

Abstract
Immune checkpoint inhibitors (ICI) have been effective in treating a subset of refractory solid tumors, but only a small percentage of treated patients benefit from these therapies. Thus, there is a clinical need for reliable tools that allow for the early assessment of response to ICIs, as well as a preclinical need for imaging tools that aid in the future development and understanding of immunotherapies. Here we demonstrate that CD69, a canonical early-activation marker expressed on a variety of activated immune cells, including cytotoxic T cells and natural killer (NK) cells, is a promising biomarker for the early assessment of response to immunotherapies. We have developed a PET probe by radiolabeling a highly specific CD69 mAb, H1.2F3, with Zirconium-89 (89Zr), [89Zr]-deferoxamine (DFO)-H1.2F3. [89Zr]-DFO-H1.2F3 detected changes in CD69 expression on primary mouse T cells in vitro and detected activated immune cells in a syngeneic tumor immunotherapy model. In vitro uptake studies with [89Zr]-DFO-H1.2F3 showed a 15-fold increase in CD69 expression for activated primary mouse T cells, relative to untreated resting T cells. In vivo PET imaging showed that tumors of ICI-responsive mice had greater uptake than the tumors of nonresponsive and untreated mice. Ex vivo biodistribution, autoradiography, and IHC analyses supported the PET imaging findings. These data suggest that the CD69 PET imaging approach detects CD69 expression with sufficient sensitivity to quantify immune cell activation in a syngeneic mouse immunotherapy model and could allow for the prediction of therapeutic immune responses to novel immunotherapies.
Introduction
Cancer immunotherapies have had transformative success and durable clinical responses in patients with intractable malignancies (1). Immune checkpoint inhibitors (ICI) have been particularly effective in treating non–small cell lung cancer (NSCLC) and melanoma, which has led to several FDA drug approvals for indications across 19 different forms of cancer over the past decade (2). Although almost 50% of U.S. patients with cancer are eligible for ICIs, approximately only 12% benefit from this type of therapy, and as eligibility rates appear to be increasing, benefit rates appear to be slowing down (3). Furthermore, ICIs may induce a wide variety of toxic side effects known as immune-related adverse events that range from mild to severe, life-threatening reactions (4, 5).
Current patient response prediction methods typically rely on tissue biopsies to assess expression of biomarkers, like PD-L1 expression and tumor mutational burden, in tumor cells. However, due to the dynamic nature of metastatic tumors and infiltrating immune cells, biomarkers tend to be spatially and temporally variable, which means that a biopsy may fail to accurately reflect the heterogeneous composition of a patient’s tumor microenvironment (TME) and their response to certain ICIs (6, 7). The invasive nature of biopsies also limits their use for serial monitoring, again highlighting the need for broadly applicable imaging biomarkers (8, 9).
The current standard of care for the noninvasive imaging of a patient’s response to immunotherapy utilizes traditional imaging approaches such as MRI, CT, and/or [18F]-FDG PET/CT to evaluate changes in tumor size over a period of several months. However, efforts have been made to image and monitor immune activity, particularly within the TME, which has the potential to assess early response to ICIs. These efforts include using radiolabeled probes to image T-cell phenotypic markers (like CD8; ref. 10). However, immune suppression by the TME and T-cell exhaustion also play a role in determining an immunologic response, and therefore assessing immune cell functional status may be highly beneficial (11). Thus, probes for T-cell activation markers (granzyme B, ICOS, IFNγ, and OX40), as indicators of T cell–mediated immune response, have also been developed (12–15). These methods show promising results in quantifying immunologic activity within tumors using PET imaging and various preclinical immunotherapy models. They also show that PET imaging is ideal for noninvasive, whole body, serial-monitoring of immune cell localization and activity within the TME (16).
We hypothesized that the global immune activation marker, CD69, could be used as a biomarker of response to immunotherapy. CD69 is a canonical early-activation marker expressed in a variety of activated immune cells, including cytotoxic T cells and natural killer (NK) cells (17, 18). Its expression is specific to activated immune cells and is markedly upregulated very quickly upon activation (<2 hours; (19). The CD69 molecule is a membrane-bound receptor with C-type lectin-like domains. It is thought to play a role in cell differentiation of certain types of T cells, T-cell migration, and retention in lymph nodes and tissues by antagonizing sphingosine 1-phosphate receptor 1 (S1PR1)-mediated egress of T cells from tissue, and it is considered a negative immune regulator because its deficiency or blocking promote a more potent immune response in murine models of inflammation, infection, and cancer (17, 20). To image CD69 expression, we have used a Zirconium-89 (89Zr)-labeled, highly specific, Armenian Hamster monoclonal anti-CD69, [89Zr]-deferoxamine (DFO)-H1.2F3, to track and monitor activated immune cells, in vivo (Fig. 1A and B). Using a validated preclinical CT26 syngeneic tumor immunotherapy model and anti–CTLA-4 and anti–PD-1 checkpoint inhibitor combination therapy (21), we demonstrated that the [89Zr]-DFO-H1.2F3 probe identifies responding tumors containing increased numbers of activated immune cells compared with nonresponding tumor and controls. Our results indicate that CD69 is a promising biomarker for assessing and monitoring response to checkpoint blockade therapy and has potential for predicting therapeutic immune response to novel immunotherapies.
Figure 1.
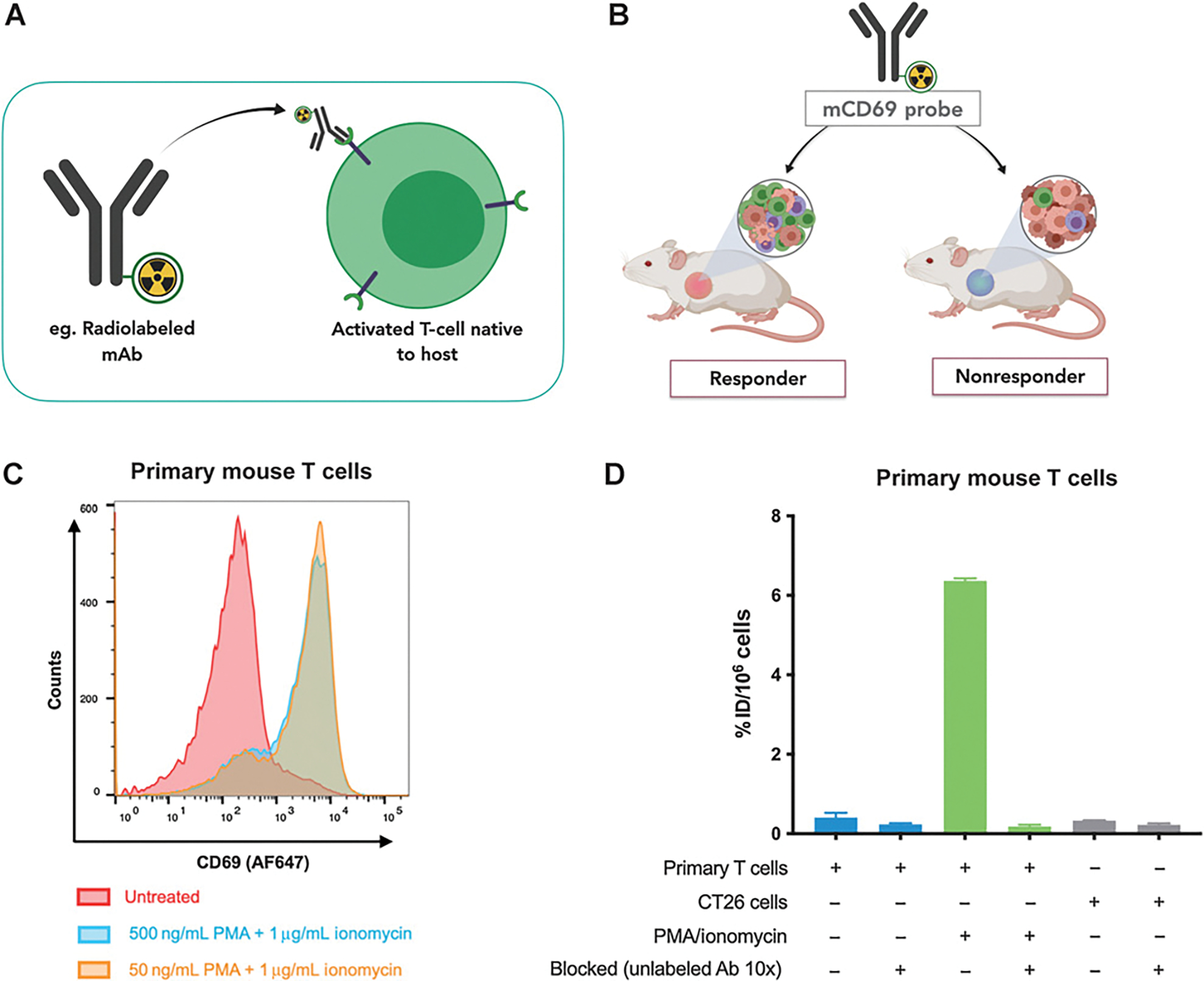
Assessing mCD69 expression in primary mouse T cells using flow cytometry and [89Zr]-DFO-H1.2F3 uptake. A, Scheme of CD69 antibody binding: H1.2F3 monoclonal anti-CD69 binds to mCD69 on the surface of activated primary mouse T cells and other immune cells. B, Scheme of CD69 imaging: CD69 can be used as a biomarker to distinguish immunologically active tumors of mice that respond to checkpoint blockade (CBP) therapy (responders) from the tumors of mice that do not respond to therapy (nonresponders). C, Ex vivo flow cytometry analysis of CD69 expression for primary mouse T cells treated with PMA/ionomycin or untreated primary mouse T cells. D, Ex vivo [89Zr]-DFO-H1.2F3 uptake for primary mouse T cells treated with 50 ng/mL PMA and 1 μg/mL ionomycin, untreated primary mouse T cells, and CT26 cells. Uptake was measured on γ-counter and normalized to percent injected dose per million cells (% ID/106 cells). Error bars, SD.
Materials and Methods
Cell line generation and culture
Murine cell culture
Primary murine T-cell isolation and culture:
The spleens of 6- to 8-week-old female BALB/c mice (Charles River Laboratories International, Inc., 6–8 weeks old) were harvested and disrupted using a 70-mm nylon mesh strainer (Fisher Scientific, catalog no. 352350). Highly purified T cells were isolated from splenocytes by negative selection using the EasySep Mouse T Cell Isolation Kit (STEMCELL Technologies Inc., catalog no. 19851). Cells were resuspended in complete RPMI-1640 medium, containing 2 mmol/L L-glutamine, 10 mmol/L HEPES, 1 mmol/L sodium pyruvate, 4,500 mg/L glucose, and 1,500 mg/L sodium bicarbonate (Gibco, catalog no. A1049101), plus 10% FBS (Gibco, catalog no. 16000044) and 1% penicillin/streptomycin (Invitogen, catalog no. 15140122), for in vitro flow cytometry assays. Cells were maintained in a humidified incubator at 37°C.
CT26.WT murine colon carcinoma cells (acquired from, authenticated by, and Mycoplasma tested by vendor: ATCC, catalog no. CRL-2638) were cultured in complete media according to the ATCC recommended formulation: RPMI-1640 Medium containing 2 mmol/L L-glutamine, 10 mmol/L HEPES, 1 mmol/L sodium pyruvate, 4,500 mg/L glucose, and 1,500 mg/L sodium bicarbonate (Gibco, catalog no. A1049101), plus 10% FBS (Gibco, catalog no. 16000044) and 1% penicillin/streptomycin (Invitogen, catalog no. 15140122). Cells were maintained in a humidified incubator at 37°C. Cells were used for in vitro and in vivo assays within 3 months of thawing, and passaged no more than approximately 10 times before use.
Human cell culture
Human HCT116 (ATCC, catalog no. CCL-247) and SKOV-3 human cell lines (ATCC, catalog no. HTB-77) were cultured in complete media: DMEM containing 10% FBS (Invitrogen), 2 mmol/L glutamine (Gibco), 1% penicillin/streptomycin (Gibco), and were maintained in a humidified incubator at 37°C. Both cell lines were acquired from, authenticated by, and Mycoplasma tested by vendor, and were cultured, cryopreserved, and stored according to ATCC protocol. Cells were cultured for less than 2 months after thawing. HCT116 and HEK293 cells were transduced with lentivirus (Origene Lentiviral Packaging Kit, catalog no. TR30037, and Lenti Concentrator, catalog no. TR30025) encoding a mGFP C-terminal–fused CD69 gene (CD69-mGFP, Origene, catalog no. MR226790L4) at a multiplicity of infection of 5. HCT116-CD69-GFP and SKOV-3-CD69-GFP transduced cell lines were selected using puromycin (7.5 μg/mL, Life Technologies Inc., catalog no. A11138-03)-treated media as well as fluorescence-activated cell sorting (expression enrichment by maximizing GFP signal per cell). HCT116, SKOV-3, and HEK293T cells were Mycoaplasma-free, as verified by the MycoAlert Mycoplasma Detection Kit, (Lonza, catalog no. LT07, tested by Cell Center Services at University of Pennsylvania, Philadelphia, PA).
H1.2F3 Armenian Hamster monoclonal anti-CD69
The H1.2F3-producing hybridoma cell line was derived in the lab of Dr. Ethan Shevach at the NIH. H1.2F3-producing hybridomas were then cultured and H1.2F3 antibodies were purified from the supernatant using a Protein G column by the Cell Center Services Facility at the University of Pennsylvania. The concentration of the antibody stock solution was adjusted to 0.5 mg/mL for in vitro, in vivo, and ex vivo studies. Commercial H1.2F3 (BioLegend, Clone: H1.2F3, catalog no. 104518) and IgG isotype control HTK888 (BioLegend, Clone: HTK888, catalog no. 400924) antibodies were used for the initial validation studies with CD69-transduced human cell lines.
H1.2F3-AF647 conjugation
Alexa Fluor 647 (AF647) was conjugated to the purified H1.2F3 antibody (~100 μg) using an antibody labeling kit (Molecular Probes by Life Technologies, catalog no. A20186). AF647-conjugated H1.2F3 antibodies were used for flow cytometry assays.
Flow cytometry
Flow cytometry was used to analyze GFP expression on CD69-transduced human cells. CD69 cell surface expression on CD69-transduced human cells and wild-type (WT) controls, as well as treated primary murine T cells and untreated control cells, were also analyzed. HCT116-CD69-GFP, HCT116-WT, SKOV-3-CD69-GFP, and SKOV-3_WT cells in suspension (2 × 106 cells in 400 μL 2% BSA/PBS) were incubated with AF647-conjugated anti-CD69 and isotype negative control antibodies (BioLegend, catalog no. 104518 and catalog no. 400924, respectively) at room temperature for 1.5 hours, then washed three times with 2% BSA/PBS (BSA, Sigma-Aldrich, catalog no. B6917; and PBS, Gibco, catalog no. 10010072). Stained cells were then analyzed using BD LSR II cytometer and data was analyzed with FlowJo V 10.6 software. Primary murine T cells were treated with 50 or 500 ng/mL phorbol 12-myristate-13-acetate (PMA, Sigma-Aldrich, catalog no. 79346) and 1 μg/mL ionomycin (Sigma-Aldrich, catalog no. I0634) for 2 hours. Both treated and untreated cells were then incubated with the AF647-conjugated monoclonal H1.2F3 anti-CD69 (0.5 mg/mL at a dilution of 1:100, Cell Center Service Facility at the University of Pennsylvania) and primary anti-CD3 (0.5 mg/mL at a dilution of 1:100, Abcam) for 30 minutes. Cells were washed three times with ice cold PBS and stained with an AF488-conjugated goat anti-Armenian Hamster polyclonal secondary antibody (2 mg/mL at 1:2,000 dilution) for 30 minutes. After three washes, cells were fixed with 1% paraformaldehyde (Electron Microscopy Sciences, catalog no. 15710) in PBS for 10 minutes, washed twice, and resuspended in 3% BSA in PBS for flow cytometry analysis.
To evaluate changes in CD69 expression over time, primary murine T cells were treated with either 50 ng/mL PMA and 1 μg/mL ionomycin for 2 hours or stimulated with CD3/CD28 Dynabeads (Gibco, catalog no. 11452D) for 2 hours. PMA/ionomycin- and CD3/CD28 Dynabead-activated cells were then stained with AF647-conjugated H1.2F3 anti-CD69 (BioLegend, Clone: H1.2F3, catalog no. 104518), a Pacific Blue-conjugated CD4 antibody (BioLegend, Clone: RM4-5, catalog no. 100531), a FITC-conjugated CD8a anitbody (BioLegend, Clone: 53–6.7, 100706), and a Zombie NIR Fixable Viability Kit (BioLegend, catalog no. 423106). An AF647-conjugated Hamster IgG negative control (BioLegend, Clone: HTK888, catalog no. 400924) was also used. The activated cells were then monitored over the course of 6 days. Compensation controls and calculations were used for cells that were stained with multiple markers at the time of acquisition using the using the BD FACSDiva Software and the BD LSR II cytometer. Data was further analyzed with FlowJo V 10.6 software
Antibody-DFO conjugation and 89Zr radiolabeling
Antibody-DFO conjugation
Antibodies were buffer exchanged from PBS to Bicarb-carb buffer, pH 9, and concentrated to 2 mg/mL using ultra-spin filters [Amicon ultra-0.5 centrifugal filters (Millipore Sigma, catalog no. UFC501096)]. Antibodies (~125 μL) were then reacted with 6 μL of the metal chelator deferoxamine (5 mmol/L in DMSO, p-SCN-Bn-deferoxamine; Macrocyclics, catalog no. B-705) at 600 rpm, at 37°C, for 3.5 hours. For matrix-assisted laser desorption/ionization–time-of-flight (MALDI-TOF) characterization, the crude product was buffer exchanged to water. MALDI-TOF was used to approximate the degree of labeling (DOL). For subsequent 89Zr-radiolabeling reactions, the crude product was buffer exchanged to HEPES using 1 M HEPES buffer (Cell Center Services at the University of Pennsylvania). All consumables were prewashed with Chelex 100 resin-treated (Sigma-Aldrich, catalog no. C7901) deionized water prior to use, where necessary. Chelex 100-treated buffers were also used.
89Zr radiolabeling
The DFO-conjugated antibodies in HEPES buffer were then incubated with 89Zr [Cyclotron Facility of Mallinckrodt Institute of Radiology, Washington University in St. Louis, St. Louis, MO) for the biodistribution study; 3D Imaging, for the PET Imaging Study] in a metal-free Eppendorf tube at room temperature for 1 hour. Product was purified using 1 M HEPES buffer Amicon ultra-0.5 centrifugal filters (Millipore Sigma, catalog no. UFC501096). Radio TLC was used to assess the presence and purity of the 89Zr-labeled product before and after purification [mobile phase: 50 mmol/L DTPA (aq), pH 5.5]. Radioactivity measurements and radiochemical yield were measured and determined using a RC-15R Dose Calibrator (Capintec). Purified product was resuspended in PBS for in vitro and in vivo studies. All consumables were prewashed with Chelex 100 resin-treated (Sigma-Aldrich, catalog no. C7901) deionized water prior to use, where necessary. Chelex 100-treated buffers were also used.
In vitro cell uptake of 89Zr-labeled anti-CD69
CD69-transduced HCT116 and SKOV-3 cells in suspension (2 × 106 cells in 400 μL 2% BSA/PBS) were incubated with purified Zr-89 labeled anti-CD69 ([89Zr]-DFO-H1.2F3; 0.5 μCi) for 1.5 hours at room temperature and washed three times with 2% BSA in PBS. Excess unlabeled anti-CD69 (1.85 mmol/L) was added as a blocking agent at the same time as adding the radiotracer to determine nonspecific binding. Total radioactive uptake was then determined by a γ-counter (Wizard Detector, Perkin Elmer) to measure the retained counts per minute (cpm), which correlates with total antibody binding. Uptake was measured by dividing counts by incubated dose of Zr-89 labeled anti-CD69. Following the same protocol, primary mouse T cells that had been treated with PMA/ionomycin for 2 hours (2 × 106 cells in 400 mL 2% BSA/PBS), were incubated with [89Zr]-DFO-H1.2F3. Uptake was measured, relative to blocked and untreated primary murine T cells.
Syngeneic CT26 colon carcinoma immunotherapy model
Fifteen 8- to 12-week-old female BALB/c mice (Charles River Laboratories) were subcutaneously xenografted on the right side, across from the spleen, with 0.5 × 106 CT26 murine colon carcinoma cells (ATCC) suspended in 1:1, v:v combination of 75 μL of RPMI complete medium and 75 μL of Matrigel Matrix (Corning, catalog no. 354234), day 0. Mice were administered checkpoint blockade therapy intraperitoneally on days 3, 6, and 9 after tumor inoculation, in the form of a multi-dose combination treatment regimen of anti–PD-1 (200 μg; Bio-X-Cell) and anti–CTLA-4 (100 mg; Bio-X-Cell) immunotherapies. This model was used for both biodistribution and PET/CT imaging studies.
PET imaging and biodistribution quantification
For the healthy mouse biodistribution study:
3 female NOD/SCID gamma (NSG) (The Jackson Laboratory) and 3 BALB/c (Charles River) mice were each given a 100-μCi dose (~20 μg) of [89Zr]-DFO-H1.2F3 via tail vein, while 2 BALB/c mice were each given a 100-μCi dose of [89Zr]-DFO-abISO (isotype control) via tail vein. Mice were sacrificed 72 hours after radiotracer injection. Major organs were harvested and total radioactive uptake per organ was determined by γ-counter (Perkin Elmer).
For studies using the syngeneic immunotherapy model:
On day 12 after tumor inoculation, each mouse was injected with either a 50-μCi (~10 μg) or 200-μCi (~40 μg) dose of [89Zr]-DFO-H1.2F3 via tail vein for biodistribution or PET imaging studies, respectively. On day 15 after tumor inoculation, i.e., 72 hours after radiotracer administration, mice were euthanized for organ harvest or imaged using small animal PET/CT. For both studies, tumor sizes were measured from days 4 through 15 after tumor inoculation. For biodistribution analysis, major organs were harvested, and uptake was quantified using γ-counter. For PET imaging ROI analysis, tumors and other major organs were manually and three-dimentionally contoured using MIM software and mean counts and volumes (Bq/mL) were generated for each mouse. For both studies, uptake values were normalized to percent injected dose per gram (% ID/g).
All animal studies were completed with University of Pennsylvania’s Institutional Animal Care and Use Committee (IACUC) approval.
Ex vivo autoradiography, histology, and IHC analyses
On day 15 of both the biodistribution and PET/CT studies, tumors were harvested from all mice, dissected, embedded in optimal cutting temperature (OCT) medium (Electron Microscopy Solutions, catalog no. 62550-12), and flash frozen by submerging embedding mold in dry ice-ethanol slush. Sections, 10-mm-thick, were placed and allowed to adhere to microscope slides, which were then exposed to a phosphor imaging plate overnight (GE). Digital autoradiography images were read and recorded using the Typhoon Phosphorimager (GE). Adjacent sections were stained with hematoxylin and eosin (Abcam, catalog no. ab245880). Anti-CD69 and anti–[89Zr]-DFO-H1.2F3 IHC were also performed. To probe for CD69, a polyclonal rabbit anti-murine CD69 was used (1:50; 1 mg/mL; Bioss, catalog no. bs-2499R) followed by a goat anti-rabbit IgG amplifier Ab and an horseradish peroxidase (HRP)-conjugated horse anti-goat Ab (ImmPRESS Excel Amplified Polymer Staining Kit, Vector Laboratories, catalog no. MP-7451-15). To probe for [89Zr]-DFO-H1.2F3 an unconjugated goat anti-hamster IgG was used (1:300; 3 mg/mL; Vector Laboratories, catalog no. AI-9100-1.5) followed by the HRP-conjugated horse anti-goat Ab (ImmPRESS Excel Amplified Polymer Staining Kit, Vector Laboratories, catalog no. MP-7451-15). Slides were imaged and analyzed using the Zeiss Axio Imager. Next, CD4 (Clone 4SM95; 1:50; 0.5 mg/mL; eBioscience, catalog no. 14-9766-82), CD8 (Clone 4SM15; 1:50; 0.5 mg/mL; eBioscience, catalog no. 14-0808-82), and CD45 (Clone D3F8Q; 1:400; Cell Signaling Technology, catalog no. 70257S) staining of 10-mm-thick frozen sections was performed at the University of Pennsylvania Perelman School of Medicine Pathology Clinical Service Center and digitized (Zeiss Axio system).
Statistical analysis
Statistical analyses were performed using GraphPad Prism version 9 software. For all comparisons between cohorts, across organs of interest, an unpaired two-tailed t test was used. A P less than 0.05 (P < 0.05) was used to determine statistical significance.
Data availability statement
Data available upon request from the corresponding authors.
Results
CD69 probe development and in vitro characterization
To confirm the specificity of H1.2F3 for murine CD69 and its potential use for detecting differences in CD69 expression, we first used human HCT116 and SKOV-3 cell lines that were transduced to express murine CD69-GFP as a surface receptor. CD69 general expression and surface expression were both quantified using flow cytometry and Western blotting with commercially available antibodies (Supplementary Fig. S1). H1.2F3 quantified differences as great as 37-fold between CD69-transduced cells and the WT counterparts. After validating the specificity of H1.2F3, we synthesized both AF647-conjugated and 89Zr-radiolabeled H1.2F3 (anti-CD69) probes. DFO, a radiometal chelating moiety, was conjugated to H1.2F3 to make DFO-H1.2F3. The DOL was determined by MALDI-TOF, and ranged from 1.8 to 3.5 DFO molecules per antibody (Supplementary Fig. S1G). [89Zr]-DFO-H1.2F3 radiochemical yield ranged from 64% to 83% with a specific activity of 5.6 to 7.1 mCi/mg (208.0–261.0 MBq/mg) and radiochemical purity of more than 99% (Supplementary Fig. S1H). We confirmed that the labeled antibody could quantify differences in CD69 expression using primary mouse T cells with flow cytometry and [89Zr]-DFO-H1.2F3 uptake studies, respectively. Immediately after harvesting and isolating T cells from the spleens of 6- to 10-week-old BALB/c mice, T cells were treated with PMA/ionomycin for 2 hours prior to incubation with the probe. Flow cytometry showed a strong upregulation in CD69 expression for activated T cells relative to resting untreated T cells (Fig. 1C). [89Zr]-DFO-H1.2F3 uptake showed a 15-fold increase in CD69 expression for activated T cells relative to resting untreated T cells (Fig. 1D). CD69 expression in primary mouse T cells, including CD4+ and CD8+ T-cell subsets, was then evaluated over time following stimulation with CD3/CD28 Dynabeads or PMA/ionomycin for 2 hours; CD69 expression was markedly increased by 2 hours after stimulation, with continued high expression for 2 days followed by a return to baseline (Supplementary Figs. S2 and S3).
Ex vivo biodistribution quantification of [89Zr]-DFO-H1.2F3 probe uptake
The biodistribution of [89Zr]-DFO-H1.2F3 was assessed using healthy BALB/c and NSG mice. [89Zr]-DFO-H1.2F3 was administered to both BALB/c and NSG mice, and a 89Zr-labeled isotype control antibody was administered to BALB/c mice (Supplementary Fig. S4). A qualitative analysis of the biodistribution data revealed decreased activity in the blood in BALB/c mice compared with both NSG mice and isotype control, and slightly increased uptake in the spleen in BALB/c mice compared with NSG mice and isotype control, consistent with mild basal CD69 expression in BALB/c mice, primarily in the spleen. No other sites of specific binding were identified in any other organs, including the bone, confirming low CD69 basal expression in a wide variety of tissues. Following the biodistribution study in healthy mice, we used [89Zr]-DFO-H1.2F3 to detect CD69+ activated immune cells in an immunocompetent ICI preclinical model (21). Biodistribution studies with CT26 tumors using other antibodies and isotype controls have been performed previously (22). 10 mice, bearing CT26 tumors, were treated with anti–PD-1 and anti–CTLA-4 combination therapy on days 3, 6, and 9 after tumor inoculation; 5 control mice were treated with saline. All 15 mice were injected with [89Zr]-DFO-H1.2F3 (50–60 μCi; 1.85–2.22 MBq) 12 days after tumor inoculation. At 72 hours after radiotracer injection (15 days after tumor inoculation), mice were euthanized and organs of interest [blood, thymus, spleen, tumor draining lymph nodes (dLN), nontumor dLNs, inguinal lymph nodes, muscle, bone, bone marrow, and the CT26 tumor] were harvested for uptake quantification (Fig. 2A). Tumor growth measurements captured the differences in tumor growth dynamics between groups that were assigned at completion of the experiment based on tumor size: responders, nonresponders, and untreated. A decrease in tumor volume between days 12 and 15, as well as a tumor volume less than 400 mm3 on day 15, was used to define response relative to nonresponse. 10 days after tumor inoculation, the tumors of mice from the responder group began to decrease in size. 15 days after tumor inoculation, responders had significantly smaller tumors than nonresponder and untreated groups (Fig. 2B and C). Analysis of the ex vivo biodistribution data showed increased tumor uptake in responders (43.1 ± 12.8% ID/g; n = 6) relative to that of nonresponders and untreated groups (10.3 ± 1.2% ID/g; n = 4, and 12.6 ± 1.8% ID/g; n = 5, respectively). The spleens of responding mice had higher uptake (34.03 ± 2.66% ID/g) than those of nonresponder and untreated groups (24.21 ± 0.52% ID/g and 22.84 ± 2.80% ID/g, respectively). The tumor dLNs of responding mice also had higher uptake than those of untreated mice (23.15 ± 6.75% ID/g and 10.54 ± 5.68% ID/g respectively; Fig. 2D; Supplementary Fig. S5A). Organ-to-blood (OTB) ratios also confirmed increased tumor uptake in responders (Supplementary Fig. S5B).
Figure 2.
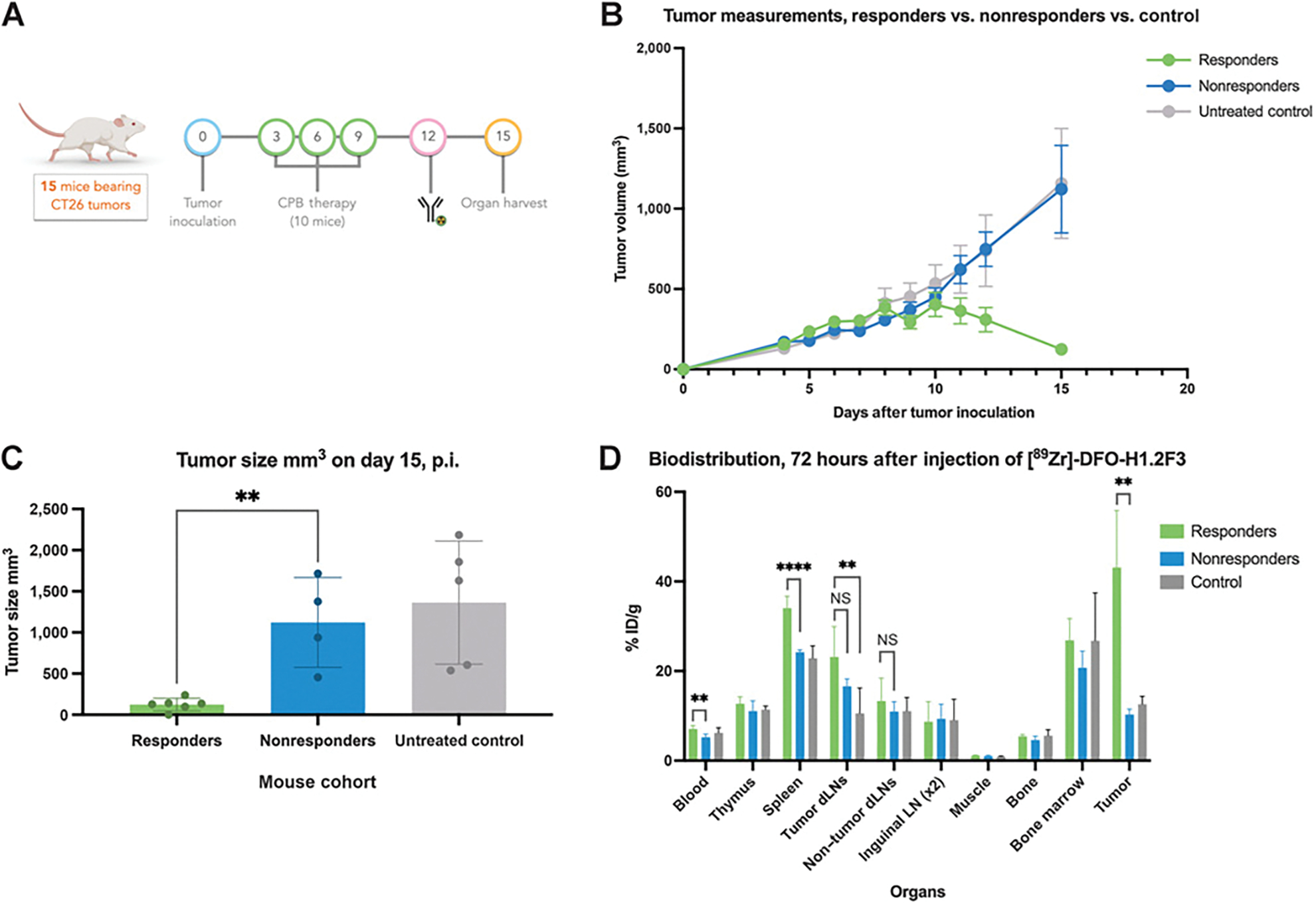
Quantifying [89Zr]-DFO-H1.2F3 ex vivo biodistribution from a CT26 syngeneic tumor immunotherapy model. A, Study design of 15-day biodistribution study for quantifying immune cell activation. B, Tumor growth monitoring by volume. C, Tumor volume measurements on day 15 for responder (n = 6), nonresponder (n = 4), and untreated (n = 5) groups. D, Biodistribution quantification at 72 hours after [89Zr]-DFO-H1.2F3 injection for major organs. Two-tailed unpaired t test was used to compare groups., P < 0.05; **, P < 0.01; ***, P < 0.001; CPB, checkpoint blockade; NS, not significant; p.i., postinjection.
PET/CT imaging of [89Zr]-DFO]H1.2F3 probe biodistribution
To test the ability of [89Zr]-DFO-H1.2F3 to detect CD69+ activated immune cells in vivo, we again used a CT26 syngeneic tumor immunotherapy model. Mice were injected 12 days after tumor inoculation with [89Zr]-DFO-H1.2F3 (200–220 μCi; 7.4–8.14 MBq). Mice were then imaged 72 hours after radiotracer injection, i.e., 15 days after tumor inoculation (Fig. 3A). Tumor growth measurements captured the differences in tumor growth dynamics between responder, non-responder, and untreated groups. A decrease in tumor volume between days 12 and 15, as well as a tumor volume less than 400 mm3 on day 15, was used to define response relative to nonresponse (Fig. 3B). PET region of interest (ROI) quantification of the PET images showed increased uptake in tumors from responders (18.0 ± 2.4% ID/g, n = 8), relative to tumors from nonresponder and untreated groups (11.8 ± 0.4% ID/g, n = 2; and 13.3 ± 0.6% ID/g, n = 5, respectively; Fig. 3C–E). OTB ratios also confirmed the significance of the increased tumor uptake in responders (Supplementary Fig. S6A–S6C). These imaging results, along with the biodistribution ex vivo study, demonstrate that uptake of [89Zr]-DFO-H1.2F3 correlates with tumor response to immunotherapy.
Figure 3.
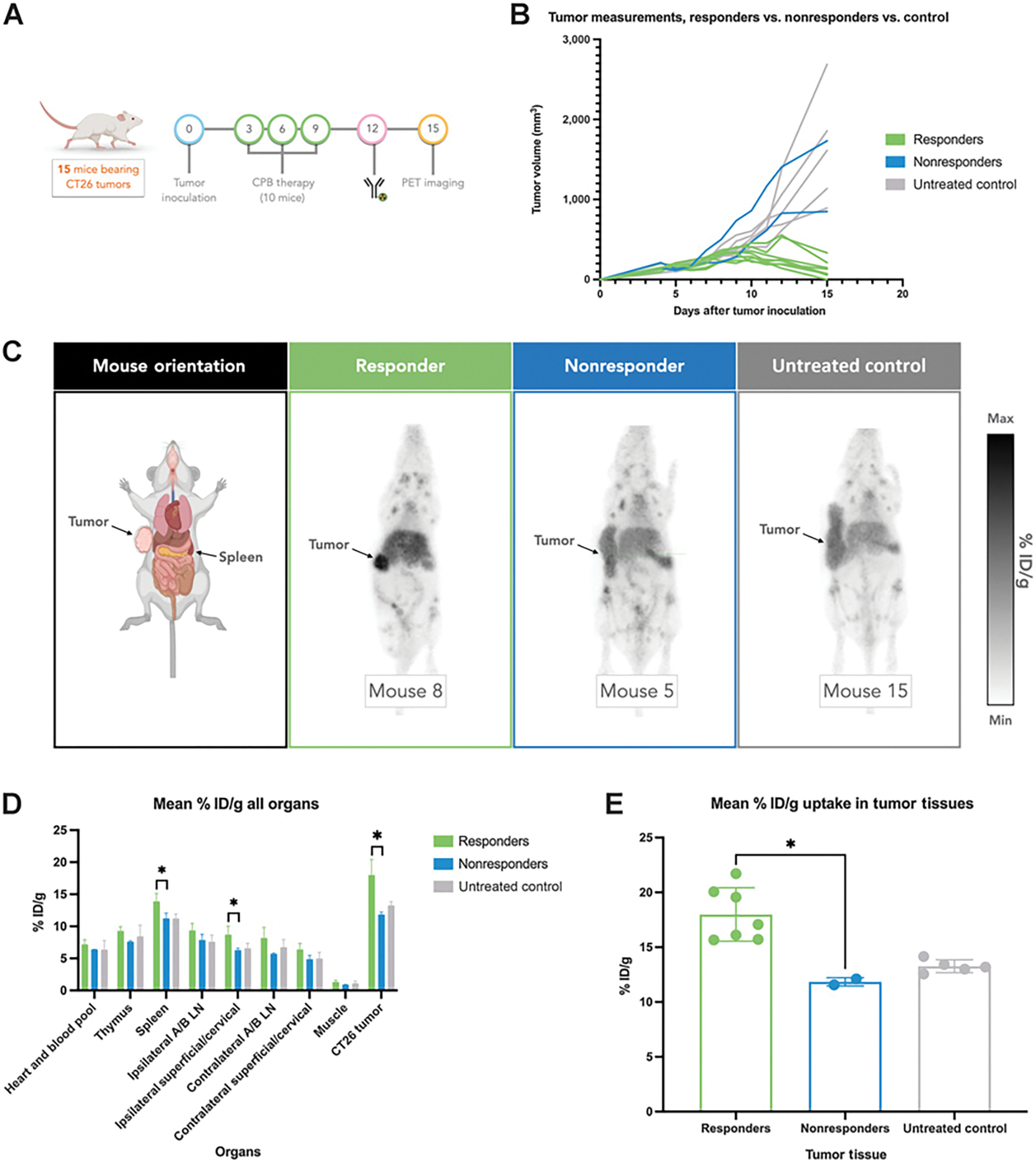
Quantifying [89Zr]-DFO-H1.2F3 in vivo using PET imaging in a CT26 syngeneic tumor immunotherapy model. A, Study design of 15-day PET imaging study for monitoring immune cell activation. B, Tumor growth monitoring by volume per mouse. C, Representative PET images taken 72 hours after intravenous injection of [89Zr]-DFO-H1.2F3 mAb of responder (n = 8), nonresponder (n = 2), and untreated (n = 5) groups, 72 hours postinjection. D, PET ROI quantification of [89Zr]-DFO-H1.2F3, in mean % ID/g, for major organs, including tumor tissues for each group. E, PET ROI quantification of [89Zr]-DFO-H1.2F3, in mean % ID/g, for tumors only. Two-tailed unpaired t test was used to compare groups., P < 0.05;, P < 0.01;, P < 0.001; Max, maximum; Min, minimum; NS, not significant; Ipsilateral A/B LN, ipsilateral axillary and brachial lymph nodes; Contralateral A/B LN, contralateral axillary and brachial lymph nodes.
Ex vivo autoradiography and IHC validation
To confirm the differences in CD69 expression and the biodistribution of [89Zr]-DFO-H1.2F3 between the tumors of responder versus nonresponder and untreated groups, we performed ex vivo autoradiography and IHC validation. Autoradiography corroborated biodistribution analyses showing increased tumor uptake (% ID/mm2) for responder relative to nonresponder and untreated mice (Fig. 4A; Supplementary Fig. S5C and S6D). For complementary validation, chromogenic IHC detection methods were used to probe for both CD69 expression and the presence of [89Zr]-DFO-H1.2F3. Anti-CD69 staining detected increased CD69-expression, which corresponded to increased anti–[89Zr]-DFO-H1.2F3 detection, for tumor sections of responding mice relative to tumor sections from nonresponders and untreated mice (Fig. 4B). In addition, lineage markers (e.g., CD45, CD4, and CD8) correlated with CD69 expression in responding mice tumors (Fig. 5). Tumor sections from nonresponding mice demonstrated uniformly low CD69 expression, in comparison with CD45, CD4, and CD8, which had low, but more variable, expression.
Figure 4.
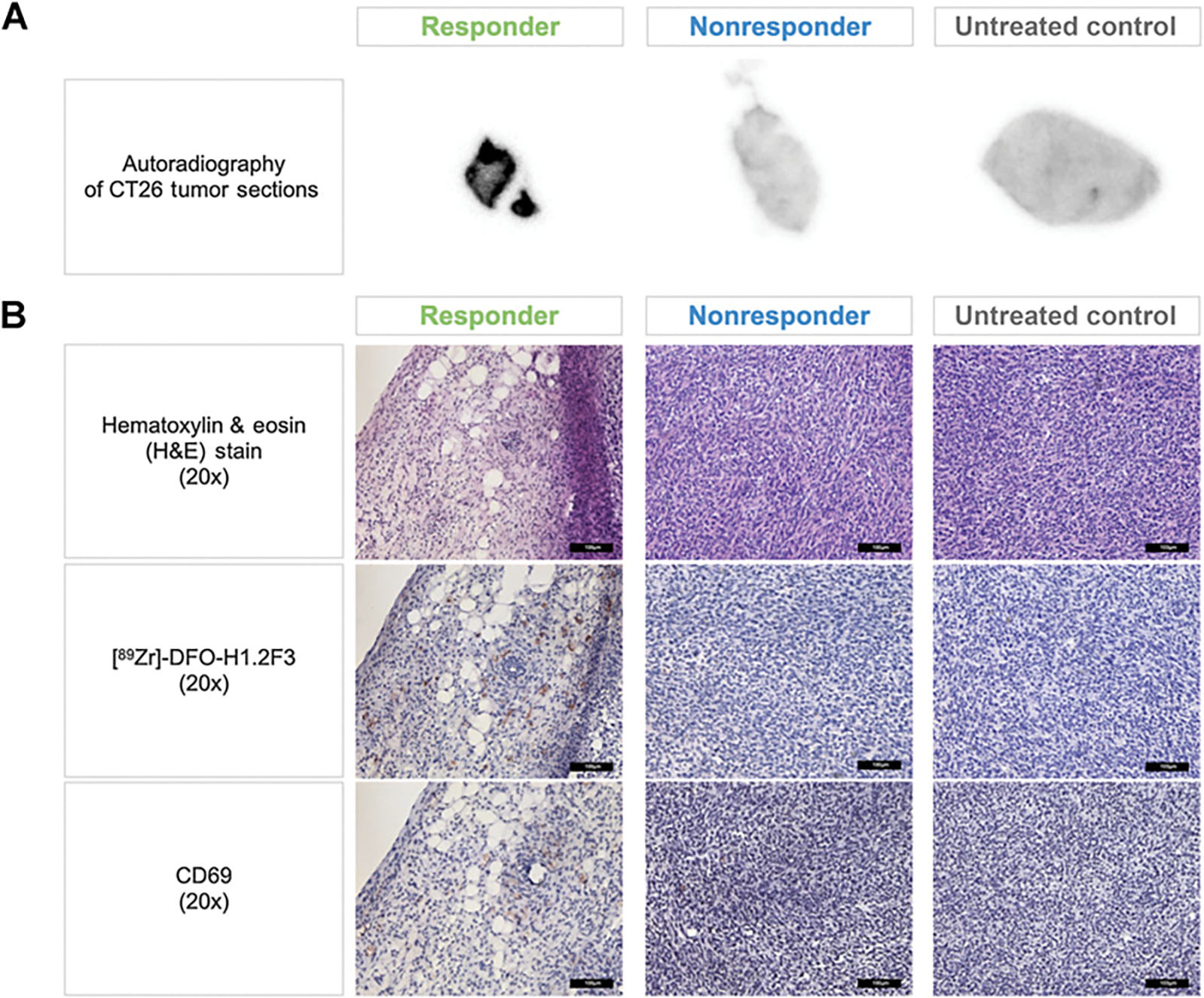
Validating CD69 expression in a CT26 syngeneic tumor immunotherapy model using ex vivo autoradiography and IHC. A, Representative autoradiography images of whole tumor sections from responders, nonresponders, and untreated control groups. B, 20x magnified histological and IHC images of CD69 expression (NovaRED) on tumors sections from responders, nonresponders, and untreated control groups. Scale bar, 100 μmol/L.
Figure 5.
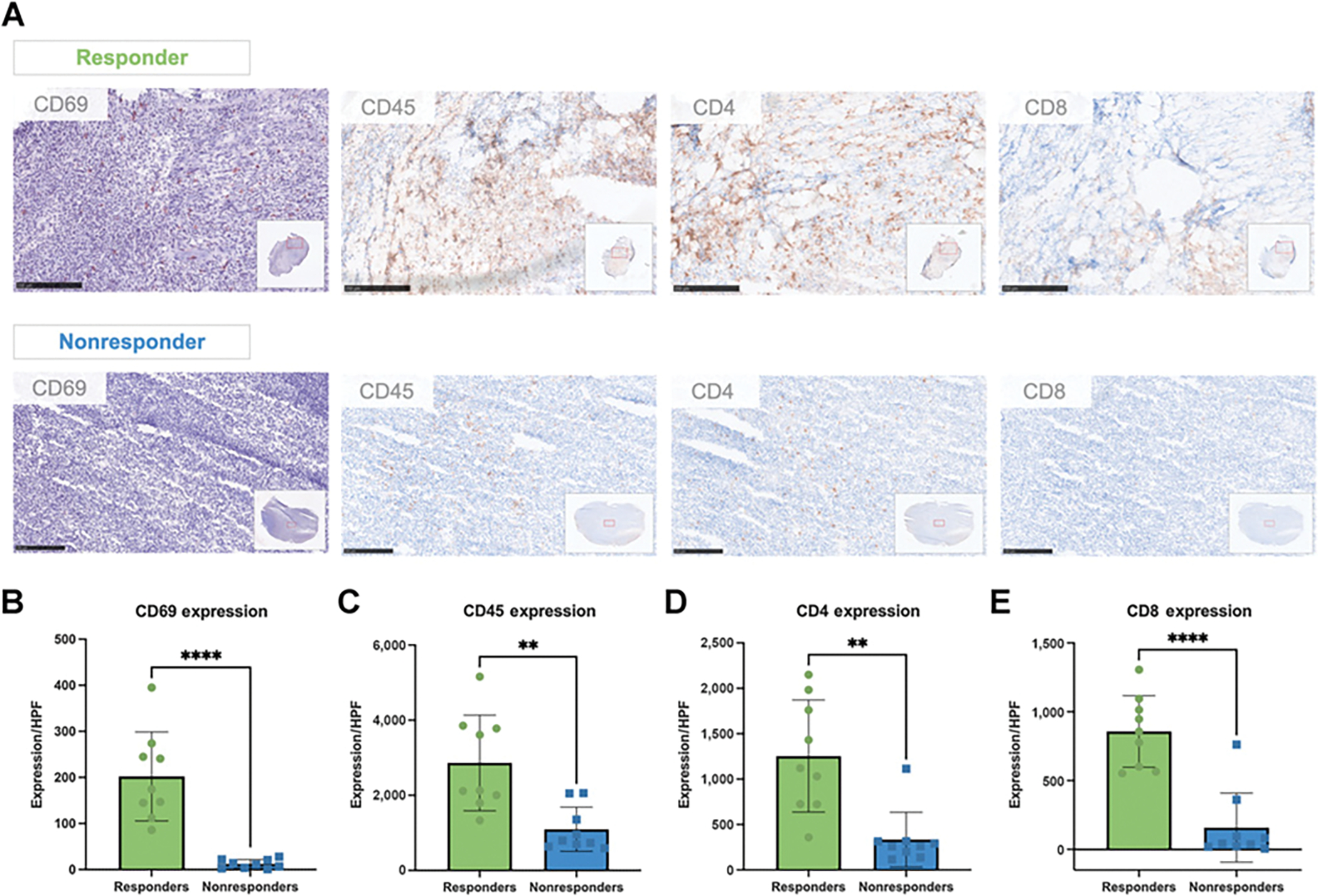
Correlating CD69 IHC with immune lineage markers. A, Representative tumor sections from responder and nonresponder mice. Tumor sections were assayed for the CD69 activation marker using NovaRED, and for CD4, CD8, and CD45 lineage markers using DAB staining. Scale bar, 250 μmol/L. B to E, Quantification of the number of positively stained cells per high power field (HPF) for responder and nonresponder tumor sections, at 10x magnification. Scale bars, 250 μm. Two-tailed unpaired t test was used to compare groups., P < 0.05; **, P < 0.01; ***, P < 0.001;, P < 0.0001. n = 3 tumors per group with three measurements per HPF per tumor.
To support CD69 PET imaging as a biomarker in the clinic, we used The Human Protein Atlas to compare the distribution of CD69 expression across normal tissues and cancer types (Supplementary Fig. S7A and S7B). CD69 expression was not detected in the majority of normal tissues analyzed (40 out of 45), with the remaining five tissues demonstrating medium CD69 expression (appendix and tonsil) or low CD69 protein expression (spleen, lymph node, and bone marrow; ref. 23). In addition, there was no CD69 expression across a wide variety of cancer types, including the majority of lymphoma patient samples (24).
Discussion
The CD69 transmembrane protein is one of the first biomarkers expressed on immune cells upon activation; it is regarded as a canonical marker of leukocyte activation and is known for its rapid surface expression on T cells in vitro ≤2 hours poststimulation (17, 18). We found that, as expected, CD69 is rapidly upregulated in vitro upon stimulation, and other groups have shown that CD69 can be upregulated for longer periods of time in the setting of chronic stimulation (25, 26). CD69 is also expressed on various other types of immune cells upon activation, including NK cells, B cells, monocytes, neutrophils, and eosinophils (27). Therefore, using CD69 as a biomarker for immune-response monitoring takes advantage of its rapid surface expression on a wide variety of immune cells postactivation. This may be benefical relative to other known approaches for capturing immune activation in tumors that may have vastly different immune cell populations. Targeting CD69 also takes advantage of the dynamic range (>15x) in CD69 expression between activated cells and unstimulated cells, a critical feature for high contrast imaging, which may be especially important given the expected heterogenetity of CD69 expression within the target tissue. In this study, we show that CD69 is a promising biomarker for early assessment of response to immunotherapy by developing a 89Zr-radiolabeled CD69-targeting mAb, [89Zr]-DFO-H1.2F3, which exploits the affinity and specificity of the mAb, as well as the noninvasive and sensitive characteristics of PET imaging.
We performed an ex vivo biodistribution experiment in BALB/c (immunocompetent) and NSG (immunodeficient) mice, which demonstrated similar [89Zr]-DFO-H1.2F3 uptake in both strains of mice across a wide variety of normal organs, with the exception of the spleen, which is known to contain CD69+ cells. This suggests that at baseline, in a healthy immunocompetent animal, basal CD69 expression is very low in most tissues other than the spleen, and similar to NSG mice that are deficient in lymphoid cells and thymic structures.
A validated syngeneic CT26 colon carcinoma cancer immunotherapy model (21) was used to compare differences in [89Zr]-DFO-H1.2F3 uptake between mice from the responder group and mice from the nonresponder group. One of the advantages of this model is a stochastic split between responders (n = 6) and nonresponders (n = 4) on treatment. [89Zr]-DFO-H1.2F3 was able to quantify and confirm differences in CD69 expression within the tumor (>4-fold between responders and nonresponders), which correlated with each mouse’s treatment response status. As also observed from the healthy BALB/c mouse ex vivo biodistribution experiment, there was low uptake in most background tissues correlating to low CD69 expression for the syngeneic CT26 colon carcinoma cancer immunotherapy BALB/c model. Hepatobiliary clearance was observed (high liver uptake) as expected for an antibody conjugate. Low blood pool activity in BALB/c mice, due to mild basal CD69 expression in the spleen and some target-mediated clearance, were also observed. Some secondary lymphoid organs (i.e., spleen and tumor dLNs) in responders had increased uptake relative to nonresponders and/or untreated mice, suggesting that responding mice have both regional and systemic increases in immune cell activation that can be detected with CD69 PET imaging. Primary lymphoid organs (i.e., thymus and bone marrow) had higher [89Zr]-DFO-H1.2F3 uptake relative to blood pool across all three groups. However, no significant difference in uptake between the groups was observed for this organ type.
Following the ex vivo biodistrubution study using the CT26 immunotherapy model, a similar study was conducted to further validate [89Zr]-DFO-H1.2F3 biodistribution through PET imaging. Standardized uptake values (SUV) were used to quantify [89Zr]-DFO-H1.2F3 uptake at 72 hours after probe injection across various tissues. The PET imaging study corroborated the ex vivo biodistribution, showing increased tumor uptake for responding mice relative to nonresponders and untreated groups. Significant differences in uptake between responders and nonresponders were also observed for the spleen and ipsilateral superficial/cervical tumor dLNs. OTB calculations were done to account for tissue uptake relative to blood pool, which further confirmed increased tumor uptake for responders relative to nonresponders. The differences in tumor uptake between responders and nonresponders were also qualitatively evident from the maximal intensity projection images alone. Future studies will assess the optimal time of imaging with respect to radiotracer uptake time, for example testing imaging at 24, 48, and 72 hours after radiotracer administration. Of note, there is a difference in radiosignal (% ID/g) from the ex vivo biodistribution and the PET imaging data from each organ. For example, the spleen in responder mice measures approximately 34% ID/g by ex vivo biodistribution whereas the PET responder spleen signal averages approximately 14% ID/g. This difference may be attributable to different unlabeled antibody masses in our experimental design (e.g., ~10 μg ex vivo and ~40 μg for PET) and technical differences in the instrumentation (e.g., gamma counter compared with the animal PET scanner, where partial volume effects can be present).
Autoradiography and IHC of tumor samples showed a correlation between [89Zr]-DFO-H1.2F3 signal and CD69 protein expression. Chromogenic detection showed marked increased [89Zr]-DFO-H1.2F3 uptake and localization within the tumors of responders relative to tumors from nonresponders and untreated groups, which related to increased CD69 protein expression in similar regions for tumors from responding mice. There was also a correlation between CD69 expression and lineage markers, which matches what was previously seen with granzyme B probes and the CT26 colon carcinoma model (28). In addition, nonresponding tumors demonstrated uniformly low CD69 expression, highlighting the specificity of CD69 for response to immunotherapy, in comparison with lineage markers (e.g., CD45, CD4, and CD8) that had more variable expression in nonresponding tumors. This data supports CD69 PET imaging as a potential biomarker for imaging activated immune cells throughout the body in patients with a variety of malignancies treated with ICIs.
It is understood that immune cell activity and composition within the tumor microenvironment varies across tumor types and from patient to patient. Some tumors may display features characteristic of immunologic ignorance, and typically contain key molecular players dampening cytotoxic antitumor immunity. Within this environment, CTLs may be constrained to the tumor border and excluded from the core of the TME. TME stressors, chronic stimulation, and checkpoint inhibitory interactions may also cause these CTLs to become dysfunctional with a marked depletion in the expression of activation markers (29). Therefore, due to a reduction in T-cell, B-cell, and dendritic cell populations, a lack of cytotoxic immune cell infiltration, and reduced antitumor cytotoxic activity, we would expect the total expression of CD69 protein, and corresponding [89Zr]-DFO-H1.2F3 uptake, to be relatively low in these tumors. On the other hand, during an effective antitumor response, immunologically active tumors allow the infiltration of activated effector T lymphocytes and other antitumor immune cells. For example, in addition to T cells, activated NK cells contribute to antitumor immunity through the direct killing of tumor cells (30). Activated dendritic cells are key to helper and cytotoxic T-cell priming, and subsequent differentiation and proliferation in response to cancer. Dendritic cells from the TME traffic to the dLNs to prime T cells for antitumor cytotoxic activity while also transferring tumor antigens to resident dendritic cells in the dLNs (31). Primed CD4+ and CD8+ T cells then traffic from the dLNs to the TME for effective tumor killing. Therefore, due to an increase in activated T-cell, NK-cell, B-cell, and dendritic cell populations, robust cytotoxic immune cell infiltration, and increased antitumor cytotoxic activity, we would expect the total expression of CD69 protein, and corresponding [89Zr]-DFO-H1.2F3 uptake, to be relatively high in immunologically active tumors and secondary lymphoid organs. This is supported by the differences observed in uptake for the tumor and spleen between responders versus nonresponders and untreated groups, and may suggest that the spleen is involved in a favorable response to ICIs. A difference in [89Zr]-DFO-H1.2F3 uptake for tumor dLNs was also observed between responders and untreated groups.
Alternative biomarkers for imaging immune cell activation have taken a more specific approach, e.g., targeting ICOS (13) and OX40 (15) expression, which are restricted to activated T cells, or granzyme B (12), which is produced by cytotoxic T cells and NK cells. While there are advantages to targeting activated T cells or cytotoxic activity specifically, we believe that there is also considerable benefit to concurrently capturing the diverse repertoire of activated immune cells throughout the body, because noncytotoxic activated cells also play crucial roles in launching effective anticancer immune responses.
Our results suggest that CD69 PET imaging has potential for whole body, noninvasive, and serial assessments of immunotherapy-induced immune activation. This CD69 PET imaging approach detected CD69 expression with sufficient sensitivity to quantify immune cell activation in a syngeneic mouse immunotherapy model. The concurrent monitoring of various subsets and populations of activated immune cells allows for the capturing of a broader range of immune activity across the tumor macroenvironment, which could be beneficial for assessing immune response at the earliest stages of response to therapy. Future preclinical experiments will address whether CD69 PET imaging can be used to assess response prior to an anatomic response, with correlative ex vivo validation of immune cell activation and infiltration. Additional studies will be designed to assess whether radiotracer doses of H1.2F3 affect immune activation and function, as has been previously shown for therapeutic doses (32). As researchers develop new immunomodulating agents which may be used concurrently with existing therapies, monitoring and understanding cancer immunotherapies will become more challenging in both preclinical and clinical realms. The long-term clinical goal of this work is to test whether CD69 imaging at the onset of immunotherapy administration is a potential key to assessing response at earlier time points. Our preclinical [89Zr]-DFO-H1.2F3 studies suggest the potential of CD69 as a biomarker for imaging patient response in the clinic and may allow monitoring of immune response to novel immunotherapies and therapeutic combinations, providing a complementary approach to existing techniques for understanding immune activation.
Supplementary Material
Acknowledgments
We thank members of the University of Pennsylvania Cell Center Service Facility (Sabine Baxter), the Small Animal Imaging Facility (Eric Blankemeyer), the Flow Cytometry and Cell Sorting Facility, and the Pathology Clinical Service Center. We thank Dr. Ethan Shevach, at the NIH, for the derived H1.2F3-producing hybridoma cell line, and Daniel Jacome for review of the manuscript. Figures 1A, 1B, 3A, 4A, and 4C (mouse orientation) were created using Biorender.com.
Project funding was provided by Bristol Myers Squibb.
Other funding sources include Presidential Ph.D. Fellowship (University of Pennsylvania; to K.J. Edwards); Radiological Society of North America (RSNA) Resident Research Award and the NIH (NIH/NIBIB grant no. T32 EB004311, to B. Chang).
Footnotes
The costs of publication of this article were defrayed in part by the payment of page charges. This article must therefore be hereby marked advertisement in accordance with 18 U.S.C. Section 1734 solely to indicate this fact.
Authors’ Disclosures
K.J. Edwards reports grants from Bristol Myers Squibb during the conduct of the study. K. Lohith reports grants from Bristol Myers Squibb during the conduct of the study. M.D. Farwell reports grants from Bristol Myers Squibb during the conduct of the study; grants from Merck; grants and personal fees from ImaginAb; and grants from Carisma Therapeutics outside the submitted work. M.A. Sellmyer reports grants from Bristol Myers Squibb during the conduct of the study; other support from Vellum Biosciences; and grants and personal fees from AstraZeneca outside the submitted work; in addition, M.A. Sellmyer has a patent for TMP related patents pending and issued to/optioned to Vellum. No disclosures were reported by the other authors.
Supplementary data for this article are available at Cancer Immunology Research Online (http://cancerimmunolres.aacrjournals.org/).
References
- 1.Galon J, Bruni D. Tumor immunology and tumor evolution: Intertwined histories. Immunity 2020;52:55–81. [DOI] [PubMed] [Google Scholar]
- 2.Twomey JD, Zhang B. Cancer immunotherapy update: FDA-approved checkpoint inhibitors and companion diagnostics. AAPS J 2021;23:39. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 3.Haslam A, Prasad V. Estimation of the percentage of US patients with cancer who are eligible for and respond to checkpoint inhibitor immunotherapy drugs. JAMA Netw Open 2019;2:e192535. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 4.Postow MA, Sidlow R, Hellmann MD. Immune-related adverse events associated with immune checkpoint blockade. N Engl J Med 2018;378:158–68. [DOI] [PubMed] [Google Scholar]
- 5.Patel RP, Parikh R, Gunturu KS, Tariq RZ, Dani SS, Ganatra S, et al. Cardiotoxicity of immune checkpoint inhibitors. Curr Oncol Rep 2021;23:79. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 6.Arfe A, Fell G, Alexander B, Awad MM, Rodig SJ, Trippa L, et al. Meta-analysis of PD-L1 expression as a predictor of survival after checkpoint blockade. JCO Precis Oncol 2020;4:1196–206. [DOI] [PubMed] [Google Scholar]
- 7.Strickler JH, Hanks BA, Khasraw M. Tumor mutational burden as a predictor of immunotherapy response: Is more always better? Clin Cancer Res 2021;27:1236–41. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 8.Russano M, Napolitano A, Ribelli G, Iuliani M, Simonetti S, Citarella F, et al. Liquid biopsy and tumor heterogeneity in metastatic solid tumors: The potentiality of blood samples. J Exp Clin Cancer Res 2020;39:95. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 9.Merker JD, Oxnard GR, Compton C, Diehn M, Hurley P, Lazar AJ, et al. Circulating tumor DNA analysis in patients with cancer: American Society of Clinical Oncology and College of American Pathologists Joint Review. J Clin Oncol 2018;36:1631–41. [DOI] [PubMed] [Google Scholar]
- 10.Tavaré R, Escuin-Ordinas H, Mok S, McCracken MN, Zettlitz KA, Salazar FB, et al. An effective immuno-PET imaging method to monitor CD8-dependent responses to immunotherapy. Cancer Res 2016;76:73–82. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 11.Wherry EJ, Kurachi M. Molecular and cellular insights into T cell exhaustion. Nat Rev Immunol 2015;15:486–99. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 12.Larimer BM, Wehrenberg-Klee E, Dubois F, Mehta A, Kalomeris T, Flaherty K, et al. Granzyme B PET imaging as a predictive biomarker of immunotherapy response. Cancer Res 2017;77:2318–27. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 13.Xiao Z, Mayer AT, Nobashi TW, Gambhir SS. ICOS is an indicator of T cell-mediated response to cancer immunotherapy. Cancer Res 2020;80:3023–32. [DOI] [PubMed] [Google Scholar]
- 14.Gibson HM, McKnight BN, Malysa A, Dyson G, Wiesend WN, McCarthy CE, et al. IFNγ PET imaging as a predictive tool for monitoring response to tumor immunotherapy. Cancer Res 2018;78:5706–17. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 15.Alam IS, Mayer AT, Sagiv-Barfi I, Wang K, Vermesh O, Czerwinski DK, et al. Imaging activated T cells predicts response to cancer vaccines. J Clin Invest 2018;128:2569–80. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 16.Wei W, Rosenkrans ZT, Liu J, Huang G, Luo Q-Y, Cai W. ImmunoPET: Concept, design, and applications. Chem Rev 2020;120:3787–851. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 17.Cibrián D, Sánchez-Madrid F. CD69: From activation marker to metabolic gatekeeper. Eur J Immunol 2017;47:946–53. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 18.Simms PE, Ellis TM. Utility of flow cytometric detection of CD69 expression as a rapid method for determining poly- and oligoclonal lymphocyte activation. Clin Diagn Lab Immunol 1996;3:301–4. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 19.de la Fuente H, Cruz-Adalia A, Martinez del Hoyo G, Cibrián-Vera D, Bonay P, Pérez-Hernández D, et al. The leukocyte activation receptor CD69 controls T cell differentiation through its interaction with Galectin-1. Mol Cell Biol 2014;34:2479–87. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 20.Kimura MY, Koyama-Nasu R, Yagi R, Nakayama T. A new therapeutic target: The CD69-Myl9 system in immune responses. Semin Immunopathol 2019;41:349–58. [DOI] [PubMed] [Google Scholar]
- 21.Duraiswamy J, Kaluza KM, Freeman GJ, Coukos G. Dual blockade of PD-1 and CTLA-4 combined with tumor vaccine effectively restores T-cell rejection function in tumors. Cancer Res 2013;73:3591–603. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 22.Kristensen LK, Fröhlich C, Christensen C, Melander MC, Poulsen TT, Galler GR, et al. CD4+ and CD8a+ PET imaging predicts response to novel PD-1 checkpoint inhibitor: Studies of Sym021 in syngeneic mouse cancer models. Theranostics 2019;9:8221–38. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 23.Tissue expression of CD69 – Summary. The Human Protein Atlas. [cited 2021 Aug 4]. Available from: https://www.proteinatlas.org/ENSG00000110848-CD69/tissue. [Google Scholar]
- 24.Expression of CD69 in cancer – Summary. The Human Protein Atlas. [cited 2021 Jul 14]. Available from: https://www.proteinatlas.org/ENSG00000110848-CD69/pathology. [Google Scholar]
- 25.Wherry EJ, Ha S-J, Kaech SM, Haining WN, Sarkar S, Kalia V, et al. Molecular signature of CD8+ T cell exhaustion during chronic viral infection. Immunity 2007;27:670–84. [DOI] [PubMed] [Google Scholar]
- 26.Simonetta F, Alam IS, Lohmeyer JK, Sahaf B, Good Z, Chen W, et al. Molecular imaging of chimeric antigen receptor T cells by ICOS-ImmunoPET. Clin Cancer Res 2021;27:1058–68. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 27.Ziegler SF, Ramsdell F, Alderson MR. The activation antigen CD69. Stem Cells 1994;12:456–65. [DOI] [PubMed] [Google Scholar]
- 28.Goggi JL, Tan YX, Hartimath SV, Jieu B, Hwang YY, Jiang L, et al. Granzyme B PET imaging of immune checkpoint inhibitor combinations in colon cancer phenotypes. Mol Imaging Biol 2020;22:1392–402. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 29.Binnewies M, Roberts EW, Kersten K, Chan V, Fearon DF, Merad M, et al. Understanding the tumor immune microenvironment (TIME) for effective therapy. Nat Med 2018;24:541–50. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 30.Ben-Shmuel A, Biber G, Barda-Saad M. Unleashing natural killer cells in the tumor microenvironment–The next generation of immunotherapy? Front Immunol 2020;11:275. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 31.Ruhland MK, Roberts EW, Cai E, Mujal AM, Marchuk K, Beppler C, et al. Visualizing synaptic transfer of tumor antigens among dendritic cells. Cancer Cell 2020;37:786–99. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 32.Karlhofer FM, Yokoyama WM. Stimulation of murine Natural Killer (NK) cells by a monoclonal antibody specific for the NK1.1 antigen. IL-2-activated NK cells possess additional specific stimulation pathways. J Immunol 1991;146:3662–73. [PubMed] [Google Scholar]
Associated Data
This section collects any data citations, data availability statements, or supplementary materials included in this article.
Supplementary Materials
Data Availability Statement
Data available upon request from the corresponding authors.