

Abstract
Domestication and improvement are important processes that generate the variation in genome and phonotypes underlying crop improvement. Unfortunately, during selection for certain attributes, other valuable traits may be inadvertently discarded. One example is the decline in fruit soluble solids content (SSC) during tomato breeding. Several genetic loci for SSC have been identified, but few reports on the underlying mechanisms are available. In this study we performed a genome-wide association study (GWAS) for SSC of the red-ripe fruits in a population consisting of 481 tomato accessions with large natural variations and found a new quantitative trait locus, STP1, encoding a sugar transporter protein. The causal variation of STP1, a 21-bp InDel located in the promoter region 1124 bp upstream of the start codon, alters its expression. STP1Insertion accessions with an 21-bp insertion have higher SSC than STP1Deletion accessions with the 21-bp deletion. Knockout of STP1 in TS-23 with high SSC using CRISPR/Cas9 greatly decreased SSC in fruits. In vivo and in vitro assays demonstrated that ZAT10-LIKE, a zinc finger protein transcription factor (ZFP TF), can specifically bind to the promoter of STP1Insertion to enhance STP1 expression, but not to the promoter of STP1Deletion, leading to lower fruit SSC in modern tomatoes. Diversity analysis revealed that STP1 was selected during tomato improvement. Taking these results together, we identified a naturally occurring causal variation underlying SSC in tomato, and a new role for ZFP TFs in regulating sugar transporters. The findings enrich our understanding of tomato evolution and domestication, and provide a genetic basis for genome design for improving fruit taste.
Introduction
Tomato (Solanum lycopersicum) is one of the most widely cultivated vegetables worldwide [1] and tomato fruit is an vital source of nutrition in the human diet [2]. The content of flavor chemicals, including volatiles, acids, and sugars largely determines consumer liking in tomato fruits [3]. The soluble solids content (SSC) is a comprehensive index to characterize soluble sugars and organic acids that make an important contribution to taste and flavor perception.
The content of soluble sugars and organic acids is a quantitative trait controlled by multiple genes. Thus far, several loci have been found to regulate accumulation of soluble sugars in tomato. The sucrose accumulator gene (sucr) from Solanum chmielewskii can confer enhanced fruit sugar composition [4]. Brix9-2-5 increased sugar yield in tomato and further analysis showed that LIN5, encoding a flower- and fruit-specific invertase, was mapped within this region. Variation of an amino acid led to a decrease in its enzyme activity [5, 6]. RNAi lines of LIN5 significantly reduced the Brix content in tomato fruits [7]. Introgression of the FgrH allele from the wild species (LA1777) into cultivated tomato reduced glucose levels and increased fructose levels and thus the fructose-to-glucose ratio of the red-ripe fruits was increased. Further research showed that the SlFgr gene is a member of the SWEET family, encoding plasma membrane-localized glucose efflux transporters [8, 9]. The above quantitative trait loci (QTLs) were characterized based on recombinant inbred lines (RILs) or near-isogenic lines (NILs). Tomato genome sequencing and comprehensive evolution analysis have provided molecular insights into gene function during evolution [10, 11].
Genome-wide association study (GWAS) is a powerful technology to deepen insights into the evolution of agronomically important traits and provides loci for genetic improvement [12]. Metabolic genome-wide association study (mGWAS) is one of the strongest tools for genetic determinants for differential accumulation of metabolites and diversity of plant metabolism. mGWAS was applied in Arabidopsis thaliana initially and subsequently extended with improvements to other species, especially important crops [13], providing deeper insights into the genetic bases of metabolic diversity [14–20]. In tomato, several genes related to SSC have been mapped through mGWAS and their natural variations have been analyzed. Typically, a 3-bp InDel in the promoter of SlALMT9 was related to high malate content in fruits [21]. This InDel prevented SlWRKY42 from binding the promoter of SlALMT9, which alleviated the expression of SlALMT9 and promoted the accumulation of high fruit malate [21]. An 8-bp InDel in the promoter of SlbHLH59 directly regulated the expression of genes related to ascorbate biosynthesis and thereby determined ascorbate content in tomato fruits [22].
In tomato fruits, soluble sugars mainly comprise glucose and fructose, in near-equimolar quantities [9, 23], which play vital roles as sources of energy and as primary metabolites [24]. Uptake of glucose and fructose is mediated by sugar transport proteins (STPs) [25]. Originally, the three STP genes LeHT1/2/3 were identified from the same family [26]. Subsequently the STP family was classified as the largest subfamily of sugar transporters in tomato [24]. RNAi lines of LeHT genes led to a 55% decrease in hexose accumulation in fruits [27]. Heterologous expression of LeHT1 in yeast can rescue a hexose transport-impaired mutant and LeHT1 showed the same transport characteristics as the high-affinity glucose/H+ cotransporter [27]. Despite the functional identification of sugar transporters in plants, the causal variation and regulatory mechanism remain largely unknown.
Many biological processes are regulated at the transcriptional level, including responses to the environment, balance in metabolism and physiology, and progression through the cell cycle [28]. Zinc finger protein transcription factors (ZFP TFs) usually show sequence-specific DNA binding and can activate and/or repress gene transcription [28]. ZFP TFs participated widely in stress responses and plant development [29, 30]. Overexpression of SlCZFP1 enhanced tolerance to cold treatments or freezing in transgenic Arabidopsis and rice [31]. Overexpression of ZFP179 increased salt tolerance in rice [32]. AtZAT10, a C2H2-EAR zinc finger protein, can elevate the expression of genes related to reactive oxygen defense and enhance the tolerance of osmotic stress, heat, and salinity in Arabidopsis plants [33]. MdZAT10 markedly accelerated leaf senescence and elevated the expression of genes related to senescence in apple [34]. In Arabidopsis, a few zinc finger proteins have been identified to participate in the regulation of the flowering time of Arabidopsis, such as JAGGED (JAD) [35] and SUPPRESSOR OF FRIGIDA4 (SUF4) [36]. The B-box (BBX) proteins, a class of ZFP TFs, are indispensable factors in controlling plant growth and development [37–40]. However, there is still a big gap between the functions of ZFP TFs in regulating sugar transporters that needs to be filled.
In this study, we measured the SSC of red-ripe fruits in 481 tomato accessions. Through mGWAS, we identified reliable QTLs that contribute to fruit SSC. Furthermore, we determined a new QTL, STP1 (Sugar Transport Protein 1) by analyzing gene expression, genetic variations, and functional verification, and confirmed its contribution to high SSC in tomato fruits. In addition, we found that ZAT10-LIKE, a ZFP TF, cannot bind to the promoter of STP1 with a 21-bp deletion, but specifically binds to the promoter of STP1 with a 21-bp insertion, and positively regulates gene expression of STP1Insertion. These findings provide genetic insights into the evolution and regulation of SSC and gene resources for fruit quality improvement.
Results
GWAS reveals a locus, STP1, influencing tomato SSC in chromosome 2
Tomato SSC is a complicated quantitative trait controlled by multiple genes. Previous studies have found several QTLs that have a significant influence on SSC in tomato. In order to discover new QTLs, we determined SSC of red-ripe fruits in 481 accessions, performed a GWAS (Supplementary Data Table S1), and identified a single-nucleotide polymorphism (SNP) (SL2.50ch02:44438607) that showed a strong correlation with the level of SSC (P = 3.45 × 10−7) (Fig. 1a and b). Two alleles based on the lead SNP (SL2.50ch02:44438607, A/G) from the association signal—high-SSC allele (A) and low-SSC allele (G),were associated with high-SSC and low-SSC phenotypes in tomato, respectively (Fig. 1f). We examined the DNA sequences within 2 Mb of the lead SNP (SL2.50ch02:44438607), and 39 genes were identified (Fig. 1c and d, Supplementary Data Table S3). The gene annotated as Sugar Transporter Protein 1, Solyc02g079220, was selected and named STP1 (Fig. 1e). To identify whether STP1 has a major regulatory role for SSC, 13 accessions with high SSC and 8 accessions with low SSC were selected to quantify STP1 expression in red-ripe fruits using quantitative real-time PCR (qRT–PCR). The expression levels of STP1 were markedly higher in the high-SSC accessions than low-SSC accessions (Fig. 1g), which indicates that STP1 is a major candidate for having an important influence on SSC in tomato.
Figure 1.
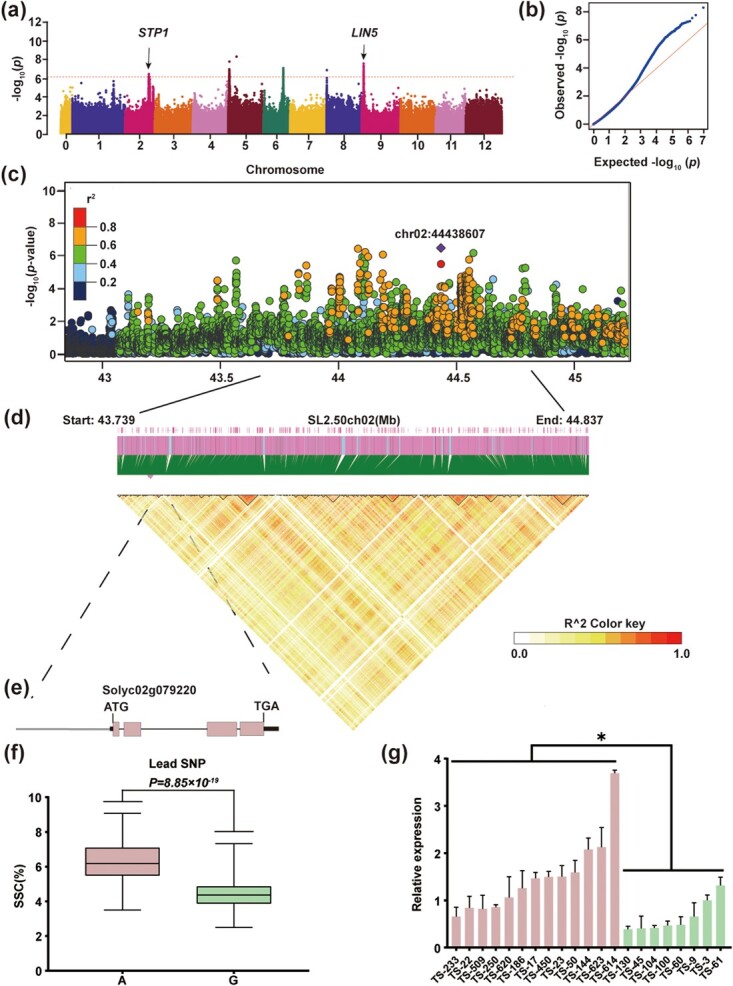
GWAS of SSC in tomato fruits. a Manhattan plot for GWAS result of fruit SSC (n = 481). The y-axis represents observed P-values transformed from minus log10. The genome-wide suggestive threshold (P = 9.93 × 10−7) is indicated by the red horizontal dashed line. b Quantile–quantile plot of expected versus observed P-values for GWAS. c Genome-wide association signals for SSC are shown in the region of 42.8–45.2 Mb on chromosome 2 (x-axis). The purple dot indicates the lead SNP and the color of each plot corresponds to the r2 value (a measure of linkage disequilibrium (LD)) according to the legend. d Representation of pairwise r2 values (a measure of LD) among all polymorphic sites in the 1.1-Mb genomic region corresponding to (c). e Gene structure of STP1 (Solyc02g079220). The pink box and thin black lines represent coding sequence and introns, respectively. The thick black lines represent the 5′-untranslated (5′-UTR) and 3′-untranslated (3′-UTR) regions. The gray line represents the promoter. f Box diagram of SSC in two sets of genotypes distinguished based on the lead SNP, Chr02:44438607. g Relative expression of STP1 in red-ripe fruits of different accessions. Data represent means ± standard deviation (n = 3). The asterisk indicates a significant difference: *P ≤ .05.
A 21-bp InDel in the promoter of STP1 alters its expression
To determine the sequences variation in STP1, the genomic regions surrounding STP1 including the introns, exons, promoters, 5′-UTR and 3′-UTR were sequenced in 10 accessions with high SSC and 10 accessions with low SSC. At 1124 bp upstream of the start codon, we found a 21-bp InDel. We further checked the presence and absence of the 21-bp InDel among 334 tomato accessions by PCR amplification and restriction enzyme digestion (Fig. 2a, Supplementary Data Table S4). The test results indicated that there was a 21-bp insertion in 47 accessions and a 21-bp deletion in 287 accessions (Fig. 2b). The 21-bp insertion occurred mainly in Solanum pimpinellifolium (PIM) and Solanum lycopersicum var. cerasiforme (CER), and the 21-bp deletion occurred mainly in S. lycopersicum (BIG). This suggests that STP1 may have been selected during tomato improvement.
Figure 2.
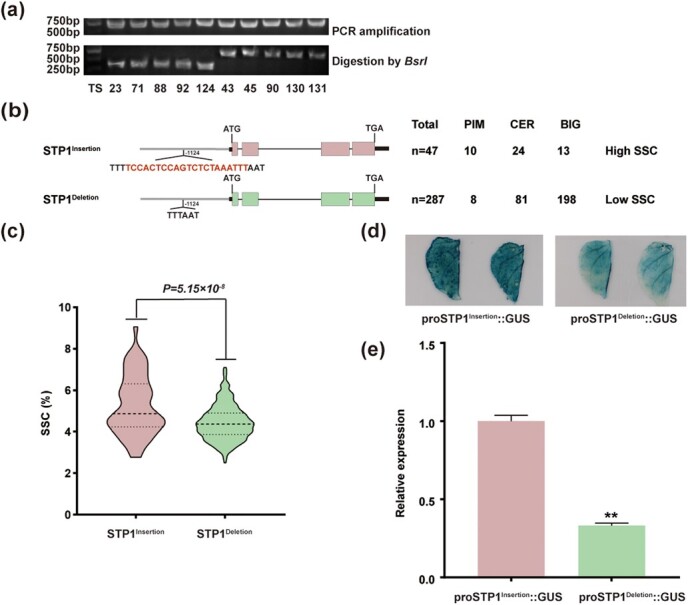
Structural variations and activity of the STP1 promoters in high-SSC and low-SSC alleles among tomato accessions. a CAPS marker after digestion by BsrI shows a 21-bp InDel variation in the STP1 promoter (partial results). Upper part is the result of PCR amplification and the lower part shows the result of digestion by BsrI. Bands around 750 bp represent accessions with STP1Deletion and bands around 380 bp represent accessions with STP1Insertion. b Structural variations of STP1 in 334 tomato accessions. Gene structure is as in Fig. 1e. The 21-bp InDel is marked in red. PIM, CER, and BIG represent subgroups covering two STP1 alleles (STP1Insertion and STP1Deletion). The numbers below Total, PIM, CER, and BIG are numbers of accessions. c SSC in red-ripe fruits of two STP1 alleles. d GUS staining of tobacco leaves infiltrated with Agrobacterium harboring proSTP1Insertion::GUS and proSTP1Deletion::GUS, respectively. The promoters of STP1Insertion and STP1Deletion were inserted into the GUS expression vector driving the GUS gene and were infiltrated into tobacco leaves. eGUS expression shown in (d) was evaluated. Asterisks indicate a significant difference: **P ≤ .01.
By examining the sequencing results and significant SNPs within STP1, two different alleles were identified and named STP1Insertion and STP1Deletion (Fig. 2b). We wondered if the SSC variation between the two alleles was attributable to the 21-bp InDel in the promoter, so the frequency distribution of STP1Insertion and STP1Deletion was analyzed (Supplementary Data Table S4). Interestingly, STP1Insertion accessions contained significantly higher SSC (5.25) than STP1Deletion accessions (4.43) (Fig. 2c).
To further illustrate that 21-bp InDel on the STP1 promoter can indeed alter its activation capacity, β-glucuronidase (GUS) staining assays were carried out. Two GUS reporter vectors, proSTP1Insertion::GUS and proSTP1Deletion::GUS, were constructed, and each was transiently expressed in tobacco leaves. Leaves infiltrated with proSTP1Insertion::GUS showed darker GUS staining than those infiltrated with proSTP1Deletion::GUS (Fig. 2d). We further quantified GUS expression and found that GUS expression driven by proSTP1Insertion was approximately twice that of GUS expression driven by proSTP1Deletion (Fig. 2e). These data confirm that possession of the 21-bp insertion enhanced the promoter activity of STP1.
STP1 mutation decreased SSC in tomato
To further study the role of STP1 in SSC of tomato fruits, CRISPR/Cas9 technology was used to generate knockout lines in the TS-23 background with high SSC. Through PCR amplification and sequencing, we obtained three homozygous lines with different types of editing, CR-5, CR-8, and CR-9. CR-5 and CR-8 showed a 1-bp deletion and CR-9 harbored a 2-bp deletion in the second target. The deletions in the three lines all caused frameshift mutations in the coding region of STP1, which would be expected to disrupt the STP1 protein structure and transport activity (Fig. 3a, Supplementary Data Fig. S1a). We also found that STP1 expression was suppressed in three STP1 knockout lines using qRT–PCR (Supplementary Data Fig. S1b). We performed SSC phenotyping of three knockout lines and wild-type (WT), and the results showed that SSC decreased significantly in three knockout lines (Fig. 3b). Previous studies have shown that SlSTP1, an STP, exhibited glucose and fructose transport activity in yeast [27]. SSC in tomato mainly comprises three soluble sugars (glucose, fructose, and sucrose) and two organic acids (malic acid and citric acid). We speculated that STP1 knockout lines may alter SSC by decreasing sugar content. We further determined the contents of glucose, fructose, sucrose, malic acid, and citric acid in red-ripe fruits of three knockout lines and WT. As expected, the contents of glucose, fructose, and sucrose significantly decreased (Fig. 3c–e), while the contents of malic acid and citric acid were unaffected and remained similar to the control (Supplementary Data Fig. S2). These data indicate that STP1 positively affects the SSC by regulating the contents of glucose, fructose, and sucrose in tomato fruits.
Figure 3.
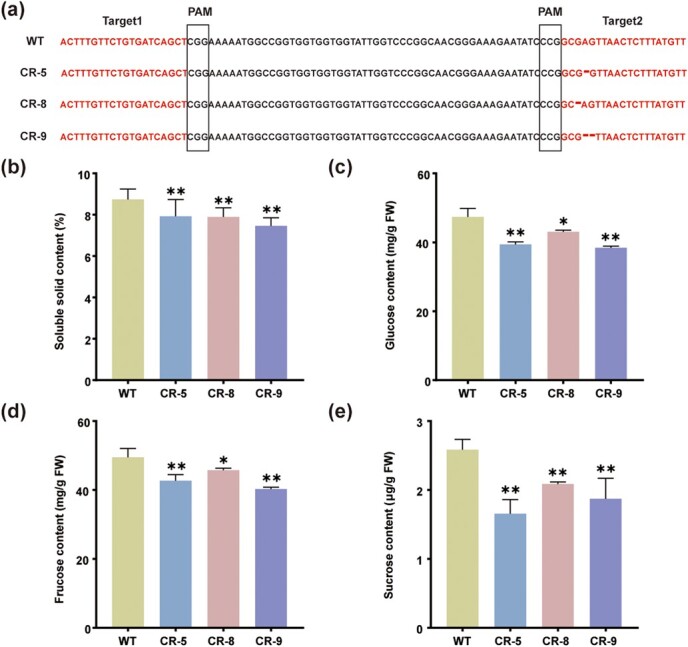
SSC, glucose, fructose, and sucrose levels in STP1 knockout lines and WT. a Different types of editing in STP1 knockout lines. WT represents background accession TS-23 with high SSC. CR-5, CR-8, and CR-9 represent knockout lines with deletions of A, G, and AG in the coding region of STP1, respectively. b–e Determination of SSC (b), glucose (c), fructose (d), and sucrose (e) in red-ripe fruits of three knockout lines and WT. Data represent means ± standard deviation (n = 3). Asterisks indicate significant differences: *P ≤ .05, **P ≤ .01.
ZAT10-LIKE, a zinc finger protein transcription factor, positively regulates gene expression of STP1Insertion
In order to study how the 21-bp InDel in the STP1 promoter regulates the expression of STP1, we carried out yeast one-hybrid screening. We took the 21-bp InDel sequence in the STP1 promoter as the center and added 9-bp on the left and right sides, yielding proSTP1Insertion and proSTP1Deletion (Supplementary Data Fig. S3). Putatively STP1 promoter binding proteins were retrieved and sequenced (Supplementary Data Table S5).
Two frequently occurring genes, Solyc12g088390 and Solyc04g077980, were retrieved as candidate genes (Supplementary Data Table S5). Evolutionary analysis showed that these two genes are homologous genes in tomato with a homology of 52% (Fig. 4a), and were named ZAT10-LIKE and ZAT10, respectively. Subcellular localization results showed that ZAT10-LIKE and ZAT10 were both located in the nucleus (Fig. 4b; Supplementary Data Fig. S4).
Figure 4.
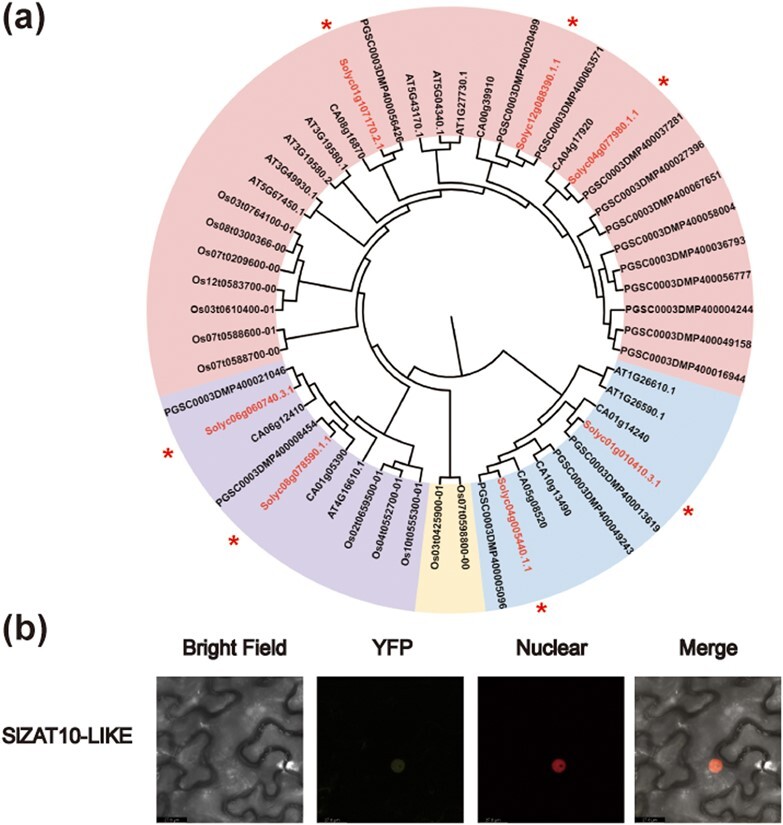
Molecular characterization of ZAT10-LIKE. a Phylogenetic tree of ZAT10-LIKE with its homologs from other plant species. Multiple sequences were aligned with the amino acid sequence of tomato ZAT10-LIKE and its homologs from Arabidopsis (Arabidopsis thaliana), rice (Oryza sativa), pepper (Capsicum annuum), and potato (Solanum tuberosum). The asterisks indicate ZAT10-LIKE (Solyc12g088390) and its homologs in tomato. b Subcellular localization of ZAT10-LIKE-YFP fusion protein. The fusion construct (ZAT10-LIKE-YFP) and a nuclear marker (StERF3-RFP) were transiently expressed in tobacco leaves. Bright-field, yellow fluorescent, red fluorescent, and merged images are shown.
To determine the regulatory effect of ZAT10-LIKE or ZAT10 on the transcription of STP1Insertion and STP1Deletion, luciferase (LUC) assays were performed. We used CaMV35S promoter-driven ZAT10-LIKE and ZAT10 effectors (pGreen II 62-SK) and LUC reporters driven by the promoters of STP1Insertion and STP1Deletion (pGreen II 0800-LUC) to perform transient transactivation assays. Both effector and reporter vectors were co-transformed into Agrobacterium GV3101 cells and then the ratios of firefly LUC to Renilla luciferase (REN) were determined. The LUC to REN ratio obtained by co-transformation of ZAT10-LIKE effector with STP1Insertion was significantly higher than the control (Fig. 5a and c), while there was no marked difference in the ratios of LUC to REN obtained in co-transformation of ZAT10-LIKE effector with STP1Deletion and the control (Fig. 5b and d). However, there was neither a marked difference in the ratios of LUC to REN obtained between co-transformation of ZAT10 effector with STP1Insertion and the control nor co-transformation of ZAT10 effector with STP1Deletion and the control (Supplementary Data Fig. S5). Collectively, our results indicate that ZAT10-LIKE, but not ZAT10, can activate the expression of STP1Insertion but does not exert a regulatory effect on the STP1Deletion promoter.
Figure 5.
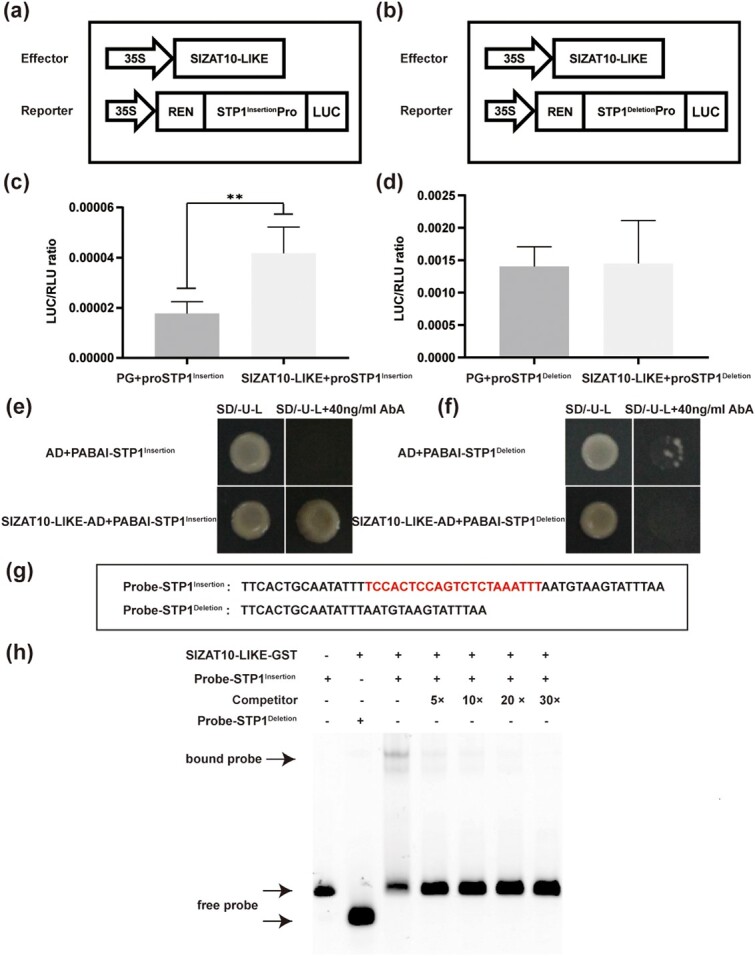
Differential binding capacity of ZAT10-LIKE to the promoters of STP1Insertion and STP1Deletion. a, b Schematic representation of constructs for the dual luciferase assays. The full-length CDS of ZAT10-LIKE was cloned into pGreen II 62-SK to generate an effector construct, pGreen II 62-SK-ZAT10-LIKE. The promoter fragments of STP1 (−1 to −1187), amplified from TS-23 and TS-9, respectively, were cloned into pGreen II 0800-LUC to create the reporter constructs pGreen II 0800-STP1Insertion-Pro (a) and pGreen II 0800-STP1Deletion-Pro (b). c, d Relative LUC/REN ratios were used to evaluate the promoter activity of STP1Insertion (c) and STP1Deletion (d) in the presence of ZAT10-LIKE. PG, the pGreenII 62-SK empty vector with pGreen II 0800-STP1Insertion-Pro or pGreen II 0800-STP1Deletion-Pro, was used as a control. Data represent means ± standard deviation (n = 6). e, f Yeast one-hybrid assays of ZAT10-LIKE binding to the promoters of STP1Insertion (e) and STP1Deletion (f). The full-length CDS of ZAT10-LIKE was cloned into pGADT7 (AD) to generate the prey vector, pGADT7-ZAT10-LIKE. The promoters of STP1Insertion (e) and STP1Deletion (f) were cloned into pAbai to generate bait vectors, pAbai-STP1Insertion (e) and pAbai-STP1Deletion (f), respectively. The bait vector and the prey vector were introduced into Y1HGold yeast. A combination introducing the bait vector and the empty vector pGADT7 was used as a control. The transformants were cultured on SD/−Ura−Leu media with different concentrations of aureobasidin A (AbA). g Diagram of probes of EMSAs. The sequence in red is the 21-bp insertion. Sequences of Probe-STP1Insertion and Probe-STP1Deletion were derived from natural accessions. h EMSA assays of ZAT10-LIKE binding to the probes of STP1Insertion and STP1Deletion. + and − represent presence and absence, respectively. 5×, 10×, 20×, and 30× represent different competition multiples.
In order to verify whether the different expression levels of STP1Insertion and STP1Deletion were caused by the differential binding capacity of ZAT10-LIKE to the STP1 promoters, we conducted yeast one-hybrid assays. This confirmed that ZAT10-LIKE could specifically bind to the promoter of STP1Insertion but not to the promoter of STP1Deletion (Fig. 5e and f).
To further study the interactions between ZAT10-LIKE and the promoters of STP1Insertion and STP1Deletion, we performed electrophoretic mobility shift assays (EMSAs). We designed two 5'-fluorescein amidite (5'-FAM) -labeled probes, a 39-bp Probe-STP1Insertion with the 21-bp insertion in the center, and an 18-bp Probe-STP1Deletion without the 21-bp insertion (Fig. 5g). EMSAs indicated that ZAT10-LIKE can bind to Probe-STP1Insertion but not to Probe-STP1Deletion (Fig. 5h).
STP1 mutation exerts broad effects on gene expression and metabolite accumulation
To study the effect of STP1 mutation on whole-genome gene expression patterns, RNA-seq was performed in STP1 knockout lines and WT. There were 2573 differentially expressed genes (DEGs), including 1089 upregulated genes and 1484 downregulated genes (Fig. 6a). GO (Gene Ontology) terms of DEGs included iron ion binding, oxidoreductase activity, tetrapyrrole binding, heme binding, extracellular region and apoplast (Fig. 6b). KEGG (Kyoto Encyclopedia of Genes and Genomes) pathways of DEGs included plant–pathogen interaction, nitrogen metabolism, phenylpropanoid biosynthesis, and MAPK signaling pathway (Fig. 6c).
Figure 6.
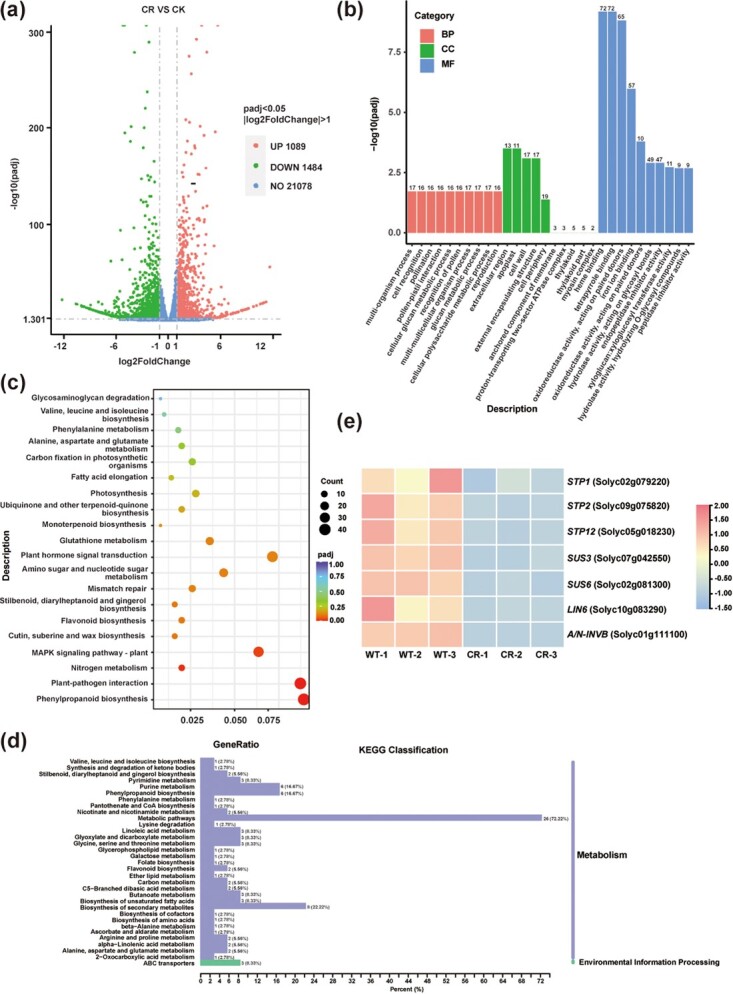
DEGs and metabolites in STP1 knockout lines and WT. a Volcano map of DEGs. b, c Significantly enriched GO terms (b) and KEGG pathways (c) of DEGs. (d) KEGG pathways of differentially expressed metabolites. e Heat map of gene expression related to sugar metabolism from RNA-seq.
We detected the content of primary metabolites between STP1 knockout lines and WT, and a total of 592 primary metabolites were identified. Further analysis revealed that 106 differentially enriched metabolites (DEMs) were identified, of which 36 were significantly enriched metabolites. The annotation results for the DEMs using KEGG indicated that ~72.22% of DEMs were involved in metabolic pathways, 22.22% of DEMs were involved in biosynthesis of secondary metabolites, and 16.67% of DEMs were involved in phenylpropanoid biosynthesis (Fig. 6d). These results are consistent with those from RNA-seq. Biological processes are complex and integrative, and the combined analysis of multi-omics data is helpful for the study of phenotypes and regulatory mechanisms of biological processes [41]. Combining metabolite and transcriptome analysis, we found that 44 DEMs and 6 DEGs were involved in phenylpropanoid biosynthesis and 28 DEMs and 2 DEGs participated in carbon metabolism (Supplementary Data Fig. S6). The results of the combined analysis suggest that these DEMs may be regulated by DEGs. Overall, our results suggest that STP1 mutation had large-scale effects on gene expression and metabolite accumulation.
More interestingly, the expression levels of genes related to sugar metabolism were significantly downregulated, such as Sugar Transporter Protein 2 (STP2), Sugar Transporter Protein 12 (STP12), Sucrose Synthase 3 (SUS3), Sucrose Synthase 6 (SUS6), Invertase 6 (LIN6), and Neutral/alkaline Invertase (A/N-INVB) (Fig. 6e). Sucrose synthase, invertase and neutral/alkaline invertase are related to conversion of sucrose to glucose and fructose [42]. These results indicate that the decreased content of glucose and fructose in STP1 knockout lines is probably due to decreased cleavage of sucrose.
STP1 InDel_21 was selected during tomato improvement
Since InDel_21 appeared to be the causal variation responsible for the differences in STP1 expression and SSC content, we investigated InDel_21 variants in a panel of 334 accessions, containing 18 PIM, 105 CER, and 211 BIG accessions. A total of 55.6% (10 of 18) of the PIM accessions carried Insertion_21, 22.9% (24 of 105) of the CER accessions carried Insertion_21 and 6.2% (13 of 211) of the BIG accessions carried Insertion_21 (Fig. 7a). Nucleotide diversity (π) in PIM, CER, and BIG accessions was measured (Fig. 7b) and the π ratios between groups were calculated to detect putative domestication (πPIM/πCER = 0.92) and improvement (πCER/πBIG = 8.50) sweeps (Fig. 7c). Our data provide proof that STP1 was selected during tomato improvement.
Figure 7.

Analysis of molecular diversity of STP1 during domestication and improvement. a Frequency of InDel_21bp allele in tomato subpopulations. n represents the number of accessions. b Distribution of nucleotide diversity (π) of PIM (red line), CER (navy blue line), and BIG (light blue line) within the 43.6–44 Mb region on chromosome 2. Red arrows, STP1. c Ratios of nucleotide diversity (π) between PIM and CER or between CER and BIG accessions on chromosome 2. Red arrows, STP1.
Discussion
Plant domestication, which can create a new plant form to meet human needs, is the genetic modification of wild species [43]. Food crops typically have enhanced determinate growth or increased apical dominance, more robust growth habit, larger fruits or grains, and a loss of natural seed dispersal for easy harvest by humans compared with their progenitors [43]. Tomato domestication has greatly increased fruit yield [44]. However, there was a negative relationship between SSC and fresh weight in fruits so that it was difficult to transfer high-SSC traits from wild species into cultivated varieties [44]. Although tomato varieties with improved traits have been generated successfully [45, 46], the efficiency of genetic improvement of SSC is still relatively limited [47].
The sequencing of the tomato genome [10] and resequencing of hundreds of natural accessions [11, 48] has accelerated the process of gene/allele discovery by GWAS, QTL sequencing and mapping-by-sequencing (MBS) [1]. Compared with traditional genetic populations, including NILs and RILs, GWAS saves time in building populations and provides richer allelic diversity. mGWAS has been widely used to study the evolution of tomato fruit quality [49–51] and a large number of genes related to tomato quality have been identified through mGWAS. Functional and evolutionary verification of candidate genes has, however, rarely been carried out, which hampers the use of natural variation to improve tomato flavor quality. Here we used a GWAS for SSC with 481 tomato accessions. Besides the previously reported locus, LIN5 [6], we also found other QTLs on chromosomes 2, 5, 6, and 8 (Fig. 1a), and further analyzed the candidate genes on the four chromosomes (Supplementary Data Tables S3 and S6–S8). Using significant SNPs, sequence amplification, and gene annotation, we identified a candidate gene on chromosome 2, STP1, attributed to the variation of SSC. This locus has also been mentioned in previous studies by GWAS on glucose [51] and SSC [22]. However, its function and natural variation have not been identified. To further test whether STP1 plays a key regulatory role in SSC, 13 accessions with high-SSC phenotypes and 8 accessions with low-SSC phenotypes were selected randomly and the expression of STP1 was quantified in the red-ripe fruits using qRT–PCR. Our data showed that the expression of STP1 in the high-SSC accessions was observably higher than that in the low-SSC accessions (Fig. 1g). We confirmed a sequence variation of a 21-bp InDel at 1124 bp upstream of the start codon in the STP1 promoter. We characterized the 21-bp InDel variation in 334 tomato accessions with the cleaved amplified polymorphic sequence (CAPS)marker (Fig. 2a). STP1 can be classified into two different alleles, STP1Insertion and STP1Deletion (Fig. 2b). The accessions with the 21-bp insertion mainly belong to PIM and CER, and the accessions without the 21-bp insertion mainly fall within BIG (Fig. 2b). Through phenotypic statistics, we found SSC of STP1Insertion accessions was significantly higher than that of STP1Deletion accessions (Fig. 2c). GUS staining showed stronger transcription activity by the promoter of STP1Insertion than by the STP1Deletion promoter (Fig. 2d and e). These results demonstrate that the 21-bp InDel does regulate the expression of STP1.
STP usually has glucose and fructose transport activity [27, 52, 53], and a related protein, MdSTP13a, also has sucrose transport activity [54]. Previous studies have shown that RNAi-mediated knockdown of LeHT genes led to a 55% decrease in fruit hexose accumulation [27] and heterologous expression of LeHT1 in yeast can rescue a hexose transport-impaired mutant [27]. CRISPR/Cas9 technology was used to construct STP1 knockout lines in TS-23 with high SSC, and the SSC decreased significantly in three knockout lines (Fig. 3b). Furthermore, the contents of glucose, fructose, and sucrose all significantly decreased, while the content of malic acid and citric acid was not altered (Fig. 3c–e; Supplementary Data Fig. S2). These data indicate that STP1 positively regulates the SSC by regulating the contents of glucose, fructose, and sucrose in tomato fruits. To explore the potential mechanism underlying the altered sugar concentration, we further analyzed RNA-seq data of the red-ripe fruits in STP1 knockout lines and WT. As expected, gene expression related to sugar metabolism was significantly downregulated, including SUS3 (Sucrose Synthase), SUS6 (Sucrose Synthase), LIN6 (Invertase 6), and A/N-INVB (Neutral/alkaline Invertase), which are involved in sucrose dissociation (Fig. 6e). These findings further support the suggestion that sucrose cleavage decreased in STP1 knockout lines, resulting in a decrease in glucose and fructose content.
In order to study the regulation by the 21-bp InDel in the promoter of the gene expression of STP1, we performed a yeast one-hybrid screening and identified a transcription factor, ZAT10-LIKE. We showed that ZAT10-LIKE can bind to the promoter of STP1Insertion but not to the promoter of STP1Deletion through LUC assays, EMSAs, and yeast one-hybrid assays (Fig. 5). Conserved domain analysis of ZAT10-LIKE revealed that it has a typical EAR motif at its C-terminus (Supplementary Data Fig. S7), which reportedly inhibits the transcription of target genes [55, 56]. However, some EAR domain-containing ZFP TFs may also activate other target genes [57, 58], and we speculate that other domains in ZAT10-LIKE may affect its transcriptional activity. ZFP TFs were involved in plant development and stress responses [29, 30], especially in salt stress [33, 59, 60], but little is known about their role in the regulation of metabolism. Our study demonstrates a novel function of ZFP TFs in regulating sugar transporters, but a detailed function of ZAT10-LIKE in regulating SSC needs to be further identified.
Finally, we investigated the InDel_21 variants among 334 accessions (Fig. 7a). We measured the level of nucleotide diversity (π) in PIM, CER, and BIG accessions and calculated the ratios of π between PIM and CER and between CER and BIG, which were suggestive of an improvement event (Fig. 7b and c). STP1 was observed in an improvement sweep, which was consistent with the previous report [11].
In summary, we identified a genetic locus, STP1, which is required for increased SSC of red-ripe fruits in tomato. During tomato improvement, the removal of InDel_21 in the STP1 promoter led to a decrease in fruit SSC. This research can help uncover the genetic basis and natural variation of primary metabolism. Our data indicate that STP1 might have been a critical factor for high SSC in undomesticated plants and its restoration and/or modification in crops may improve SSC. We have also discovered a new role for ZFP TFs in regulating sugar transporters.
Materials and methods
Tomato accessions and genome-wide association study
Four hundred and eighty-one tomato accessions, including 6 Solanum cheesmaniae (wild), 22 S. pimpinellifolium (PIM), 118 S. lycopersicum var. cerasiforme (CER), 275 S. lycopersicum (BIG) and 60 Guangxi accessions collected by our laboratory (Supplementary Data Table S1), were used to perform GWAS conducted in a greenhouse at Wuhan Agricultural Academy of Sciences, China, in Spring 2016. A total of 4 540 171 SNPs (minor allele frequency >5% and missing rate <10%) for 481 accessions from the previously study [48] were used for GWAS. Beagle software was chosen to estimate missing genotypes [61] and the efficient mixed-model association expedited (EMMAX) algorithm was chosen to perform the association analysis [62]. The effective number of independent SNPs was calculated by GEC software [63] and a total of 1 007 010 independent SNPs were obtained. The significance P threshold was determined by Bonferroni correction as P = α/n (n = 1 007 010). The suggestive and significant thresholds were imputed to be 9.930388 × 10−7 (α = 1) and 4.965194 × 10−8 (α = 0.05), respectively. The physical locations of the SNPs were based on the tomato reference genome sequence version SL2.50 (http://solgenomics.net). The results of GWAS for specific candidate segments were visualized with LocusZoom software [64]. The linkage segment of each significant locus was calculated using LDBlockShow software [65], and the linkage segment harboring significant SNPs was used as a candidate region.
RNA extraction and gene expression analysis
Total RNA was extracted using the Trizol reagent (Invitrogen, USA) and further reverse transcribed into cDNA using a HiScript II 1st Strand cDNA Synthesis Kit (Vazyme, China). The relative transcript levels of specific genes were quantified using qRT–PCR. The expression level of the Actin gene (Solyc11g005330) was used as reference [39]. Related primer sequences can be found in Supplementary Data Table S2.
Gene cloning, vector construction, and transformation
STP1 knockout lines were generated using pTX vector, a CRISPR/Cas9 binary vector. TS-23 with high SSC was transformed using Agrobacterium (strain GV3101)-mediated transformation [66]. A total of three homozygous knockout lines were chosen for further analysis.
DNA sequencing and CAPS markers for the 21-bp InDel
To detect variation in the STP1 gene, specific primers were designed to perform PCR amplification of a 2-kb STP1 promoter region and full-length genomic DNA from 10 accessions with extremely high SSC and 10 with extremely low SSC accessions, and the PCR products were confirmed by sequencing. STP1Insertion and STP1Deletion accessions were genotyped using CAPS markers. Specific primers were designed to perform PCR amplification of STP1 promoter sequences containing the 21-bp InDel. The PCR products were subsequently digested with BsrI (New England Biolabs, USA) for 3 hours at 65°C and then incubated for 20 minutes at 85°C in a reaction containing 10 μl PCR products, 7.6 μl double-distilled water, 2 μl 10 × NEBuffer 3.1, and 0.4 μl BsrI. The PCR products of STP1Insertion accessions were designed to generate bands ~380 bp in length, while the PCR products of STP1Deletion accessions could not be digested by BsrI.
GUS staining
The promoters of STP1 were amplified from TS-23 (STP1Insertion accession) and TS-9 (STP1Deletion accession), and then inserted into the pHELLSGATE8 vector without the CaMV35S promoter to generate GUS reporter constructs, proSTP1Insertion::GUS and proSTP1Deletion::GUS. These two reporter constructs were injected into tobacco leaves and the leaves were collected 48 hours after infiltration. One half of each leaf was used to determine the expression of the GUS gene and the other half was stained. GUS expression was quantified using qRT–PCR. For GUS staining, the tobacco leaves were incubated with staining buffer for 12 hours at 37°C in the dark and faded with 70% (v/v) ethanol.
Phenotyping
SSC was quantified as Brix in total fruit juice from red-ripe fruits using a digital refractometer (PAL-BX|ACID 3). Glucose, fructose, sucrose, malate, and citric acid were measured by gas chromatography (GC). The red-ripe fruits were ground with liquid nitrogen and further extracted with 80% methanol. The extracted samples were concentrated in vacuo and then derivatized with hydroxylamine hydrochloride, hexamethyldisilane (HMDS, Sigma), and trimethylchlorosilane (TMCS, Sigma). Derivatized supernatants were added to a 2-ml automatic sample injection bottle for GC-FID analysis.
Subcellular localization
The full-length coding sequences (CDSs) of ZAT10-LIKE and ZAT10 without the termination codons were amplified from the cDNA of ‘Ailsa Craig’ (AC) and cloned into 101LYFP containing the CaMV35S promoter [67]. Agrobacterium strain GV3101 containing CaMV35S: ZAT10-LIKE-YFP or CaMV35S: ZAT10-YFP and the cell nucleus marker CaMV35S: StERF3-RFP [68] was infiltrated into tobacco leaves. After 48 hours of incubation, the image of yellow fluorescent protein (YFP) and red fluorescent protein (RFP) fluorescence was captured by confocal laser scanning microscopy (Leica SP8SP8, Germany).
Yeast one-hybrid assay
The 21-bp insertion sequence in the STP1 promoter was taken as the center, and 9-bp fragments were added to the left and right sides, yielding an 18-bp STP1Deletion fragment and a 39-bp STP1Insertion fragment. The 18- or 39-bp fragments were used as repeats and fragments with five repeats in tandem were artificially synthesized. The synthesized fragments were connected to pAbai vectors and then used for yeast one-hybrid screening. The full-length CDS of ZAT10-LIKE was amplified from the cDNA of AC and cloned into a pGADT7 vector. Yeast strains were chosen and dissolved in 100 μl double-distilled water. PCR amplification was carried out with universal primers. PCR products were confirmed by sequencing. Heinz 1706 (LA3530) was used for reference.
Both the pGADT7 prey vector and the bait vector were introduced into Y1HGold yeast (Clontech, USA) and grown on SD/−Ura−Leu media. After 3 days of culture, the positive yeast strains were picked and dissolved in double-distilled water, and a suspension was spotted onto SD/−Ura−Leu media with different concentrations of aureobasidin A (AbA).
Electrophoretic mobility shift assay
We first amplified the full-length CDS of ZAT10-LIKE from the cDNA of AC, and transferred it into EcoRI and XhoI-digested PGEX-4 T-1 (GST) vector by homologous recombination. Protein with a GST tag was expressed in Escherichia coli BL21 (DE3) cells and then cultured to an OD600 of 0.6. Protein was further induced with isopropyl-β-d-thiogalactopyranoside (IPTG) for 20 hours at 16°C and purified. The STP1Insertion and STP1Deletion probes were labeled with FAM and are shown in Fig. 5g. FAM was attached to the 5′ end of the sense oligonucleotides to label the probes. The double-stranded probes were formed by annealing the single-stranded probes and then dissolved in double-distilled water to 10 μM. For competitor probes, the double-stranded probe was used directly in 100 μM after annealing. ZAT10-LIKE protein was incubated with double-stranded probes with a 20-μl reaction mixture under dark conditions. After 30 minutes at 4°C, the 6% non-denaturing polyacrylamide gels were used to separate the mixture and the image was captured using a multifunctional imaging analysis system (ProteinSample) after 1 hour of migration.
Dual luciferase transactivation assay
The STP1 promoters (−1 to 1187 bp) were amplified from TS-23 and TS-9 and then cloned into the pGreen II 0800-LUC vector to generate reporter constructs. The full-length CDSs of ZAT10-LIKE and ZAT10 were cloned into the pGreen II 62-SK vector to generate effector constructs. Agrobacterium strains containing a reporter construct and an effector construct were infiltrated into tobacco leaves. After 3 days of incubation, firefly luciferase (LUC) and Renilla luciferase (REN) activity was determined and the ratios of LUC to REN were calculated to indicate transactivation activity.
RNA-seq
Samples from STP1 knockout lines and WT were collected with three biological replicates and each replicate contained 18 red-ripe fruits from three plants. RNA was extracted and RNA-seq was performed using a HiSeq-PE150 sequencing system. We used the average FPKM (expected number of fragments per kilobase of transcript sequence per million base pairs sequenced) value as a measure of gene expression [69]. A gene yielding >2-fold expression with P ≤ .05 was classified as a DEG.
Metabolome profiling
Samples of red-ripe fruits were collected from STP1 knockout lines and WT to detect primary metabolites (Wuhan, China; http://www.metware.cn/). Metabolome profiling was carried out according to the method of Liu et al. [70].
Molecular diversity analysis
We used the nucleotide diversity (π) ratio to identify selective sweeps for the molecular diversity analysis of STP1 in tomato. VCFtools was used to measure the levels of π in PIM, CER, and BIG accessions in 10-kb windows, with a step size of 1 kb [71]. The π ratios (πPIM/πCER, and πCER/πBIG) were calculated. Windows with the top 5% of ratios were selected for further analysis (3.02 and 8.09 for domestication and improvement, respectively).
Acknowledgements
This work was supported by grants from the National Key Research & Development Plan (2021YFD1200201; 2022YFD1200502), the National Natural Science Foundation of China (31972426; 31991182; 32060685), the Wuhan Biological Breeding Major Project (2022021302024852), the International Cooperation Promotion Plan of Shihezi University (GJHZ202104), the Key Project of Hubei Hongshan Laboratory (2021hszd007), the Hubei Key Research & Development Plan (2022BBA0062; 2022BBA0066), and the Fundamental Research Funds for the Central Universities (2662022YLPY001).
Author contributions
Y.Z. designed and supervised the project. Y.W. did most of the experiments and wrote the manuscript. C.S. performed the GWAS data analysis. P.G., F.L., L.Z., Y.W., J.T., X.Z., H.D., W.G., and F.W. performed some of the experiments. W.X., D.G., Y.Z., and Z.Y. advised on experiments and revised the manuscript.
Conflict of interest statement
All authors declare no conflicts of interest.
Data availability
All data supporting the results of this study can be obtained in this article or in the supplementary materials.
Supplementary data
Supplementary data is available at Horticulture Research online.
Supplementary Material
Contributor Information
Ying Wang, National Key Laboratory for Germplasm Innovation and Utilization of Horticultural Crops, College of Horticulture and Forestry Science, Huazhong Agricultural University, Wuhan 430070, China.
Chunmei Shi, National Key Laboratory for Germplasm Innovation and Utilization of Horticultural Crops, College of Horticulture and Forestry Science, Huazhong Agricultural University, Wuhan 430070, China.
Pingfei Ge, National Key Laboratory for Germplasm Innovation and Utilization of Horticultural Crops, College of Horticulture and Forestry Science, Huazhong Agricultural University, Wuhan 430070, China.
Fangman Li, National Key Laboratory for Germplasm Innovation and Utilization of Horticultural Crops, College of Horticulture and Forestry Science, Huazhong Agricultural University, Wuhan 430070, China.
Lihui Zhu, National Key Laboratory for Germplasm Innovation and Utilization of Horticultural Crops, College of Horticulture and Forestry Science, Huazhong Agricultural University, Wuhan 430070, China.
Yaru Wang, National Key Laboratory for Germplasm Innovation and Utilization of Horticultural Crops, College of Horticulture and Forestry Science, Huazhong Agricultural University, Wuhan 430070, China.
Jinbao Tao, National Key Laboratory for Germplasm Innovation and Utilization of Horticultural Crops, College of Horticulture and Forestry Science, Huazhong Agricultural University, Wuhan 430070, China.
Xingyu Zhang, National Key Laboratory for Germplasm Innovation and Utilization of Horticultural Crops, College of Horticulture and Forestry Science, Huazhong Agricultural University, Wuhan 430070, China.
Haiqiang Dong, National Key Laboratory for Germplasm Innovation and Utilization of Horticultural Crops, College of Horticulture and Forestry Science, Huazhong Agricultural University, Wuhan 430070, China.
Wenxian Gai, National Key Laboratory for Germplasm Innovation and Utilization of Horticultural Crops, College of Horticulture and Forestry Science, Huazhong Agricultural University, Wuhan 430070, China.
Fei Wang, National Key Laboratory for Germplasm Innovation and Utilization of Horticultural Crops, College of Horticulture and Forestry Science, Huazhong Agricultural University, Wuhan 430070, China.
Zhibiao Ye, National Key Laboratory for Germplasm Innovation and Utilization of Horticultural Crops, College of Horticulture and Forestry Science, Huazhong Agricultural University, Wuhan 430070, China.
Donald Grierson, Plant Sciences Division, School of Biosciences, University of Nottingham, Sutton Bonington Campus, Loughborough LE12 5RD, UK.
Wei Xu, Key Laboratory of Special Fruits and Vegetables Cultivation Physiology and Germplasm Resources Utilization (Xinjiang Production and Construction Crops), College of Agriculture, Shihezi University, Shihezi 832003, Xinjiang, China.
Yuyang Zhang, National Key Laboratory for Germplasm Innovation and Utilization of Horticultural Crops, College of Horticulture and Forestry Science, Huazhong Agricultural University, Wuhan 430070, China; Hubei Hongshan Laboratory, Wuhan 430070, China.
References
- 1. Rothan C, Diouf I, Causse M. Trait discovery and editing in tomato. Plant J. 2019;97:73–90. [DOI] [PubMed] [Google Scholar]
- 2. Tohge T, Fernie AR. Metabolomics-inspired insight into developmental, environmental and genetic aspects of tomato fruit chemical composition and quality. Plant Cell Physiol. 2015;56:1681–96. [DOI] [PubMed] [Google Scholar]
- 3. Tieman D, Zhu G, Resende MFR Jret al. A chemical genetic roadmap to improved tomato flavor. Science. 2017;355:391–4. [DOI] [PubMed] [Google Scholar]
- 4. Chetelat RT, DeVerna JW, Bennett AB. Introgression into tomato (Lycopersicon esculentum) of the L. chmielewskii sucrose accumulator gene (sucr) controlling fruit sugar composition. Theor Appl Genet. 1995;91:327–33. [DOI] [PubMed] [Google Scholar]
- 5. Eshed Y, Zamir D. An introgression line population of Lycopersicon pennellii in the cultivated tomato enables the identification and fine mapping of yield-associated QTL. Genetics. 1995;141:1147–62. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 6. Fridman , Carrari F, Liu Y-Set al. Zooming in on a quantitative trait for tomato yield using interspecific introgressions. Science. 2004;305:1786–9. [DOI] [PubMed] [Google Scholar]
- 7. Zanor MI, Osorio S, Nunes-Nesi Aet al. RNA interference of LIN5 in tomato confirms its role in controlling Brix content, uncovers the influence of sugars on the levels of fruit hormones, and demonstrates the importance of sucrose cleavage for normal fruit development and fertility. Plant Physiol. 2009;150:1204–18. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 8. Levin I, Gilboa N, Yeselson Eet al. Fgr, a major locus that modulates the fructose to glucose ratio in mature tomato fruits. Theor Appl Genet. 2000;100:256–62. [Google Scholar]
- 9. Shammai A, Petreikov M, Yeselson Yet al. Natural genetic variation for expression of a SWEET transporter among wild species of Solanum lycopersicum (tomato) determines the hexose composition of ripening tomato fruit. Plant J. 2018;96:343–57. [DOI] [PubMed] [Google Scholar]
- 10. Tomato Genome Consortium . The tomato genome sequence provides insights into fleshy fruit evolution. Nature. 2012;485:635–41. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 11. Lin T, Zhu G, Zhang Jet al. Genomic analyses provide insights into the history of tomato breeding. Nat Genet. 2014;46:1220–6. [DOI] [PubMed] [Google Scholar]
- 12. Nordborg M, Weigel D. Next-generation genetics in plants. Nature. 2008;456:720–3. [DOI] [PubMed] [Google Scholar]
- 13. Fang C, Luo J. Metabolic GWAS-based dissection of genetic bases underlying the diversity of plant metabolism. Plant J2019;97:91–100. [DOI] [PubMed] [Google Scholar]
- 14. Dong X, Gao Y, Chen Wet al. Spatiotemporal distribution of phenolamides and the genetics of natural variation of hydroxycinnamoyl spermidine in rice. Mol Plant. 2015;8:111–21. [DOI] [PubMed] [Google Scholar]
- 15. Strauch RC, Svedin E, Dilkes Bet al. Discovery of a novel amino acid racemase through exploration of natural variation in Arabidopsis thaliana. Proc Natl Acad Sci USA. 2015;112:11726–31. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 16. Angelovici R, Batushansky A, Deason Net al. Network-guided GWAS improves identification of genes affecting free amino acids. Plant Physiol. 2017;173:872–86. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 17. Chen J, Hu X, Shi Tet al. Metabolite-based genome-wide association study enables dissection of the flavonoid decoration pathway of wheat kernels. Plant Biotechnol J. 2020;18:1722–35. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 18. Slaten ML, Yobi A, Bagaza Cet al. mGWAS uncovers Gln-glucosinolate seed-specific interaction and its role in metabolic homeostasis. Plant Physiol. 2020;183:483–500. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 19. Ying S, Su M, Wu Yet al. Trichome regulator SlMIXTA-like directly manipulates primary metabolism in tomato fruit. Plant Biotechnol J. 2020;18:354–63. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 20. Chen J, Xue M, Liu Het al. Exploring the genic resources underlying metabolites through mGWAS and mQTL in wheat: from large-scale gene identification and pathway elucidation to crop improvement. Plant Commun 2021;2:100216. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 21. Ye J, Wang X, Hu Tet al. An InDel in the promoter of Al-ACTIVATED MALATE TRANSPORTER9 selected during tomato domestication determines fruit malate contents and aluminum tolerance. Plant Cell. 2017;29:2249–68. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 22. Ye J, Li W, Ai Get al. Genome-wide association analysis identifies a natural variation in basic helix-loop-helix transcription factor regulating ascorbate biosynthesis via D-mannose/L-galactose pathway in tomato. PLoS Genet. 2019;15:e1008149. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 23. Davies JN, Hobson GE, McGlasson WB. The constituents of tomato fruit – the influence of environment, nutrition, and genotype. Crit Rev Food Sci Nutr. 1981;15:205–80. [DOI] [PubMed] [Google Scholar]
- 24. Reuscher S, Akiyama M, Yasuda Tet al. The sugar transporter inventory of tomato: genome-wide identification and expression analysis. Plant Cell Physiol. 2014;55:1123–41. [DOI] [PubMed] [Google Scholar]
- 25. Paulsen PA, Custodio TF, Pedersen BP. Crystal structure of the plant symporter STP10 illuminates sugar uptake mechanism in monosaccharide transporter superfamily. Nat Commun 2019;10:407. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 26. Gear ML, McPhillips ML, McCurdy JWPet al. Hexose transporters of tomato: molecular cloning, expression analysis and functional characterization. Plant Mol Biol. 2000;44:687–97. [DOI] [PubMed] [Google Scholar]
- 27. McCurdy DW, Dibley S, Cahyanegara Ret al. Functional characterization and RNAi-mediated suppression reveals roles for hexose transporters in sugar accumulation by tomato fruit. Mol Plant. 2010;3:1049–63. [DOI] [PubMed] [Google Scholar]
- 28. Riechmann JL, Heard J, Martin Get al. Arabidopsis transcription factors: genome-wide comparative analysis among eukaryotes. Science. 2000;290:2105–10. [DOI] [PubMed] [Google Scholar]
- 29. Wang W, Wu P, Li Yet al. Genome-wide analysis and expression patterns of ZF-HD transcription factors under different developmental tissues and abiotic stresses in Chinese cabbage. Mol Gen Genomics. 2016;291:1451–64. [DOI] [PubMed] [Google Scholar]
- 30. Khatun K, Nath UK, Robin AHKet al. Genome-wide analysis and expression profiling of zinc finger homeodomain (ZHD) family genes reveal likely roles in organ development and stress responses in tomato. BMC Genomics. 2017;18:695. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 31. Zhang X, Guo X, Lei Cet al. Overexpression of SlCZFP1, a novel TFIIIA-type zinc finger protein from tomato, confers enhanced cold tolerance in transgenic Arabidopsis and rice. Plant Mol Biol Rep. 2010;29:185–96. [Google Scholar]
- 32. Sun SJ, Guo SQ, Yang Xet al. Functional analysis of a novel Cys2/His2-type zinc finger protein involved in salt tolerance in rice. J Exp Bot. 2010;61:2807–18. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 33. Mittler R, Kim YS, Song Let al. Gain- and loss-of-function mutations in Zat10 enhance the tolerance of plants to abiotic stress. FEBS Lett. 2006;580:6537–42. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 34. Yang K, An JP, Li CYet al. The apple C2H2-type zinc finger transcription factor MdZAT10 positively regulates JA-induced leaf senescence by interacting with MdBT2. Hortic Res. 2021;8:159. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 35. Ohno CK, Reddy GV, Heisler MGet al. The Arabidopsis JAGGED gene encodes a zinc finger protein that promotes leaf tissue development. Development. 2004;131:1111–22. [DOI] [PubMed] [Google Scholar]
- 36. Kim S, Choi K, Park Cet al. SUPPRESSOR OF FRIGIDA4, encoding a C2H2-type zinc finger protein, represses flowering by transcriptional activation of Arabidopsis FLOWERING LOCUS C. Plant Cell. 2006;18:2985–98. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 37. Gangappa SN, Botto JF. The BBX family of plant transcription factors. Trends Plant Sci. 2014;19:460–70. [DOI] [PubMed] [Google Scholar]
- 38. Luo D, Xiong C, Lin Aet al. SlBBX20 interacts with the COP9 signalosome subunit SlCSN5-2 to regulate anthocyanin biosynthesis by activating SlDFR expression in tomato. Hortic Res. 2021;8:163. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 39. Xiong C, Luo D, Lin Aet al. A tomato B-box protein SlBBX20 modulates carotenoid biosynthesis by directly activating PHYTOENE SYNTHASE 1, and is targeted for 26S proteasome-mediated degradation. New Phytol. 2019;221:279–94. [DOI] [PubMed] [Google Scholar]
- 40. Datta S, Hettiarachchi C, Johansson Het al. SALT TOLERANCE HOMOLOG2, a B-box protein in Arabidopsis that activates transcription and positively regulates light-mediated development. Plant Cell. 2007;19:3242–55. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 41. Zhang C, Hao Y-J. Advances in genomic, transcriptomic, and metabolomic analyses of fruit quality in fruit crops. Hortic Plant J. 2020;6:361–71. [Google Scholar]
- 42. Wang D, Zhao J, Qin Yet al. Molecular cloning, characterization and expression profile of the sucrose synthase gene family in Litchi chinensis. Hortic Plant J. 2021;7:520–8. [Google Scholar]
- 43. Doebley JF, Gaut BS, Smith BD. The molecular genetics of crop domestication. Cell. 2006;127:1309–21. [DOI] [PubMed] [Google Scholar]
- 44. Prudent M, Causse M, Génard Met al. Genetic and physiological analysis of tomato fruit weight and composition: influence of carbon availability on QTL detection. J Exp Bot. 2009;60:923–37. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 45. Butelli E, Titta L, Giorgio Met al. Enrichment of tomato fruit with health-promoting anthocyanins by expression of select transcription factors. Nat Biotechnol. 2008;26:1301–8. [DOI] [PubMed] [Google Scholar]
- 46. Luo Z, Zhang J, Li Jet al. A STAY-GREEN protein SlSGR1 regulates lycopene and β-carotene accumulation by interacting directly with SlPSY1 during ripening processes in tomato. New Phytol. 2013;198:442–52. [DOI] [PubMed] [Google Scholar]
- 47. Fernie AR, Schauer N. Metabolomics-assisted breeding: a viable option for crop improvement? Trends Genet. 2009;25:39–48. [DOI] [PubMed] [Google Scholar]
- 48. Zhu G, Wang S, Huang Zet al. Rewiring of the fruit metabolome in tomato breeding. Cell. 2018;172:249–261.e12. [DOI] [PubMed] [Google Scholar]
- 49. Sauvage C, Segura V, Bauchet Get al. Genome-wide association in tomato reveals 44 candidate loci for fruit metabolic traits. Plant Physiol. 2014;165:1120–32. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 50. Bauchet G, Grenier S, Samson Net al. Identification of major loci and genomic regions controlling acid and volatile content in tomato fruit: implications for flavor improvement. New Phytol. 2017;215:624–41. [DOI] [PubMed] [Google Scholar]
- 51. Zhao J, Sauvage C, Zhao Jet al. Meta-analysis of genome-wide association studies provides insights into genetic control of tomato flavor. Nat Commun. 2019;10:1534. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 52. Lemonnier P, Gaillard C, Veillet Fet al. Expression of Arabidopsis sugar transport protein STP13 differentially affects glucose transport activity and basal resistance to Botrytis cinerea. Plant Mol Biol. 2014;85:473–84. [DOI] [PubMed] [Google Scholar]
- 53. Wang Z, Wei X, Yang Jet al. Heterologous expression of the apple hexose transporter MdHT2.2 altered sugar concentration with increasing cell wall invertase activity in tomato fruit. Plant Biotechnol J. 2020;18:540–52. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 54. Li C, Meng D, Piñeros MAet al. A sugar transporter takes up both hexose and sucrose for sorbitol-modulated in vitro pollen tube growth in apple. Plant Cell. 2020;32:449–69. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 55. Ciftci-Yilmaz S, Morsy MR, Song Let al. The EAR-motif of the Cys2/His2-type zinc finger protein Zat7 plays a key role in the defense response of Arabidopsis to salinity stress. J Biol Chem. 2007;282:9260–8. [DOI] [PubMed] [Google Scholar]
- 56. Hichri I, Muhovski Y, Žižková Eet al. The Solanum lycopersicum zinc Finger2 cysteine-2/histidine-2 repressor-like transcription factor regulates development and tolerance to salinity in tomato and Arabidopsis. Plant Physiol. 2014;164:1967–90. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 57. Xu S, Wang X, Chen J. Zinc finger protein 1 (ThZF1) from salt cress (Thellungiella halophila) is a Cys-2/His-2-type transcription factor involved in drought and salt stress. Plant Cell Rep. 2007;26:497–506. [DOI] [PubMed] [Google Scholar]
- 58. Jain D, Roy N, Chattopadhyay D. CaZF, a plant transcription factor functions through and parallel to HOG and calcineurin pathways in Saccharomyces cerevisiae to provide osmotolerance. PLoS One. 2009;4:e5154. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 59. Sakamoto H, Araki T, Meshi Tet al. Expression of a subset of the Arabidopsis Cys/His-type zinc-finger protein gene family under water stress. Gene. 2000;248:23–32. [DOI] [PubMed] [Google Scholar]
- 60. He F, Niu MX, Feng CHet al. PeSTZ1 confers salt stress tolerance by scavenging the accumulation of ROS through regulating the expression of PeZAT12 and PeAPX2 in Populus. Tree Physiol. 2020;40:1292–311. [DOI] [PubMed] [Google Scholar]
- 61. Browning SR, Browning BL. Rapid and accurate haplotype phasing and missing-data inference for whole-genome association studies by use of localized haplotype clustering. Am J Hum Genet. 2007;81:1084–97. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 62. Kang HM, Sul JH, Service SKet al. Variance component model to account for sample structure in genome-wide association studies. Nat Genet. 2010;42:348–54. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 63. Li MX, Yeung JM, Cherny SSet al. Evaluating the effective numbers of independent tests and significant p-value thresholds in commercial genotyping arrays and public imputation reference datasets. Hum Genet. 2012;131:747–56. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 64. Pruim RJ, Welch RP, Sanna Set al. LocusZoom: regional visualization of genome-wide association scan results. Bioinformatics2010;26:2336–7. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 65. Dong SS, He WM, Ji JJet al. LDBlockShow: a fast and convenient tool for visualizing linkage disequilibrium and haplotype blocks based on variant call format files. Brief Bioinform. 2021;22:1–6. [DOI] [PubMed] [Google Scholar]
- 66. Ouyang B, Chen YH, Li HXet al. Transformation of tomatoes with osmotin and chitinase genes and their resistance to Fusarium wilt. J Hortic Sci Biotechnol. 2005;80:517–22. [Google Scholar]
- 67. Zhang Y, Ming R, Khan Met al. ERF9 of Poncirus trifoliata (L.) Raf. undergoes feedback regulation by ethylene and modulates cold tolerance via regulating a glutathione S-transferase U17 gene. Plant Biotechnol J. 2022;20:183–200. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 68. Tian Z, He Q, Wang Het al. The potato ERF transcription factor StERF3 negatively regulates resistance to Phytophthora infestans and salt tolerance in potato. Plant Cell Physiol. 2015;56:992–1005. [DOI] [PubMed] [Google Scholar]
- 69. Trapnell C, Williams BA, Pertea Get al. Transcript assembly and quantification by RNA-Seq reveals unannotated transcripts and isoform switching during cell differentiation. Nat Biotechnol. 2010;28:511–5. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 70. Liu L, Zhang K, Bai Jet al. All-flesh fruit in tomato is controlled by reduced expression dosage of AFF through a structural variant mutation in the promoter. J Exp Bot. 2022;73:123–38. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 71. Danecek P, Auton A, Abecasis Get al. The variant call format and VCFtools. Bioinformatics. 2011;27:2156–8. [DOI] [PMC free article] [PubMed] [Google Scholar]
Associated Data
This section collects any data citations, data availability statements, or supplementary materials included in this article.
Supplementary Materials
Data Availability Statement
All data supporting the results of this study can be obtained in this article or in the supplementary materials.