

Abstract
We probed the lifecycle of Epstein‐Barr virus (EBV) on a cell‐by‐cell basis using single cell RNA sequencing (scRNA‐seq) data from nine publicly available lymphoblastoid cell lines (LCLs). While the majority of LCLs comprised cells containing EBV in the latent phase, two other clusters of cells were clearly evident and were distinguished by distinct expression of host and viral genes. Notably, both were high expressors of EBV LMP1/BNLF2 and BZLF1 compared to another cluster that expressed neither gene. The two novel clusters differed from each other in their expression of EBV lytic genes, including glycoprotein gene GP350. The first cluster, comprising GP350 – LMP1 hi cells, expressed high levels of HIF1A and was transcriptionally regulated by HIF1‐α. Treatment of LCLs with Pevonedistat, a drug that enhances HIF1‐α signaling, markedly induced this cluster. The second cluster, containing GP350 + LMP1 hi cells, expressed EBV lytic genes. Host genes that are controlled by super‐enhancers (SEs), such as transcription factors MYC and IRF4, had the lowest expression in this cluster. Functionally, the expression of genes regulated by MYC and IRF4 in GP350 + LMP1 hi cells were lower compared to other cells. Indeed, induction of EBV lytic reactivation in EBV+ AKATA reduced the expression of these SE‐regulated genes. Furthermore, CRISPR‐mediated perturbation of the MYC or IRF4 SEs in LCLs induced the lytic EBV gene expression, suggesting that host SEs and/or SE target genes are required for maintenance of EBV latency. Collectively, our study revealed EBV‐associated heterogeneity among LCLs that may have functional consequence on host and viral biology.
Keywords: EBV reactivation, Epstein‐Barr virus, lymphoblastoid, Pevonedistat and HIF1‐a, single cell RNA‐sequencing, super enhancer
1. IMPORTANCE
Epstein‐Barr virus (EBV) establishes a life‐long latency program within host cells. As such, EBV immortalized lymphoblastoid cells (LCLs) often carry the latent EBV genome and only a small percentage of LCLs contain lytic EBV. However, the cellular programs that distinguish latent from lytic cells and the heterogeneity of cells in latent or lytic phases remains poorly explored. To explore these unknowns, we reanalyzed publicly available single cell RNA‐seq data from nine LCLs. This approach permitted the simultaneous study of cells in both latent and lytic phases. We identified three cell populations with distinct lytic/latent activity and further characterized the transcriptomes of these cells. We also identified a new role of super‐enhancers (SEs) in regulating EBV lytic replication. Collectively, our studies revealed EBV‐associated heterogeneity among LCLs that contribute to EBV life cycle and biology.
2. INTRODUCTION
EBV is the first oncogenic human DNA virus discovered more than 50 years ago. 1 EBV causes ∼200 000 cases of diverse cancers every year, including lymphomas, nasopharyngeal carcinoma and gastric adenocarcinomas. 2 , 3 , 4 , 5 Most EBV infections occur early in life and are transmitted through saliva. EBV first infects oral epithelial cells and then B lymphocytes in the oral epithelium. EBV persists in memory B cells for life in a latent phase, so these cells express minimum EBV genes under host immune surveillance. However, when host immunity is impaired, for example by immunosuppressive treatment or HIV infection, EBV in infected B cells can enter type III latency where six EBV nuclear antigens (EBNAs), three latent membrane proteins, and a few noncoding RNAs and microRNAs, are expressed. 3 , 6 This can result in lymphoproliferative diseases or lymphomatous transformation. 7 EBV in infected memory B cells can also enter a lytic phase to actively produce live virus. During EBV lytic replication, immediate early genes RTA and ZTA encoded byBRLF1 and BZLF1 genes, respectively, are first expressed. These are transcription factors (TFs) that turn on the expression of genes necessary for viral DNA replication and structural proteins, including viral membrane protein gp350, that binds to human B cell EBV receptor CD21, to assemble virions. 8
In vitro, EBV infection of primary B lymphocytes leads to the establishment of LCLs. 9 LCLs express EBV type III latency genes, the same genes seen in some EBV malignancies, including post‐transplant lymphoproliferative disease and AIDS CNS lymphomas. Therefore, LCLs are an important model system to study EBV oncogenesis. Genetic studies have found that EBNA1, 2, LP, 3A, 3C, and LMP1 are essential for EBV‐mediated growth transformation. 10 , 11 EBNA1 tethers EBV episomes to host DNA. 12 , 13 , 14 EBNA2 and LP are the major EBV transcription activators that activate expression of key oncogenes, including MYC. 15 , 16 EBNA3A and 3C repress expression of p16INK4A and p14ARF, to overcome senescence and BIM to avoid apoptosis. 17 , 18 , 19 LMP1 activates NF‐κB to provide survival signals. 20
EBV infection significantly alters chromatin topology and function at EBV‐interacting genomic loci of host cells. 21 This alteration could be mediated by EBV‐encoded transcription factors 22 , 23 or via interaction between EBV episomes and the host genome, 24 and may also depend on the EBV latency program. 25 , 26 SEs are critical regions of mammalian genomes comprised of clusters of enhancers bound by arrays of transcription factors. 27 Viral transcription factors and host NF‐κB subunits can form EBV SEs 23 with markedly high and broad histone 3 lysine 27 acetylation (H3K27ac). 28 These SEs are linked to many genes essential for LCL growth and survival, including MYC and IRF4, and their perturbation pauses LCL growth and causes cell death. 29 , 30 We have previously shown that EBV episomes physically interact with SE‐containing genomic host loci in EBV‐transformed lymphoblastoid cells. 24 However, the consequences of these interactions and the effects of perturbations at these SE‐containing loci on the EBV life cycle remains unexplored.
High throughput sequencing technologies can aid to dissect mechanisms underlying host‐virus interactions. 4 , 24 , 31 , 32 However, the heterogenous nature of virally infected cells is an impediment to precisely probing the phases of virus life cycle and their effect on host genes in individual cells. Recent advances in single‐cell transcriptomics have enabled successful resolution of tissue/cell heterogeneity in several species. Since these technologies agnostically capture both host and infecting viral sequences, they have also been utilized to explore host‐virus interactions at a single cell level. 33 , 34 , 35 Leveraging this feature here, we sought to identify the determinants of EBV latency in lymphoblastoid cells. Using single cell transcriptomics, we identified distinct clusters marked by differences in expression of GP350 and LMP1. Cells expressing high levels of LMP1, but not GP350, demonstrated high HIF1‐α activity and could be induced by a HIF1‐α stabilizer. Cells co‐expressing GP350 and LMP1/BNLF2 had significantly reduced expression of SE‐containing genes compared to cells containing EBV that was clearly in the latent phase (i.e., GP350 – cells). Using proof‐of‐principle SE inactivation experiments, we found that host SEs are necessary for the maintenance of EBV latency. Collectively, our data not only highlight the heterogeneity among LCLs but also identifies common functional themes of the cells and their role in EBV‐associated biology.
3. METHODS AND MATERIALS
3.1. Cell culture
LCL‐358 (catalog no. 1038‐3754NV17, Astarte Biologics), GTEX‐UPJH‐0001‐SM‐3YRE9, GM12878, AKATA EBV positive and AKATA EBV negative cells were cultured in RPMI1640 (VWRL0105‐0500) media supplemented with 10% fetal calf serum (Gibco or Hyclone), 100 unit/ml streptomycin and 100 mg/ml penicillin (Gibco or Life Technologies). HEK293T cells purchased from ATCC were cultured in Dulbecco modified Eagle medium supplemented with 10% fetal calf serum (Gibco), 100 unit/ml streptomycin and 100 mg/ml penicillin. All the cells were maintained at 37°C in a 5% CO2 humidified chamber. Cells were routinely confirmed to be mycoplasma negative according to PCR Mycoplasma Detection Kit (ABM Inc.) and were used at low passage (<10) number but were not independently authenticated.
3.2. CRISPRi repression
Plasmid dCAS9‐KRAB‐MeCP2 (#110821) purchased from Addgene was packaged with lentiviruses and used to transduce LCLs for 2 days followed by selection with 5 ug/ml blasticidin for another 5 days. The expression of dCAS9‐KRAB‐MeCP2 was verified by western blot. sgRNAs targeting genomic sites of interest were designed with online software Benchling (www.benchling.com). sgRNAs were annealed and cloned into LentiGuide‐Puro vector according to previously published protocol. 36 LentiGuide‐Puro containing sgRNAs were packaged into lentiviruses and were used to transduce LCLs stably expressing dCAS9‐KRAB‐MeCP2. Cells were selected with 3 ug/ml puromycin for 3 days and then allowed to grow for another 2 days. The list of sgRNAs are provided in Supporting Information: Table S3
3.3. qRT‐PCR
Cells were harvested and washed once with cold PBS. Total mRNAs were extracted using PureLink RNA mini kit (Life Technologies) or Direct‐zol RNA extraction kit with DNase I treatment (Zymo Research) following manufacturer's instructions. mRNAs were then reverse transcript into cDNA with iScript™ Reverse Transcription Supermix (Bio‐rad) or OneScript Plus cDNA Synthesis SuperMix (ABM Inc.). cDNAs were amplified on an CFX96 Touch real‐time PCR detection system (Bio‐Rad) and SYBG Green (Thermo Fisher) was used to detect cDNA amplification. All experiments were performed in triplicates in total reaction volumes of 15 μL using BrightGreen 2X qPCR MasterMix‐No Dye (ABM Inc.). A housekeeping gene was used to normalize gene expression. RNA relative expression was calculated using the 2 ΔΔC T method. The value for the cells transduced with nontargeting sgRNA was set to 1. The list of all qPCR probes is provided in Supporting Information: Table S3.
3.4. ChIP‐qPCR
LCLs stably expressing dCAS9‐KRAB‐MeCP2 were transduced with lentiviruses expressing sgRNAs. Two days after transduction cells were selected with 3 ug/ml puromycin for another 3 days. Cells were then collected and fixed with 1% formaldehyde. The cells were lysed and sonicated with bioruptor (Diagenode). Sonicated chromatin was diluted and precleared with protein A beads followed by incubation with 4 ug H3K27ac (Abcam, #ab4729) or control antibodies with rotating at 4°C overnight. The next day, Protein A/salmon DNA beads (Millipore, #16‐157) were used to capture protein–DNA complexes. After precipitation, beads were washed with low salt wash buffer (1% TritonX‐100, 0.1% SDS, 2 mM EDTA (pH8.0), 150 mM NaCl, 20 mM Tris‐HCl [pH 8.0]) once, high salt wash buffer (1% TritonX‐100, 0.1% SDS, 2 mM EDTA [pH8.0], 500 mM NaCl, 20 mM Tris‐HCl [pH 8.0]) twice, Licl wash buffer (0.25 M LiCl, 1% NP‐40, 1% NaDOC, 1 mM EDTA, 10 mM Tris‐HCl [pH 8.0]) once, and TE buffer (1 mM EDTA, 10 mM Tris‐HCl [pH 8.0]) once. Each wash was performed by gently spinning down beads at 300 g for one minute and resuspend beads with 1 ml wash buffer followed by shaking at 4°C for 5 min. DNA and protein complexes were eluted with elution buffer (1% SDS, 100 mM NaHCO3). Protein–DNA complexes were reverse cross‐linked with proteinase K (Thermo Fisher, #EO0491). DNA was purified by using QIAquick Spin columns (Qiagen, #28104). qPCR was used to quantify the DNA from ChIP assay and normalize it to the percent of input DNA.
3.5. Induction of EBV
AKATA EBV positive and negative cells were treated with IgG (Agilent, # A042301‐2) at a final concentration of 0.5% followed by incubation at 37°C, 5% CO2 for 6 h. Cells were then centrifuged and re‐suspended with fresh RPMI1640 supplemented with 10% FBS and continue culture for another 48 h. mRNAs were extracted by using PureLink RNA mini kit (Life Technologies), qRT‐PCR was used to detect EBV lytic gene expression. To induce LMP1 expression, LCLs were treated with 100 nM of NEDD8 inhibitor‐MLN4924 (Pevonedistat) (A gift from Dr. Liu) or DMSO control and were collected at indicated time points for qRT‐PCR and/or Flow cytometry.
3.6. Dual CRISPR mediated DNA deletion
Dual gRNAs were designed with webtools from benchling (www.benchling.com) and were cloned into pLentiGuide‐Puro (Addgene Plasmid #52963) using the Multiplex gRNA kit (System Biosciences) according to the manufacturer protocol. The success of gRNAs insertion was verified by sequencing with U6 promoter primer. HEK293T cells were used to package lentiviruses by co‐transfecting viral packaging plasmids pCMV‐VSV‐G (Addgene #8454), psPAX2 (Addgene #12260) and the pLentiGuide‐Puro vector containing the target sgRNAs. Eighteen hours after transfection, media were changed to fresh RPMI media containing 30% of FBS. Twenty four and forty eight hours later, supernatant containing lentivirus was collected and filtered with a 0.45 micron filter. LCLs in which Cas9 was stably expressed were transduced with the filtered lentivirus (Day 0) for 2 days, and then selected with 3 ug/ml Puromycin for 3 days. On Day 5, genomic DNA was extracted using the DNeasy Blood & Tissue kit (Qiagen) and RNA was extracted with the PureLink RNA Mini kit (Ambion). Genomic deletions were verified by PCR using the PrimeSTAR polymerase (Clonetech). qRT‐PCR was done using the Power SYBR Green RNA‐to‐CT 1‐Step Kit (Applied Biosystems).
3.7. Single‐cell RNA sequencing analysis
10x Genomics Cell Ranger 6.0.2 count 37 was used to align the raw sequencing reads to a customized human (GRCh38) and EBV (NC_007605, obtained from 66 ) hybrid reference genome to generate barcode and UMI counts. Seurat (v4) 38 was applied for the downstream analysis and visualization of the data as following: Genes that were expressed in less than three cells were discarded. Cells with >20% of their unique molecular identifiers (UMIs) mapping to mitochondrial genes or cells with <250 detected genes were discarded. Only cells with >80% log10 (Genes per UMI) were retained. Cell cycle score for each cell was calculated by Seurat function CellCycleScorin using human cell cycle genes. SCTransform was then used to normalize the data set using default parameters while regressing out mitochondrial genes and cell cycle scores (S and G2M) and identify variable genes. Then, the datasets were integrated based on “anchors” identified among datasets (nfeatures = 2000, normalization.method = “SCT”) before perform linear dimensional reduction by Principal Component Analysis (PCA). The top 50 PCs were used for the Uniform Manifold Approximation and Projection (UMAP) dimensionality reduction. Clusters were identified on a shared nearest neighbor (SNN) graph of the top 50 PCs with the Louvain algorithm at three resolutions (i.e., 0.1, 0.3, 0.5). The clusters corresponding to latent EBV life cycle were combined as one cluster for the downstream analyses. Differential gene expression was determined by “FindMarkers” function on SCT normalized expression values with the default Wilcox Rank Sum test either as one versus rest or as a direct comparison with default parameters except logfc.threshold = 0. The cell annotation was based on the EBV genes and top differentially expressed genes. Gene list module scores were calculated with Seurat function AddModuleScore. 56 This calculates the average scaled expression levels of each gene list, subtracted by the expression of control feature sets (n = 100). All the displayed expression values on violin plots, feature plots and dot plots are SCT normalized expression values. The IRF4 bound genes are sourced from the ChIP‐Atlas “Target Genes” database 39 with options: “hg38” as the genome and “± 5 kb” as distance from TSS. Target genes with binding score not less than 500 in GM12878 cells are selected. All genesets used in this study are provided in Supporting Information: Table S2.
3.8. Geneset enrichment analysis (GSEA)
GSEA was performed using pre‐ranked mode and “No Collapse” options. The preranked gene lists were ranked by the SCT normalized expression fold‐change between comparison groups. EBV‐contacted and EBV‐non‐contacted genesets are curated from our previous study 24 and provided in Supporting Information: Table S2.
3.9. Statistical analysis and data visualization
Statistical analyses were performed using GraphPad PRISM 9 (La Jolla) with the method detailed in the legend.
3.10. Flow cytometry
All stained/fixed samples were acquired on Attune NxT Flow Cytometer (Thermo Fisher Scientific) with necessary internal controls to help assign gates. Fluorescence from multiple antibodies were compensated using AbC Total Compensation beads (Thermo Fisher Scientific, catalog no. A10497). In all experiments, cells were collected and stained with fixable viability dye eFluor780 (1:2000 dilution Life Technologies, catalog no. 65‐0865‐14;) followed by surface staining for PDL1 (CD274; clone 29E.2A3; BioLegend catalog no. 329714; 1:60 dilution) as per the manufacturer's instructions. Cells were then fixed with 4% methanol‐free formaldehyde (Thermo Fisher Scientific, catalog no. 28908) followed by intracellular staining for BZLF1 (Santa Cruz Biotechnology, catalog no. sc‐53904; 1:60 dilution) and LMP1 (clone LMPO24; Novus Biologicals, catalog no. NBP2‐50383; 1:60 dilution) using FoxP3/Transcription factor staining buffer set (eBioscience, catalog no. 5523) as per manufacturer's instructions. Data were analyzed using FlowJo and cumulated using GraphPad PRISM software.
4. RESULTS
4.1. Single cell RNA‐sequencing analyses resolve LCLs into three distinct populations
To better understand how EBV in infected cells spontaneously enter the lytic life cycle, we analyzed publicly available single cell RNA‐sequencing (scRNA‐seq) data from nine LCLs from several independent sources (see Section 5). 34 , 40 , 41 , 42 , 43 , 44 Briefly, we performed an unbiased integrative analysis across all these LCLs after regressing for potential batch effects, doublets and/or artifacts and known sources of heterogeneity, such as the stage of cell cycle using the Seurat platform 45 (Supporting Information: Figure S1A–C see Section 5). Unsupervised clustering of all 46 205 cells according to expressions of both host and viral genes at three different resolutions yielded several clusters (Supporting Information: Figure S1D). Further examination of these clusters based on the expression of salient EBV genes, including GP350 and LMP1/BNLF2, and separation in UMAP space revealed that these clusters fall into three major groups according to the status of EBV gene expression, namely latent, early lytic and full lytic EBV cell clusters. The multiple clusters corresponding to EBV in the latent state were recently examined thoroughly. 34 Since our focus was mainly on understanding the biology of EBV lytic life cycle, we combined all the latent cells into one cluster, resulting in three major clusters (Figure 1A). These were evident in all LCL datasets examined (Supporting Information: Figure S1A,B). These clusters contained GP350 – LMP1 lo , GP350 – LMP1 hi and GP350 + LMP1 hi cells, representing cells with EBV in the latent, early lytic and fully lytic states (Figure 1A, Supporting Information: S1E). Approximately 50–100 host genes were differentially expressed in each cluster compared to all other clusters (Figure 1B, Supporting Information: Figure S1F and Table S1). Consistently, GP350 + LMP1 hi cells was the cluster expressing the most EBV genes, while GP350 – LMP1 lo cells represented the cluster showing the lowest expression of EBV genes (Figure 1B–D).
Figure 1.
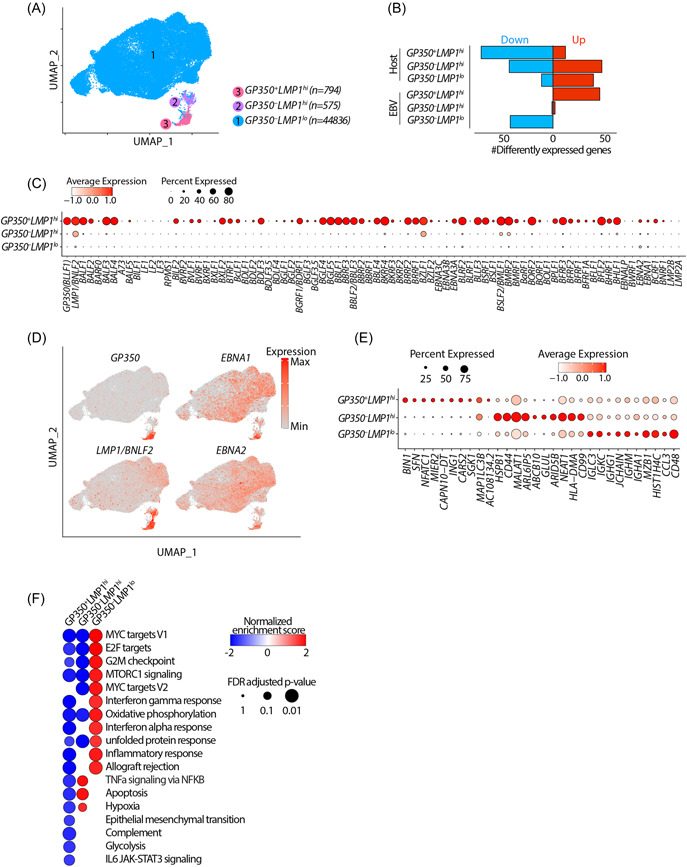
Single cell RNA‐sequencing analyses resolve LCLs into three distinct populations. (A) Integrated UMAP showing 3 major cell types w.r.t. EBV status in nine LCLs used in this study. (B) Numbers of differentially expressed genes (FC > 1.5 and adjusted p < 0.05) in indicated cluster compared to other clusters. (C, D) mRNA expression of EBV genes across all clusters shown as dot plot (C) or projected on the UMAP (D). (E) mRNA expression of top 10 human host cell defining genes across all clusters. (F) Significantly enriched hallmark pathways by geneset enrichment analysis comparing transcriptomes of cells in indicated cluster with all other cells. The positive and negative enrichment scores indicate activation and inactivation of the indicated pathway in each cell cluster, respectively. Only pathways that are enriched (FDR < 5%) in at least one of the clusters are shown. EBV, Epstein‐Barr virus; GEA, geneset enrichment analysis; LCL, lymphoblastoid cell lines; UMAP, Uniform Manifold Approximation and Projection.
GP350 – LMP1 lo cells comprised 94%–98% of all LCLs across all the samples (Figure 1A, Supporting Information: Figure S1A,B. They displayed minimal expression of LMP1/BNLF2 and minimal or no expression of EBV lytic genes, including GP350, BMRF1, BALF1, and BALF3 (Figure 1C). Additionally, these cells expressed latency genes, including EBNA1 and EBNA2, indicating that this cluster mainly consisted of transformed cells that were in the EBV latent phase (Figure 1D). This cluster was also the highest expressor of genes from immunoglobulin heavy or light chains, indicating the mature status of these transformed B cells (Figure 1E, Supporting Information: S1G Consistently, nearly a quarter of cells in this cluster expressed high levels of PRDM1, indicating that these cells might have entered plasmacytic differentiation. 46
The two lytic clusters, GP350 – LMP1 hi and GP350 + LMP1 hi cells, respectively, each accounted for 1%–5% of all LCLs (Figure 1A, Supporting Information: S1B). The salient viral feature of both clusters was the high expression of LMP1/BNLF2, a gene with well‐established contribution to oncogenic human B‐cell transformation 47 and BZLF1. GP350 + LMP1 hi cells were the highest expressors of EBV lytic genes, including GP350, BZLF1, and BMRF1, while GP350 – LMP1 hi cells express very few lytic genes (Figure 1C). Remarkably, these two clusters had distinct expressions of host genes (Figure 1E, Supporting Information: S1F Consistent with previous reports, 34 , 35 GP350 + LMP1 hi cells highly expressed host NFATC1, MIER2, SFN, and SGK1 genes and were the highest expressors of host box‐dependent myc‐interacting protein 1 (BIN1). Conversely, GP350 – LMP1 hi cells had the highest expression of host genes HSPB1, ABCB10, MALAT1, and CD44 (Figure 1E).
To obtain insights into the functional state of cells in each cluster, we performed geneset enrichment analysis (GSEA), comparing the transcriptomes of cells in each cluster against those of cells from the other two clusters (Figure 1F), and querying enrichment of all 50 hallmark genesets curated by the Molecular Signatures Database (MSigDB). 48 Genes differently regulated in GP350 – LMP1 lo cells were significantly enriched in MYC targets, MTORC1 signaling and inflammatory response. Conversely, genes differently regulated in GP350 – LMP1 hi cells were enriched in tumor necrosis factor alpha signaling, apoptosis, and hypoxia (Figure 1F). As expected, genes differently regulated in GP350 + LMP1 hi cells were significantly depleted of genesets from most hallmark pathways, including MYC targets, MTORC1 signaling and interferon responses (Figure 1F, Supporting Information: S1H. This is consistent with the fact that fully lytic EBV reactivation pauses transcription of most host genes and pathways, which is evidenced by significantly reduced numbers of total host transcripts in lytic cells (Supporting Information: Figure S1I).
Collectively these analyses indicated that LCLs are predominantly comprised of three distinct cell populations characterized by differences in expression of both host and viral genes, notably cells containing EBV in the latent phase (GP350 – LMP1 lo ), cells containing virus in the lytic phase (GP350 + LMP1 hi ) and cells that were in between lytic and latent phases (GP350 – LMP1 hi ). Furthermore, these data suggested that distinct functional states of individual LCL clusters may be related to expression of genes encoded by EBV and the host cell.
4.2. GP350–LMP1hi LCLs have a HIF1A‐associated signature
We next explored the transcriptional regulators of host gene expression. Our attention was drawn to HIF1A because GP350 – LMP1 hi cells were high expressors of several genes including HSPB1, MALAT1, and CD44 (Figure 1E) that in other settings are known to be regulated by hypoxia or HIF1‐α 49 , 50 , 51 and because our GSEA analysis had also indicated that the transcriptomes of these cells are highly enriched in the hypoxia gene set (Figure 1F). HIF1‐α is a critically important TF that is tightly regulated by oxygen tension and transactivates many genes essential for cellular responses and adaptation to hypoxia. 52 To better characterizeGP350 – LMP1 hi cells, we therefore quantified the mRNA expression of HIF1A, the gene that encodes HIF1‐α, and its classical direct target PDL1 53 in all clusters. HIF1A and PDL1 were both more highly expressed in GP350 – LMP1 hi cells compared to others (Figure 2A). This was specifically evident for HIF1A as its expression levels were significantly higher in GP350 – LMP1 hi cells (Figure 2A,Supporting Information: S2A To determine whether the changes in HIF1A expression could have any functional consequence, we next assessed the expression of HIF1‐α target genes. We sourced a public list 54 of HIF1‐α–induced (n = 110) and HIF1‐α–repressed (n = 77) genes from MSigDB 55 and assessed the expression of these two sets in all three identified LCL clusters (Figure 2B and Supporting Information: Table S2). GP350 – LMP1 hi cells had the highest and lowest expressions among all clusters for HIF1‐α–induced and HIF1‐α–repressed genes, expressed as the module score, 56 respectively (Figure 2B). We confirmed these findings using two additional independent lists of HIF1‐α–regulated genes 57 (Supporting Information: Figure S2B,C and Table S2).
Figure 2.
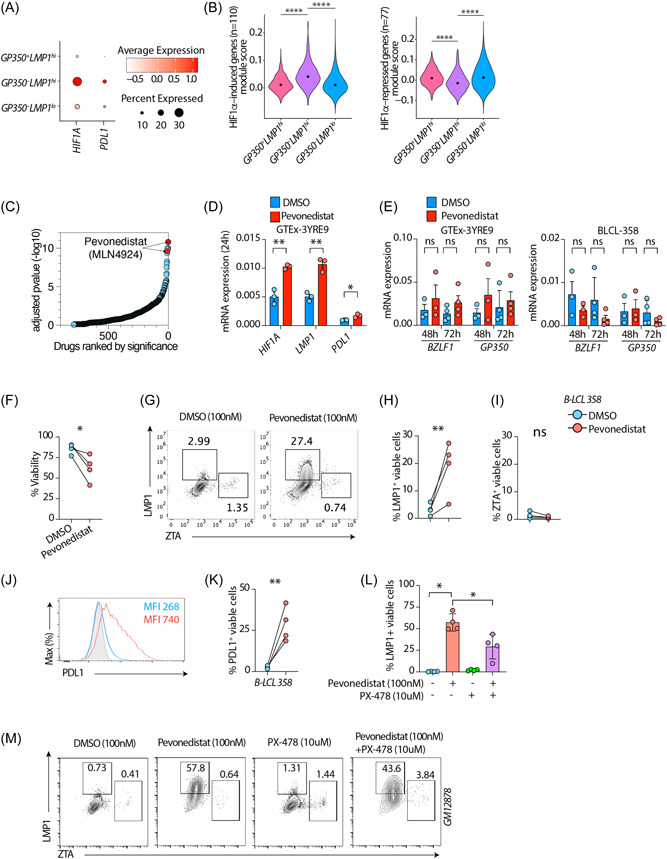
GP350–LMP1hi LCLs have a HIF1A‐associated signature. (A) mRNA expression of HIF1A or CD274 genes across all clusters as dot plot. (B) Module scores of HIF1A induced genes (left panel) or HIF1A‐repressed genes (right panel). HIF1A induced and repressed genes are sourced from MSigDB (M1308). ****p < 0.0001 by two‐tailed Wilcoxon rank‐sum test. (C) Enrichr based drugs predicted (out of 906 total drugs) to counteract genes induced in GP350–LMP1hi LCLs compared to other cells, ordered by adjusted p value. Drugs are sourced from Enrichr library “Drug_Perturbations_from_GEO_down.” (D, E) mRNA expression of indicated host (E) or EBV (E) genes in LCLs treated with 100 nM DMSO or Pevonedistat. UBC was used as a housekeeping gene. (F–K) Flow cytometry on BLCL‐358 treated with 100 nM DMSO or Pevonedistat for 72 h. Plots showing cell viability and LMP1, BZLF1, or PDL1 expression in LCLs treated with DMSO or Pevonedistat. Shown are cumulative %viability plots (F), representative flow cytometry plots (G) and cumulative data showing %LMP1+ (H) and %BZLF1+(I) in gated live LCLs. (J, K) Representative PDL1 expression as mean fluorescent intensity or cumulative %PDL1+ in gated live LCLs. (L, M) Flow cytometry on GM12878 LCLs treated with indicated drug combination for 72 h. PX‐478 treatments were done 1 h before Pevonedistat treatments. Plots showing cumulative LMP1+ (l) or representative flow cytometry plots (M) in gated live cells. Data in (D–M) are from n = 3 or n = 4 independent experiments; gating strategy is shown in Supporting Information: Figure S2D. *p < 0.05; **p < 0.01; ***p < 0.001; ****p < 0.0001 by two‐tailed paired ratio t‐test. EBV, Epstein‐Barr virus; LCL, lymphoblastoid cells.
We next predicted pharmaceutical agents that could induce the unique gene signatures of cells in the GP350 – LMP1 hi cells, using methods established previously by our group. 33 , 58 Among the topmost significant drugs predicted to enhance host gene expression pattern ofGP350 – LMP1 hi cells was Pevonedistat (MLN4924) (Figure 2C). Pevonedistat is a ubiquitin‐activating enzyme E1 inhibitor that significantly stabilizes HIF1‐α to potentiate its function. 59 Because the HIF1‐α pathway was one of the main features ofGP350 – LMP1 hi cells, we hypothesized that enhancing HIF1‐α signaling might preferentially induce this program. To test this hypothesis, we treated LCLs with Pevonedistat and measured HIF1A, LMP1, PDL1, and GP350. HIF1‐α potentiation markedly induced mRNA expression of HIF1A, LMP1, and PDL1 (Figure 2D), but not GP350 or BZLF1 (Figure 2E). To further substantiate these observations at the single cell level, and confirm expression of protein, we treated three different LCLs with Pevonedistat and performed flow cytometry. Pevonedistat reduced cell viability by nearly 30% (Figure 2F, Supporting Information: S2E The frequency of LMP1+ cells was significantly increased (Figure 2G,H,Supporting Information: S2F,G) without increasing that of ZTA (Figure 2I, Supporting Information: S2H). The frequency of PDL1+ cells and PDL1 expression were also significantly increased upon treatment (Figure 2J,K,Supporting Information: S2I Importantly, HIF1‐α inhibitor PX‐478 significantly decreased the frequency of LMP1+ cells induced by Pevonedistat (Figure 2L‐M). The presence and induction of LMP1+ cells were not due to cell clumping (Supporting Information: Figure S3A–C). We have also performed dose titration of Pevonedistat and have observed dose dependent increase of LMP1 and PD‐L1, but not BZLF1, in gated live cells (Supporting Information: Figure S4A,B), suggesting that Pevonedistat preferentially induce LMP1+ cells without increasing full lytic cycle.
4.3. GP350–LMP1lo LCLs have distinct MYC‐dependent transcriptional programs
We next focused on transcriptional regulators of GP350 – LMP1 lo LCLs, the cluster containing EBV in the latent phase. MYC‐regulated genes were among the top affected pathways when comparing transcriptomes of LCL clusters against each other (Figure 1F) and box‐dependent MYC‐interacting protein 1 (BIN1) was one of the top host genes distinguishing GP350 + from GP350 – cells (Figure 1E). Moreover, MYC has been described as a key host factor repressing EBV lytic reactivation. 60 Thus, we further examined the role of MYC in shaping the distinct LCL clusters. Because MYC is a transcription factor, we first determined the fraction of differently expressed genes in each cluster directly bound by MYC. For this, we sourced a publicly available ChIP‐seq data set for MYC in GM12878 (GSM822290, curated by ENCODE). Nearly 18%–24% of genes differently expressed in each cluster were directly bound by MYC, withGP350 + LMP1 hi cluster having the most number of MYC targets (Supporting Information: Figure S5A and Table S2). This was significantly higher than what would be expected by chance because only ∼10% of all human genes are bound by MYC in GM12878 (Supporting Information: Figure S5A).
The mRNA expression of MYC was significantly higher in GP350 – LMP1 lo LCLs than in either of the other two clusters (Supporting Information: Figure S5B). To determine whether MYC is biologically active, we looked for the signature of genes regulated by MYC. We curated a list of genes regulated by MYC in GM12878 from a publicly available data set 60 (Supporting Information: Table S2). Expression of MYC‐induced genes was significantly higher (Supporting Information: Figure S5C, left panel) and MYC‐repressed genes significantly lower (Supporting Information: Figure S5C, right panel) in GP350 – LMP1 lo cells than in the other two clusters. We also performed GSEA comparing transcriptomes of cells from each cluster against MYC targets curated by MSigDB. 48 Consistent with our earlier observation (Figure 1F), genes that were more highly expressed in GP350 – LMP1 lo cells were highly enriched in MYC targets (Supporting Information: Figure S5D, left and right panels), while there was no significant difference between LMP1hi clusters (Supporting Information: Figure S5D, middle panel). Collectively, these data indicated that MYC preferentially regulates a subset of genes that are differently expressed in GP350 – LMP1 lo LCLs.
4.4. Super‐enhancer‐regulated genes are less highly expressed in GP350±LMP1hi LCLs
EBV‐infected cells periodically enter the lytic phase to produce progeny viruses but in EBV‐immortalized lymphoblastoid cells EBV is mostly in the latent state. Earlier studies have shown that a small percentage of these cells are lytic. 31 However, due to the technical challenges at the time, it was difficult to distinguish the cells in lytic phase from cells at latency phase in a mixed population. The recent development of scRNA‐seq techniques allows us to capture the cells in lytic phase together with their transcriptome.
Nearly 10% of genes are regulated by multiple enhancers forming a complex architecture known as “super‐enhancers” (SEs). SE‐regulated genes are critically important for cell identity 28 and are associated with both Mendelian and polygenic diseases 61 , 62 as well as cancers. 63 Enhancer‐promoter interactions are the cornerstones of mammalian gene regulation. We have previously shown that EBV episomes make reproducible contacts with the human genome at SE loci. 24 To explore whether EBV disrupts modes of gene regulation in the three LCL subsets, we sourced a list of 257 annotated SE regulated genes from GM12878 27 and determined whether these genes are differently expressed in the three identified LCL clusters. Unexpectedly, expression of SE‐regulated genes, summarized as the module score, was significantly lower in GP350 + LMP1 hi cells, the cluster containing EBV in the lytic state, than in the other two subsets (Figure 3A). We observed similar results when we used an independent curated set of 187 EBV‐associated SEs 23 (Supporting Information: Figure S6A). Examples of such genes included MYC, which contains one of the largest SEs in the genome, 23 IRF4, RUNX3, PAX5, IKZF3, and DUSP22 (Figure 3B). Similarly, when through GSEA analysis we found that genes less highly expressed in GP350 + LMP1 + cells compared to the cells in the other two clusters were markedly enriched in SE‐regulated genes (Supporting Information: Figure S6B).
Figure 3.
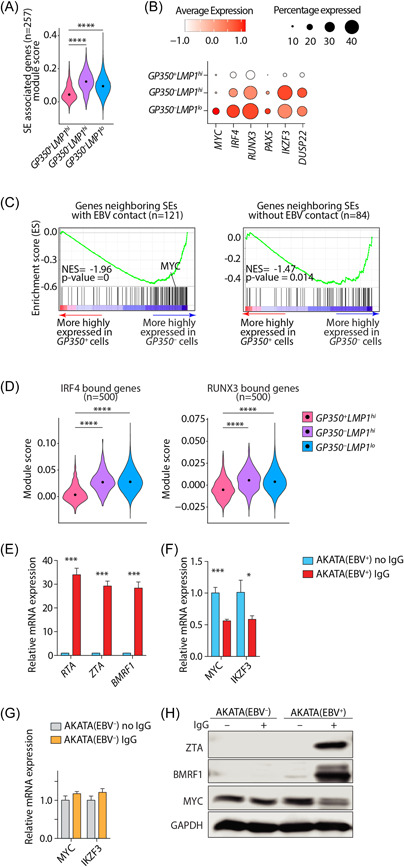
Super‐enhancer‐regulated genes are less highly expressed in GP350 + LCLs. (A) Module score of SE containing genes in GM12878. SEs and their annotation are sourced from. 27 (B) mRNA expression of select SE containing genes across all cell types. (C) geneset enrichment analysis plots comparing transcriptomes of GP350 + and GP350 – cells for enrichment in genes neighboring SEs with (left panel) or without (right panel) EBV contacts. (D) Module score of n = 500 top IRF4 (left panel) and RUNX3 (right panel) bound genes. ****p < 0.0001 by two‐tailed Wilcoxon rank‐sum test. (E, F) mRNA expression of indicated EBV (E) or host (F) genes in EBV+‐AKATA cells with or without anti‐IgG (1:200) treatment after 48 h. (G) Control EBV–‐AKATA cells are included when measuring host genes. Data are from n = 3 independent experiments. *p < 0.05; **p < 0.01; ***p < 0.001 by two‐tailed unpaired t‐test. (H) Western blots of lysates of AKATA cells treated with or without anti‐IgG after 48 h. Shown are representative (n = 2) images of ZTA (BZLF1), BMRF1 and MYC with GAPDH as loading control. EBV, Epstein‐Barr virus; LCL, lymphoblastoid cells; NES, normalized enrichment scores.
We next assessed whether genes in these clusters were differently expressed when their associated SEs physically interact with EBV episomes. To this end, we divided SE‐regulated genes into those that physically interact, or not, with EBV episomes and performed GSEA analysis comparing GP350 + cells to the cells of other two GP350 – subsets. Genes that were more highly expressed in GP350 – cells were significantly enriched in SEs that interact with EBV episomes (Figure 3C, left panel). This enrichment was less evident for SEs that do not interact with EBV episomes (Figure 3C, right panel). To determine the functional consequences of differential expression of SE‐regulated genes across LCL clusters, we focused on the transactivator IRF4 and transcription factor RUNX3 for which we could source their direct targets from ChIP‐seq experiments and assess the expression of their targets. We noted that expression of both IRF4‐ and RUNX3‐bound genes, summarized as the module score, was significantly lower in GP350 + LMP1 hi cells (Figure 3D, Supporting Information: S6C in which expression of both these TFs was also the lowest (Figure 3B).
Since GP350 + LMP1 hi cells represented the cluster in which lytic reactivation of EBV was apparent (Figure 1C), we tested whether EBV reactivation affects the expression of SE containing genes. To this end, we treated EBV+ AKATA cells with either anti‐IgG or carrier. Anti‐IgG is a potent inducer of EBV lytic reactivation in these cells. 64 After stimulation, we measured mRNA and/or protein expression of EBV lytic markers and the host SE‐regulated geneMYC (Figure 3E–H). As expected, anti‐IgG induced strong expression of the EBV lytic markers RTA, ZTA, and BMRF1 (Figure 3E). In contrast, the expression of both MYC and IKZF3 were significantly repressed following anti‐IgG‐treatment of cells (Figure 3F). This effect was specifically a predicate of EBV‐reactivation since anti‐IgG did not repress MYC or IKZF3 expression in EBV– AKATA cells (Figure 3G). These observations were further confirmed by immunoblots of ZTA, BMRF1 and MYC proteins (Figure 3H). Similarly, chemical EBV reactivation of LCLs by doxorubicin significantly reduced MYC expression (Supporting Information: Figure S6D). To test whether depletion of IRF4 can similarly reactivate EBV lytic cycle, we reanalyzed RNA‐seq from GM12878 LCLs that were subjected to IRF4 deletion via the CRISPR/Cas9 system. 30 In this setting, depletion ofIRF4 induced multiple EBV lytic genes, including GP350, RTA, ZTA, and BMRF1 (Supporting Information: Figure S6E). Collectively, our data suggest that SE‐regulated genes are less highly expressed in GP350 + LMP1 hi LCLs, which show evidence of lytic EBV reactivation, and that experimental induction of EBV lytic cycle also represses expression of these genes.
4.5. Disruption of super‐enhancers in LCLs induces EBV lytic reactivation
The reciprocal relationship between expression of SE‐regulated genes in GP350 + LMP1 hi LCLs and EBV reactivation suggested the possibility that SEs may be necessary for maintenance of EBV in the latent phase. To test this possibility, we initially selected SEs near MYC and IRF4/DUSP22 and performed CRISPR‐mediated knockout or inactivation and then measured the expression of EBV lytic markers. For these experiments, appropriate guide RNAs were situated within the SEs at sites of maximal H3K27ac signal, a marker of active regions of the genome, especially promoters and enhancers. The selected sites were bound by one or more viral transcription factors (e.g., EBNA2, LP, 3A, and 3C) and/or host NF‐κB family members (e.g., RelA, RelB, cRel, p50, and p52) and interacted within a topologically associated domain that contained the SE (Figure 4A–C). Dual sgRNAs targeting both sides of MYC SE (∼525 kb upstream) successfully deleted MYC SE from the genome (Supporting Information: Figure S7A), which led to a reduction of MYC transcription and upregulation of EBV lytic genes, namely ZTA, RTA, BGLF5, and BMRF1 expression (Figure 4B). Similarly, inactivation of IRF4 SE by CRISPR‐dcas9 tethered with a repressor consisting of KRAB and the transcription repression domain of MeCP2 successfully reduced IRF4 SE activity (Supporting Information: Figure S7b). This led to decrease in IRF4/DUSP22 mRNA expression and a significant increase of EBV lytic gene expression 2 days postinactivation (Figure 4D). Deletion of both MYC and IRF4 genes have been previously shown to induce EBV lytic phase. Unexpectedly, however, when we measured the expression of EBV lytic genes at earlier time points, we observed that EBV lytic genes were significantly induced before decrease of IRF4 (Supporting Information: Figure S7C). This suggests the possibility that SE might be also necessary for the maintenance of EBV latency. To further explore this possibility, we selected another SE in the same topological associated domain of MIR155HG (Figure 4E), using the same criteria as above and performed CRISPR‐mediated inactivation. The disruption of this SE did not significantly reduce the expression of MIR155HG; however, it significantly increased the expression of EBV lytic genes (Figure 4F), namely ZTA, RTA, BDLF5, and BMRF1. CRISPRi disruption of RUNX3 SE also had similar activity (Figure 4G,H). Collectively these data indicate that deletion of select host SEs leads to lytic reactivation of EBV and, by extension, that host SEs, or their target genes, are necessary for maintenance of EBV in the latent phase.
Figure 4.
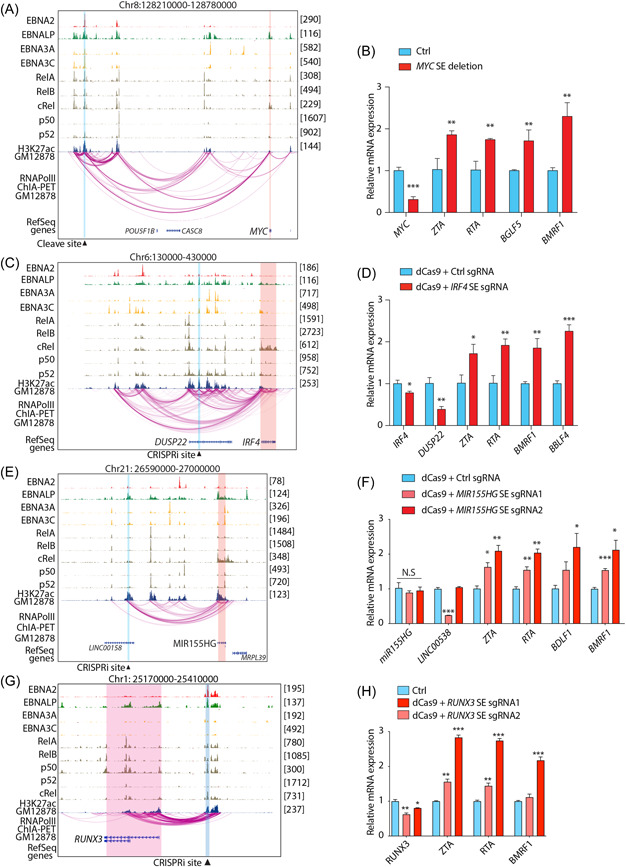
Disruption of super‐enhancers in LCLs induces EBV lytic reactivation. Genome browser tracks showing EBV transcription factors (i.e., EBNA2, EBNALP, EBNA3A, and EBNA3C), host transcription factors (i.e., RELA, RELB, c‐REL, p50, and p52) and H3K27ac at indicated loci. The CRISPR cleavage/inactivation site is highlighted with vertical blue box. The expected affected target genes are highlighted by vertical red box. Bar plots showing mRNA expression of indicated host and EBV genes after CRISPR mediated inactivation of specified loci in GM12878 cells. Shown are MYC (A, B), IRF4/DUSP22 (C, D), MIR155HG (E, F) and RUNX3 (G, H) loci. Data are from n = 3 independent experiments. *p < 0.05; **p < 0.01; ***p < 0.001 by two‐tailed unpaired t‐test. EBV, Epstein‐Barr virus; LCL, lymphoblastoid cells.
5. DISCUSSION
LCLs have been instrumental for genetic and functional studies of human diseases over the past several decades. 65 We and others have previously analyzed large numbers of LCL bulk RNA‐seq data and found that EBV lytic gene expression correlates with cellular cancer‐associated pathways, such as interferon‐alpha, WNT, and B cell receptor signaling. 4 , 31 However, these data were generated from bulk populations of cells, which biases insights towards those occurring in the largest subpopulations. While the majority of LCLs contain EBV in the latent phase of its life cycle, a small fraction (<5%) demonstrate spontaneous EBV reactivation, indicating that LCLs as a whole are heterogeneous. Important aspects of LCL heterogeneity have recently been explored using single cell RNA‐sequencing. 34 This analysis focused on heterogeneity within and across LCLs with respect to immunoglobulin isotypes, which further associated with pathways involving activation and differentiation of B cells. We have taken an integrative approach to combine the same data with several more data sets that are generated across different conditions and eliminate batch and technical effects. This integrative analysis provides a consistent representation of the data for downstream analyses and thus has the potential to uncover previously undetected biology. Specifically, we found LCLs to have higher heterogeneity in relation to the EBV status than previously appreciated. We identified three prominent clusters that were marked by the expression of the EBV genesGP350 and LMP1/BNLF2. It is important to note that in the latent phase, EBV genes are either not expressed or expressed at very low levels (e.g., EBNA2), making them highly susceptible to “dropout” in single cell RNA‐seq, a phenomenon by which a given transcript is detected in only some, but not all, of otherwise homogenous cells within a cluster. Moreover, the EBV genome has extensive numbers of overlapping genes such as LMP1 and BNLF2a/b, making the quantification of their mRNA expression more challenging. 66 This challenge could be further exacerbated by the 3‘ mRNA capture bias in some of the current single cell technologies. Nevertheless, we showed that these clusters have distinct transcriptional programs and identified MYC and HIF1‐α as transcriptional regulators of gene expression.
GP350+ LCLs had high expression of several genes. Some of these genes could be biomarkers and some could be the drivers of GP350+ cells. For example, BIN1 was identified as a tumor suppressor and was shown to inhibit cell proliferation via both Myc‐dependent and Myc‐independent mechanisms. 67 , 68 Moreover, BIN1 implicated in the apoptosis function of MYC. 69 Since BIN1 binds to MYC, it is conceivable that BIN1 may affect lytic replication by modulating MYC activity. Indeed, BIN1 inhibits activation by MYC. 68 However, whether it promotes EBV reactivation is unknown. MIER2 largely unexplored protein coding gene that is predicted to be a transcriptional repressor. Interestingly, it happens to be a “hit” in a genome‐wide CRISPR screen for candidate genes that affect EBV lytic replication, 60 suggesting that it might directly affect EBV reactivation. NFATC1 itself does not reactivate EBV but in combination with Calcium/calmodulin‐dependent protein kinase type IV promotes EBV lytic reactivation. 70 Importantly, LCLs in theGP350 + cluster expressed SE‐regulated genes at significantly lower levels compared to cells in the other two clusters. Physical interactions between SE‐containing loci and EBV episomes marked genes in GP350 + LCLs that were particularly lowly expressed. Indeed, in proof‐of‐principle experiments we found that experimental lytic reactivation of EBV disrupted expression of SE‐regulated genes and, conversely, that disruption of SEs induced EBV lytic reactivation. For IRF4 or MIR155HG associated SEs, lytic reactivation after SE disruption occurred before IRF4 downregulation, suggesting that these SEs themselves might be necessary for the maintenance of EBV latency. However, further studies including the ectopic expression of these genes are needed to fully discern this observation.
In the largest subset of LCLs, annotated as GP350 – LMP1 lo , EBV was clearly in the latent phase. This cluster showed a host gene signature enriched in MYC‐regulated targets. As an oncogene, MYC is exquisitely carefully regulated by an archetypal SE. MYC itself binds to the EBV genome origin of lytic replication and suppresses DNA looping to the promoter of the lytic cycle initiator gene BZLF1. 60 MYC depletion reactivates the lytic cycle in different cells. 60 Consistent with this, when we deleted theMYC SE, MYC expression was decreased and EBV lytic genes were induced, supporting the role of MYC as a repressor of EBV lytic activation. Thus, it appears that both the MYC gene and its associated SE have a role in maintenance of EBV latency. It is evident that these latent LCLs have high expression of multiple immunoglobulin genes suggesting their maturation and increased antibody production. The induced mTORC and unfolded protein response pathways are essential for development of antibody‐secreting cells, 71 , 72 consistent with the observed induction of these pathways.
Another GP350‐negative cluster, characterized by high expression of LMP1/BNLF2, was the highest expressor of several host genes including HSPB1, MALAT1, and CD44 that are known features of cancer stem cells, 73 , 74 , 75 inhibit apoptosis, 76 , 77 , 78 and suppress cell senescence. 79 , 80 , 81 Interestingly, LMP1 alone induces CSC features in nasopharyngeal cell lines. 82 However, such characteristics have not previously been ascribed to LCLs and warrant further investigation. LMP1 is a known oncogene and expressed in most EBV‐associated cancers 83 and has been previously associated with synthesis of HIF1‐α protein and its DNA binding activity. 84 Here, we also found thatGP350 – LMP1 hi LCLs have a prominent HIF1‐α signature, which could be preferentially induced by Pevonedistat. Moreover, Pevonedistat did not change BZLF1 expression in the LCLs, consistent with the recent observation that Pevonedistat fails to induce BZLF1 in AKATA and an LCL. 85 GP350 – LMP1 hi LCLs also expressed higher frequencies of PDL1, which were markedly increased upon Pevonedistat treatment. Interestingly, a recent study has found an association between numbers of PDL1 expressing B cells and the development of AIDS related non‐Hodgkin lymphoma. 86 Such PDL1 expressing B cells have previously been described to suppress effector function of immune cells. 87 Thus, the identification of these cells might play an important role in understanding the oncogenesis and may suggest that drugs that stabilize HIF1‐α might inadvertently induce LMP1 in diseases associated with EBV type III latency programs such as AIDS‐associated B cell lymphoma, post‐transplant lymphoproliferative disorder and diffuse large B cell lymphoma.
B cell differentiation into plasma cell has been linked to EBV lytic replication. 88 , 89 Specifically, PRDM1, a known driver of B cell differentiation into plasma cells, 90 promotes EBV lytic replication by activating the transcription from immediate early gene promoters of ZTA and RTA. 81 A recent single cell RNA‐seq analysis of LCLs have revealed a positive correlation between specific immunoglobulin isotype and cell differentiation markers. 34 However, these immunoglobin genes were not specifically characterized in lytic cells. We have found that the mRNA expression of PRDM1 and a range of immunoglobulin genes in GP350 + LMP1 hi cells was lower than in latent cells, which contrasts with previous reports about the role of PRDM1 in EBV lytic reactivation (Supporting Information: Figure S1E). It is possible that PRDM1 is important for initiation, but not maintenance, of the lytic cycle. Another possibility is that the transcription factor activity, but not the overall expression level, of PRDM1 is important for lytic replication. Further study is clearly required to delineate this relationship.
In summary, we performed integrative analysis of publicly available single cell RNA‐seq data from different LCLs to help resolve their heterogeneity. We identified a novel cluster of cells that are between lytic and latent stage, marked by LMP1 and controlled by HIF1a. We also found that the mRNA expression of super‐enhancer target genes is inversely correlated with lytic status of the cells and consistently CRISPR perturbation of super‐enhancers increased the expression of EBV lytic genes. Our studies revealed EBV‐associated heterogeneity among LCLs that contribute to EBV life cycle and biology.
AUTHOR CONTRIBUTIONS
Bingyu Yan, Zonghao Zhang, Simran D. Kadadi, Luopin Wang, and Minwoo Jung performed computational work. Chong Wang, Srishti Chakravorty, Isabella Sirit, Yuxin Zhuang, Yonghua Hu, Subhransu S. Sahoo, Kunming Shao, and Nicole L. Anderson performed experimental work. All other authors contributed significantly to computational, experimental and/or conceptual development of this work. Bo Zhao and Majid Kazemian conceptualized the study, supervised the project and wrote the manuscript.
CONFLICT OF INTEREST
The authors declare no conflict of interest.
Supporting information
Supporting information.
Supporting information.
Supporting information.
Supporting information.
Supporting information.
ACKNOWLEDGMENTS
This work was supported by extramural research programs of the NIH (R35GM138283 to M. K. and 5R01AI123420 and 5R01CA047006 to B. Z. and AI136995 to S. B.) and the Showalter Trust (research award to M. K.). This research was supported (in part) by the Intramural Research Programs of the National Institute of Diabetes and Digestive and Kidney Diseases (project number ZIA/DK075149 to B. A.). The authors also gratefully acknowledge the SIRG Graduate Research Assistantships Award to B. Y. and S. C. and support from the Purdue University Center for Cancer Research, P30CA023168, as well as Purdue college of Agriculture Bilsland Fellowship to B. Y.
Yan B, Wang C, Chakravorty S, et al. A comprehensive single cell data analysis of lymphoblastoid cells reveals the role of super‐enhancers in maintaining EBV latency. J Med Virol. 2022;95:e28362. 10.1002/jmv.28362
Bingyu Yan, Chong Wang, Srishti Chakravorty, Zonghao Zhang, and Simran D. Kadadi contributed equally to this study.
Contributor Information
Bo Zhao, Email: bzhao@bwh.harvard.edu.
Majid Kazemian, Email: kazemian@purdue.edu.
DATA AVAILABILITY STATEMENT
The single cell RNA‐seq data are sourced from GSE126321 for GM12878 and GM18502; GSE111912 for GM12891; GSE158275 for LCL777B958, LCL777M81, and LCL461B958; GSE162528 for LCL; and GSE121926 for GM22648 and GM22649. The ChIA‐PET in GM12878 is from GSE127053. The ChIP‐seq data are from EBNA2: GSE29498; EBNALP: GSE49338; EBNA3A: GSM1429820; EBNA3C: GSE52632; NF‐κB: GSE55105 and H3K27Ac: GSM733771.
REFERENCES
- 1. Epstein MA, Achong BG, Barr YM. Virus particles in cultured lymphoblasts from Burkitt's lymphoma. Lancet. 1964;283:702‐703. [DOI] [PubMed] [Google Scholar]
- 2. Cohen JI, Fauci AS, Varmus H, Nabel GJ. Epstein‐Barr virus: an important vaccine target for cancer prevention. Sci Transl Med. 2011;3:107fs7. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 3. Howley PM, Knipe DM, Cohen JL, Damania BA. Fields Virology: DNA Viruses. Wolters Kluwer Health; 2021. [Google Scholar]
- 4. Chakravorty S, Yan B, Wang C, et al. Integrated pan‐cancer map of EBV‐associated neoplasms reveals functional host‐v irus interactions. Cancer Res. 2019;79:6010‐6023. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 5. Chakravorty S, Afzali B, Kazemian M. EBV‐associated diseases: current therapeutics and emerging technologies. Front Immunol. 2022;13:13. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 6. Cameron JE, Fewell C, Yin Q, et al. Epstein‐Barr virus growth/latency III program alters cellular microRNA expression. Virology. 2008;382:257‐266. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 7. Young LS, Yap LF, Murray PG. Epstein‐Barr virus: more than 50 years old and still providing surprises. Nat Rev Cancer. 2016;16:789‐802. [DOI] [PubMed] [Google Scholar]
- 8. Lieberman PM. Epstein‐Barr virus turns 50. Science. 2014;343:1323‐1325. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 9. Pope JH, Horne MK, Scott W. Transformation of foetal human leukocytesin vitro by filtrates of a human leukaemic cell line containing herpes‐like virus. Int J Cancer. 1968;3:857‐866. [DOI] [PubMed] [Google Scholar]
- 10. Knipe DM, Howley PM. Fields Virology. 6th ed. Wolters Kluwer/Lippincott Williams & Wilkins Health; 2013. [Google Scholar]
- 11. Pei Y, Wong JH, Robertson ES. Herpesvirus epigenetic reprogramming and oncogenesis. Annu Rev Virol. 2020;7:309‐331. [DOI] [PubMed] [Google Scholar]
- 12. Frappier L. Ebna1. Curr Top Microbiol Immunol. 2015;391:3‐34. [DOI] [PubMed] [Google Scholar]
- 13. Saridakis V, Sheng Y, Sarkari F, et al. Structure of the p53 binding domain of HAUSP/USP7 bound to Epstein‐Barr nuclear antigen 1. Mol Cell. 2005;18:25‐36. [DOI] [PubMed] [Google Scholar]
- 14. Wu H, Ceccarelli DFJ, Frappier L. The DNA segregation mechanism of Epstein‐Barr virus nuclear antigen 1. EMBO Rep. 2000;1:140‐144. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 15. Kaiser C, Laux G, Eick D, Jochner N, Bornkamm GW, Kempkes B. The proto‐oncogene c‐myc is a direct target gene of Epstein‐Barr virus nuclear antigen 2. J Virol. 1999;73:4481‐4484. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 16. Portal D, Zhao B, Calderwood MA, Sommermann T, Johannsen E, Kieff E. EBV nuclear antigen EBNALP dismisses transcription repressors NCoR and RBPJ from enhancers and EBNA2 increases NCoR‐deficient RBPJ DNA binding. Proc Natl Acad Sci. 2011;108:7808‐7813. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 17. Maruo S, Zhao B, Johannsen E, Kieff E, Zou J, Takada K. Epstein‐Barr virus nuclear antigens 3C and 3A maintain lymphoblastoid cell growth by repressing p16INK4A and p14ARF expression. Proc Natl Acad Sci. 2011;108:1919‐1924. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 18. Skalska L, White RE, Parker GA, Sinclair AJ, Paschos K, Allday MJ. Induction of p16(INK4a) is the major barrier to proliferation when Epstein‐Barr virus (EBV) transforms primary B cells into lymphoblastoid cell lines. PLoS Pathog. 2013;9:e1003187. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 19. Paschos K, Smith P, Anderton E, Middeldorp JM, White RE, Allday MJ. Epstein‐barr virus latency in B cells leads to epigenetic repression and CpG methylation of the tumour suppressor gene Bim. PLoS Pathog. 2009;5:e1000492. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 20. Laherty CD, Hu HM, Opipari AW, Wang F, Dixit VM. The Epstein‐Barr virus LMP1 gene product induces A20 zinc finger protein expression by activating nuclear factor kappa B. J Biol Chem. 1992;267:24157‐24160. [PubMed] [Google Scholar]
- 21. Okabe A, Huang KK, Matsusaka K, et al. Cross‐species chromatin interactions drive transcriptional rewiring in Epstein‐Barr virus‐positive gastric adenocarcinoma. Nature Genet. 2020;52:919‐930. [DOI] [PubMed] [Google Scholar]
- 22. McClellan MJ, Wood CD, Ojeniyi O, et al. Modulation of enhancer looping and differential gene targeting by Epstein‐Barr virus transcription factors directs cellular reprogramming. PLoS Pathog. 2013;9:e1003636. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 23. Zhou H, Schmidt SCS, Jiang S, et al. Epstein‐Barr virus oncoprotein super‐enhancers control B cell growth. Cell Host Microbe. 2015;17:205‐216. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 24. Wang L, Laing J, Yan B, et al. Epstein‐Barr virus episome physically interacts with active regions of the host genome in lymphoblastoid cells. J Virol. 2020;94:94. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 25. Buschle A, Mrozek‐Gorska P, Cernilogar FM, et al. Epstein‐Barr virus inactivates the transcriptome and disrupts the chromatin architecture of its host cell in the first phase of lytic reactivation. Nucleic Acids Res. 2021;49:3217‐3241. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 26. Tempera I, Klichinsky M, Lieberman PM. EBV latency types adopt alternative chromatin conformations. PLoS Pathog. 2011;7:e1002180. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 27. Hnisz D, Abraham BJ, Lee TI, et al. Super‐enhancers in the control of cell identity and disease. Cell. 2013;155:934‐947. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 28. Whyte WA, Orlando DA, Hnisz D, et al. Master transcription factors and mediator establish super‐enhancers at key cell identity genes. Cell. 2013;153:307‐319. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 29. Jiang S, Zhou H, Liang J, et al. The Epstein‐Barr virus regulome in lymphoblastoid cells. Cell Host Microbe. 2017;22:561‐573. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 30. Ma Y, Walsh MJ, Bernhardt K, et al. CRISPR/Cas9 screens reveal Epstein‐Barr virus‐transformed B cell host dependency factors. Cell Host Microbe. 2017;21:580‐591. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 31. Arvey A, Tempera I, Tsai K, et al. An atlas of the Epstein‐Barr virus transcriptome and epigenome reveals host‐virus regulatory interactions. Cell Host Microbe. 2012;12:233‐245. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 32. Yan B, Chakravorty S, Mirabelli C, et al. Host‐virus chimeric events in SARS‐CoV‐2‐infected cells are infrequent and artifactual. J Virol. 2021;95:e0029421. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 33. Yan B, Freiwald T, Chauss D, et al. SARS‐CoV‐2 drives JAK1/2‐dependent local complement hyperactivation. Sci Immunol. 2021;6:6. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 34. SoRelle ED, Dai J, Bonglack EN, et al. Single‐cell RNA‐seq reveals transcriptomic heterogeneity mediated by host‐pathogen dynamics in lymphoblastoid cell lines. eLife. 2021;10:e62586. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 35. Bristol JA, Brand J, Ohashi M, et al. Reduced IRF4 expression promotes lytic phenotype in Type 2 EBV‐infected B cells. PLoS Pathog. 2022;18:e1010453. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 36. Sanjana NE, Shalem O, Zhang F. Improved vectors and genome‐wide libraries for CRISPR screening. Nat Methods. 2014;11:783‐784. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 37. Zheng GXY, Terry JM, Belgrader P, et al. Massively parallel digital transcriptional profiling of single cells. Nat Commun. 2017;8:14049. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 38. Hao Y, Hao S, Andersen‐Nissen E, et al. Integrated analysis of multimodal single‐cell data. Cell. 2021;184:3573‐3587. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 39. Oki S, Ohta T, Shioi G, et al. ChIP‐Atlas: a data‐mining suite powered by full integration of public ChIP‐seq data. EMBO Rep. 2018;19:e46255. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 40. Osorio D, Yu X, Yu P, Serpedin E, Cai JJ. Single‐cell RNA sequencing of a European and an African lymphoblastoid cell line. Sci Data. 2019;6:112. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 41. Ozgyin L, Horvath A, Hevessy Z, Balint BL. Extensive epigenetic and transcriptomic variability between genetically identical human B‐lymphoblastoid cells with implications in pharmacogenomics research. Sci Rep. 2019;9:4889. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 42. Sokka J, Yoshihara M, Kvist J, et al. CRISPR activation enables high‐fidelity reprogramming into human pluripotent stem cells. Stem Cell Reports. 2022;17:413‐426. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 43. Zhang X, Li T, Liu F, et al. Comparative analysis of droplet‐based ultra‐high‐throughput single‐cell RNA‐seq systems. Mol Cell. 2019;73:130‐142. [DOI] [PubMed] [Google Scholar]
- 44. Osorio D, Yu X, Zhong Y, et al. Single‐cell expression variability implies cell function. Cells. 2019;9(1):14. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 45. Hafemeister C, Satija R. Normalization and variance stabilization of single‐cell RNA‐seq data using regularized negative binomial regression. Genome Biol. 2019;20:296. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 46. Tunyaplin C, Shaffer AL, Angelin‐Duclos CD, Yu X, Staudt LM, Calame KL. Direct repression of prdm1 by Bcl‐6 inhibits plasmacytic differentiation. J Immun. 2004;173:1158‐1165. [DOI] [PubMed] [Google Scholar]
- 47. Kaye KM, Izumi KM, Kieff E. Epstein‐Barr virus latent membrane protein 1 is essential for B‐lymphocyte growth transformation. Proc Natl Acad Sci. 1993;90:9150‐9154. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 48. Liberzon A, Birger C, Thorvaldsdóttir H, Ghandi M, Mesirov JP, Tamayo P. The molecular signatures database hallmark gene set collection. Cell Systems. 2015;1:417‐425. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 49. Salle‐Lefort S, Miard S, Nolin MA, et al. Hypoxia upregulates Malat1 expression through a CaMKK/AMPK/HIF‐1alpha axis. Int J Oncol. 2016;49:1731‐1736. [DOI] [PubMed] [Google Scholar]
- 50. Liang G, Li S, Du W, Ke Q, Cai J, Yang J. Hypoxia regulates CD44 expression via hypoxia‐inducible factor‐1α in human gastric cancer cells. Oncol Lett. 2017;13:967‐972. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 51. Whitlock NA, Agarwal N, Ma JX, Crosson CE. Hsp27 upregulation by HIF‐1 signaling offers protection against retinal ischemia in rats. Investig Ophthalmol Vis Sci. 2005;46:1092‐1098. [DOI] [PubMed] [Google Scholar]
- 52. Wang GL, Jiang BH, Rue EA, Semenza GL. Hypoxia‐inducible factor 1 is a basic‐helix‐loop‐helix‐PAS heterodimer regulated by cellular O2 tension. Proc Natl Acad Sci. 1995;92:5510‐5514. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 53. Noman MZ, Desantis G, Janji B, et al. PD‐L1 is a novel direct target of HIF‐1α, and its blockade under hypoxia enhanced MDSC‐mediated T cell activation. J Exp Med. 2014;211:781‐790. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 54. Gross C, Dubois‐Pot H, Wasylyk B. The ternary complex factor Net/Elk‐3 participates in the transcriptional response to hypoxia and regulates HIF‐1α. Oncogene. 2008;27:1333‐1341. [DOI] [PubMed] [Google Scholar]
- 55. Subramanian A, Tamayo P, Mootha VK, et al. Gene set enrichment analysis: a knowledge‐based approach for interpreting genome‐wide expression profiles. Proc Natl Acad Sci. 2005;102:15545‐15550. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 56. Tirosh I, Izar B, Prakadan SM, et al. Dissecting the multicellular ecosystem of metastatic melanoma by single‐cell RNA‐seq. Science. 2016;352:189‐196. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 57. Elvidge GP, Glenny L, Appelhoff RJ, Ratcliffe PJ, Ragoussis J, Gleadle JM. Concordant regulation of gene expression by hypoxia and 2‐oxoglutarate‐dependent dioxygenase inhibition. J Biol Chem. 2006;281:15215‐15226. [DOI] [PubMed] [Google Scholar]
- 58. Chauss D, Freiwald T, McGregor R, et al. An autocrine vitamin D‐driven Th1 shutdown program can be exploited for COVID‐19. Nature Immunol. 2022;23:62‐74. 10.1101/2020.07.18.210161 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 59. Ryu JH, Li SH, Park HS, Park JW, Lee B, Chun YS. Hypoxia‐inducible factor α subunit stabilization by NEDD8 conjugation is reactive oxygen species‐dependent. J Biol Chem. 2011;286:6963‐6970. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 60. Guo R, Jiang C, Zhang Y, et al. MYC controls the Epstein‐Barr virus lytic switch. Mol Cell. 2020;78:653‐669. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 61. Afzali B, Grönholm J, Vandrovcova J, et al. BACH2 immunodeficiency illustrates an association between super‐enhancers and haploinsufficiency. Nature Immunol. 2017;18:813‐823. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 62. Vahedi G, Kanno Y, Furumoto Y, et al. Super‐enhancers delineate disease‐associated regulatory nodes in T cells. Nature. 2015;520:558‐562. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 63. Lovén J, Hoke HA, Lin CY, et al. Selective inhibition of tumor oncogenes by disruption of super‐enhancers. Cell. 2013;153:320‐334. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 64. Yuan J, Cahir‐McFarland E, Zhao B, Kieff E. Virus and cell RNAs expressed during Epstein‐Barr virus replication. J Virol. 2006;80:2548‐2565. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 65. Hussain T, Mulherkar R. Lymphoblastoid cell lines: a continuous in vitro source of cells to study carcinogen sensitivity and DNA repair. Int J Mol Cell Med. 2012;1:75‐87. [PMC free article] [PubMed] [Google Scholar]
- 66. Casco A, Gupta A, Hayes M, Djavadian R, Ohashi M, Johannsen E. Accurate quantification of overlapping herpesvirus transcripts from RNA sequencing data. J Virol. 2022;96:e0163521. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 67. Sakamuro D, Elliott KJ, Wechsler‐Reya R, Prendergast GC. BIN1 is a novel MYC‐interacting protein with features of a tumour suppressor. Nature Genet. 1996;14:69‐77. [DOI] [PubMed] [Google Scholar]
- 68. Elliott K, Sakamuro D, Basu A, et al. Bin1 functionally interacts with Myc and inhibits cell proliferation via multiple mechanisms. Oncogene. 1999;18:3564‐3573. [DOI] [PubMed] [Google Scholar]
- 69. DuHadaway JB, Sakamuro D, Ewert DL, Prendergast GC. Bin1 mediates apoptosis by c‐Myc in transformed primary cells. Cancer Res. 2001;61:3151‐3156. [PubMed] [Google Scholar]
- 70. Romero‐Masters JC, Huebner SM, Ohashi M, et al. B cells infected with Type 2 Epstein‐Barr virus (EBV) have increased NFATc1/NFATc2 activity and enhanced lytic gene expression in comparison to Type 1 EBV infection. PLoS Pathog. 2020;16:e1008365. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 71. Jones DD, Gaudette BT, Wilmore JR, et al. mTOR has distinct functions in generating versus sustaining humoral immunity. J Clin Invest. 2016;126:4250‐4261. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 72. Gass JN, Gifford NM, Brewer JW. Activation of an unfolded protein response during differentiation of antibody‐secreting B cells. J Biol Chem. 2002;277:49047‐49054. [DOI] [PubMed] [Google Scholar]
- 73. Tang D, Yang Z, Long F, et al. Long noncoding RNA MALAT1 mediates stem cell‐like properties in human colorectal cancer cells by regulating miR‐20b‐5p/Oct4 axis. J Cell Physiol. 2019;234:20816‐20828. [DOI] [PubMed] [Google Scholar]
- 74. Fan GC. Role of heat shock proteins in stem cell behavior. Prog Mol Biol Transl Sci. 2012;111:305‐322. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 75. Xiong J, Li Y, Tan X, Fu L. Small heat shock proteins in cancers: functions and therapeutic potential for cancer therapy. Int J Mol Sci. 2020;21(18):6611. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 76. Wang QM, Lian GY, Song Y, Huang YF, Gong Y. LncRNA MALAT1 promotes tumorigenesis and immune escape of diffuse large B cell lymphoma by sponging miR‐195. Life Sci. 2019;231:116335. [DOI] [PubMed] [Google Scholar]
- 77. Yasuda M, Tanaka Y, Fujii K, Yasumoto K. CD44 stimulation down‐regulates Fas expression and Fas‐mediated apoptosis of lung cancer cells. Int Immunol. 2001;13:1309‐1319. [DOI] [PubMed] [Google Scholar]
- 78. Kennedy D, Mnich K, Oommen D, et al. HSPB1 facilitates ERK‐mediated phosphorylation and degradation of BIM to attenuate endoplasmic reticulum stress‐induced apoptosis. Cell Death Dis. 2017;8:e3026. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 79. Tripathi V, Shen Z, Chakraborty A, et al. Long noncoding RNA MALAT1 controls cell cycle progression by regulating the expression of oncogenic transcription factor B‐MYB. PLoS Genet. 2013;9:e1003368. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 80. Chen SY, Jou IM, Ko PY, et al. Amelioration of experimental tendinopathy by lentiviral CD44 gene therapy targeting senescence‐associated secretory phenotypes. Mol Ther‐Methods Clin Dev. 2022;26:157‐168. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 81. Lin N, Yao Z, Xu M, et al. Long noncoding RNA MALAT1 potentiates growth and inhibits senescence by antagonizing ABI3BP in gallbladder cancer cells. J Exp Clin Cancer Res. 2019;38:244. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 82. Kondo S, Wakisaka N, Muramatsu M, et al. Epstein‐Barr virus latent membrane protein 1 induces cancer stem/progenitor‐like cells in nasopharyngeal epithelial cell lines. J Virol. 2011;85:11255‐11264. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 83. Ersing I, Bernhardt K, Gewurz B. NF‐κB and IRF7 pathway activation by Epstein‐Barr virus latent membrane protein 1. Viruses. 2013;5:1587‐1606. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 84. Wakisaka N, Kondo S, Yoshizaki T, Murono S, Furukawa M, Pagano JS. Epstein‐Barr virus latent membrane protein 1 induces synthesis of hypoxia‐inducible factor 1α. Mol Cell Biol. 2004;24:5223‐5234. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 85. Kraus RJ, Cordes BA, Sathiamoorthi S, et al. Reactivation of Epstein‐Barr virus by HIF‐1α requires p53. J Virol. 2020;94(18):e00722‐20. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 86. Epeldegui M, Conti DV, Guo Y, Cozen W, Penichet ML, Martínez‐Maza O. Elevated numbers of PD‐L1 expressing B cells are associated with the development of AIDS‐NHL. Sci Rep. 2019;9:9371. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 87. Khan AR, Hams E, Floudas A, Sparwasser T, Weaver CT, Fallon PG. PD‐L1hi B cells are critical regulators of humoral immunity. Nat Commun. 2015;6:5997. [DOI] [PubMed] [Google Scholar]
- 88. Crawford DH, Ando I. EB virus induction is associated with B‐cell maturation. Immunology. 1986;59:405‐409. [PMC free article] [PubMed] [Google Scholar]
- 89. Laichalk LL, Thorley‐Lawson DA. Terminal differentiation into plasma cells initiates the replicative cycle of Epstein‐Barr virus in vivo. J Virol. 2005;79:1296‐1307. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 90. Shaffer AL, Lin KI, Kuo TC, et al. Blimp‐1 orchestrates plasma cell differentiation by extinguishing the mature B cell gene expression program. Immunity. 2002;17:51‐62. [DOI] [PubMed] [Google Scholar]
- 91. Reusch JA, Nawandar DM, Wright KL, Kenney SC, Mertz JE. Cellular differentiation regulator BLIMP1 induces Epstein‐Barr virus lytic reactivation in epithelial and B cells by activating transcription from both the R and Z promoters. J Virol. 2015;89:1731‐1743. [DOI] [PMC free article] [PubMed] [Google Scholar]
Associated Data
This section collects any data citations, data availability statements, or supplementary materials included in this article.
Supplementary Materials
Supporting information.
Supporting information.
Supporting information.
Supporting information.
Supporting information.
Data Availability Statement
The single cell RNA‐seq data are sourced from GSE126321 for GM12878 and GM18502; GSE111912 for GM12891; GSE158275 for LCL777B958, LCL777M81, and LCL461B958; GSE162528 for LCL; and GSE121926 for GM22648 and GM22649. The ChIA‐PET in GM12878 is from GSE127053. The ChIP‐seq data are from EBNA2: GSE29498; EBNALP: GSE49338; EBNA3A: GSM1429820; EBNA3C: GSE52632; NF‐κB: GSE55105 and H3K27Ac: GSM733771.