

Abstract
The peroxisome proliferator-activated receptor γ coactivator-1 (PGC-1) family of transcriptional coactivators are regulators of mitochondrial oxidative capacity and content in skeletal muscle. Many of these conclusions are based primarily on gain-of-function studies using muscle-specific overexpression of PGC1s. We have previously reported that genetic deletion of both PGC-1α and PGC-1β in adult skeletal muscle resulted in a significant reduction in oxidative capacity with no effect on mitochondrial content. However, the contribution of PGC-1-related coactivator (PRC), the third PGC-1 family member, in regulating skeletal muscle mitochondria is unknown. Therefore, we generated an inducible skeletal muscle-specific PRC knockout mouse (iMS-PRC-KO) to assess the contribution of PRC in skeletal muscle mitochondrial function. We measured mRNA expression of electron transport chain (ETC) subunits as well as markers of mitochondrial content in the iMS-PRC-KO animals and observed an increase in ETC gene expression and mitochondrial content. Furthermore, the increase in ETC gene expression and mitochondrial content was associated with increased expression of PGC-1α and PGC-1β. We therefore generated an adult-inducible PGC-1 knockout mouse in which all PGC-1 family members are deleted (iMS-PGC-1TKO). The iMS-PGC-1TKO animals exhibited a reduction in ETC mRNA expression and mitochondrial content. These data suggest that in the absence of PRC alone, compensation occurs by increasing PGC-1α and PGC-1β to maintain mitochondrial content. Moreover, the removal of all three PGC-1s in skeletal muscle results in a reduction in both ETC mRNA expression and mitochondrial content. Taken together, these results suggest that PRC plays a role in maintaining baseline mitochondrial content in skeletal muscle.
Keywords: mitochondria, mitochondrial biogenesis, PGC-1-related coactivator, skeletal muscle
INTRODUCTION
Impaired skeletal muscle metabolic function is associated with the development of various diseases, including obesity and type 2 diabetes (1). Normal mitochondrial functionality is a critical component for maintaining normal skeletal muscle metabolic function (2). The ability to maintain or improve mitochondrial function is important to prevent the development of metabolic diseases (2). Exercise, particularly endurance exercise, is a powerful tool in both the prevention and treatment of metabolic diseases (3–5). Endurance exercise mediates many of its benefits by improving mitochondrial functionality, including both oxidative capacity (mitochondrial respiration) and mitochondrial biogenesis (6). Therefore, understanding the molecular components that are able to control mitochondrial function is imperative to stave off metabolic diseases.
The peroxisome proliferator-activated receptor gamma coactivator-1 (PGC-1) family of transcriptional coactivators (PGC-1α, PGC-1β, and PRC) have been shown to be potent regulators of oxidative capacity and mitochondrial biogenesis in a variety of cellular contexts (7–9). In skeletal muscle, PGC-1α is the most extensively studied family member, based largely on its proposed role in exercise-induced mitochondrial adaptions. In both humans and rodents, PGC-1α is induced in skeletal muscle in response to bouts of endurance exercise (10–12). However, the specific role of the other family members (PGC-1β and PRC) in skeletal muscle is not fully understood or delineated. The use of genetic models has begun to provide insight into the contributions of the different PGC-1 family members in skeletal muscle.
Muscle-specific overexpression of either PGC-1α or PGC-1β has increased exercise performance due in part to having increased mitochondrial content and oxidative capacity (13, 14). Conversely, muscle-specific loss of function of either PGC-1α or PGC-1β results in a mild effect on mitochondrial functionality and exercise performance (15, 16), which suggests functional redundancy and possible compensation. Furthermore, studies using models in which both PGC-1α and PGC-1β are absent from skeletal muscle have begun to address the actual contribution of PGC-1α and PGC-1β in skeletal muscle (16–19). We have previously reported that the deletion of skeletal muscle-specific PGC-1α and PGC-1β significantly reduced oxidative capacity with no effect on mitochondrial content (17). In addition, we have reported that in the absence of both PGC-1α and PGC-1β in skeletal muscle, mice are able to increase exercise performance in response to exercise training (18). These studies suggest an additional component in skeletal muscle that can compensate for the absence of PGC-1α and PGC-1β, possibly the third family member PRC. However, it is currently unknown what specific role PRC plays in skeletal muscle mitochondrial function.
In our current study, we sought to determine the contribution of the transcriptional coactivator PRC in adult skeletal muscle on regulating baseline mitochondrial function, by systematically deleting PRC and the other PGC-1 family members in adult skeletal muscle. Our results suggest that genetic deletion of PRC in adult skeletal muscle results in a compensation by both PGC-1α and PGC-1β via increased expression to maintain mitochondrial function (ETC subunit expression). However, genetic deletion of PRC in combination with PGC-1α and/or PGC-1β results in a reduction in mitochondrial content. Taken together, these data identify PRC as being an important regulator in maintaining mitochondrial content in skeletal muscle.
METHODS
Animal Studies
All animal experiments were performed according to procedures approved by the University of Alabama at Birmingham (UAB) Institutional Animal Care and Use Committee (IACUC). PRC floxed mice were generated by flanking exons 2 through 6 with loxP sites using a targeting vector generated by PCR amplification using BAC clone RP23-461L11 and RP23-282N19 from the C57BL/6 library as a template. PGC-1α floxed mice and PGC-1β floxed mice used were previously generated and described elsewhere (20, 21). The various PGC-1 floxed alleles were intercrossed with the tamoxifen-inducible Cre recombinase under the control of human skeletal actin promoter (HSA-MerCreMer) (22), to generate the indicated single (iMS-PRC KO), double (iMS-PGC-1α/PRC DKO and iMS-PGC-1β/PRC DKO) or triple (iMS-PGC-1 TKO) knockout animals. To induce deletion of the PGC-1 alleles, animals were administered tamoxifen (2 mg/day for 5 consecutive days) dissolved in sesame oil by intraperitoneal injections, as previously described (18, 23, 24). Female mice aged 20 to 24 wk old were used unless otherwise specified, flox-only littermates served as controls and all animals received tamoxifen injections. All assessments were performed at 8 wk post tamoxifen administration as we have previously described (18, 23).
qPCR mRNA Expression
RNA was isolated from skeletal muscle using TRIzol (Invitrogen) per the manufacturer’s instructions. Samples were subjected to reverse transcription (Thermo Fisher) and real-time qPCR with SYBR Green (Bio-Rad). The comparative cycle threshold method was used to determine the relative expression levels; all primer pairs have identical efficiencies of amplification and single peaks with melt curve analysis. The following housekeeping genes were used for normalization: ribosomal protein, large, P0 (36B4), TATA-binding protein (TBP), and hypoxanthine phosphoribosyltransferase (HPRT); they were not affected by the experimental genotypes.
Treadmill Exercise Stress Test
Exercise stress tests were performed as previously described (18, 23). Briefly, before exhaustion runs, animals were subjected to acclimation runs [10 meters(m)/min for 5 min] for two consecutive days. Exercise stress tests were performed on a 10% grade incline at a speed of 10 m/min for 5 min and then increasing by 2 m/min every 2 min until exhaustion was reached and time duration recorded.
Citrate Synthase Activity
Skeletal muscle samples were snap-frozen on liquid nitrogen and then pulverized to a fine powder before processing. Citrate synthase enzymatic activity was measured using a coupled reaction with acetyl-CoA, 5′,5-dithiobis(2-nitrobenzoic acid), and oxaloacetate followed by spectrophotometry, as previously described (25).
Mitochondrial-to-Nuclear DNA Ratio
Mitochondrial DNA (mtDNA) and nuclear DNA (nDNA) were isolated with a DNeasy kit (Qiagen) per the manufacturer’s instructions. Real-time qPCR using SYBR green with primers specific to the mitochondrial and nuclear genome to calculate mitochondrial-to-nuclear DNA (mtDNA/nDNA) ratio, as previously described (26).
Western Blotting Analysis
Protein was isolated from skeletal muscle with a modified radioimmunoprecipitation assay (mRIPA) with protease and phosphatase inhibitors (Thermo Fisher). Protein extracts of 10 μg were subjected to electrophoresis on 4%–20% gradient gels (Bio-Rad). The antibodies used were the following: VDAC (Cell Signaling Technology Cat. No. 4661), α-tubulin (Developmental Studies Hybridoma Bank Cat. No. 12G10), and total OXPHOS rodent antibody cocktail (Abcam Cat. No. ab110413). Bands were captured on a digital imager after chemiluminescence detection. Band intensities were quantified using Image J software (NIH) by normalizing bands of interest to tubulin or Ponceau S staining of the membrane.
Statistical Analysis
All data are presented as means ± SE unless otherwise specified. P values were calculated by two-tailed Student’s t test and any P values < 0.05 were considered significant.
RESULTS
Deletion of PRC from Skeletal Muscle
Here, we sought to determine the role PRC plays in skeletal muscle and the regulation of mitochondrial biogenesis. To achieve this objective, we generated inducible muscle-specific PRC knockout mice (iMS-PRC KO) by crossing our newly developed PRC floxed mice with the tamoxifen-inducible human skeletal actin (HSA) Cre recombinase mouse line (22). All assessments were performed at 8 wk post deletion based on our previous findings with this muscle-specific-inducible model, where we observed a plateau in exercise performance around 7 to 8 wk (18, 23). We observed a 75%–80% reduction in the mRNA expression of PRC in the iMS-PRC KO animals (Fig. 1A); the residual PRC transcript is likely from nonmuscle cells given PRC’s involvement in proliferating cells (27). We measured the mRNA expression of components of the electron transport chain (ETC) that are targets of PGC-1s (28, 29), and we observed a 1.5- to 2-fold significant increase in the expression of genes Nduf5b (NADH:ubiquinone oxidoreductase subunit B5), Sdhb (succinate dehydrogenase complex iron sulfur subunit B), Cycs (cytochrome c, somatic), and Cox5b (cytochrome c oxidase subunit 5B) (Fig. 1B). We also measured the protein expression of ETC subunits but did not observe any significant changes in protein levels (Fig. 1, C and D). We next tested whether loss of skeletal muscle PRC expression affected exercise performance, by subjecting the iMS-PRC KO animals to a progressive endurance exercise stress test. The iMS-PRC KO mice showed no significant difference in exercise performance compared with their littermate controls (Fig. 1E). We next determined whether loss of PRC in skeletal muscle would have an effect on mitochondrial biogenesis. We performed citrate synthase (CS) activity measurements on muscles from the iMS-PRC KO animals and observed a 20% increase in CS activity (Fig. 1F), suggesting an increase in mitochondrial content. To confirm this increase in mitochondrial biogenesis, we also measured mtDNA/nDNA ratio and VDAC protein expression. We observed a significant increase in mtDNA/nDNA ratio (∼50%) and VDAC protein expression (∼50%) (Fig. 1, G–I). Given the potential for functional compensation by the other PGC-1 members, we next measured the expression of PGC-1α and PGC-1β. We observed a two- to threefold increase in mRNA expression of PGC-1α and PGC-1β, respectively, in the iMS-PRC KO animals (Fig. 1J). Taken together, these data suggest that in the absence of skeletal muscle PRC, PGC-1α and PGC-1β are induced and presumably able to functionally compensate for PRC to increase ETC mRNA expression and mitochondrial biogenesis.
Figure 1.
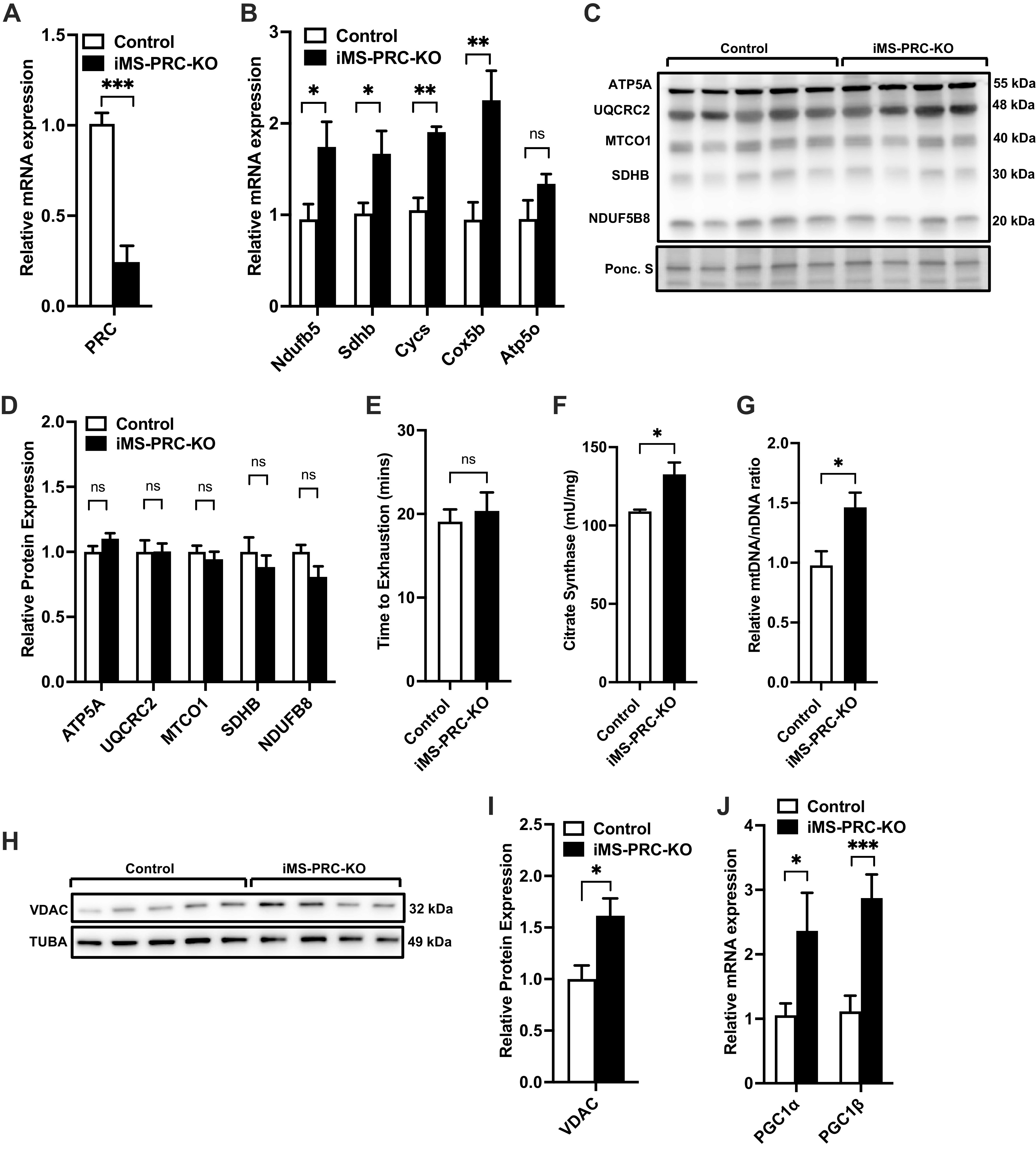
Effect of adult deletion of PRC on skeletal muscle ETC expression and mitochondrial content. PRC mRNA expression (A), nuclear-encoded ETC mRNA expression (B), protein expression of ETC subunits (C), densitometry quantification of ETC subunits protein expression to Ponceau S staining in gastrocnemius muscle (D), time to exhaustion (E), citrate synthase activity (F), mtDNA-to-nDNA (mtDNA/nDNA) ratio (G), VDAC protein expression (H), densitometry quantification of VDAC protein expression normalized to Tuba protein expression (I) and PGC-1α and PGC-1β mRNA expression in gastrocnemius muscle in adult-inducible skeletal muscle-specific PRC knockouts (iMS-PRC-KO) and control littermates (J). Data are presented as means ± SE; n = 4–5 per group; *P < 0.05, **P < 0.01, ***P < 0.001 compared with control as determined by unpaired t test. ETC, electron transport chain; KO, knockout; mtDNA, mitochondrial DNA; nDNA, nuclear DNA; PGC-1, peroxisome proliferator-activated receptor γ coactivator-1; PRC, PGC-1-related coactivator; VDAC, voltage-dependent anion channel.
Deletion of PGC-1α and PRC in Skeletal Muscle
Given that we observed an increase in both PGC-1α and PGC-1β in the iMS-PRC KO animals, we sought to dissect the individual contributions of PGC-1α and PGC-1β in the context of PRC deletion. We therefore generated an inducible skeletal muscle-specific PGC-1α/PRC double knockout animals (iMS-PGC-1α/PRC DKO), as described above with the administration of tamoxifen. We observed an 80%–90% reduction in the levels of PGC-1α mRNA expression, similar to what we have previously reported with PGC-1α deletion (18) and ∼75% reduction in PRC expression (Fig. 2A). We did not observe any changes in PGC-1β mRNA expression (Fig. 2A), suggesting changes in PGC-1β expression is not compensating in the absence of PGC-1α and PRC. We next measured the mRNA expression of ETC subunits and observed a significant reduction in expression in Nduf5b, Sdhb, and Atp5o (ATP synthase, H+ transporting, mitochondrial F1 complex, O subunit) mRNA levels (Fig. 2B). We also measured the protein expression of ETC subunits, however, did not observe any significant changes in the subunits assessed (Fig. 2, C and D). We next determined whether loss of PGC-1α and PRC in skeletal muscle affected exercise performance. We observed a 20% reduction in time to exhaustion in the iMS-PGC-1α/PRC DKO animals compared with littermate controls (Fig. 2E). We next determined whether loss of PGC-1α and PRC affected mitochondrial biogenesis. We observed a significant 25% reduction in CS activity in the iMS-PGC-1α/PRC DKO animals (Fig. 2F). We also observed a reduction in mtDNA/nDNA ratio (15%) (Fig. 2G) and a reduction in VDAC protein expression (50% reduction) in the iMS-PGC-1α/PRC DKO animals (Fig. 2, H and I). Taken together, these data suggest that deletion of both PGC-1α and PRC reduces exercise performance, a subset of ETC mRNA expression and mitochondrial biogenesis in skeletal muscle.
Figure 2.
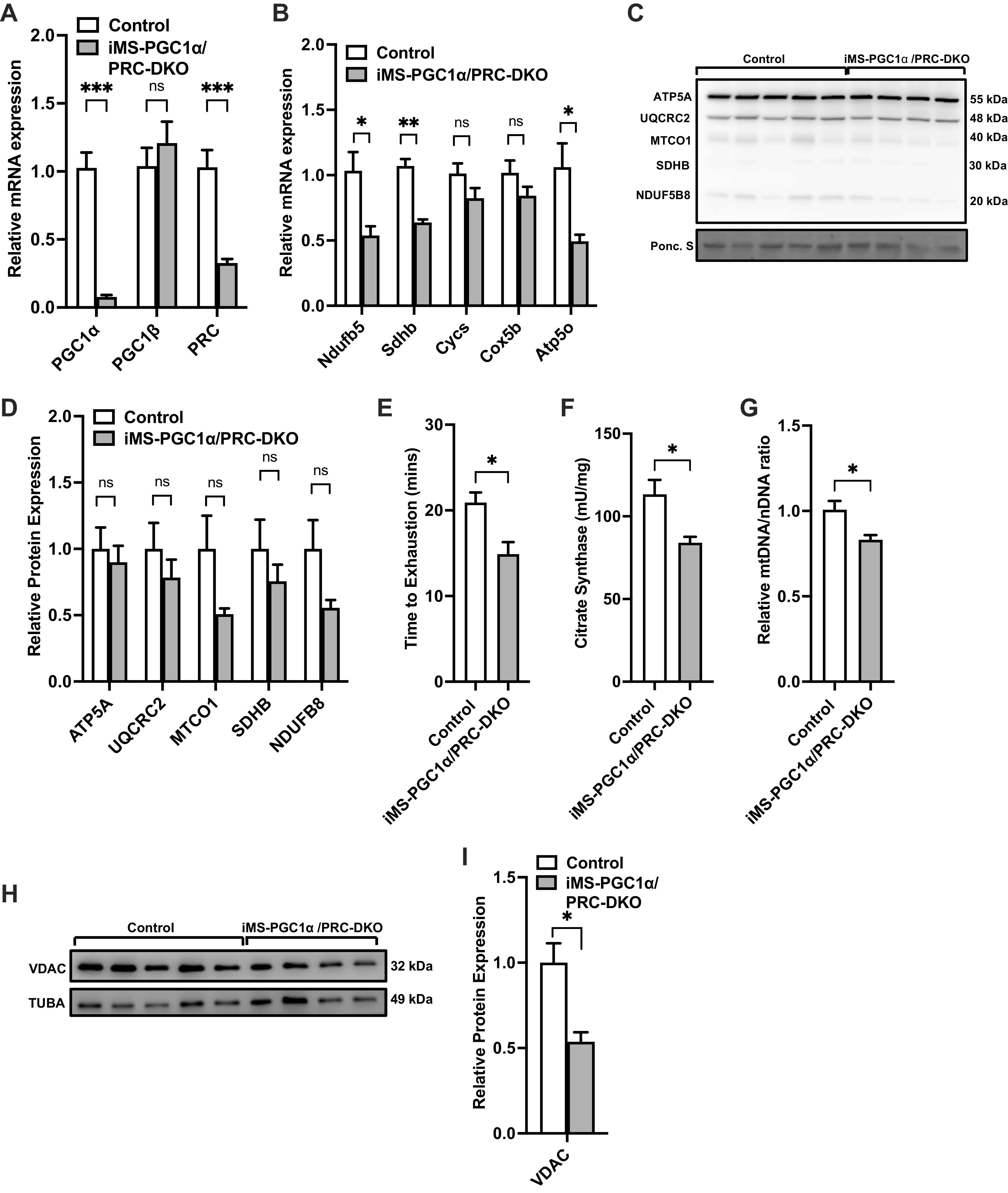
Effect of adult deletion of PRC and PGC-1α on skeletal muscle ETC expression and mitochondrial content. PGC-1α, PGC-1β, and PRC mRNA expression (A); nuclear-encoded ETC mRNA expression (B); protein expression of ETC subunits (C); densitometry quantification of ETC subunits protein expression to Ponceau S staining in gastrocnemius muscle (D); time to exhaustion (E); citrate synthase activity (F); mtDNA-to-nDNA (mtDNA/nDNA) ratio (G); VDAC protein expression (H); densitometry quantification of VDAC protein expression normalized to Tuba protein expression in gastrocnemius muscle in adult-inducible skeletal muscle-specific PGC-1α/PRC knockouts (iMS-PGC-1α/PRC-DKO) and control littermates (I). Data are presented as means ± SE; n = 4–5 per group; *P < 0.05, **P < 0.01, ***P < 0.001 compared with control as determined by unpaired t test. ETC, electron transport chain; KO, knockout; mtDNA, mitochondrial DNA; nDNA, nuclear DNA; PGC-1, peroxisome proliferator-activated receptor γ coactivator-1; PRC, PGC-1-related coactivator; VDAC, voltage-dependent anion channel.
Deletion of PGC-1β and PRC in Skeletal Muscle
We next wanted to determine whether the increased PGC-1β was a contributor to the increase in ETC gene expression and mitochondrial content observed in the iMS-PRC KO animals. To address this, we next generated inducible skeletal muscle-specific PGC-1β/PRC double knockout animals (iMS-PGC-1β/PRC DKO). We observed a significant reduction in PGC-1β and PRC with a 90% and 75% decrease, respectively (Fig. 3A). Furthermore, we did not observe a compensatory increase in PGC-1α mRNA expression in the iMS-PGC-1β/PRC DKO animals (Fig. 3A). We next measured the mRNA expression of ETC components in the muscle of the iMS-PGC-1β/PRC DKO animals and observed a significant reduction in all the measured subunits (including Ndufb5, Sdhb, Cycs, Cox5b, and Atp50) (Fig. 3B). We also measured the protein expression of ETC subunits and observed only a significant decrease in NDUFB8 protein expression (Fig. 3D). We next investigated whether loss of both PGC-1β and PRC in skeletal muscle affected exercise performance. We observed a reduction in the time to exhaustion in the iMS-PGC-1β/PRC DKO animals compared with their littermate controls albeit not statistically significant (Fig. 3E). We next determined whether loss of PGC-1β and PRC had an effect on mitochondrial biogenesis (CS activity, mtDNA/nDNA ratio, and VDAC protein expression). We did not observe any statistically significant changes in any of the mitochondrial biogenesis parameters measured (Fig. 3, F–I). These data suggest that in the absence of PGC-1β and PRC, ETC gene expression is decreased but has no significant effect on exercise performance or mitochondrial biogenesis.
Figure 3.
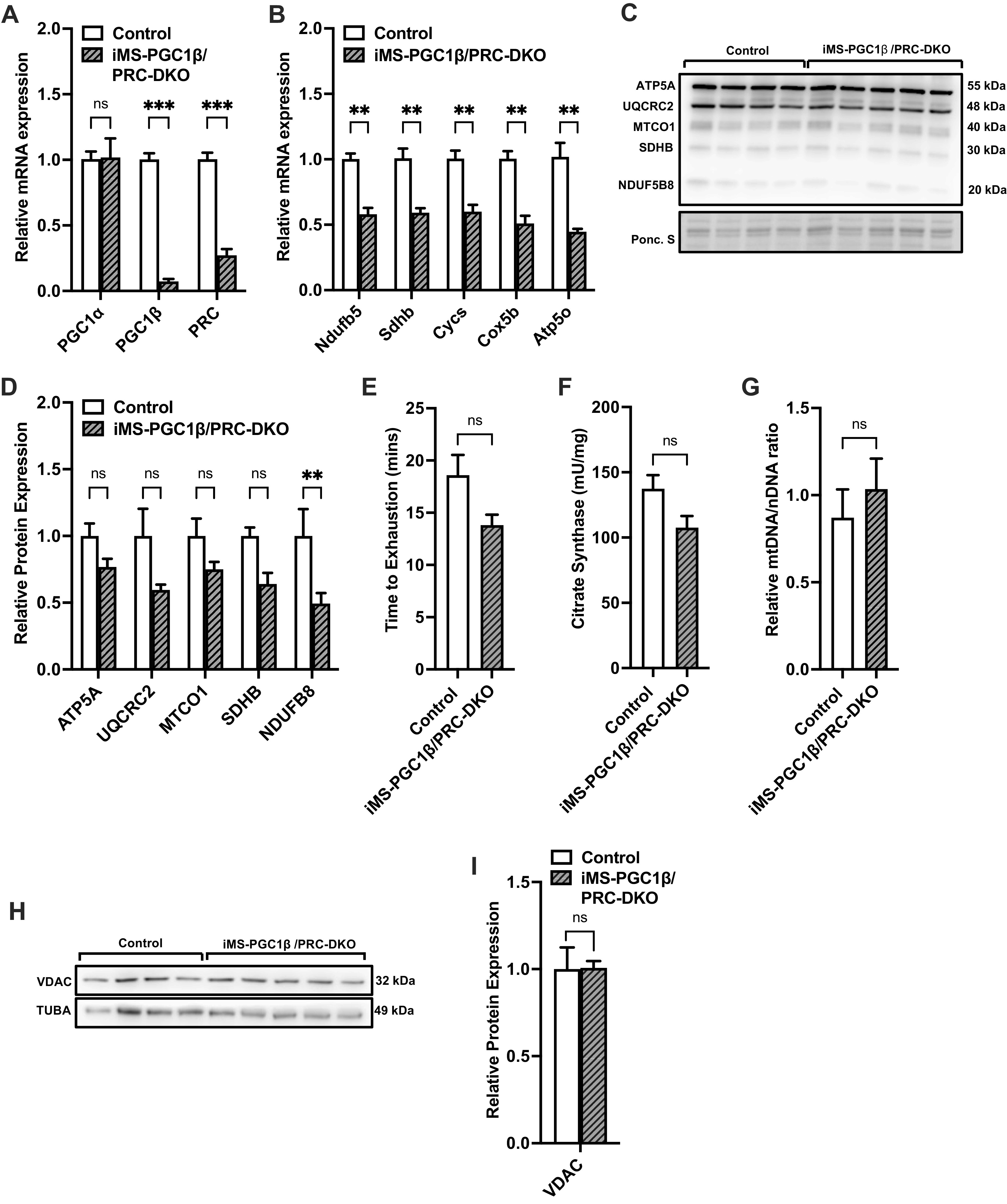
Effect of adult deletion of PRC and PGC-1β on skeletal muscle ETC expression and mitochondrial content. PGC-1α, PGC-1β, and PRC mRNA expression (A); nuclear-encoded ETC mRNA expression (B); protein expression of ETC subunits (C); densitometry quantification of ETC subunits protein expression to Ponceau S staining in gastrocnemius muscle (D); time to exhaustion (E); citrate synthase activity (F); mtDNA-to-nDNA (mtDNA/nDNA) ratio (G); VDAC protein expression (H); densitometry quantification of VDAC protein expression normalized to Tuba protein expression in gastrocnemius muscle in adult-inducible skeletal muscle-specific PGC-1β/PRC knockouts (iMS-PGC-1β/PRC-DKO) and control littermates (I). Data are presented as means ± SE; n = 4–5 per group; *P < 0.05, **P < 0.01, ***P < 0.001 compared with control as determined by unpaired t test. ETC, electron transport chain; KO, knockout; mtDNA, mitochondrial DNA; nDNA, nuclear DNA; PGC-1, peroxisome proliferator-activated receptor γ coactivator-1; PRC, PGC-1-related coactivator; VDAC, voltage-dependent anion channel.
Deletion of PGC-1α, PGC-1β, and PRC in Skeletal Muscle
We next wanted to completely exclude the possibility of compensation by any of the PGC-1 family members in regulating mitochondrial function (ETC subunit expression) and biogenesis. We therefore generated inducible skeletal muscle-specific PGC-1α/PGC-1β/PRC triple knockout animals (iMS-PGC-1 TKO). We were successfully able to genetically delete all three family members, with a 75% to 80% reduction in mRNA expression for all PGC-1 family members (Fig. 4A). We next measured the mRNA expression of ETC subunits in the iMS-PGC-1 TKO animals and observed a 75% to 80% reduction in mRNA expression of all ETC subunits measured (Fig. 4B). We also measured the protein expression of ETC subunits in the iMS-PGC-1 TKO animals and observed a 40% to 80% reduction in expression of the different subunits (Fig. 4, C and D). We next determined the effect of deletion of all PGC-1s would have on exercise performance and observed a significant reduction in time to exhaustion by 60% in the iMS-PGC-1 TKO animals (Fig. 4E). We next determined whether mitochondrial biogenesis was affected in the iMS-PGC-1 TKO animals and observed a 25% reduction in both CS activity and mtDNA/nDNA ratio as well as a ∼60% reduction in VDAC protein expression in the iMS-PGC-1 TKO animals (Fig. 4, F–I). These data suggest that in the absence of all three PGC-1s, both mitochondrial function and more interestingly biogenesis are significantly impaired, as this was not previously observed with the deletion of PGC-1α and PGC-1β in skeletal muscle (17, 18). Taken together, these data highlight the importance of PRC expression in skeletal muscle in maintaining mitochondrial content in skeletal muscle.
Figure 4.
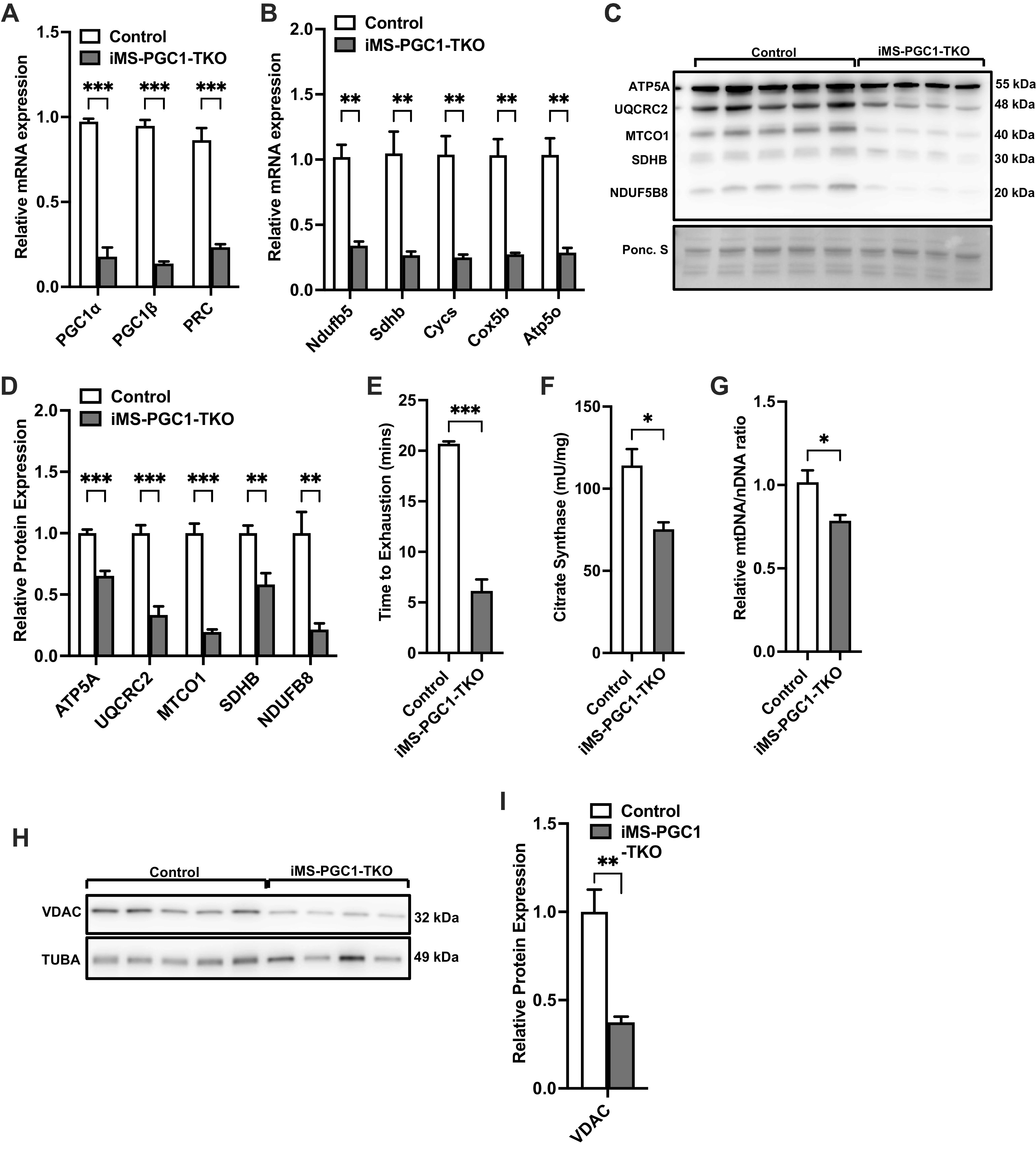
Effect of adult deletion of all three PGC-1s on skeletal muscle ETC expression and mitochondrial content. PGC-1α, PGC-1β, and PRC mRNA expression (A); nuclear-encoded ETC mRNA expression (B); protein expression of ETC subunits (C); densitometry quantification of ETC subunits protein expression to Ponceau S staining in gastrocnemius muscle (D); time to exhaustion (E); citrate synthase activity (F); mtDNA-to-nDNA (mtDNA/nDNA) ratio (G); VDAC protein expression (H); densitometry quantification of VDAC protein expression normalized to Tuba protein expression in gastrocnemius muscle in adult-inducible skeletal muscle-specific PGC-1α/PGC-1β/PRC knockouts (iMS-PGC-1-TKO) and control littermates (I). Data are presented as means ± SE; n = 4–5 per group; *P < 0.05, **P < 0.01, ***P < 0.001 compared with control as determined by unpaired t test. ETC, electron transport chain; KO, knockout; mtDNA, mitochondrial DNA; nDNA, nuclear DNA; PGC-1, peroxisome proliferator-activated receptor γ coactivator-1; PRC, PGC-1-related coactivator; VDAC, voltage-dependent anion channel.
DISCUSSION
The PGC-1 family of transcriptional coactivators has been shown to be powerful regulators of mitochondrial oxidative capacity and mitochondrial biogenesis in a variety of tissues, including skeletal muscle and myotubes (13, 14, 30–33). However, many of these reports have been based on gain-of-function studies and show that these factors are sufficient to increase mitochondrial oxidative capacity and mitochondrial biogenesis. Recent studies using loss-of-function and conditional muscle-specific knockouts have challenged whether the individual PGC-1s are required for some of their proposed functions. Previous work from our group and others have shown that PGC-1α is not required for exercise-induced adaptations in skeletal muscle (34–36). Furthermore, we have also shown that double deletion of PGC-1α and PGC-1β in skeletal muscle affects oxidative capacity with no effect on mitochondrial content (17, 18). These observations suggest that another factor is responsible for maintaining baseline mitochondrial content in skeletal muscle; we propose this factor to be PRC, the third PGC-1 family member.
Genetic deletion of PRC is embryonic lethal due to impairment in peri-implantation during embryonic development (37), therefore, making the study of its function in adult skeletal muscle largely impossible. Attempts have been made to bypass this limitation with the use of heterozygote mice in which one allele of PRC is still present (38). Male PRC heterozygotes were reportedly resistant to a high-fat diet (HFD) due to increased elevated metabolic rates (38). Analysis of soleus muscle from the PRC heterozygotes revealed a trend to increased mtDNA, albeit not statistically significant, and no changes in gene expression for fatty acid oxidation and electron transport chain. However, given the nature of whole body heterozygotes, it is difficult to fully dissect the specific role it plays in skeletal muscle with the other metabolic changes reported in the PRC heterozygotes (38). Therefore, in this study, we sought to elucidate the role of PRC in skeletal muscle and its potential contribution to maintaining mitochondrial biogenesis. To achieve this goal, we generated genetic models in which PRC was knocked out alone or in conjunction with PGC-1α and/or PGC-1β in adult skeletal muscle.
Adult deletion of skeletal muscle PRC resulted in an increase in ETC gene expression and mitochondrial biogenesis. The increase in these mitochondrial parameters was initially surprising and potentially contradictory given the proposed function of PRC based on the gain-of-function studies (32). However, the loss of skeletal muscle PRC resulted in a compensatory increase in expression of PGC-1α and PGC-1β mRNA expression. These data suggest that the absence of PRC in skeletal muscle must be functionally compensated for to maintain normal mitochondrial ETC gene expression. Functional redundancy has been suggested for the absence of the different PGC-1 isoforms, particularly PGC-1α and PGC-1β, largely based on the mild phenotypes reported with single knockout of either PGC-1α or PGC-1β (39). However, the functional compensation in the absence of PRC is lost when both PRC and PGC-1α are deleted in skeletal muscle in the iMS-PGC-1α/PRC DKO animals. These animals have a significant reduction in both ETC gene expression and mitochondrial content. We did not observe any significant effect on exercise performance or mitochondrial content when we deleted both PRC and PGC-1β in the iMS-PGC-1β/PRC knockouts, presumably due to the fact that PGC-1α is left intact. PGC-1α is well established as a potent regulator of mitochondrial oxidative capacity and mitochondrial biogenesis (7–9). However, loss of function studies by our group and others have challenged to extent to which PGC-1α is required for skeletal muscle mitochondrial oxidative capacity and biogenesis (17, 34, 36). We have previously shown that PGC-1α is dispensable for exercise-induced biogenesis (36), notably, we did not observe any significant difference in mitochondrial content in the sedentary group. In a recent study, using an adult-inducible skeletal muscle-specific PGC-1α knockout mouse, the authors reported no significant difference in mtDNA/nucDNA ratio or mitochondrial content in either the subsarcolemmal or intermyofibrillar fractions in untrained aged mice (∼15 mo) compared with controls (40). However, they observed a significant difference in mitochondrial content in both the younger and older trained groups. PGC-1α is sufficient to regulate mitochondrial biogenesis; however, the full extent of its necessity still needs to be fully elucidated.
Notwithstanding these points, the generation of the iMS-PGC-1TKO animals in which all PGC-1 family members are deleted in skeletal muscle allowed for us to remove all PGC-1-dependent compensatory mechanisms. The iMS-PGC-1TKO animals had significantly reduced ETC gene and protein expression as well as exercise performance, similar to what we had previously observed with the deletion of PGC-1α and PGC-1β in skeletal muscle (17, 18). However most notably, mitochondrial biogenesis markers are significantly reduced in the iMS-PGC-1TKO animals, something not observed with the deletion of PGC-1α and PGC-1β in skeletal muscle (17, 18). In the current study, mtDNA/nucDNA, CS activity, and VDAC protein expression are used as indirect measures of mitochondrial biogenesis. However, direct visualization and quantification of mitochondria may allow for a better comparison with our previous reports (17, 18). Notwithstanding, these data highlights that PRC is important for maintaining mitochondrial content in adult skeletal muscle.
In addition to the regulation of mitochondrial oxidative capacity and mitochondrial biogenesis, PGC-1s have been proposed to regulate skeletal muscle fiber type composition (16). However, the role that PGC-1s play in this process is not clear, given the different phenotypes reported by different groups(41). We have previously reported in a congenital skeletal muscle deletion of both PGC-1α and PGC-1β did not affect fiber composition (17). Similarly, analysis of our skeletal muscle PGC-1 deleted animal models, we did not observe any differences in myosin heavy chain gene expression (Supplemental Fig. S1, A–D: https://doi.org/10.6084/m9.figshare.21774548), suggesting that deletion of PGC-1s in adult skeletal muscle does not affect myosin heavy chain expression. However, a more detailed histological analysis would be needed to determine if the fiber distribution is altered.
These results have brought us to the following conclusions: 1) deletion of PRC in adult skeletal muscle results in increased ETC gene expression; 2) the increase in mitochondrial ETC gene expression is due in part to compensatory increases in PGC-1α and PGC-1β mRNA expression; and 3) complete deletion of all three PGC-1 family members in skeletal muscle reduces ETC gene expression and mitochondrial content, something that was not observed previously when both PGC-1α and PGC-1β were deleted (17, 18).
Perspectives and Significance
The current study demonstrates that PRC plays an important role in maintaining mitochondrial content in adult skeletal muscle. Furthermore, its absence needs to be accounted for by the upregulation of other family members in the form of PGC-1α and PGC-1β. However, identifying pathways that are specifically regulated by PRC could provide a new target for improving skeletal muscle mitochondrial function. Furthermore, identifying the mechanism by which loss of PRC results in increased PGC-1α and PGC-1β mRNA expression, potentially opens new and novel ways to activate mitochondrial content in skeletal muscle.
DATA AVAILABILITY
Data will be made available upon reasonable request.
SUPPLEMENTAL DATA
Supplemental Fig. S1: https://doi.org/10.6084/m9.figshare.21774548.
GRANTS
This work was supported in part by grants from the National Institute of Health (NIH) (DK121299 and AG069906) to G.C.R.
DISCLOSURES
No conflicts of interest, financial or otherwise, are declared by the authors.
AUTHOR CONTRIBUTIONS
G.C.R. conceived and designed research; D.B., Y.A., J.F., S.S., K.A.-P., and C.D. performed experiments; D.B., Y.A., and G.C.R. analyzed data; D.B., Y.A., and G.C.R. interpreted results of experiments; Y.A. and G.C.R. prepared figures; D.B., Y.A., and G.C.R. drafted manuscript; D.B., Y.A., and G.C.R. edited and revised manuscript; D.B., Y.A., and G.C.R. approved final version of manuscript.
REFERENCES
- 1. Goodpaster BH, Sparks LM. Metabolic flexibility in health and disease. Cell Metab 25: 1027–1036, 2017. doi: 10.1016/j.cmet.2017.04.015. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 2. Hood DA, Memme JM, Oliveira AN, Triolo M. Maintenance of skeletal muscle mitochondria in health, exercise, and aging. Annu Rev Physiol 81: 19–41, 2019. doi: 10.1146/annurev-physiol-020518-114310. [DOI] [PubMed] [Google Scholar]
- 3. Rowe GC, Safdar A, Arany Z. Running forward: new frontiers in endurance exercise biology. Circulation 129: 798–810, 2014. doi: 10.1161/CIRCULATIONAHA.113.001590. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 4. Heo JW, No MH, Park DH, Kang JH, Seo DY, Han J, Neufer PD, Kwak HB. Effects of exercise on obesity-induced mitochondrial dysfunction in skeletal muscle. Korean J Physiol Pharmacol 21: 567–577, 2017. doi: 10.4196/kjpp.2017.21.6.567. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 5. Hesselink MK, Schrauwen-Hinderling V, Schrauwen P. Skeletal muscle mitochondria as a target to prevent or treat type 2 diabetes mellitus. Nat Rev Endocrinol 12: 633–645, 2016. doi: 10.1038/nrendo.2016.104. [DOI] [PubMed] [Google Scholar]
- 6. Egan B, Zierath JR. Exercise metabolism and the molecular regulation of skeletal muscle adaptation. Cell Metab 17: 162–184, 2013. doi: 10.1016/j.cmet.2012.12.012. [DOI] [PubMed] [Google Scholar]
- 7. Finck BN, Kelly DP. PGC-1 coactivators: inducible regulators of energy metabolism in health and disease. J Clin Invest 116: 615–622, 2006. doi: 10.1172/JCI27794. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 8. Scarpulla RC. Nuclear control of respiratory chain expression by nuclear respiratory factors and PGC-1-related coactivator. Ann N Y Acad Sci 1147: 321–334, 2008. doi: 10.1196/annals.1427.006. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 9. Rowe GC, Jiang A, Arany Z. PGC-1 coactivators in cardiac development and disease. Circ Res 107: 825–838, 2010. doi: 10.1161/CIRCRESAHA.110.223818. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 10. Baar K, Wende AR, Jones TE, Marison M, Nolte LA, Chen M, Kelly DP, Holloszy JO. Adaptations of skeletal muscle to exercise: rapid increase in the transcriptional coactivator PGC-1. FASEB J 16: 1879–1886, 2002. doi: 10.1096/fj.02-0367com. [DOI] [PubMed] [Google Scholar]
- 11. Russell AP, Feilchenfeldt J, Schreiber S, Praz M, Crettenand A, Gobelet C, Meier CA, Bell DR, Kralli A, Giacobino JP, Deriaz O. Endurance training in humans leads to fiber type-specific increases in levels of peroxisome proliferator-activated receptor-gamma coactivator-1 and peroxisome proliferator-activated receptor-alpha in skeletal muscle. Diabetes 52: 2874–2881, 2003. doi: 10.2337/diabetes.52.12.2874. [DOI] [PubMed] [Google Scholar]
- 12. Pilegaard H, Saltin B, Neufer PD. Exercise induces transient transcriptional activation of the PGC-1alpha gene in human skeletal muscle. J Physiol 546: 851–858, 2003. doi: 10.1113/jphysiol.2002.034850. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 13. Lin J, Wu H, Tarr PT, Zhang CY, Wu Z, Boss O, Michael LF, Puigserver P, Isotani E, Olson EN, Lowell BB, Bassel-Duby R, Spiegelman BM. Transcriptional co-activator PGC-1 alpha drives the formation of slow-twitch muscle fibres. Nature 418: 797–801, 2002. doi: 10.1038/nature00904. [DOI] [PubMed] [Google Scholar]
- 14. Arany Z, Lebrasseur N, Morris C, Smith E, Yang W, Ma Y, Chin S, Spiegelman BM. The transcriptional coactivator PGC-1beta drives the formation of oxidative type IIX fibers in skeletal muscle. Cell Metab 5: 35–46, 2007. doi: 10.1016/j.cmet.2006.12.003. [DOI] [PubMed] [Google Scholar]
- 15. Handschin C, Chin S, Li P, Liu F, Maratos-Flier E, Lebrasseur NK, Yan Z, Spiegelman BM. Skeletal muscle fiber-type switching, exercise intolerance, and myopathy in PGC-1alpha muscle-specific knock-out animals. J Biol Chem 282: 30014–30021, 2007. doi: 10.1074/jbc.M704817200. [DOI] [PubMed] [Google Scholar]
- 16. Zechner C, Lai L, Zechner JF, Geng T, Yan Z, Rumsey JW, Collia D, Chen Z, Wozniak DF, Leone TC, Kelly DP. Total skeletal muscle PGC-1 deficiency uncouples mitochondrial derangements from fiber type determination and insulin sensitivity. Cell Metab 12: 633–642, 2010. [Erratum in Cell Metab 13: 114, 2011]. doi: 10.1016/j.cmet.2010.11.008. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 17. Rowe GC, Patten IS, Zsengeller ZK, El-Khoury R, Okutsu M, Bampoh S, Koulisis N, Farrell C, Hirshman MF, Yan Z, Goodyear LJ, Rustin P, Arany Z. Disconnecting mitochondrial content from respiratory chain capacity in PGC-1-deficient skeletal muscle. Cell Rep 3: 1449–1456, 2013. doi: 10.1016/j.celrep.2013.04.023. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 18. Ballmann C, Tang Y, Bush Z, Rowe GC. Adult expression of PGC-1alpha and -1beta in skeletal muscle is not required for endurance exercise-induced enhancement of exercise capacity. Am J Physiol Endocrinol Metab 311: E928–E938, 2016. doi: 10.1152/ajpendo.00209.2016. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 19. Fan W, He N, Lin CS, Wei Z, Hah N, Waizenegger W, He MX, Liddle C, Yu RT, Atkins AR, Downes M, Evans RM. ERRγ promotes angiogenesis, mitochondrial biogenesis, and oxidative remodeling in PGC1α/β-deficient muscle. Cell Rep 22: 2521–2529, 2018. doi: 10.1016/j.celrep.2018.02.047. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 20. Lin J, Wu PH, Tarr PT, Lindenberg KS, St-Pierre J, Zhang CY, Mootha VK, Jager S, Vianna CR, Reznick RM, Cui L, Manieri M, Donovan MX, Wu Z, Cooper MP, Fan MC, Rohas LM, Zavacki AM, Cinti S, Shulman GI, Lowell BB, Krainc D, Spiegelman BM. Defects in adaptive energy metabolism with CNS-linked hyperactivity in PGC-1alpha null mice. Cell 119: 121–135, 2004. doi: 10.1016/j.cell.2004.09.013. [DOI] [PubMed] [Google Scholar]
- 21. Lai L, Leone TC, Zechner C, Schaeffer PJ, Kelly SM, Flanagan DP, Medeiros DM, Kovacs A, Kelly DP. Transcriptional coactivators PGC-1alpha and PGC-lbeta control overlapping programs required for perinatal maturation of the heart. Genes Dev 22: 1948–1961, 2008. doi: 10.1101/gad.1661708. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 22. McCarthy JJ, Srikuea R, Kirby TJ, Peterson CA, Esser KA. Inducible Cre transgenic mouse strain for skeletal muscle-specific gene targeting. Skelet Muscle 2: 22, 2012. doi: 10.1186/2044-5040-2-22. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 23. Bell MB, Bush Z, McGinnis GR, Rowe GC. Adult skeletal muscle deletion of Mitofusin 1 and 2 impedes exercise performance and training capacity. J Appl Physiol (1985) 126: 341–353, 2019. doi: 10.1152/japplphysiol.00719.2018. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 24. Schroder EA, Harfmann BD, Zhang X, Srikuea R, England JH, Hodge BA, Wen Y, Riley LA, Yu Q, Christie A, Smith JD, Seward T, Wolf Horrell EM, Mula J, Peterson CA, Butterfield TA, Esser KA. Intrinsic muscle clock is necessary for musculoskeletal health. J Physiol 593: 5387–5404, 2015. doi: 10.1113/JP271436. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 25. Shepherd D, Garland PB. The kinetic properties of citrate synthase from rat liver mitochondria. Biochem J 114: 597–610, 1969. doi: 10.1042/bj1140597. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 26. Quiros PM, Goyal A, Jha P, Auwerx J. Analysis of mtDNA/nDNA Ratio in Mice. Curr Protoc Mouse Biol 7: 47–54, 2017. doi: 10.1002/cpmo.21. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 27. Vercauteren K, Gleyzer N, Scarpulla RC. Short hairpin RNA-mediated silencing of PRC (PGC-1-related coactivator) results in a severe respiratory chain deficiency associated with the proliferation of aberrant mitochondria. J Biol Chem 284: 2307–2319, 2009. doi: 10.1074/jbc.M806434200. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 28. Arany Z, He H, Lin J, Hoyer K, Handschin C, Toka O, Ahmad F, Matsui T, Chin S, Wu P-H, Rybkin II, Shelton JM, Manieri M, Cinti S, Schoen FJ, Bassel-Duby R, Rosenzweig A, Ingwall JS, Spiegelman BM. Transcriptional coactivator PGC-1 alpha controls the energy state and contractile function of cardiac muscle. Cell Metab 1: 259–271, 2005. doi: 10.1016/j.cmet.2005.03.002. [DOI] [PubMed] [Google Scholar]
- 29. Gali Ramamoorthy T, Laverny G, Schlagowski AI, Zoll J, Messaddeq N, Bornert JM, Panza S, Ferry A, Geny B, Metzger D. The transcriptional coregulator PGC-1beta controls mitochondrial function and anti-oxidant defence in skeletal muscles. Nat Commun 6: 10210, 2015. doi: 10.1038/ncomms10210. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 30. Wu Z, Puigserver P, Andersson U, Zhang C, Adelmant G, Mootha V, Troy A, Cinti S, Lowell B, Scarpulla RC, Spiegelman BM. Mechanisms controlling mitochondrial biogenesis and respiration through the thermogenic coactivator PGC-1. Cell 98: 115–124, 1999. doi: 10.1016/S0092-8674(00)80611-X. [DOI] [PubMed] [Google Scholar]
- 31. Shao D, Liu Y, Liu X, Zhu L, Cui Y, Cui A, Qiao A, Kong X, Liu Y, Chen Q, Gupta N, Fang F, Chang Y. PGC-1 beta-regulated mitochondrial biogenesis and function in myotubes is mediated by NRF-1 and ERR alpha. Mitochondrion 10: 516–527, 2010. doi: 10.1016/j.mito.2010.05.012. [DOI] [PubMed] [Google Scholar]
- 32. Philp A, Belew MY, Evans A, Pham D, Sivia I, Chen A, Schenk S, Baar K. The PGC-1alpha-related coactivator promotes mitochondrial and myogenic adaptations in C2C12 myotubes. Am J Physiol Regul Integr Comp Physiol 301: R864–R872, 2011. doi: 10.1152/ajpregu.00232.2011. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 33. Romanino K, Mazelin L, Albert V, Conjard-Duplany A, Lin S, Bentzinger CF, Handschin C, Puigserver P, Zorzato F, Schaeffer L, Gangloff YG, Ruegg MA. Myopathy caused by mammalian target of rapamycin complex 1 (mTORC1) inactivation is not reversed by restoring mitochondrial function. Proc Natl Acad Sci USA 108: 20808–20813, 2011. doi: 10.1073/pnas.1111448109. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 34. Leick L, Wojtaszewski JF, Johansen ST, Kiilerich K, Comes G, Hellsten Y, Hidalgo J, Pilegaard H. PGC-1alpha is not mandatory for exercise- and training-induced adaptive gene responses in mouse skeletal muscle. Am J Physiol Endocrinol Metabol 294: E463–E474, 2008. doi: 10.1152/ajpendo.00666.2007. [DOI] [PubMed] [Google Scholar]
- 35. Adhihetty PJ, Uguccioni G, Leick L, Hidalgo J, Pilegaard H, Hood DA. The role of PGC-1alpha on mitochondrial function and apoptotic susceptibility in muscle. Am J Physiol Cell Physiol 297: C217–C225, 2009. doi: 10.1152/ajpcell.00070.2009. [DOI] [PubMed] [Google Scholar]
- 36. Rowe GC, El-Khoury R, Patten IS, Rustin P, Arany Z. PGC-1alpha is dispensable for exercise-induced mitochondrial biogenesis in skeletal muscle. PLoS One 7: e41817, 2012. doi: 10.1371/journal.pone.0041817. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 37. He X, Sun C, Wang F, Shan A, Guo T, Gu W, Cui B, Ning G. Peri-implantation lethality in mice lacking the PGC-1-related coactivator protein. Dev Dyn 241: 975–983, 2012. doi: 10.1002/dvdy.23769. [DOI] [PubMed] [Google Scholar]
- 38. Zhai N, Sun C, Gu W, He X, Shan A, Sun H, Lu N, Cui B, Ning G. Resistance to high-fat diet-induced obesity in male heterozygous Pprc1 knockout mice. Endocr J 62: 633–644, 2015. doi: 10.1507/endocrj.EJ14-0383. [DOI] [PubMed] [Google Scholar]
- 39. Villena JA. New insights into PGC-1 coactivators: redefining their role in the regulation of mitochondrial function and beyond. FEBS J 282: 647–672, 2015. doi: 10.1111/febs.13175. [DOI] [PubMed] [Google Scholar]
- 40. Halling JF, Jessen H, Nohr-Meldgaard J, Buch BT, Christensen NM, Gudiksen A, Ringholm S, Neufer PD, Prats C, Pilegaard H. PGC-1alpha regulates mitochondrial properties beyond biogenesis with aging and exercise training. Am J Physiol Endocrinol Metab 317: E513–E525, 2019. doi: 10.1152/ajpendo.00059.2019. [DOI] [PubMed] [Google Scholar]
- 41. Handschin C, Spiegelman BM. PGC-1 coactivators and the regulation of skeletal muscle fiber-type determination. Cell Metab 13: 351, 2011. doi: 10.1016/j.cmet.2011.03.008. [DOI] [PubMed] [Google Scholar]
Associated Data
This section collects any data citations, data availability statements, or supplementary materials included in this article.
Supplementary Materials
Supplemental Fig. S1: https://doi.org/10.6084/m9.figshare.21774548.
Data Availability Statement
Data will be made available upon reasonable request.