

Abstract
Multi-omics analysis is a powerful and increasingly utilized approach to gain insight into complex biological systems. One major hindrance with multi-omics, however, is the lengthy and wasteful sample preparation process. Preparing samples for mass spectrometry (MS)-based multi-omics involves extraction of metabolites and lipids with organic solvents, precipitation of proteins, and overnight digestion of proteins. These existing workflows are disparate and laborious. Here, we present a simple, efficient, and unified approach to prepare lipids, metabolites, and proteins for MS analysis. Our approach, termed the Bead-enabled, Accelerated, Monophasic Multi-omics (BAMM) method, combines an n-butanol-based monophasic extraction with unmodified magnetic beads and accelerated protein digestion. We demonstrate that the BAMM method affords comparable depth, quantitative reproducibility, and recovery of biomolecules as state-of-the-art multi-omics methods (e.g., Matyash extraction and overnight protein digestion). Yet, the BAMM method only requires about 3 hours to perform, which is a savings of 11 steps and 19 hours on average compared to published multi-omics methods. Furthermore, we validate the BAMM method for multiple sample types and formats (biofluid, culture plate, pellet) and show that in all cases it produces high biomolecular coverage and data quality.
Graphical Abstract
Biological systems contain complex networks of diverse molecules that work together to modulate cellular processes. While many technologies interrogate a single biomolecule class, substantially more information can be gained from performing multi-omics studies. Integrated analysis of biomolecules, once a lofty goal, is now feasible and increasingly applied across biological disciplines,1–3 in part due to improvements in mass spectrometry (MS) technologies that enable analysis of small molecules, lipids, and proteins. Evolving MS data acquisition strategies have increased the number biomolecules surveyed;4–13 yet, the demand for faster and more efficient data collection persists. To meet this demand, recent advances have focused on improving integration of MS data acquisition and analysis strategies;14–18 however, other aspects of the MS multi-omics pipeline, such as sample preparation, are still in great need of simplification and consolidation.
Sample preparation for published multi-omics studies generally involved either splitting samples into multiple aliquots for different –omes, or relied on extensive multi-step processes to isolate multiple compound classes.19–28 These multi-step methods frequently require extracting metabolites and lipids with a biphasic organic solvent system, precipitating proteins, and digesting proteins with trypsin (Figure S1). Most commonly, metabolites and lipids are extracted with the biphasic Matyash29 (methyl tert-butyl ether [MTBE], methanol, water) or Folch/Bligh-Dyer30,31 (chloroform, methanol, water) solvent systems, which require careful pipetting of the aqueous and organic portions. While these methods are robust and reproducible, they require copious pipetting, vortexing, incubating, and centrifuging steps. Such steps are low throughput and susceptible to sample loss. After lipid and metabolites are extracted, the protein pellet is washed, dried, and resolubilized. Protein resolubilization in digestion buffer can be difficult and may require sonication or other facilitation methods. Subsequent overnight digestion of proteins adds 12–18 hours to the process, which is followed by desalting with solid phase extraction. A more streamlined sample preparation would allow for a simpler, faster, and more efficient way to process lipids, metabolites, and proteins from a single sample and, when paired with an integrated acquisition method (e.g., multi-omic single-shot technology, MOST14), would allow a single lab or researcher to produce quality multi-omics data.
Here, we aimed to develop a faster, simpler method to prepare samples for multi-omics analysis while permitting high biomolecular coverage and data quality.24–26 To simplify the preparation, we explored a monophasic extraction system leveraging n-butanol’s diverse miscibility,32 with the goal of efficiently recovering both polar and non-polar metabolites. Next, we examined pairing monophasic extraction with paramagnetic bead technology for on-bead protein aggregation. In recent years, functionalized magnetic bead-based protocols have been introduced as an effective way to improve scalability, throughput, and flexibility for proteomics sample preparation, but to our knowledge they have not been tested for compatibility with metabolite and lipid extractions.33–37 Lastly, we propose reducing proteomics sample preparation time by implementing a heated, accelerated on-bead protein digestion with trypsin. Overall, our simplified approach eliminates several manual manipulations and reduces sample preparation time from 18+ to ~3 hours. We term this approach the Bead-enabled Accelerated Monophasic Multi-omics (BAMM) sample preparation for multi-omics analysis.
METHODS
Details regarding sample types, bead preparation, and data acquisition, analysis, and availability are given in the Supporting Information.
Biomolecule extractions.
For all metabolite/lipid extraction methods, samples were removed from −80 °C conditions and immediately placed on ice to thaw (plasma) or directly extracted from frozen material (cells and tissue). All extraction solvents were chilled and of liquid chromatography (LC)-MS grade.
For monophasic extraction with beads, extraction solvent (3:1:1 n-butanol/ACN/H2O) was added to each sample along with washed bead stock to achieve a 10:1 bead-to-protein ratio (or as specified, see Figure S3) and a final water percentage of 20 ± 1%. Cultured cells were detached using a plastic cell scraper39; all other samples were vortexed for 10 s after the addition of solvents and beads. Cells and tissues were also sonicated in a chilled water bath (Qsonica) at 10 °C for a total of 5 min in increments of 20 s on/10 s off, with an amplitude of 30. Beyond facilitating cell lysis, sonication aided in the shearing of DNA40, which resulted in improved proteomics results. Samples were incubated for 5 min on ice (or as specified, see Figure S3) then placed on a magnetic rack for 20 s. The resulting unbound supernatant containing metabolites and lipids was aliquoted into separate autosampler vials and dried with a SpeedVac Vacuum Concentrator (Thermo Scientific).
For extraction without beads, many n-butanol solvent systems were tested (see Figure 2) and benchmarked against a common biphasic extraction system (Matyash29; MeOH/MTBE/H2O, 3:10:2.5 v/v/v) and a monophasic extraction system (MeOH/ACN/H2O, 2:2:1 v/v/v). Solvents were added sequentially in the proportion described. Then, samples were vortexed for 10 s, sonicated for 5 min at 14°C, incubated for 10 min at 4 °C, centrifuged at 14,000 × g for 5 min at 4 °C. The resulting supernatant layers were aliquoted into autosampler vials for metabolite and lipid analyses. For biphasic systems, the organic upper layer was used for lipid analysis and the aqueous bottom layer was used for metabolite analysis.
Figure 2. Development of a monophasic solvent system for metabolite and lipid extraction.
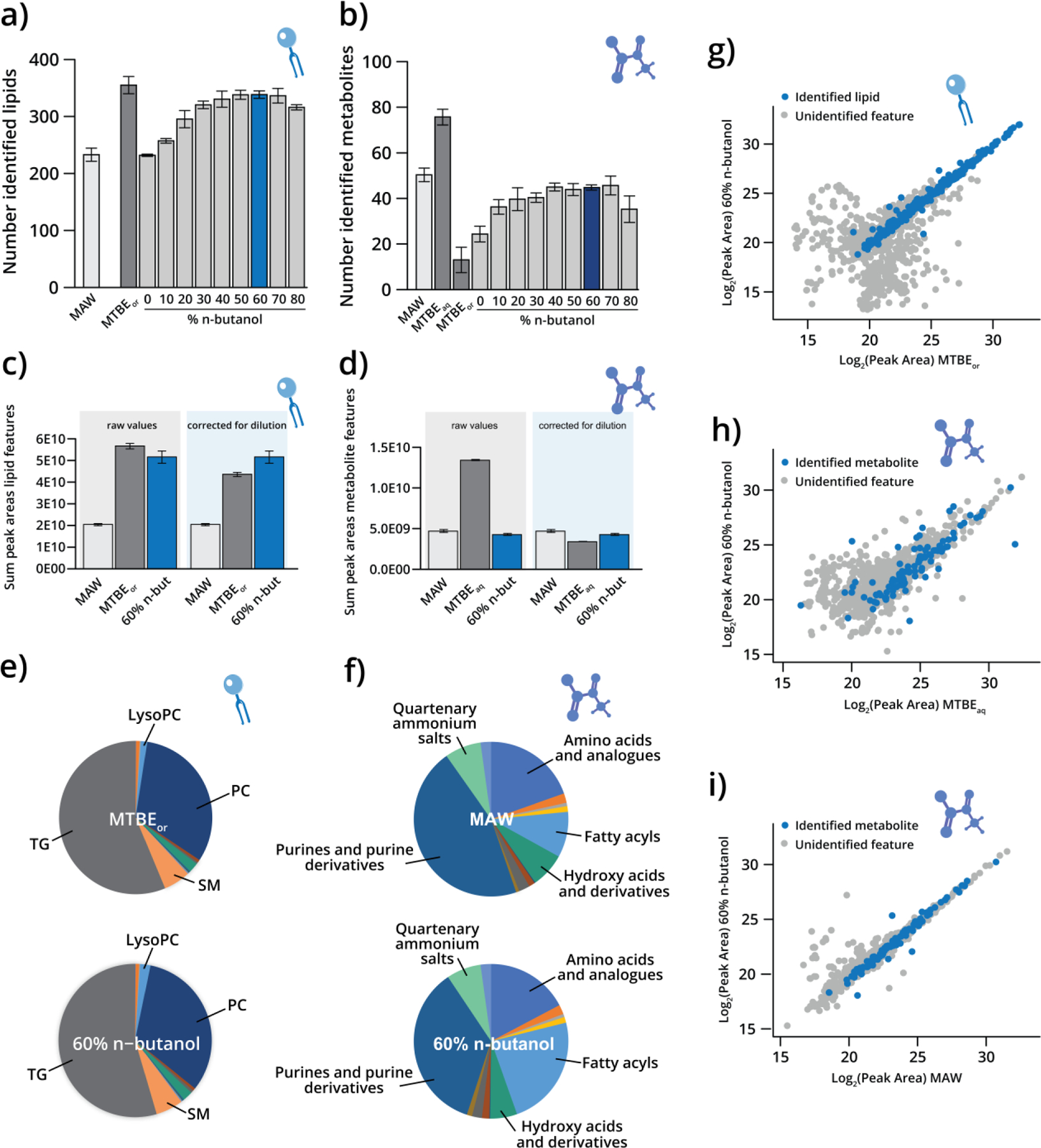
Number of identified a) lipids and b) metabolites in human plasma for the monophasic n-butanol formulations compared to MAW, MTBEaq, and MTBEor controls. The percentage of n-butanol was balanced with H2O (constant 20%) and ACN (0–80%). Sum peak areas of c) lipid features and d) metabolite features before and after dilution correction for MAW, MTBE, and 60% n-butanol/20% H2O/20% ACN (“60% n-butanol”) extractions. Data are presented as mean ± standard deviation. Class distributions for identified e) lipids and f) metabolites extracted with MAW, MTBE, and 60% n-butanol. Relative quantification in log2(peak area) correlation plots for g) lipids extracted with MTBEor and 60% n-butanol; h) metabolites extracted with MTBEaq and 60% n-butanol; and i) metabolites extracted with MAW and 60% n-butanol.
Accelerated protein digestion with paramagnetic beads.
For on-bead accelerated protein digestion (Figures 4, S4, 5, S5), after the metabolite and lipid supernatant was removed, the bead-protein mixture was reconstituted in digestion solution (Rapid Digestion Buffer [Promega] diluted to 75% from stock with spiked-in 5 mM TCEP, 20 mM CAA). Rapid Trypsin (Promega) was added in a 10:1 protein/enzyme ratio. The samples were incubated on a thermal mixer (Benchmark Scientific) at 1,000 rpm for 40 min at 60 °C (or as specified, see Figure S4). Then tubes were placed on the magnetic rack, and the supernatant was recovered and acidified with trifluoroacetic acid to pH ~2. The resulting peptides were desalted with Strata-X Polymeric Solid Phase Extraction cartridges (Phenomenex) and dried as described above.
Figure 4. Reducing overall time for proteomics sample preparation.
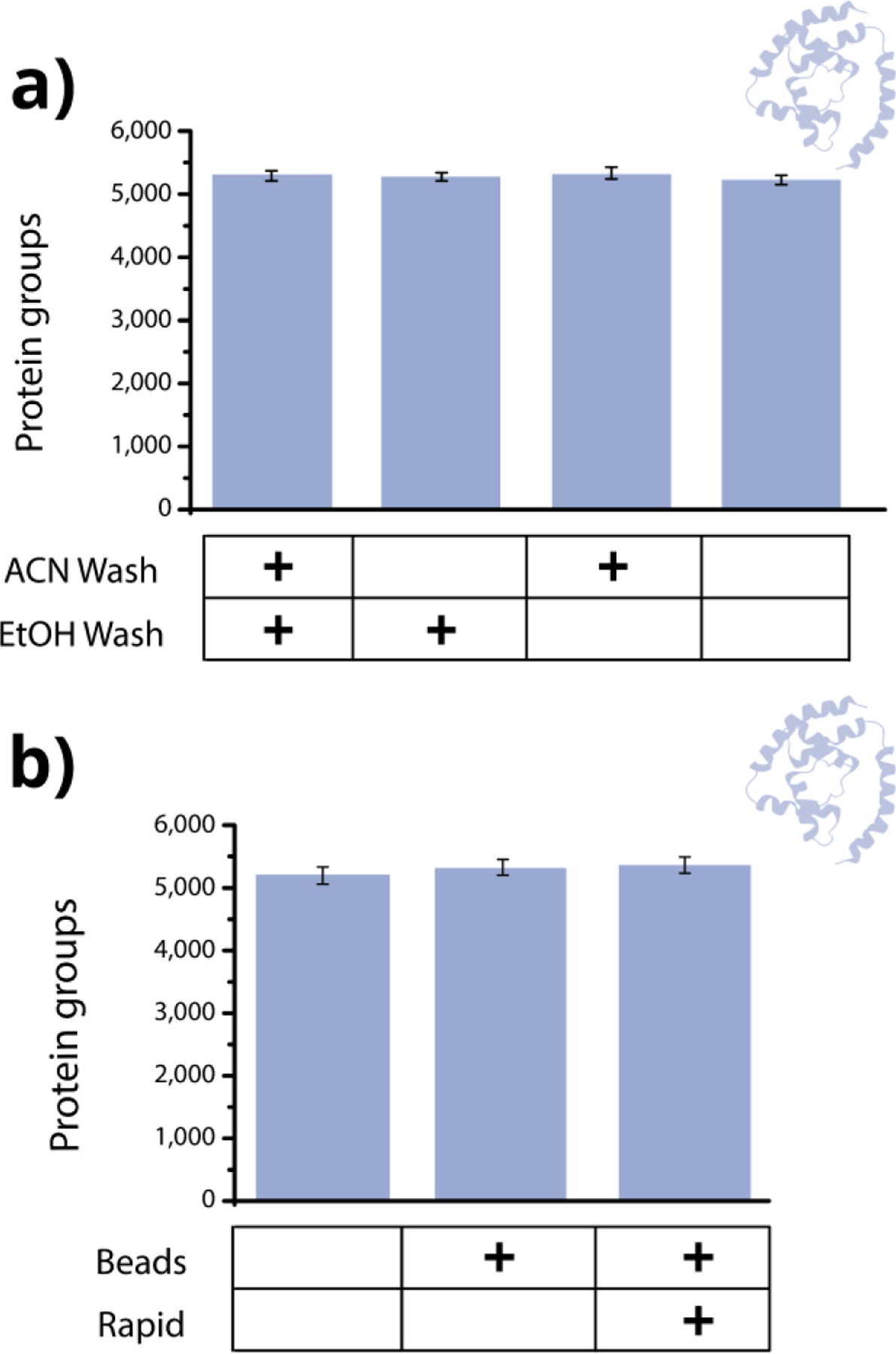
a) Number of identified proteins from a mouse brain digest when using beads and performing both the 100% acetonitrile wash and 70% ethanol wash, only the 70% ethanol wash, only the 100% acetonitrile wash, and neither wash. b) Number of identified proteins from mouse brain digested overnight without beads, overnight with beads, and at 60 °C with beads. The beads used were SeraSil-Mag 700 nm beads.
Figure 5. Versatility and performance of BAMM Prep method.
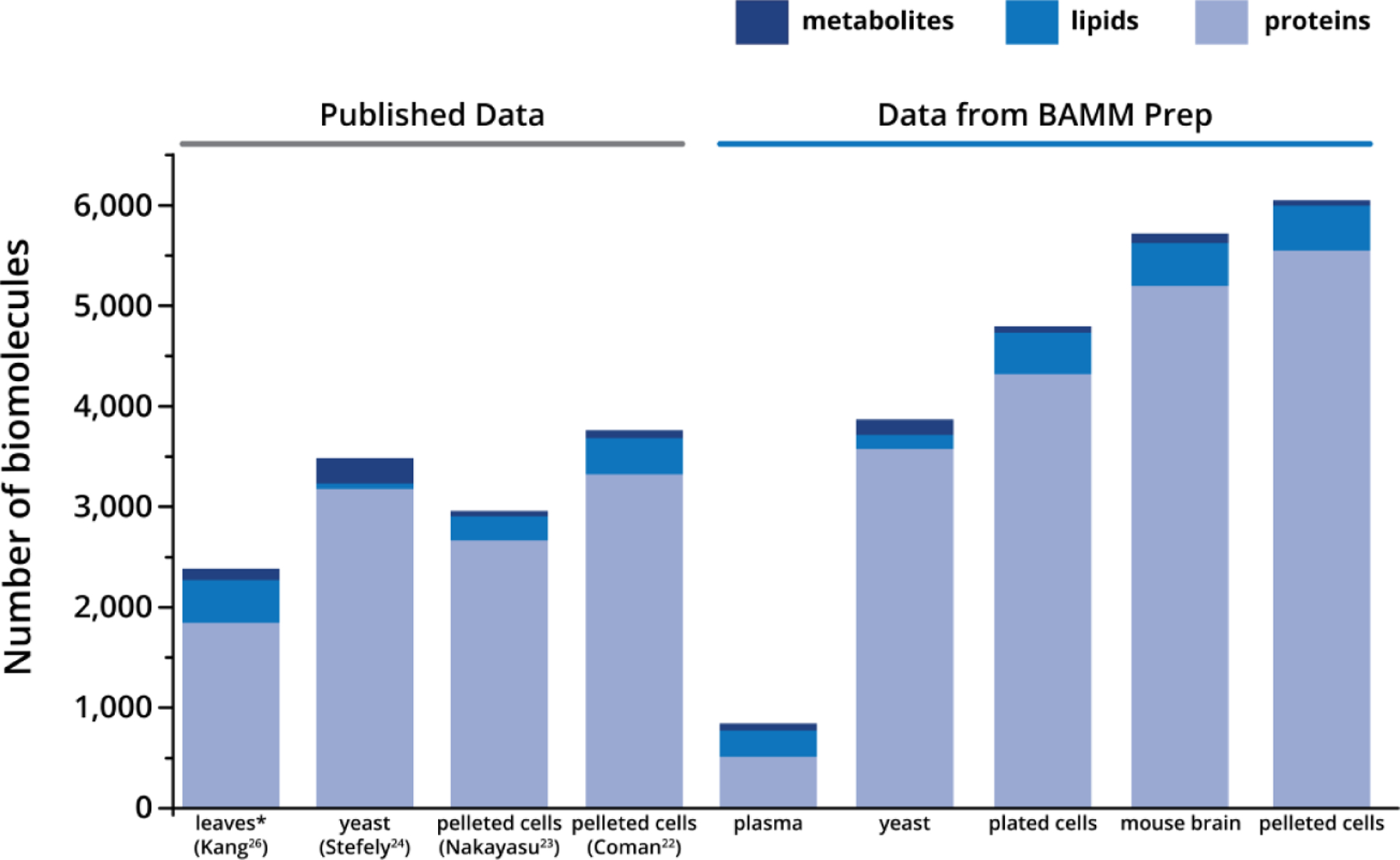
The four lefthand columns (“Published Data”) show the number of proteins, lipids, and metabolites identified from published multi-omic sample preparation workflows. All numbers were taken as reported in the results sections of the publications; however, the reported number of metabolites in Kang et al. was reduced to only those that matched the criteria for metabolite identifications used in this manuscript. The five righthand columns (“Data from BAMM Prep”) show the number of identified proteins, lipids, and metabolites from our streamlined multi-omic sample preparation (numbers are the average of three replicates). The specific samples are as follows: A. thaliana (Kang), S. cerevisiae (Stefely), Calu-3 cells (Nakayasu), mesenchymal stem cells (Coman), and NIST 1950 plasma, S. cerevisiae, cultured mouse adipocytes, C57BL/6J mouse brain, HEK293 (BAMM Prep)
Overnight protein digestion with paramagnetic beads.
When on-bead overnight protein digestion was performed (Figures 3, S3, 4, S4), the metabolite and lipid supernatant was removed, and the bead-protein mixture was reconstituted in 50 mM Tris, 10 mM TCEP, 40 mM CAA. Trypsin (Promega) was then added in an estimated 50:1 protein/enzyme ratio. The samples were incubated overnight at room temperature on a rocker and acidified, desalted, and dried as described above. Note that originally, the bead-protein mixture was washed with 100% acetonitrile and 70% ethanol immediately following supernatant removal; however, these steps were removed in the final workflow as they minimal impact on results (see Figure 4).
Figure 3. Determining the optimal type of magnetic bead for bead-based multi-omics sample preparation.
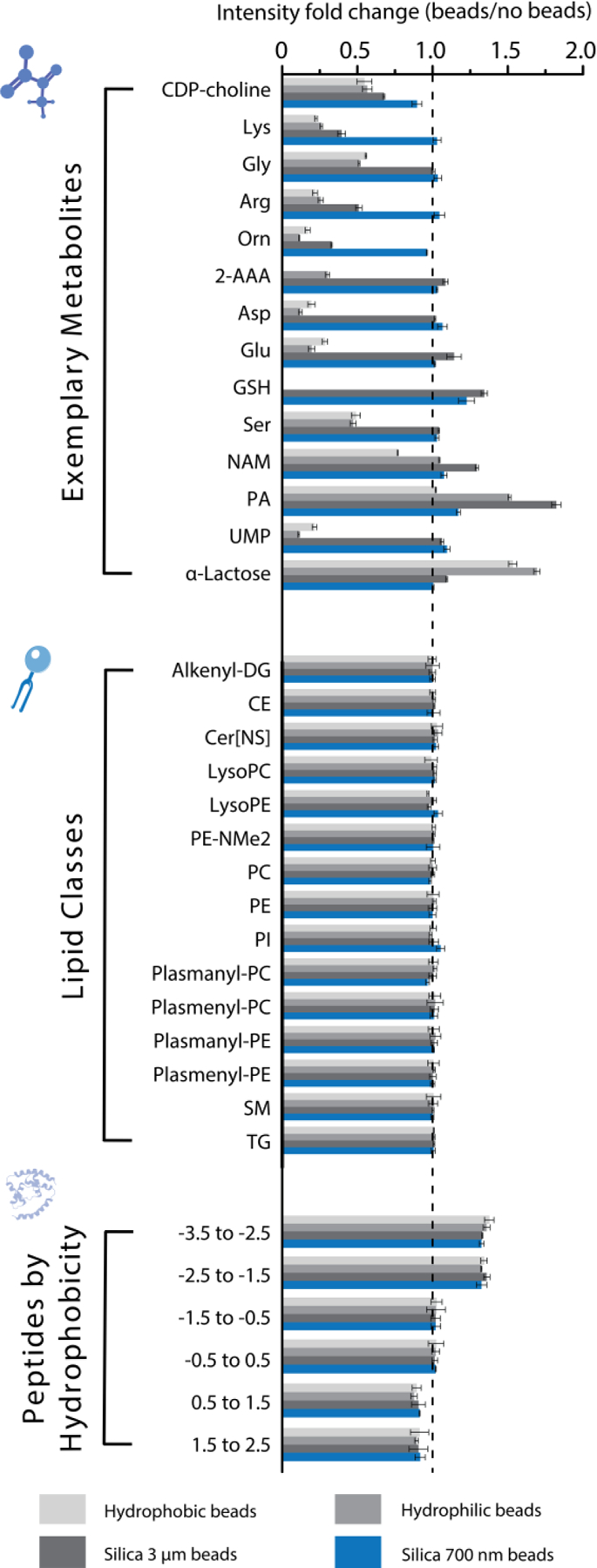
Fold change in intensity of common metabolites, lipid classes (summed), and peptide GRAVY score between each bead type and no beads (MTBE extraction) from human plasma. Each bar represents the average of three technical replicates. Bead types and venders are referenced in the text. The abbreviations used for the metabolites are as follows: cytidine 5’-diphosphocholine (CDP-choline), lysine (Lys), glycine (Gly), arginine (Arg), ornithine (Orn), 2-aminoadipic acid (2-AAA), aspartic acid (Asp), glutamic acid (Glu), glutathione (GSH), serine (Ser), nicotinamide (NAM), pantothenic acid (PA), and uridine monophosphate (UMP).
Protein digestion without magnetic beads.
For protein digestion without beads, the method was dependent on whether all –omes were analyzed (Figure 3) or the tested variables were only relevant for proteomics (Figures 4, S4). In the former case, metabolite and lipid extracts were removed from the samples, and the protein pellets were washed with acetonitrile. In the latter case, the samples were suspended in lysis buffer (6 M guanidinium hydrochloride, 100 mM Tris) after thawing (plasma) or direct removal from frozen conditions (cells and tissue). Methanol was then added to each sample (90% v/v), and then samples were centrifuged for 5 min at 10,000 × g. Proteins were resolubilized in digestion buffer (8 M urea, 10 mM TCEP, 40 mM CAA, 50 mM Tris) with 7.5 min of sonication. Before digestion, the samples were diluted to a final urea concentration of 1.5 M. Trypsin was added in an estimated 50:1 ratio of protein/enzyme, and the samples were placed on a rocker for overnight incubation at room temperature. The resulting peptides were acidified, desalted, and dried as described above.
RESULTS AND DISCUSSION
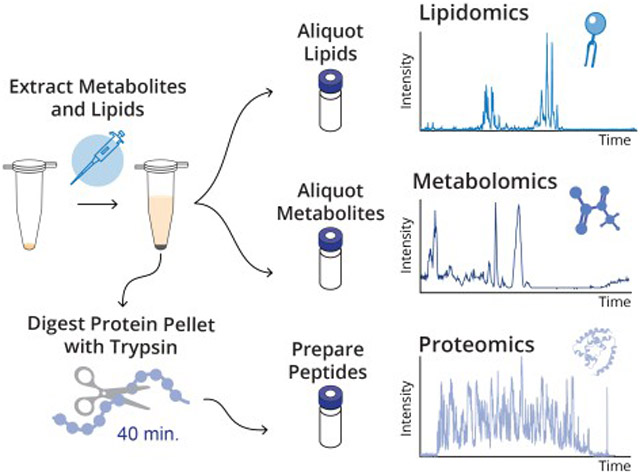
Figure 1 details our simplified BAMM workflow and summarizes the number of steps and duration of BAMM compared to several published methodologies.22,23,24,26 Here, we consider steps to include the following actions: pipetting, incubation, vortexing, centrifugation, sonication, bead beating, and freeze-thaw cycles. The average approach required 22 hours and 28 steps, while the BAMM method takes only 3 hours and 17 steps. Here, we detail the three major improvements to common multi-omics sample preparation workflows that ultimately resulted in the BAMM sample preparation method.
Figure 1. Workflow for BAMM preparation method and comparison to published workflows.
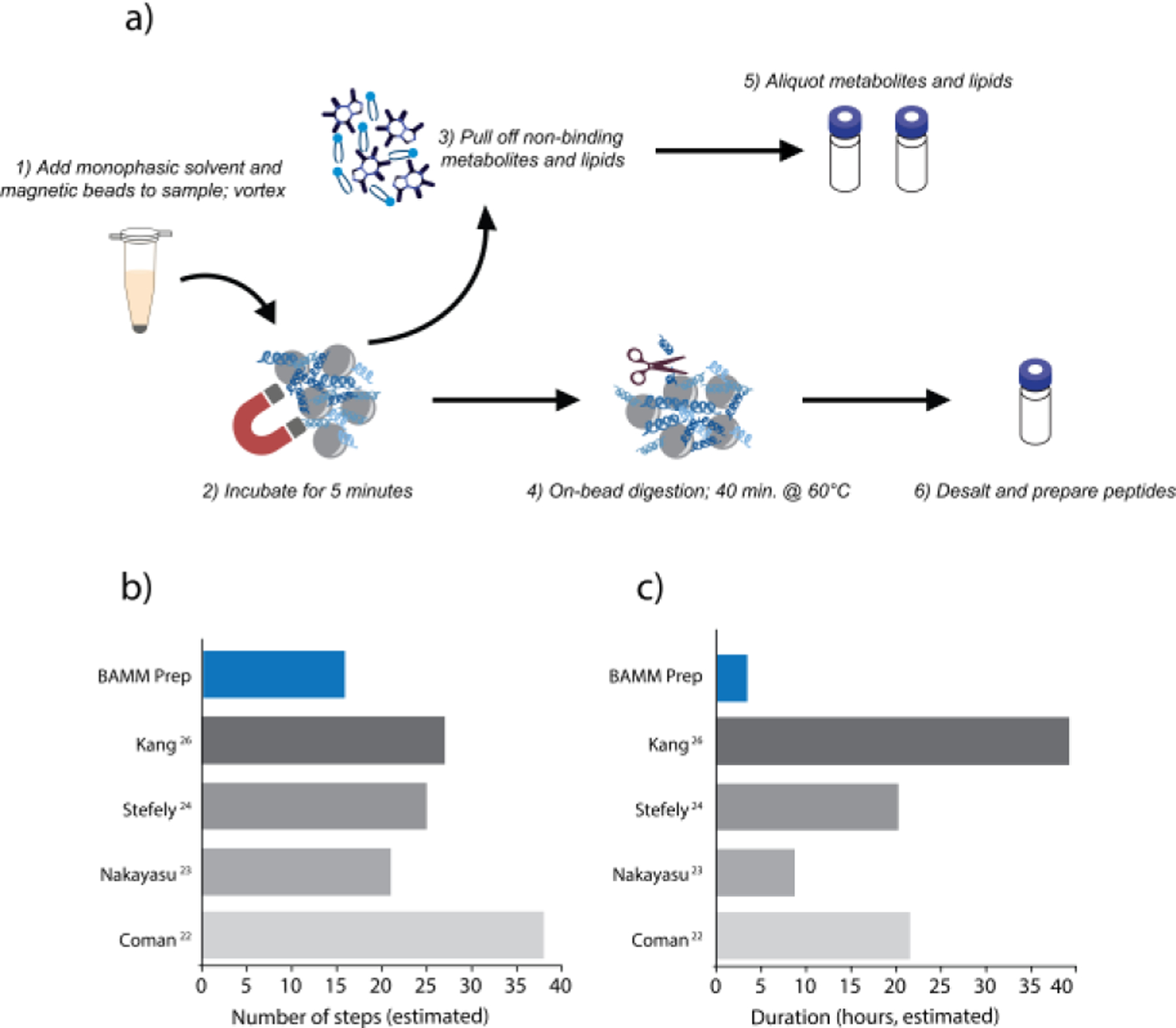
a) To perform the BAMM workflow, n-butanol-based monophasic solvent and magnetic beads are first added to sample (1). After a brief vortex, the sample is incubated on ice for 5 min (2). Unbound metabolites and lipids are then removed (3), and protein is digested for 40 min at 60 °C (4). Metabolites, lipids, and peptides are then prepared for analysis (5–6). Comparison of the estimated number of steps (b) and hours (c) necessary to prepare metabolites, lipids, and proteins with the BAMM method and various published methods.
Monophasic solvent system for lipid and polar metabolite extraction.
In biphasic solvent systems like Matyash, Folch, or Bligh-Dyer29–31, lipids partition into a strongly lipophilic solvent, while polar metabolites partition into the aqueous phase. In contrast, current monophasic extraction methods, tend to preferentially extract either lipophilic or polar metabolites.39,44–48 With the goal of developing a monophasic solvent system that recovers both lipids and small molecules with high efficiency, we looked toward aqueous n-butanol mixtures, as they have been described32 to contain properties compatible with both polar and non-polar compounds.
First, we tested a range of n-butanol formulations (0–80% n-butanol) for suitability to extract lipids and polar metabolites. The proportion of water was maintained at 20% (v/v), and acetonitrile was used to balance the proportion of n-butanol. From 0–60% n-butanol, the solvents remained miscible; however, at 70% and 80% n-butanol, slight to moderate phase separation was observed. Using 500 μL of each solvent mixture, we extracted 10 μL of human plasma, equal volumes of extract were dried by vacuum centrifugation, resuspended in either lipid or metabolite-compatible solvents for analysis, and analyzed by LC-MS/MS. For the phase-separated n-butanol extracts, the upper layer was used for both metabolite and lipid analyses. The n-butanol formulations were compared to a common metabolomics monophasic solvent48 (2:2:1 MeOH:ACN:H2O, “MAW”) and the traditional biphasic Matyash solvent system29 (10:3:2.5 MTBE:MeOH:H2O, “MTBE”), using the same ratio of plasma to solvent.
To evaluate the extraction solvents, we first assessed the number of lipids and metabolites identified (Figure 2a–b). For lipids, 60% n-butanol/20% ACN/20% H2O formulation yielded the most lipid identifications. For metabolite analysis, the 40–70% n-butanol formulations yielded similar numbers of identified metabolites, but the 60% n-butanol extraction had slightly higher sum metabolite intensity (Figure S2a). Therefore, 60% n-butanol was the best-performing monophasic solvent system when considering both lipids and metabolites. In comparison to the MAW control, the 60% n-butanol yielded markedly more lipids and a similar number of metabolites. The MTBE control recovered more identifications for both lipids and metabolites; however, this difference was due to MTBE being more highly concentrated (Figure S2b). When correcting for concentration differences, the 60% n-butanol extraction system compared favorably with the MTBE extraction for both lipids and metabolites (Figure 2c–d). Likewise, if the entire monophasic extract is used for either lipid analysis or metabolite analysis, the resulting number of identified metabolites and lipids is not different between MTBE and 60% n-butanol extractions (Figure S2c–d), and the sum feature intensities for these analyses closely mirror the dilution corrected values in Figure 2c–d (Figure S2e–f). For comparison, overlaid chromatograms of 60% n-butanol and both controls are shown in Figure S2g–h.
After determining that 60% n-butanol was optimal, we assessed class distributions of extracted lipids and metabolites (Figure 2e–f). The MTBE organic phase (MTBEor) and 60% n-butanol recovered the various lipid classes in similar proportions. Notably, the 60% n-butanol successfully extracted both hydrophilic lipids, such as lyso-phospholipids, and hydrophobic lipids, such as triglyceride lipid species. For metabolomics, we compared 60% n-butanol to MAW and the MTBE aqueous phase (MTBEaq). The distribution of metabolite classes recovered by MAW and 60% n-butanol were similar, but MAW recovered a larger percentage of purines and purine derivatives, while 60% n-butanol recovered more fatty acyls. Comparing the distribution of metabolite classes recovered between 60% n-butanol and MTBEaq (Figure S2i), we observed greater differences in the diversity of compounds extracted, which we believe is due to the difference in metabolite recoveries and affinities between the two solvent phases of the MTBE extraction system (Figure S2j). Calculating extraction recoveries revealed not only high overall recovery of both lipid (mean 85 ± 6%) and amino acid (mean 99 ± 8%) standards with 60% n-butanol, but also that the mean recovery by 60% n-butanol was higher than MTBEaq for all amino acids. Hydrophobic amino acids are disadvantageously split between the aqueous and organic layers in the biphasic system. For example, recovery of tryptophan is 51% higher with 60% n-butanol compared to MTBEaq.
Finally, we examined quantitative correlation (relative log2(peak area) across samples) between 60% n-butanol and MTBEor for lipids (Figure 2g), 60% n-butanol and MTBEaq for metabolites (Figure 2h), and 60% n-butanol and MAW for metabolites (Figure 2i). Strong correlations were observed for all, indicating that 60% n-butanol recovers lipid and metabolite species in similar proportions as the respective controls. Overall, the 60% n-butanol/20% acetonitrile/20% water monophasic system proved to efficiently recover both lipids and polar metabolites. And compared to biphasic systems, the monophasic system is simpler and does not require careful attention to the phase layers when pipetting extracts. After successfully streamlining this portion of multi-omics sample preparation, we progressed to further aspects of the workflow.
Magnetic beads to facilitate integrated sample preparation.
Expanding on the simplicity of the monophasic solvent for metabolomics and lipidomics sample preparation, we sought to integrate this extraction with paramagnetic bead technology to expedite proteomics preparation. The use of magnetic beads for proteomics (termed the SP3 approach33–36) was introduced in recent years as a streamlined sample preparation platform. The SP3 protocol uses carboxylate-coated hydrophilic magnetic beads in the presence of high organic solvent to induce protein-bead aggregation. Once proteins are immobilized on the surface of the beads, they can be rinsed of contaminants (e.g. chaotropes, detergents), released, and digested. Here, we envisioned a modified SP3 approach that would be amenable to multi-omics. First, magnetic beads would be added to monophasic solvent and sample, and proteins would be allowed to aggregate around the beads during a short incubation period. Unbound metabolites and lipids would be removed for further analysis, and bead-bound proteins would be rinsed, digested, and desalted. Overall, our goal was to eliminate centrifugation and protein resolubilization by combining a bead-based protocol with our monophasic solvent extraction. However, because this SP3 method has not been demonstrated for compatibility with metabolite or lipid analyses, we hypothesized that different functional groups on the beads may influence extractions of metabolites and/or lipids.
We obtained four different types of magnetic beads to test with multi-omics extractions: 1 μm hydrophilic carboxylate, functionalized beads (Cytiva), 1 μm hydrophobic carboxylate functionalized beads (Cytiva), 3 μm unmodified silica beads (G-Biosciences), and 700 nm unmodified silica beads (Cytiva). Even though the SP3 protocol is typically performed with carboxylate functionalized beads, it has been shown33 that proteins are not influenced by specific bead properties and thus aggregate on any available surface upon conditions known to induce aggregation. To examine the performance of the four bead types, we extracted metabolites, lipids, and proteins from plasma with each bead and without beads. We compared the fold changes in intensity between each bead type and the no-bead control for common metabolites, lipid classes, and peptide GRAVY (grand average of hydropathicity index; a measure of hydrophobicity) score range49 (Figure 3). Lipid and peptide identifications were thoroughly unaffected by the type of bead used, as their fold changes for each bead type displayed only minimal fluctuations. Metabolites, on the other hand, were quite affected by the type of bead used; recovery of the most polar metabolites was hindered with the functionalized beads. Recovery improved somewhat with the 3 μm unmodified beads, but the best-performing type was the 700 nm unmodified beads. Extracting metabolites in the presence of the 700 nm unmodified beads led to similar recovery as without beads for all metabolites. Similar results were observed when these experiments were performed with mouse brain for lipids, metabolites, and peptides (data available in repository).
The reduction in the recovery of certain metabolites when using functionalized beads is likely due to inadvertent capture of those metabolites by the bead surface. Interestingly, the 700 nm nonfunctionalized beads avoid this problem, but the 3 μm nonfunctionalized beads avoid it only partially. Some metabolites likely still have a partial interaction with the silica surface50 of the 3 μm beads, and size may potentially play a role. Regardless, the 700 nm unmodified beads were clearly optimal over the other bead types for metabolites; therefore, these beads were chosen for subsequent experiments and our final multi-omics workflow. After establishing the bead type, we verified that bead surface interaction with biomolecules was not time-dependent, as we saw little to no difference in metabolite, lipid, and peptide recovery from plasma when varying the incubation period of the beads with the sample from 5 to 60 min (Figure S3a). We also confirmed that the ratio of bead-to-protein had minimal effect on plasma peptide yields; bead-to-protein ratios of 1:1, 5:1, 10:1, and 20:1 yielded similar results (Figure S3b; identical results in yeast not shown). SP3 protocols recommend a 10:1 bead-to-protein ratio36; therefore, we used this ratio for our final workflow.
These experiments confirmed that the SP3 method for proteomics can be expanded for multi-omics with a nonfunctionalized bead. A bead-based multi-omics workflow not only consolidates the preparation but also eliminates the need for centrifugation and protein resolubilization, as aggregated proteins are digested directly on-bead.
Reducing overall preparation time by accelerating proteomics preparation.
As a final simplification to the multi-omics workflow, we looked for opportunities to reduce the overall time taken for proteomics sample preparation, which is the lengthiest portion of the process. First, we aimed to remove any unnecessary steps, such as wash steps between removal of the metabolite and lipid supernatant and addition of digestion buffer. In current SP3 protocols, it is typical to perform 2–3 wash steps (often with acetonitrile and/or ethanol) before digestion.36,37 The intent of the washes is to remove detergents and contaminants; however, detergents are largely incompatible with MS-based metabolomics and lipidomics, and therefore we reasoned that wash steps are not as necessary with multi-omics samples. We performed the experiments in Figure 3 and Figure S3 with a 100% acetonitrile wash followed by a 70% ethanol wash. However, as shown in Figure 4a, removing both wash steps for a mouse brain digest resulted in only 3% fewer protein identifications (identical result in plasma, not shown). Therefore, the washes did not appear to be necessary and were removed for the final workflow.
Second, we were interested in enabling same-day analysis of all three –omes. Metabolite and lipid samples can typically be prepared and analyzed on the instrument within the same day; however, the typical overnight digestion of proteins stalls peptide analysis until at least the following day. To expedite digestion, we looked to integrate Promega Rapid Trypsin with on-bead protein digestion. The Rapid Trypsin platform reduces protein digestion time to one hour or less by heating samples up to 70 °C, which requires a specialized non-urea buffer and thermostable trypsin.51 We optimized the Rapid Trypsin protocol to be compatible with magnetic beads, which required the sample to be shaken throughout the digestion period (~35% increase vs. not shaken) and the temperature to be lowered to 60 °C from 70 °C (Figure S4a). With these modifications, we reduced the digestion step from 12+ hours to 40 min without loss of depth or quality as compared to both a no-bead and bead overnight digestion (Figure 4b, Figure S4b). Despite this reduced duration, for both the rapid bead and overnight no-bead workflows, semi-tryptic cleavage rates and sequence coverage were comparable (<1% and ~30%, respectively). About 10–20% fewer missed cleavages were observed with the rapid bead workflow compared to the no-bead overnight digestion (which has previously been shown37 for SP3 carboxylate bead workflows).
Interestingly, while the peptide GRAVY score distributions of the rapid bead and overnight no-bead workflows were similar, the bead workflow appears to extract hydrophilic peptides to a greater extent than without beads (Figure S4b). This difference is due to the presence of beads and is not resulting from the n-butanol extraction, as we observed similar increases in hydrophilic peptide intensities when comparing n-butanol extractions with and without beads (data available in repository). The difference appears to be largely driven by the unique peptides identified in the bead workflow versus the non-bead workflow: peptides solely identified with SP3 tended to be more hydrophilic, while those solely identified with in-solution digestion tended to be more hydrophobic. However, the overlap in protein identifications was high (~80% between two replicates without match-between-runs and ~95% with match-between-runs). Previous SP3 datasets33,37 display this bias toward hydrophilicity when using the traditional carboxylate-coated beads, and our data demonstrate that this is also true with unmodified beads. Notably, all bead types that were tested showed similar bias toward hydrophilic peptides.
Final streamlined multi-omics workflow for mass spectrometry analysis. Overall, these three improvements led to a significantly streamlined multi-omics sample preparation workflow (Figure 1). The first step of our streamlined workflow is adding an n-butanol-based monophasic extraction solvent with unmodified magnetic beads to sample. After a brief vortex, samples are sonicated and then incubated on ice for 5 min. The incubation period allows proteins to aggregate onto the beads, while metabolites and lipids remain in the supernatant. The unbound metabolites and lipids are then removed for further analysis. Buffer and trypsin are subsequently added for protein digestion at 60 °C for 40 min, and the resulting peptides are acidified, desalted, and dried. At the conclusion of the workflow, all three –omes can be analyzed by the instruments within the same day as preparation. Our method, termed the BAMM sample preparation method, is simpler and more consolidated than classic methodologies, saving an average of 11 steps and 19 hours when compared to published workflows.
After developing the simplified and consolidated BAMM method, we validated it for multiple sample types and formats (cell pellets, biofluids, cell culture plates) for widespread use (Figure 5, Figure S5). Regardless of the sample type, the BAMM method generates metabolomics, lipidomics, and proteomics data of comparable depth as published multi-omics studies, but at a fraction of the required effort (Figure 5). For plasma, yeast, mouse adipocytes from culture plates, mouse brain, and human cell line pellets, we identified (on average, three technical replicates) 67, 152, 61, 90, 51 metabolites (respectively), 260, 139, 412, 430, 445 lipids (respectively), and 515, 3578, 4322, 5197, 5552 proteins (respectively). Though the metabolite yields achieved with the BAMM method may appear somewhat lower than can be achieved with other methods, our applied filters were strict in confident identification of metabolites (e.g., threshold of 80 for cosine similarity score).
Overall, our approach is highly versatile and will generate quality multi-omics data from any biological system or sample type.
CONCLUSION
Here, we describe a simple and consolidated method to prepare metabolites, lipids, and proteins from a single sample—BAMM. BAMM combines an n-butanol-based monophasic extraction with paramagnetic bead technology, expediting small molecule extraction and protein digestion. Our new strategy produces quality multi-omics data comparable to classic methodologies at a fraction of the time and effort. Prepared metabolites, lipids, and peptides are ready for MS analysis in about 3 hours, compared to about a day on average for current workflows. We also note that due to the use of magnetic beads, BAMM is potentially more amenable to robotic automation and multi-well plate formats for increased throughput.
As with all analytical methods, sample preparation, data acquisition, and data processing will inherently introduce bias, and the resuspension solvents and concentrations would need to be tailored for each analysis (based on solvent compatibility, method sensitivity, etc.). The BAMM sample preparation has been optimized for LC-MS analysis and would likely need to be further tailored for other methods such as gas chromatography or direct infusion MS. However, given widespread use of LC-MS for –omics analyses,18–29 we anticipate the BAMM preparation offers significant advantages over more laborious methods.
Furthermore, the components of this workflow can also be adapted as necessary and used individually. For example, after validating our monophasic solvent extraction, we applied it to a large-scale COVID-19 study for fast lipidomics sample preparation.25 We envision that this expedient method or its individual components may be particularly beneficial for specific applications wherein turnaround time is an important consideration, such as clinical screening, iterative process optimization, rapid process analytics, and large sample screens. These benefits could be amplified even further when pairing this streamlined multi-omics sample preparation with an integrated acquisition method such as MOST.14
Supplementary Material
ACKNOWLEDGMENT
We would like to thank Edrees Rashan and David Pagliarini for the yeast pellets, Hiroyuki Mori and Ormond MacDougald for the cultured adipocytes, and Frances Hundley and Wade Harper for the HEK293 pellets that were included in this manuscript. We would like to thank Kathy Krentz and the University of Wisconsin Animal Models Core for assistance with mouse brain tissue. We would also like to thank Aaron Simmons and Sean Palecek for graciously providing stem cell plates for method development. We gratefully acknowledge funding from the National Institutes of Health (P41GM108538 to J.J.C.).
Footnotes
Supporting Information
The Supporting Information is available free of charge on the ACS Publications website.
Supplemental Materials (PDF) Contains Supplementary Methods; Figures S1–S5, and Table S1.
The authors declare the following competing interest(s): J.J.C. is a consultant for Thermo Fisher Scientific, and J.J.C., L.K.M., K.A.O., A.J., and Y.Z. have filed a provisional patent application on portions of this work through the Wisconsin Alumni Research Foundation.
REFERENCES
- (1).Zhang B; Kuster B Proteomics Is Not an Island: Multi-omics Integration Is the Key to Understanding Biological Systems. Molecular and Cellular Proteomics 2019, 18, DOI: 10.1074/mcp.E119.001693. [DOI] [PMC free article] [PubMed] [Google Scholar]
- (2).Brademan DR; Miller IJ; Kwiecien NW; Pagliarini DJ; Westphall MS; Coon JJ; Shishkova E Argonaut: A Web Platform for Collaborative Multi-omic Data Visualization and Exploration. Patterns 2020, 1 (7), DOI: 10.1016/j.patter.2020.100122. [DOI] [PMC free article] [PubMed] [Google Scholar]
- (3).Krassowski M; Das V; Sahu SK; Misra BB State of the Field in Multi-Omics Research: From Computational Needs to Data Mining and Sharing. Frontiers in Genetics 2020, 11, DOI: 10.3389/fgene.2020.610798. [DOI] [PMC free article] [PubMed] [Google Scholar]
- (4).Hebert AS; Richards AL; Bailey DJ; Ulbrich A; Coughlin EE; Westphall MS; Coon JJ The one hour yeast proteome. Molecular and Cellular Proteomics 2014, 13 (1), 339–347. [DOI] [PMC free article] [PubMed] [Google Scholar]
- (5).Shishkova E; Hebert AS; Westphall MS; Coon JJ Ultra-High Pressure (>30,000 psi) Packing of Capillary Columns Enhancing Depth of Shotgun Proteomic Analyses. Analytical Chemistry 2018, 90 (19), 11503–11508. [DOI] [PMC free article] [PubMed] [Google Scholar]
- (6).Meier F; Geyer PE; Virreira Winter S; Cox J; Mann M BoxCar acquisition method enables single-shot proteomics at a depth of 10,000 proteins in 100 minutes. Nature Methods 2018, 15 (6), 440–448. [DOI] [PubMed] [Google Scholar]
- (7).Hebert AS; Thöing C; Riley NM; Kwiecien NW; Shiskova E; Huguet R; Cardasis HL; Kuehn A; Eliuk S; Zabrouskov V; Westphall MS; McAlister GC; Coon JJ Improved Precursor Characterization for Data-Dependent Mass Spectrometry. Analytical Chemistry 2018, 90 (3), 2333–2340. [DOI] [PMC free article] [PubMed] [Google Scholar]
- (8).Meier F; Brunner AD; Koch S; Koch H; Lubeck M; Krause M; Goedecke N; Decker J; Kosinski T; Park MA; Bache N; Hoerning O; Cox J; Räther O; Mann M Molecular and Cellular Proteomics 2018, 17 (12), 2534–2545. [DOI] [PMC free article] [PubMed] [Google Scholar]
- (9).Kelstrup CD; Bekker-Jensen DB; Arrey TN; Hogrebe A; Harder A; Olsen JV Online Parallel Accumulation-Serial Fragmentation (PASEF) with a Novel Trapped Ion Mobility Mass Spectrometer. Journal of Proteome Research 2018, 17 (1), 727–738.29183128 [Google Scholar]
- (10).Senko MW; Remes PM; Canterbury JD; Mathur R; Song Q; Eliuk SM; Mullen C; Earley L; Hardman M; Blethrow JD; Bui H; Specht A; Lange O; Denisov E; Makarov A; Horning S; Zabrouskov V Novel parallelized quadrupole/linear ion trap/Orbitrap tribrid mass spectrometer improving proteome coverage and peptide identification rates. Analytical Chemistry 2013, 85 (24), 11710–11714. [DOI] [PubMed] [Google Scholar]
- (11).Danne-Rasche N; Coman C; Ahrends R Nano-LC/NSI MS Refines Lipidomics by Enhancing Lipid Coverage, Measurement Sensitivity, and Linear Dynamic Range. Analytical Chemistry 2018, 90 (13), 8093–8101. [DOI] [PubMed] [Google Scholar]
- (12).Perez de Souza L; Alseekh S; Scossa F; Fernie AR Ultra-high-performance liquid chromatography high-resolution mass spectrometry variants for metabolomics research. Nature Methods 2021. 18 (7), 733–746. [DOI] [PubMed] [Google Scholar]
- (13).Hutchins PD; Russell JD; Coon JJ Cell Systems 2018, 6 (5), 621–625.e5. [DOI] [PMC free article] [PubMed] [Google Scholar]
- (14).He Y; Rashan EH; Linke V; Shishkova E; Hebert AS; Jochem A; Westphall MS; Pagliarini DJ; Overmyer KA; Coon JJ LipiDex: An Integrated Software Package for High-Confidence Lipid Identification. Analytical Chemistry 2021, 93 (9), 4217–4222. [DOI] [PMC free article] [PubMed] [Google Scholar]
- (15).Ding J; Blencowe M; Nghiem T; Ha SM; Chen YW; Li G; Yang X Mergeomics 2.0: a web server for multi-omics data integration to elucidate disease networks and predict therapeutics. Nucleic Acids Research 2021, 49 (W1), W375–W387. [DOI] [PMC free article] [PubMed] [Google Scholar]
- (16).Rohart F; Gautier B; Singh A; Lê Cao KA mixOmics: An R package for ‘omics feature selection and multiple data integration. PLoS Computational Biology 2017, 13 (11), DOI: 10.1371/journal.pcbi.1005752. [DOI] [PMC free article] [PubMed] [Google Scholar]
- (17).Lê Cao KA; González I; Déjean S integrOmics: an R package to unravel relationships between two omics datasets Bioinformatics 2009, 25 (21), 2855–2856. [DOI] [PMC free article] [PubMed] [Google Scholar]
- (18).Dugourd A; Kuppe C; Sciacovelli M; Gjerga E; Gabor A; Emdal KB; Vieira V; Bekker‐Jensen DB; Kranz J; Bindels Eric. M. J.; Costa ASH; Sousa A; Beltrao P; Rocha M; Olsen J. v.; Frezza C; Kramann R; Saez-Rodriguez J Causal integration of multi-omics data with prior knowledge to generate mechanistic hypotheses. Molecular Systems Biology 2021, 17 (1), DOI: 10.15252/msb.20209730. [DOI] [PMC free article] [PubMed] [Google Scholar]
- (19).Li X; Zhou X; Teng T; Fan L; Liu X; Xiang Y; Jiang Y; Xie P; Zhu D Multi-omics Analysis of the Amygdala in a Rat Chronic Unpredictable Mild Stress Model of Depression. Neuroscience 2021, 463, 174–183. [DOI] [PubMed] [Google Scholar]
- (20).Xu L; Zhao Q; Luo J; Ma W; Jin Y; Li C; Hou Y; Feng M; Wang Y; Chen J; Zhao J; Zheng Y; Yu D Integration of proteomics, lipidomics, and metabolomics reveals novel metabolic mechanisms underlying N, N-dimethylformamide induced hepatotoxicity. Ecotoxicology and Environmental Safety 2020, 205, DOI: 10.1016/j.ecoenv.2020.111166. [DOI] [PubMed] [Google Scholar]
- (21).Rampler E; Egger D; Schoeny H; Rusz M; Pacheco MP; Marino G; Kasper C; Naegele T; Koellensperger G The Power of LC-MS Based Multiomics: Exploring Adipogenic Differentiation of Human Mesenchymal Stem/Stromal Cells. Molecules 2019, 24 (19), DOI: 10.3390/molecules24193615. [DOI] [PMC free article] [PubMed] [Google Scholar]
- (22).Coman C; Solari FA; Hentschel A; Sickmann A; Zahedi RP; Ahrends R Simultaneous Metabolite, Protein, Lipid Extraction (SIMPLEX): A Combinatorial Multimolecular Omics Approach for Systems Biology. Molecular and Cellular Proteomics 2016, 15 (4), 1453–1466. [DOI] [PMC free article] [PubMed] [Google Scholar]
- (23).Nakayasu ES; Nicora CD; Sims AC; Burnum-Johnson KE; Kim Y-M; Kyle JE; Matzke MM; Shukla AK; Chu RK; Schepmoes AA; Jacobs JM; Baric RS; Webb-Robertson B-J; Smith RD; Metz TO MPLEx: a Robust and Universal Protocol for Single-Sample Integrative Proteomic, Metabolomic, and Lipidomic Analyses. mSystems 2016, 1 (3), DOI: 10.1128/msystems.00043-16. [DOI] [PMC free article] [PubMed] [Google Scholar]
- (24).Stefely JA; Kwiecien NW; Freiberger EC; Richards AL; Jochem A; Rush MJP; Ulbrich A; Robinson KP; Hutchins PD; Veling MT; Guo X; Kemmerer ZA; Connors KJ; Trujillo EA; Sokol J; Marx H; Westphall MS; Hebert AS; Pagliarini DJ; Coon JJ Mitochondrial protein functions elucidated by multi-omic mass spectrometry profiling. Nature Biotechnology 2016, 34 (11), 1191–1197. [DOI] [PMC free article] [PubMed] [Google Scholar]
- (25).Overmyer KA; Shishkova E; Miller IJ; Balnis J; Bernstein MN; Peters-Clarke TM; Meyer JG; Quan Q; Muehlbauer LK; Trujillo EA; He Y; Chopra A; Chieng HC; Tiwari A; Judson MA; Paulson B; Brademan DR; Zhu Y; Serrano LR; Linke V; Drake LA; Adam AP; Schwartz BS; Singer HA; Swanson S; Mosher DF; Stewart R; Coon JJ; Jaitovich A Large-Scale Multi-omic Analysis of COVID-19 Severity. Cell Systems 2021, 12 (1), 23–40.e7. [DOI] [PMC free article] [PubMed] [Google Scholar]
- (26).Kang J; David L; Li Y; Cang J; Chen S Three-in-One Simultaneous Extraction of Proteins, Metabolites and Lipids for Multi-Omics. Frontiers in Genetics 2021, 12, DOI: 10.3389/fgene.2021.635971. [DOI] [PMC free article] [PubMed] [Google Scholar]
- (27).Lapointe CP; Stefely JA; Jochem A; Hutchins PD; Wilson GM; Kwiecien NW; Coon JJ; Wickens M; Pagliarini DJ Multi-omics Reveal Specific Targets of the RNA-Binding Protein Puf3p and Its Orchestration of Mitochondrial Biogenesis. Cell Systems 2018, 6 (1), 125–135.e6. [DOI] [PMC free article] [PubMed] [Google Scholar]
- (28).Overmyer K; Rhoads T; Merrill A; Ye Z; Westphall M; Acharya A; Shukla S; Coon J Proteomics, Lipidomics, Metabolomics, and 16S DNA Sequencing of Dental Plaque From Patients With Diabetes and Periodontal Disease. Molecular and Cellular Proteomics 2021, 20, DOI: 10.1016/j.mcpro.2021.100126. [DOI] [PMC free article] [PubMed] [Google Scholar]
- (29).Matyash V; Liebisch G; Kurzchalia T.v.,Shevchenko A; Schwudke D. Lipid extraction by methyl-tert-butyl ether for high-throughput lipidomics. Journal of Lipid Research 2008, 49 (5), 1137–1146. [DOI] [PMC free article] [PubMed] [Google Scholar]
- (30).Folch J; Lees M; Sloane Stanley GH A simple method for the isolation and purification of total lipides from animal tissues. Journal Biological Chemistry 1957, 226 (1), 497–509. [PubMed] [Google Scholar]
- (31).Bligh E; Dyer W A rapid method of total lipid extraction and purification. The Canadian Journal of Biochemistry and Physiology 1959, 37 (8), 911–917. [DOI] [PubMed] [Google Scholar]
- (32).König G; Reetz MT; Thiel W 1-Butanol as a Solvent for Efficient Extraction of Polar Compounds from Aqueous Medium: Theoretical and Practical Aspects. Journal of Physical Chemistry B 2018, 122 (27), 6975–6988. [DOI] [PubMed] [Google Scholar]
- (33).Hughes CS; Foehr S; Garfield DA; Furlong EE; Steinmetz LM; Krijgsveld J Ultrasensitive proteome analysis using paramagnetic bead technology. Molecular Systems Biology 2014, 10 (10), 757. [DOI] [PMC free article] [PubMed] [Google Scholar]
- (34).Virant-Klun I; Leicht S; Hughes C; Krijgsveld J Identification of Maturation-Specific Proteins by Single-Cell Proteomics of Human Oocytes. Molecular and Cellular Proteomics 2016, 15 (8), 2616–2627. [DOI] [PMC free article] [PubMed] [Google Scholar]
- (35).Moggridge S; Sorensen PH; Morin GB; Hughes CS Extending the Compatibility of the SP3 Paramagnetic Bead Processing Approach for Proteomics. Journal of Proteome Research 2018, 17 (4), 1730–1740. [DOI] [PubMed] [Google Scholar]
- (36).Hughes CS; Moggridge S; Müller T; Sorensen PH; Morin GB; Krijgsveld J Single-pot, solid-phase-enhanced sample preparation for proteomics experiments. Nature Protocols 2019, 14 (1), 68–85. [DOI] [PubMed] [Google Scholar]
- (37).Batth TS; Tollenaere MAX; Rüther P; Gonzalez-Franquesa A; Prabhakar BS; Bekker-Jensen S; Deshmukh AS; Olsen JV Protein Aggregation Capture on Microparticles Enables Multipurpose Proteomics Sample Preparation. Molecular and Cellular Proteomics 2019, 18 (5), 1027–1035. [DOI] [PMC free article] [PubMed] [Google Scholar]
- (38).Mori H; Dugan CE; Nishii A; Benchamana A; Li Z; Cadenhead TS; Das AK; Evans CR; Overmyer KA; Romanelli SM; Peterson SK; Bagchi DP; Corsa CA; Hardij J; Learman BS; el Azzouny M; Coon JJ; Inoki K; MacDougald OA The molecular and metabolic program by which white adipocytes adapt to cool physiologic temperatures. PLoS Biology 2021, 19 (5), DOI: 10.1371/journal.pbio.3000988. [DOI] [PMC free article] [PubMed] [Google Scholar]
- (39).Lorenz MA; Burant CF; Kennedy RT Reducing Time and Increasing Sensitivity in Sample Preparation for Adherent Mammalian Cell Metabolomics. Analytical Chemistry 2011, 83 (9), 3406–3414. [DOI] [PMC free article] [PubMed] [Google Scholar]
- (40).Sambrook J; Russell DW Fragmentation of DNA by Sonication. Cold Spring Harbor Protocols 2017, 2006 (23), DOI: 10.1101/pdb.prot4538. [DOI] [PubMed] [Google Scholar]
- (41).Cox J; Mann M MaxQuant enables high peptide identification rates, individualized p.p.b.-range mass accuracies and proteome-wide protein quantification. Nature Biotechnology 2008, 26 (12), 1367–1372. [DOI] [PubMed] [Google Scholar]
- (42).Cox J; Neuhauser N; Michalski A; Scheltema RA; Olsen JV; Mann M Andromeda: A Peptide Search Engine Integrated into the MaxQuant Environment. Journal of Proteome Research 2011, 10 (4), 1794–1805. [DOI] [PubMed] [Google Scholar]
- (43).Cox J; Hein MY; Luber CA; Paron I; Nagaraj N; Mann M Accurate proteome-wide label-free quantification by delayed normalization and maximal peptide ratio extraction, termed MaxLFQ. Molecular & Cellular Proteomics 2014, 13, 2513–2526. [DOI] [PMC free article] [PubMed] [Google Scholar]
- (44).Löfgren L; Forsberg GB; Ståhlman M The BUME method: a new rapid and simple chloroform-free method for total lipid extraction of animal tissue. Scientific Reports 2016, 6, DOI: 10.1038/srep27688. [DOI] [PMC free article] [PubMed] [Google Scholar]
- (45).Teng Q; Huang W; Collette TW; Ekman DR; Tan C A direct cell quenching method for cell-culture based metabolomics. Metabolomics 2009, 5 (2), 199–208. [Google Scholar]
- (46).Sitnikov DG; Monnin CS; Vuckovic D Systematic Assessment of Seven Solvent and Solid-Phase Extraction Methods for Metabolomics Analysis of Human Plasma by LC-MS. Scientific Reports 2016, 6, DOI: 10.1038/srep38885. [DOI] [PMC free article] [PubMed] [Google Scholar]
- (47).Liu Z; Rochfort S; Cocks BG Optimization of a single phase method for lipid extraction from milk. Journal of Chromatography A 2016, 1458, 145–149. [DOI] [PubMed] [Google Scholar]
- (48).Rabinowitz JD; Kimball E Acidic acetonitrile for cellular metabolome extraction from Escherichia coli. Analytical Chemistry 2007, 79 (16), 6167–6173. [DOI] [PubMed] [Google Scholar]
- (49).Kyte J; Doolittle RF A simple method for displaying the hydropathic character of a protein. Journal of Molecular Biology 1982, 157 (1), 105–132. [DOI] [PubMed] [Google Scholar]
- (50).Bag S; Rauwolf S; Suyetin M; Schwaminger SP; Wenzel W; Berensmeier S Buffer Influence on the Amino Acid Silica Interaction. ChemPhysChem 2020, 21 (20), 2347–2356. [DOI] [PMC free article] [PubMed] [Google Scholar]
- (51).Gutierrez DB; Gant-Branum RL; Romer CE; Farrow MA; Allen JL; Dahal N; Nei YW; Codreanu SG; Jordan AT; Palmer LD; Sherrod SD; McLean JA; Skaar EP; Norris JL; Caprioli RM An Integrated, High-Throughput Strategy for Multiomic Systems Level Analysis. Journal of Proteome Research 2018, 17 (10), 3396–3408. [DOI] [PMC free article] [PubMed] [Google Scholar]
Associated Data
This section collects any data citations, data availability statements, or supplementary materials included in this article.