

Abstract
Image-based profiling quantitatively assesses the effects of perturbations on cells by capturing a breadth of changes via microscopy. Here we provide two complementary protocols to help explore and interpret data from image-based profiling experiments. In the first protocol, we examine the similarity among perturbed cell samples using data from compounds that cluster by their mechanism of action (MOAs). The protocol includes steps to examine feature-driving differences between samples and how to visualize correlations between features and treatments to create interpretable heatmaps using an open-source web tool, Morpheus. In the second protocol, we show how to interactively explore images together with the numerical data, while we provide scripts to create visualizations of representative single cells and image sites to understand how changes in features are reflected in the images. Together, these two tutorials help biologists and researchers interpret their image-based data to speed up research.
Basic Protocol 1: Exploratory analysis of profile similarities and driving features
Basic Protocol 2: Image and single-cell visualization following profile interpretation
Keywords: profiling, single-cell visualization, Morpheus, morphological analysis, high-dimensional data, image-based profiling
INTRODUCTION
Automated microscopy allows biologists to acquire thousands of images from cells perturbed with drugs, small interfering RNA (siRNA), CRISPR-Cas9, and more. In a typical quantitative microscopy experiment, biologists select fluorescent biomarkers, such as antibodies or dyes for specific proteins or cell compartments, and measure only the features they hypothesize will be perturbed in their experiment. By contrast, in image-based profiling, the aim is to let the cells speak for themselves. Diverse stains are used, as in the Cell Painting assay, which stains eight components of the cells (Bray et al., 2016; Cimini et al., 2022). Then, image analysis software segments the cells and measures all possible morphological features from the single cells. The collection of features for a cell is called a “profile” (sometimes described as a “morphological profile” or an “image-based profile”), and typically a thousand or more features are measured per cell. It is then possible to analyze whether features are modified in a treated sample of cells compared to controls. Afterward, samples can be grouped into clusters based on their image-based profiles (Figure 1). However, the biological meaning behind clusters is difficult to interpret because there are thousands of features in the profile. This leads to a common bottleneck: given a sample or cluster of samples, how do you interpret what a given profile means biologically?
Figure 1 -.
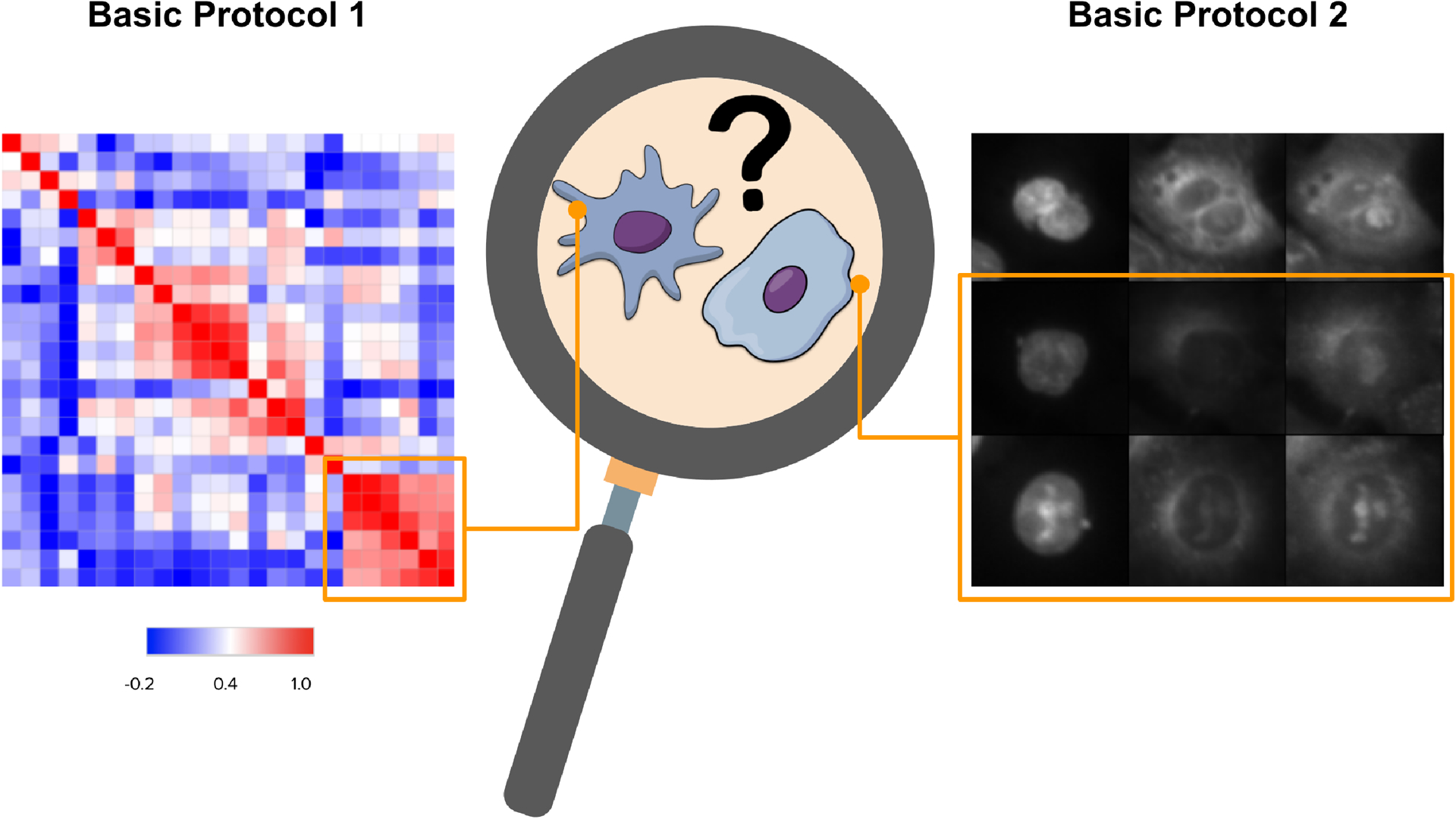
In Basic Protocol 1, based on sample clustering, biologists can understand the underlying morphology that makes certain samples cluster in a certain way. In Basic Protocol 2, biologists can examine representative cells from each sample.
Here, we present two basic protocols: Exploratory analysis using the Morpheus software (Basic Protocol 1) and image and single-cell visualization following profile interpretation (Basic Protocol 2). In Basic Protocol 1, we show researchers how to explore the overall large-scale associations of the data (after feature extraction and cleaning) using the free, web-based software Morpheus. There, the data can be grouped in different ways, revealing how features and samples are correlated. Exploring the data is essential to gain insights into the biological interpretation of the profiles. Then, in Basic Protocol 2, the goal is to help biologists create intuitions about the differences between treatments by examining example cells. This notebook contains Python scripts to help researchers crop representative or random single cells from each treatment and group the cropped images based on correlations of interest. In addition, representative images of each sample can be retrieved to understand how the cells are distributed across representative fields of view (e.g., those captured from different sites (locations) within a sample well), which can give insights into treatment toxicity and or growth-stimulating effects. In the Understanding Results section, we provide insights on how visualizing example cells from the samples and linking them to the correlations between the samples will provide the biologist with extensive information to formulate new hypotheses and interpretations from their data. While these approaches are powerful, we do note that they require high-dimensional image measurements and as such require using CellProfiler or a similar tool to generate; they unfortunately also do not always lead to easily-interpretable conclusions (see Understanding Results for further discussion).
The two basic protocols yield a similarity matrix, hierarchical clustering for the samples, and representative example cells from their data. These outputs can be easily used for reports and publications. For the input data for both protocols, we use a dataset of images processed by CellProfiler, to identify cells and extract features (Stirling et al., 2021) and pycytominer, to normalize and aggregate single-cell profiles into population-averaged profiles (Way et al., 2022a). Extensive documentation is available online on feature extraction with CellProfiler (https://github.com/CellProfiler/tutorials), and for data aggregation, normalization, and feature selection with pycytominer (https://github.com/cytomining/pipeline-examples). In addition, we provide an example dataset in our GitHub repository, including comma-separated value (CSV) spreadsheets to be processed on Morpheus (https://github.com/ciminilab/2022_Garcia-Fossa_submitted). In our example dataset, each compound is annotated with its mechanism of action (MOA). However, these protocols can be used without having the MOA for every compound in their dataset, and instead comparing treated cells with the negative and/or positive controls, or comparing multiple perturbed samples with each other.
BASIC PROTOCOL 1
Exploratory analysis of profile similarities and driving features
The main goal of this tutorial is to examine the correlations between the samples to check for their replicability, to explore the correlations among them, to discern how the features drive the differences between samples or groups, and to enable the interpretation of the biology behind the data.
After cell treatment, imaging, and feature extraction, some profiles are dramatic in only one or a few features and the feature names have obvious meanings (nucleus area or integrated intensity of the mitochondria channel in the cytoplasm, which corresponds to the total amount of staining in that channel); in these cases, looking at the feature names will help to discern their connection to the biological meaning. However, some individual features have meanings that are more difficult to translate into plain language. Furthermore, the challenge is even greater to interpret a collection of feature names that all contribute strongly to a more complex morphological phenotype. For example, a collection of features from a channel stained for actin and wheat germ agglutinin together with DNA granularity was particularly important to predict 70 specific cell health phenotypes from Cell Painting data (Way et al., 2021). Even phenotypes that are visually obvious and distinctive by the eye, such as cells stalled in a particular stage of the cell cycle, are often difficult to predict just by examining a list of distinctive features; the problem is even more acute for samples without a visual discernible phenotype yet quite distinguishable using image metrics.
To help us in the exploration and interpretation process, we often use Morpheus, available at https://software.broadinstitute.org/morpheus/ (Morpheus), is a free web-based open-source software that allows matrix visualization, analysis, clustering, filtering, and displaying charts. The tool can be readily used by those without extensive computational or statistical experience. Morpheus allows for quick visualization of an entire dataset in its different ways, so the researcher can identify patterns in their data that could lead to new biological insights, or even use it as a data quality control step by examining replicates replicability. While Morpheus was originally designed at the Broad Institute for the exploration of mRNA profiling data, it accepts a variety of matrix files from multiple formats (CSV, GCT, GMT, text file) to be imported. While raw CellProfiler (Stirling et al., 2021) outputs tables can be input into Morpheus, here, we provide notebooks to preprocess the outputs from CellProfiler so the data can undergo aggregation and normalization (both of which are also possible to perform in Morpheus) followed by multiple feature reduction steps (some of which are not available in Morpheus).
More information can be found in Morpheus’ documentation (https://software.broadinstitute.org/morpheus/documentation.html), as well as a two-part series of video tutorials for the Center for Open Bioimage Analysis YouTube channel: a beginner’s guide to morphological profiling (part 1), and a practical exploration of morphological profiling data with a demonstration of Morpheus software (part 2).
During this tutorial, the researcher will start by examining how similar each sample is to the other samples using per-well similarity matrices, sorting the data in a way that allows for interpretation. We provide a sample data set in which drugs with known mechanisms of action (MOAs) have been added at various dose points prior to Cell Painting. To observe how MOAs are grouped, and if technical artifacts such as batch or plate-layout effects are playing a role in the distribution of the groups, we use hierarchical clustering. In the end, the researcher will be able to identify if drugs with similar MOAs have similar morphological profiles and the positive and negative connections between various MOA profiles. The researcher will also learn how to determine what features drive the differences between the groups. We emphasize that this is just one of the data-exploration approaches that can be used to interpret image-based profiles, and produces comparative results rather than hard distinctions between “similar” and “not”.
Materials
Laptop or desktop computer with at least 2 GB of RAM and a suitable web browser such as Google Chrome. Access to the internet to use Morpheus at https://software.broadinstitute.org/morpheus/.
Data and Jupyter Notebooks (Kluyver et al., 2016) are available in our GitHub repo (https://github.com/ciminilab/2022_Garcia-Fossa_submitted). The data is in a GCT format, a tab-separated value table containing the extracted features aggregated by well in a Cell Painting assay. In this assay, 1,571 compounds were tested across 6 doses in A549 cells (Way et al., 2022b).
We randomly selected a plate map from this experiment (C-7161-01-LM6-011 plate map), and then downloaded the CSV (comma-separated value) files for five of its replicate plates (SQ00015195, SQ00015218, SQ00015219, SQ00015220, SQ00015221), from the cpg0004-lincs dataset (Way et al., 2022b) available from the Cell Painting Gallery on the Registry of Open Data on AWS (cellpainting-gallery). Then, we added annotations to the data (labels for each MOA, compound, and concentration), and normalized the features to the negative control (DMSO) in a Jupyter Notebook (Kluyver et al., 2016) using the pandas’ library (Reback et al., 2020) and pycytominer (Way et al., 2022a). Then, we performed feature selection to exclude features with low variance (frequency cut = 0.05), high correlation to another feature in the profile (threshold of 0.9), features that have more than 5% of NA (not available) values, excluded blocklisted features, and removed outliers by excluding a feature if its minimum or maximum absolute value is greater than the threshold (threshold = 500). These parameters serve as useful starting values but may be adjusted as needed; see the data preparation notebook and the pycytominer documentation (https://pycytominer.readthedocs.io/en/latest/) for more details. These steps are available in the basic_protocol_1/notebooks/data_processing folder using the Data_preparation.ipynb notebook in our GitHub repository (https://github.com/ciminilab/2022_Garcia-Fossa_submitted/blob/main/basic_protocol_1/notebooks/data_processing/Data_preparation.ipynb). We opened the CSV file obtained using Data_preparation.ipynb in Morpheus and clicked on Tools > Transpose, allowing the CSV table to be better visualized in Morpheus. For the researcher to apply the protocol to their own data rather than our example dataset, we recommend using CellProfiler (Stirling et al., 2021) to extract features and pycytominer (Way et al., 2022a) for data preparation.
We calculated the average precision based on https://github.com/niranjchandrasekaran/profiling-workflow-demo/blob/master/analysis/0.calculate-ap.ipynb to enable us to remove weakly correlated pairs (defined as less than a <0 mean average precision between replicates) before analysis; no such profiles were found for this data set. To reproduce our results, follow the instructions for creating an environment at https://github.com/niranjchandrasekaran/profiling-workflow-demo, and use our notebook WeakProfiles_Replicability.ipynb available on our GitHub repository at https://github.com/ciminilab/2022_Garcia-Fossa_submitted/tree/main/basic_protocol_1/notebooks to calculate the replicability between the samples in our Morpheus_Example_FeatureSelected.csv dataset. See the Critical Parameters section for more information about removing weak profiles.
Protocol steps with step annotations
-
1
Clone the GitHub repository into your computer or download the repository using https://minhaskamal.github.io/DownGit/#/home?url=https://github.com/ciminilab/2022_Garcia-Fossa_submitted, to have the dataset for this protocol.
-
2
Access the website https://software.broadinstitute.org/morpheus/ and click on “Select File” on the main screen, and select the Morpheus_Example_FeatureSelected.gct file you downloaded from GitHub. On the current tab, you will see a heatmap. Notice all the Columns displayed for Compound, Concentration, etc.
If using your own data or the provided GCT file instead of the example JSON file (which bypasses this step), click on Options (gear symbol), select the Annotations tab, and change Column annotations. Deselect id and select Compound, Concentration, MOA, Wells, and Plate. This will enable the visualization of metadata information within each column.
-
3
Click on Options (gear symbol) -> Annotations -> Columns annotations and click All to select all columns. Right-click on the column labels (Compound, Concentration, MOA, Wells, and Plate) and enable “Show color” for all the columns, to color-code the columns.
You can avoid these steps by using the Morpheus_Example_FeatureSelected.json file instead.
-
4
Click on Options (gear symbol) -> Color Scheme, and de-select the Relative color scheme. Change the minimum to −1000 and the maximum to 1000. Try also with −100 and 100. While the Relative color scheme converts values to colors based on each feature’s minimum and maximum values (making every row range from blue to red based on their own min and max), overriding and changing the color scheme to these new values allows seeing raw feature’s values distributed within this new feature range. In this way, extreme feature values became visible.
Setting the relative color scheme in this way highlights outlier features (with higher or lower values when compared to the other features). Trying with different minimum and maximum values will highlight features within different ranges. This is just a quality control step to guarantee that feature selection was performed correctly.
-
5
Close the Option window and then click the zoom tool and select “Fit To Window”.
-
6
Use the mouse pointer to scroll throughout the rows’ names in the right corner of the screen, highlighting the features’ names. Where you see any values colored in red or blue - these are the unusual features that have high or low values when compared to the rest of the features.
Notice that if feature selection was performed, features that have points with abnormal distance from other values in the distribution will often be removed during this process. Removing outlier features is recommended because they could mean errors during image acquisition like artifacts, which could affect post-analysis (e.g., interfering with logistic regressions, or other learning methods). But remove them carefully, and try to understand why they appeared. We give more details about feature selection in the Critical Parameters section, but the pycytominer tool (Way et al., 2022a) has a function to drop outliers based on their absolute minimum and maximum values, allowing the change of the outlier cutoff value.
-
7
Open Options (gear symbol) -> Color Scheme, and select “Relative color scheme” to use the minimum and maximum values in each row to convert values to colors.
Note that the results will be quite different if you use the minimum and maximum values in each column.
-
8
Select “Tools > Similarity Matrix > Pearson correlation” on the Rows. This will calculate the correlation between the wells for all pairs of features in the dataset and generate a similarity matrix for them. Click on Options (gear symbol), Display, and select Link rows and columns.
-
9
Create a Hierarchical Clustering by selecting Tools -> Hierarchical Clustering. In “Metric”, select “Matrix vales (for a pre-computed similarity matrix)”. Change “Cluster” to Rows and Columns. Click OK. This step will group the features depending on how similar their profiles are using the correlation metric you have chosen.
Steps 8 and 9 will create correlations between the features, and cluster them by their similarity. The square blocks of red color along the diagonal denote high correlation, meaning the features in those rows and columns look similar to each other. Scroll down and look for the red squares, checking their names. You can see interesting clusters when looking for different feature groups: for example, check “Nuclei_AreaShape_Extent” (next to the Help menu, in the white box, type id: Nuclei_AreaShape_Extent and hit Enter. Click on the arrows next to it to redirect to the feature, and see the name highlighted). “Nuclei_AreaShape_Extent” and “Nuclei_AreaShape_Solidity”, both related to how circular or protruded an object is and thus unsurprisingly tightly correlated, are grouped right next to Cells and Nuclei Intensity in the endoplasmic reticulum (ER), which would not be expected a priori. Since nuclei and ER are physically linked through the nuclear outer membrane which is continuous with the endoplasmic reticulum membrane (Hirano et al., 2020), we could hypothesize how these features’ correlations could translate into biology. Seeing that a couple of unexpected features group together might point at interesting unexpected biology.
-
10
Go back to the first tab “Morpheus_Example_FeatureSelected” and select “Tools > Similarity Matrix > Pearson correlation” on the Columns. This will calculate the correlation between features for all pairs of samples in the dataset and generate a similarity matrix for them.
Pearson’s coefficient is one of the many ways to calculate the correlation. There are other available methods in Morpheus (e.g., Spearman, Kendall, Cosine, etc). We give more details about Pearson’s coefficient in the Background section below.
-
11
Click on Options (gear symbol), Display, and select Link rows and columns. Linking rows and columns helps navigate the large matrix, showing the respective correlations.
-
12
While holding the shift key, click on the MOA, Compound, and Concentration columns (in this order) to sort them by value. This step will display the samples in order, based on those categories of metadata (rather than based on the profile similarity itself). The scientist should focus on the MOAs and the different compounds inside each MOA - can you see if compounds belonging to the same MOA have a similar morphological profile (Figure 2)?
Figure 2 -.
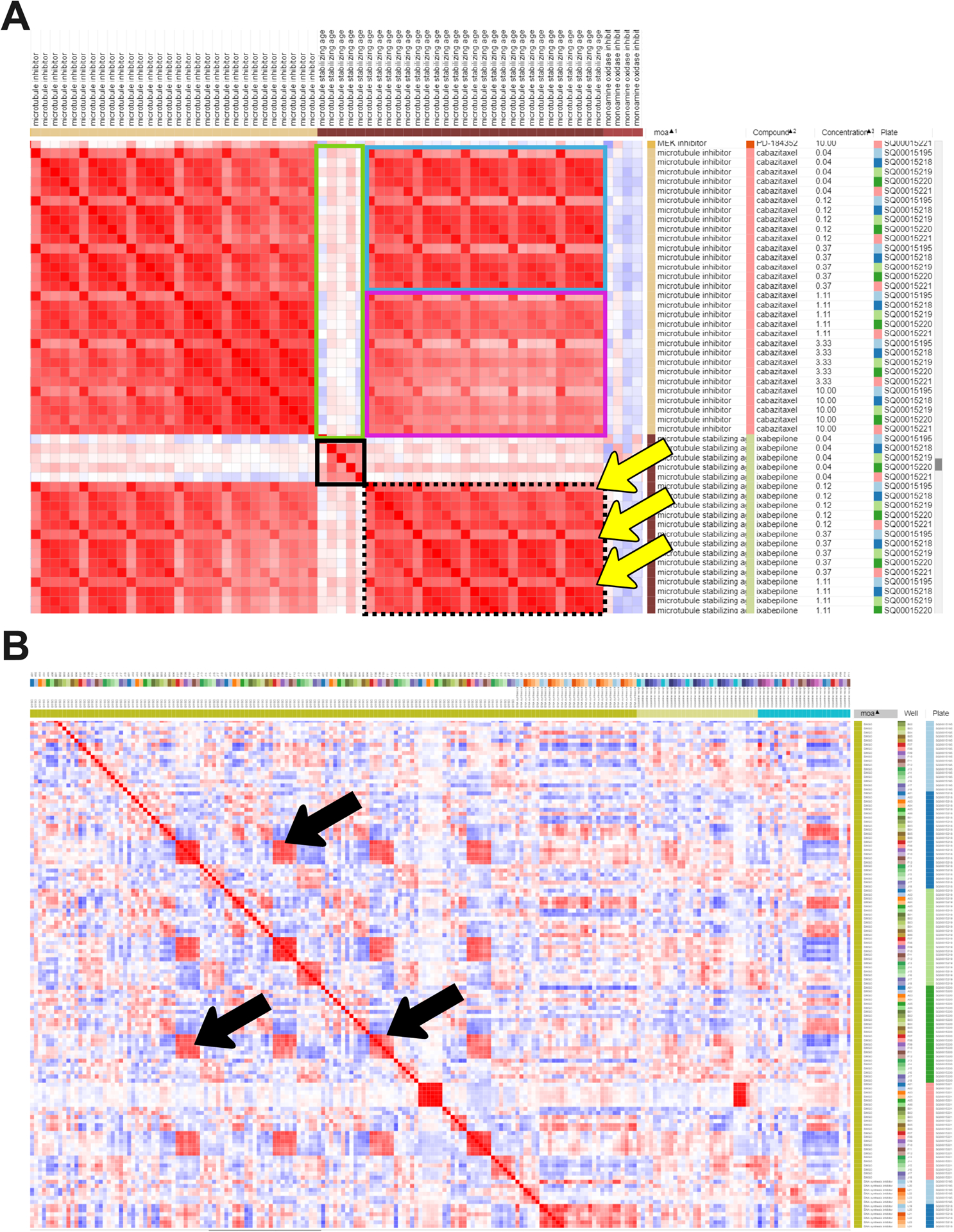
Similarity matrix generated in Morpheus. Columns were sorted by MOA, Compound, and then Concentration. (A) A subset of the similarity matrix showing the MOAs “Microtubule inhibitor” and “Microtubule stabilizing agent”. The top left and bottom right large red blocks show similarity of various doses on various plates within the same MOA class; the blocks on the top right and lower left are identical except for rotation, and show the similarity across classes. The small solid black box in the center shows the lowest dose of microtubule-stabilizing agent clusters well across replicates; its relatively poor correlation with the tightly clustered replicates at higher doses (black-dashed box) or any concentrations of microtubule inhibitor (green box) shows it might be below the effective dose of this drug. Higher doses of a microtubule-stabilizing agent, cluster well within and across doses, though a subtle recurring pattern within this block (highlighted by the yellow arrows) indicates that one of the five replicates shows a somewhat different profile than the other four, indicating a possible batch effect or technical anomaly. The effective concentration of a drug is highlighted by the lowest dose of ixabepilone clustering together (black box) but having weak correlations with the highest doses of ixabepilone. The higher doses of the microtubule-stabilizing agent are extremely similar to low concentrations of microtubule inhibitor (blue box) but less similar to higher concentrations of microtubule inhibitor (purple box). (B) Negative control (DMSO) correlation pattern, zoom out view of the similarity matrix. Black arrows highlight artifacts from plate-layout effects; treatments plated in the same or very similar well positions still can show significant similarity even after normalization. This can be alleviated at the experimental level by scrambling positions across plates and/or plating the same treatment in multiple positions spread across an individual plate.
Square blocks of red color along the diagonal denote high correlation, meaning the compounds in those rows and columns look similar to each other. Going all the way towards the end of the dataset, on the RNA polymerase inhibitor MOA, you can see how the two compounds of this MOA (favipiravir and PSI-6130) have a similar morphological profile by looking at the higher correlations between them. At the top left of the heatmap, the user should check the adrenergic receptor antagonist MOA and the compounds that define this MOA. Can you see how there is not much correlation between the different compounds? Each compound, even belonging to the same MOA, seems to have a different morphological profile.
-
13
Having the same configuration as the step before (columns sorted by MOA > Compound > Concentration), continue to explore the similarity matrix and observe: are there different MOAs that have similar morphological profiles?
Go to the microtubule inhibitor and microtubule stabilizing agent MOAs (Figure 2A). See how there are large squares colored in red when comparing the correlation coefficient for both groups, meaning these two MOA are closely correlated, and they produce similar morphological profiles. One can even distinguish subtle effects of drug concentration: the lowest concentration of microtubule stabilizer (green box) is relatively dissimilar from all concentrations of microtubule inhibitors; at higher doses of microtubule stabilizer, the phenotype is indistinguishable from low concentrations of microtubule inhibitor (blue box) but less similar to higher concentrations of microtubule inhibitor (magenta box). Look for more unusual correlations.
-
14
Sort the collapsed similarity matrix by MOA and by Plate by holding the shift key. Zoom out (pressing the minus - key) to have a broader view of the matrix.
-
15
Roll over to the DMSO MOA (the negative control in this dataset).
Observe how DMSO samples are distributed and do not have universally high correlations with each other (Figure 2B). Some are correlated by Well position. This indicates that we performed plate normalization with DMSO as the baseline (meaning DMSO is not a phenotype in this dataset and all DMSO profiles should have feature values of zero, apart from technical noise). For this reason, some of the DMSO samples in similar positions (same Wells but in different plates) have some correlation, because the Well position within the plate is one of the strongest technical artifacts in cell-based experiments (Figure 2B, black arrows indicate the Well position clusters). Normalization performed in relation to the negative control of your plate is the most reliable way to ensure that you can compare your samples even if they come from different plates, acquired on different days. Yet we can see some correlations between DMSO samples that are related to DMSO samples positioning within the plates, highlighting the importance of having the controls scattered to avoid plate position effects. We give more background about normalization in the Critical Parameters section below.
-
16
Create a Hierarchical Clustering by selecting Tools -> Hierarchical Clustering. In “Metric”, select “Matrix values (from a pre-computed similarity matrix). Change “Cluster” to Rows and Columns. Click OK. This step will group the samples depending on how similar their profiles are (using the correlation metric you have chosen). The scientist can identify different groups and try to make sense of the groups.
In this step, observe that the samples do not primarily cluster based on their location on particular plates. Instead, plates are usually spread evenly inside the grouped samples, meaning there’s no strong plate effect. Normalization is important to achieve that, and more information is available in the Critical Parameters section below.
-
17
Use the Zoom out (minus key) to have a broader view of the clustering. Scroll through and find large squares of red color in the matrix, and observe which MOAs are clustering.
The displayed color labels of the columns are useful to find patterns in this broader view. Two groups (proteasome inhibitor and NFkb pathway inhibitor|proteasome inhibitor) are clustered together, meaning these two MOAs have similar morphological profiles. Look for strongly clustered phenotypes (such as mTOR inhibitor|PI3k inhibitor) and how a few DMSO samples are still clustering with the strong phenotype. This should not be interpreted as the mTOR inhibitor phenotype is not strong or that they are close to DMSO, this is just an effect caused by the normalization of the data to the negative control (DMSO). See more details in the Critical parameters section below.
-
18
Return to the tab containing the feature value rather than the similarity matrices. Go to Tools -> Marker Selection. Choose the T-test as the metric, MOA as the field, class A as DMSO, and class B as the tubulin polymerization inhibitor. Leave the default values for “Number of Markers” and “Permutations”. This step reveals what features are driving the differences between these two groups (Figure 3).
Figure 3 -.
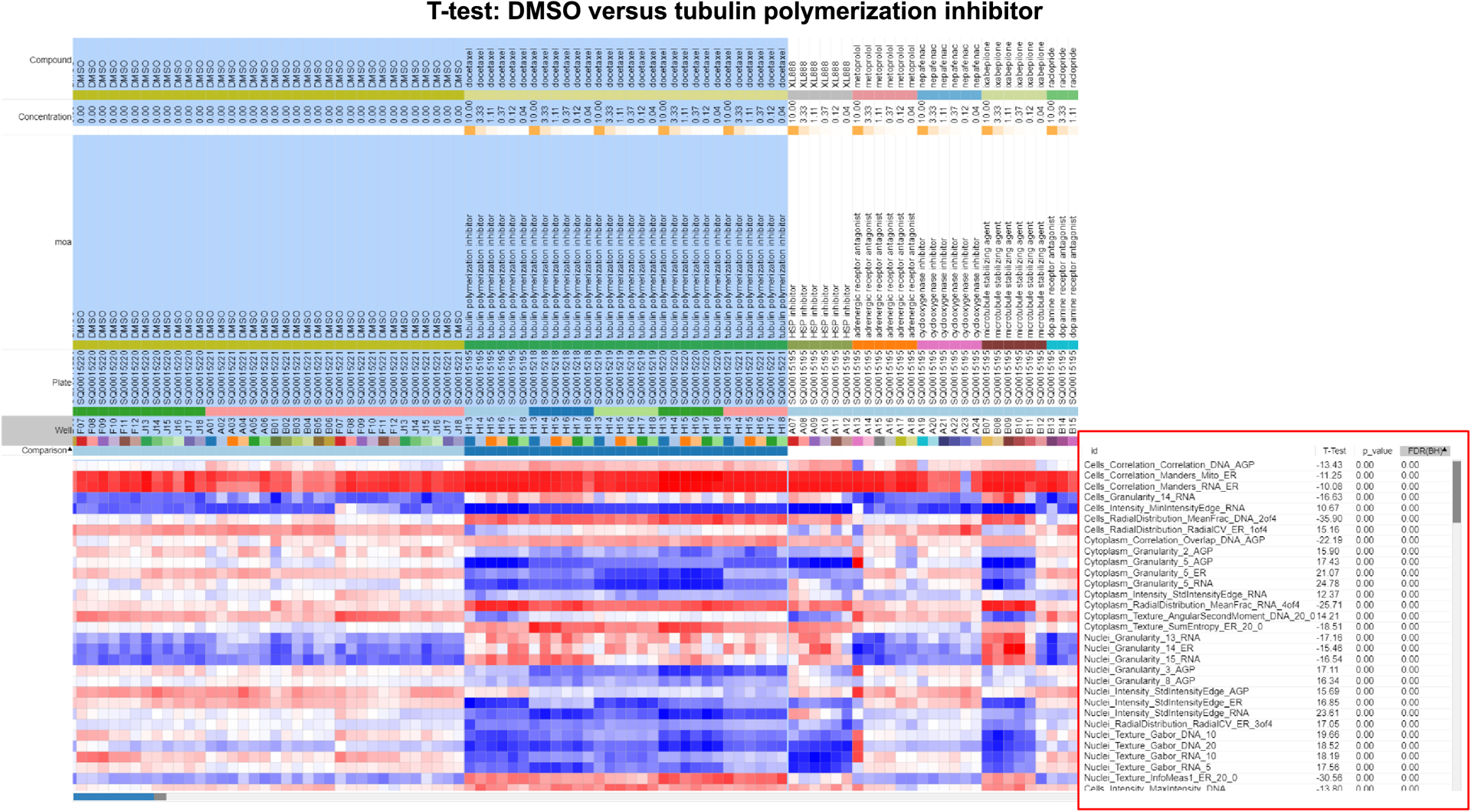
Features that are driving the differences between the groups. A T-test was performed on DMSO versus tubulin polymerization inhibitor classes using the Marker Selection tool in Morpheus in Step 20. Features that differentiate between DMSO and a tubulin polymerization inhibitor are highlighted using a red box. Highlighted in blue are the columns of the two groups being compared (DMSO and tubulin polymerization inhibitor).
The “Number of Markers” will depend on the number of features you have and want to use for this analysis. “Permutations” will depend on the number of samples in each class. For more information about Marker Selection, see the Understanding Results section and (Gould et al., 2006).
-
19
Sort the p-value column by right-clicking on the p-value column. Explore the names of the features that determine the difference between DMSO and tubulin polymerization inhibitors. If you have a large number of features with a p-value of 0.00, these will continue to be sorted alphabetically and not by strength; in this case, you can sort to find the highest and lowest T-test values, which should represent the strongest features.
The features that differentiate between DMSO and tubulin polymerization inhibitors belong to all three cell compartments: Cells, Nuclei, and Cytoplasm, (where the Cytoplasm of each cell is defined as the region identified as Cell, excluding the region identified as the nucleus. Texture and granularity (both measures of whether the stain is smooth or not) are frequent features altered by this perturbation. Tubulin is not stained in this assay but since tubulin interacts with actin, it seems reasonable that the profile would be altered in Phalloidin (F actin) staining. Another frequent feature group is Nuclei_StdIntensityEdge, the standard deviation in intensity at the edge in several channels; this likely indicates differences in the variation in staining of various organelles just outside the nuclei.
-
20
Go back to the similarity matrix and go to File > Save Dataset, write a name for your File, click Ok to save as GCT version 1.3, and save the table, allowing it to be opened again in Morpheus when needed.
BASIC PROTOCOL 2
Image and single-cell visualization following profile interpretation
With large datasets, it often becomes challenging to retrieve images of sites or single cells for visualization to perform quality control, validate a pipeline, and, most importantly, interpret any morphological changes detected in the profiles explored during the data analysis and exploration (visualized with heatmaps, UMAPs, etc). Along with visualizing sample and feature correlations as in Basic Protocol 1, it is also important to think biologically about organelle distribution, general morphology such as cell and nuclei shape, and intensities of each stain. Connecting the numbers (Pearson coefficients, T-tests, morphological feature values in the profiles, etc.), with how the cells look in the images can help the researcher to decipher a complex profile.
In Basic Protocol 2, we describe how to use a script we created to retrieve random or representative images from the dataset and plot them together, allowing the scientist to choose which samples to observe and how to group and display them. While random images are often helpful, especially in cases of high heterogeneity it can be helpful to computationally determine which cells’ phenotypes are the most representative in your sample, and compare them to control cells. This is not a trivial step but can sometimes provide critical insight into the morphological change. In this protocol, we use Jupyter Notebook to derive representative cells by performing a clustering analysis on the morphological space of the population of single cells and sampling from the subpopulation closest to the center of the sample(s) of interest. This notebook can also be used to compute similarity matrices as in Morpheus; but for large-scale experiments, we recommend examining the experiment as in Basic Protocol 1 using the per-well aggregated information. Once a few treatments of interest are identified, the user can then visualize single cells in Basic Protocol 2 and order the images by most correlated to the negative control.
From the Jupyter Notebook, the scientist will obtain representative or random image sites and single cells, enabling comparison of the images with the correlation coefficient values obtained in the similarity matrix. By establishing the relationship between the images and the heatmaps, the user can start hypothesizing about the biological processes and morphological profiles that are significant, which could lead to more specific biological questions and assays. As in Basic Protocol 1, we provide some hints and interpretations for each step, but the major discussions of biological interpretations are presented in the Understanding Results section below.
Materials:
Laptop or desktop computer with at least 2 GB of RAM and a suitable web browser such as Google Chrome. Internet and access to a Gmail account if using Google Colab.
This protocol assumes the use of a web browser to run Google Colab. Open our Google Colab notebook (link https://github.com/ciminilab/2022_Garcia-Fossa_submitted/blob/main/basic_protocol_2/notebook/Basic_Protocol_2.ipynb) and create a copy on your own Google Drive, enabling you to run Basic Protocol 2. To adapt this protocol to their own data, users should either download the Jupyter Notebook to their local computer and install the requirements based on the requirements.txt file or use Google Colab and mount their Google Drive (https://colab.research.google.com/notebooks/io.ipynb) to enable access to data they have stored within their Google Drive. In both cases, the scientist must adapt the pathnames and filenames within Section 2 of the Notebook to point to their dataset.
Our dataset table is in a CSV format, a comma-separated value table containing the extracted features for single cells in a Cell Painting assay. In this assay, 1,571 compounds were tested across 6 doses in A549 cells (Way et al., 2022a). Notice that we use the same dataset from Basic Protocol 1, but here we require information about single cells, and each row of the table must have cell features, and X and Y locations within the image, to enable single-cell image retrieval. We also provide all the images of where these single cells are located. For this purpose, we selected only a subset of samples within the dataset to minimize the memory requirements needed for the user to explore the data. We also performed normalization and feature selection with this dataset using pycytominer (Way et al., 2022a). The Jupyter Notebooks required to create this dataset from publicly available data sets (1_Samples_retrieval.ipynb and 2_Generate_Profiles.ipynb) that are available on our GitHub under the basic_protocol_2/notebook folder. We additionally provide an alternate code in the sample retrieval notebook to allow the loading of entire plates, when experiment size and memory permit.
The Jupyter Notebook functions were written using Python 3.9 (Van Rossum and Drake, 2009). Data processing was performed using pycytominer (Way et al., 2022a) tools for normalization, feature selection, and data annotation. Check pycytominer documentation (https://pycytominer.readthedocs.io/en/latest/) for details on how to change parameters and inputs depending on your dataset.
The GitHub repository contains the following files relevant to Basic Protocol 2:
util folder with .py files containing functions written to be used on this notebook. These functions are installed onto the notebook using pip install and then imported from utils.correlations import *;
basic_protocol_2/Images folder, which contains the subset of images downloaded from https://github.com/broadinstitute/cellpainting-gallery. We provide PNG images that were compressed from the original TIFF images; PNG is a lossless format that requires less storage space.
basic_protocol_2/data folder, which contains the “BasicProtocols2_Example.zip” with a CSV file. To use this notebook with their data, the user could extract the features using CellProfiler (Stirling et al., 2021), and export the information to a spreadsheet, which can be read in the Jupyter Notebook. Or, if using a database file, the scientist could transform it into a CSV file using our available Samples_retrieval.ipynb Jupyter Notebook. The notebook will perform annotation, normalization, and feature selection if you have not already run those steps; they can also be bypassed if these steps have already been done (such as by notebook 2_Generate_Profiles.ipynb)
Protocol steps with step annotations
-
1
Open the Google Colab notebook “Basic Protocol 2_Visualize cells and images.ipynb” available in the link on our GitHub page (https://github.com/ciminilab/2022_Garcia-Fossa_submitted/blob/main/basic_protocol_2/notebook/Basic_Protocol_2.ipynb). Be sure to access the notebook from our GitHub repository, allowing you to check for any recent updates.
-
2
Click the Copy to Drive button, and the notebook will be available on your Google Drive in the Colab Notebooks folder.
This step allows the researcher to have their copy of the notebook, and if they wish, perform any modifications to run the notebook using their own data.
-
3
Run the first three cells in the notebook Section 1 - Import Libraries by clicking on the start button at the top left (or hit Ctrl + Enter). The first line will clone the GitHub repository and install the functions, the second line will install the required libraries to run this notebook (this process takes ~5 min to run), and finally, import the libraries to allow their use inside the notebook. Run the lines of code in the order that they appear in the notebook.
The Python packages required to run this notebook are also available on GitHub under the requirements.txt file. This file can be used to install packages via pip or to generate an environment using Anaconda or miniconda to run this Jupyter Notebook locally.
-
4
Run only the first cell inside Section 2 - Define Inputs. This will define the inputs required for running the cells in the notebook. The script requires the filename and pathname to access the CSV table and read it as a DataFrame. It also needs the pathname for the images directory.
The pathnames are all based on the ones available in the GitHub repository for this project. If you clone the repository in the first step, there is no need to change these inputs. To run this notebook with new data, mount the notebook inside Google Drive and provide the inputs for the variables (running the second code cell inside Section 2 instead of the first cell).
-
5
Run the cells inside Section 3a, which will import the dataset and perform annotation, normalization, and feature selection. The table contains all the features measured for every single cell, and also metadata information about the mechanism of action (MOA) of each compound, compound names, and concentrations tested. For more information about feature selection, see the Critical Parameters section.
If your data set has already been annotated, normalized, and feature selected, skip section 3A and proceed directly to section 3B to load it in with no adjustments. If running section 3A with your own data, you will need to have already annotated your data with Metadata (such as in CellProfiler’s Metadata module) or provide a table here that provides the ability to map the measurement data to treatment metadata. The user can run the normalization on the whole dataset, or choose to run normalization relative to the negative control. More details are in the Critical Parameters section.
-
6
Run the first three cells in Section 4 (through cell 4.1.1) and choose “Metadata_Compound_Concentration” for this demonstration. These options were generated based on the name of the columns with the “Metadata_” prefix. This choice will impact the information visualized on the plots for the next steps. If the choice is “Metadata_Compound_Concentration”, then you will see values such as DMSO 0.0, etc. When using new data, the researcher should add the “Metadata_” prefix to any such columns before loading it into the notebook, as it will appear under this dropdown and be used for aggregation (Figure 4 A).
Figure 4 -.
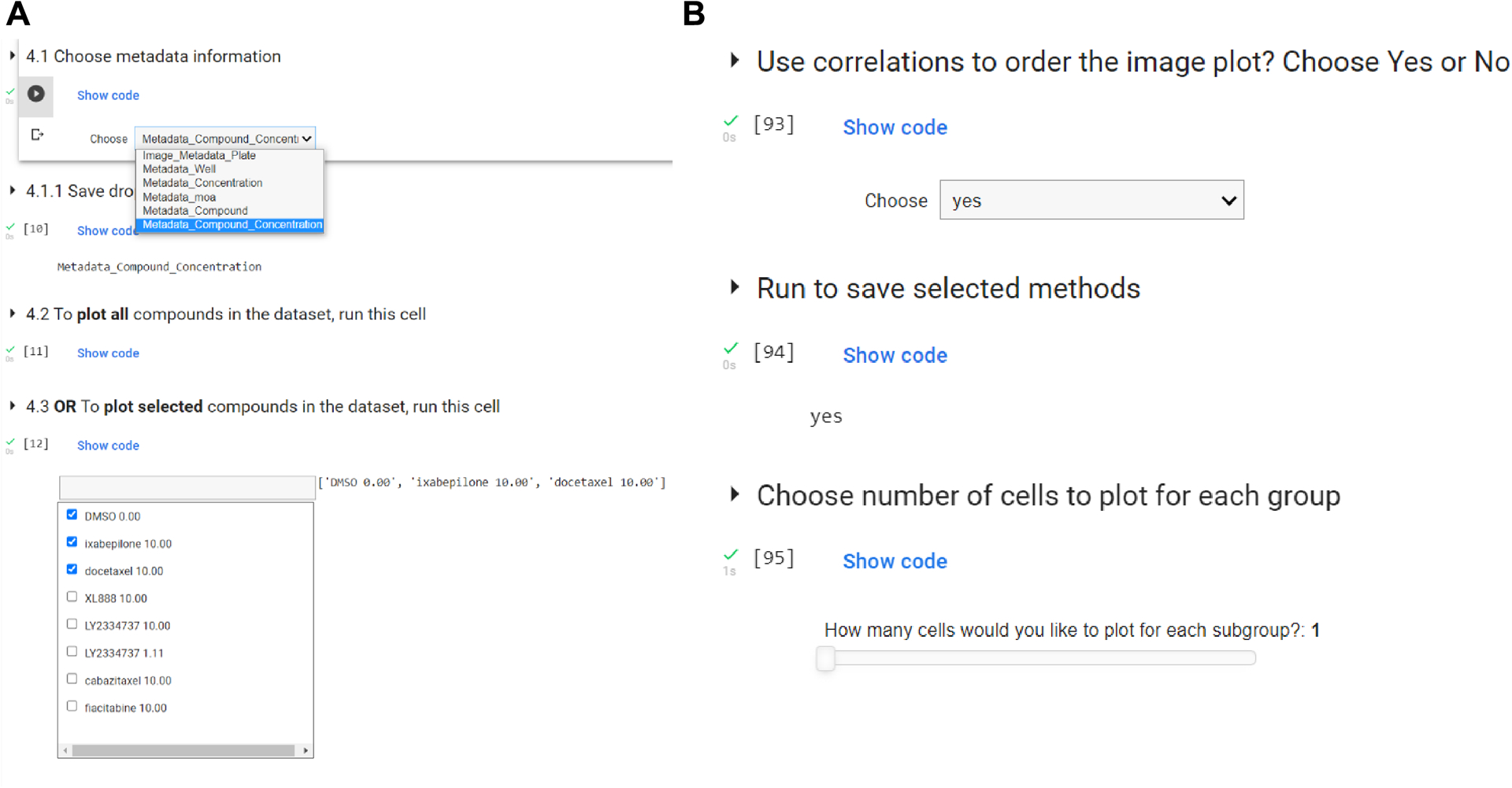
User’s interactions with the Jupyter Notebook. (A) Demonstration of the dropdown options, and choice box to choose only a subset of the compounds (step 6 of Basic protocol 2). To use new data, add the “Metadata_” prefix to the label columns. (B) More examples of interaction through dropdowns and sliders to choose the number of cells to plot.
We use dropdown interaction to allow the researcher different choices based on the DataFrame. This is because they might be interested in looking at the data from a mechanism of action perspective, or based on the compound’s names. Be aware that if using new data, the tables must have columns containing metadata information with the “Metadata_” prefix.
-
7
Run the cell in Section 4.2 to choose all the compounds available on the dataset to visualize. This step will select all the compounds in the dataset.
To select just a few compounds of interest to be visualized, run Section 4.3. This piece of code will create an interactive checkbox with the compound names for you to choose only a few options (Figure 4 A).
-
8
Run the cells in Section 5 to generate and graph the correlation between the compounds. Choose a column to be the labels for the correlation matrix using the dropdown. Then, we use pycytominer (Way et al., 2022a) to return a per-well aggregated DataFrame, and a correlation matrix is generated. There is an option to export the matrix as an image, type the name, and press “Enter/return”.
In Section 5, the function that applies pycytominer operations aggregates the data and then performs a Pearson correlation analysis in the dataset. If the user wants to visualize the matrix with different labels, they can choose a different column and rerun the notebook from that cell onward; their dataset will then be re-aggregated and a new correlation matrix will be calculated based on the new column.
-
9
Run the three cells in section 5.1 to insert the correlation values calculated in the previous step inside the initial DataFrame as a new column. This function will get the chosen compound and find the correlation values for every other compound related to the first. Choose “DMSO 0.0” for comparison because for this dataset, the aim is to evaluate what compounds have morphological profiles more similar to the control.
Choose whichever compound to have as a point of reference to be added to the DataFrame. This choice will depend on their biological question.
-
10
Run all of the cells inside section 5.2, and choose “DMSO 0.0”. This choice reflects the biological question of what compounds are closely correlated to the negative control (DMSO, in our case). However, this is a dynamic Jupyter Notebook where the user could be interested in other compounds or MOAs.
-
11
In Section 6 - Visualize Cells, run the first cell to choose whether to visualize randomly selected or representative single cells. You can choose the “random” method to select random samples for each treatment/group you have or choose the “representative” method to select the most representative cell within each subgroup. Many cells in this section rely on correlation to the reference compound selected in section 5.1; if you want to change reference compounds, rerun those cells before returning to section 6 and running all cells there.
Choosing the representative method uses the KMeans algorithm with the scikit-learn package (Pedregosa et al., 2011) to cluster data and find the most representative cell (the cell(s) closest to the mean of the subgroup) within each subgroup. The random method will instead return a random sample of one cell for each subgroup (Reback et al., 2020). The representative method allows you to evaluate average change, while the random method is often helpful for quality control to check for out-of-focus or unusual cells.
-
12
Run the next cell and select how many cells you would like to display from each subgroup and whether or not you would like the images shown in order of subgroup correlation to the reference compound.
Answering “Yes” to “Would you like to use the correlations to order your image plot?” will order the dataset based on the correlation values to the reference compound selected in step 9, starting at 1.0 and descending; answering “No” will keep the DataFrame in the original order. The second question is about how many cells (c) to plot for each group. The generated image will have (c * the number of subgroups) rows. Looking at one cell per subgroup creates a compact visualization, especially for many subgroups; looking at several per subgroup can increase confidence in the overall visual appearance of each subgroup, especially when displaying random cells.
-
13
Choose whether each image should be rescaled to the minimum and maximum before being displayed, or whether raw intensity values should be plotted instead. Raw intensities are typically more comparable across conditions (see below for caveats), but may be harder to see when the signal is dim and thus may require external rescaling after saving.
While in general raw images are more comparable than individually rescaled images, caution should be taken especially in comparing images from treatments imaged on different plates or different plate batches. Each plate is stained, imaged, and has feature normalization run independently; plates from different batches may additionally have other differences such as e.g. individual reagent lots used. Thus, a treatment that induces “2x negative-control-mean-intensity” in channel X from Plate 1 may be overall dimmer in raw pixel intensity values than a different treatment that induces “0.5x negative-control-mean-intensity” in channel X from Plate 2 if the plate means intensities in channel X are quite different. Any conclusions drawn based on looking at images should be subsequently checked against normalized feature data.
-
14
Insert the pixel size value. This is necessary for adding a scale bar in your images. Just type the value “0.29898” in this example to add the pixel size for this example dataset in μm/pixel. Each microscope and lens will have its configuration.
Some microscopes (such as the Opera Phenix microscope used in this experiment (Way et al., 2022b) record the effective pixel size in a file such as an XML (eXtensible Markup Language). Other microscopes record this information in the file metadata; one easy way to check this is by opening the image in a tool such as Fiji (Schindelin et al., 2012) and look at the Properties menu. Embedded metadata is sometimes missing or unreliable; when in doubt, consult the local expert on the microscope in question and/or calculate the effective pixel size based on the camera specifications and magnifications used.
-
15
Plot the selected single cells in random order by running the first cell of section 6.1. This step allows a first view of the cells without the labels, so the researcher can explore the images before knowing to which group the cells belong. Once you have explored the data, run the rest of the cells in section 6.1 to append labels to see if your hypotheses were correct, to create an un-shuffled version of the image, and to save the image to disk.
Looking at cells without labels allows the researcher to formulate new hypotheses without bias about what they believe each treatment “should look like”. Parameters to examine might include the organelles’ distribution within the cells; how the mitochondria, endoplasmic reticulum, or the Golgi apparatus are organized; changes in overall intensity of individual stains, or overall cell structure changes. This can be quite valuable for unbiased hypothesis generation!
-
16
Run section 6.2 to display the full images from which the single-cell crops have been pulled (Figure 5 B). Looking at the entire field of view (FOV) may provide insights into additional biological aspects.
Figure 5 -.
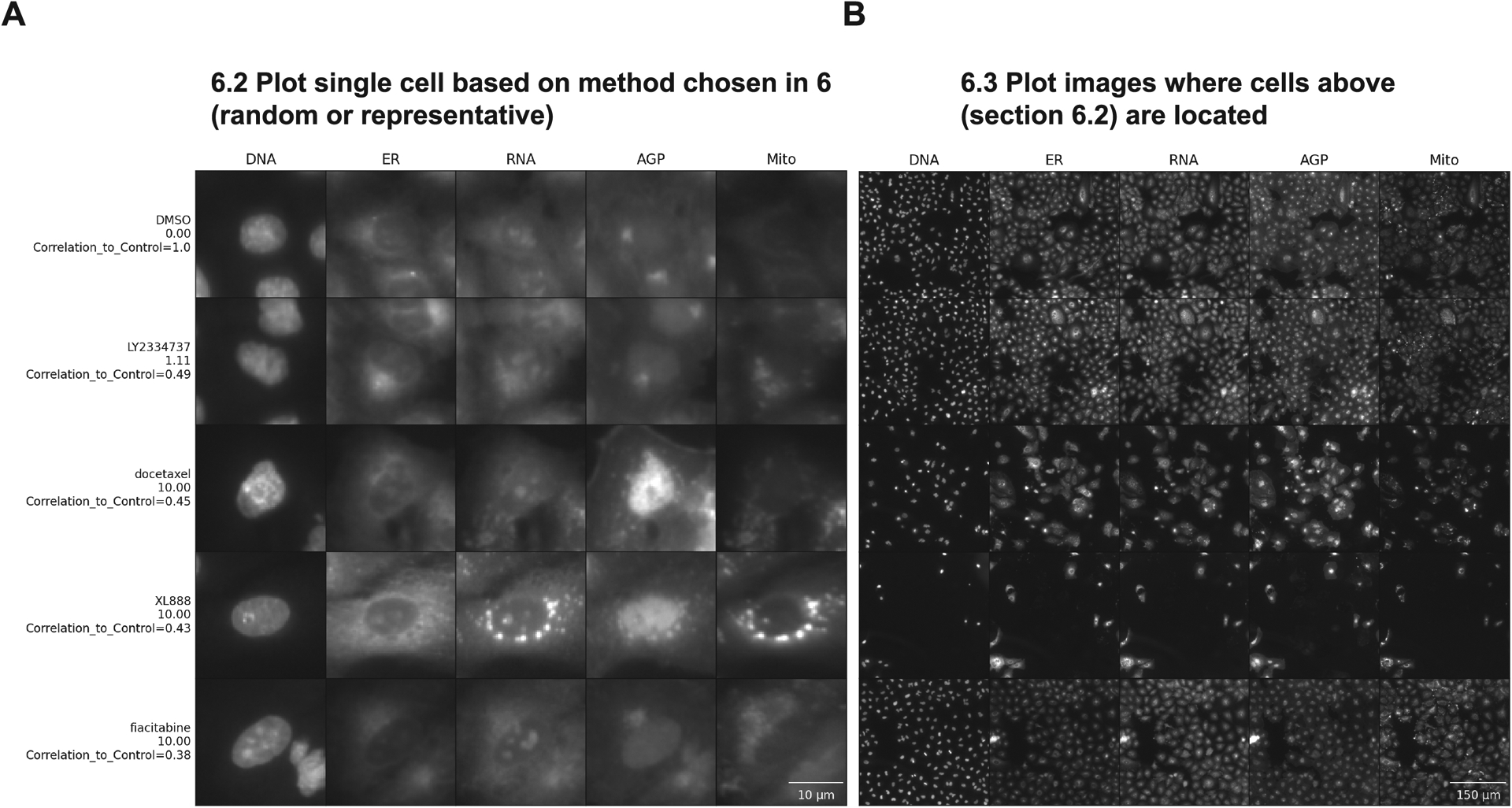
Steps to plot single cells and representative images in order of correlation values. Both images were plotted with rescaled intensity, using the representative method, 1 cell per subgroup, and ordered top to bottom by the correlation values. On the Y-axis we have the compound’s names and concentrations in μM and the X-axis above has the names of stained structures, showing the different fluorescence channels available in this experiment: DNA (nucleus stained with Hoechst 33342, excitation/emission 405/450 nm), ER (endoplasmic reticulum stained with Concanavalin A, excitation/emission 488/525 nm), RNA (nucleoli and cytoplasmic RNA stained with SYTO 14, excitation/emission 488/600 nm), AGP (actin stained with phalloidin, Golgi and plasma membrane stained with wheat germ agglutinin, both acquired with excitation/emission 561/600 nm), and Mito (mitochondria stained with MitoTracker Deep Red, excitation/emission 640/750 nm). For complete details about the Cell Painting procedure, see more at (Bray et al., 2016). (A) Shows a single representative cell for each group in this dataset. Scale bar = 10 μm (B) shows the field of view where each representative cell is located. Scale bar = 150 μm
Looking at whole images allows a more holistic view of the overall cell shape and size distributions present in the images (e.g., how were the cells affected by the treatment: do they look larger or smaller when compared to the negative control? Does their shape change, are they more elongated, or rounder?). It also allows examination of cell density (e.g., how are the cells distributed within the FOV? Can you see more cells in the FOV between perturbations and the control, meaning the perturbation might induce some proliferative signaling pathway? Or are there fewer cells in the FOV, meaning the perturbation might reduce cell viability?).
COMMENTARY
Background Information
Image-based profiling typically starts with using fluorescent markers to stain different targets and/or compartments of the cell. In our example data for both protocols, we used Cell Painting data. Cell Painting is a morphological profiling assay that multiplexes six fluorescent dyes, imaged in five channels, to reveal eight relevant cellular components. The experiment’s aim was to characterize chemical perturbations in cells by measuring the morphological changes after cells were exposed. Briefly, cells were plated in multiwell plates, perturbed with the treatments to be tested, then stained, fixed, and imaged on a high-throughput microscope. Images were acquired for DNA, RNA, endoplasmic reticulum, mitochondria, and AGP (actin, Golgi, and plasma membrane).
Software such as CellProfiler (Stirling et al., 2021) makes it easy to obtain and extract information from these images, extracting thousands of morphological features distributed into categories relating to the compartment measured (Nucleus, Cell, Cytoplasm) and types of metrics (measurements of size, shape, texture, intensity, granularity, and more) to produce a feature profile that enables the detection of subtle phenotypes. To facilitate understanding of the features, CellProfiler feature name outputs are organized as follows: [Compartment]_[FeatureGroup]_[Feature]_[Channel]_[Parameters]. Not all features have Channel information - for example, shape features relate only to the outlines of the chosen cellular compartments. From a Cell Painting assay, the Nuclei are identified by the DNA channel, the Cells by the RNA or AGP channel, and the Cytoplasm is the Cell, excluding the Nuclei object. FeatureGroups are associated with the measurements made on the Compartments (e.g., AreaShape, Intensity, Texture, Granularity, and more). To understand how each module works to extract information from the images, check the latest documentation available for CellProfiler (https://broad.io/cellprofilermanual). You can check a list of all the features extracted from one particular analysis of a Cell Painting assay at https://github.com/carpenterlab/2022_Cimini_NatureProtocols/blob/main/CellProfiler_features.csv, but note that the names of the features will vary based on the parameters used to analyze the assay.
The essential steps after extraction of the features are aggregation, normalization, and feature selection. These are the steps we describe in our Jupyter Notebooks using pycytominer (Basic protocol 1 support notebook and in the main notebook used for Basic protocol 2). Then, profiles of cells treated with different experimental perturbations are compared to identify the phenotypic impact of chemical or genetic perturbations, grouping compounds and/or genes into functional pathways, and identifying signatures of disease. We demonstrate these last two steps using Morpheus software and scripts on Jupyter Notebooks in the Basic Protocols above.
Pearson correlation coefficient
Understanding the correlation coefficients calculated for the samples on both protocols is important for this protocol paper. A Pearson correlation coefficient is a way to represent the measurement of similarity, where it measures the strength of the linear relationship between two variables (in our case, the relationship between two wells across a large set of features or for two features across a large set of wells). A Pearson coefficient of 1 means a perfect positive correlation, 0 means no correlation was found, and −1 represents a perfect negative correlation (Pearson and Galton, 1895). A similarity matrix is a way to assess the covariance in features between all pairs of columns or rows. In each square of the matrix, a Pearson correlation coefficient was calculated for all features in the data set between each pair of samples. The squares at the intersection of those two samples are set as the value of that correlation coefficient, and so on for each pair of wells. This allows us to see at a high level how similar the overall phenotype is between any pairs of samples in our experiment, and therefore how phenotypically distinct our treatments are.
Critical Parameters
We reiterate that normalizing the features is fundamental before executing the steps in this paper. Normalization is usually performed on all of the features to fix range issues and allow comparison between the features (Caicedo et al., 2017). Normalization is also recommended to increase the signal-to-noise ratio (Chandrasekaran et al., 2021). A normalization performed on a plate level is recommended because this also corrects to some degree for plate-to-plate batch effects. Where sufficient negative controls exist, we recommend normalizing the features to the negative control. Check the profiling recipe for more information about how to process single-cell morphological profiles and details on how to normalize Cell Painting data for more information.
In data normalized to the negative controls, negative control samples (or samples with otherwise weak phenotypes, here defined as a mean average precision across replicates of <0) will show limited similarity to one another and thus will show minimal clustering after step 16 from Basic Protocol 1 (hierarchical clustering). Somewhat unintuitively, this means that these samples will be spread across the entire data set post-clustering, and it is therefore expected, after hierarchical clustering and exploration (step 17 of Basic Protocol 1), to see one or a small number of “random” negative controls or weak perturbations clustering with a strong, consistent perturbation; this should not be taken as a sign that the strong perturbation in question is weak or similar to negative controls. Weak replicate correlation for any given phenotype can be checked during Basic Protocol 1 step 12; if the replicate inconsistency looks possibly driven by technical issues (such as well position - see Figure 2B), one may consider performing another experiment to attempt to confirm if a profile is truly weak, but in general, profiles with weak replicate correlation should not be used to draw biological conclusions.
Proper reduction of the feature space is also an essential step to perform before analyzing new data in our protocols; this step will be automatically performed when following the profiling recipe (Chandrasekaran et al.). If performing these steps on your own, a common starting point is looking for correlated features: when two features are too correlated, only one should be kept for further analysis. Since Pearson correlations are sensitive to large absolute feature values, we also recommend screening for unusual feature values; we provide guidance on performing this in Morpheus in Basic Protocol 1 steps 3–6. Some feature reduction algorithms, such as support vector machines, give weights for each feature and remove the ones with fewer weights (Caicedo et al., 2017). We typically perform feature reduction in pycytominer (Way et al., 2022a), which provides six options for reducing the feature space: removing based on variance threshold (removing features that have relatively few unique feature values and/or a single value that is far more common than the rest of the feature values), correlation threshold (removing features that are highly correlated to other features and thus redundant), drop NA columns (removing features where a large number of values are missing), drop outliers (removing features with aberrantly large absolute values), remove noise (removing features which tend to have a high variance across replicates), and blocklisting (removing features thought to not typically add useful biological information to Cell Painting profiles) (Way, 2019). Many of these feature removal methods have tunable parameters which ultimately guide the fraction of features removed; as such, it is critically important to check that the threshold values are appropriate for your data and to adjust them when necessary.
Profiles should be assessed for their quality before data interpretation, to remove treatments with no apparent phenotype and, in some applications, to exclude compounds that are too toxic to the cells (Rezvani et al., 2022). One method to perform profile quality assessment is to measure the precision with which one can correctly retrieve replicate wells. This approach was used in the example data we provide to check for the replicability of the profiles; more details are provided in (Way et al., 2022b).
Troubleshooting:
See Table 1 for problems, possible causes, and solutions.
Table 1.
Troubleshooting Guide for Basic Protocols 1 and 2.
| Problem | Possible Cause | Solution |
|---|---|---|
| All/almost all samples have a correlation value close to 1 (Morpheus after generating Similarity Matrix) | Features are not normalized | Check if the data were normalized, meaning all features must be in a range from 0 – 1 |
| Cells on Google Colab notebook can’t run | Notebook was not copied to user’s Google Drive | Add a copy of the Notebook to your own Google Drive by clicking on “Copy to Drive” |
| UserWarning: KMeans is known to have a memory leak on Windows with MKL (Math Kernel Library) when there are less chunks than available threads. You can avoid it by setting the environment variable OMP_NUM_THREADS=2 | Memory leak | Set the environment variable OMP_NUM_THREADS = x, being x the value specified on your error output. Follow the solution in this thread on stack_over_flow |
Understanding Results
When analyzing results, you may find a profile of interest shows a dramatic difference from controls or other samples based on only a small number of similarly-named features (such as a large number of features that fall within the nucleus or many changes in the texture of a particular stain), and the feature names have obvious meanings (e.g., nucleus area or integrated intensity of the mitochondria channel in the cytoplasm). In this scenario, interpretation may be straightforward, though you may need to look up the meaning of the feature names in the CellProfiler manual (https://broad.io/cellprofilermanual) to understand them better and discern their connection to the biological meaning. Some caution is warranted here; for example, DNA-damaging drugs could affect actin features, because F-actin plays a role in DNA repair. Damage induced to the DNA induces nuclear actin formation (Belin et al., 2015), and these nuclear actin structures play a role in double-stranded-break (DSB) repair, such as recruitment of proteins to enable repair of the heterochromatin through homologous recombination, and assisting on DSB movement in euchromatin repair (Caridi et al., 2019). There may not be a straight line from a feature name to the biological function because cells are deeply interconnected systems, and changes that start in a single genetic pathway can ripple throughout other pathways in the cell. Nevertheless, feature names can often create insights.
Instead of a few, easily interpretable features, you may find there are many dominant features in the profile and their collective meaning is not obvious. In such cases, an expert might be able to stare at the list and derive some meaning. For example, an expert might realize that many different stains showing increased correlation may actually be related to a decreased x-y cell size (because in a rounded-up cell, organelles are more likely to overlap one another on the x-y plane and may be either truly colocalized or merely spread across the z-dimension). If you’ve looked at your feature list but need some backup, consider sharing your data on forum.image.sc to get some experts to weigh in. An example of this can be found in the morphological profile induced by the microtubule inhibitor (cabazitaxel) and microtubule stabilizing agent (ixabepilone) in this data set. To understand what are the features that differentiate between our negative control (DMSO) and the microtubule perturbations, we performed marker selection using a T-test. Marker selection comes from genome analysis, but it could be defined also as a feature selection. The model takes the features belonging to two classes as input, and a T-test is calculated to assess marker features that discriminate between the two classes (DMSO vs Microtubule) (Gould et al., 2006). While individual T-tests performed in Morpheus do attempt to correct for sample number with a false discovery rate, it does not and cannot control for how many tests the users choose to run; these tests are therefore appropriate for gaining qualitative insight into the relative importance of various stains and/or feature classes in distinguishing a phenotype, but the values returned should not be directly reported and any attempt to quantify these differences should be performed through standard statistical approaches. Our results show that many important features (Figure 6A) belong to Granularity and Texture feature groups across a number of different stains, which makes sense in the context of induction of massive cytoskeletal disruption. Since microtubule disruption perturbs cell division, the presence of “Nuclei_AreaShape_FormFactor” (a measure of shape uniformity in which linear and/or irregular shapes have values near 0 and a perfect circle is 1) helps indicate that we are looking not just at cytoskeletal disruption generally, but specifically disruption of the microtubules. This result highlights that the aggregate of different features is important for connecting profiles to perturbations.
Figure 6 -.
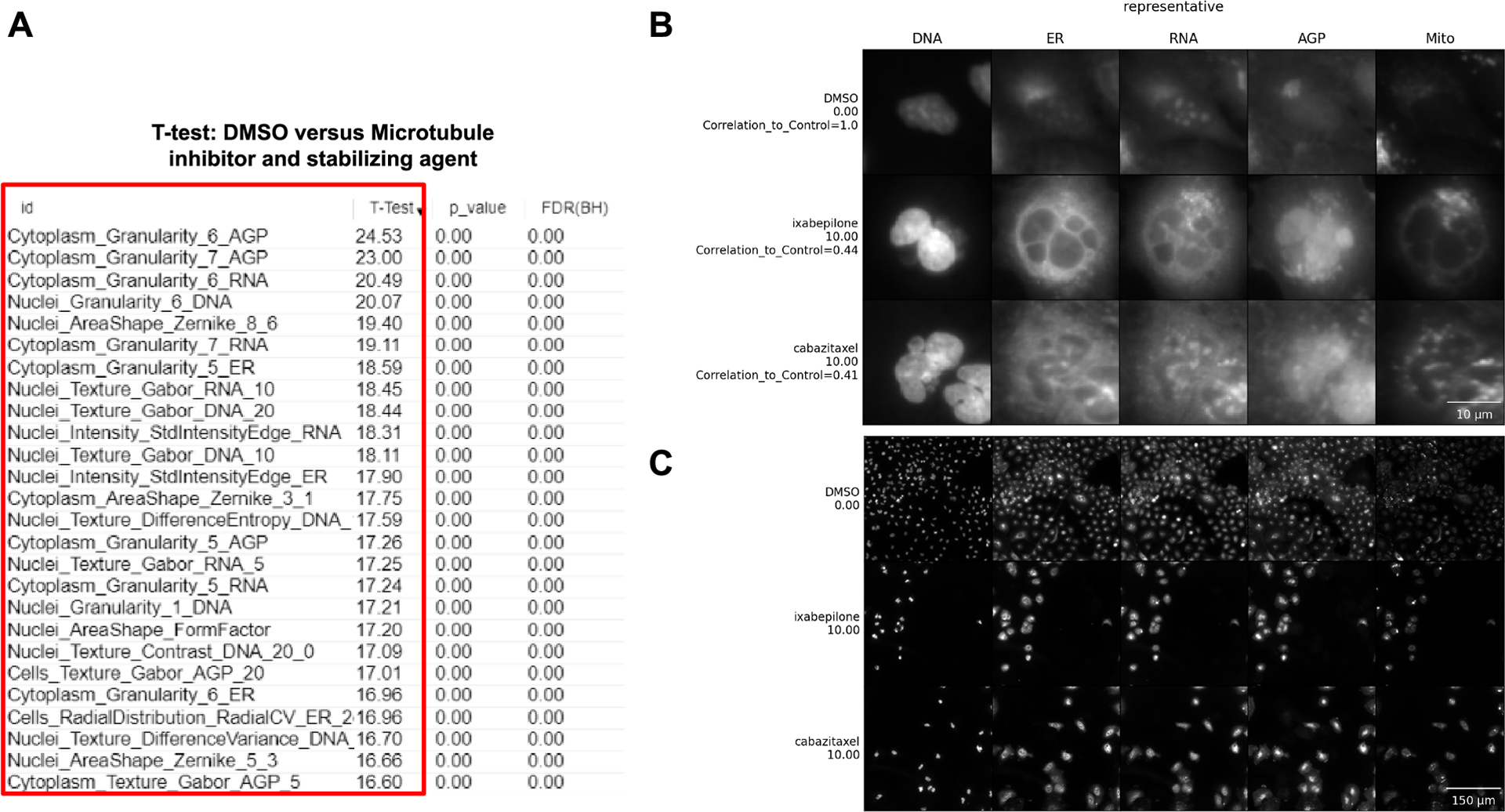
Interpretation of data using our Basic Protocols 1 and 2. (A) A marker selection was performed to test what are the features that differentiate DMSO vs Microtubule inhibitors (cabazitaxel 10 μM) and Microtubule stabilizing agents (ixabepilone 10 μM). The red box highlights the features. (B) Single cells are cropped based on an algorithm to retrieve representative cells. Scale bar = 10 μm. (C) Field-of-view where representative single cells are located. DNA (nucleus stained with Hoechst 33342, excitation/emission 405/450 nm), ER (endoplasmic reticulum stained with Concanavalin A, excitation/emission 488/525 nm), RNA (nucleoli and cytoplasmic RNA stained with SYTO 14, excitation/emission 488/600 nm), AGP (actin stained with phalloidin, Golgi and plasma membrane stained with wheat germ agglutinin, both acquired with excitation/emission 561/600 nm), and Mito (mitochondria stained with MitoTracker Deep Red, excitation/emission 640/750 nm). For complete details about the Cell Painting procedure, see more at (Bray et al., 2016). Scale bar = 150 μm.
Examining example images directly alongside the list of important features can also help decipher a complex profile. An example where looking at features and images could help uncover the biological meaning of an event is during an assay to identify cells in different phases of the cell cycle, using fluorescent markers such as DAPI to measure DNA content (Ferro et al., 2017). Based on significant changes in the feature space where the minor axis of the Nuclei and Cell area are low, and DNA staining intensity is high, the researcher could look at single cells and realize these feature changes relate to cells that actually are going through metaphase. Basic Protocol 2 facilitates displaying single cells and images, which can otherwise be challenging to locate and access in large-scale experiments. In our example images of cells treated with microtubule-related drugs, we observe that both drugs interfere with the cell cycle to produce similar morphologies, disrupting the overall appearance of every channel. As seen in Figure 6B, both treatments induce multinucleation (Figure 6 B, DNA column), as has been previously described for microtubule inhibitors (Azarenko et al., 2014); disruption of the cell cycle is also likely apparent in the lower overall cell count in the treated vs control cells (Figure 6C). The Golgi localization and distribution are visually quite distinct as compared to DMSO (Figure 6 B, AGP column), which could be related to microtubule’s function in vesicular trafficking and their role in modeling the shape of organelles, including Golgi (Fourriere et al., 2020; Thyberg and Moskalewski, 1985). We can therefore relate these morphological features and observations to the mechanism of actions of these drugs, providing a useful pattern to follow for investigators examining their own data and formulating their hypotheses. Sometimes, though the most important differences are not human visible; image-based profiling approaches have sometimes outperformed human expert image analysis for precisely such reasons (Gibson et al., 2015; Zhou et al., 2021).
Finally, we should note that in some situations, following the procedures provided still does not allow you to make much headway in truly understanding the induced phenotype. If so, one can still use profile data in other ways, such as by simply using the profile as a signature of the sample and trying to use drugs to revert this disease phenotype to a healthy-associated phenotype. If one has access to computational experts, one can also try to query their data against publicly available data sets (Rohban et al., 2022), though these approaches are currently still experimental. The interpretation of complex profiles is a challenge but when successful can propel research in new directions to uncover exciting new mechanisms.
Time Considerations
For Basic Protocol 1, supposing that the tables were pre-processed for normalization and feature selection before input into Morpheus, the total time to explore the data is approximately 1 hour. Basic Protocol 2 could take up to 2.5–3 hours if running the protocol with different settings and taking time to evaluate the images and create hypotheses.
ACKNOWLEDGEMENTS:
We thank Rebecca Senft and Erin Weisbart for suggestions in the manuscript text. We also thank Srinivas Niranj Chandrasekaran for helping with average precision concepts and data availability.
Funding was provided by the National Institutes of Health (NIH COBA P41 GM135019 to BAC and AEC and MIRA R35 GM122547 to AEC). This project has been made possible in part by grant number 2020-225720 to BAC from the Chan Zuckerberg Initiative DAF, an advised fund of the Silicon Valley Community Foundation.
Funding was also provided by São Paulo Research Foundation (FAPESP) #2022/01483-4, #2019/24033-1, and #2020/01218-3.
Footnotes
CONFLICT OF INTEREST STATEMENT:
SS and AEC serve as scientific advisors for companies that use image-based profiling and Cell Painting (AEC: Recursion, SS: Waypoint Bio, Dewpoint Therapeutics) and receive honoraria for occasional talks at pharmaceutical and biotechnology companies.
INTERNET RESOURCES:
https://software.broadinstitute.org/morpheus/ Versatile matrix visualization and analysis software.
https://forum.image.sc Forum for image analysis questions and discussions.
https://cellprofiler.org/ Download and learn how to use CellProfiler.
https://github.com/CellProfiler/tutorials Beginners and advanced tutorials for CellProfiler.
https://www.youtube.com/c/COBACenterforOpenBioimageAnalysis/videos Video tutorials for CellProfiler, Morpheus, and other many tools on the Center for Open Bioimage Analysis (COBA) channel.
https://github.com/cytomining/pycytominer Data processing functions for profiling perturbations. More information on how to use pycytominer, documentation, and workflows.
DATA AVAILABILITY STATEMENT:
The data that support the protocol are openly available at https://github.com/ciminilab/2022_Garcia-Fossa_submitted.git
LITERATURE CITED:
- Azarenko O, Smiyun G, Mah J, Wilson L, and Jordan MA 2014. Antiproliferative mechanism of action of the novel taxane cabazitaxel as compared with the parent compound docetaxel in MCF7 breast cancer cells. Molecular cancer therapeutics 13:2092–2103. [DOI] [PubMed] [Google Scholar]
- Belin BJ, Lee T, and Mullins RD 2015. DNA damage induces nuclear actin filament assembly by Formin −2 and Spire-½ that promotes efficient DNA repair. [corrected]. eLife 4:e07735. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Bray M-A, Singh S, Han H, Davis CT, Borgeson B, Hartland C, Kost-Alimova M, Gustafsdottir SM, Gibson CC, and Carpenter AE 2016. Cell Painting, a high-content image-based assay for morphological profiling using multiplexed fluorescent dyes. Nature protocols 11:1757–1774. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Caicedo JC, Cooper S, Heigwer F, Warchal S, Qiu P, Molnar C, Vasilevich AS, Barry JD, Bansal HS, Kraus O, et al. 2017. Data-analysis strategies for image-based cell profiling. Nature methods 14:849–863. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Caridi CP, Plessner M, Grosse R, and Chiolo I 2019. Nuclear actin filaments in DNA repair dynamics. Nature cell biology 21:1068–1077. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Chandrasekaran SN, Ceulemans H, Boyd JD, and Carpenter AE 2021. Image-based profiling for drug discovery: due for a machine-learning upgrade? Nature reviews. Drug discovery 20:145–159. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Chandrasekaran SN, Weisbart E, Way G, Carpenter A, and Singh S Broad Institute Imaging Platform Profiling Recipe. Available at: https://github.com/cytomining/profiling-recipe.
- Cimini BA, Chandrasekaran SN, Kost-Alimova M, Miller L, Goodale A, Fritchman B, Byrne P, Garg S, Jamali N, Logan DJ, et al. 2022. Optimizing the Cell Painting assay for image-based profiling. bioRxiv:2022.07.13.499171. Available at: https://www.biorxiv.org/content/10.1101/2022.07.13.499171v1 [Accessed July 15, 2022]. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Ferro A, Mestre T, Carneiro P, Sahumbaiev I, Seruca R, and Sanches JM 2017. Blue intensity matters for cell cycle profiling in fluorescence DAPI-stained images. Laboratory investigation; a journal of technical methods and pathology 97:615–625. [DOI] [PubMed] [Google Scholar]
- Fourriere L, Jimenez AJ, Perez F, and Boncompain G 2020. The role of microtubules in secretory protein transport. Journal of cell science 133. Available at: 10.1242/jcs.237016. [DOI] [PubMed] [Google Scholar]
- Gibson CC, Zhu W, Davis CT, Bowman-Kirigin JA, Chan AC, Ling J, Walker AE, Goitre L, Delle Monache S, Retta SF, et al. 2015. Strategy for identifying repurposed drugs for the treatment of cerebral cavernous malformation. Circulation 131:289–299. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Gould J, Getz G, Monti S, Reich M, and Mesirov JP 2006. Comparative gene marker selection suite. Bioinformatics 22:1924–1925. [DOI] [PubMed] [Google Scholar]
- Hirano Y, Kinugasa Y, Osakada H, Shindo T, Kubota Y, Shibata S, Haraguchi T, and Hiraoka Y 2020. Lem2 and Lnp1 maintain the membrane boundary between the nuclear envelope and endoplasmic reticulum. Communications biology 3:276. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Kluyver T, Ragan-Kelley B, Pérez F, Granger B, Bussonnier M, Frederic J, Kelley K, Hamrick J, Grout J, Corlay S, et al. 2016. Jupyter Notebooks – a publishing format for reproducible computational workflows. In Positioning and Power in Academic Publishing: Players, Agents and Agendas (Loizides F and Scmidt B, eds.) pp. 87–90. IOS Press. [Google Scholar]
- Morpheus Available at: https://software.broadinstitute.org/morpheus/ [Accessed August 9, 2022].
- Pearson K, and Galton F 1895. VII. Note on regression and inheritance in the case of two parents. Proceedings of the Royal Society of London 58:240–242. [Google Scholar]
- Pedregosa F, Varoquaux G, Gramfort A, Michel V, Thirion B, Grisel O, Blondel M, Prettenhofer P, Weiss R, Dubourg V, et al. 2011. Scikit-learn: Machine Learning in Python. Journal of machine learning research: JMLR 12:2825–2830. [Google Scholar]
- Reback J, McKinney W, jbrockmendel, Van den Bossche J, Augspurger T, Cloud P, gfyoung, Hawkins S, Sinhrks, Roeschke M, et al. 2020. pandas-dev/pandas: Pandas 1.2.0. Available at: https://zenodo.org/record/4394318.
- Rezvani A, Bigverdi M, and Rohban MH 2022. Image-based cell profiling enhancement via data cleaning methods. PloS one 17:e0267280. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Rohban MH, Fuller AM, Tan C, Goldstein JT, Syangtan D, Gutnick A, DeVine A, Nijsure MP, Rigby M, Sacher JR, et al. 2022. Virtual screening for small-molecule pathway regulators by image-profile matching. Cell systems 13:724–736.e9. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Schindelin J, Arganda-Carreras I, Frise E, Kaynig V, Longair M, Pietzsch T, Preibisch S, Rueden C, Saalfeld S, Schmid B, et al. 2012. Fiji: an open-source platform for biological-image analysis. Nature methods 9:676–682. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Stirling DR, Swain-Bowden MJ, Lucas AM, Carpenter AE, Cimini BA, and Goodman A 2021. CellProfiler 4: improvements in speed, utility and usability. BMC bioinformatics 22:433. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Thyberg J, and Moskalewski S 1985. Microtubules and the organization of the Golgi complex. Experimental cell research 159:1–16. [DOI] [PubMed] [Google Scholar]
- Van Rossum G, and Drake FL 2009. Python 3 Reference Manual. CreateSpace, Scotts Valley, CA. [Google Scholar]
- Way GP 2019. Blocklist Features - Cell Profiler. Available at: https://figshare.com/articles/dataset/Blacklist_Features_-_Cell_Profiler/10255811.
- Way GP, Chandrasekaran SN, Bornholdt M, Fleming S, Tsang H, Adeboye A, Cimini B, Weisbart E, Ryder P, Stirling D, et al. 2022a. Pycytominer: Data processing functions for profiling perturbations. Github; Available at: https://github.com/cytomining/pycytominer [Accessed September 9, 2022]. [Google Scholar]
- Way GP, Kost-Alimova M, Shibue T, Harrington WF, Gill S, Piccioni F, Becker T, Shafqat-Abbasi H, Hahn WC, Carpenter AE, et al. 2021. Predicting cell health phenotypes using image-based morphology profiling. Molecular biology of the cell 32:995–1005. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Way GP, Natoli T, Adeboye A, Litichevskiy L, Yang A, Lu X, Caicedo JC, Cimini BA, Karhohs K, Logan DJ, et al. 2022b. Morphology and gene expression profiling provide complementary information for mapping cell state. Cell systems 13:911–923.e9. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Zhou W, Yang Y, Yu C, Liu J, Duan X, Weng Z, Chen D, Liang Q, Fang Q, Zhou J, et al. 2021. Ensembled deep learning model outperforms human experts in diagnosing biliary atresia from sonographic gallbladder images. Nature communications 12:1259. [DOI] [PMC free article] [PubMed] [Google Scholar]
Associated Data
This section collects any data citations, data availability statements, or supplementary materials included in this article.
Data Availability Statement
The data that support the protocol are openly available at https://github.com/ciminilab/2022_Garcia-Fossa_submitted.git