

Background and Aims:
Receptor‐interacting protein kinase 3 (RIPK3) mediates NAFLD progression, but its metabolic function is unclear. Here, we aimed to investigate the role of RIPK3 in modulating mitochondria function, coupled with lipid droplet (LD) architecture in NAFLD.
Approach and Results:
Functional studies evaluating mitochondria and LD biology were performed in wild‐type (WT) and Ripk3 −/− mice fed a choline‐deficient, amino acid‐defined (CDAA) diet for 32 and 66 weeks and in CRISPR‐Cas9 Ripk3‐null fat‐loaded immortalized hepatocytes. The association between hepatic perilipin (PLIN) 1 and 5, RIPK3, and disease severity was also addressed in a cohort of patients with NAFLD and in PLIN1‐associated familial partial lipodystrophy. Ripk3 deficiency rescued impairment in mitochondrial biogenesis, bioenergetics, and function in CDAA diet–fed mice and fat‐loaded hepatocytes. Ripk3 deficiency was accompanied by a strong upregulation of antioxidant systems, leading to diminished oxidative stress upon fat loading both in vivo and in vitro. Strikingly, Ripk3 −/− hepatocytes displayed smaller size LD in higher numbers than WT cells after incubation with free fatty acids. Ripk3 deficiency upregulated adipocyte and hepatic levels of LD‐associated proteins PLIN1 and PLIN5. PLIN1 upregulation controlled LD structure and diminished mitochondrial stress upon free fatty acid overload in Ripk3 −/− hepatocytes and was associated with diminished human NAFLD severity. Conversely, a pathogenic PLIN1 frameshift variant was associated with NAFLD and fibrosis, as well as with increased hepatic RIPK3 levels in familial partial lipodystrophy.
Conclusions:
Ripk3 deficiency restores mitochondria bioenergetics and impacts LD dynamics. RIPK3 inhibition is promising in ameliorating NAFLD.
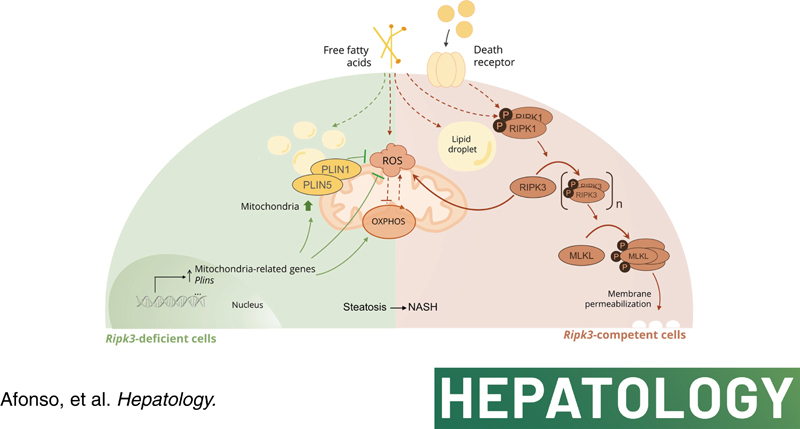
INTRODUCTION
NAFLD is a complex multifactorial chronic liver disease that emerges as a result of intricate interactions of several hits, including metabolic unbalances, lipotoxicity, organelle dysfunction, and inflammation, on the top of an eventual pernicious genetic and epigenetic landscape, ultimately leading to hepatocellular injury and cell death.1 Necroptosis is activated in the liver of human and experimental NAFLD, but the therapeutic relevance of targeting necroptosis is unclear.2–8 Necroptosis is an immunogenic‐regulated cell‐death modality that critically relies on mixed lineage kinase domain‐like protein (MLKL) and receptor‐interacting protein kinase 3 (RIPK3) activity.2 Considering that necroptosis ultimately results in regulated cell membrane permeabilization, it is tempting to hypothesize that lipids could be involved in the regulation and execution of necroptotic signaling pathway. We recently showed that Ripk3‐deficient mice with NASH displayed decreased hepatic infiltration of inflammatory cells, fibrosis, and preneoplastic nodules, impacting on the hepatic lipidome, likely through upregulation of peroxisome proliferator‐activated receptor γ (PPARγ).9 Particularly, Ripk3 deficiency increased lipogenesis and hepatic lipid accumulation to buffer excessive detrimental lipid species but reduced very long‐chain fatty acids that are functionally implicated in plasma membrane disruption during necroptosis.9–11.
PPARγ, a ligand‐activated transcription factor, plays a broad range of biological functions, including the control of energy metabolism as well as mitochondrial biogenesis and function. Indeed, proliferator‐activated receptor gamma coactivator 1 alpha (PGC1α), a master regulator of mitochondria turnover and bioenergetics, coactivates PPARγ, which in turn upregulates PGC1α in an autoregulatory circuit.12,13 Thus, the protective role of Ripk3 deletion in NASH9 may also rely on an improved metabolic balance related to the maintenance of mitochondrial function. Accordingly, we have shown that the improvement of high‐fat diet–induced experimental NASH achieved by pharmacological inhibition of RIPK1 is MLKL‐dependent and, at least in part, comprises a boost in mitochondrial respiration.5 Still, the association between necroptosis signaling and mitochondria has been controversial. In particular, RIPK3 increases aerobic respiration upon necroptotic stimuli, leading to increased generation of mitochondrial reactive oxygen species (ROS),14,15 which in turn could activate RIPK1 autophosphorylation in a feed‐forward feedback loop that ensures effective and sustained induction of necroptosis.16 However, the role of ROS in necroptosis execution might be cell‐ and context‐dependent,17 and mitochondrial depletion via mitophagy does not halt necroptotic cell death.18 Although most of these studies used relevant in vitro systems, the pathophysiological role of RIPK3 in vivo, which integrates bioenergetics, lipid metabolism, oxidative stress, and cell death, remains unknown. Here, we aimed to evaluate the role of RIPK3 in the interaction between mitochondria and lipid metabolism in NAFLD.
MATERIALS AND METHODS
Detailed materials and methods are included in Supporting Information.
Patient cohort
Samples of liver tissue were obtained from 22 individuals with risk factors for NAFLD and a patient with familial partial lipodystrophy (FPLD) type 4 carrying a heterozygous PLIN1 frameshift variant,19 who underwent diagnostic liver biopsy. All subjects gave written informed consent before taking part in this study. The procedure was approved by the Ethics Committee of the Pitié‐Salpêtrière Hospital (Paris, France), and performed following the ethical guidelines of the 1975 Declaration of Helsinki.
Animals, diets, and sample collection
Seven‐ to eight‐week‐old male C57BL/6 wild‐type (WT) and Ripk3 −/− mice were fed a choline‐deficient, amino acid‐defined (CDAA; Envigo) diet for 32 or 66 weeks (n = 6–7 mice per experimental group)9 with subsequent total RNA/protein extraction, quantitative real time reverse transcriptase‐polymerase chain reaction, immunoblotting, and mitochondrial respiratory chain enzymatic assays. All animal experiments were carried out with the permission of the local Animal Welfare Organ in accordance with the EU Directive (2010/63/EU), Portuguese laws (DL 113/2013, 2880/2015, 260/2016, and 1/2019) for the use and care of animals in research and all relevant legislation. The experimental protocol was approved by the competent national authority, Direção Geral de Alimentação e Veterinária, Portugal. Animals received humane care in a temperature/humidity‐controlled environment with 12 hours light–dark cycles, complying with the Institute’s guidelines, and as outlined in the “Guide for the Care and Use of Laboratory Animals” prepared by the National Academy of Sciences and published by the National Institutes of Health (NIH publication 86‐23 revised 1985). All experiments were performed by investigators accredited for directing animal experiments (FELASA level C).
Cell culture
CRISPR/Cas9‐mediated deletion of Ripk3 was performed in AML‐12 hepatocytes (LGC Standards) and in 3T3‐L1 preadipocytes (LGC Standards), followed by cell culture and treatments and subsequent cellular oxygen consumption rate (OCR) measurements, viability assays, and cell staining.
Statistical analysis
Regular statistical analysis was performed with GraphPad Prism 7 software (GraphPad Software, Inc.). The Shapiro–Wilk test was used for testing the normality of data. Student's t test was applied when two groups were analyzed. According to normality of values distribution, analysis of variance followed by Tukey's multiple comparison or Krustal–Wallis followed by Dunn's multiple comparison test were performed if three or more groups were compared. Association between two variables was assessed by Pearson or Spearman correlation coefficient for normally and nonnormally distributed data, respectively. Values of p < 0.05 were considered statistically significant.
RESULTS
Ripk3 deficiency rescues mitochondrial biogenesis and function in CDAA diet–induced experimental NASH
We have recently shown that Ripk3 deficiency induces PPARγ expression in the liver of mice fed a CDAA diet or an isocaloric choline‐sufficient L‐amino acid‐defined (CSAA) control diet for 32 and 66 weeks.9 Because PPARγ is coactivated by PGC1α, a master regulator of mitochondrial biogenesis and function, we investigated the role of Ripk3 deletion in reversing mitochondrial dysfunction in experimental NASH. Ripk3 deficiency reduced the severity of CDAA diet–induced experimental NASH (Figure 1A) while increasing hepatic mRNA and protein levels of PGC1α (a significance level of at least, p < 0.05) (Figure 1B and Figure S1A) and its target mitochondrial transcription factor A (at least, p < 0.05) (Figure 1B and Figure S1A) after mid‐ and long‐term CDAA feeding. CDAA diet‐fed WT mice also displayed decreased mitochondrial DNA (mtDNA) copy number compared with CSAA diet‐fed mice at both 32 and 66 weeks (p < 0.05), an outcome that was abrogated by Ripk3 deficiency (p < 0.05) (Figure 1C), suggesting that the absence of Ripk3 rescued impaired mitochondria biogenesis induced by the CDAA diet. Accordingly, the activity of citrate synthase, a tricarboxylic acid cycle enzyme that is also an indirect marker of mitochondrial mass, tended to increase in Ripk3 −/− mice compared with WT mice, reaching statistical significance in mice fed the CDAA diet for 32 weeks and the CSAA diet for 66 weeks (p < 0.01) (Figure 1C).
FIGURE 1.
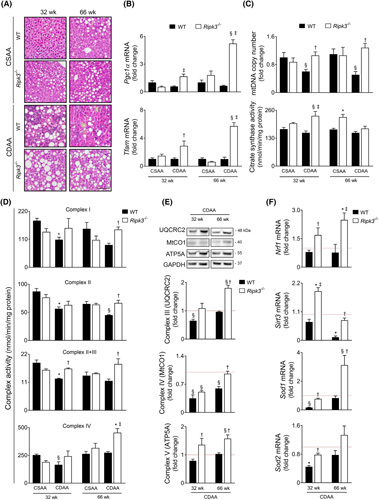
Ripk3 deficiency rescues mitochondrial biogenesis, mitochondria respiratory chain (MRC) activity, and reactive oxygen species scavenging systems in choline‐deficient, amino acid‐defined (CDAA) diet–induced experimental NASH. C57BL/6 wild‐type (WT) or Ripk3 −/− mice were fed a CDAA or an isocaloric control choline‐sufficient L‐amino acid‐defined (CSAA) diet for 32 or 66 weeks. (A) Representative images of hematoxylin and eosin–stained liver sections. Scale bar = 100 μM. (B) qRT‐PCR analysis of Pgc1α and Tfam in mouse liver. (C) Quantification of relative mitochondrial DNA copy number assessed by qPCR analysis of mitochondria‐encoded gene mt‐Co1. Nuclear Rn18s was used as loading control (top); and citrate synthase activity determined as described in Supplementary Materials and Methods (bottom). (D) MRC activity determined as described in Supplementary Materials and Methods. (E) Immunoblotting and densitometry of oxidative phosphorylation protein complexes in mouse liver, namely UQCRC2 (CIII), mtCO1 (CIV), and ATP5A (CV). Blots were normalized to endogenous GAPDH. F. qRT‐PCR analysis of Nrf1, Sod1, Sod2, and Sirt3 in mouse liver. Results are expressed as mean ± SEM arbitrary units or fold change of 6–7 individual mice. Red dashed line represents the CSAA diet‐fed WT mice levels. § p < 0.05 and *p < 0.01 from CSAA diet‐fed WT mice; † p < 0.05 and ‡ p < 0.01 from CDAA diet‐fed WT mice.
Then, we hypothesized that Ripk3 deficiency mitigates the dysfunction of mitochondrial oxidative phosphorylation (OXPHOS) induced by the CDAA diet. In both WT and Ripk3 −/− mice, CDAA diet led to decreased hepatic protein content of NADH dehydrogenase (ubiquinone) 1b subcomplex 8 at 32 and 66 weeks and succinate dehydrogenase (ubiquinone) iron–sulfur subunit at 32 weeks (p < 0.05; Figure S1B), which are nuclear DNA‐coded subunits of mitochondria respiratory chain (MRC) complexes I and II, respectively. Still, the activities of MRC complexes I and II were restored to basal levels in Ripk3 −/− mice at 66 weeks upon CDAA feeding (p < 0.05) (Figure 1D). Activities of complex II + III at 32 and 66 weeks and complex IV at 66 weeks were also rescued by Ripk3 deficiency in CDAA diet‐fed mice (p < 0.05; Figure 1D), following the increase in mtDNA encoded ubiquinol‐cytochrome c reductase core protein II from complex III and cytochrome c oxidase subunit I (MTCO1) from complex IV in Ripk3 −/− mice compared with WT (p < 0.05) (Figure 1E). Finally, although CDAA diet did not impact ATP synthase subunit a (ATP5A) protein levels from complex V, which uses the proton‐motive force created by MRC complexes I, III, and IV to generate ATP, Ripk3 deficiency increased ATP5A protein irrespective of diet and time point (at least, p < 0.05) (Figure 1E). Collectively, RIPK3 may contribute to impairment of OXPHOS machinery in experimental NAFLD.
PGC1α also increases the expression of nuclear respiratory factors (NRFs), which are transcription factors that regulate the expression of nuclear DNA genes encoding subunits of MRC complexes and regulate oxidative stress.13 In line with MRC complex expression and activity, liver NRF1 mRNA and protein were increased in Ripk3 −/− mice compared with WT mice under a CDAA diet, most notably at 66 weeks (at least, p < 0.05; Figure 1F and Figure S1C). Similarly, sirtuin 3 (SIRT3), a major mitochondria NAD+‐dependent deacetylase that participates in the antioxidant defense and interacts with subunits of MRC complexes I and II, increasing their activity,20 was upregulated in CDAA diet‐fed Ripk3 −/− mice (at least, p < 0.05) (Figure 1F and Figure S1C), particularly at 32 weeks. The expression of superoxide dismutase 1 and 2 was also increased in the liver of CDAA diet‐fed Ripk3 −/− mice (p < 0.05) (Figure 1F and Figure S1C). Thus, although mitochondria, particularly the MRC, play a major role in perpetuating oxidative stress under pathological conditions,21 several enzymes that detoxify ROS are induced by Ripk3 deficiency during CDAA feeding for 32 and 66 weeks. In agreement, we have previously shown that Ripk3 −/− mice fed a CDAA diet for 66 weeks displayed lower hepatic levels of ROS comparing with WT mice.9 Finally, mitofusin 2 (MFN2) promotes outer mitochondrial membrane fusion while also regulating cellular energy and mitochondrial metabolism.22 Accordingly, MFN2 protein levels were significantly increased in CDAA diet‐fed Ripk3 −/− mice, compared with WT counterparts (p < 0.05) (Figure S1D). Conversely, mitochondrial fission is controlled mainly by the dynamin‐related protein‐1, which was upregulated in CDAA diet‐fed WT mice at both 32 and 66 weeks (p < 0.05), an outcome that is prevented by Ripk3 deficiency at 32 weeks (p < 0.05) (Figure S1D). Overall, Ripk3 deficiency contributes to metabolic homeostasis in response to NASH‐related hepatic stress, improving mitochondria biogenesis, metabolic reprogramming, and antioxidant defense mechanisms.
Ripk3 deficiency impacts hepatocyte transcriptome improving mitochondrial bioenergetics
We next asked whether the recovery of hepatic MRC subunit expression and activity observed in Ripk3 −/− mice results from an overall improvement of NAFLD pathology and/or from specific modulation by RIPK3‐dependent signaling pathways. CRISPR/Cas9‐mediated deletion of Ripk3 was conducted in the immortalized murine hepatocyte cell line AML‐12 (Figure S2A), in which lipid metabolism and expression of mitochondrial complexes mimic that of primary mouse hepatocytes.23 The nonsupervised principal component analysis of the transcriptome data of WT and Ripk3 −/− AML‐12 cells in the presence or absence of 125 μM palmitate (PA) bound to bovine serum albumin for 24 h showed four distinct clusters according to genotype and stimulus (Figure 2A). A similar range and distribution of reads across all samples was observed confirming a comparable transcriptomic coverage. A total of 2894 differentially expressed genes (DEGs; padj ≤ 0.05, |log2FoldChange| ≥ 0) were detected between unchallenged WT and Ripk3 −/− hepatocytes, 4333 DEGs were detected between PA‐loaded WT and Ripk3 −/− hepatocytes, 2174 DEGs were detected between vehicle‐ and PA‐treated WT hepatocytes, and 1316 DEGs were detected between vehicle‐ and PA‐treated Ripk3 −/− hepatocytes (Figure 2B). In addition, by comparing the transcriptomes of both control AML‐12 cells, 207 and 178 genes were only expressed in Ripk3 −/− and WT hepatocytes, respectively (Figure 2C). Similarly, 243 and 208 transcripts were elevated in PA‐loaded Ripk3 −/− and WT hepatocytes, respectively (Figure 2D). The DEGs were clustered using a hierarchical clustering algorithm (Figure 2B), and Kyoto Encyclopedia of Genes and Genomes pathway enrichment analysis unveiled that transcripts associated with OXPHOS and thermogenesis were markedly upregulated in unchallenged Ripk3 −/− hepatocytes compared with WT cells (Figure 2C), an effect that was aggravated by the PA treatment (Figure 2D). Similarly, gene ontology enrichment analysis showed that mitochondria respiration‐related proteins were overrepresented among the cellular components that were upregulated in Ripk3 −/− hepatocytes compared with WT cells (Figure S3). Those include several subunits of cytochrome c oxidase and ATP synthase (Figure 2E), which were also increased in the liver of CDAA diet‐fed Ripk3 −/− mice (Figure 1E).
FIGURE 2.
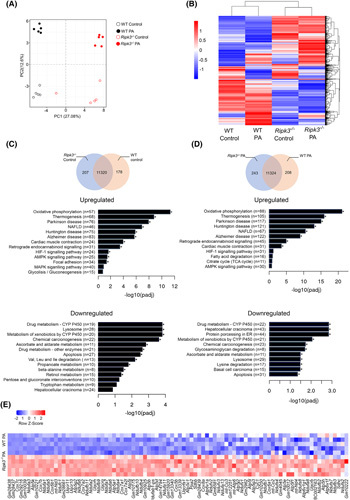
Ripk3 deficiency shifts the hepatocyte transcriptome. Wild‐type (WT) and Ripk3 −/− AML‐12 cells were treated with 125 μM palmitate (PA) for 24 h. (A) Score scatter plot corresponding to a principal component analysis (PCA) of the RNA sequencing data. Each individual is represented by one dot. (B) Heatmap of RNA‐Seq expression z scores computed for all genes that are differentially expressed between (padj ≤ 0.05, |log2FoldChange| ≥ 0), where red indicates overexpressed transcripts and blue represents underexpressed transcripts. (C) Venn diagrams showing genes expressed in unchallenged Ripk3 −/− versus WT hepatocytes. Kyoto Encyclopedia of Genes and Genomes (KEGG) pathway enrichment analysis of upregulated and downregulated differentially expressed genes (DEGs). (D) Venn diagrams showing genes expressed in PA‐treated Ripk3 −/− versus WT hepatocytes. KEGG pathway enrichment analysis of upregulated and downregulated DEGs. (E) Heatmap of z scores of DEGs associated with the KEGG pathways oxidative phosphorylation relative to the comparison Ripk3 −/− versus WT hepatocytes treated with PA. Each row in the heatmap is an individual sample. Red indicates overexpressed transcripts and blue represents underexpressed transcripts. *p < 0.05 from respective control.
Next, we assessed the mitochondrial respiration of WT and Ripk3 −/− AML‐12 cells exposed to 62.5 μM PA for 48 h (Figure 3A), a concentration that did not impact cell viability as shown by resazurin metabolism (Figure S2B). During mitochondrial respiration assays, ATP synthesis‐linked respiration is determined by adding the ATP synthase inhibitor oligomycin, whereas remaining basal respiration not coupled to ATP generation is considered the proton leak. Maximal OCR is revealed by injecting the uncoupler p‐trifluoromethoxy carbonyl cyanide phenyl hydrazone. Spare respiratory capacity representing the ability of a cell to respond to increased metabolic demands is calculated as the difference between maximal and basal respiration. Finally, the injection of rotenone and antimycin, inhibitors of MRC complexes I and III, respectively, allow for the determination of nonmitochondrial OCR. Our results showed that the presence of PA resulted in an approximately 30%–40% reduction of basal (p < 0.01), ATP‐linked (p < 0.01), maximal (p < 0.05), and spare (p < 0.05) OCR only in WT AML‐12 cells, whereas Ripk3 −/− hepatocytes maintained mitochondrial bioenergetics after PA exposure (Figure 3B). Proton leak and nonmitochondrial respiration remained unchanged in both PA‐loaded WT and Ripk3 −/− AML‐12 cells (Figure 3B). Altogether, the absence of Ripk3 impacts on hepatocyte transcriptomic profile by increasing the expression of genes implicated in mitochondria bioenergetics and function and rescuing mitochondria respiration after free fatty acid overload.
FIGURE 3.
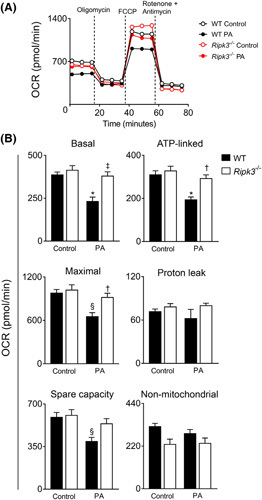
Ripk3 deficiency improves hepatocyte mitochondrial bioenergetics following palmitate (PA) treatment in AML‐12 cells. Wild‐type (WT) and Ripk3 −/− AML‐12 cells were treated with 62.5 μM PA for 48 h. (A) Respiratory flux profiles of cells were determined using a Seahorse extracellular flux analyzer as described in Supplementary Materials and Methods. (B) Quantification of bioenergetic parameters including oxygen consumption rate (OCR) associated with basal respiration, ATP‐linked, maximal respiration capacity, proton leak, spare capacity, and nonmitochondrial respiration. Results are expressed as mean ± SEM arbitrary units of four independent experiments. § p < 0.05 and *p < 0.01 from control; † p < 0.05 from and ‡ p < 0.01 from respective control.
Ripk3 deficiency dampens mitochondrial dysfunction and oxidative stress
Free fatty acids can modulate mitochondria ROS generation by uncoupling or by interference with MRC.24 Mitochondrial ROS were increased in WT AML‐12 cells treated with 125 μM PA for 24 h (p < 0.05) (Figure 4A), whereas transcripts for Nrf1 (p < 0.01), Sod2 (p < 0.01), and Sirt3 (p < 0.01) (Figure 4B) were also increased as an adaptative cellular mechanism. In turn, Ripk3 −/− AML‐12 cells were protected from mitochondrial ROS induced by PA (p < 0.05; Figure 4A), at least in part, by a more robust upregulation of radical scavenger systems (p < 0.01) (Figure 4B). We also evaluated mitochondrial network morphology and mitochondrial membrane potential using the fluorescent probe tetramethylrhodamine methyl ester perchlorate (TMRM). A decrease in TMRM fluorescence intensity was observed in PA‐loaded WT cells (p < 0.01), indicating decreased mitochondrial membrane polarization, which was restored by Ripk3 deficiency (p < 0.05) (Figure 4C,D). TMRM labelling also showed that PA treatment reduced the number and area of polarized mitochondria only in WT AML‐12 cells (p < 0.05) (Figure 4C,D), whereas the maintenance of the mitochondrial network in PA‐treated Ripk3 −/− cells (p < 0.05) (Figure 4D) was accompanied by a robust increase in Pgc1α mRNA levels (p < 0.01) (Figure S4A) downstream to PPARγ activation (at least p < 0.05) (Figure S5). In addition, although the average size of mitochondria was not significantly impacted by the presence of PA and RIPK3 (Figure 4D), transcripts for optic atrophy protein (Opa1) and Mfn2, mitochondrial fusion‐related genes, were increased in PA‐treated Ripk3 −/− cells (at least p < 0.05) (Figure S4A). Collectively, Ripk3 deficiency dampens mitochondrial dysfunction induced by PA in hepatocytes.
FIGURE 4.
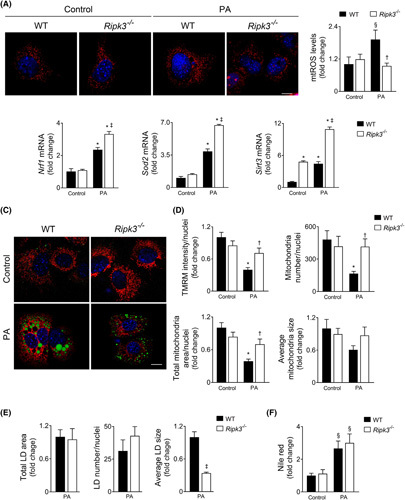
Ripk3 deficiency dampens hepatocyte mitochondrial reactive oxygen species (ROS) following palmitate treatment in AML‐12 cells. Wild‐type (WT) and Ripk3 −/− AML‐12 cells were treated with 125 μM palmitate (PA) for 24 h. (A) Representative staining for MitoSOX Red (red). Nuclei were counterstained with Hoechst 33258 (blue). Scale bar = 85 μM. Histogram shows the quantification of mitochondrial ROS. (B) qRT‐PCR analysis of Nfr1, Sod2, and Srit3 in AML‐12 cells. (C) Representative staining for tetramethylrhodamine methyl ester perchlorate (TMRM) (red), LipidTOX Green (green), and Hoechst 33342 (blue) for mitochondrial network, neutral LD, mitochondrial superoxide, and nuclei, respectively. Scale bar = 85 μM. (D) Histograms show the quantification of TMRM intensity and number, area, and average size of mitochondria. (E) Histograms show the quantification of total area, average size, and number of lipid droplets. (F) Fluorometric measurement of Nile red staining normalized by SRB method. Results are expressed as mean ± SEM fold change or percentage of four independent cultures from each genotype § p < 0.05 and *p < 0.01 from control; † p < 0.05 from and ‡ p < 0.01 from respective control.
Ripk3 deficiency upregulates lipid droplet coat proteins in hepatocytes in experimental NAFLD and in patients
Channeling PA into triglyceride droplet formation has a protective effect by preventing the generation of lipotoxic intermediates.25 Indeed, while reducing lipidosis, inhibition of triglyceride synthesis exacerbates liver damage and fibrosis in obese mice with NASH.26 We have shown that Ripk3 −/− hepatocytes display increased hepatic lipid content under both hypercaloric CSAA and CDAA diets, as well as after PA exposure in vitro.9 In agreement, here we showed that the 125‐μM PA‐induced intracellular fat accumulation, as measured by Nile red fluorometric quantification, was higher in Ripk3 −/− cells than in WT AML‐12 cells at 48 h (Figure S2C). However, although the total area of lipid droplets (LDs) (Figure 4C,E) and intracellular neutral fat accumulation (Figure 4F) were similar between PA‐loaded WT and Ripk3 −/− AML‐12 cells at 24 h, a higher number of smaller (p < 0.01) LDs were found in Ripk3 −/− cells (Figure 4C,E). Rescue experiments revealed that prior to necroptotic‐associated cell swelling, overexpression of GFP‐tagged murine RIPK3 enlarged LDs after free fatty acid exposure when compared with neighboring Ripk3 −/− AML‐12 cells (p < 0.05) (Figure S4B), cosubstantiating the role of RIPK3 on LD dynamics. The impact of Ripk3 deficiency on LD structure was independent of the modulation of lipophagy at this time point, as the decreased microtubule‐associated protein LC3‐II levels and increased p62 levels indicated a decrease in autophagy in PA‐exposed hepatocytes regardless of genotype (Figure S4C).
LDs are not stable lipid depots, and their size and protein composition are cell context‐dependent. The perilipins (PLINs) are a major family of five LD‐associated proteins that regulate lipid storage and hydrolysis. Here, we evaluated the expression of the most well‐known PLINs (Plin1, Plin2, and Plin5) in AML‐12 cells and in the CDAA model. Our results showed that basal mRNA levels of Plin1 and Plin5 were already significantly increased in Ripk3 −/− AML‐12 cells compared with WT cells (at least p < 0.05) and further increased with PA treatment at 24 h (p < 0.01) (Figure 5A). In turn, Plin2 mRNA levels were increased by PA in a dose‐dependent manner in both WT and Ripk3 −/− cells, although Plin2 upregulation was more pronounced in Ripk3 −/− AML‐12 cells treated with the highest dose of PA (at least p < 0.05) (Figure 5A). The transcription of Plin1 and Plin5 was also markedly increased in Ripk3 −/− mice fed a CDAA diet compared with WT mice (p < 0.05), whereas Plin2 mRNA levels were only modestly affected by Ripk3 deficiency (Figure 5B). PLIN1 and PLIN5 protein levels were also increased in CDAA diet‐fed Ripk3 −/− mice at both 32 and 66 weeks of feeding (p < 0.05) and even slightly augmented in CSAA diet‐fed Ripk3 −/− mice compared with WT mice (Figure 5C). Similarly, in the adipose tissue, in which PLIN1 is highly expressed, Plin1 mRNA levels were increased in Ripk3 −/− mice fed either CSAA or CDAA diet at both time points when compared with WT mice (Figure 5D). Plin1 transcript levels also increased in CRISPR‐Cas9 Ripk3‐null 3 T3‐L1 cells differentiated in adipocytes (Figure S6) compared with WT cells (p < 0.05; Figure 5E), whereas the cell death associated with the differentiation process was reduced (p < 0.01; Figure 5F).
FIGURE 5.
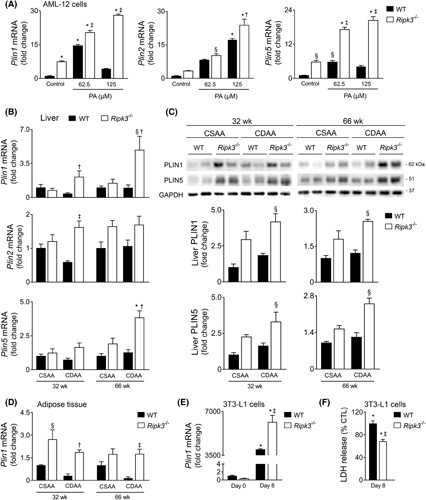
Ripk3 deficiency upregulates lipid droplets coat proteins in hepatocytes in vitro and in vivo. (A) qRT‐PCR analysis of Plin1, Plin2, Plin5 in wild‐type (WT) and Ripk3 −/− AML‐12 cells treated with 62.5 or 125 μM palmitate for 24 h. (B) qRT‐PCR analysis of Plin1, Plin2, Plin5 in the liver of C57BL/6 WT or Ripk3 −/− mice fed a choline‐deficient, amino acid‐defined (CDAA) or an isocaloric control choline‐sufficient L‐amino acid‐defined (CSAA) diet for 32 or 66 weeks. (C) Immunoblotting and densitometry of PLIN1 and PLIN5 in the liver of C57BL/6 WT or Ripk3 −/− mice fed a CDAA or CSAA diet for 32 or 66 weeks. Blots were normalized to endogenous GAPDH. Representative immunoblots are shown. (D) qRT‐PCR analysis of Plin1 in the white adipose tissue of C57BL/6 WT or Ripk3 −/− mice fed a CDAA or an isocaloric control CSAA diet for 32 or 66 weeks. (E) qRT‐PCR analysis of Plin1 in WT and Ripk3 −/− 3 T3‐L1 cells before (day 0) and after differentiation in adipocytes (day 8). (F) Percentage of released LDH in differentiated adipocytes relative to undifferentiated 3 T3‐L1 cells, as surrogate of general cells death. Results are expressed as mean ± SEM fold change or percentage to control of three to four independent in vitro experiments or six to seven individual mice. § p < 0.05 and *p < 0.01 from CSAA diet‐fed WT mice; † p < 0.05 and ‡ p < 0.01 from CDAA diet‐fed WT mice.
A negative correlation between protein levels of PLIN1 or PLIN5 and RIPK3 was found in the liver of patients from an NAFLD cohort (p < 0.01) (Figure 6A). PLIN5 levels were not differentially modulated by the NAFLD stage but were inversely correlated with circulating levels of liver injury (aspartate aminotransferase, p < 0.01; alkaline phosphatase, p = 0.07; γ‐glutamyl transferase, p = 0.07) (Figure S7A) and slightly decreased in higher fibrosis scores (p = 0.07) (Figure 6B). In turn, although PLIN1 levels were increased in patients with NAFLD with more hepatic large or medium‐sized LDs (steatosis score 3 vs. 2; p < 0.05) (Figure 6C), patients with NAFLD with active disease (histological activity score ≥ 2 vs. <2) display diminished protein levels of PLIN1 (p < 0.05; Figure 6C) and alkaline phosphatase (p < 0.05) (Figure S7B). Furthermore, despite the lack of effect on fibrosis score, hepatic PLIN1 levels were negatively correlated with liver stiffness measurement (p < 0.05) and Fibrosis‐4 index (p = 0.054) (Figure 6D). Hepatic PLIN1 levels were also decreased in diabetic and hypertensive patients with NAFLD (p < 0.01) and with increased circulating hemoglobin A1C (p < 0.06) (Figure S7B, C). Strikingly, a patient with FPLD type 4 carrying a 4‐bp duplication in exon 8 (c.1202_1205dup), leading to the synthesis of a p. (Pro403Argfs*164) mutant PLIN1 protein with an altered interaction domain with 1‐acylglycerol‐3‐phosphate O‐acyltransferase (ABHD5),19 presented severe NAFLD and liver fibrosis diagnosed at early adulthood (Table S1), without splenomegaly or biliary defect. In the liver of a patient with PLIN1‐associated FPLD, only part of the LDs colocalized with PLIN1 (Figure S8), whereas RIPK3 protein levels increased in injured liver tissue (Figure 6E), suggesting a bidirectional functional link between PLIN1 and RIPK3 in NAFLD severity. Taken together, Ripk3 deficiency strongly associates with upregulation of liver Plin1 and Plin5 in experimental NAFLD and in human NAFLD, likely playing a role in LD dynamics and disease pathogenesis.
FIGURE 6.
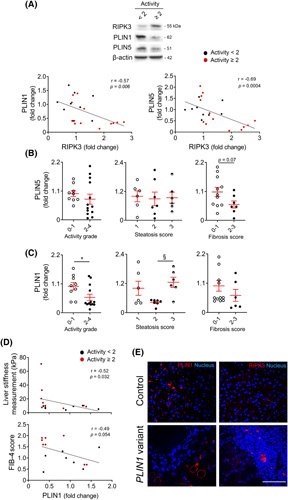
Ripk3 deficiency associates with augmented hepatic perilipin (PLIN)1 and PLIN5 in patients with NAFLD. (A) Representative immunoblots of RIPK3 and PLIN1 and PLIN5 in the liver of patients from an NAFLD cohort (n = 22) and their Spearman correlation scatter plot. Blots were normalized to endogenous β‐actin. Densitometry of PLIN1 (B) and PLIN5 (C) according to activity grade, steatosis score, and fibrosis score. Activity score in patients with NAFLD corresponds to the unweighted addition of hepatocyte ballooning and lobular inflammation as described in Supplementary Materials and Methods. (D) Spearman correlation scatter plot of hepatic PLIN1 with liver stiffness measurement assessed by FribroScan and Fibrosis‐4 (FIB4) score in patients with NAFLD. Each individual is represented by one dot. Data are expressed as mean ± SEM arbitrary units or fold change. § p < 0.05 and *p < 0.01 compared with respective controls. (E) Representative immunostaining of PLIN1 (left) and RIPK3 (right) in liver tissue from a healthy control (top) and a patient with familial partial lipodystrophy type 4 carrying a PLIN1 frameshift variant (bottom). Nuclei were counterstained with Hoechst 33258 (blue). Scale bar = 100 μM.
PLIN1 upregulation in Ripk3‐deficient hepatocytes controls LD structure and diminishes mitochondrial stress upon free fatty acid overload
To evaluate the functional role of PLIN1 in fatty acid‐loaded hepatocytes, AML‐12 cells were transfected with an siRNA‐targeting Plin1, leading to an approximately 60%–75% decrease in Plin1 mRNA levels, compared with siRNA‐control transfected cells (p < 0.05) (Figure 7A). The absence of Ripk3 protected AML‐12 cells from 125‐μM PA‐induced cell membrane permeabilization (Figure 7B), whereas the cosilencing of Plin1 had no impact on cell viability or on neutral lipid storage in hepatocytes at 24 h (Figure 7B,C). Still, Plin1 resulted in fewer (p < 0.01) and enlarged LD in Ripk3 −/− AML‐12 cells at 24 h (p < 0.05) (Figure 7D), corroborating that the upregulation of Plin1 observed in PA‐loaded Ripk3 −/− hepatocytes shape LD morphology at early time points.
FIGURE 7.
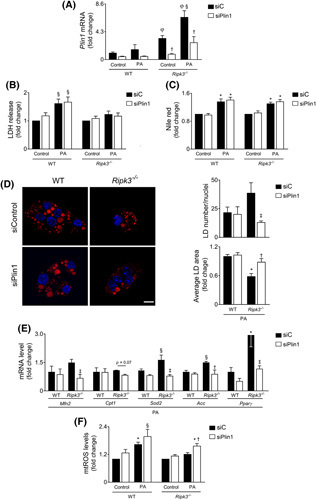
PLIN1 upregulation in Ripk3‐deficient hepatocytes controls lipid droplet structure and diminished mitochondrial stress upon free fatty acid overload. Wild‐type (WT) and Ripk3 −/− AML‐12 hepatocytes were transfected with an siRNA‐targeting Plin1 (siPlin1) or a scrambled control (siC) for 24 h. Then, cells were loaded with 125 μM palmitate (PA) or vehicle control for 24 h. (A) qRT‐PCR analysis of Plin1 in AML‐12 cells. (B) General cell death as assessed by the lactate dehydrogenase (LDH) release assay. (C) Fluorometric measurement of Nile red staining normalized by SRB method. § p < 0.05 and *p < 0.01 from control; † p < 0.05 from respective control. φ p < 0.01 from WT control. Data are expressed as mean ± SEM fold change for WT control of four independent experiments. (D) Representative staining for Nile Red (red) and Hoechst 33342 (blue) for neutral lipid droplet (LD) and nuclei, respectively. Scale bar = 15 μM. Histograms show the quantification of number and area of LD. (E) qRT‐PCR analysis of Mfn2, Cpt1a, Sod2, Acc, and Pparγ in AML‐12 cells. Data are expressed as mean ± SEM fold change for WT PA of four independent experiments. (F) Mitochondrial reactive oxygen species as determined by the fluorometric measurement of MitoSOX Red. Data are expressed as mean ± SEM fold change for WT control of four independent experiments. § p < 0.05 and *p < 0.01 from WT control; † p < 0.05 from and ‡ p < 0.01 from respective control.
PLIN1 interacts with MFN2 in the outer mitochondrial membrane, which is key for transferring fatty acids to mitochondria in brown adipose tissue.22 Here, we found that the knockdown of Plin1 diminished Mfn2 mRNA levels only in Ripk3 −/− AML‐12 cells after PA exposure (p < 0.01), which was accompanied by a slight decrease in the mRNA levels of carnitine palmitoyltransferase I (p = 0.07) (Figure 7E). Curiously, Plin1 downregulation in PA‐loaded Ripk3 −/− AML‐12 cells also restored Sod2 mRNA levels to those of PA‐loaded WT cells (Figure 7E), abolishing protection against PA‐induced mitochondrial ROS generation that was achieved by Ripk3 deficiency (p < 0.05) (Figure 7F). The contacts between mitochondria and LD have also been functionally implicated in triacylglycerol synthesis and LD expansion,27 whereas adipose‐specific Mfn2 knockout mice display reduced transcription of acetyl‐CoA carboxylase (Acc) and PPARγ, implicated in lipogenesis.22 Similarly, upon PA exposure, the siRNA‐mediated silencing of Plin1 abrogated the Ripk3 −/− ‐dependent upregulation of Acc (p < 0.01) and Pparγ (p < 0.01) (Figure 7E). In murine adipocytes, the expression of the Plin1 gene is regulated by PPARγ28; however, although Pparγ knockdown decreased the basal expression of Plin1 in both WT and Ripk3 −/− AML‐12 cells (p < 0.01), the upregulation of Plin1 in response to PA exposure was not affected by Pparγ silencing (Figure S5). Overall, Ripk3 deficiency in hepatocytes led to an upregulation of Plin1 that modulates LD structure and metabolism, together with the protection against mitochondrial stress upon free fatty acid overload, possibly by impacting on LD‐mitochondria interaction.
DISCUSSION
Metabolic coupling between MRC activity and mitochondrial fatty acid oxidation is crucial to curb hepatic fat accumulation and restrain ROS overabundance. In turn, the decline of mitochondrial function aggravates metabolic disturbances and may potentially contribute to NAFLD progression. On the other hand, free fatty acid overload leads to impaired mitochondria function, with consequent ROS overproduction and defective MRC activity.21 RIPK3 acts as a lipid metabolism regulator, and its deficiency attenuates liver damage, inflammation, oxidative stress fibrosis, and carcinogenesis, both in mouse models and human NAFLD.3–5,8,9 However, the role of RIPK3 in mitochondrial bioenergetics and function remain unclear. Here, we found that Ripk3 deficiency strongly impacts the hepatocyte transcriptome by increasing genes associated with OXPHOS and rescues mitochondrial biogenesis, bioenergetics, and function in experimental NASH and fat‐loaded hepatocytes. Moreover, Ripk3 deficiency was accompanied by a strong upregulation of the ROS scavenging systems, leading to diminished oxidative stress upon fat loading both in vivo and in vitro. Altogether, although accumulating evidence indicates that RIPK3/MLKL could increase mitochondrial respiration and oxidative stress in response to necroptotic stimuli,14–16 the absence of RIPK3 also prevents NAFLD‐related mitochondria damage and ROS, coupled with increased mitochondria content and MRC activity. The improved mitochondria fitness was likely triggered, at least in part, by increased activation of PGC1α/PPARγ axis in Ripk3 −/− cells, which is a pivotal pathway in lipid and metabolic regulation, redox balance, and immune responses in NAFLD.13,29
LD are multifunctional and highly dynamic intracellular organelles implicated in the packaging and management of body fat stores, critical for energy metabolism homeostasis and NAFLD development.30 The PLIN family comprises some of the most abundant LD proteins, playing a key role in LD formation, structural remodeling, and lipolysis. PLIN1 and 2 are considered as LD resident proteins that are subjected to degradation when in the cytosol, whereas PLIN3, 4, and 5 are either bound to LDs or located in the cytoplasm associated with other organelles.31 We showed that increased expression of Plin1 in PA‐loaded Ripk3 −/− AML‐12 cells was accompanied by more and smaller LDs at early time points, whereas the amount and size of LDs were recovered to WT levels upon Plin1 loss in Ripk3 −/− cells. Importantly, the increase in LD size can cause activation of hepatic stellate cells,30 which we found previously decreased upon RIPK3 knockdown.9 Likewise, it has been shown that AML‐12 cells lacking both Plin2 and Plin3 exhibited fewer and larger LDs, together with decreased AKT phosphorylation as a surrogate of insulin resistance.32 This phenotype could reflect a specific function of PLINs in controlling LD life cycling and/or suggest that some PLINs present surfactant properties that minimize the contact of LD surface area with the surrounding aqueous cytosol, counteracting LD fusion. Of note, small multilocular LDs and large unilocular LDs likely show distinct profiles of interaction with their binding partners through membrane contact sites, including mitochondria. Indeed, it has been shown that the overexpression of PLIN5, which mediates the interaction between LDs and mitochondria, recruits mitochondria to LDs in multiple cell types, including AML‐12 cells.33 In adipocytes, it has been described that LD are anchored to mitochondria via a PLIN1–MFN2 interaction.22 Here, we found that the loss‐of‐function of Plin1 in Ripk3 −/− hepatocytes led to a diminished expression of Mfn2, together with decreased expression of Pparγ and Acc, after free fatty acid exposure. Furthermore, the absence of Ripk3 in hepatocytes treated with free fatty acids at longer time points and in livers of mice fed hypercaloric CSAA/CDAA diets9 led to an augmented intracellular fat accumulation, accompanied by increased Acc, Plin1, and Plin5 expression, and enhanced mitochondrial OXPHOS and citrate synthase activities. Of note, it has been reported that a higher level of mitochondria–LD contact could fuel de novo lipogenesis by increasing OXPHOS and tricarboxylic acid cycle capacity that provides ATP and citrate, respectively, which ultimately promotes the ACC‐mediated production of new fatty acids27,34. It has also been shown that PLIN1 overexpression increases the expression of genes associated with fatty acid and triglyceride synthesis in bovine adipocytes,35 which is also increased in the livers of Ripk3 −/− mice from the CDAA model9 and in PA‐loaded Ripk3 −/− AML‐12 cells. Overall, these findings suggest that PLIN1 may ultimately regulate the amount of fat produced and the interaction between LD and mitochondria in Ripk3 −/− hepatocytes.
Increased hepatic expression of PLIN1 has been reported in patients with NAFLD.36,37 Whether its abundance is causal or consequential to the disease is unclear. We have previously reported that RIPK3 is increased in patients with more severe NAFLD, including those who are carriers of the rs738409 variant of the PNPLA3 gene.9 Here, we found that PLIN1 protein levels were negatively correlated with RIPK3 levels in human livers and increased in patients with NAFLD with marked hepatic accumulation of large or medium‐sized LDs. More importantly, PLIN1 levels were decreased in patients with more severe disease. Thus, LD growth associated with higher PLIN1 levels in hepatocytes of patients with NAFLD is likely an adaptive, beneficial, regulated phenomenon rather than a consequence of impaired utilization. Indeed, LD sustain energy and cellular homeostasis in stressed cells by controlling free fatty acid mobilization for optimal energy production and sequestering toxic lipids into their neutral lipid core.31 Accordingly, in heterozygous PLIN1 frameshift variant associated with FPLD19 and severe liver fibrosis and NAFLD, only some hepatic LDs contain PLIN1, whereas RIPK3 levels increased in injured liver tissue. Curiously, physical exercise was shown to promote an intramyocellular increase of PLIN1 and triglycerides in both control and high‐fat diet–fed mice,38 whereas the number of LDs and mitochondria and their physical interaction were also augmented in the muscle of individuals subjected to endurance exercise training.39 Thus, we cannot exclude that upregulation of PLIN1 in other cell types could also contribute to a better phenotype in Ripk3 −/− mice subjected to diet‐induced experimental NAFLD.
Epigenetic mechanisms contribute to the regulation of PPARγ, PGC‐1α, and PLIN1 during metabolic disease.40,41 Importantly, the necroptosis‐associated hepatic cytokine microenvironment is associated with lineage commitment in liver cancer by impacting on epigenome.42 Noteworthy, we found that SIRT3, a nuclear NAD+‐dependent histone deacetylase that translocates to the mitochondria upon cellular stress,43 was augmented in Ripk3 −/− hepatocytes both in vitro and in vivo, even at basal conditions. In line, it has been described that necroptosis is preceded by nuclear translocation of RIPK344 that directly phosphorylates and, subsequently, activates the pyruvate dehydrogenase complex,15 which in turn converts pyruvate into acetyl‐CoA. Future studies should explore whether RIPK3 might impact the metabolic‐epigenetic landscape and subsequent mitochondrial‐related gene expression. In addition, RIPK3 might also affect mitochondrial function by directly phosphorylating critical regulators of cellular metabolism, such as pyruvate dehydrogenase complex or AMP‐activated protein kinase.45
In conclusion, the absence of RIPK3 associates with increased activation of PGC1α/PPARγ/PLIN signaling pathways in the liver, coupled with a rescue of mitochondria content, OXPHOS machinery, and LD remodeling, as well as improved ROS detoxification capacity in experimental NAFLD. Furthermore, we uncovered a protective role of PLIN1 upregulation in the liver of patients with NAFLD. Importantly, accumulating evidence points to the role of mitochondrial maladaptation in the progression of NAFLD toward cancer. Similarly, large LDs allow rapid dynamics between lipid synthesis and consumption to sustain cell proliferation in cancer, being PLIN1 downregulated during early hepatocarcinogenesis in patients.46 Thus, our findings may contribute to a better understanding of the mechanisms underlying the progression of NAFLD/NASH and highlight that targeting RIPK3 might provide a better metabolic adaptative response to NASH‐related stress.
Supplementary Material
Acknowledgments
AUTHOR CONTRIBUTIONS
Marta B. Afonso, Tawhidul Islam, and Julie Magusto performed most of the experiments and analyzed and interpreted results. Marta B. Afonso wrote the manuscript. Ricardo Amorim, Véronique Lenoir, Rui Simões, José Teixeira, and Liana C. Silva helped in confocal imaging and mitochondrial functional assays. Dominique Wendum, Isabelle Jéru, Corinne Vigouroux, and Vlad Ratziu provided human samples and clinical data. Rui E. Castro, Paulo J. Oliveira, Carina Prip‐Buus, Vlad Ratziu, and Jérémie Gautheron provided intellectual input. Jérémie Gautheron was involved in planning and organization of the study. Cecília M. P. Rodrigues was the principal investigator of the study responsible for study concept and design. All authors critically revised the manuscript.
ACKNOWLEDGMENTS
We thank Dr. Vishva Dixit, Molecular Oncology Department, Genentech, Inc. (San Fransisco, California) and Dr. Miguel Soares, Instituto Gulbenkian de Ciência (Oeiras, Portugal) for kindly providing WT and Ripk3 −/− from the same genetic background. We also thank Andreia Barateiro and Catarina Barros (Fernandes Lab), iMed.ULisboa (Lisboa, Portugal) for technical support in confocal microscopy. Finally, we thank all members of Rodrigues Lab for insightful discussions.
FUNDING INFORMATION
Main funding is provided by FEDER funds through the COMPETE programme and by national funds through Fundação para a Ciência e a Tecnologia to CMPR (grants SAICTPAC/0019/2015 – LISBOA‐01‐0145‐FEDER‐016405 and PTDC/MED‐FAR/29097/2017 – LISBOA‐01‐0145‐FEDER‐029097). Additional funding comes from the European Union's Horizon 2020 Research and Innovation programme under the Marie Skłodowska‐Curie Grant Agreement No. 722619 to CMPR, CP‐B and PJO. JG is funded by the Fondation pour la Recherche Médicale (ARF20170938613 & EQU202003010517), the Agence Nationale de la Recherche (ANR‐21‐CE18‐0002‐01), the Mairie de Paris (Emergences – R18139DD) and La Caixa Foundation (LCF/PR/HR21/52410028).
CONFLICT OF INTEREST
The authors have no conflicts to report.
DATA AVAILABILITY STATEMENT
RNAseq data are deposited on the NCBI Sequence Read Archive (SRA) under the accession number GEO Submission GSE199155.
Footnotes
Abbreviations: Acc, acetyl‐CoA carboxylase; CDAA, choline‐deficient, amino acid‐defined; CSAA, choline‐sufficient L‐amino acid‐defined; DEG, differentially expressed gene; FPLD, familial partial lipodystrophy; LD, lipid droplet; MFN2, mitofusin 2; MLKL, mixed lineage kinase domain‐like protein; MRC, mitochondria respiratory chain; OCR, oxygen consumption rate; OXPHOS, oxidative phosphorylation; PA, palmitate; PGC1α, proliferator‐activated receptor gamma coactivator 1 alpha; PLIN, perilipin; PPARγ, peroxisome proliferator‐activated receptor γ; RIPK3, receptor‐interacting protein kinase 3; ROS, reactive oxygen species; RT‐PCR, reverse transcriptase‐polymerase chain reaction; TMRM, tetramethylrhodamine methyl ester perchlorate; WT, wild‐type.
Marta B. Afonso, Tawhidul Islam, and Julie Magusto contributed equally to this work.
Supplemental Digital Content is available for this article. Direct URL citations appear in the printed text and are provided in the HTML and PDF versions of this article on the journal's website, www.hepjournal.com.
Funding information Agence Nationale de la Recherche, Grant/Award Number: ANR-21-CE18-0002-01; Fondation pour la Recherche Médicale, Grant/Award Number: ARF20170938613 & EQU202003010517; Fundação para a Ciência e a Tecnologia, Grant/Award Number: PTDC/MED-FAR/29097/2017 -LISBOA-01-0145-FEDER-029 and SAICTPAC/0019/2015-LISBOA-01-0145-FEDER-016405; H2020 Marie Skłodowska-Curie Actions, Grant/Award Number: 722619; Mairie de Paris, Grant/Award Number: Emergences -R18139DD; La Caixa Foundation, Grant/Award Number: LCF/PR/HR21/52410028.
REFERENCES
- 1.Tilg H, Moschen AR. Evolution of inflammation in nonalcoholic fatty liver disease: the multiple parallel hits hypothesis. Hepatology. 2010;52:1836–46. [DOI] [PubMed] [Google Scholar]
- 2.Galluzzi L, Vitale I, Aaronson SA, Abrams JM, Adam D, Agostinis P, et al. Molecular mechanisms of cell death: recommendations of the Nomenclature Committee on Cell Death 2018. Cell Death Differ. 2018;25:486–541. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 3.Afonso MB, Rodrigues PM, Carvalho T, Caridade M, Borralho P, Cortez‐Pinto H, et al. Necroptosis is a key pathogenic event in human and experimental murine models of non‐alcoholic steatohepatitis. Clin Sci (Lond). 2015;129:721–39. [DOI] [PubMed] [Google Scholar]
- 4.Gautheron J, Vucur M, Reisinger F, Vargas Cardenas D, Roderburg C, Koppe C, et al. A positive feedback loop between RIP3 and JNK controls non‐alcoholic steatohepatitis. EMBO Mol Med. 2014;6:1062–74. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 5.Majdi A, Aoudjehane L, Ratziu V, Islam T, Afonso MB, Conti F, et al. Inhibition of receptor‐interacting protein kinase 1 improves experimental non‐alcoholic fatty liver disease. J Hepatol. 2020;72:627–35. [DOI] [PubMed] [Google Scholar]
- 6.Roychowdhury S, McCullough RL, Sanz‐Garcia C, Saikia P, Alkhouri N, Matloob A, et al. Receptor interacting protein 3 protects mice from high‐fat diet‐induced liver injury. Hepatology. 2016;64:1518–33. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 7.Gautheron J, Vucur M, Schneider AT, Severi I, Roderburg C, Roy S, et al. The necroptosis‐inducing kinase RIPK3 dampens adipose tissue inflammation and glucose intolerance. Nat Commun. 2016;7:11869. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 8.Gautheron J, Gores GJ, Rodrigues CMP. Lytic cell death in metabolic liver disease. J Hepatol. 2020;73:394–408. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 9.Afonso MB, Rodrigues PM, Mateus‐Pinheiro M, Simao AL, Gaspar MM, Majdi A, et al. RIPK3 acts as a lipid metabolism regulator contributing to inflammation and carcinogenesis in non‐alcoholic fatty liver disease. Gut. 2021;70:2359–72. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 10.Parisi LR, Sowlati‐Hashjin S, Berhane IA, Galster SL, Carter KA, Lovell JF, et al. Membrane disruption by very long chain fatty acids during necroptosis. ACS Chem Biol. 2019;14:2286–94. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 11.Parisi LR, Li N, Atilla‐Gokcumen GE. Very long chain fatty acids are functionally involved in necroptosis. Cell Chem Biol. 2017;24:1445–54 e1448. [DOI] [PubMed] [Google Scholar]
- 12.Corona JC, Duchen MR. PPARgamma as a therapeutic target to rescue mitochondrial function in neurological disease. Free Radic Biol Med. 2016;100:153–63. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 13.Piccinin E, Villani G, Moschetta A. Metabolic aspects in NAFLD, NASH and hepatocellular carcinoma: the role of PGC1 coactivators. Nat Rev Gastroenterol Hepatol. 2019;16:160–74. [DOI] [PubMed] [Google Scholar]
- 14.Zhang DW, Shao J, Lin J, Zhang N, Lu BJ, Lin SC, et al. RIP3, an energy metabolism regulator that switches TNF‐induced cell death from apoptosis to necrosis. Science. 2009;325:332–6. [DOI] [PubMed] [Google Scholar]
- 15.Yang Z, Wang Y, Zhang Y, He X, Zhong CQ, Ni H, et al. RIP3 targets pyruvate dehydrogenase complex to increase aerobic respiration in TNF‐induced necroptosis. Nat Cell Biol. 2018;20:186–97. [DOI] [PubMed] [Google Scholar]
- 16.Zhang Y, Su SS, Zhao S, Yang Z, Zhong CQ, Chen X, et al. RIP1 autophosphorylation is promoted by mitochondrial ROS and is essential for RIP3 recruitment into necrosome. Nat Commun. 2017;8:14329. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 17.Degterev A, Huang Z, Boyce M, Li Y, Jagtap P, Mizushima N, et al. Chemical inhibitor of nonapoptotic cell death with therapeutic potential for ischemic brain injury. Nat Chem Biol. 2005;1:112–9. [DOI] [PubMed] [Google Scholar]
- 18.Tait SW, Oberst A, Quarato G, Milasta S, Haller M, Wang R, et al. Widespread mitochondrial depletion via mitophagy does not compromise necroptosis. Cell Rep. 2013;5:878–85. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 19.Jeru I Vantyghem MC Bismuth E Cervera P Barraud S, Group PL‐S, et al. Diagnostic challenge in PLIN1‐associated familial partial lipodystrophy. J Clin Endocrinol Metab. 2019;104:6025–32. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 20.Ahn BH, Kim HS, Song S, Lee IH, Liu J, Vassilopoulos A, et al. A role for the mitochondrial deacetylase Sirt3 in regulating energy homeostasis. Proc Natl Acad Sci U S A. 2008;105:14447–52. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 21.Mansouri A, Gattolliat CH, Asselah T. Mitochondrial dysfunction and signaling in chronic liver diseases. Gastroenterology. 2018;155:629–47. [DOI] [PubMed] [Google Scholar]
- 22.Boutant M, Kulkarni SS, Joffraud M, Ratajczak J, Valera‐Alberni M, Combe R, et al. Mfn2 is critical for brown adipose tissue thermogenic function. EMBO J. 2017;36:1543–58. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 23.Nagarajan SR, Paul‐Heng M, Krycer JR, Fazakerley DJ, Sharland AF, Hoy AJ. Lipid and glucose metabolism in hepatocyte cell lines and primary mouse hepatocytes: a comprehensive resource for in vitro studies of hepatic metabolism. Am J Physiol Endocrinol Metab. 2019;316:E578–89. [DOI] [PubMed] [Google Scholar]
- 24.Hodson L, Gunn PJ. The regulation of hepatic fatty acid synthesis and partitioning: the effect of nutritional state. Nat Rev Endocrinol. 2019;15:689–700. [DOI] [PubMed] [Google Scholar]
- 25.Listenberger LL, Han X, Lewis SE, Cases S, Farese RV, Jr, Ory DS, et al. Triglyceride accumulation protects against fatty acid‐induced lipotoxicity. Proc Natl Acad Sci U S A. 2003;100:3077–82. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 26.Yamaguchi K, Yang L, McCall S, Huang J, Yu XX, Pandey SK, et al. Inhibiting triglyceride synthesis improves hepatic steatosis but exacerbates liver damage and fibrosis in obese mice with nonalcoholic steatohepatitis. Hepatology. 2007;45:1366–74. [DOI] [PubMed] [Google Scholar]
- 27.Benador IY, Veliova M, Mahdaviani K, Petcherski A, Wikstrom JD, Assali EA, et al. Mitochondria bound to lipid droplets have unique bioenergetics, composition, and dynamics that support lipid droplet expansion. Cell Metab. 2018;27:869–85 e866. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 28.Dalen KT, Schoonjans K, Ulven SM, Weedon‐Fekjaer MS, Bentzen TG, Koutnikova H, et al. Adipose tissue expression of the lipid droplet‐associating proteins S3‐12 and perilipin is controlled by peroxisome proliferator‐activated receptor‐gamma. Diabetes. 2004;53:1243–52. [DOI] [PubMed] [Google Scholar]
- 29.Wang W, Xu MJ, Cai Y, Zhou Z, Cao H, Mukhopadhyay P, et al. Inflammation is independent of steatosis in a murine model of steatohepatitis. Hepatology. 2017;66:108–23. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 30.Scorletti E, Carr RM. A new perspective on NAFLD: focusing on lipid droplets. J Hepatol. 2022;76:934–45. [DOI] [PubMed] [Google Scholar]
- 31.Olzmann JA, Carvalho P. Dynamics and functions of lipid droplets. Nat Rev Mol Cell Biol. 2019;20:137–55. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 32.Bell M, Wang H, Chen H, McLenithan JC, Gong DW, Yang RZ, et al. Consequences of lipid droplet coat protein downregulation in liver cells: abnormal lipid droplet metabolism and induction of insulin resistance. Diabetes. 2008;57:2037–45. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 33.Wang H, Sreenivasan U, Hu H, Saladino A, Polster BM, Lund LM, et al. Perilipin 5, a lipid droplet‐associated protein, provides physical and metabolic linkage to mitochondria. J Lipid Res. 2011;52:2159–68. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 34.Benador IY, Veliova M, Liesa M, Shirihai OS. Mitochondria bound to lipid droplets: where mitochondrial dynamics regulate lipid storage and utilization. Cell Metab. 2019;29:827–35. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 35.Zhang S, Liu G, Xu C, Liu L, Zhang Q, Xu Q, et al. Perilipin 1 mediates lipid metabolism homeostasis and inhibits inflammatory cytokine synthesis in bovine adipocytes. Front Immunol. 2018;9:467. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 36.Straub BK, Stoeffel P, Heid H, Zimbelmann R, Schirmacher P. Differential pattern of lipid droplet‐associated proteins and de novo perilipin expression in hepatocyte steatogenesis. Hepatology. 2008;47:1936–46. [DOI] [PubMed] [Google Scholar]
- 37.Pawella LM, Hashani M, Eiteneuer E, Renner M, Bartenschlager R, Schirmacher P, et al. Perilipin discerns chronic from acute hepatocellular steatosis. J Hepatol. 2014;60:633–42. [DOI] [PubMed] [Google Scholar]
- 38.Morton TL, Galior K, McGrath C, Wu X, Uzer G, Uzer GB, et al. Exercise increases and browns muscle lipid in high‐fat diet‐fed mice. Front Endocrinol (Lausanne). 2016;7:80. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 39.Tarnopolsky MA, Rennie CD, Robertshaw HA, Fedak‐Tarnopolsky SN, Devries MC, Hamadeh MJ. Influence of endurance exercise training and sex on intramyocellular lipid and mitochondrial ultrastructure, substrate use, and mitochondrial enzyme activity. Am J Physiol Regul Integr Comp Physiol. 2007;292:R1271–8. [DOI] [PubMed] [Google Scholar]
- 40.Kramer AI, Handschin C. How epigenetic modifications drive the expression and mediate the action of PGC‐1alpha in the regulation of metabolism. Int J Mol Sci. 2019;20:5449. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 41.Bialesova L, Kulyte A, Petrus P, Sinha I, Laurencikiene J, Zhao C, et al. Epigenetic Regulation of PLIN 1 in obese women and its relation to lipolysis. Sci Rep. 2017;7:10152. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 42.Seehawer M, Heinzmann F, D'Artista L, Harbig J, Roux PF, Hoenicke L, et al. Necroptosis microenvironment directs lineage commitment in liver cancer. Nature. 2018;562:69–75. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 43.Scher MB, Vaquero A, Reinberg D. SirT3 is a nuclear NAD + ‐dependent histone deacetylase that translocates to the mitochondria upon cellular stress. Genes Dev. 2007;21:920–8. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 44.Yoon S, Bogdanov K, Kovalenko A, Wallach D. Necroptosis is preceded by nuclear translocation of the signaling proteins that induce it. Cell Death Differ. 2016;23:253–60. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 45.Wu W, Wang X, Sun Y, Berleth N, Deitersen J, Schlutermann D, et al. TNF‐induced necroptosis initiates early autophagy events via RIPK3‐dependent AMPK activation, but inhibits late autophagy. Autophagy. 2021;17:3992–4009. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 46.Straub BK, Herpel E, Singer S, Zimbelmann R, Breuhahn K, Macher‐Goeppinger S, et al. Lipid droplet‐associated PAT‐proteins show frequent and differential expression in neoplastic steatogenesis. Mod Pathol. 2010;23:480–92. [DOI] [PubMed] [Google Scholar]
Associated Data
This section collects any data citations, data availability statements, or supplementary materials included in this article.
Data Availability Statement
RNAseq data are deposited on the NCBI Sequence Read Archive (SRA) under the accession number GEO Submission GSE199155.