

Abstract
Astrocytes respond to all central nervous system (CNS) insults with reactive changes that influence disorder outcome1–4. These changes include differentially expressed genes (DEGs) whose contextual diversity and regulation are poorly understood. Here, we combined biological and informatic analyses including RNAseq, protein detection, ATACseq and conditional gene deletion, to predict transcriptional regulators (TRs) that differentially control over 12,000 potential astrocyte reactivity DEGs across diverse CNS disorders in mice and humans. Astrocyte reactivity DEGs exhibited pronounced heterogeneity across disorders. TRs also exhibited disorder-specific differences, but a core group of 61 TRs was identified as common across multiple disorders in both species. We show experimentally that DEG diversity is determined by combinatorial, context-specific TR interactions, notably: the same reactivity TRs can regulate markedly different DEG cohorts in different disorders; TR DNA-binding motif access changes differ markedly across disorders; DEG changes can crucially require multiple reactivity TRs. We show that modulating reactivity TRs can powerfully alter disorder outcome, implicating them as therapeutic targets. We provide searchable resources of disorder-related reactive astrocyte DEGs and their predicted TRs. Our findings show that transcriptional changes associated with astrocyte reactivity are exquisitely heterogeneous and are customized from vast numbers of potential DEGs via context-specific combinatorial TR interactions.
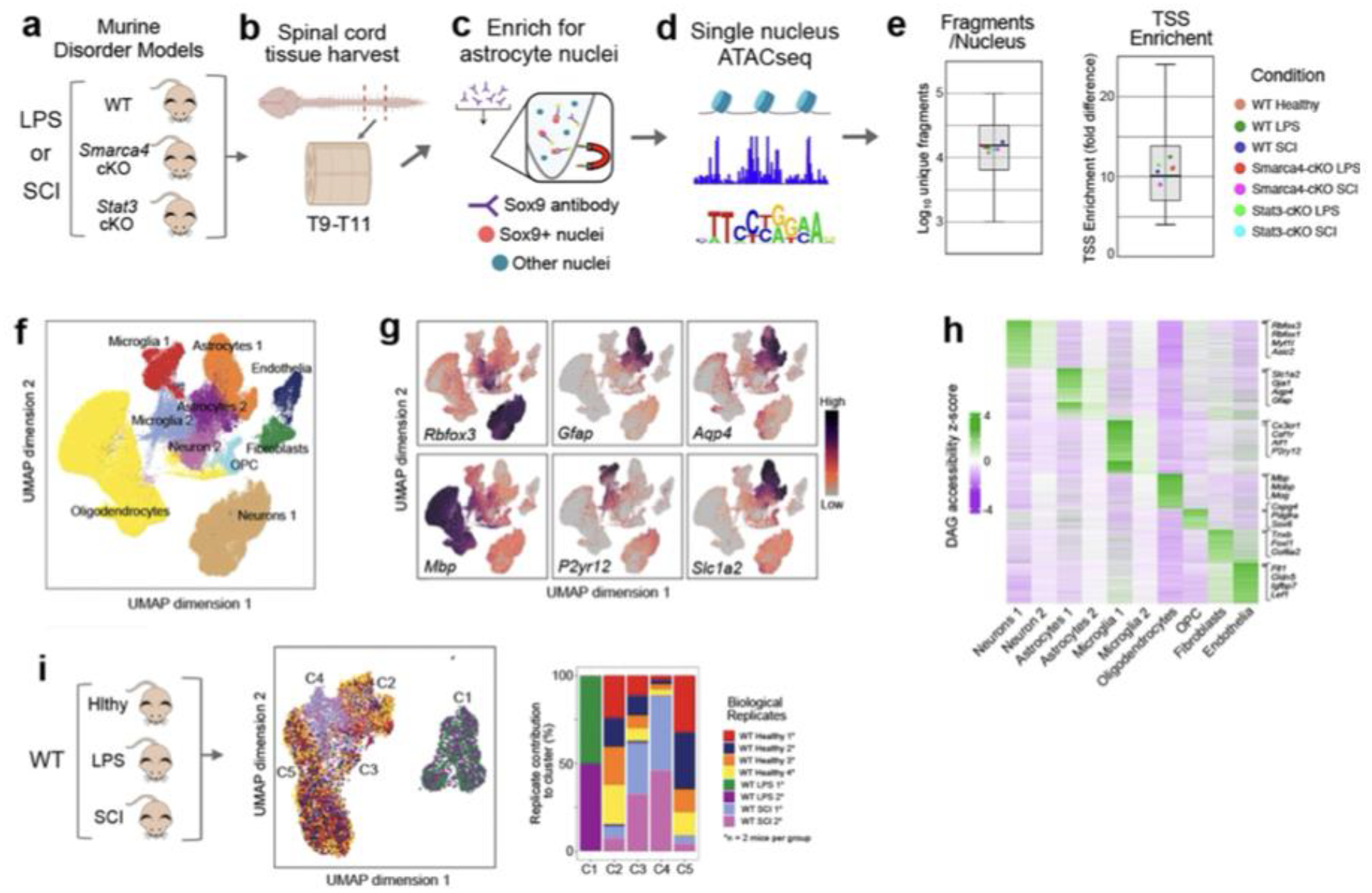
Astrocytes contiguously tile the central nervous system (CNS) and provide activities essential for healthy CNS function, ranging from homeostatic control of extracellular fluids, ions, and transmitters, to regulating synapse formation and modulating circuit functions5–7. Astrocytes also exhibit an evolutionarily ancient response to CNS injury and disease known as astrocyte reactivity1–4,8. Although regarded for decades as homogenous and functionally passive, astrocyte reactivity is emerging as a heterogeneous, context-specific and powerful outcome-determining factor in CNS disorders that can modulate CNS inflammation and preserve tissue1,2,9, contribute to circuit reorganization5,6,10,11 and axon regeneration12, and in some contexts exert detrimental effects through loss of healthy functions or gain of detrimental effects9,13,14. There is mounting interest in manipulating astrocyte reactivity as treatment strategies for CNS disorders. Doing so will require a detailed understanding of what astrocyte reactivity is, how it differs across disorders and how these differences are regulated. Here, we integrated findings from experimental biological and computational approaches to broadly identify fundamental principles of transcriptional regulation of astrocyte reactivity across disorders with different reactivity-triggering stimuli. To so we used: (i) RNAseq to identify differentially expressed genes (DEGs) associated with astrocyte reactivity, (ii) computational analysis of DEGs to predict astrocyte reactivity transcriptional regulators (TRs), (iii) single nucleus assay for transposase-accessible chromatin with high-throughput sequencing (ATACseq) to profile chromatin accessibility of reactivity DEGs and of DNA-binding motifs of predicted TRs, (iv) immunohistochemical detection of TR protein, and (v) astrocyte-specific conditional gene deletion (cKO) of certain test-case TRs.
Reactivity DEGs and TRs vary across disorders
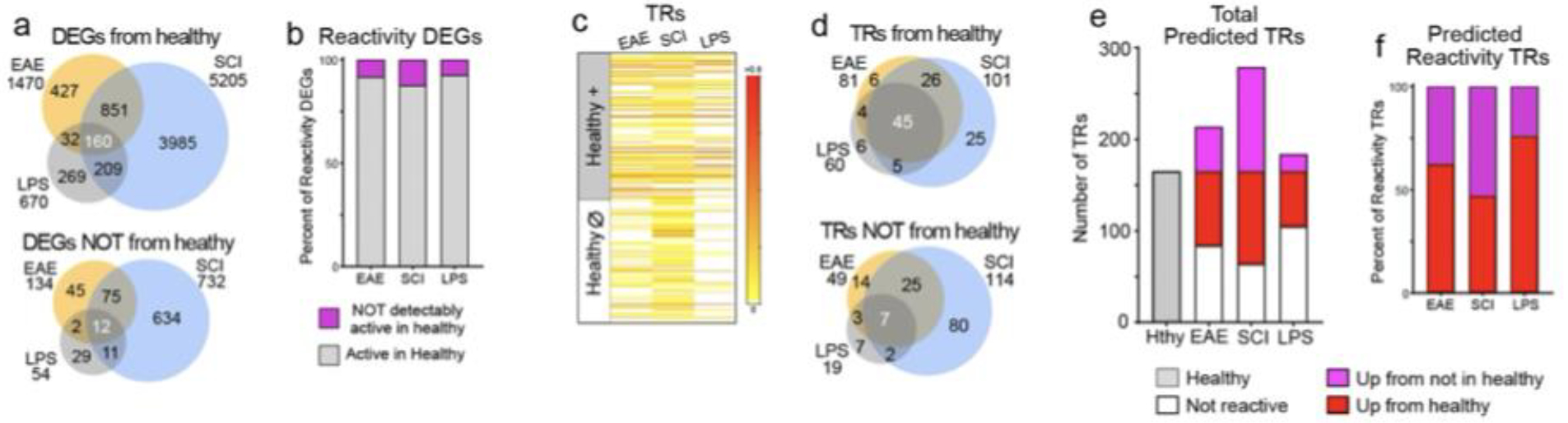
We first compared astrocyte reactivity DEGs and their predicted TRs in three experimental disorders triggered by different stimuli: (i) permanent traumatic tissue damage after spinal cord injury (SCI), (ii) transient neuroinflammation triggered by acute systemic injection of microbial antigen, lipopolysaccharide (LPS), and (iii) chronic neuroinflammation induced in experimental autoimmune encephalomyelitis (EAE) (Fig. 1a; Extended Data Fig.1a–c). Healthy spinal cord astrocytes exhibited 11,260 expressed genes (EGs) above threshold criteria, of which 13%, 46% or 6% significantly changed either up or down after EAE, SCI or LPS respectively at timepoints evaluated (Fig. 1b). In addition, DEGs not detectably expressed in healthy astrocytes were upregulated in a disorder selective manner (Fig. 1b). Remarkably, of the 6741 different astrocyte reactivity DEGs identified in at least one of the three disorders, only 2.6% (172/6741) were shared by all three disorders and principal component analysis (PCA) of reactivity DEG log-fold changes showed near orthogonal differences (Fig. 1c).
Fig. 1. Astrocyte reactivity DEGs and TRs vary across disorders.
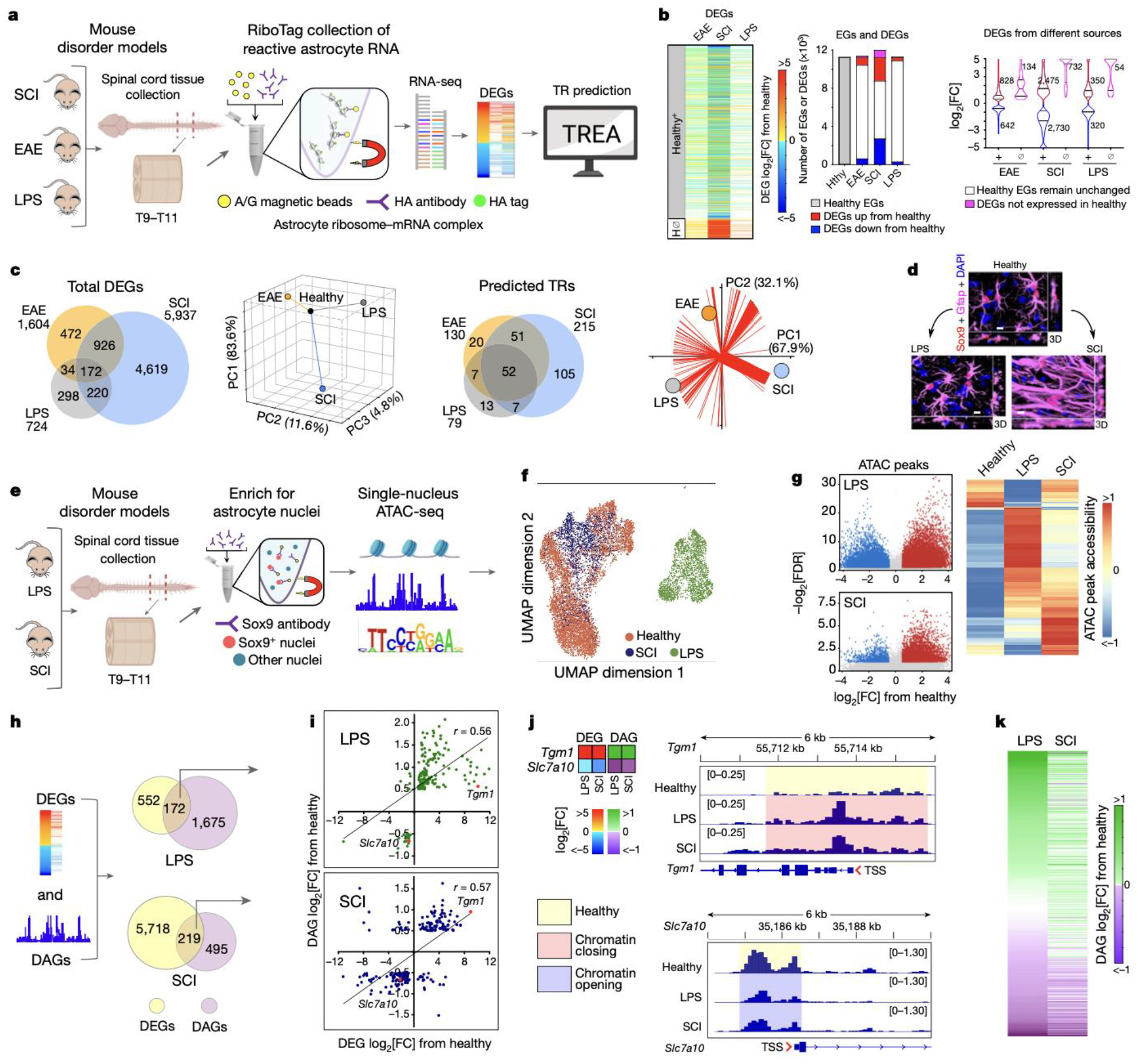
a. TR prediction schematic. b. Heatmap, bar graph and violin plot comparing astrocyte reactivity DEGs that either are, or are not (H⊘), expressed in healthy astrocytes. c. Venn diagrams and PCA of DEGs or predicted TRs in SCI, EAE or LPS compared with healthy (Hlth). Red lines show vectors of TR contributions to disorders. d. Immunohistochemistry comparing astrocytes in Healthy, LPS and SCI. e. Astrocyte ATACseq schematic. f. UMAP clustering of astrocyte nuclei based on differential ATAC peaks. g. Volcano plots and hierarchical clustering of ATAC peaks in LPS or SCI relative to healthy. h. Schematic and Venn diagrams comparing DEGs and DAGs in LPS or SCI. i. Correlation of congruence between DEGs and DAGs. j. Heatmaps and ATAC peak opening or closing in two examples of up- or down-regulated DEGs. k. Heatmaps comparing chromatin accessibility of the same genes in LPS and SCI.
To begin identifying astrocyte reactivity TR candidates, we first predicted potential TRs from DEG profiles by applying a conservative, multi-step upstream transcriptional regulator enrichment analysis (TREA) that draws on both computationally- and biologically-derived regulator-target gene interaction data available in resource databases with the ability to identify approximately 1350 TRs including: (i) DNA-binding transcription factors and their cofactors, (ii) modifiers of chromatin structure and epigenetic tags, and (iii) regulators that act via protein-protein interactions or by RNA-binding (Extended Data Fig.1d). TREA predicted 255 TRs controlling reactivity DEGs across these three disorders, of which 52 TRs (20%) were common to all three disorders and 138 (54%) were unique to one disorder (Fig. 1c). PCA of TR contributions weighted according to their fraction of the total number of reactivity DEGs regulated per condition, also showed pronounced differences among EAE, SCI and LPS (Fig. 1c).
TREA predicted many novel astrocyte reactivity TRs. To probe their accuracy, we used multiple approaches. First, we noted that of the 62 published astrocyte reactivity TRs identified across diverse disorders, 94% (58/62) were included among TRs predicted by TREA in either EAE, SCI or LPS (Extended Data Fig. 1e). We then experimentally tested TREA predictions by (i) genome-wide ATACseq profiling of chromatin accessibility15, (ii) immunohistochemical detection of TR protein, and (iii) astrocyte selective gene deletion (cKO) of test-case TRs. For these studies, we focused on the experimental models of LPS and SCI that exhibit different forms of transient or permanent astrocyte reactivity that differ fundamentally in transcriptional regulation (Fig. 1b,c), cell morphology (Fig. 1d) and function, such that after SCI, reactive astrocytes proliferate to form permanent borders around lesions to corral inflammation and fibrosis16, whereas after LPS, reactive astrocytes exhibit only moderate structural changes (Fig. 1d) with no detectable increase in proliferation, and transcriptional changes that return to baseline within two weeks17.
Disorders diversely alter astrocyte chromatin landscape
We used single nucleus ATACseq to examine chromatin landscape changes during astrocyte reactivity after LPS or SCI in WT mice and mice with astrocyte-specific conditional gene deletion (cKO) of test-case TRs (Fig. 1e; Extended data Fig. 2a–h). Unbiased clustering of 11,970 spinal cord astrocyte nuclei from healthy, LPS- or SCI-treated WT mice showed separation of treatment groups with some overlap of SCI and healthy and essentially no overlap of LPS with either healthy or SCI (Fig. 1f; Extended data Fig. 2i). Comparison of differentially accessible peaks15 showed substantial chromatin remodelling during astrocyte reactivity, with more chromatin opening than closing and pronounced differences in LPS and SCI (Fig. 1g). Although LPS treatment exhibited proportionately more chromatin changes (Fig. 1g), it exhibited fewer DEGs (Fig. 1b,c), consistent with evidence that extent of chromatin remodelling need not reflect proportional changes in gene expression18,19.
We identified differentially accessible genes (DAGs) based on chromatin accessibility associated with specific gene loci20. Consistent with previous reports18,19, we found partial overlap of changes in chromatin and gene expression (Fig. 1h). Nevertheless, genes with significant changes in both chromatin accessibility and RNA expression exhibited congruence between these changes, such that increased or decreased RNA expression correlated with chromatin opening or closing respectively (Fig. 1h,i). Examination of individual DEG loci confirmed specific examples where chromatin opening (Tgm1) or closing (Slc7a10) correlated with up or down changes in gene expression respectively, particularly in regulatory regions around transcription start sites (TSS) (Fig. 1j). Notably, direct comparison of ATAC peaks associated with the same DEGs significantly regulated in LPS and SCI revealed both similarities and differences in chromatin accessibility changes across the two disorders (Fig. 1k).
These findings showed that in WT mice: (i) astrocyte reactivity is associated with genome-wide chromatin remodelling that differed markedly across disorders; (ii) up or down changes in expression of many DEGs during astrocyte reactivity correlated with significant opening or closing of chromatin respectively, but many other DEGs were not associated with detectable chromatin changes; (iii) significant chromatin changes can occur across genes that do not display detectable changes in gene expression and (iv) there are no default reactive astrocyte subtypes with common chromatin signatures in both LPS and SCI despite the pronounced neuroinflammation associated with both.
TR motif access changes during astrocyte reactivity
ATACseq can predict potential TR activity by examining chromatin accessibility of TR DNA-binding motif sequences21. We quantified DNA-binding motifs of TREA-predicted astrocyte reactivity TRs on a per-nucleus basis21 (Fig. 2a–d). As discussed above, not all TRs exhibit DNA-binding motifs but interact with proteins or RNA. In LPS 87% (69/79), and in SCI 74% (159/215), of TREA-predicted TRs had DNA-binding motifs identifiable by the profiling software. In LPS 90% (62/69), and in SCI 89% (142/159), of TREA-predicted TRs with detectable motif sequences exhibited significant differences in chromatin accessibility of their motif sequences compared to healthy (Fig. 2d). Of these, 46 were identified in both LPS and SCI, and 16 and 96 were identified preferentially in LPS or SCI, so that altogether 158 TREA-identified TRs also exhibited significant differences in accessibility of their DNA binding motif in either LPS or SCI. In addition, immunohistochemistry demonstrated protein expression by reactive astrocytes for 53 of the newly identified TRs with motifs, providing a third independent method of experimental confirmation (Fig. 2d,f, Extended Data Fig. 3–5).
Fig. 2. Astrocyte reactivity TRs identified by multiple approaches: DEG analysis, chromatin accessibility changes at DNA-binding motifs and immunohistochemistry (IHC).
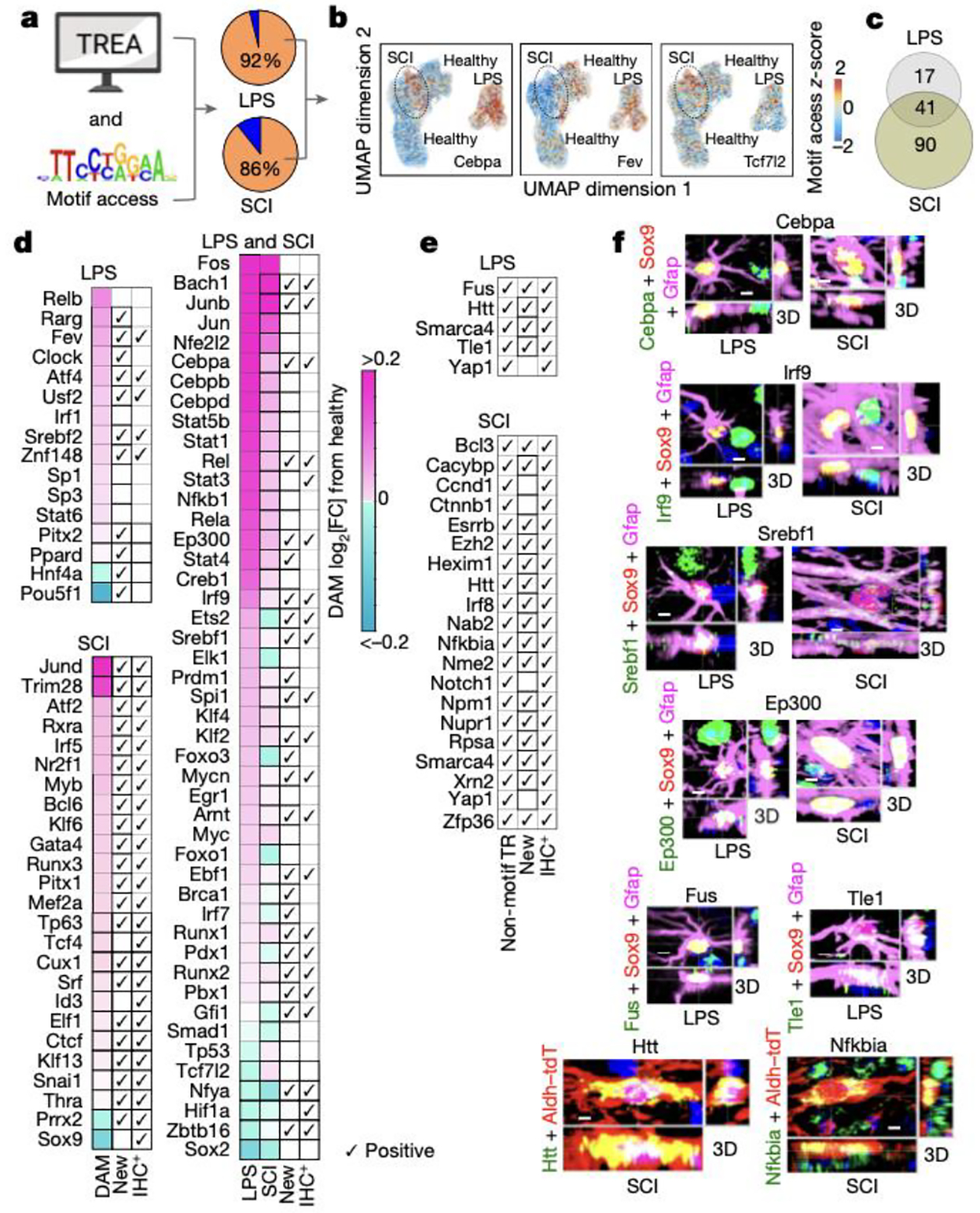
a. Schematic and pie charts of TREA-identified TRs with significant changes in motif access (orange). b. UMPA clusters of representative TRs with motif access differences in healthy, LPS and SCI. c. Venn diagram of TREA-TRs with significant changes in motif access. d. Heatmaps of TREA-predicted TRs with differentially accessible motif (DAM) z-scores. All such TRs are shown for LPS or both, with 25 selected TRs for SCI. For all SCI TRs see Extended Data figure 4b. e. TREA-predicted TRs without known DNA-binding motifs confirmed by IHC of TR-protein. f,g. IHC of representative new TRs in reactive astrocytes; for individual channels of all IHC+ TRs see Extended Data figures 3–5.
Immunohistochemistry also confirmed protein expression in reactive astrocytes for 18 TREA-predicted TRs without known DNA-binding sites, including TRs that act via protein-protein interactions (Htt, Nfkbia), via chromatin remodelling (Smarca4, Nupr1), or by targeting RNA degradation (Zfp36) (Fig. 2e,g; Extended Data Figs. 3–5). Thus, a total of 71 newly identified reactivity TRs were confirmed by immunohistochemistry of protein in reactive astrocytes. Interestingly, many disorder-activated astrocyte reactivity TRs were also expressed by nearby non-astrocytes (Fig. 2f,g; Extended Data Figs. 3–5). Altogether, 89% (70/79) of TREA-predicted TRs in LPS, and 81% (174/215) of TREA-predicted TRs predicted in SCI, were co-identified by either ATACseq motif analysis, immunohistochemistry, previous publication, or combinations of these, providing strong validation of TREA predictions (Extended data Fig. 3b,5b).
These findings showed that: (i) nearly all TREA-predicted TRs that had detectable DNA-binding motifs exhibited changes in genome-wide motif accessibility during astrocyte reactivity; (ii) TR motif access changes differ markedly across different disorders; and (iii) TR protein was confirmed in reactive astrocytes for many newly identified astrocyte reactivity TRs.
TR control of reactivity is combinatorial and disorder divergent
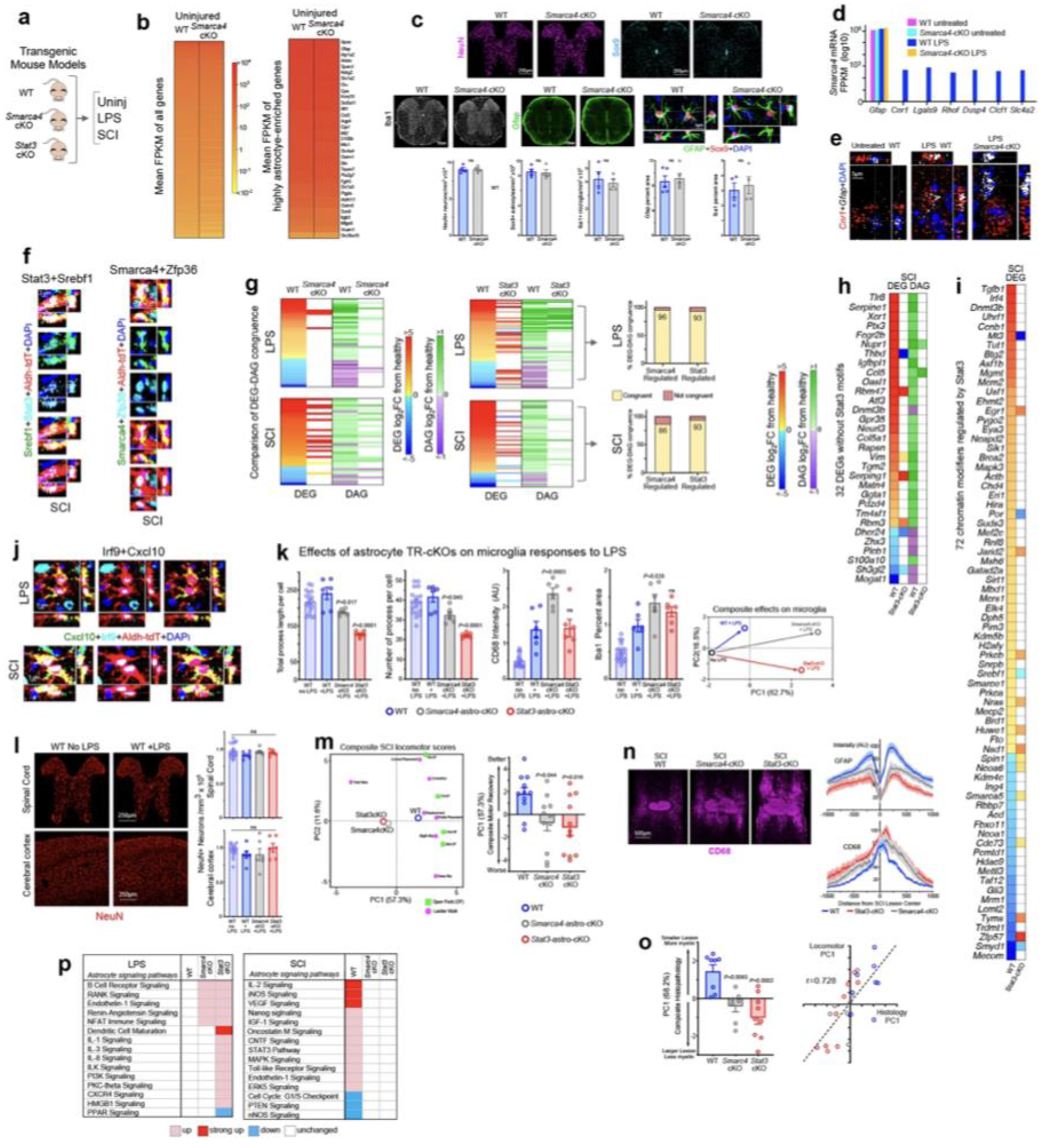
We next examined how a limited number of TRs might control a vast number of potential astrocyte reactivity DEGs diversely across different disorders. We integrated results from RNAseq, TREA predictions, cKO experiments, ATACseq, and immunohistochemistry. To compare individual TR contributions to transcriptional changes in different disorders, we conducted astrocyte-specific loss-of-function experiments with two test-case TRs, Smarca4 and Stat3, in two disorders models, LPS and SCI. Stat3 (Stat3-astro-cKO) or Smarca4 (Smarca4-astro-cKO) were selectively deleted from astrocytes in well-characterized transgenic mice12,16 (Fig. 3a,b; Extended data Fig. 6a–e). In Smarca4-astro-cKO mice, 94% (165/175) in LPS and 87% (1067/1226) in SCI of TREA-predicted Smarca4-regulated DEGs were either not regulated at all or were regulated differently (Fig. 3c). In Stat3-astro-cKO mice 62% (235/378) in LPS and 80% (2242/2820) in SCI of TREA-predicted Stat3-regulated DEGs were either not regulated at all or were regulated differently (Fig. 3c). Using these TR-cKO models, we asked: (1) Do TRs modulate the same DEGs similarly or differently in different disorders? (2) Can different TRs crucially regulate the same DEGs? (3) Can TRs regulate other TRs? (4) Does TR regulation of chromatin remodelling contribute to DEG regulation?
Fig. 3. TR regulation of astrocyte reactivity DEGs, chromatin accessibility and disorder outcome.

a. Experimental models. b. Smarca4 immunohistochemistry. c. Smarca4-astro-cKO or Stat3-astro-cKO effects on Smarca4- or Stat3-regulated genes. d. TREA-predicted Smarca4- or Stat3-regulated DEGs in both LPS and SCI, and astro-cKO effects. e,f. DEGs regulated in the same (e) or in opposite (f) directions by Smarca4 or Stat3. g. Stat3 and Smarca4 in same reactive astrocyte. h. TREA-predicted Smarca4- and Stat3-regulated DEGs and astro-cKO effects on those DEGs. i. TRs significantly regulated as DEGs; Smarca4 and Pitx1 in same reactive astrocyte. j. UMAP clustering of astrocyte nuclei from WT, Smarca4-astro-cKO or Stat3-astro-cKO mice based on chromatin accessibility after LPS or SCI. k. Stat3-cKO effects on DEG and DAG. l. Smarca4-astro-cKO or Stat3-astro-cKO effects on Serpine1 DEG, DAG; and ATAC peaks in Serpine1 TSS. m. Stat3-astro-cKO effects on DEG and DAG of 10 genes without Stat3 DNA-binding motifs. n. Stat3-astro-cKO effects on 10 chromatin regulators. o. Cxcl10 DEG and DAG; ATAC peaks in Cxcl10 TSS and Irf9 DNA-binding motif; Irf9 and Cxcl10 in same reactive astrocyte. p. Timp1 DEG and DAG; ATAC peaks in Timp1 TSS and DNA-binding motifs of 7 predicted TRs; Runx1 and Srebf1 in same reactive astrocyte. q. Sickness behavior after LPS. r. Microglia immunohistochemistry after LPS. s. PCA of composite histopathology after LPS. t. Open field (OF) locomotor recovery after SCI. u,v. Images and graph of mean±sem staining intensity for Gfap, CD13 and myelin basic protein (MBP). w. ETS family TRs DEG and differential motif access (DAM) after SCI. x. PU.1 (Spi1) in reactive astrocytes. y. Fcgr2b DEG and DAG; ATAC peaks in Fcgr2b TSS and PU.1 DNA-binding motif. Quantitative values in q,s,t are mean ± sem analyzed by one-way (q,s) or two-way (t) ANOVA with Bonferroni’s test, P values are cKO versus WT LPS or SCI, NS non-significant. n = mice.
We first examined TR effects on the same DEGs in different disorders. Resource databases (Extended data Fig. 1d) indicate that Smarca4 and Stat3 can regulate over 4200 and 12,500 DEGs respectively in mammalian cells. In reactive astrocytes, of these potential DEGs, Smarca4 was predicted by TREA and demonstrated by Smarca4-astro-cKO to significantly regulate only 165 in LPS and 1092 in SCI, with 59 regulated in both disorders (Fig. 3d). Stat3 was predicted by TREA and demonstrated by Stat3-astro-cKO to significantly regulate only 235 in LPS and 2263 in SCI, with 73 regulated in both disorders (Fig. 3d). Comparison of the same DEGs showed that Smarca4 or Stat3 each regulated many DEGs in the same direction in both disorders in WT mice (Fig. 3d,e). Remarkably, this analysis also revealed that Smarca4 or Stat3 could each crucially regulate the same reactivity DEGs in opposite directions in LPS or SCI, such that TR-cKO prevented a gene expression increase in one disorder and a decrease in the other disorder, or vice versa (Fig. 3d,f). For example, Smarca4 and Stat3 were each required to up-regulate Slc14a2 and Rhof in LPS and to down-regulate them in SCI (Fig. 3f).
We next compared the effects of different TRs on the same DEGs. Immunohistochemistry demonstrated Smarca4 and Stat3 proteins in the same reactive astrocytes (Fig. 3g). TREA predicted that in WT mice, Smarca4 and Stat3 both regulated the same 99 DEGs in LPS and the same 763 DEGs in SCI. We determined TR-cKO effects on these DEGs. In LPS 61% (60/99) and in SCI 66% (502/763) of the same DEGs either failed to be regulated or were regulated differently after either Smarca4- astro-cKO or Stat3-astro-cKO (Fig. 3h), demonstrating essential regulation by both TRs.
To determine whether TRs regulate the expression of other TRs, we focused on reactivity TRs confirmed by TREA plus ATACseq motif analysis or immunohistochemistry or all three (Extended data Fig. 3b,5b). In LPS, 17 of such verified astrocyte reactivity TRs were significantly regulated as DEGs, and of these, 94% (16/17) or 53% (9/17) either failed to be regulated or were regulated differently after Smarca4-astro-cKO or Stat3-astro-cKO respectively. In SCI, 107 of such verified reactivity TRs were significantly regulated as DEGs, and of these, 93% (100/107) or 81% (87/107) either failed to be regulated or were regulated differently after Smarca4-astro-cKO or Stat3-astro-cKO, including TRs regulated by both Smarca4 and Stat3 (Fig. 3i), demonstrating TR regulation of other TRs. Immunohistochemistry confirmed examples of co-expression of Smarca4 or Stat3 in the same WT reactive astrocytes together with TRs whose expression they regulated (Fig. 3i; Extended Data Fig. 6f).
We next examined TR-cKO effects on chromatin remodelling. UMAP clustering based on differential ATAC peaks across 21,825 astrocyte nuclei showed discrete clusters of WT, Smarca4-astro-cKO or Stat3-astro-cKO nuclei after both LPS and SCI, with no appreciable overlap of reactive astrocyte nuclei from LPS or SCI treated mice (Fig. 3j). DEGs demonstrated by cKO to be regulated by Smarca4 or Stat3 showed 86 to 96% congruence between changes in gene expression and changes in chromatin accessibility after LPS or SCI (Extended Data Fig. 6g). Interestingly, although transcriptional regulation by Stat3 via DNA-binding motifs is well established22, Stat3-astro-cKO was also associated with pronounced changes in chromatin accessibility in many Stat3-regulated DEGs, particularly after SCI (Fig. 3k,l). Moreover, we found 31 DEGs that lacked detectable known Stat3-motifs within their genomic regulatory regions but were nonetheless significantly regulated by Stat3 after SCI, as demonstrated by Stat3-astro-cKO prevention of changes in both gene expression and chromatin accessibility (Fig. 3l,m; Extended Data Fig. 6h). We also found 72 TREA-predicted TRs with chromatin modifying activity whose expression after SCI was significantly regulated by Stat3 as demonstrated by Stat3-astro-cKO (Fig. 3n; Extended Data Fig. 6i), consistent with Stat3 regulating reactivity DEGs indirectly by regulating chromatin remodelling as reported in other tissues22.
We used ATACseq and IHC to test specific examples of newly identified TRs regulating specific DEGs. Both TREA and ATAC motif analysis predicted Irf9 as a reactivity TR (Fig. 2d) and TREA predicted Irf9 regulation of Cxcl10. Supporting these predictions, we found that (i) immunoreactive Cxcl10 and Irf9 protein were both detectable within the same astrocytes in both LPS and SCI, (ii) chromatin on the Cxcl10 locus was significantly more accessible in both LPS and SCI and (iii) regulatory regions around the Cxcl10 transcription start site (TSS) contained an Irf9 binding motif (Fig. 3o; Extended Data Fig. 6j). In support of multiple TRs regulating the same DEGs, we found that the TSS of upregulated genes such as Timp1 contained motifs for multiple TRs identified by both TREA and ATAC motif analysis including newly identified TRs such as Runx1, Srebf1, Pitx1, Cebpa, Fli1 and Junb, and that different TRs were immunohistochemically confirmed in the same reactive astrocytes (Fig. 3g,i,p; Extended Data Fig. 6f).
These findings showed that (i) the same TR can regulate markedly different cohorts of astrocyte reactivity DEGs in different disorders; (ii) the same TR can regulate specific DEGs in the same or different directions in different disorders; (iii) the same TR can exert different effects on reactive astrocyte chromatin landscapes in different disorders; (iv) TR-dependent changes in chromatin accessibility can be congruent with and contribute to TR-mediated changes in gene expression; (v) TRs can regulate the expression of chromatin regulators and thereby influence chromatin accessibility; and (vi) TRs can regulate the expression of other reactivity TRs and thereby indirectly modulate reactivity DEGs.
Astrocyte reactivity TRs influence disorder outcome
We examined TR loss-of-function effects on disorder outcome. Systemic LPS induces acute sickness behavior and neuroinflammation23. Stat3-astro-cKO significantly exacerbated LPS-induced sickness behavior, whereas Smarca4-astro-cKO did not, but both cKOs significantly increased microglial activation and neuroinflammation (Figs. 3q–s; Extended Data Fig. 6k). Nevertheless, in agreement with previous findings in cerebral cortex17, we found no evidence for loss of spinal cord neurons under any condition (Extended Data Fig. 6l), arguing against the notion that neuroinflammation per se causes microglia to induce neurotoxic astrocytes in vivo. After a partial crush SCI, both Smarca4-astro-cKO and Stat3-astro-cKO significantly impaired naturally occurring locomotor recovery and significantly exacerbated lesion size, phagocytic inflammation and myelin loss (Figs. 3t–v; Extended Data Fig. 6m–o), consistent with previous reports for Stat324. DEG functional pathway analysis showed Smarca4- or Stat3-loss-of-function leads to loss of multiple astrocyte regulatory pathways and increased pro-inflammatory signaling in LPS and SCI (Extended Data Figs. 6p). These findings show that modulating reactivity TRs can powerfully alter disorder outcome.
Reactive astrocytes can adopt novel and unexpected signaling
Many astrocyte reactivity DEGs in LPS, SCI or EAE were not detectably expressed in healthy astrocytes (Fig. 1b; Extended Data Fig. 7a,b). Similarly, many TREA-predicted TRs were not identified in healthy astrocytes (Extended Data Fig. 7c–f) suggesting that reactive astrocytes might exhibit new and unexpected molecular signatures and functions. Supporting this notion, Spi1 (PU.1) expression was highly upregulated in reactive astrocytes in WT SCI and was further upregulated by Smarca4 or Stat3 cKO (Fig. 3i). Spi1/PU.1 is an innate immune regulator considered restricted to macrophages or microglial in CNS, but its recent identification as a regulator of fibroblast wound responses25 and mounting evidence that astrocytes are essential components of CNS innate immunity26 prompted further examination. Both TREA and ATAC motif analysis identified Spi1/PU.1 as an astrocyte reactivity TR (Fig. 2d,3w) and immunohistochemistry demonstrated Spi1/PU.1 protein in reactive astrocytes (Fig. 3x). Immunohistochemistry also confirmed the absence of RiboTag from microglia (Extended data Fig. 1b). Nine other ETS family TRs were identified as reactivity TRs, but motif access decreased for seven of these, whereas both gene expression and motif access markedly increased for Spi1 (Fig. 3w). Ehf, the only other ETS TR with increased expression and motif access (Fig. 3w) possesses a PNT domain that specifies functions via protein-protein interactions, whereas Spi1 does not. Specific molecules predicted by TREA to be regulated by Spi1/PU.1, such as IgG receptor, Fcgr2b, exhibited increased expression and chromatin access in reactive astrocytes and an Spi1/PU.1 motif in its TSS region (Fig. 3y). These findings argue that reactive astrocytes, like fibroblasts25, can be regulated by Spi1/PU.1 during wound responses and that Spi1/PU.1 signaling cannot be assumed to be unique to microglia in CNS disorders.
Reactivity DEGs and TRs diverge across multiple disorders
We next compared DEGs and their predicted TRs in reactive astrocytes across a broad spectrum of fifteen CNS disorders, experimental models and aging in mice and humans by using our and archival data (Fig. 4; Extended Data Table 1). Over 12,300 different astrocyte DEGs were identified in one or more disorder, with remarkable heterogeneity and low overlap across disorders, and TREA predicted 449 different TRs regulating these DEGs in one or more disorder, with markedly greater overlap of TRs than DEGs (Fig. 4b; Extended Data Figs. 8a,b). Notably, disorders with genetic mutations or polymorphisms of genes influencing astrocyte functions, Htt27, Sod128 and Apoe429, (i) exhibited vanishingly small overlaps with disorders having WT astrocytes, (ii) failed to upregulate the top 15 DEGs upregulated in 7 of 8 disorders with WT astrocytes, (iii) clustered together with pronounced separation away from disorders with WT astrocytes upon unsupervised clustering of PCA DEG and TR vectors, and (iv) failed to activate a broad cross-section of functional pathways activated by disorders with WT astrocytes (Fig. 4b–e; Extended Data Figs. 8a,c).
Fig. 4. Astrocyte reactivity DEGs and TRs diverge across CNS disorders in mice and humans.
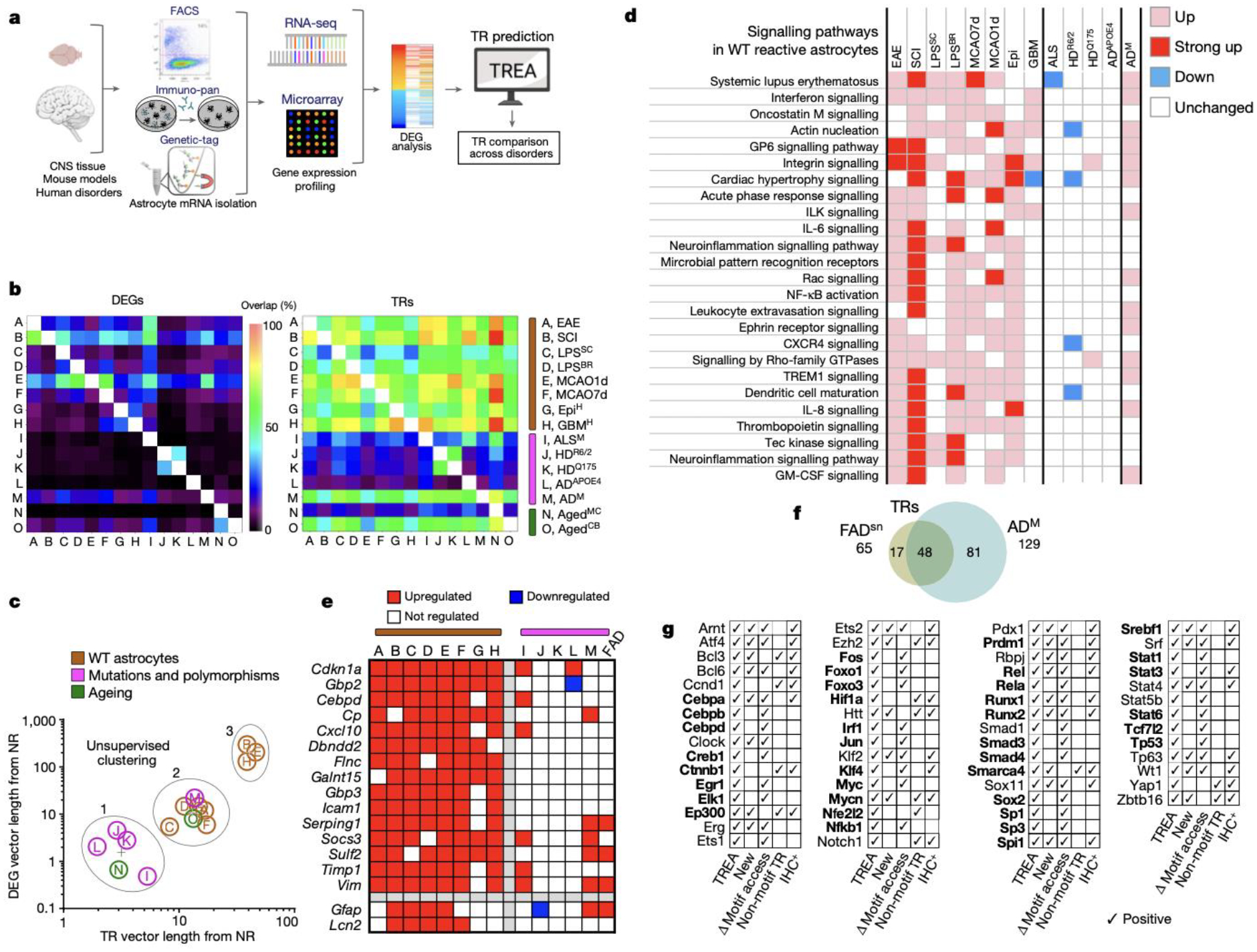
a. Experimental procedures. b. Heatmaps of pairwise overlap of DEGs or TRs. c. Unsupervised clustering of disorders based on DEG and TR PCA vector lengths. d. DEG-associated functional pathways in disorders with WT astrocytes or genetic mutations and polymorphisms. e. Heatmap comparing DEGs upregulated in disorders with WT astrocytes. f. Venn diagram of TRs predicted in FADsn and ADM. g. Astrocyte reactivity TRs predicted in all eight (bold) or seven of eight mouse or human disorders with WT astrocytes, plus confirmation of motif access changes or protein immunohistochemistry in LPS or SCI.
We also applied TREA to archival data obtained by single nucleus RNAseq of astrocytes from transgenic mice with familial Alzheimer’s disease (FAD) Aβover-expression30. TREA predicted 65 TRs controlling reactivity DEGs in FADsn-associated astrocytes (cluster 430) compared with healthy control astrocytes (cluster 130). Remarkably, 74% (48/65) of reactivity TRs predicted based on 250 DEGs identified by single nucleus RNAseq in FADsn-associated astrocytes, overlapped with astrocyte reactivity TRs predicted based on nearly 2000 DEGs identified by bulk gene array analysis of a different cohort of transgenic mice (ADM) with familial Alzheimer’s disease Aβ over-expression31, indicating a strong concordance of TREA predictions derived from different DEG datasets collected using different techniques from similar disorder models (Fig. 4f; Extended Data Fig. 8d).
Astrocytes in aging motor cortex (AgedMC)32 compared with young adult healthy exhibited only 72 DEGs with only 26 predicted TRs, whereas astrocytes in aging cerebellum (AgedCB) exhibited 1308 DEGs with 126 predicted TRs, levels comparable to disorders with WT astrocytes (Extended Data Fig. 8b). Unsupervised clustering of PCA DEG and TR vectors showed that AgedMC astrocytes clustered significantly closer to young adult non-reactive states, whereas AgedCB astrocytes clustered among young adult WT astrocytes responding to non-cell autonomous reactivity triggers (Fig. 4a–c; Extended Data Fig. 8c).
Lastly, we sought to identify astrocyte reactivity DEGs and TRs that might be common across different disorders. Because disease-associated genetic mutations or polymorphisms cell-autonomously could modify responses to non-cell-autonomous triggers of astrocyte reactivity, we separately evaluated disorders with WT astrocytes and those with mutations. In the six primary neurodegenerative disorders and models with mutations of genetic polymorphisms examined here, no DEG was upregulated in more than three and only one TR, Tp53, was common across all but was also shared by all other disorders (Extended Data Fig. 8d,e). Thus, we found no evidence for a common gain-of-function degeneration-provoking transcriptional signature that might be shared by astrocytes in different neurodegenerative disorders. Instead, neurodegenerative disorders with mutations and polymorphisms failed to upregulate a majority of the DEGs common across disorders with WT astrocytes (Fig. 4b–e) in a manner consistent with reports that disorder-selective disruption of normal reactive astrocyte transcription and functions contributes to neurodegeneration13,33,34. In eight disorders with WT astrocytes in both mice and humans, we identified over 10,500 astrocyte reactivity DEGs with surprisingly low DEG overlap across disorders and very different disorder distributions in PC space (Fig. 4c; Extended Data Figs. 8c). Remarkably, despite the large number of DEGs identified across all disorders, we found only two DEGs common across all eight disorders and only a further eight DEGs common across seven of eight (Fig. 4e). In striking contrast to DEGs, 37 predicted TRs were shared across all eight disorders with WT astrocyte in both species, and a further 24 TRs were shared by at least seven of eight (Fig. 4g). Notably, all 61 of these TRs, including 28 newly identified TRs, were also confirmed experimentally as TRs in LPS or SCI either by significant changes in motif access demonstrated by ATACseq or by immunohistochemistry or by both (Fig. 4g), implicating them as core astrocyte reactivity TRs.
Discussion
Astrocyte reactivity was long regarded as a uniform response to tissue damage that was of little functional importance and was of interest primarily as a marker of tissue pathology1. Recent studies have changed this perception by showing that attenuating or augmenting astrocyte reactivity can alter disorder outcome9,24,33–35. Nevertheless, defining basic attributes of reactive astrocytes and their regulation has been elusive and sometimes controversial4. Our findings across multiple disorders in mice and humans show that transcriptional changes associated with astrocyte reactivity are exquisitely heterogeneous and are customized from a vast number of potential DEGs via context-specific TR interactions. We experimentally demonstrate multiple ways in which a limited number of interacting TRs could achieve substantial diversity of astrocyte reactivity DEGs. Our findings point towards a parsimonious working model of astrocyte reactivity regulation in which a core set of TRs is active across many if not most forms of astrocyte reactivity, but that these TRs can regulate different cohorts of DEGs in different disorders and contexts via complex and interdependent combinatorial interactions that also involve disorder- or context-selective TRs (Extended Data Fig. 9). These TR interactions can be modified by disease-associated genetic mutations or polymorphisms, or by experimental interventions, which not only alter astrocyte reactivity DEGs, but can powerfully change disorder histopathology and neurological outcome, thus implicating TRs as potential therapeutic targets. Dissecting how to beneficially modulate reactive astrocyte TRs and their target DEG networks has the potential to reveal a broad range of novel therapeutic strategies for CNS disorders. Towards this end, we provide a resource database of reactive astrocyte DEGs and their predicted TRs across a broad spectrum of CNS disorders and conditions. Most studies to date have examined the roles only of single TRs individually in reactive astrocytes. Developmental studies indicate that understanding the diversity of neural differentiation will require understanding the interaction of multiple TRs18,36. Our findings similarly indicate that understanding how the molecular diversity of context dependent astrocyte reactivity is achieved will require deconstructing the regulatory grammar governing the interaction of multiple TRs.
METHODS
Mice.
Young adult male and female 657Bl6 mice were used between two and four months of age at the time of experimental procedure. Astrocyte-conditional transgenic mice were obtained by crossing the well-characterized, astrocyte-specific Cre-driver line, mGfap-Cre 73.1237 to various floxed gene lines: (1) mGfap-Cre-RiboTag mice (mGfap-RiboTag) were generated by crossing to loxP-STOP-loxP-Rpl22-HA (RiboTag) mice38; (2) mGfap-Cre-Ribotag-Stat3-loxP mice (Stat3-astro-cKO) were generated by crossing to Stat3-loxP mice24 and RiboTag mice; (3) mGfap-Cre-Ribotag-Smacra4-loxP mice (Smarca4-astro-cKO) were generated by backcrossing Smarca4-loxP mice (B6;129S2-Smarca4tm1.2cn)39 provided by Dr. Trevor Archer and mGfap-RiboTag mice. All mice were housed in a facility with 12-h light/dark cycle, controlled temperature and humidity, and were allowed free access to food and water. All experiments were conducted according to protocols approved by the Institutional Animal Care and Use Committee at University of California, Los Angeles.
Spinal cord injury (SCI).
All surgeries were performed under general anesthesia with isoflurane in oxygen-enriched air using an operating microscope (Zeiss, Oberkochen, Germany), and rodent stereotaxic apparatus (David Kopf, Tujunga, CA). Laminectomy of a single vertebra was performed. Partial crush SCI was made at cord level T10 using No. 5 Dumont forceps (Fine Science Tools, Foster City, CA) with a 0.4 mm spacer and with a tip width of 0.5 mm. Complete crush SCI was made as described12. All animals received the opiate analgesic, buprenorphine, subcutaneously before surgery and every 12 h for at least 48 h post-injury. Animals were evaluated thereafter blind to genotype and experimental condition. Daily bladder expression was performed for the duration of the study or until voluntary voiding returned.
LPS-mediated neuroinflammation.
E.coli-derived lipopolysaccharide (LPS) (Sigma-Aldrich L5024, O127:B8; Lot number 125M4091V with >500,000 endotoxin units per mg) was dissolved and diluted to 1 mg/ml in sterile PBS and stored in aliquots at −80°C. LPS (5 mg/kg) was administered to mice by intraperitoneal injection at 11 am. Mice were euthanised 24 hours later.
Mouse behavior analyses.
6-Point locomotor score: Partial crush mice were scored at 3, 7, 14, and 28 dpi for hindlimb stepping ability as described24. Mice with no hindlimb movement received a score of 0, and mice with normal walking were given a score of 5. Locomotor score uninjured baselines were taken one day before crush injury. Ladder rung test: Mice that received partial crush SCI were tested on 28 days post injury using the Ladder Walk Apparatus as described40. Briefly, metal rungs were inserted at random distances between two clear plastic sheets. The sheets were approximately 2” apart to prevent mice from turning around. A camera was placed on a slider, and as the mice moved through the apparatus, the camera was moved to follow. 4. A score of 0–6 was given for each hind paw for every step, and the average of 3 trials was used for each animal at every time point. Open Field (OF) test. LPS or no-LPS control mice were placed individually in opaque plastic containers measuring 12”X7”X10” inside a dimly-lit room. Mice underwent OF testing every day at 5 PM, starting on the day of injection, until sacrifice. Mice were allowed to acclimate for 3 minutes, and then recorded the following 6 minutes. A white noise generator was used to reduce ambient sound interference. AnyMaze video analysis software (Stoelting Co., Wood Dale, IL) was used to quantify distance traveled, time immobile, and time spent in the center of the arena as measures of sickness behavior41,42.
Harvesting of fresh spinal cord tissue for RiboTag analysis of astrocyte mRNA or for precipitation of astrocyte nuclei for single nuclei ATAC-sequencing (snATACseq).
Fourteen days after complete crush SCI or 24 hours following IP injection of LPS, spinal cords of wild-type (mGfap-RiboTag), Stat3-astro-cKO or Smarca4-astro-cKO mice were perfused with ice cold saline and spinal cords rapidly dissected and frozen on dry ice. These time points were selected on a disorder-specific basis based on evidence from previous studies that indicated that peak changes in transcriptome profiles could be observed at those specific times. For SCI samples, the central 3 mm of the lower thoracic lesion including the lesion core and 1 mm rostral and caudal were then rapidly removed and snap frozen in liquid nitrogen. Anatomically equivalent regions of spinal cord were taken from mice with treated with LPS or from genotype-matched uninjured controls. Frozen tissue samples were processed either for RiboTag analysis of mRNA or for precipitation of nuclei for snATACseq.
Isolation, sequencing and analysis of astrocyte mRNA using RiboTag technology.
Using fresh frozen spinal cord tissue harvested as described above, haemagglutinin (HA) immunoprecipitation (HA-IP) and purification of astrocyte ribosome-associated mRNA was carried out as described previously12. To avoid potential batch effects, frozen tissue samples were collected from entire experiments were then processed at the same time. Astrocyte RNA Integrity was analysed by a 2100 Bioanalyzer (Agilent) using the RNA Pico chip, RIN ≥ 8.9 for all samples. RNA concentration determined by RiboGreen RNA Assay kit (Life Technologies). cDNA was generated from 10 ng of RNA using the Universal plus mRNA-Seq Kit (Nugen). The workflow consisted of poly(A) RNA selection, RNA fragmentation and double-stranded cDNA synthesis using a mixture of random and oligo(dT) priming, followed by end repair to generate blunt ends, adaptor ligation, strand selection, and PCR amplification to produce the final library. Multiplexed sequencing was performed with a NovaSeq 6000 sequencer (Illumina) on a NovaSeq S2 flow cell to produce 50 base-pair paired-end reads. The number of reads obtained per sample ranged from 43.3 to 77.4 million (average 61.8 million). Data quality was assessed by Illumina SAV and demultiplexing was performed by Illumina Bcl2fastq2 v2.17. Sequences were aligned to mouse mm10 genome using STAR aligner (v.2.4.0j). The number of uniquely aligned reads per sample ranged from 54.2 to 84.7% (average 73.4%). Read counts were determined using HT-seq (v.0.6.0).). Four samples were removed from the analysis due to poor complexity (fewer than 7500 genes with >100 counts). At least 3, and in most cases 4, samples were evaluated per experimental condition. Differential expression analysis was conducted using Bioconductor EdgeR package (v.3.28.0) after removal of low count genes (five counts for all replicates in at least one condition). The threshold for significance of differential gene expression was defined as an adjusted p<0.1 calculated with EdgeR using Benjamini-Hochberg’s procedure for multiple comparisons adjustment and referred to as False Discovery Rate (FDR<0.1). This FDR<0.1 threshold was used for all samples to enable direct comparisons with our two previously published studies examining spinal cord astrocyte RNAseq after SCI or EAE using FDR<0.1 (Extended Data Figure 8), which had yielded biologically important information validated by additional means. To test whether use of a more conservative FDR altered our basic observations, we applied a FDR<0.05 to our EAE, SCI and LPS DEG data and then determined TRs. Although fewer DEGs were detected, their relative overlaps across the disorders were unchanged such that 2.6% DEGs were shared by all 3 disorders at FDR<0.1 and 2.1% were shared by all 3 disorders at FDR<0.05 and other comparisons also remained the same (Extended Data Fig. 1c). Principle component analysis (PCA) of DEGs showed that the differences in locations of disorders in PC space relative to non-reactive astrocytes as reflected in the percent of total vector length and the angles between vectors were precisely the same for all 3 disorders using either FDR<0.1 or FDR<0.05 and other comparisons also remained the same (Extended Data Fig. 1c). Similarly, although the TRs predicted at FDR<0.05 was reduced somewhat in comparison with a FDR<0.1, the relative overlaps of TRs across the disorders was unchanged such that 20% of TRs were shared by all 3 disorders at FDR<0.1 and 23% were shared by all 3 disorders at FDR<0.05 (Extended Data Fig. 1c). Thus, our basic conclusions regarding low overlap of DEGs across disorders and a significantly and substantially greater overlap of TRs were not changed by using the more conservative FDR. In addition, all of the TRs that we predicted as being common across all eight datasets from disorders with WT astrocytes continued to be predicted in our EAE and SCI data using the FDR<0.05, one of these core WT TRs, Rel, was no longer predicted in our LPS data at FDR<0.05, even though it was predicted from the Barres LPS data, and even though it was also predicted from ATACseq data in both LPS and SCI on the basis of significant changes in motif access (Fig. 1d). We considered this a type 2 error (failure to identify) introduced by the FDR<0.05, which provided further support for using the FDR<0.1 threshold to identify candidate TRs.
Threshold criteria for gene expression in healthy astrocytes.
Genes expressed by healthy astrocyte were identified by establishing a threshold for the minimum FPKM value from which a significant down-regulation in expression from a healthy state could be determined by differential gene expression analysis with an FDR<0.1. Genes with an FPKM value at or above this threshold in healthy astrocytes were considered healthy expressed genes (“EGs”). These criteria were then applied over three separate healthy astrocyte gene expression datasets acquired from separate sequencing runs from the SCI, EAE and LPS experiments to achieve a representative consensus healthy EG dataset for comparison with these datasets.
Transcriptional Regulator Enrichment Analysis (TREA).
Overview of methodology:
To identify TRs of astrocyte gene expression, we applied a conservative, multi-step methodology that draws on both computationally- and biologically-derived regulator-target gene interaction data from multiple resource databases: i) ChEA43: transcription factor regulation inferred from integrating genome-wide ChIP-X experiments. ChEA is a curated and continually updated database containing information on over 200,000 TR-gene interactions manually extracted from over 100 experimental studies involving ChIP-chip, ChIP-seq, ChIP-PET and DamID (together referred to as ChIP-X), from both mouse and human cells and tissues. Chea contains experimentally-determined binding of over 300 transcriptional regulatory molecules to over 30,000 target gene sites. ii) JASPAR44, and iii) TRANSFAC45 transcription factor DNA-binding preference position weight matrices: JASPAR and TRANSFAC are curated and continually updated collections of transcriptional regulator DNA-binding preferences, arranged as position weighted matrices. Transcriptional regulator DNA binding site information is derived from a combination of experimental biological and computational modelling, and iv) Ingenuity Pathway Analysis Upstream Regulator Analytic (IPA®, Qiagen, Valencia, CA): the Upstream Regulator Analytic references IPA’s expansive knowledgebase of interactions between TRs and their target genes, which are derived from published, peer-reviewed literature. By examining a user’s gene expression dataset, IPA Upstream Regulator analytic can determine the involvement of a transcriptional regulator based on dataset enrichment for known target genes. While details surrounding the IPA knowledge base are proprietary, Upstream Regulator results are based on findings from published peer-reviewed biological experiments.
Using these diverse resource databases, TREA identifies TRs implicated in regulating DEGs by interrogating multiple forms of TR-target gene regulatory interactions, which include findings from experimental studies involving techniques such as chromatin immunoprecipitation and genetic loss-of-function studies, as well as well-validated predictive computational DNA binding ‘motif’ analytics. As such, TR-target gene interactions considered by TREA include traditional direct TR-DNA binding mechanisms, as well as indirect forms of gene expression regulation wherein a TR may act through different types of intermediaries to effect expression of downstream target genes, including chromatin modifiers and other forms of epigenetic regulators. Together, these resource databases allow for interrogation of gene expression datasets for enrichment of downstream targets of ~1350 TRs.
Examining an astrocyte gene expression profile by TREA:
Universal gene ID formatted astrocyte gene expression datasets were first evaluated independently by each database analytic. Chea and JASPAR-TRANSFAC databases were accessed through the online open-access Enrichr platform46. IPA Upstream Regulator Analytic was accessed through subscription to the IPA software package. Resource database output files were then streamlined to contain only statistically enriched TR IDs (Enrichr adj. P < 0.05; IPA Upstream Regulator P < 0.01) and their associated downstream astrocyte target gene IDs. Resource database output files were subsequently processed for consensus analysis by TREA using in-house python scripts (see GitHub link below) in order to generate a final TREA library containing IDs of significantly enriched TRs and associated astrocyte target genes. To be included in a final TREA library, TRs must meet ‘type I’ or ‘type II’ TREA criteria: type I) TR is predicted by IPA® and Chea or JASPAR-TRANSFAC; type II) TR is predicted by IPA® or Chea or JASPAR-TRANSFAC and exhibits a significant fold-change (up or down) in the respective astrocyte gene expression profile (Extended Data Fig. 1d). When the same TR was predicted across two or more resource databases (e.g. IPA and Chea), target gene ID lists were merged into a single TR-target gene signature in the final TREA library for that gene expression profile. In-house python scripts for TREA, protocols, an example gene expression dataset and TREA resource database output files can be found in the TREA GitHub repository (https://github.com/burdalab/TREA).
Isolation and precipitation of astrocyte nuclei for single nuclei ATAC-sequencing.
Using fresh frozen spinal cord tissue harvested as described above, astrocyte-enriched nuclei were isolated by antibody-binding and magnet-assisted nuclear immunoprecipitation (MAN-IP) using well characterized procedures47–49.To avoid potential batch effects, frozen tissue samples were collected from entire experiments were then processed at the same time. Tissue samples were first gently dissociated by trituration, trypsinized and pelleted by centrifugation. Nuclei were extracted from cell pellets by gentle resuspension in ice-cold lysis buffer (10mM Tris buffer, 10mM NaCl, 3 mM MgCl2, 0.1% Nonidet P40 Substitute). Nuclei were pelleted by centrifugation (500RPM (Model 5415R, Eppendorf) for 5 min at 4°C) and then resuspended in Nuclei Wash and Resuspension Buffer (NWRB) (1xPBS, 1% BSA, 0.2U/uL RNAse inhibitor) before being washed once more in NWRB and then filtered using a 5 mL Polystyrene Round-Bottom Tube with 35 μm Cell-Strainer Cap and concentrated to a nuclei concentration of 1000 nuclei/μL (1 × 106 nuclei/mL). Nuclei quality was assessed by trypan blue staining. Nuclei were resuspended and incubated with Sox9 rabbit monoclonal antibody (ThermoFisher, Cat#72016) for 30 minute and then centrifuged at 700g for 10 minutes. The pellet was resuspended in 80μl of MACS Buffer composed of 1X PBS (Tissue Culture grade; Ca2+, Mg2+ free), 0.5% Nuclease free Bovine Serum Albumin (BSA), and 2mM EDTA, and then incubated with anti-Rabbit IgG Microbeads (Miltenyi, Cat# 130-048-602) for 20 minutes at 4°C. After washing with 1ml of MACS buffer at 300g for 10 minutes at 4 °C, immunolabeled nuclei were enriched by magnetic separation using MACS MS columns (Miltenyi Cat# 30-042-201, 130-042-102 and 130-042-303). After isolation and washing, dissociated single nuclei were suspended in 1 x Nuclei Buffer and concentrated by centrifugation to 3000 – 7000 nuclei/μL. and immediately processed for ATAC-seq by the UCLA Technology Center for Genomics & Bioinformatics (TCGB). Single nuclei partitioning, barcoding and library preparation was performed using the 10X Genomics Chromium Controller and Chromium™ Single Cell ATAC Solution, and sequenced using Novaseq S2 (illumina) with pair end reads (2×50 – 50bp length) and >300 M read per sample.
Single nuclei ATAC-sequencing analysis.
For all libraries, demultiplexed FASTQs were obtained from the UCLA Technology Center for Genomics and Bioinformatics (TCGB). Each library was processed individually through the 10x Cellranger-ATAC pipeline using Cellranger-ATAC count with default settings and aligned to the mm10 reference genome (version 2020-A-2.0.0). The output fragments.tsv files for all libraries (Extended Data Fig. 2e) were loaded into R and processed together using Archr v.1.0.120. After filtering low quality nuclei and doublets, iterative Latent Semantic Indexing (LSI)50 was used to perform dimensionality reduction and clustered using Uniform Manifold Approximation and Projection (UMAP) (Extended Data Fig. 2f). Using Archr, a “gene score” was calculated for every gene on a per-nuclei basis, which integrates accessibility to the gene body with activity of putative distal regulatory elements to generate a prediction of how highly a gene may be expressed based on chromatin accessibility20. Differentially Accessible Genes (DAGs) were identified through comparison of gene scores based on the question being asked by Wilcoxon Ranked Sum test with a False Discovery Rate p value correction. Gene scores enabled the assignment of nuclei identity to clusters generated in the UMAP based on putative marker genes(Extended Data Fig. 2g). Magnetic-bead isolated nuclei were enriched for astrocytes but also contained nuclei from other lineages(Extended Data Fig. 2g). Astrocytes were identified through high scores for known astrocyte genes including Slc1a2, Slc4a4, Aqp4, S100b, Gfap, Aqp4, Aldh1l1, Gja1 etc and subsetted to be used for the rest of the analysis in this paper (Extended Data Fig 2g,h). To identify peaks, astrocytes were first pseudobulked by condition and the ‘addReproduciblePeakset’ function was used in Archr. First 501 bp,fixed-width peaks were called using MACS2 v2.27.151. This peak set was then used as input to an iterative overlap peak merging procedure implemented in Archr20 which both reduces “daisy-chaining” peaks and improves identification of smaller peaks. Differentially Accessible Peaks (DAPs) were identified using Wilcoxon ranked sum test, accounting for bias due to number of reads per nuclei and TSS enrichment. This peakset was then annotated for the presence of Transcription Factor motifs using Position Weight Matrices (PWMs) from Archr’s CISBP database20, the CISBP website52, or ENCODE53. Motif enrichment was calculated using Chromvar21, to generate an enrichment of ‘deviation’ score for each transcription factor on a per-nuclei basis. Differentially Accessible Motifs (DAM) were identified through comparison of deviation scores by using Wilcoxon Rank-Sum Test with a False Discovery Rate p value correction based on the question being asked. For visualization of ATAC peaks as track diagrams, the Archr generated peakset for each condition was outputted as a BigWig file and visualized using the IGV Genome Browser.
Histology, immunohistochemistry and in situ hybridization.
After terminal anaesthesia by barbiturate overdose, mice were perfused transcardially with 4% paraformaldehyde (Electron Microscopy Sciences, Hatfield, PA). Spinal cords were removed, post-fixed overnight, and cryoprotected in buffered 30% sucrose for 48 hours. Frozen sections (30 μm horizontal) were prepared using a cryostat microtome (Leica) and processed for immunofluorescence as described12. All antibodies used were sourced from commercial vendors and were selected because they had previously been validated for fluorescent immunohistochemistry (IHC) in mouse tissue and had manufacturer provided demonstration of specificity based on Western Blots and in most cases validation in peer-reviewed publications. Primary antibodies used: rabbit Aldh1l1 (1:1000; Ab87117; Abcam, Cambridge, MA, USA); rabbit anti-Arnt (1:200; NB100–479SS; Novus Biologicals, Centennial, CO, USA); rabbit anti-Atf2 (1:200; 14834–1-AP; ProteinTech, Rosemont, IL, USA); rabbit anti-Atf4 (1:200; NBP2–67766; Novus Biologicals); rabbit anti-Bach1 (1:500; NBP2–55133; Novus Biologicals); rabbit anti-Bcl3 (1:200; 23959–1-AP; ProteinTech); rabbit anti-Bcl6 (1:200; NBP2–59786; Novus Biologicals); rabbit anti-Ccnd1 (1:200; NBP2–24695; Novus Biologicals); goat anti-CD13 (1:600, AF2335; R&D Systems, USA); rat anti-CD68 (1:1000, MCA1957; Biorad, USA); sheep anti-Cebpa (1:200; AF7094; Novus Biologicals); rabbit anti-Ctcf (1:200; HPA004122; Sigma, St. Louis, MO, USA); rabbit anti-Ctnnb1 (1:200; 8480T; Cell Signaling, Boston, MA, USA); rabbit anti-Cux1 (1:200; 11733–1-AP; ProteinTech); rabbit anti-Ebf1 (1:400; 14159–1-AP; ProteinTech); rabbit anti-Elf1 (1:300; 22565–1-AP; ProteinTech); rabbit anti-Ep300 (1:800; 86377S; Cell Signaling); rabbit anti-Essrb (1:300; 22644–1-AP; ProteinTech); rabbit anti-Ets2 (1:200; NBP3–04749; Novus Biologicals); rabbit anti-Ezh2 (1:200; A11085; Abclonal, MA, USA); goat anti-Ezh2 (1:200; AF4767; Novus Biologicals); rabbit anti-Fev (1:200; 25058-AP; ProteinTech); rabbit anti-Fus (1:200; 11570–1-AP; ProteinTech); rabbit anti-Fli1 (1:200; BP2–57756; Novus Biologicals); rabbit anti-Gata2 (1:200; 11103–1-AP; ProteinTech); rabbit anti-Gata4 (1:200; 19530–1-AP; ProteinTech); rabbit anti-GFAP (1:1,000; GA524; Dako/Aligent, Carpinteria, CA, USA); rat anti-GFAP (1:1,000, 13–0300; Thermofisher, USA); rabbit anti-Gfi1 (1:200; 14198–1-AP, ProteinTech); rabbit anti-hemagglutinin (HA) (1:1000, H6908, Sigma); goat anti-HA (1:800, NB600–362, Novus Biologicals); rabbit anti-Hexim1 (1:600;15676–1-AP, ProteinTech); rabbit anti-Hmga1 (1:200; NBP3–03907; Novus Biologicals); rabbit anti-Hoxc8 (1:200; 15448–1-AP; ProteinTech); rabbit anti-Htt (1:200; ab109115; Abcam); guinea pig anti-Iba1 (1:1000, 234004; Synaptic Systems, USA); rabbit anti-Iba-1 (1:800, 019–19741; Wako, Osaka, Japan); rabbit anti-Id3 (1:200; 9837; Cell Signaling); sheep anti-Irf5 (1:200; AF4508; Novus Biologicals); rabbit anti-Irf8 (1:200; NBP2–99595; Novus Biologicals); rabbit anti-Irf9 (1:200; 14167–1-AP; Proteintech); rabbit anti-JunB (1:200; ab128878; Abcam); rabbit anti-JunD (1:1000; ab181615; Abcam); rabbit anti-Klf2 (1:200; A16480; Abclonal); rabbit anti-Klf13 (1:200; 18352–1-AP; Proteintech); rat anti-MBP (1:300, MAB386; Milipore); rabbit anti-Mef2a (1:200; A7911; Abclonal); rabbit anti-Myb (1:200; NBP2–98885; Novus Biologicals); rabbit anti-Mycn (1:200;10159–2-AP; Proteintech); rabbit anti-Myod1(1:200;18943–1-AP; Proteintech); rabbit anti-Nab2 (1:400; NMP1–82804; Novus Biologicals); guinea pig anti-NeuN (1:1000, 266004; Synaptic Systems); rabbit anti NeuN (1:1000, ab177487, Abcam); sheep anti-Nfkbia (1:200; AF4299; Novus Biologicals); rabbit anti-Nfya (1:200; 12981–1-AP; Proteintech); rabbit anti-Nme2 (1:200; NBP3–05624; Novus Biologicals); rabbit anti-Notch1 (1:200; 20687–1-AP; Proteintech); rabbit anti-Npm1 (1:400; 10306–1-AP; Proteintech); rabbit anti-Nr2f1 (1:200; 24573–1-AP; Proteintech); rabbit anti-Nupr1 (1:200; NBP1–98280; Novus Biologicals); rabbit anti-Pbx1 (1:200, 18204–1-AP; Proteintech); goat anti-Pdx1 (1:200, AF2419; R&D Systems); rabbit anti-Pitx1 (1:200; 10873–1-AP; Proteintech); rabbit anti-Ppard (1:200; NBP2–22468; Novus Biologicals); rabbit anti-Prxx2 (1:200; NBP1–80893; Novus Biologicals); rabbit anti-PU.1 (1:300; 2266S; Cell Signaling); rabbit anti-RAR (1:200; NBP2–47314; Novus Biologicals); rabbit anti-Rel (1:200; NBP2–34064; Novus Biologicals); rabbit anti-Rpsa (1:200; NBP2–41246; Novus Biologicals); rabbit anti-Runx1 (1:200; NBP1–89105; Novus Biologicals); rabbit anti-Runx2 (1:200; A2851; Abclonal); rabbit anti-Runx3 (1:200; NBP2–38863; Novus Biologicals); rabbit anti-Rxra (1:200; 21218–1-AP; Proteintech); goat anti-Smarca4 (1:500; AF5738; Novus Biologicals); mouse anti-Smarca4 (1:100, SC-17796; Santa Cruz, CA, USA); rabbit anti-Snai1 (1:200; 13099–1-AP; Proteintech); rabbit anti-Snd1 (1:400; 10760–1-AP; Proteintech); goat anti-Sox9 (1:500, AF3075; R&D Systems); rabbit anti-Sox9 (1:600, 702016; ThermoFisher); rabbit anti-Srebf1 (1:400; 14088–1-AP; Proteintech); rabbit anti-Srebf2 (1:200; NB100–74543; Novus Biologicals); rabbit anti-Srp (1:200; NBP1–87814; Novus Biologicals); rabbit anti-Stat3 (1:400;10253–2-AP; Proteintech); rabbit anti-Stat4 (13028–1-AP; Proteintech); sheep anti-Tcf3 (AF6116; Novus Biologicals); rabbit anti-Tcf4 (1:200; 22337–1-AP; Proteintech); rabbit anti-Tgm1 (1:200; 12912–3-AP; Proteintech); rabbit anti-Thra (1:200; NBP2–22523; Novus Biologicals); sheep anti-Tle1 (1:200; AF5947; R&D Systems); rabbit anti-Tp63 (1:200; 12143–1-AP; Proteintech); rabbit anti-Trim28 (1:200; A2245; Abclonal); rabbit anti-Xrn2 (1:200; 11267–1-AP; Proteintech); rabbit anti-Yap1 (1:200; 13584–1-AP; Proteintech); goat anti-Zbtb16 (1:200, AF2944; R&D Systems); rabbit anti-Zfp36 (1:200; SAB4301804; Sigma).
Fluorescence secondary antibodies were conjugated to: Alexa 488 (green) or Alexa Cy3 (550, red) or Cy5 (649, far red) all from (Jackson Immunoresearch Laboratories, USA). Mouse primary antibodies were visualized using the Mouse- on-Mouse detection kit (M.O.M., Vector). Nuclear stain: 4′,6′-diamidino-2-phenylindole dihydrochloride (DAPI, blue; 2 ng ml−1; Molecular Probes, USA). Sections were cover-slipped using ProLong Gold anti-fade reagent (Invitrogen, Grand Island, NY). Sections were examined and photographed with an epifluorescence microscope using structured illumination hardware and deconvolution software (Zeiss, Oberkochen, Germany). In situ hybridization was carried out on fixed-frozen mouse spinal cord sections using proprietary RNAscope® probes (Mm-Cnr1, Mm-Gfap) and Fluorescent Multiplex Detection kit per manufacturer protocols (Advanced Cell Diagnostics, Inc. Newark, CA).
Morphometric quantification procedures.
SCI lesions. Horizontal spinal cord sections at the level of the central canal were stained by immunohistochemistry for GFAP, CD13 and MBP, whole slides were imaged using Leica Aperio Versa at 20X magnification and staining-intensity quantification was performed on raw/uncorrected images taken using standardized exposure times for each fluorescent channel. Average staining intensity values across the width of the spinal cord as a function of longitudinal distance were plotted using NIH ImageJ software and were centered at the epicenter of the lesion core which was defined as the minimum of the GFAP intensity trace. Total staining values were determined by taking the integral (Area under the curve (AUC)) of the intensity profile for each IHC stain extending 1000 μm rostral and caudal from the injury epicenter. Effects of systemic LPS. To evaluate microglia, horizontal spinal cord sections were stained by immunohistochemistry for Iba1 and CD68 and photographed using constant exposure settings. Numbers and lengths of Iba1-stained microglial cell processes were evaluated by using Imaris image analysis software (Oxford Instruments, Concord, MA). Staining intensity of CD68 and area of Iba1 were quantified by using NIH ImageJ software. To evaluate neurons, horizontal spinal cord sections and transverse sections of brain neuron density, were stained by immunohistochemistry for NeuN and photographed using constant exposure settings, and numbers of neurons were quantified by using NIH ImageJ software.
Statistics and Reproducibility.
Statistical evaluations of repeated measures were conducted by one-way or two-way ANOVA with post hoc, independent pairwise analysis as per Bonferroni or Tukey tests, or t tests using Prism 8 (GraphPad Software Inc, San Diego, CA). P-values, F-values and degrees of freedom are reported in the figures, figure legends and within the source data files. Differences with p<0.05 were considered statistically significant. Power calculations were performed using G*Power Software V 3.1.9.2. All immunohistochemistry and in situ hybridization shown (Main figures 1d, 2f–g; 3b, g, i, o–p, r, u, x; Extended Data Figures 1b; 3a, c; 4a; 5a; 6e–f, j) were repeated at least three times with similar results.
Principle Component analysis (PCA).
PCA of DEG, TR, histopathological and mouse behavioral assessment datasets was performed using the XLStat Add-in in Excel (Addinsoft Inc, Long Island City, NY). PCA was used to reduce multi-dimensional data sets that had a total number of variables of three or more such that each experimental condition could be satisfactorily described by 1, 2 or 3 variables only and represented as 1-, 2-, or 3- dimensional projections. PCA was performed using Pearson’s correlation methodology as we have described previously54 and data was graphically represented using distance and correlation biplots where appropriate with the percentage variability of the total system described by each principal component indicated. PCA vector magnitudes were used to define the Euclidean distance or the length of the line segment between individual conditions.
Extended Data
Extended Data Fig. 1. RNA sequencing, Transcriptional Regulator Enrichment Analysis (TREA) and previously published TRs.
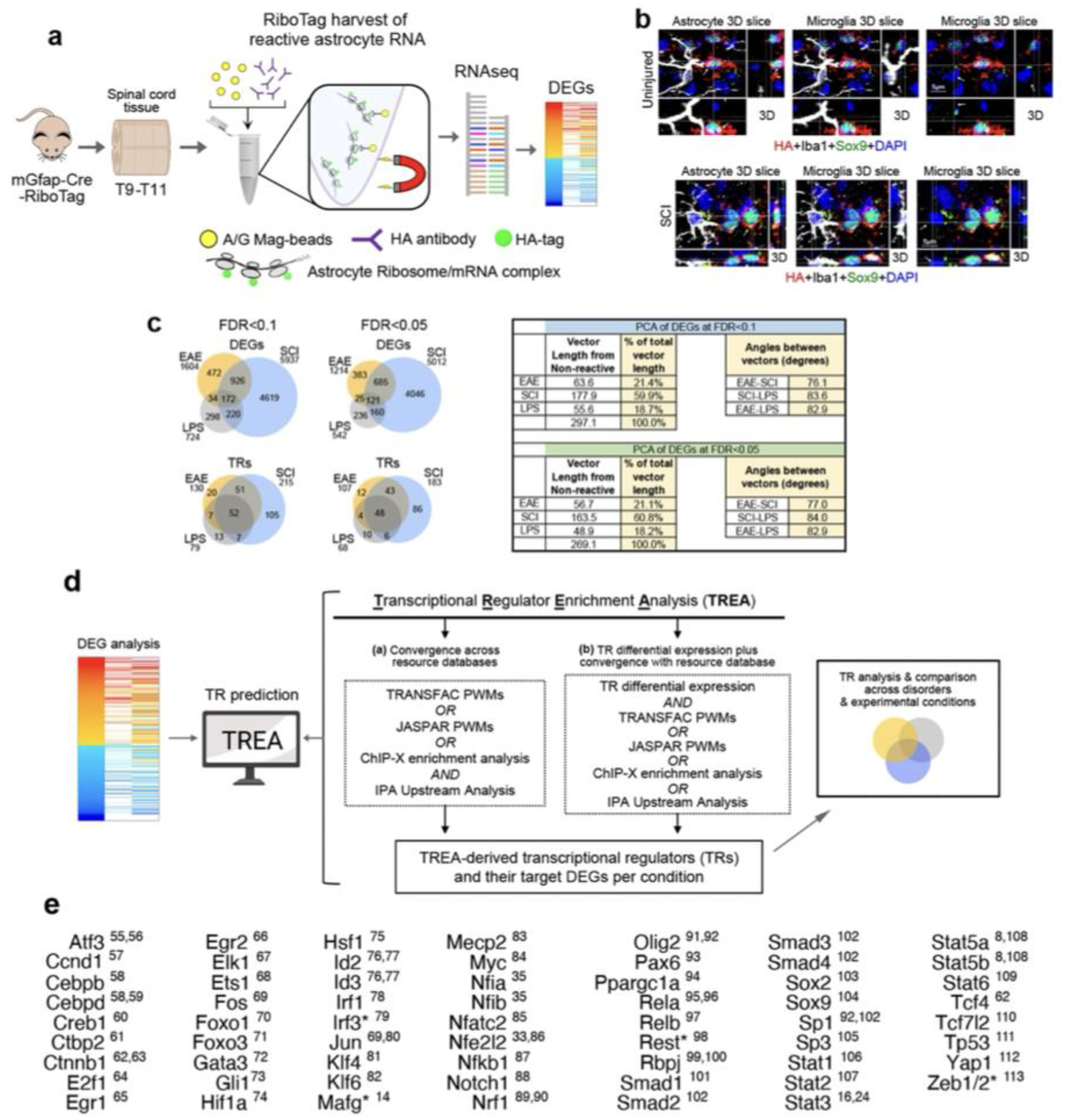
a. Flow diagram of RNAseq procedure. To minimize technical differences, we used mice of similar age and genetic background for all experimental disorder models and examined the same anatomical region (thoracic spinal cord). Spinal cord tissue from different experiments were frozen until all experiments were completed. All tissue was then processed at the same time to limit the potential for technical variations. The RiboTag procedure was used to harvest ribosome-associated RNA transcripts specifically from reactive astrocytes12. RiboTag hemagglutinin (HA) was transgenically-targeted specifically to astrocytes by using well-characterized mGFAP-Cre12. RNA-sequencing and analysis were conducted under identical conditions. b. Specificity of RiboTag-HA targeting to astrocytes and not microglia or other cells. Two sets of orthogonal (3D) scans from uninjured spinal cord or after spinal cord injury (SCI) with multichannel immunofluorescence for HA targeted to astrocytes plus Sox9 and Iba1 as markers of astrocytes or microglia respectively. The same areas are shown with different fluorescent wavelength filters and different orthogonal slices that demonstrate 3D staining associated with astrocytes or microglia. HA is robustly present in Sox9-positive astrocytes but is not detectable in Iba1-postive microglia in either uninjured cord or after SCI. Absence of HA-targeting to neurons or oligodendrocytes has been demonstrated previously12. These immunohistochemical comparisons were repeated independently three times with similar results. c. Venn diagrams show that the relative proportions of shared astrocyte reactivity DEGs and TRs identified in spinal cord astrocytes after EAE, SCI or LPS do not detectably differ when using thresholds of FDR<0.1 or FDR<0.05. Table shows PCA analysis of DEGs identified in spinal cord astrocytes after EAE, SCI or LPS using thresholds of FDR<0.1 or FDR<0.05. The relative locations of the three disorders in PC space when compared to non-reactive astrocytes do not detectably differ when using thresholds of FDR<0.1 or FDR<0.05 as reflected in the percent of total vector length and the angles between vectors. d. Flow diagram of Transcriptional Regulator Enrichment Analysis (TREA) procedure for TR identification by upstream analysis of DEGs in reactive astrocytes. To identify TRs of astrocyte gene expression, we applied a conservative, multi-step algorithm that draws on both computationally- and biologically-derived regulator-target gene interaction data from multiple resource databases: i) ChEA43: transcription factor regulation inferred from integrating genome-wide ChIP-X experiments, ii) JASPAR44 and iii) TRANSFAC45 transcription factor DNA-binding preferences as position weight matrices, and iv) Ingenuity Pathway Analysis Upstream Regulator Analytic (IPA®, Qiagen, Valencia, CA). Using these resource databases, TREA identifies TRs implicated in regulating DEGs by interrogating multiple forms of TR-target gene regulatory interactions that include findings from experimental studies involving techniques such as chromatin immunoprecipitation and genetic loss-of-function studies, as well as well-validated predictive computational DNA binding ‘motif’ analytics. In this manner, TR-target gene interactions considered by TREA include traditional direct TR-DNA binding mechanisms, as well as indirect forms of gene expression regulation wherein a TR may act through different types of intermediaries to effect expression of downstream target genes, including chromatin modifiers and other forms of epigenetic regulators. TRs were included if they met either of two criteria: (1) convergence across resource databases; (2) differential gene expression of the TR plus convergence with at least one resource database. Together, these databases allow for interrogation of gene expression datasets for enrichment of downstream targets for approximately 1350 TRs. Resource database output files containing statistically enriched TRs and their downstream astrocyte target gene IDs were processed for TREA using in-house python scripts available at GitHub repository (https://github.com/burdalab/TREA). Final TREA libraries containing significantly enriched TRs and associated astrocyte target genes were then generated for each condition’s DEG dataset. TREA libraries were used for all comparisons of astrocyte TRs and gene expression profiles across disorders and experimental conditions and to generate a resource database of reactive astrocyte TRs and the DEGs that they regulate across a broad spectrum of CNS disorders and conditions. This database can be accessed via an open-source website http://tr.astrocytereactivity.com and has multiple search parameters according to TR, DEG or condition. e. Published astrocyte reactivity TRs plus literature references. * 4 of 62 published TRs not predicted in EAE, LPS or SCI.
Extended Data Fig. 2. Single nucleus ATAC sequencing.
a. Experimental models: All ATACseq experiments examining LPS or SCI treatments or healthy controls used wild-type or transgenic mice of 657Bl6 background strain. Transgenic mice expressing mGFAP-Cre12 were used for astrocyte-specific deletion (cKO) of Smarca4 or Stat3. b. The same region of thoracic spinal cord at T9-T10 was harvested for all evaluations. Spinal cord tissue from different experiments were frozen until all experiments were completed. All tissue was then processed at the same time to limit technical variations. c. Two spinal cords from the same experimental group were pooled to prepare suspensions of nuclei. These suspensions were enriched for astrocyte nuclei with a Sox9 antibody and magnetic beads precipitation. Two such suspensions were prepared from a total of n=4 mice per experimental condition. d. Two biological replicates, each consisting of nuclei from two mice, were used for single nucleus ATACseq. All biological replicates from all experimental conditions were sequenced at the same time to avoid batch effects. e. Box plots compare the distribution of the per-nucleus averages of unique DNA fragments per nucleus or TSS enrichment per nucleus for the seven experimental conditions listed, with each dot representing the average value of all nuclei collected from each experimental group (whiskers show range, box encompass 25–75% quartiles and the centre line indicates the median; n=8 WT healthy mice and n=4 mice for all other experimental conditions). f. UMAP clustering based on differential ATAC peaks across 145,973 high quality nuclei isolated across all conditions showed separation of nuclei into multiple distinct clusters. g. UMAP distribution of examples of specific DAGs used to identify the dominant cell types in different clusters. h. Heatmap of DAGs used to identify specific cell types. Each line represents the per-nucleus z-score for a given gene averaged across all nuclei in a cluster. i. UMAP and stacked bar graph show relative contribution of biological replicates for different experimental conditions. Because experiments for LPS and SCI were conducted at different times, groups of healthy control mice were collected for each experiment. All biological replicates showed similar contributions to their respective clusters, confirming that separation of treatment groups with essentially no overlap of LPS with either healthy or SCI was due to biological variation and not due to technical artifacts. * each biological replicate consisted of nuclei from two mice.
Extended Data Fig. 3. Identification of TR proteins in reactive astrocytes by immunohistochemistry.

For immunohistochemical (IHC) detection of TR protein we focused on previously unpublished, TREA-predicted astrocyte reactivity TRs for which sensitive antibodies were commercially available whose specificity was supported by western blots. a. Newly identified TRs co-localized by IHC in reactive astrocytes in thoracic spinal cord (T9-T-10) after LPS treatment. TRs are show in alphabetical sequence. b. Summary of all (newly identified and previously published) astrocyte reactivity TRs after LPS treatment identified here by at least two experimental procedures, either prediction from DEGs by TREA, or prediction based on significant change in motif access determined by ATACseq or by IHC or by all three. c. Newly identified TRs co-localized by IHC in reactive astrocytes in thoracic spinal cord (T9-T-10) after SCI (continued in Extended data figures 4,5). TRs are show in alphabetical sequence. Each immunohistochemical evaluation was repeated at least three times with similar results.
Extended Data Fig. 4. . Immunohistochemistry and motif analysis of TR proteins in reactive astrocytes after SCI.

a. Newly identified TRs co-localized by IHC in reactive astrocytes in thoracic spinal cord (T9-T-10) after SCI (see also Extended data figures 3,5). Each immunohistochemical evaluation was repeated at least three times with similar results. b. Summary and heatmap of all (newly identified and previously published) astrocyte reactivity TRs after SCI that have detectable DNA-binding motifs and were identified here as exhibiting significant change in motif access determined by ATACseq.
Extended Data Fig. 5. Immunohistochemistry of TR proteins in reactive astrocytes after SCI.

a. Newly identified TRs co-localized by IHC in reactive astrocytes in thoracic spinal cord (T9-T-10) after SCI (see also Extended data figures 3,4). b. Summary of all (newly identified and previously published) astrocyte reactivity TRs after LPS treatment identified here by at least two experimental procedures, either prediction from DEGs by TREA, or prediction based on significant change in motif access determined by ATACseq or by IHC or by all three. c. Nine examples of previously published TRs co-localized by IHC in reactive astrocytes in thoracic spinal cord (T9-T-10) after SCI show that these exhibit similar staining patterns to TRs newly identified here. Each immunohistochemical evaluation was repeated at least three times with similar results.
Extended Data Fig. 6. Astrocyte-specific TR deletion (cKO).
a. Experimental models: Transgenic mice expressing mGFAP-Cre12 were used for astrocyte-specific deletion (cKO) of Smarca4 or Stat3. Smarca4 was selected as a newly identified TREA-predicted reactivity TR that lacks DNA binding motifs and acts as a chromatin regulator via protein-protein interactions. Stat3 is a well-established astrocyte reactivity TR that acts via DNA-binding motifs12,24 and that was predicted by both TREA and ATACseq motif analysis as a reactivity TR in both LPS and SCI. b. Heatmaps show that Smarca4-astro-cKO had minimal effects on overall gene expression or on the expression of highly enriched astrocyte genes under basal conditions in untreated mice, shown as mean FPKM (fragments per kilobase of transcript sequence per million mapped fragments). c. Immunohistochemistry images and graphs (mean ± sem) of various staining parameters show that Smarca4-astro-cKO had no visibly detectable or quantifiably significant effects on the appearance or number of astrocytes, neurons or microglia in untreated mice. Unpaired t tests, ns non-significant WT versus Smarca4-cKO, n=4 mice per group. d. Effects of Smarca4-astro-cKO on RNAseq reads of Gfap and seven predicted Smarca4-regulated DEGs that are not expressed in WT untreated and are upregulated by LPS in WT but not Smarca4-astro-cKO mice. e. In situ hybridization shows predicted Smarca4-regulated gene, Cnr1, expresed in WT, but not Smarca4-astro-cKO mice after LPS. f. Multichannel immunofluorescence demonstration of Stat3 and Srebf1, or of Smarca4 and Zfp36, in the same reactive astrocytes after SCI. g. Heatmaps compare changes from healthy in differential gene expression (DEG) or differential chromatin accessibility (DAG) across the same genes after LPS or SCI in WT, Smarca4-cKO or Stat3-cKO mice, and graphs show an 86 to 96% congruence between changes in gene expression and changes in chromatin accessibility in both Smarca4- and Stat3-regulated DEGs after LPS or SCI respectively. h. Heatmaps show that Stat3-cKO significantly alters the changes from healthy normally observed after SCI in both differential gene expression (DEG) and differential chromatin accessibility (DAG) across the same 31 genes that lack Stat3-binding motifs. i. Heatmaps show that Stat3-cKO significantly alters the changes from healthy normally observed after SCI in differential gene expression (DEG) of 71 chromatin regulators. j. Multichannel immunofluorescence of Irf9 and Cxcl10 in the same reactive astrocyte after SCI. k. Graphs (mean ± sem) show effects of Smarca4-astro-cKO and Stat3-astro-cKO on various microglial histopathological responses to LPS, P values are cKO+LPS versus WT+LPS, one-way ANOVA with Bonferroni’s test, ns nonsignificant; for all graphs WT with no LPS n = 18 mice, all other conditions n = 6 mice. PCA shows composite microglia histopathology score derived from histopathological quantifications in the four graphs. l. Immunohistochemistry and graphs of mean ± sem (n ≥ 6) cell counts of the neuronal marker, NeuN, shows that high dose systemic LPS sufficient to cause pronounced microglial activation and behavioral effects did not lead to detectable neuronal loss in either spinal cord or brain. One-way ANOVA with Bonferroni’s test, ns nonsignificant; for both graphs WT with no LPS n = 18 mice, all other conditions n = 4 mice. m. Distance biplot of PCA for the effects of Smarca4-astro-cKO and Stat3-astro-cKO on composite locomotor scores after SCI. The locations of values for each individual locomotor parameter, from either the longitudinal observer scored Open Field (OF) evaluations or ladder walk testing at 28 days after SCI, indicates their contributions to defining the PC space. Graph (mean ± sem) shows composite SCI locomotor score derived from PCA of all OF and ladderwalk locomotor parameters recorded over 28 days of recovery; P values are cKO versus WT, one-way ANOVA with Bonferroni’s test, n = 11 mice per group. n. Immunofluorescence images and graph (mean ± sem) of staining intensity show effects of Smarca4-astro-cKO and Stat3-astro-cKO on CD68 and Gfap 28 days after SCI. o. Bar graph (mean ± sem) shows composite histology scores derived from PCA of Gfap, CD13, MBP and CD68 quantifications at 28 days after SCI; P values are cKO versus WT, one-way ANOVA with Bonferroni’s test, WT n = 8, Smarca4cKO n = 7, Stat3cKO n = 9 mice. Line graph shows correlation analysis of composite locomotor and histological scores. p. DEG-associated functional signaling pathway analysis shows that deletion of either Smarca4 or Stat3 alters many functions normally associated with WT astrocyte reactivity after LPS or SCI. Each immunohistochemical or in situ hybridization evaluation shown (c,e,f,j,l,n) was repeated at least three times with similar results.
Extended Data Fig. 7. DEGs and TRs in reactive astrocytes compared with astrocytes in healthy tissue.
a,b. Venn diagrams and graph compare proportions of astrocyte reactivity DEGs that derive from genes that either are, or are not, expressed in healthy astrocytes in EAE, LPS or SCI. c-f. Heatmap, Venn digrams and graphs compare proportion of TREA-predicted reactivity TRs that derive from TRs that either are, or are not, predicted in healthy astrocytes in EAE, LPS or SCI. In all three disorders much higher proportions of TRs than DEGs were recruited from those not detectably active in healthy tissue. Notably, of the TRs shared by all three disorders 87% were derived from TRs already active in healthy states, whereas 70%, 76% and 54% of disorder unique TRs respectively in EAE, SCI or LPS, were derived from TRs not detectably active in healthy states.
Extended Data Fig. 8. Comparison of DEGs and TRs across disorders.

8. a. Heatmap and graph show that in every disorder examined, TRs overlapped more with other disorders than did DEGs. b. Bar graphs show numbers of astroctye DEGs or TRs across all 15 disorders and conditions examined here. c. 3D distance plots of PCA for DEGs and TRs from disorders with WT astrocytes. Graphs on left show PCA of all 8 disorders, and on right show PCA of 5 disorders with three most divergent disorders removed. d. TRs predicted in three or more of six neurodegenerative disorders with genetic mutations or polymorphisms, compared with their predictions in disorders with WT astrocytes. No TRs are unique to multiple neurodegenerative disorders. e. DEGs upregulated in three or more of six neurodegenerative disorders with genetic mutations or polymorphisms, compared with their expression levels in disorders with WT astrocytes. No DEGs are unique to multiple neurodegenerative disorders.
Extended Data Fig. 9. Schematic of a working model of astrocyte reactivity transcriptional regulation.
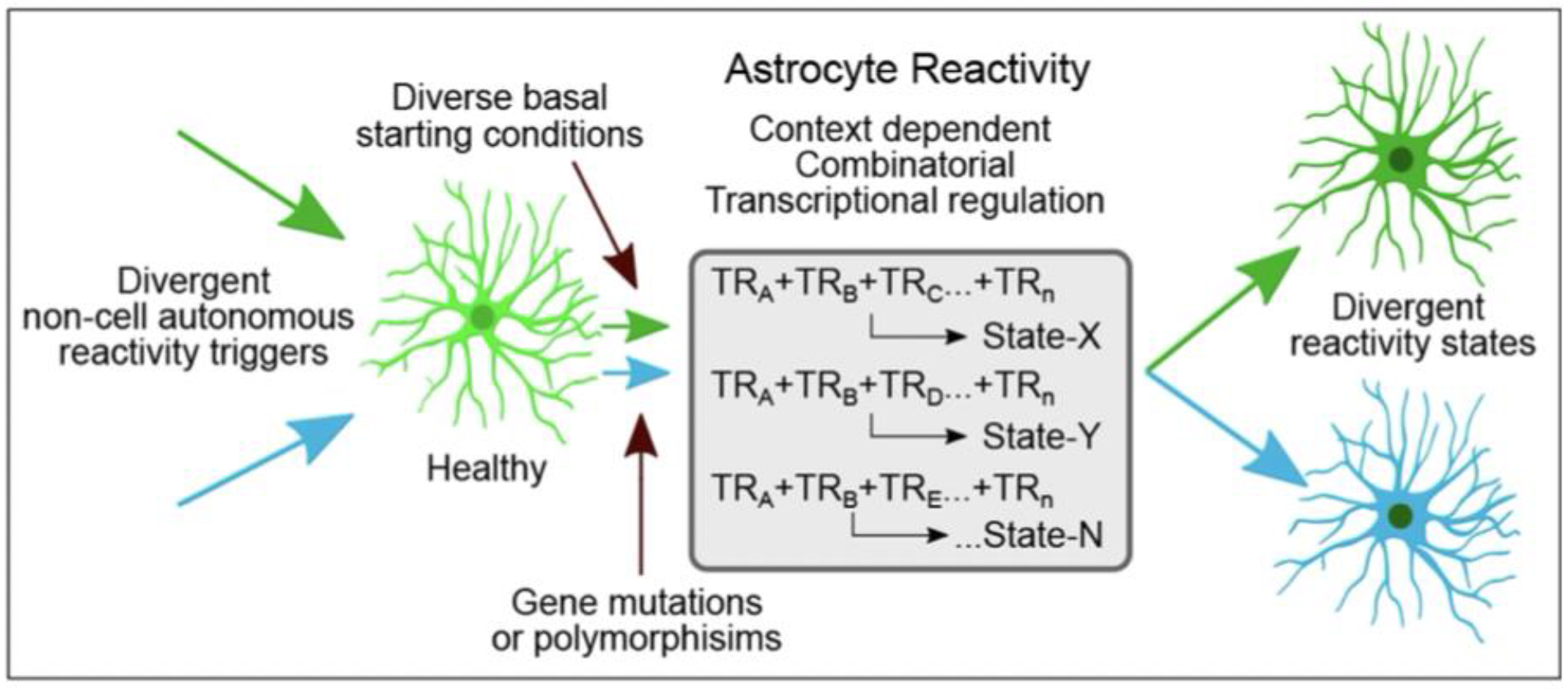
Divergent non-cell autonomous, disorder-selective reactivity triggers lead to context-dependent and combinatorial TR interactions in which a core set of TRs (TRA & TRB) is active across many if not most forms of astrocyte reactivity. These core TRs can nevertheless regulate different cohorts of DEGs in different disorders and contexts via complex and interdependent combinatorial interactions that also involve disorder- or context-selective TRs (TRC - TRn). These TR interactions can be influenced by astrocyte cell autonomous factors such as mutations, polymorphisms, or by differing basal starting conditions that can vary with regional or local astrocyte heterogeneity or with other factors such as exposure to previous insults. These TR interactions give rise to the exquisitely heterogenous DEG profiles associated with different astrocyte reactivity states in different disorders and different contexts.
Extended Data Table 1. RNA Archival data sources.
Summaries and literature references of the sources of genome-wide expression data used for the informatic analyses in this study. Datasets were obtained from NCBI and DEGs with orthologue Refseq ID matches were evaluated.

|
Acknowledgements
This work was supported by US National Institutes of Health (NS084030 to M.V.S., F32NS096858, K99NS105915 and R00NS105915 to J.E.B.); Dr. Miriam and Sheldon G. Adelson Medical Foundation (M.V.S., R.K.); Paralyzed Veterans Research Foundation of America (to J.E.B., T.M.O’S. and M.V.S.); American Australian Fellowship (T.M.O’S.); Microscopy Core Resource of UCLA Broad Stem Cell Research Center; and Wings for Life (M.V.S., J.E.B., and T.M.O’S.).
Footnotes
The authors declare no competing financial interests.
Data Availability
Raw and normalized genomic data have been deposited in NCBI’s Gene Expression Omnibus under the SuperSeries accession number GSE199482. Genomics data are also available via a searchable, open-access website: http://tr.astrocytereactivity.com.
References
- 1.Sofroniew MV & Vinters HV Astrocytes: biology and pathology. Acta Neuropathol 119, 7–35, doi: 10.1007/s00401-009-0619-8 (2010). [DOI] [PMC free article] [PubMed] [Google Scholar]
- 2.Burda JE & Sofroniew MV Reactive gliosis and the multicellular response to CNS damage and disease. Neuron 81, 229–248, doi: 10.1016/j.neuron.2013.12.034 (2014). [DOI] [PMC free article] [PubMed] [Google Scholar]
- 3.Linnerbauer M, Wheeler MA & Quintana FJ Astrocyte Crosstalk in CNS Inflammation. Neuron 108, 608–622, doi: 10.1016/j.neuron.2020.08.012 (2020). [DOI] [PMC free article] [PubMed] [Google Scholar]
- 4.Escartin C et al. Reactive astrocyte nomenclature, definitions, and future directions. Nat Neurosci 24, 312–325, doi: 10.1038/s41593-020-00783-4 (2021). [DOI] [PMC free article] [PubMed] [Google Scholar]
- 5.Allen NJ & Eroglu C Cell Biology of Astrocyte-Synapse Interactions. Neuron 96, 697–708, doi: 10.1016/j.neuron.2017.09.056 (2017). [DOI] [PMC free article] [PubMed] [Google Scholar]
- 6.Haim LB & Rowitch DH Functional diversity of astrocytes in neural circuit regulation. Nat Rev Neurosci 18, 31–41, doi: 10.1038/nrn.2016.159 (2017). [DOI] [PubMed] [Google Scholar]
- 7.Khakh BS & Deneen B The Emerging Nature of Astrocyte Diversity. Annu Rev Neurosci 42, 187–207, doi: 10.1146/annurev-neuro-070918-050443 (2019). [DOI] [PubMed] [Google Scholar]
- 8.Lu TY et al. Axon degeneration induces glial responses through Draper-TRAF4-JNK signalling. Nature communications 8, doi:ARTN 14355 10.1038/ncomms14355 (2017). [DOI] [PMC free article] [PubMed] [Google Scholar]
- 9.Sofroniew MV Astrocyte barriers to neurotoxic inflammation. Nat Rev Neurosci 16, 249–263, doi: 10.1038/nrn3898 (2015). [DOI] [PMC free article] [PubMed] [Google Scholar]
- 10.Khakh BS & Sofroniew MV Diversity of astrocyte functions and phenotypes in neural circuits. Nat Neurosci 18, 942–952, doi: 10.1038/nn.4043 (2015). [DOI] [PMC free article] [PubMed] [Google Scholar]
- 11.Yu X et al. Context-Specific Striatal Astrocyte Molecular Responses Are Phenotypically Exploitable. Neuron 108, 1146–1162 e1110, doi: 10.1016/j.neuron.2020.09.021 (2020). [DOI] [PMC free article] [PubMed] [Google Scholar]
- 12.Anderson MA et al. Astrocyte scar formation aids central nervous system axon regeneration. Nature 532, 195–200, doi: 10.1038/nature17623 (2016). [DOI] [PMC free article] [PubMed] [Google Scholar]
- 13.Schreiner B et al. Astrocyte Depletion Impairs Redox Homeostasis and Triggers Neuronal Loss in the Adult CNS. Cell reports 12, 1377–1384, doi: 10.1016/j.celrep.2015.07.051 (2015). [DOI] [PubMed] [Google Scholar]
- 14.Wheeler MA et al. MAFG-driven astrocytes promote CNS inflammation. Nature 578, 593–599, doi: 10.1038/s41586-020-1999-0 (2020). [DOI] [PMC free article] [PubMed] [Google Scholar]
- 15.Buenrostro JD, Giresi PG, Zaba LC, Chang HY & Greenleaf WJ Transposition of native chromatin for fast and sensitive epigenomic profiling of open chromatin, DNA-binding proteins and nucleosome position. Nature methods 10, 1213–1218, doi: 10.1038/nmeth.2688 (2013). [DOI] [PMC free article] [PubMed] [Google Scholar]
- 16.Wanner IB et al. Glial scar borders are formed by newly proliferated, elongated astrocytes that interact to corral inflammatory and fibrotic cells via STAT3-dependent mechanisms after spinal cord injury. J Neurosci 33, 12870–12886, doi: 10.1523/JNEUROSCI.2121-13.2013 (2013). [DOI] [PMC free article] [PubMed] [Google Scholar]
- 17.Diaz-Castro B, Bernstein AM, Coppola G, Sofroniew MV & Khakh BS Molecular and functional properties of cortical astrocytes during peripherally induced neuroinflammation. Cell reports 36, 109508, doi: 10.1016/j.celrep.2021.109508 (2021). [DOI] [PMC free article] [PubMed] [Google Scholar]
- 18.Inoue F, Kreimer A, Ashuach T, Ahituv N & Yosef N Identification and Massively Parallel Characterization of Regulatory Elements Driving Neural Induction. Cell Stem Cell 25, 713–727 e710, doi: 10.1016/j.stem.2019.09.010 (2019). [DOI] [PMC free article] [PubMed] [Google Scholar]
- 19.Lattke M et al. Extensive transcriptional and chromatin changes underlie astrocyte maturation in vivo and in culture. Nature communications 12, 4335, doi: 10.1038/s41467-021-24624-5 (2021). [DOI] [PMC free article] [PubMed] [Google Scholar]
- 20.Granja JM et al. ArchR is a scalable software package for integrative single-cell chromatin accessibility analysis. Nat Genet 53, 403–411, doi: 10.1038/s41588-021-00790-6 (2021). [DOI] [PMC free article] [PubMed] [Google Scholar]
- 21.Schep AN, Wu B, Buenrostro JD & Greenleaf WJ chromVAR: inferring transcription-factor-associated accessibility from single-cell epigenomic data. Nature methods 14, 975–978, doi: 10.1038/nmeth.4401 (2017). [DOI] [PMC free article] [PubMed] [Google Scholar]
- 22.Wingelhofer B et al. Implications of STAT3 and STAT5 signaling on gene regulation and chromatin remodeling in hematopoietic cancer. Leukemia 32, 1713–1726, doi: 10.1038/s41375-018-0117-x (2018). [DOI] [PMC free article] [PubMed] [Google Scholar]
- 23.Henry CJ et al. Minocycline attenuates lipopolysaccharide (LPS)-induced neuroinflammation, sickness behavior, and anhedonia. J Neuroinflamm 5, 15, doi: 10.1186/1742-2094-5-15 (2008). [DOI] [PMC free article] [PubMed] [Google Scholar]
- 24.Herrmann JE et al. STAT3 is a critical regulator of astrogliosis and scar formation after spinal cord injury. J. Neurosci 28, 7231–7243, doi:28/28/7231 [pii] 10.1523/JNEUROSCI.1709-08.2008 (2008). [DOI] [PMC free article] [PubMed] [Google Scholar]
- 25.Wohlfahrt T et al. PU.1 controls fibroblast polarization and tissue fibrosis. Nature 566, 344–349, doi: 10.1038/s41586-019-0896-x (2019). [DOI] [PMC free article] [PubMed] [Google Scholar]
- 26.Sofroniew MV Astrocyte Reactivity: Subtypes, States, and Functions in CNS Innate Immunity. Trends Immunol 41, 758–770, doi: 10.1016/j.it.2020.07.004 (2020). [DOI] [PMC free article] [PubMed] [Google Scholar]
- 27.Diaz-Castro B, Gangwani MR, Yu X, Coppola G & Khakh BS Astrocyte molecular signatures in Huntington’s disease. Sci Transl Med 11, doi: 10.1126/scitranslmed.aaw8546 (2019). [DOI] [PubMed] [Google Scholar]
- 28.Sun SY et al. Translational profiling identifies a cascade of damage initiated in motor neurons and spreading to glia in mutant SOD1-mediated ALS. P Natl Acad Sci USA 112, E6993–E7002, doi: 10.1073/pnas.1520639112 (2015). [DOI] [PMC free article] [PubMed] [Google Scholar]
- 29.Sekar S et al. Alzheimer’s disease is associated with altered expression of genes involved in immune response and mitochondrial processes in astrocytes. Neurobiol Aging 36, 583–591, doi: 10.1016/j.neurobiolaging.2014.09.027 (2015). [DOI] [PMC free article] [PubMed] [Google Scholar]
- 30.Habib N et al. Disease-associated astrocytes in Alzheimer’s disease and aging. Nat Neurosci 23, 701–706, doi: 10.1038/s41593-020-0624-8 (2020). [DOI] [PMC free article] [PubMed] [Google Scholar]
- 31.Kamphuis W et al. GFAP and vimentin deficiency alters gene expression in astrocytes and microglia in wild-type mice and changes the transcriptional response of reactive glia in mouse model for Alzheimer’s disease. Glia 63, 1036–1056, doi: 10.1002/glia.22800 (2015). [DOI] [PubMed] [Google Scholar]
- 32.Boisvert MM, Erikson GA, Shokhirev MN & Allen NJ The Aging Astrocyte Transcriptome from Multiple Regions of the Mouse Brain. Cell reports 22, 269–285, doi: 10.1016/j.celrep.2017.12.039 (2018). [DOI] [PMC free article] [PubMed] [Google Scholar]
- 33.Rojo AI et al. Deficiency in the transcription factor NRF2 worsens inflammatory parameters in a mouse model with combined tauopathy and amyloidopathy. Redox Biol 18, 173–180, doi: 10.1016/j.redox.2018.07.006 (2018). [DOI] [PMC free article] [PubMed] [Google Scholar]
- 34.Oksanen M et al. NF-E2-related factor 2 activation boosts antioxidant defenses and ameliorates inflammatory and amyloid properties in human Presenilin-1 mutated Alzheimer’s disease astrocytes. Glia 68, 589–599, doi: 10.1002/glia.23741 (2020). [DOI] [PMC free article] [PubMed] [Google Scholar]
- 35.Laug D et al. Nuclear factor I-A regulates diverse reactive astrocyte responses after CNS injury. J Clin Invest 129, 4408–4418, doi: 10.1172/JCI127492 (2019). [DOI] [PMC free article] [PubMed] [Google Scholar]
- 36.Venkatesh I et al. Co-occupancy identifies transcription factor co-operation for axon growth. Nature communications 12, 2555, doi: 10.1038/s41467-021-22828-3 (2021). [DOI] [PMC free article] [PubMed] [Google Scholar]
- 37.Garcia ADR, Doan NB, Imura T, Bush TG & Sofroniew MV GFAP-expressing progenitors are the principle source of constitutive neurogenesis in adult mouse forebrain. Nature Neurosci. 7, 1233–1241 (2004). [DOI] [PubMed] [Google Scholar]
- 38.Sanz E et al. Cell-type-specific isolation of ribosome-associated mRNA from complex tissues. Proc Natl Acad Sci U S A 106, 13939–13944, doi: 10.1073/pnas.0907143106 (2009). [DOI] [PMC free article] [PubMed] [Google Scholar]
- 39.Sumi-Ichinose C, Ichinose H, Metzger D & Chambon P SNF2beta-BRG1 is essential for the viability of F9 murine embryonal carcinoma cells. Mol Cell Biol 17, 5976–5986, doi: 10.1128/mcb.17.10.5976 (1997). [DOI] [PMC free article] [PubMed] [Google Scholar]
- 40.Metz GA & Whishaw IQ The ladder rung walking task: a scoring system and its practical application. J Vis Exp, doi: 10.3791/1204 (2009). [DOI] [PMC free article] [PubMed] [Google Scholar]
- 41.Dantzer R, O’Connor JC, Freund GG, Johnson RW & Kelley KW From inflammation to sickness and depression: when the immune system subjugates the brain. Nat Rev Neurosci 9, 46–56, doi: 10.1038/nrn2297 (2008). [DOI] [PMC free article] [PubMed] [Google Scholar]
- 42.Sukoff Rizzo SJ et al. Evidence for sustained elevation of IL-6 in the CNS as a key contributor of depressive-like phenotypes. Translational psychiatry 2, e199, doi: 10.1038/tp.2012.120 (2012). [DOI] [PMC free article] [PubMed] [Google Scholar]
- 43.Lachmann A et al. ChEA: transcription factor regulation inferred from integrating genome-wide ChIP-X experiments. Bioinformatics 26, 2438–2444, doi: 10.1093/bioinformatics/btq466 (2010). [DOI] [PMC free article] [PubMed] [Google Scholar]
- 44.Fornes O et al. JASPAR 2020: update of the open-access database of transcription factor binding profiles. Nucleic Acids Res 48, D87–D92, doi: 10.1093/nar/gkz1001 (2020). [DOI] [PMC free article] [PubMed] [Google Scholar]
- 45.Matys V et al. TRANSFAC: transcriptional regulation, from patterns to profiles. Nucleic Acids Res 31, 374–378, doi: 10.1093/nar/gkg108 (2003). [DOI] [PMC free article] [PubMed] [Google Scholar]
- 46.Kuleshov MV et al. Enrichr: a comprehensive gene set enrichment analysis web server 2016 update. Nucleic Acids Res 44, W90–97, doi: 10.1093/nar/gkw377 (2016). [DOI] [PMC free article] [PubMed] [Google Scholar]
- 47.Krishnaswami SR et al. Using single nuclei for RNA-seq to capture the transcriptome of postmortem neurons. Nature protocols 11, 499–524, doi: 10.1038/nprot.2016.015 (2016). [DOI] [PMC free article] [PubMed] [Google Scholar]
- 48.Bhattacharyya S, Sathe AA, Bhakta M, Xing C & Munshi NV PAN-INTACT enables direct isolation of lineage-specific nuclei from fibrous tissues. PLoS One 14, e0214677, doi: 10.1371/journal.pone.0214677 (2019). [DOI] [PMC free article] [PubMed] [Google Scholar]
- 49.Batiuk MY et al. An immunoaffinity-based method for isolating ultrapure adult astrocytes based on ATP1B2 targeting by the ACSA-2 antibody. J Biol Chem 292, 8874–8891, doi: 10.1074/jbc.M116.765313 (2017). [DOI] [PMC free article] [PubMed] [Google Scholar]
- 50.Cusanovich DA et al. Multiplex single cell profiling of chromatin accessibility by combinatorial cellular indexing. Science 348, 910–914, doi: 10.1126/science.aab1601 (2015). [DOI] [PMC free article] [PubMed] [Google Scholar]
- 51.Deschamps S et al. Chromatin loop anchors contain core structural components of the gene expression machinery in maize. BMC Genomics 22, 23, doi: 10.1186/s12864-020-07324-0 (2021). [DOI] [PMC free article] [PubMed] [Google Scholar]
- 52.Weirauch MT et al. Determination and inference of eukaryotic transcription factor sequence specificity. Cell 158, 1431–1443, doi: 10.1016/j.cell.2014.08.009 (2014). [DOI] [PMC free article] [PubMed] [Google Scholar]
- 53.Kheradpour P & Kellis M Systematic discovery and characterization of regulatory motifs in ENCODE TF binding experiments. Nucleic Acids Res 42, 2976–2987, doi: 10.1093/nar/gkt1249 (2014). [DOI] [PMC free article] [PubMed] [Google Scholar]
- 54.Courtine G et al. Transformation of nonfunctional spinal circuits into functional states after the loss of brain input. Nat. Neurosci 12, 1333–1342, doi:nn.2401 [pii] 10.1038/nn.2401 (2009). [DOI] [PMC free article] [PubMed] [Google Scholar]
- 55.Neve LD, Savage AA, Koke JR & Garcia DM Activating transcription factor 3 and reactive astrocytes following optic nerve injury in zebrafish. Comp Biochem Physiol C Toxicol Pharmacol 155, 213–218, doi: 10.1016/j.cbpc.2011.08.006 (2012). [DOI] [PubMed] [Google Scholar]
- 56.Kim KH, Jeong JY, Surh YJ & Kim KW Expression of stress-response ATF3 is mediated by Nrf2 in astrocytes. Nucleic Acids Res 38, 48–59, doi: 10.1093/nar/gkp865 (2010). [DOI] [PMC free article] [PubMed] [Google Scholar]
- 57.Koyama Y et al. Endothelin-1 stimulates expression of cyclin D1 and S-phase kinase-associated protein 2 by activating the transcription factor STAT3 in cultured rat astrocytes. J Biol Chem 294, 3920–3933, doi: 10.1074/jbc.RA118.005614 (2019). [DOI] [PMC free article] [PubMed] [Google Scholar]
- 58.Cardinaux JR, Allaman I & Magistretti PJ Pro-inflammatory cytokines induce the transcription factors C/EBPbeta and C/EBPdelta in astrocytes. Glia 29, 91–97 (2000). [PubMed] [Google Scholar]
- 59.Ko CY et al. Glycogen synthase kinase-3beta-mediated CCAAT/enhancer-binding protein delta phosphorylation in astrocytes promotes migration and activation of microglia/macrophages. Neurobiol Aging 35, 24–34, doi: 10.1016/j.neurobiolaging.2013.07.021 (2014). [DOI] [PubMed] [Google Scholar]
- 60.Pardo L et al. Targeted activation of CREB in reactive astrocytes is neuroprotective in focal acute cortical injury. Glia 64, 853–874, doi: 10.1002/glia.22969 (2016). [DOI] [PubMed] [Google Scholar]
- 61.Zou F et al. Different functions of HIPK2 and CtBP2 in traumatic brain injury. J Mol Neurosci 49, 395–408, doi: 10.1007/s12031-012-9906-2 (2013). [DOI] [PubMed] [Google Scholar]
- 62.Robinson KF, Narasipura SD, Wallace J, Ritz EM & Al-Harthi L Negative regulation of IL-8 in human astrocytes depends on beta-catenin while positive regulation is mediated by TCFs/LEF/ATF2 interaction. Cytokine 136, doi:ARTN 155252 10.1016/j.cyto.2020.155252 (2020). [DOI] [PMC free article] [PubMed] [Google Scholar]
- 63.Yang C et al. beta-Catenin signaling initiates the activation of astrocytes and its dysregulation contributes to the pathogenesis of astrocytomas. Proc Natl Acad Sci U S A 109, 6963–6968, doi: 10.1073/pnas.1118754109 (2012). [DOI] [PMC free article] [PubMed] [Google Scholar]
- 64.Wu JF et al. Ablation of the transcription factors E2F1–2 limits neuroinflammation and associated neurological deficits after contusive spinal cord injury. Cell Cycle 14, 3698–3712, doi: 10.1080/15384101.2015.1104436 (2015). [DOI] [PMC free article] [PubMed] [Google Scholar]
- 65.Beck H, Semisch M, Culmsee C, Plesnila N & Hatzopoulos AK Egr-1 regulates expression of the glial scar component phosphacan in astrocytes after experimental stroke. American Journal of Pathology 173, 77–92, doi: 10.2353/ajpath.2008.070648 (2008). [DOI] [PMC free article] [PubMed] [Google Scholar]
- 66.Mayer SI, Rossler OG, Endo T, Charnay P & Thiel G Epidermal-growth-factor-induced proliferation of astrocytes requires Egr transcription factors. Journal of cell science 122, 3340–3350, doi: 10.1242/jcs.048272 (2009). [DOI] [PubMed] [Google Scholar]
- 67.Wang HH, Hsieh HL, Wu CY & Yang CM Oxidized low-density lipoprotein-induced matrix metalloproteinase-9 expression via PKC-delta/p42/p44 MAPK/Elk-1 cascade in brain astrocytes. Neurotox Res 17, 50–65, doi: 10.1007/s12640-009-9077-2 (2010). [DOI] [PubMed] [Google Scholar]
- 68.Gerhauser I, Alldinger S & Baumgartner W Ets-1 represents a pivotal transcription factor for viral clearance, inflammation, and demyelination in a mouse model of multiple sclerosis. J Neuroimmunol 188, 86–94, doi: 10.1016/j.jneuroim.2007.05.019 (2007). [DOI] [PubMed] [Google Scholar]
- 69.Hashimoto K et al. Long-term activation of c-Fos and c-Jun in optic nerve head astrocytes in experimental ocular hypertension in monkeys and after exposure to elevated pressure in vitro. Brain Research 1054, 103–115, doi: 10.1016/j.brainres.2005.06.050 (2005). [DOI] [PubMed] [Google Scholar]
- 70.Yang CC, Hsiao LD & Yang CM Galangin Inhibits LPS-Induced MMP-9 Expression via Suppressing Protein Kinase-Dependent AP-1 and FoxO1 Activation in Rat Brain Astrocytes. J Inflamm Res 13, 945–960, doi: 10.2147/JIR.S276925 (2020). [DOI] [PMC free article] [PubMed] [Google Scholar]
- 71.Cui M, Huang Y, Tian C, Zhao Y & Zheng J FOXO3a inhibits TNF-alpha- and IL-1beta-induced astrocyte proliferation:Implication for reactive astrogliosis. Glia 59, 641–654, doi: 10.1002/glia.21134 (2011). [DOI] [PMC free article] [PubMed] [Google Scholar]
- 72.Kizil C et al. Regenerative neurogenesis from neural progenitor cells requires injury-induced expression of Gata3. Developmental cell 23, 1230–1237, doi: 10.1016/j.devcel.2012.10.014 (2012). [DOI] [PubMed] [Google Scholar]
- 73.Garcia AD, Petrova R, Eng L & Joyner AL Sonic hedgehog regulates discrete populations of astrocytes in the adult mouse forebrain. J Neurosci 30, 13597–13608, doi:30/41/13597 [pii] 10.1523/JNEUROSCI.0830-10.2010 (2010). [DOI] [PMC free article] [PubMed] [Google Scholar]
- 74.Du F et al. Hyperthermic preconditioning protects astrocytes from ischemia/reperfusion injury by up-regulation of HIF-1 alpha expression and binding activity. Biochimica et biophysica acta 1802, 1048–1053, doi: 10.1016/j.bbadis.2010.06.013 (2010). [DOI] [PubMed] [Google Scholar]
- 75.Choi K, Ni L & Jonakait GM Fas ligation and tumor necrosis factor alpha activation of murine astrocytes promote heat shock factor-1 activation and heat shock protein expression leading to chemokine induction and cell survival. J Neurochem 116, 438–448, doi: 10.1111/j.1471-4159.2010.07124.x (2011). [DOI] [PubMed] [Google Scholar]
- 76.Tzeng SF, Kahn M, Liva S & De Vellis J Tumor necrosis factor-alpha regulation of the Id gene family in astrocytes and microglia during CNS inflammatory injury. Glia 26, 139–152 (1999). [DOI] [PubMed] [Google Scholar]
- 77.Aronica E et al. Expression of Id proteins increases in astrocytes in the hippocampus of epileptic rats. Neuroreport 12, 2461–2465 (2001). [DOI] [PubMed] [Google Scholar]
- 78.Jarosinski KW & Massa PT Interferon regulatory factor-1 is required for interferon-gamma-induced MHC class I genes in astrocytes. J Neuroimmunol 122, 74–84, doi: 10.1016/s0165-5728(01)00467-2 (2002). [DOI] [PubMed] [Google Scholar]
- 79.Tarassishin L et al. Interferon regulatory factor 3 inhibits astrocyte inflammatory gene expression through suppression of the proinflammatory miR-155 and miR-155*. Glia 59, 1911–1922, doi: 10.1002/glia.21233 (2011). [DOI] [PMC free article] [PubMed] [Google Scholar]
- 80.Gadea A, Schinelli S & Gallo V Endothelin-1 regulates astrocyte proliferation and reactive gliosis via a JNK/c-Jun signaling pathway. J. Neurosci 28, 2394–2408, doi:28/10/2394 [pii] 10.1523/JNEUROSCI.5652-07.2008 (2008). [DOI] [PMC free article] [PubMed] [Google Scholar]
- 81.Park JH et al. Induction of Kruppel-like factor 4 expression in reactive astrocytes following ischemic injury in vitro and in vivo. Histochem Cell Biol 141, 33–42, doi: 10.1007/s00418-013-1134-5 (2014). [DOI] [PubMed] [Google Scholar]
- 82.Jeong KH, Lee KE, Kim SY & Cho KO Upregulation of Kruppel-like factor 6 in the mouse hippocampus after pilocarpine-induced status epilepticus. Neuroscience 186, 170–178, doi: 10.1016/j.neuroscience.2011.02.046 (2011). [DOI] [PubMed] [Google Scholar]
- 83.Liu F, Ni JJ, Huang JJ, Kou ZW & Sun FY VEGF overexpression enhances the accumulation of phospho-S292 MeCP2 in reactive astrocytes in the adult rat striatum following cerebral ischemia. Brain Res 1599, 32–43, doi: 10.1016/j.brainres.2014.12.014 (2015). [DOI] [PubMed] [Google Scholar]
- 84.Wang F et al. 2-Arachidonylglycerol Protects Primary Astrocytes Exposed to Oxygen-Glucose Deprivation Through a Blockade of NDRG2 Signaling and STAT3 Phosphorylation. Rejuvenation Res 19, 215–222, doi: 10.1089/rej.2015.1703 (2016). [DOI] [PubMed] [Google Scholar]
- 85.Perez-Ortiz JM et al. Mechanical lesion activates newly identified NFATc1 in primary astrocytes: implication of ATP and purinergic receptors. Eur J Neurosci 27, 2453–2465, doi: 10.1111/j.1460-9568.2008.06197.x (2008). [DOI] [PubMed] [Google Scholar]
- 86.Yang Y et al. Hemoglobin pretreatment endows rat cortical astrocytes resistance to hemin-induced toxicity via Nrf2/HO-1 pathway. Experimental cell research 361, 217–224, doi: 10.1016/j.yexcr.2017.10.020 (2017). [DOI] [PubMed] [Google Scholar]
- 87.Brambilla R et al. Inhibition of astroglial nuclear factor kappaB reduces inflammation and improves functional recovery after spinal cord injury. J. Exp. Med 202, 145–156 (2005). [DOI] [PMC free article] [PubMed] [Google Scholar]
- 88.LeComte MD, Shimada IS, Sherwin C & Spees JL Notch1-STAT3-ETBR signaling axis controls reactive astrocyte proliferation after brain injury. Proc Natl Acad Sci U S A 112, 8726–8731, doi: 10.1073/pnas.1501029112 (2015). [DOI] [PMC free article] [PubMed] [Google Scholar]
- 89.Chen XL et al. Effects of interleukin-6 and IL-6/AMPK signaling pathway on mitochondrial biogenesis and astrocytes viability under experimental septic condition. Int Immunopharmacol 59, 287–294, doi: 10.1016/j.intimp.2018.04.020 (2018). [DOI] [PubMed] [Google Scholar]
- 90.Gutbier S et al. Prevention of neuronal apoptosis by astrocytes through thiol-mediated stress response modulation and accelerated recovery from proteotoxic stress. Cell Death Differ 25, 2101–2117, doi: 10.1038/s41418-018-0229-x (2018). [DOI] [PMC free article] [PubMed] [Google Scholar]
- 91.Chen Y et al. The basic helix-loop-helix transcription factor olig2 is critical for reactive astrocyte proliferation after cortical injury. J. Neurosci 28, 10983–10989, doi:28/43/10983 [pii] 10.1523/JNEUROSCI.3545-08.2008 (2008). [DOI] [PMC free article] [PubMed] [Google Scholar]
- 92.Koyama Y Signaling molecules regulating phenotypic conversions of astrocytes and glial scar formation in damaged nerve tissues. Neurochem Int 78, 35–42, doi: 10.1016/j.neuint.2014.08.005 (2014). [DOI] [PubMed] [Google Scholar]
- 93.Steliga A et al. Transcription factor Pax6 is expressed by astroglia after transient brain ischemia in the rat model. Folia Neuropathol 51, 203–213 (2013). [DOI] [PubMed] [Google Scholar]
- 94.Guo X et al. The AMPK-PGC-1alpha signaling axis regulates the astrocyte glutathione system to protect against oxidative and metabolic injury. Neurobiol Dis 113, 59–69, doi: 10.1016/j.nbd.2018.02.004 (2018). [DOI] [PubMed] [Google Scholar]
- 95.Diehl JA, Tong W, Sun G & Hannink M Tumor necrosis factor-alpha-dependent activation of a RelA homodimer in astrocytes. Increased phosphorylation of RelA and MAD-3 precede activation of RelA. J Biol Chem 270, 2703–2707, doi: 10.1074/jbc.270.6.2703 (1995). [DOI] [PubMed] [Google Scholar]
- 96.Yoo KY et al. Time-course alterations of Toll-like receptor 4 and NF-kappaB p65, and their co-expression in the gerbil hippocampal CA1 region after transient cerebral ischemia. Neurochem Res 36, 2417–2426, doi: 10.1007/s11064-011-0569-0 (2011). [DOI] [PubMed] [Google Scholar]
- 97.Gupta AS et al. RelB controls adaptive responses of astrocytes during sterile inflammation. Glia 67, 1449–1461, doi: 10.1002/glia.23619 (2019). [DOI] [PMC free article] [PubMed] [Google Scholar]
- 98.Li H et al. The deficiency of NRSF/REST enhances the pro-inflammatory function of astrocytes in a model of Parkinson’s disease. Biochim Biophys Acta Mol Basis Dis 1866, 165590, doi: 10.1016/j.bbadis.2019.165590 (2020). [DOI] [PubMed] [Google Scholar]
- 99.Marumo T et al. Notch signaling regulates nucleocytoplasmic Olig2 translocation in reactive astrocytes differentiation after ischemic stroke. Neuroscience research 75, 204–209, doi: 10.1016/j.neures.2013.01.006 (2013). [DOI] [PubMed] [Google Scholar]
- 100.Tanigaki K & Honjo T Two opposing roles of RBP-J in Notch signaling. Curr Top Dev Biol 92, 231–252, doi: 10.1016/S0070-2153(10)92007-3 (2010). [DOI] [PubMed] [Google Scholar]
- 101.Wong JK et al. Attenuation of Cerebral Ischemic Injury in Smad1 Deficient Mice. PLoS One 10, e0136967, doi: 10.1371/journal.pone.0136967 (2015). [DOI] [PMC free article] [PubMed] [Google Scholar]
- 102.Law AKT et al. TGF-beta 1 induction of the adenine nucleotide translocator 1 in astrocytes occurs through Smads and Sp1 transcription factors. BMC neuroscience 5, doi: 10.1186/1471-2202-5-1 (2004). [DOI] [PMC free article] [PubMed] [Google Scholar]
- 103.Chen C et al. Astrocyte-Specific Deletion of Sox2 Promotes Functional Recovery After Traumatic Brain Injury. Cereb Cortex 29, 54–69, doi: 10.1093/cercor/bhx303 (2019). [DOI] [PMC free article] [PubMed] [Google Scholar]
- 104.Song W et al. Immunohistochemical staining of ERG and SOX9 as potential biomarkers of docetaxel response in patients with metastatic castration-resistant prostate cancer. Oncotarget 7, 83735–83743, doi: 10.18632/oncotarget.13407 (2016). [DOI] [PMC free article] [PubMed] [Google Scholar]
- 105.Mao X, Moerman-Herzog AM, Wang W & Barger SW Differential transcriptional control of the superoxide dismutase-2 kappaB element in neurons and astrocytes. J Biol Chem 281, 35863–35872, doi: 10.1074/jbc.M604166200 (2006). [DOI] [PMC free article] [PubMed] [Google Scholar]
- 106.Haroon F et al. Gp130-dependent astrocytic survival is critical for the control of autoimmune central nervous system inflammation. J Immunol 186, 6521–6531, doi:jimmunol.1001135 [pii] 10.4049/jimmunol.1001135 (2011). [DOI] [PubMed] [Google Scholar]
- 107.Khorooshi R, Babcock AA & Owens T NF-kappaB-driven STAT2 and CCL2 expression in astrocytes in response to brain injury. J Immunol 181, 7284–7291 (2008). [DOI] [PubMed] [Google Scholar]
- 108.Doherty J et al. PI3K signaling and Stat92E converge to modulate glial responsiveness to axonal injury. PLoS Biol 12, e1001985, doi: 10.1371/journal.pbio.1001985 (2014). [DOI] [PMC free article] [PubMed] [Google Scholar]
- 109.Park SJ et al. Astrocytes, but Not Microglia, Rapidly Sense H2O2 via STAT6 Phosphorylation, Resulting in Cyclooxygenase-2 Expression and Prostaglandin Release. Journal of Immunology 188, 5132–5141, doi: 10.4049/jimmunol.1101600 (2012). [DOI] [PubMed] [Google Scholar]
- 110.Lurbke A et al. Limited TCF7L2 expression in MS lesions. PLoS One 8, e72822, doi: 10.1371/journal.pone.0072822 (2013). [DOI] [PMC free article] [PubMed] [Google Scholar]
- 111.Chung YH et al. Enhanced expression of p53 in reactive astrocytes following transient focal ischemia. Neurol Res 24, 324–328, doi: 10.1179/016164102101199828 (2002). [DOI] [PubMed] [Google Scholar]
- 112.Huang ZH et al. YAP Is a Critical Inducer of SOCS3, Preventing Reactive Astrogliosis. Cerebral Cortex 26, 2299–2310, doi: 10.1093/cercor/bhv292 (2016). [DOI] [PMC free article] [PubMed] [Google Scholar]
- 113.Vivinetto AL et al. Zeb2 Is a Regulator of Astrogliosis and Functional Recovery after CNS Injury. Cell reports 31, 107834, doi: 10.1016/j.celrep.2020.107834 (2020). [DOI] [PMC free article] [PubMed] [Google Scholar]
- 114.Itoh N et al. Cell-specific and region-specific transcriptomics in the multiple sclerosis model: Focus on astrocytes. Proc Natl Acad Sci U S A 115, E302–E309, doi: 10.1073/pnas.1716032115 (2018). [DOI] [PMC free article] [PubMed] [Google Scholar]
- 115.Zamanian JL et al. Genomic analysis of reactive astrogliosis. J Neurosci 32, 6391–6410, doi: 10.1523/JNEUROSCI.6221-11.2012 (2012). [DOI] [PMC free article] [PubMed] [Google Scholar]
- 116.Zhang Y et al. Purification and Characterization of Progenitor and Mature Human Astrocytes Reveals Transcriptional and Functional Differences with Mouse. Neuron 89, 37–53, doi: 10.1016/j.neuron.2015.11.013 (2016). [DOI] [PMC free article] [PubMed] [Google Scholar]
Associated Data
This section collects any data citations, data availability statements, or supplementary materials included in this article.
Data Availability Statement
Raw and normalized genomic data have been deposited in NCBI’s Gene Expression Omnibus under the SuperSeries accession number GSE199482. Genomics data are also available via a searchable, open-access website: http://tr.astrocytereactivity.com.