

SKYLIGHT 4 (Study to Find Out How Safe Long-term Treatment With Fezolinetant is in Women With Hot Flashes Going Through Menopause) demonstrates the 52-week safety and tolerability of fezolinetant and its use for the treatment of vasomotor symptoms associated with menopause.
OBJECTIVE:
To evaluate the safety, tolerability, and effect of fezolinetant on endometrial health over 52 weeks.
METHODS:
We conducted a phase 3, randomized, double-blind, 52-week safety study (SKYLIGHT 4 [Study to Find Out How Safe Long-term Treatment With Fezolinetant is in Women With Hot Flashes Going Through Menopause]) of placebo, fezolinetant 30 mg, and fezolinetant 45 mg once daily (1:1:1). Participants were postmenopausal and seeking treatment for vasomotor symptoms associated with menopause. Primary endpoints were treatment-emergent adverse events, percentage of participants with endometrial hyperplasia, and percentage with endometrial malignancy. Endometrial hyperplasia or malignancy was evaluated according to U.S. Food and Drug Administration guidance (point estimate of 1% or less with an upper bound of one-sided 95% CI of 4% or less). Secondary endpoints included change in bone mineral density (BMD) and trabecular bone score. A sample size of 1,740 was calculated to enable observation of one or more events (≈80% probability for events with background rate of less than 1%).
RESULTS:
A total of 1,830 participants were randomized and took one or more medication dose (July 2019–January 2022). Treatment-emergent adverse events occurred in 64.1% (391/610) of the placebo group, 67.9% (415/611) of the fezolinetant 30-mg group, and 63.9% (389/609) of the fezolinetant 45-mg group. Treatment-emergent adverse events leading to discontinuation were similar across groups (placebo, 26/610 [4.3%]; fezolinetant 30 mg, 34/611 [5.6%]; fezolinetant 45 mg, 28/609 [4.6%]). Endometrial safety was assessed in 599 participants. In the fezolinetant 45-mg group, 1 of 203 participants had endometrial hyperplasia (0.5%; upper limit of one-sided 95% CI 2.3%); there were no cases in the placebo (0/186) or fezolinetant 30 mg (0/210) group. Endometrial malignancy occurred in 1 of 210 in the fezolinetant 30-mg group (0.5%; 95% CI 2.2%) with no cases in the other groups. Liver enzyme elevations more than three times the upper limit of normal occurred in 6 of 583 placebo, 8 of 590 fezolinetant 30 mg, and 12 of 589 fezolinetant 45 mg participants; no Hy's law cases were reported (ie, no severe drug-induced liver injury with alanine aminotransferase or aspartate aminotransferase more than three times the upper limit of normal and total bilirubin more than two times the upper limit of normal, with no elevation of alkaline phosphatase and no other reason to explain the combination). Changes in BMD and trabecular bone score were similar across groups.
CONCLUSION:
Results from SKYLIGHT 4 confirm the 52-week safety and tolerability of fezolinetant and support its continued development.
FUNDING SOURCE:
Astellas Pharma Inc.
CLINICAL TRIAL REGISTRATION:
Worldwide, most women aged 40–64 years old experience vasomotor symptoms (eg, hot flushes, night sweats) associated with menopause.1 Moderate-to-severe vasomotor symptoms are reported by 34% of postmenopausal individuals in the United States, 40% in Europe, and 16% in Japan2; 9.6% of Latin American3 and 11.4% of Australian4 women report severe or bothersome vasomotor symptoms. Vasomotor symptoms can also have substantial effects on quality of life by affecting sleep, mood, and cognitive functioning and contributing to social impairment, work-related difficulties, anxiety, and fatigue.5–8
Menopausal hormone therapy is an effective choice for vasomotor symptoms but is not appropriate for everyone.9,10 Fezolinetant, in development for the treatment of moderate-to-severe vasomotor symptoms associated with menopause, is a nonhormonal, selective neurokinin 3 receptor antagonist that blocks neurokinin B binding on the kisspeptin/neurokinin B/dynorphin neurons in the hypothalamus to moderate neuronal activity in the thermoregulatory center.11–14 Phase 2 studies demonstrated a reduction in vasomotor symptom frequency and severity after fezolinetant treatment, with improvements in health-related quality of life.11,12,15 Results from two replicate double-blind, placebo-controlled phase 3 studies (SKYLIGHT [Study to Find Out if Fezolinetant Helps Reduce Moderate to Severe Hot Flashes in Women Going Through Menopause] 1 [NCT04003155] and SKYLIGHT 2 [NCT04003142]) showed that fezolinetant 30 and 45 mg met statistical significance in reducing vasomotor symptom frequency and severity at weeks 4 and 12 compared with placebo,16,17 with reduction persisting over 52 weeks. Over the 12-week double-blind period, there was a low incidence of individual treatment-emergent adverse events, and few serious treatment-emergent adverse events were noted with fezolinetant treatment. SKYLIGHT 4 (Study to Find Out How Safe Long-term Treatment With Fezolinetant is in Women With Hot Flashes Going Through Menopause) was conducted to provide 52-week, placebo-controlled safety data in a large cohort, with a focus on endometrial safety.
METHODS
SKYLIGHT 4 was a randomized, placebo-controlled, double-blind, parallel-group, phase 3, multicenter, 52-week study (Fig. 1). The screening period included at least one visit during which a physical examination, clinical laboratory testing, urinalysis, electrocardiogram (ECG), Pap test, mammogram, transvaginal ultrasonogram, and endometrial biopsy (in participants with a uterus) were conducted. Demographic data (age, sex, self-identified race and ethnicity, smoking status, and prior hormone therapy use) were collected at screening.
Fig. 1. Study design. *Screening period of up to 5 weeks. At the discretion of the medical monitor, participants could be rescreened once, and an extra 15 screening days were allowed for repeat endometrial biopsy if needed.
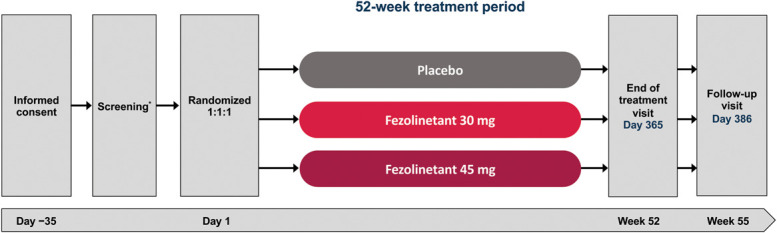
Neal-Perry. Fifty-Two–Week Fezolinetant Safety. Obstet Gynecol 2023.
At study visit 2 (day 1), participants were randomized according to the schedule and stratified by smoking status (current vs former or never) using Interactive Response Technology. The investigator, sponsor's study management team, clinical staff, and participants were blinded to treatment allocation. Participants were assigned 1:1:1 to fezolinetant 30 mg, fezolinetant 45 mg, or matching placebo given orally as two tablets, indistinguishable in size and shape, once daily for 52 weeks. Additional study visits occurred at week 2 and every 4 weeks from weeks 4 to 52, or the early discontinuation visit, with one final follow-up visit at week 55.
The study was conducted in accordance with the Declaration of Helsinki, Good Clinical Practice, and International Council for Harmonisation guidelines. An Independent Ethics Committee or IRB reviewed the ethical, scientific, and medical appropriateness of the study at each site before data collection. Signed informed consent forms were obtained before any study-related procedures were performed.
Participants were aged 40–65 years, inclusive, seeking treatment for vasomotor symptoms associated with menopause, had body mass index (BMI, calculated as weight in kilograms divided by height in meters squared) between 18 and 38, inclusive, and were confirmed to be postmenopausal by having spontaneous amenorrhea for 12 consecutive months or longer, spontaneous amenorrhea for 6 months or longer with follicle-stimulating hormone level higher than 40 international units/L, or bilateral oophorectomy 6 or more weeks before the screening visit. Use of strong or moderate cytochrome P450 1A2 inhibitors, hormone therapy, hormonal contraceptives, or any prescription, over-the-counter, or herbal treatment for vasomotor symptoms precluded participation unless treatment was discontinued with drug-specific washout periods based on the known half-lives of the medications. Other exclusion criteria included having an endometrial biopsy that confirmed disordered proliferative endometrium, endometrial hyperplasia, endometrial malignancy, or other clinically significant findings. Additional exclusion criteria are shown in Appendix 1, available online at http://links.lww.com/AOG/D63.
Primary endpoints were frequency and severity of treatment-emergent adverse events, percentages of participants with endometrial hyperplasia, and percentages of participants with endometrial malignancy. Secondary endpoints were change from baseline to 12 months in endometrial thickness, percentage of participants with disordered proliferative endometrium, change from baseline to 12 months in bone mineral density (BMD) and trabecular bone score at the hip and spine, vital signs, clinical laboratory tests, Columbia Suicide Severity Rating Scale score, and ECG parameters.
Treatment-emergent adverse events were collected throughout the study, including certain treatment-emergent adverse events of special interest. Events were rated as mild, moderate, or severe, and relatedness of events to the study drug was assessed by the investigator. Serious treatment-emergent adverse events were defined as those resulting in death, persistent or significant disability or incapacity, congenital anomaly, or birth defect; life-threatening; requiring or prolonging inpatient hospitalization; discontinuation attributable to increased liver enzymes; or other medically important events. Some events were always categorized as serious according to a prespecified list.
For participants with a uterus, transvaginal ultrasonograms and suction endometrial biopsies were performed at screening and at the week 52 or early discontinuation visit. In addition, biopsies were to be taken in all cases of uterine bleeding during treatment. Endometrial biopsies were read concurrently by three independent pathologists at three companies (LabCorp Expanded Laboratory Management Solutions [Los Angeles, California], Interpace Biosciences [Parsippany, New Jersey], and NeoGenomics [Fort Myers, Florida]). Each pathologist was blinded to the treatment group and to the readings of other pathologists using criteria outlined in the 2003 U.S. Food and Drug Administration (FDA) draft guidance.18,19 Dual-energy X-ray absorptiometry was used to assess BMD and trabecular bone score at screening and week 52 or the end of treatment and reviewed by a central reader.
The sample size of 1,740 participants was calculated to provide a final number of participants to observe at least one endometrial biopsy event (eg, endometrial hyperplasia within a treatment group), with about 80% probability for events with a background rate of less than 1%. This calculation assumes that 60% of participants did not have an evaluable biopsy at baseline or refused an endometrial biopsy at early discontinuation or end of treatment.
The safety analysis set comprised all randomized participants who took at least one dose of study intervention. The endometrial health set was a safety analysis subset of participants who had a postbaseline biopsy within 30 days after the last dose of study intervention, an acceptable biopsy at baseline (at least one endometrial biopsy with satisfactory tissue and no read of hyperplasia, disordered proliferative pattern, or malignancy), and a satisfactory endometrial biopsy result on day 326 or later or had a postbaseline final diagnosis of hyperplasia, disordered proliferative pattern, or malignancy before day 326.
The full analysis set consisted of all randomized participants who took at least one dose of study treatment. The randomized treatment for each participant was used for summaries by treatment group based on the full analysis set.
In general, summary statistics were generated for all safety parameters. For the percentage of participants with a final diagnosis of endometrial hyperplasia, endometrial malignancy, or disordered proliferative endometrium, the exact (Clopper-Pearson) upper one-sided 95% CI was provided. This analysis was based on the final diagnosis evaluated by the three pathologists. The final diagnosis was the concordance of the three pathologists' diagnoses: if at least two pathologists agreed, the result was included; if none of them agreed, the worst result was used. Treatment comparisons were performed for the endometrial thickness- and bone density-related endpoints. Changes from baseline in endometrial thickness and bone densities were analyzed, and each fezolinetant group was compared with placebo using an analysis of covariance model, with treatment and strata smoking status (current vs former or never) as factors and baseline measurement and baseline weight as covariates.
RESULTS
The study was conducted between July 2019 and January 2022. Overall, 1,831 participants were randomized and 1,830 took at least one dose of study drug (safety analysis set; Fig. 2). Demographic data were balanced across the treatment groups (Table 1). The rate of withdrawal was higher in the placebo group (119/610 [19.5%]) than the fezolinetant groups and similar between the two study drug doses (79/611 [12.9%] for fezolinetant 30 mg; 85/609 [14.0%] for fezolinetant 45 mg; Fig. 2). Median duration of treatment was similar across all groups (364.0 days). The endometrial health set included 599 participants (placebo, n=186; fezolinetant 30 mg, n=210; and fezolinetant 45 mg, n=203).
Fig. 2. Patient disposition. *The participant who died (fezolinetant 30-mg group) had a treatment-emergent adverse event (considered unrelated to study drug) of cardiac arrest and anoxic brain injury leading to treatment discontinuation and was counted under the adverse event category. This event subsequently resulted in death.
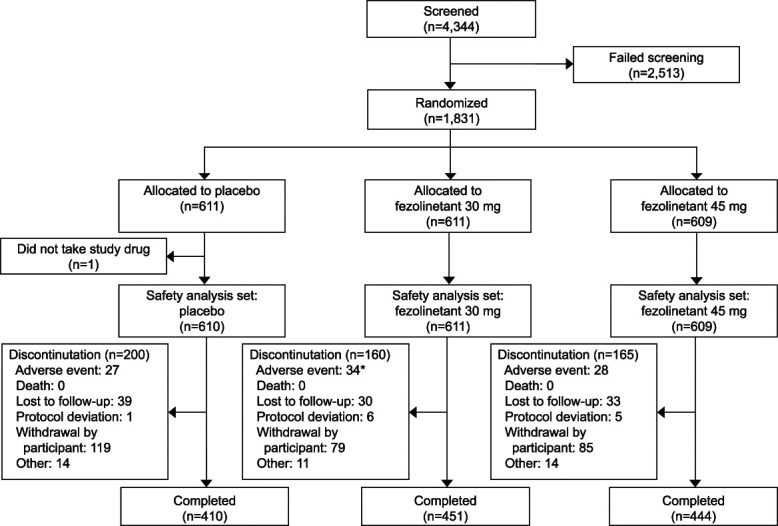
Neal-Perry. Fifty-Two–Week Fezolinetant Safety. Obstet Gynecol 2023.
Table 1.
Demographics and Baseline Characteristics (Safety Analysis Set)

In terms of the primary endpoints, treatment-emergent adverse event incidences were comparable between treatment groups. The most common treatment-emergent adverse events (5% or greater) by preferred term were headache and coronavirus disease 2019 (COVID-19), with a similar incidence across treatment groups (Table 2). A low incidence of serious treatment-emergent adverse events was reported in 2.3% of the placebo group (14 participants), 3.3% of the fezolinetant 30-mg group (20 participants), and 3.8% of the fezolinetant 45-mg group (23 participants). Most of the events were mild or moderate in severity, and occurrences were typically similar between groups, although a slightly higher incidence of mild events was noted in the fezolinetant 30-mg group (215 participants [35.2%]) compared with the placebo group (180 participants [29.5%]). One death was reported in the study. This participant, in the fezolinetant 30-mg group, had an out-of-hospital cardiac arrest, delayed airway access, and anoxic brain injury that were not considered to be related to study treatment. Treatment-emergent adverse events leading to withdrawal of study treatment were experienced by 4.3% of the placebo group (26 participants), 5.6% of the fezolinetant 30-mg group (34 participants), and 4.6% of the fezolinetant 45-mg group (28 participants).
Table 2.
Summary of Treatment-Emergent Adverse Events (Safety Analysis Set)

The primary endpoint of endometrial safety, as conducted in the endometrial health set, was met. Endometrial hyperplasia or malignancy in fezolinetant-treated participants, as assessed by centrally read endometrial biopsies in the endometrial health set, was within the FDA’s prespecified limits of 1% or less, with an upper limit of one-sided 95% CI of 4% or less.18 Endometrial hyperplasia that was determined by the final biopsy diagnosis was reported for none of the 186 participants in the placebo group (0%; upper limit of one-sided 95% CI 1.6%), 0 of 210 in the fezolinetant 30-mg group (0%; 1.4%), and 1 of 203 in the fezolinetant 45-mg group (0.5%; 2.3%). Endometrial malignancy as determined by the final biopsy diagnosis was reported for 0 of 186 participants in the placebo group (0%; upper limit of one-sided 95% CI 1.6%), 1 of 210 participants in the fezolinetant 30-mg group (0.5%; 2.2%), and 0 of 203 in the fezolinetant 45-mg group (0%; 1.5%).
Transvaginal ultrasonogram data were generally unremarkable. There was no significant difference in endometrial thickness as measured by transvaginal ultrasonography over 1 year between fezolinetant- and placebo-treated participants. Mean (SD) thickness at baseline and week 52 were as follows: 3.59 (2.19) and 3.52 (2.59) mm for placebo, 3.52 (2.15) and 3.26 (2.03) mm for fezolinetant 30 mg, and 3.51 (2.59) and 3.21 (2.01) mm for fezolinetant 45 mg. The least-squares mean change (SE) from baseline was −0.09 (0.11) for placebo, −0.17 (0.11) for fezolinetant 30 mg, and −0.26 (0.11) for fezolinetant 45 mg. The difference in least-squares mean compared with placebo was −0.08 (SE 0.14, 95% CI −0.36 to 0.21) for fezolinetant 30 mg and −0.17 (0.14; −0.45 to 0.11) for fezolinetant 45 mg. The prevalence of uterine bleeding was slightly higher in the placebo group (30 participants [4.9%]) than in the fezolinetant groups (30 mg: 20 participants [3.3%]; 45 mg: 19 participants [3.1%]; Table 3).
Table 3.
Treatment-Emergent Adverse Events of Special Interest by Preferred Term as Reported by the Investigator (Safety Analysis Set)

A disordered proliferative pattern as determined by the final biopsy diagnosis was reported for 4 of 186 participants in the placebo group (2.2%; upper limit of one-sided 95% CI 4.9%), 3 of 210 in the fezolinetant 30-mg group (1.4%; 3.7%), and 0 of 203 in the fezolinetant 45-mg group (0%; 1.5%).
Treatment-emergent adverse events of special interest, as reported by the investigator, in the category of endometrial hyperplasia or malignancy or disordered proliferative endometrium occurred in 1.0% or less across the groups (Table 3).
A low incidence of bone fractures was reported, with similar incidences across groups (9 participants [1.5%] for fezolinetant 30 mg and 10 participants [1.6%] for placebo and fezolinetant 45 mg; Table 3). Changes in BMD and trabecular bone score at hip and spine were consistent between placebo- and fezolinetant-treated participants (Appendices 2 and 3, available online at http://links.lww.com/AOG/D63).
The incidence of elevations of laboratory assessments of liver function was low (Fig. 3 and Table 4).20,21 Elevations of alanine aminotransferase (ALT) or aspartate aminotransferase (AST) more than three times the upper limit of normal were observed in six participants (1.0%) who received placebo, eight (1.4%) who received fezolinetant 30 mg, and 12 (2.0%) who received fezolinetant 45 mg. No Hy's law cases were reported (ie, no severe drug-induced liver injury with ALT or AST more than three times the upper limit of normal and total bilirubin more than two times the upper limit of normal, with no elevation of alkaline phosphatase and no other reason to explain the combination).21 Increases in ALT or AST were generally asymptomatic and isolated, intermittent, or transient and generally returned to baseline while on treatment or soon after discontinuation. Increased liver transaminases cases were independent of COVID-19. Treatment-emergent adverse events of liver test elevations were reported at similar rates for each group: 4.9% (30 participants), 5.7% (35 participants), and 5.3% (32 participants) for placebo, fezolinetant 30 mg, and fezolinetant 45 mg, respectively (Table 3).
Fig. 3. Evaluation of study drug–induced serious hepatotoxicity: maximum aspartate aminotransferase (AST) or alanine aminotransferase (ALT) vs maximum total bilirubin values* (safety analysis set). *Plotting peak ALT or AST against peak total bilirubin allows visualization of data by quadrant and easy identification of any cases that fall outside of the normal range (bottom left quadrant). Potential cases of Hy's law (ALT or AST more than three times the upper limit of normal [ULN] and total bilirubin more than two times the ULN, with no elevation of alkaline phosphatase and no other reason to explain the combination)21 would be located in the upper right-hand quadrant of the graph. Cases of Temple's corollary would be located in the bottom right-hand quadrant of the graph. Temple's corollary states that, for every 10 cases of ALT more than 10 times the ULN in a clinical trial, there would be one instance of Hy's law.20 The elevated bilirubin in the upper left-hand quadrant represents an individual with pre-existing Gilbert's disease.
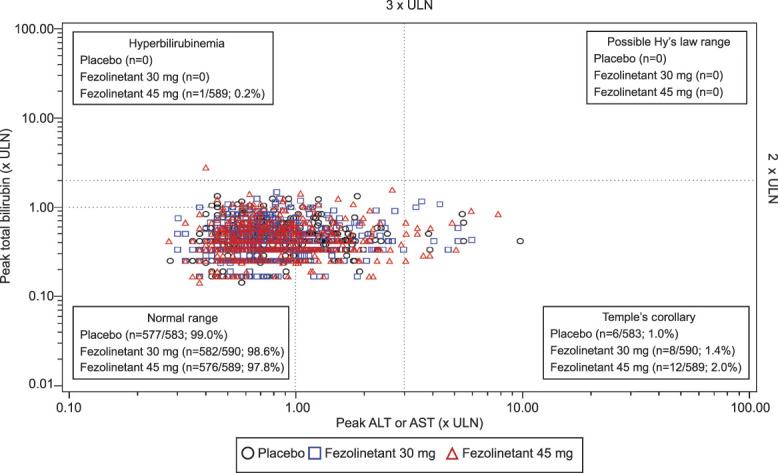
Neal-Perry. Fifty-Two–Week Fezolinetant Safety. Obstet Gynecol 2023.
Table 4.
Liver Safety Assessments (Safety Analysis Set)

Treatment-emergent adverse events were no different when analyzed by BMI category. Assessments of other secondary endpoints, including vital signs and ECG parameters, showed no notable findings.
Based on a numeric imbalance for serious treatment-emergent adverse events of the System Organ Class of “neoplasm benign, malignant, and unspecified” (including cysts and polyps; Appendix 4, available online at http://links.lww.com/AOG/D63), an in-depth post hoc investigation was conducted for each identified malignant neoplasm. Per the sponsor's process, all malignancies are designated as serious independent of the investigator report. No signal of increased risk for nonbenign neoplasms overall or at specific sites was identified in this investigation on the basis of various considerations, such as short latency, apparent preexisting neoplasm at study entry, alternative etiology, medical history, and known risk factors.
DISCUSSION
These data over 52 weeks in 1,830 randomized and treated participants experiencing vasomotor symptoms associated with menopause demonstrate the safety and tolerability of fezolinetant 30 mg and 45 mg and support its use over a 1-year period. No apparent safety signals for fezolinetant were observed over the study. Furthermore, the incidence of endometrial hyperplasia or malignancy in fezolinetant-treated participants, as assessed by blinded, centrally read endometrial biopsies, was within the prespecified limits and aligned with the FDA communication or draft guidance.18 In addition, there was no significant difference in change from baseline in endometrial thickness between fezolinetant- and placebo-treated participants and no effect on bone health (Appendix 5, available online at http://links.lww.com/AOG/D64).
The treatment-emergent adverse events reported in the study were generally mild or moderate in severity, and the most common were headache and COVID-19, with fezolinetant consistent with placebo. Treatment-emergent adverse events leading to discontinuation were also similar between fezolinetant 30 mg and 45 mg.
Demonstrating endometrial safety is required for any treatment used to alleviate menopause symptoms according to the FDA draft guidance.18 The risk for endometrial hyperplasia or malignancy in individuals with a uterus is increased by unopposed estrogen exposure22; hence, with hormonal therapy, progestogen is frequently used in combination for endometrial protection.10
Fezolinetant is a nonhormonal treatment that acts through the neurokinin B pathway.14 Although the mechanism of action of fezolinetant is different from that of estrogen and estrogen plus progestin products, assessment with the same FDA clinical trial criteria18 provides important endometrial safety information on fezolinetant use. Given the risk and benefits associated with different treatment choices, some menopausal individuals may benefit from nonhormonal therapy that does not affect the endometrium.
Treatment-emergent adverse event reporting by the investigators also included events of endometrial hyperplasia, malignancy, or disordered proliferative endometrium, which are distinct from the events reported for the primary endpoints.
Incidences of elevations of liver tests were low. Overall, 98.2% of participants who received fezolinetant had liver test results within the normal range compared with 99.0% of those receiving placebo, and there was no evidence of liver function impairment or liver-associated symptoms, including no Hy's law cases. Hepatic safety is important for all pharmaceutical therapies in development.21 This study collected robust hepatic assessments to further inform the safety profile of fezolinetant, which, if approved, would be a potential therapeutic option for a large number of menopausal individuals experiencing vasomotor symptoms. Given the hepatic safety profile of the neurokinin 3 receptor antagonist candidate pavinetant,23,24 we ensured that this safety study included participants with a variety of potential risk factors (eg, obesity, with inclusion of BMIs up to 38) and no restrictions for those with nonalcoholic fatty liver disease or nonalcoholic steatohepatitis to establish hepatic safety in a broad population.25
A limited number of malignancies were reported (as treatment-emergent adverse events of special interest or in the system organ class of neoplasms) during the treatment or follow-up periods, but no evidence of an increased risk for nonbenign neoplasms overall or at specific sites was identified, and only one occurrence (fezolinetant 45-mg group) was reported by the investigators as treatment related.
Strengths of this study are the large number of participants randomized and with up to 1 year of exposure, participant diversity, inclusion of individuals with BMIs up to 38, placebo-controlled protocol, and low rate of withdrawal. The study also involved a comprehensive assessment of bone health in participants with vasomotor symptoms associated with menopause over a 52-week treatment period.
The endometrial health set included 599 of 1,830 participants in the safety analysis set. For those with a uterus (n=1,489), willingness to have a biopsy at screening and the end of study was an inclusion criterion. Compliance for end-of-study biopsies was difficult because some participants declined to have a second biopsy as a result of the invasiveness of the procedure. However, other end-of-study data were collected from these participants, including any reported treatment-emergent adverse events. The initial protocol allowed participants to enroll after hysterectomy (n=341) or without a biopsy if the transvaginal ultrasonogram demonstrated an endometrial thickness of less than 4 mm (n=1,045). To increase the number of participants eligible for inclusion in the endometrial health set, subsequent protocol amendments required all participants to have a screening and end-of-study biopsy and specified hysterectomy as an exclusion criterion.
Phase 3 data obtained from SKYLIGHT 1 and 2 demonstrated the efficacy of fezolinetant in reducing the frequency and severity of vasomotor symptoms and provided information on the safety profile of fezolinetant compared with placebo over 12 weeks.16,17 Results from SKYLIGHT 4 provide additional evidence confirming the longer-term safety during a 52-week treatment period, and the data support the continued development of fezolinetant as a novel nonhormonal treatment option for moderate-to-severe vasomotor symptoms associated with menopause.
Authors' Data Sharing Statement
Will individual participant data be available (including data dictionaries)? Access may be requested to anonymized participant-level data, trial-level data, and protocols.
What data in particular will be shared? For the Astellas criteria on data sharing, see https://clinicalstudydatarequest.com/Study-Sponsors/Study-Sponsors-Astellas.aspx.
What other documents will be available? For the Astellas criteria on data sharing, see https://clinicalstudydatarequest.com/Study-Sponsors/Study-Sponsors-Astellas.aspx.
When will data be available (start and end dates)? For the Astellas criteria on data sharing, see https://clinicalstudydatarequest.com/Study-Sponsors/Study-Sponsors-Astellas.aspx.
By what access criteria will data be shared (including with whom, for what types of analyses, and by what mechanism)? For the Astellas criteria on data sharing, see https://clinicalstudydatarequest.com/Study-Sponsors/Study-Sponsors-Astellas.aspx.
Footnotes
This study was funded by Astellas Pharma Inc.
Financial Disclosure: Genevieve Neal-Perry is a member of the scientific advisory board for Astellas and Ferring and has received research funding from Merck and NICHD. She is also Vice President of Diversity, Equity and Structural Change for the Society for Gynecological Investigations, Programing Committee Chair for the American Society of Reproductive Medicine, holds Committee Membership in the Endocrine Society, and serves as an Associate Editor for the Journal of Clinical Endocrinology and Metabolism. Antonio Cano is past President of the European Menopause and Andropause Society and a consultant for Astellas, Theramex, and ItalFarmaco. Samuel Lederman has received honoraria from AbbVie and research funding from AbbVie, Amgen, Aspira, Estetra, and Janssen. Rossella E. Nappi has past financial relationships (lecturer, member of advisory boards, or consultant) with Boehringer Ingelheim, Eli Lilly, Endoceutics, Merck Sharp & Dohme, Novo Nordisk, Palatin Technologies, Pfizer Inc., Procter & Gamble Co., TEVA Women's Health Inc., and Zambon SpA; ongoing relationships with Astellas, Bayer HealthCare AG, Exeltis, Fidia, Gedeon Richter, HRA Pharma, Organon & Co., Shionogi Limited, and Theramex; serves as General Secretary of the International Menopause Society. Nanette Santoro is a study investigator and member of the Scientific Advisory Board and consultant for Astellas; a member of the Scientific Advisory Board for Amazon (Project Ember), MenoGeniX Inc., and QUE Oncology; and is a consultant for Ansh Labs. Nanette Santoro is also on the Program Committee for the North American Menopause Society and is a past President of the Society for Reproductive Investigation. Wendy Wolfman is an advisory board member, consultant, and speaker for Astellas, BioSyent, Knight, Merck, Pfizer, and Searchlight, and President of the Canadian Menopause Society. Marci English, Udaya Valluri, and Faith D. Ottery are employees of Astellas Pharma Inc. Catherine Franklin was an employee of Astellas Pharma Inc. at the time of the study.
Presented at the North American Menopause Society Annual Meeting, October 12–15, 2022, Atlanta, Georgia; and at IMS 18, October 26–29, 2022, Lisbon, Portugal.
The authors thank the study investigators and staff and all participants who took part in the study. Medical writing support was provided by Becky Ayles, PhD, and Sue Cooper, PhD, CMPP, of Excel Scientific Solutions, Horsham, UK, and funded by Astellas Pharma Inc.
Researchers may request access to anonymized participant-level data, trial-level data, and protocols from Astellas-sponsored clinical trials at www.clinicalstudydatarequest.com. For the Astellas criteria on data sharing, see https://clinicalstudydatarequest.com/study-sponsors/study-sponsors-astellas.aspx.
Each author has confirmed compliance with the journal's requirements for authorship.
Peer reviews are available at http://links.lww.com/AOG/D65.
Figure.

No available caption
REFERENCES
- 1.Makara-Studzińśka MT, Kryś-Noszczyk KM, Jakiel G. Epidemiology of the symptoms of menopause: an intercontinental review. Menopausal Rev 2014;3:203–11. doi: 10.5114/pm.2014.43827 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 2.Nappi RE, Kroll R, Siddiqui E, Stoykova B, Rea C, Gemmen E, et al. Global cross-sectional survey of women with vasomotor symptoms associated with menopause: prevalence and quality of life burden. Menopause 2021;28:875–82. doi: 10.1097/GME.0000000000001793 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 3.Blümel JE, Chedraui P, Baron G, Belzares E, Bencosme A, Calle A, et al. A large multinational study of vasomotor symptom prevalence, duration, and impact on quality of life in middle-aged women. Menopause 2011;18:778–85. doi: 10.1097/gme.0b013e318207851d [DOI] [PubMed] [Google Scholar]
- 4.Gartoulla P, Bell RJ, Worsley R, Davis SR. Moderate-severely bothersome vasomotor symptoms are associated with lowered psychological general wellbeing in women at midlife. Maturitas 2015;81:487–92. doi: 10.1016/j.maturitas.2015.06.004 [DOI] [PubMed] [Google Scholar]
- 5.Whiteley J, DiBonaventura MD, Wagner JS, Alvir J, Shah S. The impact of menopausal symptoms on quality of life, productivity, and economic outcomes. J Womens Health (Larchmt) 2013;22:983–90. doi: 10.1089/jwh.2012.3719 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 6.English M, Stoykova B, Slota C, Doward L, Siddiqui E, Crawford R, et al. Qualitative study: burden of menopause-associated vasomotor symptoms (VMS) and validation of PROMIS Sleep Disturbance and Sleep-Related Impairment measures for assessment of VMS impact on sleep. J Patient Rep Outcomes 2021;5:37. doi: 10.1186/s41687-021-00289-y [DOI] [PMC free article] [PubMed] [Google Scholar]
- 7.Utian WH. Psychosocial and socioeconomic burden of vasomotor symptoms in menopause: a comprehensive review. Health Qual Life Outcomes 2005;3:47. doi: 10.1186/1477-7525-3-47 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 8.Whiteley J, Wagner JS, Bushmakin A, Kopenhafer L, Dibonaventura M, Racketa J. Impact of the severity of vasomotor symptoms on health status, resource use, and productivity. Menopause 2013;20:518–24. doi: 10.1097/GME.0b013e31827d38a5 [DOI] [PubMed] [Google Scholar]
- 9.de Villiers TJ, Hall JE, Pinkerton JV, Pérez SC, Rees M, Yang C, et al. Revised global consensus statement on menopausal hormone therapy. Maturitas 2016;91:153–5. doi: 10.1016/j.maturitas.2016.06.001 [DOI] [PubMed] [Google Scholar]
- 10.“The 2022 Hormone Therapy Position Statement of the North American Menopause Society” Advisory Panel. The 2022 hormone therapy position statement of the North American Menopause Society. Menopause 2022;29:767–94. doi: 10.1097/GME.0000000000002028 [DOI] [PubMed] [Google Scholar]
- 11.Depypere H, Timmerman D, Donders G, Sieprath P, Ramael S, Combalbert J, et al. Treatment of menopausal vasomotor symptoms with fezolinetant, a neurokinin 3 receptor antagonist: a phase 2a trial. J Clin Endocrinol Metab 2019;104:5893–905. doi: 10.1210/jc.2019-00677 [DOI] [PubMed] [Google Scholar]
- 12.Fraser GL, Lederman S, Waldbaum A, Kroll R, Santoro N, Lee M, et al. A phase 2b, randomized, placebo-controlled, double-blind, dose-ranging study of the neurokinin 3 receptor antagonist fezolinetant for vasomotor symptoms associated with menopause. Menopause 2020;27:382–92. doi: 10.1097/GME.0000000000001510 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 13.Fraser GL, Hoveyda HR, Clarke IJ, Ramaswamy S, Plant TM, Rose C, et al. The NK3 receptor antagonist ESN364 interrupts pulsatile LH secretion and moderates levels of ovarian hormones throughout the menstrual cycle. Endocrinology 2015;156:4214–25. doi: 10.1210/en.2015-1409 [DOI] [PubMed] [Google Scholar]
- 14.Depypere H, Lademacher C, Siddiqui E, Fraser GL. Fezolinetant in the treatment of vasomotor symptoms associated with menopause. Expert Opin Investig Drugs 2021;30:681–94. doi: 10.1080/13543784.2021.1893305 [DOI] [PubMed] [Google Scholar]
- 15.Santoro N, Waldbaum A, Lederman S, Kroll R, Fraser GL, Lademacher C, et al. Effect of the neurokinin 3 receptor antagonist fezolinetant on patient-reported outcomes in postmenopausal women with vasomotor symptoms: results of a randomized, placebo-controlled, double-blind, dose-ranging study (VESTA). Menopause 2020;27:1350–6. doi: 10.1097/GME.0000000000001621 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 16.Johnson KA, Martin N, Nappi RE, Neal-Perry G, Shapiro M, Stute P, et al. Efficacy and safety of fezolinetant in moderate-to-severe vasomotor symptoms associated with menopause: a phase 3 RCT. J Clin Endocrinol Metab 2023. doi: 10.1210/clinem/dgad058. Online ahead of print. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 17.Lederman S, Shapiro CMM, Stute P, Lee M, Wang X, Neal-Perry G. Phase 3 study of fezolinetant for treatment of moderate-to-severe vasomotor symptoms associated with menopause [abstract A132]. Obstet Gynecol 2022;139:39S. doi: 10.1097/01.aog.0000825808.38519.3b [DOI] [Google Scholar]
- 18.U.S. Food and Drug Administration. Draft guidance document. Estrogen and estrogen/progestin drug products to treat vasomotor symptoms and vulvar and vaginal atrophy symptoms—recommendations for clinical evaluation. 2003. Accessed April 28, 2022. https://www.fda.gov/regulatory-information/search-fda-guidance-documents/estrogen-and-estrogenprogestin-drug-products-treat-vasomotor-symptoms-and-vulvar-and-vaginal-atrophy
- 19.Kurman RJ, Ronnett BM, Hedrick Ellenson L. Blaustein's pathology of the female genital tract. 6th ed. In: Kurman RJ, Ronnett BM, Ellenson LH. editors. Springer; 2011. [Google Scholar]
- 20.Lisi DM. Drug-induced liver injury: an overview. U.S. Pharmacist 2016;41:30–4. [Google Scholar]
- 21.U.S. Department of Health and Human Services, Food and Drug Administration, Center for Drug Evaluation and Research (CDER), Center for Biologics Evaluation and Research (CBER) . Guidance for industry: drug-induced liver injury: premarketing clinical evaluation. Accessed November 19, 2021. https://www.fda.gov/media/116737/download
- 22.Furness S, Roberts H, Marjoribanks J, Lethaby A. Hormone therapy in postmenopausal women and risk of endometrial hyperplasia. The Cochrane Database of Systematic Reviews 2012, Issue 8. Art. No.: CD000402. doi: 10.1002/14651858.CD000402.pub4 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 23.Prague JK, Roberts RE, Comninos AN, Clarke S, Jayasena CN, Nash Z, et al. Neurokinin 3 receptor antagonism as a novel treatment for menopausal hot flushes: a phase 2, randomised, double-blind, placebo-controlled trial. Lancet 2017;389:1809–20. doi: 10.1016/S0140-6736(17)30823-1 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 24.Modi M, Dhillo WS. Neurokinin 3 receptor antagonism: a novel treatment for menopausal hot flushes. Neuroendocrinology 2019;109:242–8. doi: 10.1159/000495889 [DOI] [PubMed] [Google Scholar]
- 25.DiStefano JK. NAFLD and NASH in postmenopausal women: implications for diagnosis and treatment. Endocrinology 2020;161:bqaa134. doi: 10.1210/endocr/bqaa134 [DOI] [PMC free article] [PubMed] [Google Scholar]