

Abstract
Finding effective treatments for cancer remains a challenge. Recent studies have found that the mechanisms of tumor evasion are becoming increasingly diverse, including abnormal expression of immune checkpoint molecules on different immune cells, in particular T cells, natural killer cells, macrophages and others. In this review, we discuss the checkpoint molecules with enhanced expression on these lymphocytes and their consequences on immune effector functions. Dissecting the diverse roles of immune checkpoints in different immune cells is crucial for a full understanding of immunotherapy using checkpoint inhibitors.
Keywords: immune checkpoint, immunotherapy, T cell, NK cell, macrophage
1. Introduction
It now appears that immunotherapies can elicit durable antitumor responses in metastatic cancer. These immunotherapies include adoptive cell therapy (ACT) and checkpoint inhibitor therapies (1). In particular, recent studies have confirmed that targeting immune checkpoint pathways has remarkable clinical efficiency across several tumor types (2). Immune checkpoint molecules are mainly expressed on immune cells and can maintain immunological homeostasis. Under normal physiological conditions, they can inhibit and prevent immune cells from killing tumor cells (3). In the past few years, studies have mainly focused on finding new immune checkpoint molecules expressed on T cells, which can effectively restore the exhaustion of T cells when blocked. The immune checkpoint targets that have been validated clinically include CTLA4 and PD-1, and many new candidates are being discovered and will undergo clinical evaluation (4). In addition to T cells, Nature Killer cells also express immune checkpoints, but the consequences of these checkpoints on NK cells’ functions are much less explored (5). Recently, literature has shown that macrophage-centered blockade of immune checkpoints represents promising therapeutic avenues (6). In this review, we will discuss recent advances in knowledge regarding the diversity of immune checkpoints expressed on different immune cells and their relationships with cancer immunotherapy ( Figure 1 ).
Figure 1.
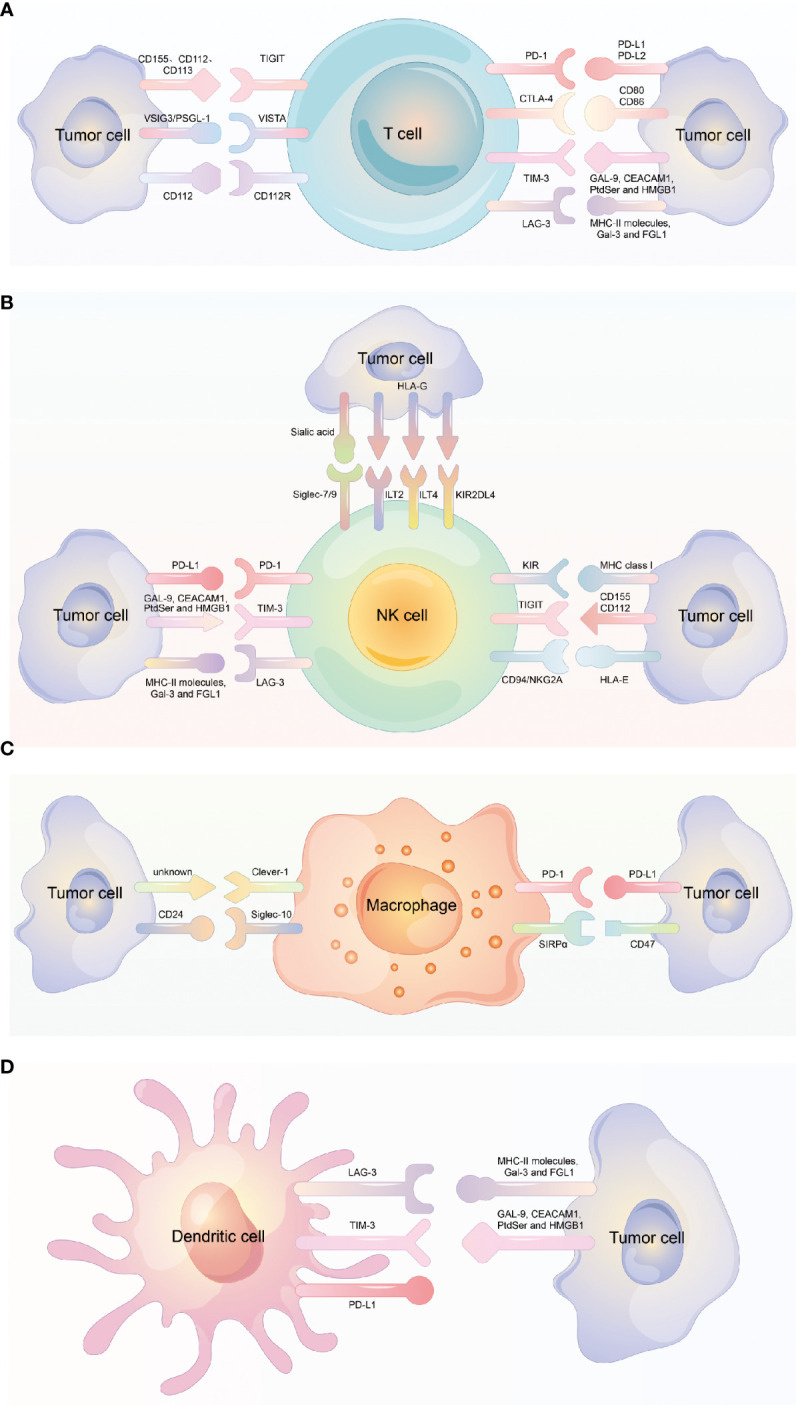
Different immune checkpoint molecules expressed on different immune cells. (A) Different immune checkpoint molecules expressed on T cell and the corresponding ligand molecules expressed on tumor cells. (B) Different immune checkpoint molecules expressed on NK cell and the corresponding ligand molecules expressed on tumor cells. (C) Different immune checkpoint molecules expressed on Macrophage and the corresponding ligand molecules expressed on tumor cells. (D) Different immune checkpoint molecules expressed on dendritic cell and the corresponding ligand molecules expressed on tumor cells.
2. Checkpoint immunotherapy based on T cells
In the last few decades, the function of tumor-infiltrating lymphocytes (TILs), especially the cytotoxic CD8+ T cells and other subgroups of T cells, such as CD4+ T cells and Tregs on tumor progression and patient prognosis have been deeply explored (7–9).
In immunological homeostasis, the engagement of T-cell antigen receptors (TCRs) with antigenic peptides can result in the activation and proliferation of T cells (10). To prevent overreaction and autoimmunity, inhibitory receptors are upregulated on T cells and other immune cells. These inhibitory receptors are also called immune checkpoints. Because of the presence of the immunoreceptor tyrosine-based inhibitory motif (ITIM), immune checkpoints can induce inhibitory signals in inhibitory receptor-expressing immune cells (11).
In the immunosuppressive tumor microenvironment, tumor cells make use of the overexpression of inhibitory receptors on immune cells to avoid immune clearance (12). The expression of immune checkpoints can lead to T-cell exhaustion, which is defined by a decline in T-cell proliferation and reduced T-cell function. To date, immune checkpoints that have been explored for their expression by T cells include PD-1 (programmed cell death protein-1), CTLA-4 (cytotoxic T-lymphocyte-associated protein-4), TIM-3 (mucin-domain containing-3), LAG-3 (lymphocyte-activation gene-3), and T cell immunoglobulin and ITIM domain (TIGIT), among others (13).
2.1. PD-1
PD-1 (CD279) is a coinhibitory receptor that is extensively expressed on T cells, NK cells (natural killer cells), and B cells. In particular, PD-1 is expressed on activated T cells at high levels and is considered to be involved in immune tolerance (14). There are two ligands for PD-1, known as PD-L1 and PD-L2, which have low expression in normal tissue but abnormal expression in some tumor types. For example, it has been reported that the expression of PD-L1 is upregulated in melanoma, non-small-cell lung cancer, breast cancer, and squamous cell head and neck cancer (15).
PD-1+ T-cell exhaustion was originally studied in murine models and then extended to human infection and cancer (16). In chronic viral infections, CD8+ T cells are in a state of dysfunction and have abnormal expression of PD-1. Se Jin Im et al. found that in a mouse model chronically infected with lymphocytic choriomeningitis virus, a population of virus-specific CD8+ T cells proliferated after PD-1 blockade, and this proliferative burst occurred only in this type of CD8+ T cell (17). Tim Wartewig et al. found that mono- and biallelic deletions of PDCD1, which encodes PD-1, are recurrently observed in human T-cell lymphomas with frequencies of up to 30%, indicating high clinical relevance; these findings imply that PD-1 is a potent haploinsufficient tumor suppressor in T-cell lymphomas (18). In a study of colorectal cancer, Xiao Albert Zhou et al. identified a major PD-1-associated protein, KLHL22, that can mediate the degradation of PD-1 before its transport to the cell surface. They found that the expression of KLHL22 was markedly decreased in tumor-infiltrating T cells from colorectal cancer patients and suggested the therapeutic potential of 5-FU (which could increase PD-1 expression by inhibiting the transcription of KLHL22) in combination with anti-PD-1 in colorectal cancer patients (19).
Based on previous research, new strategies have emerged that target PD-1 or PD-L1 and block them; as a result, T-cell function is successfully reinvigorated (20). Along these lines, antibodies targeting the PD-1/PD-L1 axis have been used for various tumors. For example, Alexander C Huang et al. found that neoadjuvant anti-PD-1 treatment is effective against high-risk resectable stage III/IV melanoma (21). Edward B Garon et al. assessed the efficacy and safety of PD-1 inhibition with pembrolizumab in patients with advanced non-small-cell lung cancer enrolled in a phase 1 study and found that a blocking antibody targeting PD-1 had an obvious antitumor effect in NSCLC patients and an acceptable side-effect profile (22). Fan Zhang et al. performed scRNA-seq analysis on 3110 peripheral T cells of NSCLC patients before and after the initiation of PD-1 blockade and found a higher cytotoxic activity in tumor-related CD4+ T-cell clones than in CD8+ T-cell clones (23). In a prognostic analysis of advanced renal cell carcinoma, the investigator assessed the efficacy and safety of nivolumab treatment versus everolimus treatment over a 3-year follow-up and found that nivolumab treatment was more effective and safer than everolimus (24). Two phase III clinical trials (CheckMate 141 and KEYNOTE 040) analyzed the overall survival (OS) of patients with recurrent or metastatic head-and-neck squamous cell carcinoma (HNSCC) and found that anti-PD-1 monotherapy improved the therapeutic effects of platinum chemotherapy (25, 26).
Although a promising therapeutic effect using a PD-1 blocking antibody was observed in those tumor patients, some patients did not respond to this blocking antibody blocking, or it had limited effects. This implies that there are other inhibitory pathways involved in T-cell dysfunction.
2.2. CTLA-4
CTLA-4 (cytotoxic T-lymphocyte-associated protein 4), also known as CD152, is a protein receptor mainly expressed on T cells that was first identified as a second receptor for the T-cell costimulatory legend B7 and later discovered to be a negative regulator of T-cell activation (27–29). In naïve T cells, the expression of CTLA4 is low, but in phases of TCR engagement and activation, CTLA4 can be rapidly upregulated in both CD4 helper T cells and CD8 effector T cells, while its upregulation is obvious in helper T cells (30). CTLA4 has two ligands, CD80 and CD86, also called B7-1 and B7-2, which can also be recognized by CD28, a T-cell costimulatory protein that is homologous to CTLA4. However, for both ligands, CTLA4 has higher affinity and avidity than CD28, implying that it is an antagonist of CD28-mediated costimulation (31, 32). This mechanism suggests that the CD28/CTLA4 regulatory form can act as a rheostat in T-cell activation.
In mouse models, anti-CTLA4 antibody treatment initially resulted in the rejection of tumors, including preestablished tumors; furthermore, the rejection resulted in immunity to a secondary exposure to tumor cells (33). During the subsequent development of clinical immunotherapy, two CTLA-4 blockade antibodies, ipilimumab and tremelimumab, have been tested in many types of human tumors, and their treatment efficacy has been reported in melanoma (34, 35), non-small-cell lung cancer (36), mesothelioma (37), prostate cancer (38), breast cancer (39) and urothelial cancer (40). Despite the promising therapeutic effects, a broad range of immune-related adverse events (irAEs) occurring in the skin, gastrointestinal tract, liver and endocrine organs have been reported in some trials, with an incidence of 60-65% (41). A landmark clinical trial called the CheckMate 067 clinical trial (ClinicalTrials.gov NCT01844505) used a combination CPI therapy with an anti-CTLA-4 antibody and an anti-PD-1 antibody This study was carried out on 945 patients with stage III or IV melanoma and evaluated the median overall survival under treatment with nivolumab plus ipilimumab or with nivolumab or ipilimumab monotherapy. Although the results showed that the OS appeared to be improved in the combination treatment cohort compared with the single-treatment cohorts, the trial did not have sufficient power to show a significant difference between the two nivolumab-containing groups, and the incidence of adverse events was increased in the combination therapy cohort in this trial (42, 43).
Additionally, while CTLA4 is expressed at high levels on Tregs and although an important role of conventional T-cell CTLA4 in self-tolerance has been reported, CTLA4 blockade therapy combined with Treg depletion has led to considerable success in tumor treatment as well as autoimmune disease treatment (41). Therefore, more research should be conducted to reveal the pros and cons of CTLA4 blockade immunotherapy.
2.3. TIM-3
TIM-3 (T-cell immunoglobulin and mucin domain containing-3), a member of the TIM family is a coinhibitory receptors. It is expressed on IFN-γ-producing T helper 1 CD4+ and CD8+ T cells and Th17 cells (44). The expression of TIM-3 is regulated by antigenic stimulation and proinflammatory cytokines (45). In early studies, TIM-3 was reported to have an inhibitory function, suppressing effector Th1 responses in EAE and type I diabetes in a mouse model, and the use of an anti-TIM-3 antibody was reported to lead to disease exacerbation in EAE (46). In subsequent studies, the overexpression of TIM-3 has been found to be correlated with T-cell dysfunction and T-cell exhaustion (47). The role of TIM-3 as a suppressive receptor that regulates T-cell activity in some chronic viral infections, such as HIV-1, HBV and HCV infections, has been reported (48–50). In the tumor microenvironment, TIM-3 has also been found to be expressed on CD8+ TILs (tumor-infiltrating leukocytes), which is closely associated with PD-1 expression. Specifically, the expression patterns of TIM-3 and PD-1 indicate the degree of T-cell exhaustion; for example, in mice bearing solid tumors, TIM-3+PD-1+ TILs exhibit the most severely exhausted phenotype, as defined by failure to proliferate and produce cytokines. Additionally, high expression of TIM-3 on CD8+ T cells has been found to be correlated with poor prognosis in certain types of cancers, and blockade of TIM-3 combined with anti-PD-1 antibody treatment has been confirmed to be more effective than blockade of either molecule alone in antitumor immunotherapy (51–53). In a study on medullary thyroid carcinoma (MTC) in 200 MTC patients, TIM-3 positivity was 48%, and TIM-3 expression was positively correlated with PD-1 and CTLA-4 expression. Log-rank tests and multivariate Cox analyses both indicated that TIM-3, CTLA-4 and PD-1/PD-L1 coexpression were associated with poor structural recurrence-free survival (54).
2.4. LAG-3
LAG-3, lymphocyte activation gene 3, is a cell surface protein belonging to the immunoglobulin superfamily that is expressed on CD4+ and CD8+ T cells (55), NK cells (56), B cells and plasmacytoid dendritic cells (57). It is a coinhibitory transmembrane receptor whose ligands are MHC class II and FGL1, and interaction with the ligands can negatively regulate the activation of T cells (58, 59), similar to the case for CTLA4 and PD-1 (60, 61). In particular, LAG-3 has a synergistic effect with PD-1 to regulate immune responses (62). In clinical immunotherapy, a LAG-3 Ig fusion protein named IMP321 was first used in advanced renal cell carcinoma patients and resulted in reduced tumor growth and improved progression-free survival (63). When LAG-3 blockade antibody (BMS-986016) and nivolumab (a PD-1 antibody) were used in combination in melanoma patients, the initial resistance when only blocking of the PD-1/PD-L1 axis was converted (64). In addition, many types of human tumors present aberrant expression of LAG-3, which correlates with poor outcomes (65–69). Kosaku Mimura et al. evaluated the distribution of different inhibitory ligands in 365 GC patients and found coexpression of inhibitory ligands for PD-1, Tim-3 and Lag-3 in the largest proportion (34.7%). Their findings suggest that the expression of inhibitory ligands for Tim-3 and Lag-3 on GC cells serve as potential predictive biomarkers of the response to anti-PD-1 therapy (70).
2.5. TIGIT
TIGIT, T-cell immunoglobulin and ITIM domain, belongs to the immunoglobulin superfamily and is also a T-cell coinhibitory receptor. It is expressed on CD4+ memory and regulatory T cells, CD8+ T cells and NK cells. To date, the ligands that have been discovered to be recognized by TIGIT are CD155 (PVR or poliovirus receptor), CD112 (PVRL2) and CD113 (PVRL3, NECTIN-3), of which CD155 has the highest affinity for TIGIT (71). TIGIT has been implicated in tumor immunosurveillance, and its role is analogous to that of PD-1 in tumor immunosuppression because it is overexpressed in tumor antigen-specific CD8+ T cells and CD8+ TILs and is often coexpressed with PD-1. Therefore, co-blockade of the two checkpoint molecules can enhance the antitumor efficacy of single blockade (72).
2.6. VISTA
VISTA, V-domain Ig-containing suppressor of T-cell activation, also belongs to the transmembrane Ig superfamily (73). It is part of the B7 family and is mainly expressed on T cells and CD11b+ antigen-presenting cells (APCs)/myeloid cells (74). It has been reported that VISTA can act as both a receptor and a ligand on T cells and that it functions as an inhibitor to maintain immune tolerance (75). In tumor-infiltrating lymphocytes, VISTA is overexpressed, especially in myeloid-derived suppressor cells and regulatory T cells. Recently, it has been reported to be highly expressed in human ovarian and endometrial cancers. The abnormal expression of VISTA in tumor cells suppresses T-cell proliferation and cytokine production in vitro and decreases the tumor infiltration of CD8+ T cells in vivo. VISTA blockade prolongs the survival of tumor-bearing mice (76). In a study on oropharyngeal squamous cell carcinoma (OPSCC) including 241 tumor tissues aiming to describe the expression of LAG-3, Tim-3, and VISTA in the TME of OPSCC, immunohistochemistry showed that 168 OPSCC samples stained positive for VISTA. The results also revealed that CD8+ T cells were significantly associated with LAG-3, Tim-3 and VISTA expression (p < 0.001, p < 0.001, p = 0.007), so immune checkpoint therapy targeting LAG-3, Tim-3, and/or VISTA could be a promising treatment strategy, especially for HPV-related OPSCC (77).
2.7. Siglec-15
Siglec-15, short for sialic acid-binding immunoglobulin-like lectin 15, belongs to the Siglec gene family because of its sialic acid-binding immunoglobulin-type lectin structure (78). Originally, Siglec-15 was mainly reported to play roles in osteoclast differentiation and bone remodeling (79, 80). Recently, Wang et al. identified Siglec-15 as a potent immunosuppressive molecule. In their study, using a newly developed genome-scale T-cell Activity Array, they identified that the expression of Siglec-15 was upregulated in many human cancer cells and tumor-infiltrating myeloid cells, while under normal physiological conditions, it was limited to cells in the myeloid lineage. In particular, its expression was mutually exclusive with that of B7-H1 in cancer cells and could be regulated by M-CSF and IFN-γ. In thorough in vitro and in vivo experiments, Siglec-15 was confirmed to suppress antigen-specific T-cell responses and impair antitumor immunity. Conversely, a Siglec-15-blocking mAb reversed T-cell suppression and promoted tumor immunity in multiple tumor models (81). Siglec-15 has unique molecular features compared with those of many other known checkpoint inhibitory ligands; it shows mutually exclusive expression with PD-L1, which suggests that it plays a key role in tumor escape in PD-L1-negative patients. As a new player in cancer immunotherapy, siglec-15 may have potential applications in anti-PD-1/PD-L1-resistant patients (82). Collectively, the evidence suggests that Siglec-15 is an attractive target for cancer immunotherapy.
2.8. CD112R
CD112R is a poliovirus receptor-like protein and has been described as a new coinhibitory receptor for human T cells that can interact with CD112 with higher affinity than CD226 and TIGIT. Recently, it has also been reported to be expressed in subpopulations of NK cells (83). Zhu et al. reported that CD112 is expressed on DCs and many tumor cells and mediates the interaction of CD112R with DCs and tumor cells. When the interaction between CD112R and CD112 is disrupted, human T-cell function is enhanced. These results imply that the CD112R/CD112 axis is a new checkpoint in human T cells (84).
3. Checkpoint immunotherapy based on NK cells
Natural killer (NK) cells are involved in innate immunity and play a significant role in immunological surveillance against various infections and malignant transformation. Unlike that of T cells, the activation of NK cells does not require prior sensitization, and the NK cell function is determined by the balance of a series of activated and inhibitory receptors expressed on the cell surface (85, 86). In the tumor microenvironment, tumor cells often downregulate the expression of major histocompatibility complex (MHC) class I to escape killing by T cells, nevertheless, these “missing self” tumor cells become more susceptible to the immunosurveillance executed by NK cells. Based on these intrinsic properties and accumulating evidence that defects in NK-cell function and number are often associated with viral infections and tumorigenesis (87), increased attention has been given to NK-cell-based immunotherapy to compensate for the lack of T cell immunotherapy.
3.1. KIRs
Killer-cell immunoglobulin-like receptors (KIRs) are a family of type I transmembrane glycoproteins that are expressed on NK cells and a minority of T cells (88). KIRs have dual functions: they can inhibit NK-cell cytotoxicity by interacting with MHC class I molecules but can also activate cytotoxic activity as activating receptors (89). KIR family members have many haplotypes because of their polymorphic genes, such as KIR2DL1 and KIR3DL2, which are named by the number of extracellular immunoglobulin domains and by the length of the cytoplasmic domain they express (90). KIR inhibitory receptors conduct inhibitory signals through the ITIM, which is located in their long cytoplasmic domain. Based on the “missing self” theory, the humanized antagonistic antibody lirilumab (IPH2102), which can target inhibitory KIRs such as KIR2DL1-3 and KIR2DS1-2, has been used in clinical immunotherapy studies (91). Although the use of lirilumab has been shown to promote NK-cell cytotoxicity toward multiple myeloma, lymphoma and leukemia in preclinical studies, its efficacy in some phase I or II trials on multiple myeloma and acute myeloid leukemia was not as good as expected (92–94). Another mAb targeting KIR2DL1/2/3, IPH2102, has failed to exert impressive clinical effects in patients with multiple myeloma (MM) as monotherapy, but when combined with lenalidomide in a dual immunotherapy for MM patients, it has been reported to achieve a median progression-free survival of 24 months, suggesting the promise of combination therapy (95).
3.2. NKG2A
NKG2 belongs to the C-type lectin-like receptor superfamily and has seven types, NKG2A, NKG2B, NKG2C, NKG2D, NKG2E, NKG2F and NKG2H. NKG2 is expressed on NK cells and acts as an activating receptor or inhibitory receptor when dimerized with other molecules. CD94/NKG2A forms a heterodimeric receptor and plays an inhibitory role on both T cells and NK cells by interacting with HLA-E, which is upregulated in many tumors (96, 97). Pascale André et al. reported that the use of an NKG2A blocking antibody, monalizumab, can enhance NK-cell effector functions against various tumor cells and can rescue CD8+ T-cell function in combination with PD-x axis blockade (98). Takahiro Kamiya et al. constructed NKG2A-null NK cells in which NKG2A expression was abrogated and found that they had increased cytotoxicity against HLA-E-expressing tumor cells. In immunodeficient mice, NKG2A-null NK cells showed an enhanced antitumor effect against HLA-E-expressing tumors (99). In an in vivo study on cancer vaccination using mouse tumor models, the impact of therapeutic vaccines was greatly potentiated by disruption of the NKG2A/Qa-1b (conserved ortholog of HLA-E) axis even in a PD-1-refractory mouse model. However, in this research, the blockade therapy affected CD8 T cells, not NK cells. These findings indicate that NKG2A-blocking antibodies might improve clinical responses to therapeutic cancer vaccines (100). Overall, blockade of the NKG2A axis represents a promising therapeutic approach, but monalizumab monotherapy or combination therapy with another blocking antibody (cetuximab or durvalumab) is still under investigation, and more trials are needed.
3.3. TIGIT
As mentioned above, TIGIT is expressed on some NK cells and can interact with its ligands CD155 and CD112, which are expressed on many tumor cells (71). The binding of TIGIT with its ligands has been reported to result in an inhibitory signal and downregulate NK-cell functions. Qing Zhang et al. reported that TIGIT was associated with NK-cell exhaustion in mouse models and in patients with colon cancer. In mice bearing tumors, including colon tumors, breast tumors and chemically induced fibrosarcomas, treatment with an mAb to TIGIT induced tumor growth inhibition and tumor volume reduction and prevented NK-cell exhaustion. In addition, blockade of TIGIT resulted in potent tumor-specific T-cell immunity in an NK-cell-dependent manner and exerted a synergistic effect with an mAb blocking PD-1 (101).
3.4. PD-1
In addition to being expressed in T cells as mentioned above, PD-1 has also been reported to be expressed in human NK cells from healthy donors and cancer patients and to have an inhibitory effect on NK-cell function (102, 103). Joy Hsu et al. reported that blockade of the PD-1/PD-L1 axis can elicit a strong NK-cell response, which is essential for the therapeutic effect, and implied the importance of PD-1 in inhibiting NK-cell responses in vivo and of the coordinating roles of T cells in PD-1/PD-L1 blockade immunotherapy (104). Wenjuan Dong et al. found that some tumors can induce PD-L1 expression on NK cells via AKT signaling and that an anti-PD-L1 mAb can directly act on PD-L1+ NK cells to combat PD-L1- tumors via a p38 pathway. Their findings reveal a PD-1-independent mechanism of antitumor efficacy through PD-L1+ NK cells that is activated with an anti-PD-L1 mAb (105).
3.5. TIM-3
The expression of TIM-3 is extensive in immune cells, as mentioned above. In addition to T cells, TIM-3 is constitutively expressed on resting human NK cells and is upregulated upon activation (106). The transcriptional levels of TIM-3 are higher in NK cells than in other lymphocytes, and TIM-3 can serve as a maturation marker. Antibodies that crosslink TIM-3 suppress NK-cell-mediated cytotoxicity, indicating that the function of NK cells may be negatively regulated by the interaction of TIM-3 with its cognate ligands, which are expressed on target cells (107). TIM-3 is upregulated in peripheral NK cells of patients with gastric cancer, lung adenocarcinoma and melanoma, while it is upregulated in tumor-infiltrating NK cells of gastrointestinal stromal tumors. This abnormal expression of TIM-3 on NK cells often predicts a poor prognosis, especially in melanoma and lung adenocarcinoma, but blockade of TIM-3 reverses NK-cell exhaustion and improves NK-cell-mediated cytotoxicity (108–111).
3.6. LAG-3
LAG-3 is an inhibitory receptor that is upregulated on activated T cells and NK cells, as mentioned above. It is homologous to CD4 but has a greater affinity for MHC class II molecules; additionally, LAG-3 can bind to LSECtin and FGL1, which are expressed by some tumor cells (112). Unlike in T cells, the function of LAG-3 in NK cells is not clear. Although previous studies have not found that blockade of LAG-3 on human NK cells can influence NK-cell cytotoxicity (113), one study reported that patients with HIV have lower expression of LAG-3 along with other inhibitory molecules involved in viral control, such as PD-1 and TIM-3, than individuals in a low-risk population or progressors (114). IMP321, a soluble recombinant LAG-3-Ig fusion protein, has been reported to induce NK cells to produce IFN-γ and/or TNF-α in healthy donors in an ex vivo short-term experiment, but in metastatic cancer patients, the values are reduced (5, 115). In clinical trials, many anti-LAG-3 monoclonal antibodies have been analyzed either as monotherapies or in combination with other checkpoint-blocking antibodies, such as anti-PD-1 mAb, for the immunotherapy of solid tumors and hematologic malignancies. Two examples are relatlimab (BMS-986016) (NCT01968109) and LAG525 (NCT02460224). However, further work on the effect of LAG-3 on NK cells needs to be explored (116).
3.7. Siglec-7/9
Siglecs, sialic acid-binding immunoglobulin-type lectins, are a subset of the I-type lectins that bind sialic acid and are mainly expressed on the surfaces of immune cells, including neutrophils, eosinophils, monocytes, macrophages, NK cells, dendritic cells, mast cells, B cells and T cells (117). To date, the siglec receptor family comprises 15 members that vary in their expression patterns and in the specificity of ligand binding. Among the family members, siglec-7 and siglec-9 are reported to be mainly expressed on NK cells and to transport inhibitory signals through the ITIM motifs in their cytoplasmic tails (118). Many studies have reported that changes in sialic acid are correlated with tumorigenesis and cancer progression (119). Therefore, siglec-sialic acid interactions may play an important role in modulating the immune response and can be targeted as useful checkpoints (120). In human cancer, siglec-9 has been found to be upregulated in peripheral NK cells, mainly in CD56dimCD16+ NK cells. In an in vitro study, blockade of siglec-7 and siglec-9 using Fab fragments increased the cytotoxicity of NK cells against tumor cells, and in an in vivo mouse model, sialoglycan-dependent NK-cell inhibition led to the killing of tumor cells (118). In a recent study, Itziar Ibarlucea-Benitez et al. investigated the impacts of siglec-7 and siglec-9 on tumor progression using a humanized immunocompetent murine model and found reduced tumor burden when using Fc-engineered anti–Siglec-7 and anti–Siglec-9 blocking antibodies. This effect may have been mediated by prevention of macrophage polarization into tumor-associated macrophages and thus reprogramming of the immune-suppressive tumor microenvironment (121). In addition, Siglec-9 has been found to be upregulated on tumor-infiltrated CD8+ T cells in non-small-cell lung cancer and ovarian and colorectal cancers, and other inhibitory receptors, such as PD-1, are also coexpressed by T cells expressing siglec-9, implying that combination with other immune checkpoint inhibitors could be used for coinhibition in immunotherapy (122).
3.8. HLA-G
Human leukocyte antigen (HLA)-G is a nonclassical MHC-I molecule that was initially found to be expressed in pregnancies by cells of the trophoblast at the maternal–fetal interface and acts as a mediator of immune tolerance because it protects the fetus from NK-cell-mediated lysis (123, 124). To date, seven isoforms have been found, including HLA-G1 to HLA-G7, some of which are membrane-bound molecules and some of which are soluble forms. Under normal physiological conditions, the expression of HLA-G is restricted to immune-privileged organs, but it is upregulated in some immune-mediated diseases, such as viral infections and cancer. By interacting with different receptor molecules on different immune cells, HLA-G exerts several immunomodulatory effects. In NK cells, the inhibitory receptors ILT2 and ILT4 are responsible for the HLA-G-mediated inhibitory effect (125). One study has found that these two inhibitory receptors are broadly expressed on T cells, B cells and dendritic cells, implying the immunosuppressive effect of HLA-G on these cells (126). The abnormal expression of HLA-G in different cancers is associated with poor clinical outcomes in patients, so increasing attention has been given to HLA-G as an immune checkpoint in cancer (127). Numerous studies have reported that the expression of HLA-G in ovarian carcinoma, hepatocellular carcinoma, glioma and renal cell carcinoma inhibits NK cell-mediated cytolysis of these cancer cells but that this inhibition can be reversed by the use of specific antibodies targeting HLA-G or its receptors. In addition, the modulation of cytokine secretion by sHLA-G/ILT2 binding and the different immunosuppressive functions of HLA-G on T cells, B cells, macrophages, dendritic cells, and neutrophils have been deeply discussed (128–132). Chia-Ing Jan et al. designed and tested a CAR strategy to target HLA-G in solid tumors, and the results showed that HLA-G CAR-transduced NK cells effectively cytolyzed breast, brain, pancreatic and ovarian cancer cells in vitro and resulted in reduced xenograft tumor growth with extended median survival in orthotopic mouse models (133). In our study, we found that HLA-G desensitizes breast cancer cells to trastuzumab by binding to the NK-cell receptor KIR2DL4 and the blockade of HLA-G/KIR2DL4 axis improves the vulnerability of HER2-positive breast cancer to trastuzumab treatment in vivo (134).
4. Checkpoint immunotherapy based on macrophage
As an essential innate immune population, macrophages are also important components of the tumor microenvironment (TME). Tumor-associated macrophages (TAMs) have been found to be the most abundant immune cell type in solid tumors and to play an important role in orchestrating the immunosuppressive mechanism of the TME (135). Macrophages are highly plastic and generally can be classified into two polarized cell types: classically activated M1 cells and alternatively activated M2 cells. M1 cells have an antitumor function with a proinflammatory phenotype, and M2 cells can promote tumor progression as immunosuppressive cells. The specific phenotype or polarization type a macrophage assumes is dependent on factors released from TME (136). Many studies have revealed that macrophages play key roles in homeostasis and tumor development; thus, they have been regarded as promising targets for immunotherapy in a variety of diseases.
4.1. PD-1
In addition to T cells and NK cells, PD-1 has been found to be expressed in macrophages, and its expression increases over time and with disease progression (137, 138). Previous studies focused on blockade of the PD-1/PD-L1 axis have demonstrated the promising role of PD-1 in rejuvenating T cells, but the influence of axis blockade on macrophages has not been fully revealed. A recent study has reported that the expression of PD-L1 on macrophages is correlated with clinical responses to anti-PD-L1 therapy; moreover, macrophage polarization can have an effect on the suppression of tumor metastasis (139). Genevieve P Hartley et al. used PD-L1 antibodies to treat mouse and human macrophages and found that the treatment increased spontaneous macrophage proliferation, survival and activation, as indicated by evidence including costimulatory molecule expression and cytokine production. In an in vivo model, the use of a PD-L1 antibody increased tumor infiltration by activated macrophages and triggered macrophage-mediated antitumor activity (140). On the other hand, macrophages may be regulators participating in the mechanism of PD1/PD-L1 treatment resistance. Arlauckas et al. found that PD-1+ CD8+ T cells bound PD-1 antibody in a transient period, and then the antibody was seized within minutes from the T-cell surface by PD-1- macrophages, which led to the failure of reactivation of exhausted T cells (141). Therefore, consideration of the macrophage effect and phenotype in checkpoint immunotherapy is very important.
4.2. CTLA-4
In a study analyzing the action of ipilimumab, a CTLA-4 blocking mAb, Emanuela Romano et al. found that unlike nonresponder patients, patients who respond to ipilimumab treatment display higher peripheral frequencies of nonclassical monocytes at baseline and enrichment of tumor-infiltrating CD68+CD16+ macrophages (142). Previously, Tyler R Simpson et al. explored the activity of an anti-CTLA-4 antibody in the treatment of metastatic melanoma and found that blocking CTLA-4 resulted in selective depletion of Treg cells within tumor lesions; remarkably, this depletion was dependent on Fcγ receptor-expressing macrophages in the TME (143). TAM-mediated elimination of anti-CTLA4-sensitized Tregs resulted in effective antitumor immunity. These results suggest that macrophages in the tumor microenvironment may contribute to the action of anti-CTLA-4 antibodies in tumor treatment.
4.3. CD47-SIRPα
Signal regulatory protein alpha (SIRPα) is a receptor expressed on macrophages that can interact with CD47, which is upregulated on some tumor cells, and thus transmit a “don’t eat me” signal. This is a strategy that is used by tumor cells to avoid phagocytosis. Based on this, anti-CD47 antibodies or engineered SIRPα-Fc fusion proteins have been used to prevent the immunosuppressive signal and restore macrophage phagocytic ability. Inhibition of the CD47/SIRPα axis can reduce tumor size and metastasis in many tumor models (144, 145). In clinical trials, anti-CD47 antibodies such as Hu5F9-G4 and CC-90002 and engineered high-affinity SIRPα and SIRPα-Fc fusion proteins (ALX148 and TTI-621) have been investigated for their therapeutic effects. However, this strategy has a defect: because of the ubiquitous expression of CD47 on red blood cells, anti-CD47 therapy can also lead to transient anemia (146). However, an alternative method has emerged involving a bispecific antibody that can target CD47 and tumor-associated antigens at the same time (147). Moreover, researchers have found that SIRPα is upregulated in NK cells upon IL-2 stimulation and interacts with target cell CD47 in a threshold-dependent manner. SIRPα deficiency or antibody blockade increases the killing capacity of NK cells, so disruption of the SIRPα-CD47 immune checkpoint may augment NK-cell antitumor responses, and elevated expression of CD47 may prevent NK-cell-mediated killing of allogeneic and xenogeneic tissues (148).
4.4. SFRs
In the study of phagocytic responses of different tumor cells to phagocytic cells when using SIRPα-CD47 blackade, Chen et al. found that phagocytosis of haematopoietic tumor cells during SIRPα–CD47 blockade was strictly dependent on SLAM (signalling lymphocytic activation molecule) family receptors (SFRs) in vitro and in vivo in mouse model. As the same results obtained in mouse, they also confirmed that this dependence required SLAMF7 (CD319 or CRACC), a SLAM family member which expressed on macrophages and tumor cell targets in human cells. Unlike other SLAM receptors, whose phagocytosis function are dependent on signalling lymphocyte activation molecule-associated protein (SAP) adaptors, SLAMF7 depended on its interaction with integrin Mac-1 and signals involving immunoreceptor tyrosine-based activation motifs. What counts is, their findings suggest that maybe the SIRPα–CD47 blockade therapy are more effective in patients with SLAMF7 expressing (149). Recently, Li et al. reported a critical role of the other two members of SFRs, SLAMF3 and SLAMF4, in constraining macrophage phagocytosis. Because of their ubiquitous expression on hematopoietic cells, the authors knockout SLAMF3 and SLAMF4 and found that the SFRs deficiency increased the ability of macrophages to phagocytose hematopoietic cells. In mouse model, the SFRs knockout lead to hematopoietic tumor rejection. Importantly, in CAR-macrophage therapy of hematopoietic cancer, the SFRs deletion also enhanced the efficacy. Together, their finding pointing to a potential therapeutic target for hematopoietic cancers (150).
4.5. Clever-1
The full name of Clever-1 is common lymphatic endothelial and vascular endothelial receptor-1, and it is also called Stabilin-1 or Feel-1. It is a conserved, multifunctional adhesion and scavenger receptor that is expressed by some endothelial cells and immunosuppressive macrophages and TAMs. Recent studies have found that Clever-1 can promote tumor progression (151–153). Miro Viitala et al. found that removal of Clever-1 from macrophages can significantly impair tumor growth in multiple solid tumor models, and a lack of Clever-1 in macrophages is associated with an increasingly immunostimulatory phenotype and enhanced signaling through the inflammatory mTOR pathway. Then, anti-Clever-1 treatment displays outcomes comparable to those of PD-1 blockade, implying Clever-1 as a novel target in clinical cancer evaluation and immunotherapy (154).
4.6. CD24/Siglec-10
CD24, a surface protein that is also called heat-stable antigen (HSA) or small cell lung carcinoma cluster 4 antigen, can interact with Siglec-10 and elicit inhibitory signals. CD24 has been reported to be expressed in several solid cancer cells (155, 156). As a member of the Siglec family, siglec-10 bears an ITIM within its cytoplasmic domain and can conduct inhibitory signals. Amira A Barkal et al. reported that many tumors overexpress CD24 and that TAMs express high levels of siglec-10. They found that the phagocytosis of all CD-24-expressing human tumors tested was augmented when CD24 or Siglec-10 was ablated genetically or when an antibody was used to block the CD24/Siglec-10 axis. In an in vivo study, ablation and blockade of CD24 resulted in both a macrophage-dependent reduction in tumor growth and extension of survival. These findings reveal the CD24/Siglec-10 axis as a promising new therapeutic target in cancer immunotherapy (157).
5. Checkpoint immunotherapy based on DCs
5.1. LAG-3
LAG-3 was found to be expressed on a subset of circulating human plasmacytoid dendritic cells (pDCs), and its interaction with MHC II can induce TLR-independent activation of pDCs with limited IFN-α and enhanced IL-6 production. The same study also found LAG-3+ pDCs in melanoma-invaded lymph nodes that were IL-6 positive. These results suggest that activation of pDCs induced by LAG-3 could be involved in creating a suppressive environment in tumor sites (158).
5.2. TIM-3
In addition to T cells, TIM-3 is expressed by multiple other cell types, including dendritic cells, and the expression of TIM-3 may inhibit nucleic acid sensing through TLRs (159). A recent study identified TIM-3, which is expressed by intratumoral CD103+ dendritic cells, as a target for therapy in a murine model of breast cancer. In that study, the use of an anti-TIM-3 antibody improved the response to paclitaxel chemotherapy in models of triple-negative and luminal B disease, with no evidence of toxicity. Anti-TIM-3 antibody administration led to enhanced granzyme B expression by CD8+ T cells and increased CXCR3 chemokine ligand expression by tumor conventional dendritic cells (160). Karen O. Dixon et al. demonstrated that loss of TIM-3 on dendritic cells, but not on CD4+ or CD8+ T cells, promotes strong antitumor immunity; moreover, it prevents dendritic cells from expressing a regulatory program and facilitates the maintenance of CD8+ effector and stem-like T cells. Conditional deletion of TIM-3 in dendritic cells leads to increased accumulation of reactive oxygen species, resulting in NLRP3 inflammasome activation, which underscores the potential of TIM-3 blockade for promoting antitumor immunity by regulating inflammasome activation (161). Overall, the immunomodulatory function mediated by TIM-3 is complex because of the broad expression of TIM-3 in different immune cells and the different interactions of this molecule with multiple ligands. Although promising therapeutic results have been reported in patients with anti-PD1-refractory disease in whom TIM-3 is co-blocked with other checkpoint receptors, the potential of TIM-3 as a drug target in different pathological conditions needs further study (162).
5.3. PD-L1
In a study investigating the anti-tumor mechanism of anti–PD-1 or PD-L1 antibodies, Mayoux et al. characterized various ligands on the surface of dendritic cells and found that PD-L1 is expressed much more abundantly than B7.1 on peripheral and tumor-associated dendritic cells in patients with cancer. PD-L1 expressed on dendritic cells can bind B7.1 on the same cell. This binding potentially prevent PD-1 ligation on T cells or B7.1 ligation of its partner CD28. Blocking PD-L1 on DCs relieves B7.1 sequestration in cis by PD-L1, which allows the B7.1/CD28 interaction to enhance T cell priming. This finding revealed that PD-L1 blockade reinvigorates DC function to generate potent anticancer T cell immunity (163).
6. Discussion
Complex communications between different cells and between cells and their surrounding microenvironment manipulate tumor oncogenesis and progression. In the tumor microenvironment, tumor cells create favorable conditions for cancer progression and avoid immunological surveillance through many strategies. For example, they can reduce neoantigen expression and alter the expression of immunoregulatory molecules on themselves. In addition, other extrinsic factors in the TME, such as the composition of tumor-infiltrating lymphocytes (TILs) and the inhibitory receptors expressed by TILs, all determine the ultimate direction of tumor development (164). Based on this, cancer immunotherapy, which mainly includes adoptive cell transfer (ACT) and immune checkpoint (IC) inhibitor (ICI) therapy, has revolutionized cancer treatment. In this review, we mainly discussed the diversity of immune checkpoints which have been found to be widely distributed in different immune cells and play different regulatory role. With the research and application of immunotherapy based on immune checkpoints in various malignant tumors ( Figure 2 and Table 1 ), their anti-tumor prospects are exciting, but there are still many problems in clinical application. The first question is that most patients exhibit primary or acquired resistance, one possible reason is due to compensatory mechanisms, such as upregulation of alternative immune checkpoints in addition to the widely noted PD-1 and CTLA-4, such as TIM-3 and VISTA, or the influence of many factors in the tumor immune microenvironment on T cell function. To explore the diversity of IC and their different effects on different lymphocytes, as well as to identify new therapeutic targets in the tumor microenvironment, will help guide the application of multi-ICI combination in clinical tumor therapy. To explore the key immunosuppressive pathways in different tumor types and different patient populations is particularly important for selecting the right immunotherapy (165). In addition, studies have found that in some refractory tumors (immunologically cold), the combination of antibodies targeting reverse inhibitory immune microenvironment and anti-PD-1 antibody can often improve the therapeutic effect (154). The second question, there is currently no effective method to distinguish ICI responders from non-responders. But with further research, the discovery of more immune checkpoints and their ligands may help predict the PD-1 therapeutic response in some tumors. For example, it has been found that the expression of inhibitory ligands for Tim-3 and Lag-3 on GC cells serve as potential biomarkers to predict the response to anti-PD-1 therapy and the combinatorial immunotherapy with ICIs targeting for PD-1, Tim-3, and Lag-3 has a therapeutic potential for GC patients (70). Third question, the irAEs present in the clinical ICI treatment is a huge problem, including systemic toxicity, dermotoxicity, gastrointestinal toxicity, endocrine toxicity, pulmonary toxicity, rheumatism, nervous system toxicity, ocular toxicity, renal toxicity, cardiotoxicity and hematological toxicity (166). These side effects will seriously affect the therapeutic effect and prognosis of patients. What’s worse, studies have found that the combined use of ICI may lead to a higher incidence of irAEs than single ICI therapy, depending on the type of malignancy and ICI used (167). At present, the cause of irAEs is not clear, but possible causes include non-specific immune stimulation of organ-specific inflammation, tissue damage and autoimmunity (168). Studies have found that the use of some immune checkpoint antibodies can affect the normal immune function of other normal tissues at the same time. For example, the use of CTLA-4 monoclonal antibodies can simultaneously produce an inhibitory effect on Treg cells expressing CTLA-4, leading to the destruction of immune tolerance, and thus an increase in the frequency and severity of irAEs was observed in some cases (169, 170). In view of the wide expression of immune checkpoints in various lymphocytes listed in this paper and the wide distribution of the same immune checkpoint in different lymphocytes ( Table 1 ), the immune response caused by the application of ICI in the whole immune system should be fully considered. It will be an urgent topic for ICI treatment in the future to consider avoiding severe irAEs caused by the breakdown of autoimmune balance while achieving good anti-tumor efficacy.
Figure 2.

Different ICs expressing on different lymphocytes and and the targeted blocking antibody.
Table 1.
The description of IC molecules、targeted monoclonal antibody drugs and indications.
| IC | Expressing cells | Targeted monoclonal antibody | Indications |
|---|---|---|---|
| PD-1 | Activated T cell, B cell, NK cell, myeloid cells | nivolumab, pembrolizumab, cemiplimab, sintilimab, camrelizumab, toripalimab, tislelizumab, zimberelimab, prolgolimab, dostarlimab | Melanoma, NSCLC, RCC, HCC, Hodgkin's lymphoma, primary mediastinal large B cell lymphoma, SCC of the head and neck, urothelial carcinoma, gastric cancer, solid tumors with high MSI, or MRD, Cutaneous squamous cell carcinoma |
| PD-L1 | various malignancies, dendritic cells | atezolizumab durvalumab avelumab |
NSCLC, urothelial carcinoma, bladder cancer, Merkel cell carcinoma |
| CTLA-4 | Activated T cell and B cells, Treg, NK cells | Ipilimumab | malignant melanoma, NSCLC, mesothelioma, prostate cancer, breast cancer, urothelial cancer |
| Tremelimumab | |||
| Tim-3 | T cell, NK cell and DC | Sabatomimab | Advanced Malignancies |
| cobolimab | |||
| LAG-3 | Activated T cell and NK cell, B cell, Treg and pDC | relatlimab | unresectable or metastatic melanoma |
| fianlimab | |||
| TIGIT | T cell and NK cell | tiragolumab | Melanoma, liver cancer, cervical cancer, prostate cancer, ESCC, breast cancer, NSCLC, NHL/DLBCL/B-cell malignancies |
| domvanalimab | |||
| ociperlimab | |||
| vibostolimab | |||
| VISTA | T cells and CD11b+ antigen-presenting cells, myeloid cells |
JNJ-61,610,588 | NSCLC, small-cell lung cancer, head and neck, pancreatic, colorectal, cervical cancer |
| Siglec-15 | tumor-associated macrophages and dendritic cells, human cancer cells cells | NC318 | advanced solid tumors |
| CD112R | T cell and NK cell | COM701 | Breast cancer, Melanoma, pancreatic cancer |
| GSK4381562 | |||
| KIR | NK cells, CD8+ T cells | lirilumab | MM, AML, relapsed/refractory lymphomas |
| NKG2A | NK cells, CD8+ T cells | monalizumab | oral squamous cell carcinoma, gynecological malignancies, relapsed hematological malignancies |
| Siglec-7/9 | T cell, NK cell and monocytes | none | NSCLC, ovarian, colorectal cancers, melanoma |
| HLA-G | various malignancies | none | Breast cancer |
| ILT2/4,KIR2DL4 | ILT2/4(T cell, NK cell, DC), KIR2DL4(NK cell) | MK-4830 (anti-ILT2) | solid malignancies and hematological malignancies |
| SIRPα | macrophages | KWAR23 | Burkitt's lymphoma, Melanoma |
| 1H9 | |||
| CD47 | many tumor cells | letaplimab | Melanoma, AML stem cells, Breast cancer |
| magrolimab | |||
| SFRs | Macrophages, NK cell | elotuzumab | MM |
| Clever-1 | Endothelial cells and TAMs | Clevegen | cutaneous and uveal melanoma, hepatobiliary, pancreatic, ovarian, oestrogen-receptor-positive breast, colorectal, gastric, gallbladder cancer and cholangiocarcinoma |
| Siglec-10 | Macrophages, B cells, activated T cells and monocytes | ONC-781(anti-CD24) | Advanced Solid Tumors, Unresectable or metastatic melanoma, Resected HCC |
HCC, hepatocellular carcinoma; MRD, minimal residual disease; MSI, microsatellite instability; NSCLC, nonsmall-cell lung carcinoma; RCC, renal cell carcinoma; SCC, squamous cell carcinoma. MM, multiple myeloma; AML, acute myeloid leukemia; ESCC, esophageal squamous cell carcinoma.
Author contributions
Conceptualization, funding acquisition: ZG. Writing original draft, review and editing: GZ、RZ and A-GY. All authors contributed to the article and approved the submitted version.
Funding Statement
This work was supported by the National Natural Sciences Foundation of China (No. 81872326 to ZG, 82203675 to GZ), the Wu Jieping Medical Foundation (320.6750.2022-19-2 to GZ), the Fund of Air Force Medical University (2022KXKT002 to GZ) and the Fund of State Key Laboratory of Cancer Biology (CBSKL2022ZZ46 to GZ).
Conflict of interest
The authors declare that the research was conducted in the absence of any commercial or financial relationships that could be construed as a potential conflict of interest.
Publisher’s note
All claims expressed in this article are solely those of the authors and do not necessarily represent those of their affiliated organizations, or those of the publisher, the editors and the reviewers. Any product that may be evaluated in this article, or claim that may be made by its manufacturer, is not guaranteed or endorsed by the publisher.
References
- 1. Pardoll DM. The blockade of immune checkpoints in cancer immunotherapy. Nat Rev Cancer (2012) 12(4):252–64. doi: 10.1038/nrc3239 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 2. Bagchi S, Yuan R, Engleman EG. Immune checkpoint inhibitors for the treatment of cancer: Clinical impact and mechanisms of response and resistance. Annu Rev Pathol (2021) 16:223–49. doi: 10.1146/annurev-pathol-042020-042741 [DOI] [PubMed] [Google Scholar]
- 3. Zhang Y, Zheng J. Functions of immune checkpoint molecules beyond immune evasion. Adv Exp Med Biol (2020) 1248:201–26. doi: 10.1007/978-981-15-3266-5_9 [DOI] [PubMed] [Google Scholar]
- 4. Qin S, Xu L, Yi M, Yu S, Wu K, Luo S. Novel immune checkpoint targets: Moving beyond pd-1 and ctla-4. Mol Cancer (2019) 18(1):155. doi: 10.1186/s12943-019-1091-2 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 5. Khan M, Arooj S, Wang H. Nk cell-based immune checkpoint inhibition. Front Immunol (2020) 11:167. doi: 10.3389/fimmu.2020.00167 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 6. Brom VC, Burger C, Wirtz DC, Schildberg FA. The role of immune checkpoint molecules on macrophages in cancer, infection, and autoimmune pathologies. Front Immunol (2022) 13:837645. doi: 10.3389/fimmu.2022.837645 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 7. Bremnes RM, Busund LT, Kilvaer TL, Andersen S, Richardsen E, Paulsen EE, et al. The role of tumor-infiltrating lymphocytes in development, progression, and prognosis of non-small cell lung cancer. J Thorac Oncol (2016) 11(6):789–800. doi: 10.1016/j.jtho.2016.01.015 [DOI] [PubMed] [Google Scholar]
- 8. Kroeger DR, Milne K, Nelson BH. Tumor-infiltrating plasma cells are associated with tertiary lymphoid structures, cytolytic T-cell responses, and superior prognosis in ovarian cancer. Clin Cancer Res (2016) 22(12):3005–15. doi: 10.1158/1078-0432.CCR-15-2762 [DOI] [PubMed] [Google Scholar]
- 9. Schalper KA, Velcheti V, Carvajal D, Wimberly H, Brown J, Pusztai L, et al. In situ tumor pd-L1 mrna expression is associated with increased tils and better outcome in breast carcinomas. Clin Cancer Res (2014) 20(10):2773–82. doi: 10.1158/1078-0432.CCR-13-2702 [DOI] [PubMed] [Google Scholar]
- 10. Thaxton JE, Li Z. To affinity and beyond: Harnessing the T cell receptor for cancer immunotherapy. Hum Vaccin Immunother (2014) 10(11):3313–21. doi: 10.4161/21645515.2014.973314 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 11. Davoodzadeh Gholami M, Kardar GA, Saeedi Y, Heydari S, Garssen J, Falak R. Exhaustion of T lymphocytes in the tumor microenvironment: Significance and effective mechanisms. Cell Immunol (2017) 322:1–14. doi: 10.1016/j.cellimm.2017.10.002 [DOI] [PubMed] [Google Scholar]
- 12. Schreiber RD, Old LJ, Smyth MJ. Cancer immunoediting: Integrating immunity's roles in cancer suppression and promotion. Science (2011) 331(6024):1565–70. doi: 10.1126/science.1203486 [DOI] [PubMed] [Google Scholar]
- 13. Blackburn SD, Shin H, Haining WN, Zou T, Workman CJ, Polley A, et al. Coregulation of Cd8+ T cell exhaustion by multiple inhibitory receptors during chronic viral infection. Nat Immunol (2009) 10(1):29–37. doi: 10.1038/ni.1679 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 14. Keir ME, Butte MJ, Freeman GJ, Sharpe AH. Pd-1 and its ligands in tolerance and immunity. Annu Rev Immunol (2008) 26:677–704. doi: 10.1146/annurev.immunol.26.021607.090331 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 15. Mahoney KM, Freeman GJ, McDermott DF. The next immune-checkpoint inhibitors: Pd-1/Pd-L1 blockade in melanoma. Clin Ther (2015) 37(4):764–82. doi: 10.1016/j.clinthera.2015.02.018 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 16. Zajac AJ, Blattman JN, Murali-Krishna K, Sourdive DJ, Suresh M, Altman JD, et al. Viral immune evasion due to persistence of activated T cells without effector function. J Exp Med (1998) 188(12):2205–13. doi: 10.1084/jem.188.12.2205 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 17. Im SJ, Hashimoto M, Gerner MY, Lee J, Kissick HT, Burger MC, et al. Defining Cd8+ T cells that provide the proliferative burst after pd-1 therapy. Nature (2016) 537(7620):417–21. doi: 10.1038/nature19330 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 18. Wartewig T, Kurgyis Z, Keppler S, Pechloff K, Hameister E, Ollinger R, et al. Pd-1 is a haploinsufficient suppressor of T cell lymphomagenesis. Nature (2017) 552(7683):121–5. doi: 10.1038/nature24649 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 19. Zhou XA, Zhou J, Zhao L, Yu G, Zhan J, Shi C, et al. Klhl22 maintains pd-1 homeostasis and prevents excessive T cell suppression. Proc Natl Acad Sci U.S.A. (2020) 117(45):28239–50. doi: 10.1073/pnas.2004570117 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 20. Day CL, Kaufmann DE, Kiepiela P, Brown JA, Moodley ES, Reddy S, et al. Pd-1 expression on hiv-specific T cells is associated with T-cell exhaustion and disease progression. Nature (2006) 443(7109):350–4. doi: 10.1038/nature05115 [DOI] [PubMed] [Google Scholar]
- 21. Huang AC, Orlowski RJ, Xu X, Mick R, George SM, Yan PK, et al. A single dose of neoadjuvant pd-1 blockade predicts clinical outcomes in resectable melanoma. Nat Med (2019) 25(3):454–61. doi: 10.1038/s41591-019-0357-y [DOI] [PMC free article] [PubMed] [Google Scholar]
- 22. Garon EB, Rizvi NA, Hui R, Leighl N, Balmanoukian AS, Eder JP, et al. Pembrolizumab for the treatment of non-Small-Cell lung cancer. N Engl J Med (2015) 372(21):2018–28. doi: 10.1056/NEJMoa1501824 [DOI] [PubMed] [Google Scholar]
- 23. Zhang F, Bai H, Gao R, Fei K, Duan J, Zhang Z, et al. Dynamics of peripheral T cell clones during pd-1 blockade in non-small cell lung cancer. Cancer Immunol Immunother (2020) 69(12):2599–611. doi: 10.1007/s00262-020-02642-4 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 24. Tomita Y, Fukasawa S, Shinohara N, Kitamura H, Oya M, Eto M, et al. Nivolumab versus everolimus in advanced renal cell carcinoma: Japanese subgroup 3-year follow-up analysis from the phase iii checkmate 025 study. Jpn J Clin Oncol (2019) 49(6):506–14. doi: 10.1093/jjco/hyz026 [DOI] [PubMed] [Google Scholar]
- 25. Ferris RL, Blumenschein G, Jr., Fayette J, Guigay J, Colevas AD, Licitra L, et al. Nivolumab for recurrent squamous-cell carcinoma of the head and neck. N Engl J Med (2016) 375(19):1856–67. doi: 10.1056/NEJMoa1602252 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 26. Cohen EEW, Soulieres D, Le Tourneau C, Dinis J, Licitra L, Ahn MJ, et al. Pembrolizumab versus methotrexate, docetaxel, or cetuximab for recurrent or metastatic head-and-Neck squamous cell carcinoma (Keynote-040): A randomised, open-label, phase 3 study. Lancet (2019) 393(10167):156–67. doi: 10.1016/S0140-6736(18)31999-8 [DOI] [PubMed] [Google Scholar]
- 27. Waterhouse P, Penninger JM, Timms E, Wakeham A, Shahinian A, Lee KP, et al. Lymphoproliferative disorders with early lethality in mice deficient in ctla-4. Science (1995) 270(5238):985–8. doi: 10.1126/science.270.5238.985 [DOI] [PubMed] [Google Scholar]
- 28. Tivol EA, Borriello F, Schweitzer AN, Lynch WP, Bluestone JA, Sharpe AH. Loss of ctla-4 leads to massive lymphoproliferation and fatal multiorgan tissue destruction, revealing a critical negative regulatory role of ctla-4. Immunity (1995) 3(5):541–7. doi: 10.1016/1074-7613(95)90125-6 [DOI] [PubMed] [Google Scholar]
- 29. Stamper CC, Zhang Y, Tobin JF, Erbe DV, Ikemizu S, Davis SJ, et al. Crystal structure of the B7-1/Ctla-4 complex that inhibits human immune responses. Nature (2001) 410(6828):608–11. doi: 10.1038/35069118 [DOI] [PubMed] [Google Scholar]
- 30. Perkins D, Wang Z, Donovan C, He H, Mark D, Guan G, et al. Regulation of ctla-4 expression during T cell activation. J Immunol (1996) 156(11):4154–9. doi: 10.4049/jimmunol.156.11.4154 [DOI] [PubMed] [Google Scholar]
- 31. Greene JL, Leytze GM, Emswiler J, Peach R, Bajorath J, Cosand W, et al. Covalent dimerization of Cd28/Ctla-4 and oligomerization of Cd80/Cd86 regulate T cell costimulatory interactions. J Biol Chem (1996) 271(43):26762–71. doi: 10.1074/jbc.271.43.26762 [DOI] [PubMed] [Google Scholar]
- 32. Walker LS, Sansom DM. The emerging role of Ctla4 as a cell-extrinsic regulator of T cell responses. Nat Rev Immunol (2011) 11(12):852–63. doi: 10.1038/nri3108 [DOI] [PubMed] [Google Scholar]
- 33. Leach DR, Krummel MF, Allison JP. Enhancement of antitumor immunity by ctla-4 blockade. Science (1996) 271(5256):1734–6. doi: 10.1126/science.271.5256.1734 [DOI] [PubMed] [Google Scholar]
- 34. Hodi FS, Mihm MC, Soiffer RJ, Haluska FG, Butler M, Seiden MV, et al. Biologic activity of cytotoxic T lymphocyte-associated antigen 4 antibody blockade in previously vaccinated metastatic melanoma and ovarian carcinoma patients. Proc Natl Acad Sci U.S.A. (2003) 100(8):4712–7. doi: 10.1073/pnas.0830997100 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 35. Hodi FS, O'Day SJ, McDermott DF, Weber RW, Sosman JA, Haanen JB, et al. Improved survival with ipilimumab in patients with metastatic melanoma. N Engl J Med (2010) 363(8):711–23. doi: 10.1056/NEJMoa1003466 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 36. Lynch TJ, Bondarenko I, Luft A, Serwatowski P, Barlesi F, Chacko R, et al. Ipilimumab in combination with paclitaxel and carboplatin as first-line treatment in stage Iiib/Iv non-Small-Cell lung cancer: Results from a randomized, double-blind, multicenter phase ii study. J Clin Oncol (2012) 30(17):2046–54. doi: 10.1200/JCO.2011.38.4032 [DOI] [PubMed] [Google Scholar]
- 37. Calabro L, Morra A, Fonsatti E, Cutaia O, Amato G, Giannarelli D, et al. Tremelimumab for patients with chemotherapy-resistant advanced malignant mesothelioma: An open-label, single-arm, phase 2 trial. Lancet Oncol (2013) 14(11):1104–11. doi: 10.1016/S1470-2045(13)70381-4 [DOI] [PubMed] [Google Scholar]
- 38. Slovin SF, Higano CS, Hamid O, Tejwani S, Harzstark A, Alumkal JJ, et al. Ipilimumab alone or in combination with radiotherapy in metastatic castration-resistant prostate cancer: Results from an open-label, multicenter phase I/Ii study. Ann Oncol (2013) 24(7):1813–21. doi: 10.1093/annonc/mdt107 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 39. Vonderheide RH, LoRusso PM, Khalil M, Gartner EM, Khaira D, Soulieres D, et al. Tremelimumab in combination with exemestane in patients with advanced breast cancer and treatment-associated modulation of inducible costimulator expression on patient T cells. Clin Cancer Res (2010) 16(13):3485–94. doi: 10.1158/1078-0432.CCR-10-0505 [DOI] [PubMed] [Google Scholar]
- 40. Carthon BC, Wolchok JD, Yuan J, Kamat A, Ng Tang DS, Sun J, et al. Preoperative ctla-4 blockade: Tolerability and immune monitoring in the setting of a presurgical clinical trial. Clin Cancer Res (2010) 16(10):2861–71. doi: 10.1158/1078-0432.CCR-10-0569 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 41. Boutros C, Tarhini A, Routier E, Lambotte O, Ladurie FL, Carbonnel F, et al. Safety profiles of anti-Ctla-4 and anti-Pd-1 antibodies alone and in combination. Nat Rev Clin Oncol (2016) 13(8):473–86. doi: 10.1038/nrclinonc.2016.58 [DOI] [PubMed] [Google Scholar]
- 42. Hodi FS, Chiarion-Sileni V, Gonzalez R, Grob JJ, Rutkowski P, Cowey CL, et al. Nivolumab plus ipilimumab or nivolumab alone versus ipilimumab alone in advanced melanoma (Checkmate 067): 4-year outcomes of a multicentre, randomised, phase 3 trial. Lancet Oncol (2018) 19(11):1480–92. doi: 10.1016/S1470-2045(18)30700-9 [DOI] [PubMed] [Google Scholar]
- 43. Willsmore ZN, Coumbe BGT, Crescioli S, Reci S, Gupta A, Harris RJ, et al. Combined anti-Pd-1 and anti-Ctla-4 checkpoint blockade: Treatment of melanoma and immune mechanisms of action. Eur J Immunol (2021) 51(3):544–56. doi: 10.1002/eji.202048747 [DOI] [PubMed] [Google Scholar]
- 44. Hastings WD, Anderson DE, Kassam N, Koguchi K, Greenfield EA, Kent SC, et al. Tim-3 is expressed on activated human Cd4+ T cells and regulates Th1 and Th17 cytokines. Eur J Immunol (2009) 39(9):2492–501. doi: 10.1002/eji.200939274 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 45. Sakuishi K, Jayaraman P, Behar SM, Anderson AC, Kuchroo VK. Emerging Tim-3 functions in antimicrobial and tumor immunity. Trends Immunol (2011) 32(8):345–9. doi: 10.1016/j.it.2011.05.003 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 46. Monney L, Sabatos CA, Gaglia JL, Ryu A, Waldner H, Chernova T, et al. Th1-specific cell surface protein Tim-3 regulates macrophage activation and severity of an autoimmune disease. Nature (2002) 415(6871):536–41. doi: 10.1038/415536a [DOI] [PubMed] [Google Scholar]
- 47. Wherry EJ, Kurachi M. Molecular and cellular insights into T cell exhaustion. Nat Rev Immunol (2015) 15(8):486–99. doi: 10.1038/nri3862 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 48. Jones RB, Ndhlovu LC, Barbour JD, Sheth PM, Jha AR, Long BR, et al. Tim-3 expression defines a novel population of dysfunctional T cells with highly elevated frequencies in progressive hiv-1 infection. J Exp Med (2008) 205(12):2763–79. doi: 10.1084/jem.20081398 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 49. Golden-Mason L, Palmer BE, Kassam N, Townshend-Bulson L, Livingston S, McMahon BJ, et al. Negative immune regulator Tim-3 is overexpressed on T cells in hepatitis c virus infection and its blockade rescues dysfunctional Cd4+ and Cd8+ T cells. J Virol (2009) 83(18):9122–30. doi: 10.1128/JVI.00639-09 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 50. Wu W, Shi Y, Li J, Chen F, Chen Z, Zheng M. Tim-3 expression on peripheral T cell subsets correlates with disease progression in hepatitis b infection. Virol J (2011) 8:113. doi: 10.1186/1743-422X-8-113 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 51. Sakuishi K, Apetoh L, Sullivan JM, Blazar BR, Kuchroo VK, Anderson AC. Targeting Tim-3 and pd-1 pathways to reverse T cell exhaustion and restore anti-tumor immunity. J Exp Med (2010) 207(10):2187–94. doi: 10.1084/jem.20100643 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 52. Xu Y, Zhang H, Huang Y, Rui X, Zheng F. Role of Tim-3 in ovarian cancer. Clin Transl Oncol (2017) 19(9):1079–83. doi: 10.1007/s12094-017-1656-8 [DOI] [PubMed] [Google Scholar]
- 53. Li H, Wu K, Tao K, Chen L, Zheng Q, Lu X, et al. Tim-3/Galectin-9 signaling pathway mediates T-cell dysfunction and predicts poor prognosis in patients with hepatitis b virus-associated hepatocellular carcinoma. Hepatology (2012) 56(4):1342–51. doi: 10.1002/hep.25777 [DOI] [PubMed] [Google Scholar]
- 54. Shi X, Li CW, Tan LC, Wen SS, Liao T, Zhang Y, et al. Immune Co-inhibitory receptors pd-1, ctla-4, Tim-3, lag-3, and tigit in medullary thyroid cancers: A Large cohort study. J Clin Endocrinol Metab (2021) 106(1):120–32. doi: 10.1210/clinem/dgaa701 [DOI] [PubMed] [Google Scholar]
- 55. Huard B, Gaulard P, Faure F, Hercend T, Triebel F. Cellular expression and tissue distribution of the human lag-3-Encoded protein, an mhc class ii ligand. Immunogenetics (1994) 39(3):213–7. doi: 10.1007/BF00241263 [DOI] [PubMed] [Google Scholar]
- 56. Triebel F, Jitsukawa S, Baixeras E, Roman-Roman S, Genevee C, Viegas-Pequignot E, et al. Lag-3, a novel lymphocyte activation gene closely related to Cd4. J Exp Med (1990) 171(5):1393–405. doi: 10.1084/jem.171.5.1393 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 57. Workman CJ, Wang Y, El Kasmi KC, Pardoll DM, Murray PJ, Drake CG, et al. Lag-3 regulates plasmacytoid dendritic cell homeostasis. J Immunol (2009) 182(4):1885–91. doi: 10.4049/jimmunol.0800185 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 58. Huard B, Prigent P, Tournier M, Bruniquel D, Triebel F. Cd4/Major histocompatibility complex class ii interaction analyzed with Cd4- and lymphocyte activation gene-3 (Lag-3)-Ig fusion proteins. Eur J Immunol (1995) 25(9):2718–21. doi: 10.1002/eji.1830250949 [DOI] [PubMed] [Google Scholar]
- 59. Wang J, Sanmamed MF, Datar I, Su TT, Ji L, Sun J, et al. Fibrinogen-like protein 1 is a major immune inhibitory ligand of lag-3. Cell (2019) 176(1-2):334–47 e12. doi: 10.1016/j.cell.2018.11.010 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 60. Workman CJ, Vignali DA. The Cd4-related molecule, lag-3 (Cd223), regulates the expansion of activated T cells. Eur J Immunol (2003) 33(4):970–9. doi: 10.1002/eji.200323382 [DOI] [PubMed] [Google Scholar]
- 61. Huang CT, Workman CJ, Flies D, Pan X, Marson AL, Zhou G, et al. Role of lag-3 in regulatory T cells. Immunity (2004) 21(4):503–13. doi: 10.1016/j.immuni.2004.08.010 [DOI] [PubMed] [Google Scholar]
- 62. Woo SR, Turnis ME, Goldberg MV, Bankoti J, Selby M, Nirschl CJ, et al. Immune inhibitory molecules lag-3 and pd-1 synergistically regulate T-cell function to promote tumoral immune escape. Cancer Res (2012) 72(4):917–27. doi: 10.1158/0008-5472.CAN-11-1620 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 63. Brignone C, Escudier B, Grygar C, Marcu M, Triebel F. A phase I pharmacokinetic and biological correlative study of Imp321, a novel mhc class ii agonist, in patients with advanced renal cell carcinoma. Clin Cancer Res (2009) 15(19):6225–31. doi: 10.1158/1078-0432.CCR-09-0068 [DOI] [PubMed] [Google Scholar]
- 64. Ascierto PA, Ignacio M, Bhatia S, Bono P, Sanborn RE, Lipson EJ, et al. Initial efficacy of anti-lymphocyte activation gene-3 (Anti-Lag-3; bms-986016) in combination with nivolumab (Nivo) in pts with melanoma (Mel) previously treated with anti-Pd-1/Pd-L1 therapy. J Clin Oncol (2017) 35(15_suppl):9520. doi: 10.1200/JCO.2017.35.15_suppl.9520 [DOI] [Google Scholar]
- 65. Shapiro M, Herishanu Y, Katz BZ, Dezorella N, Sun C, Kay S, et al. Lymphocyte activation gene 3: A novel therapeutic target in chronic lymphocytic leukemia. Haematologica (2017) 102(5):874–82. doi: 10.3324/haematol.2016.148965 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 66. Li FJ, Zhang Y, Jin GX, Yao L, Wu DQ. Expression of lag-3 is coincident with the impaired effector function of hbv-specific Cd8(+) T cell in hcc patients. Immunol Lett (2013) 150(1-2):116–22. doi: 10.1016/j.imlet.2012.12.004 [DOI] [PubMed] [Google Scholar]
- 67. Takaya S, Saito H, Ikeguchi M. Upregulation of immune checkpoint molecules, pd-1 and lag-3, on Cd4+ and Cd8+ T cells after gastric cancer surgery. Yonago Acta Med (2015) 58(1):39–44. [PMC free article] [PubMed] [Google Scholar]
- 68. Yang ZZ, Kim HJ, Villasboas JC, Chen YP, Price-Troska T, Jalali S, et al. Expression of lag-3 defines exhaustion of intratumoral pd-1(+) T cells and correlates with poor outcome in follicular lymphoma. Oncotarget (2017) 8(37):61425–39. doi: 10.18632/oncotarget.18251 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 69. He Y, Yu H, Rozeboom L, Rivard CJ, Ellison K, Dziadziuszko R, et al. Lag-3 protein expression in non-small cell lung cancer and its relationship with pd-1/Pd-L1 and tumor-infiltrating lymphocytes. J Thorac Oncol (2017) 12(5):814–23. doi: 10.1016/j.jtho.2017.01.019 [DOI] [PubMed] [Google Scholar]
- 70. Mimura K, Kua LF, Xiao JF, Asuncion BR, Nakayama Y, Syn N, et al. Combined inhibition of pd-1/Pd-L1, lag-3, and Tim-3 axes augments antitumor immunity in gastric cancer-T cell coculture models. Gastric Cancer Off J Int Gastric Cancer Assoc Japanese Gastric Cancer Assoc (2021) 24(3):611–23. doi: 10.1007/s10120-020-01151-8 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 71. Harjunpaa H, Guillerey C. Tigit as an emerging immune checkpoint. Clin Exp Immunol (2020) 200(2):108–19. doi: 10.1111/cei.13407 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 72. Chauvin JM, Pagliano O, Fourcade J, Sun Z, Wang H, Sander C, et al. Tigit and pd-1 impair tumor antigen-specific Cd8(+) T cells in melanoma patients. J Clin Invest (2015) 125(5):2046–58. doi: 10.1172/JCI80445 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 73. Wang L, Rubinstein R, Lines JL, Wasiuk A, Ahonen C, Guo Y, et al. Vista, a novel mouse ig superfamily ligand that negatively regulates T cell responses. J Exp Med (2011) 208(3):577–92. doi: 10.1084/jem.20100619 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 74. Le Mercier I, Chen W, Lines JL, Day M, Li J, Sergent P, et al. Vista regulates the development of protective antitumor immunity. Cancer Res (2014) 74(7):1933–44. doi: 10.1158/0008-5472.CAN-13-1506 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 75. Le Mercier I, Lines JL, Noelle RJ. Beyond ctla-4 and pd-1, the generation z of negative checkpoint regulators. Front Immunol (2015) 6:418. doi: 10.3389/fimmu.2015.00418 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 76. Mulati K, Hamanishi J, Matsumura N, Chamoto K, Mise N, Abiko K, et al. Vista expressed in tumour cells regulates T cell function. Br J Cancer (2019) 120(1):115–27. doi: 10.1038/s41416-018-0313-5 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 77. Wuerdemann N, Putz K, Eckel H, Jain R, Wittekindt C, Huebbers CU, et al. Lag-3, Tim-3 and vista expression on tumor-infiltrating lymphocytes in oropharyngeal squamous cell carcinoma-potential biomarkers for targeted therapy concepts. Int J Mol Sci (2020) 22(1):379. doi: 10.3390/ijms22010379 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 78. Angata T, Tabuchi Y, Nakamura K, Nakamura M. Siglec-15: An immune system siglec conserved throughout vertebrate evolution. Glycobiology (2007) 17(8):838–46. doi: 10.1093/glycob/cwm049 [DOI] [PubMed] [Google Scholar]
- 79. Hiruma Y, Hirai T, Tsuda E. Siglec-15, a member of the sialic acid-binding lectin, is a novel regulator for osteoclast differentiation. Biochem Biophys Res Commun (2011) 409(3):424–9. doi: 10.1016/j.bbrc.2011.05.015 [DOI] [PubMed] [Google Scholar]
- 80. Shimizu T, Takahata M, Kameda Y, Endo T, Hamano H, Hiratsuka S, et al. Sialic acid-binding immunoglobulin-like lectin 15 (Siglec-15) mediates periarticular bone loss, but not joint destruction, in murine antigen-induced arthritis. Bone (2015) 79:65–70. doi: 10.1016/j.bone.2015.05.029 [DOI] [PubMed] [Google Scholar]
- 81. Wang J, Sun J, Liu LN, Flies DB, Nie X, Toki M, et al. Siglec-15 as an immune suppressor and potential target for normalization cancer immunotherapy. Nat Med (2019) 25(4):656–66. doi: 10.1038/s41591-019-0374-x [DOI] [PMC free article] [PubMed] [Google Scholar]
- 82. Sun J, Lu Q, Sanmamed MF, Wang J. Siglec-15 as an emerging target for next-generation cancer immunotherapy. Clin Cancer Res (2021) 27(3):680–8. doi: 10.1158/1078-0432.CCR-19-2925 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 83. Buckle I, Guillerey C. Inhibitory receptors and immune checkpoints regulating natural killer cell responses to cancer. Cancers (2021) 13(17):4263. doi: 10.3390/cancers13174263 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 84. Zhu Y, Paniccia A, Schulick AC, Chen W, Koenig MR, Byers JT, et al. Identification of Cd112r as a novel checkpoint for human T cells. J Exp Med (2016) 213(2):167–76. doi: 10.1084/jem.20150785 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 85. Vivier E, Tomasello E, Baratin M, Walzer T, Ugolini S. Functions of natural killer cells. Nat Immunol (2008) 9(5):503–10. doi: 10.1038/ni1582 [DOI] [PubMed] [Google Scholar]
- 86. Long EO, Kim HS, Liu D, Peterson ME, Rajagopalan S. Controlling natural killer cell responses: Integration of signals for activation and inhibition. Annu Rev Immunol (2013) 31:227–58. doi: 10.1146/annurev-immunol-020711-075005 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 87. Orange JS. Natural killer cell deficiency. J Allergy Clin Immunol (2013) 132(3):515–25. doi: 10.1016/j.jaci.2013.07.020 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 88. Bashirova AA, Martin MP, McVicar DW, Carrington M. The killer immunoglobulin-like receptor gene cluster: Tuning the genome for defense. Annu Rev Genomics Hum Genet (2006) 7:277–300. doi: 10.1146/annurev.genom.7.080505.115726 [DOI] [PubMed] [Google Scholar]
- 89. Rajalingam R. Overview of the killer cell immunoglobulin-like receptor system. Methods Mol Biol (2012) 882:391–414. doi: 10.1007/978-1-61779-842-9_23 [DOI] [PubMed] [Google Scholar]
- 90. Lanier LL. Nk cell receptors. Annu Rev Immunol (1998) 16:359–93. doi: 10.1146/annurev.immunol.16.1.359 [DOI] [PubMed] [Google Scholar]
- 91. Benson DM, Jr., Bakan CE, Zhang S, Collins SM, Liang J, Srivastava S, et al. Iph2101, a novel anti-inhibitory kir antibody, and lenalidomide combine to enhance the natural killer cell versus multiple myeloma effect. Blood (2011) 118(24):6387–91. doi: 10.1182/blood-2011-06-360255 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 92. Korde N, Carlsten M, Lee MJ, Minter A, Tan E, Kwok M, et al. A phase ii trial of pan-Kir2d blockade with Iph2101 in smoldering multiple myeloma. Haematologica (2014) 99(6):e81–3. doi: 10.3324/haematol.2013.103085 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 93. Carlsten M, Korde N, Kotecha R, Reger R, Bor S, Kazandjian D, et al. Checkpoint inhibition of Kir2d with the monoclonal antibody Iph2101 induces contraction and hyporesponsiveness of nk cells in patients with myeloma. Clin Cancer Res (2016) 22(21):5211–22. doi: 10.1158/1078-0432.CCR-16-1108 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 94. Vey N, Bourhis JH, Boissel N, Bordessoule D, Prebet T, Charbonnier A, et al. A phase 1 trial of the anti-inhibitory kir mab Iph2101 for aml in complete remission. Blood (2012) 120(22):4317–23. doi: 10.1182/blood-2012-06-437558 [DOI] [PubMed] [Google Scholar]
- 95. Benson DM, Jr., Cohen AD, Jagannath S, Munshi NC, Spitzer G, Hofmeister CC, et al. A phase I trial of the anti-kir antibody Iph2101 and lenalidomide in patients with Relapsed/Refractory multiple myeloma. Clin Cancer Res (2015) 21(18):4055–61. doi: 10.1158/1078-0432.CCR-15-0304 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 96. Braud VM, Allan DS, O'Callaghan CA, Soderstrom K, D'Andrea A, Ogg GS, et al. Hla-e binds to natural killer cell receptors Cd94/Nkg2a, b and c. Nature (1998) 391(6669):795–9. doi: 10.1038/35869 [DOI] [PubMed] [Google Scholar]
- 97. Sivori S, Vacca P, Del Zotto G, Munari E, Mingari MC, Moretta L. Human nk cells: Surface receptors, inhibitory checkpoints, and translational applications. Cell Mol Immunol (2019) 16(5):430–41. doi: 10.1038/s41423-019-0206-4 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 98. Andre P, Denis C, Soulas C, Bourbon-Caillet C, Lopez J, Arnoux T, et al. Anti-Nkg2a mab is a checkpoint inhibitor that promotes anti-tumor immunity by unleashing both T and nk cells. Cell (2018) 175(7):1731–43 e13. doi: 10.1016/j.cell.2018.10.014 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 99. Kamiya T, Seow SV, Wong D, Robinson M, Campana D. Blocking expression of inhibitory receptor Nkg2a overcomes tumor resistance to nk cells. J Clin Invest (2019) 129(5):2094–106. doi: 10.1172/JCI123955 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 100. van Montfoort N, Borst L, Korrer MJ, Sluijter M, Marijt KA, Santegoets SJ, et al. Nkg2a blockade potentiates Cd8 T cell immunity induced by cancer vaccines. Cell (2018) 175(7):1744–55 e15. doi: 10.1016/j.cell.2018.10.028 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 101. Zhang Q, Bi J, Zheng X, Chen Y, Wang H, Wu W, et al. Blockade of the checkpoint receptor tigit prevents nk cell exhaustion and elicits potent anti-tumor immunity. Nat Immunol (2018) 19(7):723–32. doi: 10.1038/s41590-018-0132-0 [DOI] [PubMed] [Google Scholar]
- 102. Pesce S, Greppi M, Tabellini G, Rampinelli F, Parolini S, Olive D, et al. Identification of a subset of human natural killer cells expressing high levels of programmed death 1: A phenotypic and functional characterization. J Allergy Clin Immunol (2017) 139(1):335–46 e3. doi: 10.1016/j.jaci.2016.04.025 [DOI] [PubMed] [Google Scholar]
- 103. Liu Y, Cheng Y, Xu Y, Wang Z, Du X, Li C, et al. Increased expression of programmed cell death protein 1 on nk cells inhibits nk-Cell-Mediated anti-tumor function and indicates poor prognosis in digestive cancers. Oncogene (2017) 36(44):6143–53. doi: 10.1038/onc.2017.209 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 104. Hsu J, Hodgins JJ, Marathe M, Nicolai CJ, Bourgeois-Daigneault MC, Trevino TN, et al. Contribution of nk cells to immunotherapy mediated by pd-1/Pd-L1 blockade. J Clin Invest (2018) 128(10):4654–68. doi: 10.1172/JCI99317 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 105. Dong W, Wu X, Ma S, Wang Y, Nalin AP, Zhu Z, et al. The mechanism of anti-Pd-L1 antibody efficacy against pd-L1-Negative tumors identifies nk cells expressing pd-L1 as a cytolytic effector. Cancer Discovery (2019) 9(10):1422–37. doi: 10.1158/2159-8290.CD-18-1259 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 106. Gleason MK, Lenvik TR, McCullar V, Felices M, O'Brien MS, Cooley SA, et al. Tim-3 is an inducible human natural killer cell receptor that enhances interferon gamma production in response to galectin-9. Blood (2012) 119(13):3064–72. doi: 10.1182/blood-2011-06-360321 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 107. Ndhlovu LC, Lopez-Verges S, Barbour JD, Jones RB, Jha AR, Long BR, et al. Tim-3 marks human natural killer cell maturation and suppresses cell-mediated cytotoxicity. Blood (2012) 119(16):3734–43. doi: 10.1182/blood-2011-11-392951 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 108. Wang Z, Zhu J, Gu H, Yuan Y, Zhang B, Zhu D, et al. The clinical significance of abnormal Tim-3 expression on nk cells from patients with gastric cancer. Immunol Invest (2015) 44(6):578–89. doi: 10.3109/08820139.2015.1052145 [DOI] [PubMed] [Google Scholar]
- 109. Xu L, Huang Y, Tan L, Yu W, Chen D, Lu C, et al. Increased Tim-3 expression in peripheral nk cells predicts a poorer prognosis and Tim-3 blockade improves nk cell-mediated cytotoxicity in human lung adenocarcinoma. Int Immunopharmacol (2015) 29(2):635–41. doi: 10.1016/j.intimp.2015.09.017 [DOI] [PubMed] [Google Scholar]
- 110. da Silva IP, Gallois A, Jimenez-Baranda S, Khan S, Anderson AC, Kuchroo VK, et al. Reversal of nk-cell exhaustion in advanced melanoma by Tim-3 blockade. Cancer Immunol Res (2014) 2(5):410–22. doi: 10.1158/2326-6066.CIR-13-0171 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 111. Komita H, Koido S, Hayashi K, Kan S, Ito M, Kamata Y, et al. Expression of immune checkpoint molecules of T cell immunoglobulin and mucin protein 3/Galectin-9 for nk cell suppression in human gastrointestinal stromal tumors. Oncol Rep (2015) 34(4):2099–105. doi: 10.3892/or.2015.4149 [DOI] [PubMed] [Google Scholar]
- 112. Xu F, Liu J, Liu D, Liu B, Wang M, Hu Z, et al. Lsectin expressed on melanoma cells promotes tumor progression by inhibiting antitumor T-cell responses. Cancer Res (2014) 74(13):3418–28. doi: 10.1158/0008-5472.CAN-13-2690 [DOI] [PubMed] [Google Scholar]
- 113. Huard B, Tournier M, Triebel F. Lag-3 does not define a specific mode of natural killing in human. Immunol Lett (1998) 61(2-3):109–12. doi: 10.1016/s0165-2478(97)00170-3 [DOI] [PubMed] [Google Scholar]
- 114. Taborda NA, Hernandez JC, Lajoie J, Juno JA, Kimani J, Rugeles MT, et al. Short communication: Low expression of activation and inhibitory molecules on nk cells and Cd4(+) T cells is associated with viral control. AIDS Res Hum Retroviruses (2015) 31(6):636–40. doi: 10.1089/AID.2014.0325 [DOI] [PubMed] [Google Scholar]
- 115. Brignone C, Grygar C, Marcu M, Schakel K, Triebel F. A soluble form of lymphocyte activation gene-3 (Imp321) induces activation of a Large range of human effector cytotoxic cells. J Immunol (2007) 179(6):4202–11. doi: 10.4049/jimmunol.179.6.4202 [DOI] [PubMed] [Google Scholar]
- 116. Andrews LP, Marciscano AE, Drake CG, Vignali DA. Lag3 (Cd223) as a cancer immunotherapy target. Immunol Rev (2017) 276(1):80–96. doi: 10.1111/imr.12519 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 117. Macauley MS, Crocker PR, Paulson JC. Siglec-mediated regulation of immune cell function in disease. Nat Rev Immunol (2014) 14(10):653–66. doi: 10.1038/nri3737 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 118. Jandus C, Boligan KF, Chijioke O, Liu H, Dahlhaus M, Demoulins T, et al. Interactions between siglec-7/9 receptors and ligands influence nk cell-dependent tumor immunosurveillance. J Clin Invest (2014) 124(4):1810–20. doi: 10.1172/JCI65899 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 119. Adams OJ, Stanczak MA, von Gunten S, Laubli H. Targeting sialic acid-siglec interactions to reverse immune suppression in cancer. Glycobiology (2018) 28(9):640–7. doi: 10.1093/glycob/cwx108 [DOI] [PubMed] [Google Scholar]
- 120. Lubbers J, Rodriguez E, van Kooyk Y. Modulation of immune tolerance Via siglec-sialic acid interactions. Front Immunol (2018) 9:2807. doi: 10.3389/fimmu.2018.02807 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 121. Ibarlucea-Benitez I, Weitzenfeld P, Smith P, Ravetch JV. Siglecs-7/9 function as inhibitory immune checkpoints in vivo and can be targeted to enhance therapeutic antitumor. Proc Natl Acad Sci USA (2021) 118(26):e2107424118. doi: 10.1073/pnas.2107424118 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 122. Stanczak MA, Siddiqui SS, Trefny MP, Thommen DS, Boligan KF, von Gunten S, et al. Self-associated molecular patterns mediate cancer immune evasion by engaging siglecs on T cells. J Clin Invest (2018) 128(11):4912–23. doi: 10.1172/JCI120612 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 123. Kovats S, Main EK, Librach C, Stubblebine M, Fisher SJ, DeMars R. A class I antigen, hla-G, expressed in human trophoblasts. Science (1990) 248(4952):220–3. doi: 10.1126/science.2326636 [DOI] [PubMed] [Google Scholar]
- 124. Hunt JS, Petroff MG, McIntire RH, Ober C. Hla-G and immune tolerance in pregnancy. FASEB J (2005) 19(7):681–93. doi: 10.1096/fj.04-2078rev [DOI] [PubMed] [Google Scholar]
- 125. Pazmany L, Mandelboim O, Vales-Gomez M, Davis DM, Reyburn HT, Strominger JL. Protection from natural killer cell-mediated lysis by hla-G expression on target cells. Science (1996) 274(5288):792–5. doi: 10.1126/science.274.5288.792 [DOI] [PubMed] [Google Scholar]
- 126. Liu L, Wang L, Zhao L, He C, Wang G. The role of hla-G in tumor escape: Manipulating the phenotype and function of immune cells. Front Oncol (2020) 10:597468. doi: 10.3389/fonc.2020.597468 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 127. Carosella ED, Rouas-Freiss N, Tronik-Le Roux D, Moreau P, LeMaoult J. Hla-G: An immune checkpoint molecule. Adv Immunol (2015) 127:33–144. doi: 10.1016/bs.ai.2015.04.001 [DOI] [PubMed] [Google Scholar]
- 128. Lin A, Yan WH, Xu HH, Gan MF, Cai JF, Zhu M, et al. Hla-G expression in human ovarian carcinoma counteracts nk cell function. Ann Oncol (2007) 18(11):1804–9. doi: 10.1093/annonc/mdm356 [DOI] [PubMed] [Google Scholar]
- 129. Lin A, Chen HX, Zhu CC, Zhang X, Xu HH, Zhang JG, et al. Aberrant human leucocyte antigen-G expression and its clinical relevance in hepatocellular carcinoma. J Cell Mol Med (2010) 14(8):2162–71. doi: 10.1111/j.1582-4934.2009.00917.x [DOI] [PMC free article] [PubMed] [Google Scholar]
- 130. Wiendl H, Mitsdoerffer M, Hofmeister V, Wischhusen J, Bornemann A, Meyermann R, et al. A functional role of hla-G expression in human gliomas: An alternative strategy of immune escape. J Immunol (2002) 168(9):4772–80. doi: 10.4049/jimmunol.168.9.4772 [DOI] [PubMed] [Google Scholar]
- 131. Bukur J, Rebmann V, Grosse-Wilde H, Luboldt H, Ruebben H, Drexler I, et al. Functional role of human leukocyte antigen-G up-regulation in renal cell carcinoma. Cancer Res (2003) 63(14):4107–11. [PubMed] [Google Scholar]
- 132. Lin A, Yan WH. Heterogeneity of hla-G expression in cancers: Facing the challenges. Front Immunol (2018) 9:2164. doi: 10.3389/fimmu.2018.02164 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 133. Jan CI, Huang SW, Canoll P, Bruce JN, Lin YC, Pan CM, et al. Targeting human leukocyte antigen G with chimeric antigen receptors of natural killer cells convert immunosuppression to ablate solid tumors. J Immunother Cancer (2021) 9(10):e003050. doi: 10.1136/jitc-2021-003050 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 134. Zheng G, Guo Z, Li W, Xi W, Zuo B, Zhang R, et al. Interaction between hla-G and nk cell receptor Kir2dl4 orchestrates Her2-positive breast cancer resistance to trastuzumab. Signal Transduct Target Ther (2021) 6(1):236. doi: 10.1038/s41392-021-00629-w [DOI] [PMC free article] [PubMed] [Google Scholar]
- 135. Solinas G, Schiarea S, Liguori M, Fabbri M, Pesce S, Zammataro L, et al. Tumor-conditioned macrophages secrete migration-stimulating factor: A new marker for M2-polarization, influencing tumor cell motility. J Immunol (2010) 185(1):642–52. doi: 10.4049/jimmunol.1000413 [DOI] [PubMed] [Google Scholar]
- 136. Najafi M, Hashemi Goradel N, Farhood B, Salehi E, Nashtaei MS, Khanlarkhani N, et al. Macrophage polarity in cancer: A review. J Cell Biochem (2019) 120(3):2756–65. doi: 10.1002/jcb.27646 [DOI] [PubMed] [Google Scholar]
- 137. Huang X, Venet F, Wang YL, Lepape A, Yuan Z, Chen Y, et al. Pd-1 expression by macrophages plays a pathologic role in altering microbial clearance and the innate inflammatory response to sepsis. Proc Natl Acad Sci U.S.A. (2009) 106(15):6303–8. doi: 10.1073/pnas.0809422106 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 138. Gordon SR, Maute RL, Dulken BW, Hutter G, George BM, McCracken MN, et al. Pd-1 expression by tumour-associated macrophages inhibits phagocytosis and tumour immunity. Nature (2017) 545(7655):495–9. doi: 10.1038/nature22396 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 139. Zou W, Wolchok JD, Chen L. Pd-L1 (B7-H1) and pd-1 pathway blockade for cancer therapy: Mechanisms, response biomarkers, and combinations. Sci Transl Med (2016) 8(328):328rv4. doi: 10.1126/scitranslmed.aad7118 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 140. Hartley GP, Chow L, Ammons DT, Wheat WH, Dow SW. Programmed cell death ligand 1 (Pd-L1) signaling regulates macrophage proliferation and activation. Cancer Immunol Res (2018) 6(10):1260–73. doi: 10.1158/2326-6066.CIR-17-0537 [DOI] [PubMed] [Google Scholar]
- 141. Arlauckas SP, Garris CS, Kohler RH, Kitaoka M, Cuccarese MF, Yang KS, et al. In vivo imaging reveals a tumor-associated macrophage-mediated resistance pathway in anti-Pd-1 therapy. Sci Transl Med (2017) 9(389):eaal3604. doi: 10.1126/scitranslmed.aal3604 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 142. Romano E, Kusio-Kobialka M, Foukas PG, Baumgaertner P, Meyer C, Ballabeni P, et al. Ipilimumab-dependent cell-mediated cytotoxicity of regulatory T cells ex vivo by nonclassical monocytes in melanoma patients. Proc Natl Acad Sci U.S.A. (2015) 112(19):6140–5. doi: 10.1073/pnas.1417320112 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 143. Simpson TR, Li F, Montalvo-Ortiz W, Sepulveda MA, Bergerhoff K, Arce F, et al. Fc-dependent depletion of tumor-infiltrating regulatory T cells Co-defines the efficacy of anti-Ctla-4 therapy against melanoma. J Exp Med (2013) 210(9):1695–710. doi: 10.1084/jem.20130579 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 144. Willingham SB, Volkmer JP, Gentles AJ, Sahoo D, Dalerba P, Mitra SS, et al. The Cd47-signal regulatory protein alpha (Sirpa) interaction is a therapeutic target for human solid tumors. Proc Natl Acad Sci U.S.A. (2012) 109(17):6662–7. doi: 10.1073/pnas.1121623109 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 145. Weiskopf K, Ring AM, Ho CC, Volkmer JP, Levin AM, Volkmer AK, et al. Engineered sirpalpha variants as immunotherapeutic adjuvants to anticancer antibodies. Science (2013) 341(6141):88–91. doi: 10.1126/science.1238856 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 146. Liu J, Wang L, Zhao F, Tseng S, Narayanan C, Shura L, et al. Pre-clinical development of a humanized anti-Cd47 antibody with anti-cancer therapeutic potential. PloS One (2015) 10(9):e0137345. doi: 10.1371/journal.pone.0137345 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 147. Dheilly E, Moine V, Broyer L, Salgado-Pires S, Johnson Z, Papaioannou A, et al. Selective blockade of the ubiquitous checkpoint receptor Cd47 is enabled by dual-targeting bispecific antibodies. Mol Ther (2017) 25(2):523–33. doi: 10.1016/j.ymthe.2016.11.006 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 148. Deuse T, Hu X, Agbor-Enoh S, Jang MK, Alawi M, Saygi C, et al. The sirpalpha-Cd47 immune checkpoint in nk cells. J Exp Med (2021) 218(3):e20200839. doi: 10.1084/jem.20200839 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 149. Chen J, Zhong MC, Guo HJ, Davidson D, Mishel S, Lu Y, et al. Slamf7 is critical for phagocytosis of haematopoietic tumour cells Via mac-1 integrin. Nature (2017) 544(7651):493–+. doi: 10.1038/nature22076 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 150. Li D, Xiong W, Wang YD, Feng J, He YX, Du J, et al. Slamf3 and Slamf4 are immune checkpoints that constrain macrophage phagocytosis of hematopoietic tumors. Sci Immunol (2022) 7(67):eabj5501. doi: 10.1126/sciimmunol.abj5501 [DOI] [PubMed] [Google Scholar]
- 151. Kzhyshkowska J, Gratchev A, Goerdt S. Stabilin-1, a homeostatic scavenger receptor with multiple functions. J Cell Mol Med (2006) 10(3):635–49. doi: 10.1111/j.1582-4934.2006.tb00425.x [DOI] [PMC free article] [PubMed] [Google Scholar]
- 152. Algars A, Irjala H, Vaittinen S, Huhtinen H, Sundstrom J, Salmi M, et al. Type and location of tumor-infiltrating macrophages and lymphatic vessels predict survival of colorectal cancer patients. Int J Cancer (2012) 131(4):864–73. doi: 10.1002/ijc.26457 [DOI] [PubMed] [Google Scholar]
- 153. Karikoski M, Marttila-Ichihara F, Elima K, Rantakari P, Hollmen M, Kelkka T, et al. Clever-1/Stabilin-1 controls cancer growth and metastasis. Clin Cancer Res (2014) 20(24):6452–64. doi: 10.1158/1078-0432.CCR-14-1236 [DOI] [PubMed] [Google Scholar]
- 154. Viitala M, Virtakoivu R, Tadayon S, Rannikko J, Jalkanen S, Hollmen M. Immunotherapeutic blockade of macrophage clever-1 reactivates the Cd8(+) T-cell response against immunosuppressive tumors. Clin Cancer Res an Off J Am Assoc Cancer Res (2019) 25(11):3289–303. doi: 10.1158/1078-0432.CCR-18-3016 [DOI] [PubMed] [Google Scholar]
- 155. Kristiansen G, Winzer KJ, Mayordomo E, Bellach J, Schluns K, Denkert C, et al. Cd24 expression is a new prognostic marker in breast cancer. Clin Cancer Res (2003) 9(13):4906–13. [PubMed] [Google Scholar]
- 156. Tarhriz V, Bandehpour M, Dastmalchi S, Ouladsahebmadarek E, Zarredar H, Eyvazi S. Overview of Cd24 as a new molecular marker in ovarian cancer. J Cell Physiol (2019) 234(3):2134–42. doi: 10.1002/jcp.27581 [DOI] [PubMed] [Google Scholar]
- 157. Barkal AA, Brewer RE, Markovic M, Kowarsky M, Barkal SA, Zaro BW, et al. Cd24 signalling through macrophage siglec-10 is a target for cancer immunotherapy. Nature (2019) 572(7769):392–6. doi: 10.1038/s41586-019-1456-0 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 158. Camisaschi C, De Filippo A, Beretta V, Vergani B, Villa A, Vergani E, et al. Alternative activation of human plasmacytoid dcs in vitro and in melanoma lesions: Involvement of lag-3. J Invest Dermatol (2014) 134(7):1893–902. doi: 10.1038/jid.2014.29 [DOI] [PubMed] [Google Scholar]
- 159. Chiba S, Baghdadi M, Akiba H, Yoshiyama H, Kinoshita I, Dosaka-Akita H, et al. Tumor-infiltrating dcs suppress nucleic acid-mediated innate immune responses through interactions between the receptor Tim-3 and the alarmin Hmgb1. Nat Immunol (2012) 13(9):832–42. doi: 10.1038/ni.2376 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 160. de Mingo Pulido A, Gardner A, Hiebler S, Soliman H, Rugo HS, Krummel MF, et al. Tim-3 regulates Cd103(+) dendritic cell function and response to chemotherapy in breast cancer. Cancer Cell (2018) 33(1):60–74 e6. doi: 10.1016/j.ccell.2017.11.019 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 161. Dixon KO, Tabaka M, Schramm MA, Xiao S, Tang R, Dionne D, et al. Tim-3 restrains anti-tumour immunity by regulating inflammasome activation. Nature (2021) 595(7865):101–6. doi: 10.1038/s41586-021-03626-9 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 162. Wolf Y, Anderson AC, Kuchroo VK. Tim3 comes of age as an inhibitory receptor. Nat Rev Immunol (2020) 20(3):173–85. doi: 10.1038/s41577-019-0224-6 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 163. Mayoux M, Roller A, Pulko V, Sammicheli S, Chen S, Sum E, et al. Dendritic cells dictate responses to pd-L1 blockade cancer immunotherapy. Sci Trans Med (2020) 12(534):eaav7431. doi: 10.1126/scitranslmed.aav7431 [DOI] [PubMed] [Google Scholar]
- 164. Bilotta MT, Antignani A, Fitzgerald DJ. Managing the tme to improve the efficacy of cancer therapy. Front Immunol (2022) 13:954992. doi: 10.3389/fimmu.2022.954992 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 165. Kraehenbuehl L, Weng CH, Eghbali S, Wolchok JD, Merghoub T. Enhancing immunotherapy in cancer by targeting emerging immunomodulatory pathways. Nat Rev Clin Oncol (2022) 19(1):37–50. doi: 10.1038/s41571-021-00552-7 [DOI] [PubMed] [Google Scholar]
- 166. Naimi A, Mohammed RN, Raji A, Chupradit S, Yumashev AV, Suksatan W, et al. Tumor immunotherapies by immune checkpoint inhibitors (Icis); the pros and cons. Cell commun Signaling CCS (2022) 20(1):44. doi: 10.1186/s12964-022-00854-y [DOI] [PMC free article] [PubMed] [Google Scholar]
- 167. Larkin J, Chiarion-Sileni V, Gonzalez R, Grob JJ, Cowey CL, Lao CD, et al. Combined nivolumab and ipilimumab or monotherapy in untreated melanoma. New Engl J Med (2015) 373(1):23–34. doi: 10.1056/NEJMoa1504030 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 168. Poto R, Troiani T, Criscuolo G, Marone G, Ciardiello F, Tocchetti CG, et al. Holistic approach to immune checkpoint inhibitor-related adverse events. Front Immunol (2022) 13:804597. doi: 10.3389/fimmu.2022.804597 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 169. Lisi L, Lacal PM, Martire M, Navarra P, Graziani G. Clinical experience with ctla-4 blockade for cancer immunotherapy: From the monospecific monoclonal antibody ipilimumab to probodies and bispecific molecules targeting the tumor microenvironment. Pharmacol Res (2022) 175:105997. doi: 10.1016/j.phrs.2021.105997 [DOI] [PubMed] [Google Scholar]
- 170. Larkin J, Hodi FS, Wolchok JD. Combined nivolumab and ipilimumab or monotherapy in untreated melanoma. New Engl J Med (2015) 373(13):1270–1. doi: 10.1056/NEJMc1509660 [DOI] [PubMed] [Google Scholar]