Proper use of gloves and laboratory coats is required when working in the tissue culture hood.
Fixative should be freshly prepared by diluting 16% PFA (EMS) in PBS.
hTERT-RPE1 cells can be transfected using both chemical and electroporation approaches [22, 23]. In our hands, the electroporation using the neon transfection system gave the highest efficiency of transfection and most reproducible results.
Do not trypsinize the cells for a longer duration. Prolonged incubation of trypsin will damage the cell membrane and kill the cells.
Any cell counting method (such as hemocytometers) can be used instead of the Beckman Coulter Vi-Cell XR cell counter.
The volume of DNA plasmid added to the transfection reaction should not exceed 10% of the total transfection volume.
Optimize the electroporation parameters for specific cell types.
Avoid bubbles during the transfection by electroporation process because it causes arcing during electroporation resulting in decrease or failure of transfection.
Any compatible FACS instrument can be used for sorting of single cells.
Next day after the single cells sorting analyze wells using inverted microscope with 5× objective and discard the wells that have more than one clone. Only single clones should be used.
HEK293FT cells can detach easily from cell culture dishes. Therefore, it is suggested to coat the dish with 0.01% poly-L-lysine (0.1 mg/mL in water) for at least 2 h in sterile cell culture hood. Careful handling while changing the medium of the cells needs to be done if the poly-L-lysine coating is not done.
Proper biosafety training is mandatory before performing such experiments. Safe handling of the virus and proper disposal of the waste in 100% bleach and sealed container are required.
Ratio of reagents in the transfection reaction mixture should be optimized. For the transfection of HEK293FT cells in 10-cm dish we used the following mixture: 750 μL of Opti-MEM, 30 μL of lipofectamine 3000; 750 μL of Opti-MEM, 15 μg plasmid, 30 μL of P3000.
Addition of sodium butyrate (0.5–2 mM) can enhance transfection efficiency. Also, addition of polybrene (2–20 μg/mL) during virus transduction can increase lentiviral transduction efficiency.
After the collection of the lentiviruses, they can be stored at 4 °C for 24 h.
Be cautious when handling lentiviruses and adding them to cells. High concentration of lentiviral particles can be toxic to the cell and can result in cell death. Therefore, concentration of viral particles for transduction should be optimized.
Avoid repeated freezing and thawing of the virus. Aliquot it and use it only for single use.
Perform all experiments with proper positive and negative controls. For the positive control, we are using lentiviruses expressing GFP.
Any protein concentration measurement method compatible with SDS other than the bicinchoninic acid assay can be used.
Any other homemade or precast gradient or linear (10% acrylamide) gel can be used.
Instead of the nitrocellulose membrane, polyvinylidene difluoride (PVDF) membranes can also be used.
Any blocking buffer that does not interfere with a method of imaging can be used to block non-specific binding of antibodies in the blot—for instance, 3% BSA in PBS.
Some of the primary antibodies can bind to the protein of interest during 1-h incubation while other require longer (overnight or several days) incubation. Therefore, incubation time for each antibody used should be optimized.
Round glass coverslips can be sterilized at 110 °C for 5 h in the oven before plating the cells.
For IF microscopy experiments, 70–80% confluent cells are best for imaging.
Do not let the coverslips with cells dry during IF procedure.
Be careful while washing the cells on coverslips; some cells may be semi- or non-adherent and can be easily detached from coverslips. Therefore, collagen or poly-L-lysine coating is recommended.
Some antibodies might not work for the staining of the cell. Such problems can be overcome by tagging the “protein of interest” with small peptides such as “myc.” In our experiments, VPS54 antibody was not commercially available for cell staining. Therefore, we tagged our protein of interest with myc (VPS54-13myc) and used myc antibody to confirm its presence in VPS54-KO-R cells.
It is suggested to cover the coverslips from the sunlight when incubating with antibodies that are labeled with fluorescence probes. For instance, secondary antibodies such as Alexa Fluor labeled DAR-647, DAR-488, DAR-555.
Different mutant cell clones have different growth rates. For instance, KO cells might have a slow growth rate compared to wild type cells. Growth parameters should be considered when plating the cells. Special attention should be given when performing the cathepsin D secretion assay.
References
- 1.Wei J, Zhang Y-Y, Luo J, Wang J-Q, Zhou Y-X, Miao H-H, Shi X-J, Qu Y-X, Xu J, Li B-L (2017) The GARP complex is involved in intracellular cholesterol transport via targeting NPC2 to lysosomes. Cell Rep 19(13):2823–2835 [DOI] [PubMed] [Google Scholar]
- 2.Pérez-Victoria FJ, Mardones GA, Bonifacino JS (2008) Requirement of the human GARP complex for mannose 6-phosphate-receptor-dependent sorting of cathepsin D to lysosomes. Mol Biol Cell 19(6):2350–2362 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 3.Hirata T, Fujita M, Nakamura S, Gotoh K, Motooka D, Murakami Y, Maeda Y, Kinoshita T (2015) Post-Golgi anterograde transport requires GARP-dependent endosome-to-TGN retrograde transport. Mol Biol Cell 26(17):3071–3084 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 4.Fröhlich F, Petit C, Kory N, Christiano R, Hannibal-Bach H-K, Graham M, Liu X, Ejsing CS, Farese RV Jr, Walther TC (2015) The GARP complex is required for cellular sphingolipid homeostasis. elife 4:e08712. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 5.Conibear E, Stevens TH (2000) Vps52p, Vps53p, and Vps54p form a novel multisubunit complex required for protein sorting at the yeast late Golgi. Mol Biol Cell 11(1):305–323 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 6.Gershlick DC, Ishida M, Jones JR, Bellomo A, Bonifacino JS, Everman DB (2019) A neuro-developmental disorder caused by mutations in the VPS51 subunit of the GARP and EARP complexes. Hum Mol Genet 28(9):1548–1560 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 7.Schmitt-John T (2015) VPS54 and the wobbler mouse. Front Neurosci 9:381. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 8.Yan R-T, Li X, Huang J, Guidry C, Wang S-Z (2013) Photoreceptor-like cells from reprogramming cultured mammalian RPE cells. Mol Vis 19:1178. [PMC free article] [PubMed] [Google Scholar]
- 9.Khakurel A, Kudlyk T, Bonifacino JS, Lupashin VV (2021) The Golgi-associated retrograde protein (GARP) complex plays an essential role in the maintenance of the Golgi glycosylation machinery. Mol Biol Cell 32(17):1594–1610 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 10.Doudna JA, Charpentier E (2014) The new frontier of genome engineering with CRISPR-Cas9. Science 346(6213) [DOI] [PubMed] [Google Scholar]
- 11.Higashijima Y, Nangaku M (2020) The Nobel Prize in chemistry in 2020: genome editing tools and their immeasurable applications for humankind. Kidney Int 98(6):1367–1369 [DOI] [PubMed] [Google Scholar]
- 12.Ran FA, Hsu PD, Wright J, Agarwala V, Scott DA, Zhang F (2013) Genome engineering using the CRISPR-Cas9 system. Nat Protoc 8(11):2281–2308 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 13.Cui Y, Xu J, Cheng M, Liao X, Peng S (2018) Review of CRISPR/Cas9 sgRNA design tools. Interdiscip Sci 10(2):455–465 [DOI] [PubMed] [Google Scholar]
- 14.Jiang F, Doudna JA (2017) CRISPR–Cas9 structures and mechanisms. Annu Rev Biophys 46:505–529 [DOI] [PubMed] [Google Scholar]
- 15.Loureiro A, da Silva GJ (2019) Crispr-cas: converting a bacterial defence mechanism into a state-of-the-art genetic manipulation tool. Antibiotics 8(1):18. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 16.Wong L-F, Goodhead L, Prat C, Mitrophanous KA, Kingsman SM, Mazarakis ND (2006) Lentivirus-mediated gene transfer to the central nervous system: therapeutic and research applications. Hum Gene Ther 17(1):1–9 [DOI] [PubMed] [Google Scholar]
- 17.Federico M (2003) From lentiviruses to lentivirus vectors. In: Lentivirus gene engineering protocols. Springer, pp 3–15 [DOI] [PubMed] [Google Scholar]
- 18.Hughes SM, Parr-Brownlie LC, Bosch-Bouju C, Schoderboeck L, Sizemore R, Abraham W (2015) Lentiviral vectors as tools to understand central nervous system biology in mammalian model organisms. Front Mol Neurosci 8:14. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 19.Neschadim A, McCart JA, Keating A, Medin JA (2007) A roadmap to safe, efficient, and stable lentivirus-mediated gene therapy with hematopoietic cell transplantation. Biol Blood Marrow Transplant 13(12):1407–1416 [DOI] [PubMed] [Google Scholar]
- 20.Joglekar AV, Sandoval S (2017) Pseudotyped lentiviral vectors: one vector, many guises. Hum Gene Ther Methods 28(6):291–301 [DOI] [PubMed] [Google Scholar]
- 21.Dull T, Zufferey R, Kelly M, Mandel R, Nguyen M, Trono D, Naldini L (1998) A third-generation lentivirus vector with a conditional packaging system. J Virol 72(11):8463–8471 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 22.Alex A, Piano V, Polley S, Stuiver M, Voss S, Ciossani G, Overlack K, Voss B, Wohlgemuth S, Petrovic A (2019) Electroporated recombinant proteins as tools for in vivo functional complementation, imaging and chemical biology. elife 8:e48287. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 23.Gupta GD, Coyaud É, Gonçalves J, Mojarad BA, Liu Y, Wu Q, Gheiratmand L, Comartin D, Tkach JM, Cheung SW (2015) A dynamic protein interaction landscape of the human centrosome-cilium interface. Cell 163(6):1484–1499 [DOI] [PMC free article] [PubMed] [Google Scholar]