

Abstract
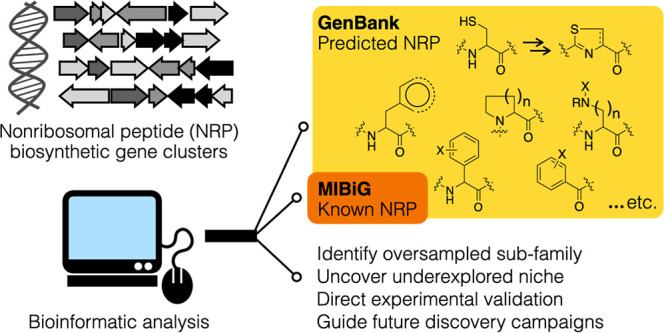
The traditional natural product discovery approach has accessed only a fraction of the chemical diversity in nature. The use of bioinformatic tools to interpret the instructions encoded in microbial biosynthetic genes has the potential to circumvent the existing methodological bottlenecks and greatly expand the scope of discovery. Structural prediction algorithms for nonribosomal peptides (NRPs), the largest family of microbial natural products, lie at the heart of this new approach. To understand the scope and limitation of the existing prediction algorithms, we evaluated their performances on NRP synthetase biosynthetic gene clusters. Our systematic analysis shows that the NRP biosynthetic landscape is uneven. Phenylglycine and its derivatives as a group of NRP building blocks (BBs), for example, have been oversampled, reflecting an extensive historical interest in the glycopeptide antibiotics family. In contrast, the benzoyl BB, including 2,3-dihydroxybenzoate (DHB), has been the most underexplored, hinting at the possibility of a reservoir of as yet unknown DHB containing NRPs with functional roles other than a siderophore. Our results also suggest that there is still vast unexplored biosynthetic diversity in nature, and the analysis presented herein shall help guide and strategize future natural product discovery campaigns. We also discuss possible ways bioinformaticians and biochemists could work together to improve the existing prediction algorithms.
Introduction
Microbial natural products have been a fruitful source of therapeutic small molecules.1 The vast majority of natural products we know to date were found by scientists examining the extracts of microbial fermentation broths; they were produced by cultured microorganisms actively expressing biosynthetic gene clusters (BGC). However, this tried-and-true approach is faced with increasingly higher rediscovery rates, such that the return-on-investments for this approach is no longer justified.2,3 This is because the natural product BGC amenable to this approach represent only a small fraction of the natural biosynthetic diversity, which has been nearly exhausted by decades of repeated screening.4,5 To circumvent this challenge, many scientists have begun to use bioinformatic algorithms to interpret the immense biosynthetic information encoded in cryptic BGC. These bioinformatically predicted natural products can then be examined in silico by virtual screening6,7 or converted into real molecules via chemical synthesis for wet lab studies.8−11 As bioinformatic analysis is not constrained by our inability to culture and express the microorganism and BGC of interest, it has the potential to greatly expand the scope of natural product studies.
A bioinformatic algorithm, once trained, can interpret the instructions encoded in biosynthetic genes and predict the structure of the resulting natural product. The current training set is a subcollection of known natural products and their corresponding BGC sequences. However, due to the two requisites of the traditional discovery approach mentioned above, culture and gene expression, most known natural products come from a few highly productive bacteria genera, for example, Pseudomonas and Streptomyces species, which constitute the training set for the existing algorithms. Since the scope and limitation of the aforementioned bioinformatics-based new approaches depend on accurate predictions, it is reasonable to ask whether the existing algorithms perform differently across major bacterial phyla in light of such a phylogenetically biased training set. We decided to address this question by looking into nonribosomal peptide synthetase (NRPS) BGC, the largest family of microbial natural products. Our results suggest that these algorithms have not been overtrained to suit A domains associated with actinobacteria, the bacterial phylum most thoroughly examined in modern natural product research, and that actinobacteria still have a lot of biosynthetic diversity yet to be explored. Our analysis also identified both underexplored and oversampled niches, which could help guide and strategize future natural product discovery and algorithm improvement endeavors.
NRPs are the largest family of microbial natural products,12 displaying extremely diverse structures and functions.13−15 Many NRPs (or their derivatives) have been used in clinical applications as therapeutic agents and in basic research as molecular probes (Figure 1a).16,17 We therefore decided to focus on NRPs in this study. The NRP biosynthesis machinery has been extensively studied and was the target of early efforts aimed at building a bioinformatic algorithm capable of predicting natural product structures based solely on the primary sequences of their biosynthetic genes. The first NRP prediction algorithm became available in 1999,18 and many more have been reported since.19−27 Today, these tools are packaged into a software suite—antibiotics and secondary metabolite analysis shell (antiSMASH)—that accepts whole genome sequences as the input, identifies BGC, parses out individual enzymatic domains, and finally outputs the predicted NRP structure. It is currently the most advanced and widely used web server that provides this analysis free of charge to the research community.28
Figure 1.

Predict NRP structures. (a) Examples of famous NRP. The genes grsA, entF, and cssA9 (d) encode A domains responsible for the activation of Phe (red), Ser (green), and Val (blue) in gramicidin S, enterobactin, and cyclosporine biosynthesis, respectively. Gramicidin S and enterobactin have 2- and 3-fold symmetry, wherein three Ser and two Phe were activated by the same A domain, respectively. (b) Crystal structure of GrsA with Phe, AMP, and Mg(II) bound in its active site (PDB: 1amu). (c) Ten residues that constitute the active site (red), termed the “nonribosomal code”, were identified by A domain sequence alignment. (d) Strong correlation between the nonribosomal code and the identity of its substrate BB provide the framework for the development of a prediction algorithm that requires only the A domain primary sequence.
As the name suggests, NRPs are peptides that are not biosynthesized by the ribosome; they are instead constructed via an enzymatic assembly line.29 Each module in the assembly line is responsible for incorporating a building block (BB), in most cases an amino acid (AA), into the NRP backbone. A module typically contains multiple semi-autonomously folded domains, each with its own function, including most commonly the condensation (C), adenylation (A), and thiolation (T) domains. The A domain is an enzyme that catalyzes the activation of a substrate BB to form an aminoacyl-adenylate, which is then attached via a thioester bond onto the phosphopantetheine arm of the T domain. Peptide bond formation between the BB on the neighboring T domains is catalyzed by the C domain in between, wherein the amino group of the BB on the NRP intermediate attacks the activated BB on its N-terminal side. This reaction extends the peptide intermediate by one residue and effectively moves it down the assembly line from the Nth to the N + 1st module (see Figure S1 for a graphical illustration). The resulting NRP is colinear to the biosynthetic gene sequences due to such an arrangement. The genetically encoded biosynthetic instructions can therefore be “translated” into a NRP structure by cracking the “nonribosomal code”, that is, correlation between the primary sequence of an A domain and its substrate BB specificity. This correlation lies at the heart of NRP structure prediction.
Methods
The raw data for analyses were obtained from sources available to all researchers. The dataset for training A domain substrate BB prediction algorithms is obtained from the supplementary information associated with the SANDPUMA study (https://bitbucket.org/chevrm/sandpuma). The minimum information about a biosynthetic gene cluster (MIBiG) dataset is part of the Genomic Standards Consortium project that is freely available to researchers worldwide (https://mibig.secondarymetabolites.org). Bacterial genomes were downloaded from the GenBank FTP site (https://ftp.ncbi.nlm.nih.gov/genomes/). Genomes were analyzed by antiSMASH 5.0 (https://antismash.secondarymetabolites.org). The predicted substrate BB information in the antiSMASH output file were extracted using an automated script and tabulated for further analyses and graphical presentations (Table S1).
The 10-residue nonribosomal code, sometimes referred to as the Stachelhaus code, were aligned head-to-tail and clustered at 70% AA identity using CD-HIT v4.6 (Figure S2).20,30,31 Incomplete Stachelhaus codes, that is, those with “–” in the code, were excluded from clustering analysis. The rarefaction curve was then estimated by sampling the nonribosomal codes without replacement and cumulatively calculating the number of clusters belonging to the nonribosomal codes. Random sampling was repeated 10 times to obtain the averaged cumulative cluster numbers. The curve was then plotted using R 4.1.1 (https://www.r-project.org/). A Heaps’ law model was adopted to determine whether or not the rarefaction curve at hand is saturated.32,33 The formula for this model is as follows
where n is the average number of clusters, N is the number of sampled nonribosomal codes, and both κ and γ were obtained by fitting the above equation. The curve is deemed “open” (unsaturated growth) when γ > 0 and closed if otherwise.32,33
Results and Discussion
An A domain substrate prediction algorithm must be trained before it becomes operational. NRPS A domains are highly conserved in terms of both structure and sequence, except for the nonribosomal codes, which refer to the 10 residues encompassing the active sites. The nonribosomal codes are diverse and define the binding pockets that accommodate a wide range of substrate BBs (Figure 1b).34 Sequence analysis indicates that the nonribosomal code correlates highly with the identity of the substrate BB an A domain recognizes and activates (Figure 1c). This correlation provides the framework for algorithm training and A domain substrate BB prediction (Figure 1d). The NORINE database documents 1,740 structurally characterized NRPs,35 and the MIBiG database links 606 known NRPs to their BGC sequences.36 The latest training set selects 434 non-redundant representative A domains paired to their corresponding BB.27 While this collection of 434 substrate BB/A domain pairs is the culmination of decades of research, most of which are derived from actinobacterial and proteobacterial NRPS BGC, it is hardly a large dataset for algorithm training. Whether or not such an uneven phylogenetic representation skews algorithm training remains an unanswered question. Specifically, the two most fruitful NRP producing genera, that is, Pseudomonas and Streptomyces, together account for approximately 4 out of every 10 entries in MIBiG (41%) and the training set (40%, Table S1).27,36 These considerations prompted us to assess systematically the performance of the existing A domain prediction algorithms.
Compile a Phylogenetically Uniform Genome Collection
We began by compiling a collection of microbial genomes that uniformly represent the genetic and microbial biosynthetic diversity humans have accessed thus far. We first downloaded all prokaryote genomes deposited in GenBank of scaffold or better assembly quality. Note that the data associated with a few extensively studied model organisms make up a sizeable portion of this database, for example, GenBank contains more than 26,000 and 14,000 genome assembly reports for Escherichia coli and Staphylococcus aureus, respectively. To represent genome sequences uniformly, only one copy is included in our curated collection when a microorganism has been sequenced multiple times. We termed this curated collection GB1, which contains a total of 19,150 prokaryotic genome sequences (Figure S3). The entire GB1 was subjected to antiSMASH analysis, yielding 5,352 NRPS BGC and 20,794 A domains, each associated with a substrate BB prediction. All 606 NRPS entries in the MIBiG database were also analyzed in the same way to yield 2,822 substrate BB predictions.
Predictability and Usage Pattern Differ across Bacterial Phyla
The most direct way to evaluate the performance of existing algorithms is to use biochemical assays to experimentally validate (or refute) the predicted substrate BB of an A domain. Unfortunately, this task is unattainable at a large enough scale for a statistically meaningful evaluation. An alternative is to compare the predicted substrate BB that come from NRPS BGC associated with GB1 and MIBiG sequences. GB1 can be viewed as a microcosm of the biosynthetic diversity in nature accessed by humans thus far. MIBiG, on the other hand, is a database of annotated BGC of characterized natural products and can be viewed as a collection that reflects the requisites imposed by the traditional discovery approach and the associated phylogenetic biases. As mentioned above, the traditional discovery approach is applicable only to actively express BGC belonging to microorganisms that are readily cultured in the laboratory. Microbial sequences deposited in GenBank are not nearly as biased since these requisites (culture and gene expression) do not apply to DNA sequencing. While whether the substrate BB of a particular A domain is predicted correctly or not can only be definitively determined by biochemical assays, a bioinformatic analysis that compares GB1 versus MIBiG substrate BB predictions shall be able to unveil algorithm performance biases, if any, when performed systematically at a large scale.
Actinobacteria Have the Most Unpredictable A Domains
The predicted substrate BBs were first grouped based on their phylogenetic origin. As expected, among the 20,794 substrate BB predictions of GB1, actinobacteria contributed the most (9,399, 45%); proteobacteria, firmicutes, cyanobacteria, and species from other phyla contributed 34, 10, 4.8, and 5.5%, respectively (Figure 2a). Some A domains yielded no prediction because the algorithms failed to align the primary sequences at hand or, if well aligned, failed to identify a matching or comparable nonribosomal code in the training set. The output may also be displayed as “no prediction” when conflicting calls were made by the two algorithms consulted by antiSMASH [support vector machine (SVM) and Stachelhaus code].18,20 Regardless of the scenario, these “unpredictable” A domains are less similar to the training set and bioinformatically more difficult to handle. They are expectedly more prevalent among A domains from GB1 (42%) than MIBiG (30%). More than half of all A domains from actinobacteria (51%) are unpredictable, followed by proteobacteria (35%), firmicutes (27%), and cyanobacteria (33%) (Figure 2a). This came as a surprise as actinobacteria have contributed the most characterized natural products to date. The fact that they also have the largest fraction of intractable A domains suggests that existing algorithms were not overtrained to suit A domains associated with actinobacteria and that enormous actinobacterial biosynthetic novelty still awaits our exploration.
Figure 2.

NRP predictions categorized by bacterial phyla. (a) Actinobacteria contributed the highest number of A domains. It also has the largest fraction of “unpredictable” A domains, followed by proteobacteria, cyanobacteria, and firmicutes. (b) The top five most frequently used substrate BB in NRP biosynthesis by bacterial phyla. Four BB (Ser, Thr, Val, and Gly) are among the top five most frequently used BB across the four major phyla.
Overall Substrate Composition Is More Skewed in Actinobacterial and Proteobacterial NRPs
Aside from unpredictable A domains, we asked which substrate BBs are the most prevalent and whether they differ by phyla. Glycine (Gly), serine (Ser), threonine (Thr), and valine (Val) turn out to be four of the five most frequently predicted substrate BBs in NRPs across all phyla (Figure 2b). However, this is not to say that all NRPs have a similar BB composition. NRPs produced by actinobacteria and proteobacteria are predicted to be overrepresented by Ser, accounting for 15.1 and 18.9% of their BBs, followed by Thr at 15.0 and 10.4%, respectively. Together, these two AAs with a side-chain OH group account for approximately 3 out of every 10 BBs in actinobacterial and proteobacterial NRPs. In comparison, the predicted composition of firmicutes and cyanobacterial NRPs is not nearly as skewed, wherein the top two BBs combined account for ∼20% of the total. Firmicutes most frequently use Leu (10.8%) and Gly (10.0%), and cyanobacteria most frequently use Gly (11.1%) and Ser (10.2%), to construct their NRPs.
NRP Substrate BBs Show Oversampled and Underexplored Niches
The physical and chemical properties of NRP BBs, for example, hydrophobicity, nucleophilicity, geometry, and flexibility, influence those of the NRPs which in turn affect their bioactivity (Figure 3a). An analysis of the predicted BB based on these properties may therefore reveal useful insights. Since natural products are often discovered as groups of congeners with similar, if not identical, bioactivities, we decided to perform this analysis in groups of BBs with similar properties. The “default” BB, alanine (ala), has an unsubstituted β-carbon and was placed in a group of its own. Gly lacks the β-carbon altogether and, as the smallest and the only achiral AA, confers flexibility to the NRP backbone. In contrast, cysteine (cys) residues in NRPs often undergo cyclodehydration to form thiazol(in)es to rigidify the NRP backbone; the side-chain free thiol is otherwise a strong nucleophile under physiological conditions. As such, gly and cys were also each placed in a group of their own due to these unique features. All other groups encompassed more than one type of BB. Secondary AA (sec) and those with a β-sp2 carbon (sp2) were two other groups of BBs that limit NRP backbone flexibility. The former included mainly proline and piperidine, and the latter included phenylglycine and its hydroxylated and/or halogenated derivatives. The benzoyl group included various hydroxylated benzoic acid derivatives that often serve as metal cation chelators in siderophore NRPs. The acidic, aliphatic, amido, aromatic, basic, and hydroxyl groups are self-explanatory and included AA with side chains that have the indicated functionalities. Finally, others included BBs that do not belong to any of the groups mentioned above (see Table S1 for the full list of BBs in each group).
Figure 3.

Usage pattern of NRP BB differs across bacterial phyla. (a) Common substrate BBs were grouped based on the physical and chemical properties of their side chains. Shown here were only representative BBs; see Table S1 for the full list of BBs in each group. (b) The Ω values were calculated for a group of BBs for all predictions (overall) and for each bacteria phyla; “n/a” indicates that no predictions fall in that group.
As described above, GB1 is a microcosm of the biosynthetic diversity of nature, and MIBiG is a phylogenetically uneven collection that reflects the natural product BGC that are amenable to the traditional discovery approach. As such, a comparison between MIBiG and GB1 can be viewed as a proxy of comparing the “known” to the entire natural products space. Unpredictable A domains were excluded from this analysis, and all others were compared based on the aforementioned groups of BBs. We defined the parameter Ω as follows
wherein Mi and Gi are the fractions of group i BB (%) predicted from MIBiG and GB1, respectively. A group of BBs predicted less frequently from MIBiG than GB1 would manifest a negative value (Ω < 0), suggesting that they have been underexplored by the traditional discovery approach (Figure 3b, shades of blue). In contrast, Ω > 0 indicates a more frequent occurrence in MIBiG than GB1, suggesting that the traditional discovery approach has been finding NRPs that contain this group of BBs at a rate that outpaces the overall NRP discovery rate; that is, this group of BBs is relatively oversampled (Figure 3b, shades of green).
Most Oversampled and Underexplored Groups of NRP BBs are sp2 and Benzoyl, Respectively
Two groups of BBs stood out in this analysis. First, the sp2 group is highly oversampled (Ωsp2 = +2.39, entry 1). Only 0.8% of the predicted BBs associated with GB1 belong to this group; the corresponding number is 4.2% for MIBiG. This group of BB is characteristic of the glycopeptide antibiotics, wherein vancomycin and teicoplanin are among the most famous members of this family.37 In glycopeptide antibiotics, the aromatic moieties of the sp2 BB undergo oxidative coupling to constrain free rotation, and the resulting atropisomerism restriction is key to their tight binding to cell wall biosynthesis intermediates. This class of NRP was once an intense research focus in both industry and academia, and this historical backdrop offers a likely explanation for the oversampling of sp2 group BBs. At the other end of the spectrum are the most underexplored benzoyl group BBs (Ωsp2 = −1.98, entry 13). They often serve as dentate(s) for iron chelation in siderophores. Many microorganisms produce siderophores to extract soluble iron from the environment.38,39 Since soluble iron is an extremely scarce resource on Earth, it is no surprise that microorganisms have evolved siderophores of diverse structures. Our analysis has identified benzoyl BB as a greatly underexplored group. This suggests that either there remains lots of new siderophores to be discovered or that NRPs with benzoyl BB may possess functions unbeknown to scientists.
Oversampled/Underexplored Pattern Differs across Bacterial Phyla
We calculated Ω for MIBiG versus GB1 predictions for each phylum (Figure 3b). The sp2 and benzoyl remain the most oversampled and underexplored BBs, respectively. We noticed a number of other noteworthy features. For example, while cys is overall an underexplored group, it is slightly oversampled outside the four major bacteria phyla (Ωcys,OTHERS = +0.63, entry 11). In contrast, while the sec BB is overall oversampled, it is slightly underexplored in cyanobacteria (Ωsec,CYA = −0.18, entry 3). Furthermore, some groups of BB show significant differences across phyla. For example, proteobacteria and firmicutes use ala BB more frequently in the predicted NRP than in the structurally characterized NRP (Ωala = +0.16 and 1.40, respectively, entry 6), whereas they are predicted to use acidic BB relatively less frequently (Ωacidic = −0.40 and −0.52, respectively, entry 8). Perhaps, the most unexpected finding was that basic BB showed opposite relative usage frequencies in the two most common phyla (entry 10). Specifically, it is highly oversampled in proteobacteria (Ωbasic,PRO = +1.59) and underexplored by actinobacteria (Ωbasic,ACT = −0.77). The above analysis points to different usage patterns across bacterial phyla and suggests that decades of natural product research by using the traditional discovery approach, based on extracting and screening of microbial fermentation broths, has resulted in inadvertent oversampled/underexplored niches in bacterial NRPs.
Much NRP Biosynthetic Diversity Remains Unexplored
A rarefaction analysis was then performed to assess the extent of biosynthetic diversity covered by A domains characterized thus far. Our analysis was based on the 10-residue nonribosomal code of each A domain. The suitable clustering identity was determined by checking the “cleanliness” at various cutoffs and set at 70% (Figure S2), yielding 1,894 clusters and a rarefaction curve that shows no sign of saturation (Figure 4a). The parameter γ has been established as a proxy of growth tendency, and higher γ values are correlated with more unidentified entities.33 Fitting the rarefaction curve to a Heaps’ law model confirmed that the growth has not saturated (γ = 0.4). This result supports the notion that there are many unexplored nonribosomal codes, which belong to A domains that likely activate new BB for NRP biosynthesis. We then conducted separate analyses on the four major bacterial phyla, and none of which showed signs of saturation either (γ = 0.41, 0.45, 0.47, and 0.30 for actinobacteria, proteobacteria, firmicutes, and cyanobacteria, respectively, Figure 4b). The rarefaction curve is similar to previous studies.27 Interestingly, with the highest growth tendency (γ = 0.47) among the major bacteria phyla, Firmicutes appear to harbor the most unexplored A domain diversity in NRP biosynthesis.33
Figure 4.

Rarefaction analysis suggests the presence of more unexplored NRPs. Rarefaction analyses were performed (a) on nonribosomal codes of all A domains and (b) separately on A domains associated with each of the four major bacterial phyla. The x-axis represents the numbers of codes sampled, and the y-axis represents the number of clusters based on 70% identity clustering. None of these curves have saturated as judged by the Heaps’ law growth model. The γ values are 0.41, 0.45, 0.47, and 0.30 for actinobacteria (green), proteobacteria (blue), firmicutes (yellow), and cyanobacteria (red) rarefaction curves, respectively. Data and extrapolations were plotted as solid and dashed lines, respectively; a solid datapoint marks the transition from data to extrapolation.
Many scientists believe that bioinformatic analysis and chemical synthesis could join forces to play a pivotal role in natural product research. Instead of examining fermentation culture extracts for new molecules, the future “discovery” process may begin with choosing a BGC of interest, followed by interpreting in silico the encoded biosynthetic instructions to predict the probable structure of the end product. The predicted structure can then be subjected to virtual screening6,7 or chemically synthesized for experimental evaluation.8−11 As the largest family of natural products with countless applications in both the clinics and basic research, NRP has been studied as a proof of principle of this new approach, whose scope and limitation depend critically on the performance of NRP structure prediction algorithms. Herein, we report our assessment of the performance of existing algorithms.
The training set for substrate prediction algorithms is a collection of select known A domains, enzymes responsible for activating a BB, usually an AA, so that it can be incorporated into a growing NRP chain. Actinobacteria have been the biggest contributor of natural products known to date, and the training set is likewise overrepresented by their A domains. After examining more than 20,000 A domain predictions, our bioinformatic analysis showed that actinobacteria, more than any other phyla, have the largest fraction of A domains that are intractable to the trained algorithms (51%). This suggests that, despite being the most extensively studied bacteria phylum, current algorithms have not been overtrained to suit actinobacterial A domains and that actinobacteria still has an enormous amount of unexplored biosynthetic diversity. This notion is corroborated by a rarefaction analysis of the nonribosomal code, the 10 residues that form the specificity conferring substrate binding pocket of an A domain, wherein all rarefaction curves appear to be unsaturated for the four major bacterial phyla (actinobacteria, cyanobacteria, firmicutes, and proteobacteria). In particular, nonribosomal codes associated with firmicutes are the least explored, showing the highest γ value (0.47) based on a Heaps’ law growth model.
Strategize Future Studies
Algorithm improvement ultimately rests on expanding the training set to establish more well-characterized substrate BB/A domain pairs. While there are assays to biochemically validate (or refute) the predicted substrate BB, they are not designed for high-throughput experimental evaluation.40−46 Furthermore, because thousands of A domains remain uncharacterized, assaying nonselectively will not provide the impetus for efficient algorithm improvement. Our analysis herein points to two complementary strategies for the experimental characterization of A domains. The first is to investigate A domains with conflicting predictions from the two most commonly used algorithms, SVM and Stachelhaus code.18,20 The second is to investigate intractable A domains that neither algorithm can predict. The former helps to improve the internal consistency and the latter expands the scope of existing algorithms.
We also examined genome sequences in MIBiG, which is the largest database that links known natural products to their BGC. A comparison of the predicted BBs from MIBiG versus GB1 informed us the differences between known NRPs and the entire NRP space. Such a comparison helped determine whether a certain group of BBs, categorized based on their physical and chemical properties, have been oversampled or underexplored. The benzoyl BBs as a group, which includes 2,3-dihydroxybenzoate, are the most underexplored according to our analysis (Ωbenzoyl = −1.98). We hypothesize that metal chelation is only one of many functional roles they can potentially take on. The fact that known NRP containing benzoyl BB are all siderophores suggests that, despite a large presence in the literature, we are far from fully exploring these NRPs. Formalizing a discovery campaign based on this notion, that is, a systematic search for NRP with benzoyl BBs, may lead to the discovery of a reservoir of NRPs with new functions. Alternatively, BGC harboring oversampled A domains are good starting points if one wishes to find congeners of a known NRP. While they will be highly similar to known NRP, minor structural and functional tweaks are often critical in improving pharmacological properties in drug development.
Last, we argue that algorithm improvement shall entail a process analogous to the scientific method, which requires scientists to experimentally test predictions derived from a theory. The theory is then refined in accordance to the experimental observations. We hope that the two strategies proposed herein will inspire bioinformatic and experimental scientists to work together to close the prediction–validation cycle for the study of NRP bioinformatics and biosynthesis, thereby advancing a tool that has the potential to revolutionize the future of natural product discovery, drug development, as well as many other fields of research.
Acknowledgments
We thank C.-H. Hsu for setting up the computer infrastructure in the Chu laboratory that enabled the large-scale analysis presented herein.
Glossary
Abbreviations
- A
adenylation
- AA
amino acid
- BB
building block
- BGC
biosynthetic gene cluster
- C
condensation
- MIBiG
minimum information about a biosynthetic gene cluster
- NRP
nonribosomal peptide
- NRPS
nonribosomal peptide synthetase
- T
thiolation standard three-letter abbreviations for canonical amino acids
- antiSMASH
antibiotics and secondary metabolite analysis shell
Supporting Information Available
The Supporting Information is available free of charge at https://pubs.acs.org/doi/10.1021/acschembio.2c00761.
This work was supported by the Ministry of Science and Technology (MOST), Taiwan, College Student Research Scholarship MOST 110-2813-C-002-030-M (SLC) and Research Project Grants MOST 109-2113-M-002-005-MY3 (JC), MOST 111-2113-M-002-019-MY2 (JC), MOST 110-2221-E-038-019-MY3 (YWW), and MOST 111-2221-E-038-023-MY3 (YWW).
The authors declare no competing financial interest.
Supplementary Material
References
- Newman D. J.; Cragg G. M. Natural products as sources of new drugs over the nearly four decades from 01/1981 to 09/2019. J. Nat. Prod. 2020, 83, 770–803. 10.1021/acs.jnatprod.9b01285. [DOI] [PubMed] [Google Scholar]
- Pye C. R.; Bertin M. J.; Lokey R. S.; Gerwick W. H.; Linington R. G. Retrospective analysis of natural products provides insights for future discovery trends. Proc. Natl. Acad. Sci. U.S.A. 2017, 114, 5601–5606. 10.1073/pnas.1614680114. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Li J. W.; Vederas J. C. Drug discovery and natural products: end of an era or an endless frontier?. Science 2009, 325, 161–165. 10.1126/science.1168243. [DOI] [PubMed] [Google Scholar]
- Handelsman J.; Rondon M. R.; Brady S. F.; Clardy J.; Goodman R. M. Molecular biological access to the chemistry of unknown soil microbes: a new frontier for natural products. Chem. Biol. 1998, 5, R245–R249. 10.1016/s1074-5521(98)90108-9. [DOI] [PubMed] [Google Scholar]
- Milshteyn A.; Schneider J. S.; Brady S. F. Mining the metabiome: identifying novel natural products from microbial communities. Chem. Biol. 2014, 21, 1211–1223. 10.1016/j.chembiol.2014.08.006. [DOI] [PMC free article] [PubMed] [Google Scholar]
- McInnes C. Virtual screening strategies in drug discovery. Curr. Opin. Chem. Biol. 2007, 11, 494–502. 10.1016/j.cbpa.2007.08.033. [DOI] [PubMed] [Google Scholar]
- Rester U. From virtuality to reality - Virtual screening in lead discovery and lead optimization: a medicinal chemistry perspective. Curr. Opin. Drug Discov. Dev. 2008, 11, 559. [PubMed] [Google Scholar]
- Chu J.; Vila-Farres X.; Inoyama D.; Ternei M.; Cohen L. J.; Gordon E. A.; Reddy B. V.; Charlop-Powers Z.; Zebroski H. A.; Gallardo-Macias R.; Jaskowski M.; Satish S.; Park S.; Perlin D. S.; Freundlich J. S.; Brady S. F. Discovery of MRSA active antibiotics using primary sequence from the human microbiome. Nat. Chem. Biol. 2016, 12, 1004–1006. 10.1038/nchembio.2207. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Vila-Farres X.; Chu J.; Inoyama D.; Ternei M. A.; Lemetre C.; Cohen L. J.; Cho W.; Reddy B. V.; Zebroski H. A.; Freundlich J. S.; Perlin D. S.; Brady S. F. Antimicrobials inspired by nonribosomal peptide synthetase gene clusters. J. Am. Chem. Soc. 2017, 139, 1404–1407. 10.1021/jacs.6b11861. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Chu J.; Vila-Farres X.; Brady S. F. Bioactive synthetic-bioinformatic natural product cyclic peptides inspired by nonribosomal peptide synthetase gene clusters from the human microbiome. J. Am. Chem. Soc. 2019, 141, 15737–15741. 10.1021/jacs.9b07317. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Chu J.; Koirala B.; Forelli N.; Vila-Farres X.; Ternei M. A.; Ali T.; Colosimo D. A.; Brady S. F. Synthetic-bioinformatic natural product antibiotics with diverse modes of action. J. Am. Chem. Soc. 2020, 142, 14158–14168. 10.1021/jacs.0c04376. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Blin K.; Shaw S.; Kautsar S. A.; Medema M. H.; Weber T. The antiSMASH database version 3: increased taxonomic coverage and new query features for modular enzymes. Nucleic Acids Res. 2021, 49, D639–D643. 10.1093/nar/gkaa978. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Caboche S.; Leclère V.; Pupin M.; Kucherov G.; Jacques P. Diversity of monomers in nonribosomal peptides: towards the prediction of origin and biological activity. J. Bacteriol. 2010, 192, 5143–5150. 10.1128/jb.00315-10. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Condurso H. L.; Bruner S. D. Structure and noncanonical chemistry of nonribosomal peptide biosynthetic machinery. Nat. Prod. Rep. 2012, 29, 1099–1110. 10.1039/c2np20023f. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Walsh C. T.; O’Brien R. V.; Khosla C. Nonproteinogenic amino acid building blocks for nonribosomal peptide and hybrid polyketide scaffolds. Angew. Chem., Int. Ed. 2013, 52, 7098–7124. 10.1002/anie.201208344. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Carlson E. E. Natural products as chemical probes. ACS Chem. Biol. 2010, 5, 639–653. 10.1021/cb100105c. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Lee A. A.; Chen Y. C.; Ekalestari E.; Ho S. Y.; Hsu N. S.; Kuo T. F.; Wang T. S. Facile and versatile chemoenzymatic synthesis of enterobactin analogues and applications in bacterial detection. Angew. Chem., Int. Ed. 2016, 55, 12338–12342. 10.1002/anie.201603921. [DOI] [PubMed] [Google Scholar]
- Stachelhaus T.; Mootz H. D.; Marahiel M. A. The specificity-conferring code of adenylation domains in nonribosomal peptide synthetases. Chem. Biol. 1999, 6, 493–505. 10.1016/s1074-5521(99)80082-9. [DOI] [PubMed] [Google Scholar]
- Challis G. L.; Ravel J.; Townsend C. A. Predictive, structure-based model of amino acid recognition by nonribosomal peptide synthetase adenylation domains. Chem. Biol. 2000, 7, 211–224. 10.1016/s1074-5521(00)00091-0. [DOI] [PubMed] [Google Scholar]
- Rausch C.; Weber T.; Kohlbacher O.; Wohlleben W.; Huson D. H. Specificity prediction of adenylation domains in nonribosomal peptide synthetases (NRPS) using transductive support vector machines (TSVMs). Nucleic Acids Res. 2005, 33, 5799–5808. 10.1093/nar/gki885. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Minowa Y.; Araki M.; Kanehisa M. Comprehensive analysis of distinctive polyketide and nonribosomal peptide structural motifs encoded in microbial genomes. J. Mol. Biol. 2007, 368, 1500–1517. 10.1016/j.jmb.2007.02.099. [DOI] [PubMed] [Google Scholar]
- Röttig M.; Medema M. H.; Blin K.; Weber T.; Rausch C.; Kohlbacher O. NRPSpredictor2-a web server for predicting NRPS adenylation domain specificity. Nucleic Acids Res. 2011, 39, W362–W367. 10.1093/nar/gkr323. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Khayatt B. I.; Overmars L.; Siezen R. J.; Francke C. Classification of the adenylation and acyl-transferase activity of NRPS and PKS systems using ensembles of substrate specific hidden Markov models. PLoS One 2013, 8, e62136 10.1371/journal.pone.0062136. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Baranašić D.; Zucko J.; Diminic J.; Gacesa R.; Long P. F.; Cullum J.; Hranueli D.; Starcevic A. Predicting substrate specificity of adenylation domains of nonribosomal peptide synthetases and other protein properties by latent semantic indexing. J. Ind. Microbiol. Biotechnol. 2014, 41, 461. 10.1007/s10295-013-1322-2. [DOI] [PubMed] [Google Scholar]
- Lee T. V.; Johnson R. D.; Arcus V. L.; Lott J. S. Prediction of the substrate for nonribosomal peptide synthetase (NRPS) adenylation domains by virtual screening. Proteins 2015, 83, 2052–2066. 10.1002/prot.24922. [DOI] [PubMed] [Google Scholar]
- Knudsen M.; Søndergaard D.; Tofting-Olesen C.; Hansen F. T.; Brodersen D. E.; Pedersen C. N. Computational discovery of specificity-conferring sites in non-ribosomal peptide synthetases. Bioinformatics 2016, 32, 325–329. 10.1093/bioinformatics/btv600. [DOI] [PubMed] [Google Scholar]
- Chevrette M. G.; Aicheler F.; Kohlbacher O.; Currie C. R.; Medema M. H. SANDPUMA: ensemble predictions of nonribosomal peptide chemistry reveal biosynthetic diversity across Actinobacteria. Bioinformatics 2017, 33, 3202–3210. 10.1093/bioinformatics/btx400. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Blin K.; Shaw S.; Kloosterman A. M.; Charlop-Powers Z.; van Wezel G. P.; Medema M. H.; Weber T. antiSMASH 6.0: improving cluster detection and comparison capabilities. Nucleic Acids Res. 2021, 49, W29–W35. 10.1093/nar/gkab335. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Fischbach M. A.; Walsh C. T. Assembly-line enzymology for polyketide and nonribosomal peptide antibiotics: logic, machinery, and mechanisms. Chem. Rev. 2006, 106, 3468–3496. 10.1021/cr0503097. [DOI] [PubMed] [Google Scholar]
- Fu L.; Niu B.; Zhu Z.; Wu S.; Li W. CD-HIT: accelerated for clustering the next-generation sequencing data. Bioinformatics 2012, 28, 3150–3152. 10.1093/bioinformatics/bts565. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Wei Z. W.; Niikura H.; Morgan K. D.; Vacariu C. M.; Andersen R. J.; Ryan K. S. Free piperazic acid as a precursor to nonribosomal peptides. J. Am. Chem. Soc. 2022, 144, 13556–13564. 10.1021/jacs.2c03660. [DOI] [PubMed] [Google Scholar]
- Tettelin H.; Riley D.; Cattuto C.; Medini D. Comparative genomics: the bacterial pan-genome. Curr. Opin. Microbiol. 2008, 11, 472–477. 10.1016/j.mib.2008.09.006. [DOI] [PubMed] [Google Scholar]
- Yang M.-R.; Wu Y.-W. Enhancing predictions of antimicrobial resistance of pathogens by expanding the potential resistance gene repertoire using a pan-genome-based feature selection approach. BMC Bioinf. 2022, 23, 131. 10.1186/s12859-022-04666-2. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Conti E.; Stachelhaus T.; Marahiel M. A.; Brick P. Structural basis for the activation of phenylalanine in the non-ribosomal biosynthesis of gramicidin S. EMBO J. 1997, 16, 4174–4183. 10.1093/emboj/16.14.4174. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Flissi A.; Ricart E.; Campart C.; Chevalier M.; Dufresne Y.; Michalik J.; Jacques P.; Flahaut C.; Lisacek F.; Leclère V.; Pupin M. Norine: update of the nonribosomal peptide resource. Nucleic Acids Res. 2020, 48, D465–D469. 10.1093/nar/gkz1000. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Kautsar S. A.; Blin K.; Shaw S.; Navarro-Muñoz J. C.; Terlouw B. R.; van der Hooft J. J. J.; van Santen J. A.; Tracanna V.; Suarez Duran H. G.; Pascal Andreu V.; Selem-Mojica N.; Alanjary M.; Robinson S. L.; Lund G.; Epstein S. C.; Sisto A. C.; Charkoudian L. K.; Collemare J.; Linington R. G.; Weber T.; Medema M. H. MIBiG 2.0: a repository for biosynthetic gene clusters of known function. Nucleic Acids Res. 2020, 48, D454–D458. 10.1093/nar/gkz882. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Reynolds P. E. Structure, biochemistry and mechanism of action of glycopeptide antibiotics. Eur. J. Clin. Microbiol. Infect. Dis. 1989, 8, 943–950. 10.1007/bf01967563. [DOI] [PubMed] [Google Scholar]
- Neilands J. B. Siderophores: structure and function of microbial iron transport compounds. J. Biol. Chem. 1995, 270, 26723–26726. 10.1074/jbc.270.45.26723. [DOI] [PubMed] [Google Scholar]
- Hider R. C.; Kong X. Chemistry and biology of siderophores. Nat. Prod. Rep. 2010, 27, 637–657. 10.1039/b906679a. [DOI] [PubMed] [Google Scholar]
- Mootz H. D.; Marahiel M. A. The tyrocidine biosynthesis operon of Bacillus brevis: complete nucleotide sequence and biochemical characterization of functional internal adenylation domains. J. Bacteriol. 1997, 179, 6843–6850. 10.1128/jb.179.21.6843-6850.1997. [DOI] [PMC free article] [PubMed] [Google Scholar]
- McQuade T. J.; Shallop A. D.; Sheoran A.; DelProposto J. E.; Tsodikov O. V.; Garneau-Tsodikova S. A nonradioactive high-throughput assay for screening and characterization of adenylation domains for nonribosomal peptide combinatorial biosynthesis. Anal. Biochem. 2009, 386, 244–250. 10.1016/j.ab.2008.12.014. [DOI] [PubMed] [Google Scholar]
- Phelan V. V.; Du Y.; McLean J. A.; Bachmann B. O. Adenylation Enzyme Characterization Using γ -18O4-ATP Pyrophosphate Exchange. Chem. Biol. 2009, 16, 473–478. 10.1016/j.chembiol.2009.04.007. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Wilson D. J.; Aldrich C. C. A continuous kinetic assay for adenylation enzyme activity and inhibition. Anal. Biochem. 2010, 404, 56–63. 10.1016/j.ab.2010.04.033. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Lee T. V.; Johnson L. J.; Johnson R. D.; Koulman A.; Lane G. A.; Lott J. S.; Arcus V. L. Structure of a eukaryotic nonribosomal peptide synthetase adenylation domain that activates a large hydroxamate amino acid in siderophore biosynthesis. J. Biol. Chem. 2010, 285, 2415–2427. 10.1074/jbc.m109.071324. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Hara R.; Suzuki R.; Kino K. Hydroxamate-based colorimetric assay to assess amide bond formation by adenylation domain of nonribosomal peptide synthetases. Anal. Biochem. 2015, 477, 89–91. 10.1016/j.ab.2015.01.006. [DOI] [PubMed] [Google Scholar]
- Kasai S.; Konno S.; Ishikawa F.; Kakeya H. Functional profiling of adenylation domains in nonribosomal peptide synthetases by competitive activity-based protein profiling. Chem. Commun. 2015, 51, 15764. 10.1039/c5cc04953a. [DOI] [PubMed] [Google Scholar]
Associated Data
This section collects any data citations, data availability statements, or supplementary materials included in this article.