

Abstract

The use of a phenylthio group (SPh) as a dummy ligand at the 6-position to control the side-chain conformation of a series of hexopyranosyl donors is described. The SPh group limits side-chain conformation in a configuration-specific manner, which parallels that seen in the heptopyranosides, and so influences glycosylation selectivity. With both d- and l-glycero-d-galacto-configured donors, the equatorial products are highly favored as they are with an l-glycero-d-gluco donor. For the d-glycero-d-gluco donor, on the other hand, modest axial selectivity is observed. Selectivity patterns are discussed in terms of the side-chain conformation of the donors in combination with the electron-withdrawing effect of the thioacetal group. After glycosylation, removal of the thiophenyl moiety and hydrogenolytic deprotection is achieved in a single step with Raney nickel.
Introduction
In the hepto- and higher carbon pyranosides (and hexo- and higher carbon furanosides), the conformation of the exocyclic C–C bond, hereinafter the side chain, is controlled by the relative configuration of the C4–C6 stereotriad (and of the C3–C5 triad in the furanosides).1−3 Compounds with the arabino configuration in this location predominantly take up the tg conformation,4−7 while those with the ribo and xylo configurations adopt the gt conformation; finally, compounds with the local lyxo configuration mainly populate the gg conformation (Figure 1). This pattern, which arises from a combination of steric, gauche, and dipolar interactions,1,8,9 differs substantially from that found in the simple hexopyranosides (and pentofuranosides),5−7,10 where the side-chain conformation is mainly dictated by the configuration at C4, with the gluco series adopting a near-equimolar mixture of the gg and gt conformations and the galacto series a 15:55:30 tg:gt:gg mixture (Figure 1).5−7
Figure 1.

Influence of relative configuration on the side-chain conformation of heptopyranosides and hexopyranosides.
In typical glycosylation reactions in which an electrophilic glycosyl donor undergoes nucleophilic substitution of a leaving group by an acceptor alcohol,11−13 systems in which the donor side chain predominantly occupies the tg conformation are less reactive and more equatorially selective than those with a predominant gg conformation of the side chain, whereas those whose side chain mainly populates the gt conformation have intermediate reactivity and selectivity. This is because (i) the tg conformation maximizes the electron-withdrawing effect of the C6–O6 bond, thereby destabilizing nascent positive charge at the anomeric center, so favoring bimolecular mechanisms, and (ii) the gg conformation is best placed to electrostatically stabilize the developing positive charge at the anomeric center, while (iii) the gt conformation has intermediate behavior.14,15 Glycosylation reactivity and selectivity in the hepto- and higher carbon pyranosyl donors is therefore an inherent function of the relative configuration of C4–C6 stereotriad and of the so-imposed side-chain conformations. In the hexopyranosides, donor reactivity can be correspondingly influenced by locking the side-chain conformation with the aid of 4,6-O-benzylidene acetals or related cyclic protecting groups. However, as only one conformation is available in this manner for the gluco (tg) and galacto (gg) series, such options are limited.16
We now address the question of the control of side-chain conformation and glycosylation reactivity and selectivity in hexopyranosyl donors without the use of rigid bicyclic protecting groups at the 4,6-position. To this end, we envisaged that side-chain conformation in the hexopyranosides could be achieved by installation of a temporary or dummy ligand at the 6-position to mimic the C6–C7 bond in the hepto and higher carbon sugars. To be effective, such a ligand would need to be easily installed and removed, of comparable electronegativity and steric bulk to a methyl group, and stable under typical glycosylation conditions, causing us to settle on the thioether moiety.
Results and Discussion
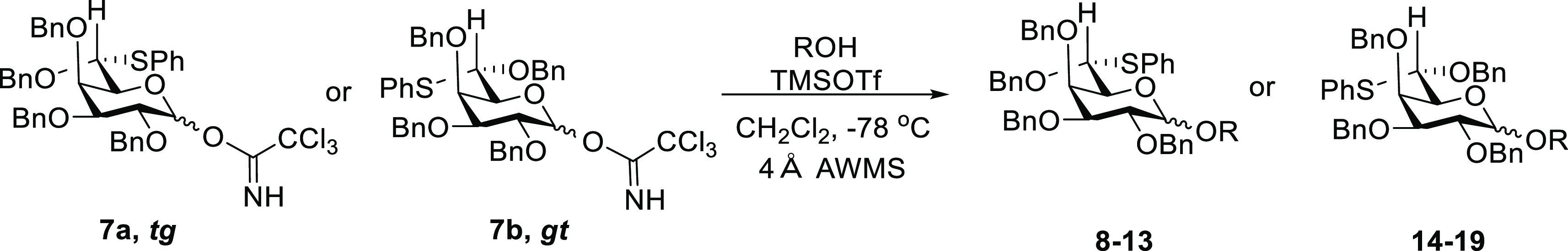
The preparation of the thioether derivatives 5a–d began with the synthesis of alcohols 1 and 2,17−19 which were oxidized to the corresponding aldehydes using the Dess–Martin periodinane20,21 followed by the formation of dibenzylacetals 3 and 4 with tribenzyl orthoformate and a catalytic amount of p-TSA in 71 and 65% yields, respectively.22 These acetals were treated with 1.5 equiv of BF3·OEt2 and 1.5 equiv of thiophenol at −78 to −20 °C to obtain the S-phenyl-O-benzyl monothioacetals 5a and 5b in the galacto series and 5c and 5d in the gluco series (Scheme 1). Provided the temperature is carefully controlled to avoid over-reaction and dithioacetal formation, the diastereomeric ratios in the formation of monothioacetals 5a and 5b and 5c and 5d were reproducible over multiple runs. At the present time, however, we do not have a rationale for the observed selectivities.
Scheme 1. Synthesis of Trichloroacetimidate Donors.

[a] 1.5 equiv of Dess–Martin periodinane (DMP), CH2Cl2, r.t. [b] 2 equiv of CH(OBn)3, 0.2 equiv of p-TSA, Na2SO4, CH3CN, r.t. [c] 1.5 equiv of PhSH, 1.5 equiv of BF3·OEt2, toluene, −78 °C to −20 °C. [d] 10 mol % RuCl2(PPh3)3, 1 equiv of DBU, EtOH, reflux. [e] cat. OsO4, 3 equiv of NMO, dioxane:H2O 4:1, r.t. [f] 5 equiv of Cl3CCN, cat. DBU, CH2Cl2, 0 °C.
Both pairs of isomers were purified by silica gel chromatography and their structures were confirmed by NMR spectroscopy. For each isomer, the side-chain configurations and conformations were determined with a combination of coupling constant analysis and NOE measurements. For both 5a and 5b, a 3J5,6 coupling constant of 8.8 Hz requires an antiperiplanar relationship between H5 and H6 (Figure 2). The heteronuclear multiple bond coherence (HMBC) correlations of H6 located the corresponding methylene carbon signals for O6-CH2-Ph and the ipso carbons for the SPh groups in both compounds. An NOE correlation between the ortho-protons of the thiophenyl moiety and the methylene protons in O1-CH2CH=CH2 confirmed the d-glycero-d-galacto configuration23 and tg conformation of 5a, for which additionally no measurable NOE correlation was found between H4 and the ortho-protons of the SPh group. For 5b, an NOE correlation revealed the spatial proximity of H4 and the ortho-protons of the SPh moiety indicating a predominant gt conformation and the l-glycero-d-galacto configuration.
Figure 2.

Key NOESY correlation. Red arrows: NOE correlation. Blue arrows: HMBC correlation. Nomenclature based on heptopyranoside series.
For both isomers in the gluco series, an NOE correlation linking H4 and H6 and the 3J5,6 coupling constants together require H6 to be predominantly antiperiplanar to O5. HBMC correlations with H6 identified the methylene carbon of the O6CH2Ph moiety whose attached protons showed an NOE correlation with H4, indicating the l-glycero-d-gluco configuration and a predominant gg conformation for 5c (Figure 2). Finally, for 5d, the ipso carbon of the SPh group, and so its respective ortho-protons, was identified by HMBC correlation to H6.
The NOE correlation between H4 and these ortho-SPh protons finally confirms the d-glycero-d-gluco configuration and gt conformation of 5d. The predominant conformations assigned to 5a–d are fully consistent with those found for the corresponding heptopyranosides (Figure 1) and support the central hypothesis of the use of a phenylthio group as a side-chain conformation-controlling dummy ligand in the hexopyranosides.
The allyl groups were removed from each of 5a–d by isomerization with 10 mol % of RuCl2(PPh3)3 followed by treatment with catalytic OsO4 and N-methylmorpholine N-oxide to give the corresponding hemiacetals 6a–d as α:β mixtures.24,25 Finally, the trichloroacetimidate donors 7a–d were obtained by treatment with 5 equiv of Cl3CCN and catalytic amount of DBU at 0 °C in the form of α:β mixtures.26−29
Glycosylation reactions were carried out by stirring 0.15 M CH2Cl2 solutions of donors 7a–d (1.0 equiv) with the acceptor (1.2 equiv) and 4 Å AWMS for 1 h, followed by cooling to −78 °C and stirring for another 0.5 h. TMSOTf (0.2 equiv) then was added to the mixture as a 10% solution in CH2Cl2.11,30−32 All glycosylations were maintained at −78 °C and quenched with 0.2 mL of triethylamine at that temperature before the mixtures were warmed to room temperature, filtered through celite, diluted with CH2Cl2, and washed with saturated aqueous NaHCO3. The resulting glycosides were then isolated by silica gel chromatography, leading to the results presented in Tables 1 and 2.
Table 1. Glycosylation Reactions with d-glycero-d-galacto Donor 7a and l-glycero-d-galacto Donor 7b.

All reactions were carried out at −78 °C with activation by 0.2 equiv of TMSOTf. The donor-to-acceptor ratio is 1.0:1.2 with a 0.15 M concentration of donor.
Isolated yield.
Anomeric ratios were determined by integration of the anomeric signals in the 1H NMR spectra of the crude reaction mixtures.
Table 2. Glycosylation Reactions with l-glycero-d-gluco Donor 7c and d-glycero-d-gluco Donor 7d.

All reactions were carried out at −78 °C with activation by 0.2 equiv of TMSOTf. The donor-to-acceptor ratio is 1.0:1.2 with a 0.15 M concentration of donor.
Isolated yield.
Anomeric ratios were determined by integration of the anomeric signals in the 1H NMR spectra of the crude reaction mixtures.
When this reaction was conducted on a scale of ∼1 g, 21 was obtained in a 92% isolated yield with an α:β ratio of 1:3.
Both the d-glycero-d-galacto and l-glycero-d-galacto donors (Table 1) showed good yields and high equatorial selectivity with primary alcohol acceptors. The 1-adamantyl glycosides were also formed with high equatorial selectivity in the case of 7a with its tg conformation and modest equatorial selectivity for 7b with the gt conformation. For such examples, this new method could be considered as complementary to the use of acetonitrile for the formation of β-galactopyranosides in the absence of neighboring group participation33 and also to the Kiso method using a 4,6-O-silylene acetal for the formation of α-galactopyranosides.34 Secondary alcohol acceptors showed good yields and typically modest equatorial selectivity, with the exception of the coupling of 7b to the less reactive35,36 secondary alcohol methyl 2,3,6-tri-O-benzyl-α-d-glucopyranoside.
In the gluco series, equatorially selective couplings were observed for the l-glycero-d-gluco donor 7c with the gg conformation with the exception of coupling to the less reactive methyl 2,3,6-tri-O-benzyl-α-d-glucopyranoside that shows modest axial selectivity. For the d-glycero-d-gluco configured donor 7d, coupling to two of the three acceptors studied, 1-adamantanol and the relatively reactive alcohol methyl 2,3-O-isopropylidene-α-l-rhamnopyranoside showed modest axial selectivity, while the third example, with 1,2;3,4-di-O-isopropylidene-α-d-galactopyranose, displayed modest equatorial selectivity (Table 2).
For the two galacto donors 7a and 7b, the observed selectivities are consistent with those found earlier with the corresponding 7-deoxy d- and l-glycero-d-galactopyranosyl donors,1,3,37 further reinforcing the design hypothesis of employing a phenylthio substituent as a surrogate for a methyl group. It is noteworthy, however, that for donor 7a, with its tg side-chain conformation, the selectivities are better than those seen for the corresponding all-carbon donor. We attribute this phenomenon to a combination of the different coupling methods used and to the additional electron-withdrawing effect of the thioacetal moiety reinforcing that due to the imposition of the tg conformation (Figure 3).
Figure 3.

Influence of the side-chain conformation in the selectivity of glycosylations.
In the gluco series, the selectivities observed with donors 7c and 7d were again grossly consistent with those observed previously with the corresponding d- and l-glycero-d-gluco-7-deoxy heptopyranosyl donors,1 again in line with the overall hypothesis. However, donor 7c was somewhat less equatorially selective than its all-carbon analog. As with the galactopyranosides, the β-selective l-glycero-d-gluco donor 7c can be considered as complementary to a simple per-O-benzyl glucopyranosyl trichloroacetimidate employed in the presence of acetonitrile.33
As we had attributed the high equatorial selectivity seen with the l-glycero-d-gluco-6-deoxy heptopyranosyl donor in the all-carbon series to stereodirecting hydrogen bonding between the gg-disposed O6 in the donor and the acceptor OH,37 the reduced selectivity seen with donor 7c can again be ascribed to the presence of the electron-withdrawing hemiacetal, which reduces the H-bond acceptor capabilities of O6 (Figure 3).
Tables 3 and 4 provide illustrative examples of desulfurization with Raney nickel achieved concomitantly with hydrogenolysis of the benzyl groups.38−41 The reactions were carried out in ethanol under 1 atm of hydrogen at room temperature and gave the desired products in good yields.
Table 3. Desulfurization with Raney Nickel for the d-glycero-d-galacto and l-glycero-d-galacto Series.


All reactions were carried out at room temperature.
Isolated yield.
Table 4. Desulfurization with Raney Nickel for the l-glycero-d-gluco and d-glycero-d-gluco Series.


All reactions were carried out at room temperature.
Isolated yield.
On a scale of 600 mg, desulfurization of 21β gave 88% of 36.
In these reactions, typically the thiophenyl ether moiety is cleaved rapidly while the O-benzyl groups are removed more slowly. Self-evidently, desulfurization and debenzylation of both side-chain isomers in either the galacto or gluco series of products give a single galacto- or gluco-pyranoside.
Conclusions
Overall, the use of a dummy ligand is an effective strategy to bias the side-chain conformation of hexopyranosyl donors and provides a conceptually novel means of achieving moderate to good stereocontrol in the coupling of galacto and glucopyranosyl donors to relatively reactive donors. Acetimidates 7a–d are obtained in a minimum of steps from readily available allyl d-galacto and glucopyranosides, and the final deprotection by desulfurization and debenzylation is achieved in a single step. Overall, the deprotected glycosides are obtained in eight steps from alcohols 1 and 2.
Experimental Section
General Experimental Details
All reactions were carried out under argon unless otherwise stated. 1-Adamantanol and 1,2:5,6-di-O-isopropylidene-α-d-glucofuranose were purchased from commercial suppliers. Solvents used for column chromatography were of analytical grade and were purchased from commercial suppliers. Thin-layer chromatography was carried out with 250 μm glass-backed silica (XHL) plates. Detection of compounds was achieved by UV absorption (254 nm) and by staining with 10% sulfuric acid in ethanol. Purification of crude residues was performed by silica gel chromatography using a 230–400 mesh-grade 60 silica. Specific rotations [deg cm3 g–1 dm–1] were measured in chloroform on an automatic polarimeter with a path length of 10 cm. NMR spectra were recorded in C6D6, CD2Cl2, or CDCl3 at either 500, 600, or 900 MHz as indicated. High-resolution mass spectra (HRMS) were recorded in the electrospray mode using an orbitrap mass analyzer (Thermo Fisher ESI-Orbitrap). Chemical shifts (δ) are recorded in ppm, and multiplicities are abbreviated as follows: s (singlet), m (multiplet), br (broad), d (doublet), t (triplet), and q (quartet). Structural assignments were made with additional information from gCOSY, gHSQC, and gHMBC experiments. All manipulations and reactions using osmium tetroxide should be conducted in a well-ventilated fume hood.
Preparation of Acceptors
Methyl 2,3,4-Tri-O-benzyl-α-d-glucopyranoside
This colorless oil was prepared according to the literature method and had spectral data consistent with the literature (1.4 g, 98%).42−44
1,2:3,4-Di-O-isopropylidene-α-d-galactopyranose
This colorless syrup was prepared according to the literature method and had spectral data consistent with the literature (0.80 g, 74%).45
Methyl 2,3-O-Isopropylidene-α-l-rhamnopyranoside
This colorless oil was prepared according to the literature method and had spectral data consistent with the literature (1.1 g, 87%).46,47
Methyl 2,3,6-Tri-O-benzyl-α-d-glucopyranoside
This colorless syrup was prepared according to the literature method and had spectral data consistent with the literature (2.95 g, 98%).42−44
Allyl 2,3,4-Tri-O-benzyl-β-d-galactopyranoside (1)
This compound, a colorless oil, was prepared from β-d-galactose pentaacetate, starting with the synthesis of the allyl glycoside and deacetylation,17 followed by trityl group protection of O6, benzylation, and the final trityl group removal (3.67 g, 51%).18,19 The spectral data is consistent with the literature.481H NMR (500 MHz, CDCl3) δ 7.40–7.26 (m, 15H, Ar-H), 6.04–5.90 (m, 1H, =CH), 5.33 (dq, J = 17.4, 1.7 Hz, 1H, =CH2), 5.19 (dq, J = 10.4, 1.5 Hz, 1H, =CH2), 5.02–4.92 (m, 2H, Ph-CH2-), 4.82 (d, J = 11.8 Hz, 1H, Ph-CH2-), 4.79 (d, J = 10.9 Hz, 1H, Ph-CH2-), 4.75 (d, J = 11.8 Hz, 1H, Ph-CH2-), 4.67 (d, J = 11.8 Hz, 1H, Ph-CH2-), 4.47–4.35 (m, 2H, CH), 4.14 (ddt, J = 13.0, 5.9, 1.5 Hz, 1H, -OCH2a-), 3.88 (dd, J = 9.8, 7.7 Hz, 1H, H2), 3.80–3.73 (m, 2H, CH), 3.57–3.47 (m, 2H, CH), 3.37 (dd, J = 6.8, 5.6 Hz, 1H, H5), 1.64 (s, 1H, OH). 13C{1H} NMR (126 MHz, CDCl3) δ 138.7 (C), 138.5 (C), 138.35 (C), 134.3 (CH), 128.7 (Ar), 128.5 (Ar), 128.4 (Ar), 128.3 (Ar), 128.0 (Ar), 127.8 (Ar), 127.7 (Ar), 127.6 (Ar), 117.2 (=CH2), 103.2 (CH1), 82.3 (CH3), 79.7 (CH2), 75.3 (CH5), 74.6 (CH6), 74.2 (CH4), 73.5 (CH2), 73.0 (CH2), 70.4 (OCH2), 62.1 (CH2). HRMS (ESI) m/z: [M + Na]+ calcd for C30H34O6Na 513.2247; found 513.2242.
Allyl 2,3,4-Tri-O-benzyl-β-d-glucopyranoside (2)
This compound, an amorphous white solid, was prepared from β-d-glucose pentaacetate, starting with the synthesis of the allyl glycoside and deacetylation,17 followed by trityl group protection of O6, benzylation, and the final trityl group removal (3.52 g, 31%).18,19 The spectral data is consistent with the literature.491H NMR (500 MHz, CDCl3) δ 7.36–7.24 (m, 15H; Ar-H), 5.95 (ddt, J = 17.2, 10.5, 5.6 Hz, 1H; =CH), 5.34 (dq, J = 17.2, 1.6 Hz, 1H; =CH2a), 5.21 (dq, J = 10.5, 1.4 Hz, 1H; =CH2b), 4.98–4.89 (m, 2H; Ph-CH2-), 4.86 (d, J = 10.9 Hz, 1H; Ph-CH2-), 4.80 (d, J = 10.9 Hz, 1H; Ph-CH2-), 4.72 (d, J = 10.9 Hz, 1H; Ph-CH2-), 4.63 (d, J = 10.9 Hz, 1H; Ph-CH2-), 4.49 (d, J = 7.8 Hz; 1H, H1), 4.39 (ddt, J = 12.9, 5.6, 1.6 Hz, 1H; -OCH2a-), 4.15 (ddt, J = 12.9, 5.6, 1.4 Hz, 1H; -OCH2b-), 3.86 (d, J = 12.0 Hz, 1H; H-6a), 3.70 (d, J = 12.0 Hz, 1H; H-6b), 3.66 (t, J = 9.1 Hz, 1H; H3), 3.56 (app t, J = 9.1 Hz, 1H; H4), 3.44 (dd, J = 9.1, 7.8 Hz, 1H; H2), 3.35 (ddd, J = 9.1, 4.7, 2.8 Hz, 1H; H5), 1.87 (s, 1H; OH). 13C{1H} NMR (126 MHz, CDCl3) δ 138.6 (Ar-H), 138.4 (Ar-H), 138.1 (Ar-H), 134.0 (=CH), 128.6 (Ar-H), 128.5 (Ar-H), 128.3 (Ar-H), 128.2 (Ar-H), 128.0 (Ar-H), 127.97 (Ar-H), 127.8 (Ar-H), 127.7 (Ar-H), 117.6 (CH2), 102.9 (CH1), 84.6 (CH4), 82.4 (CH3), 77.7 (CH2), 75.8 (CH5), 75.2 (CH2-6), 75.12 (CH2), 75.07 (CH2), 70.8 (CH2), 62.2 (CH2). HRMS (ESI) m/z: [M + Na]+ calcd for C30H34O6Na 513.2247; found 513.2240.
General Procedure for the Syntheses of Dibenzylacetals, (GP1)
To a stirred solution of alcohol (1 or 2, 1 equiv, 0.5 M) in anhydrous CH2Cl2 was added Dess–Martin periodinane (1.5 equiv). Stirring was continued for 1 h at room temperature. After the completion of the reaction as observed by thin-layer chromatography (TLC), saturated aqueous Na2S2O3 (50 mL) and saturated aqueous NaHCO3 (50 mL) were added to the reaction mixture, which was stirred for another 0.25 h. The organic layer was separated, and the aqueous layer was extracted with CH2Cl2 (3 × 50 mL). The combined organic layers were collected, dried over anhydrous Na2SO4, and concentrated under reduced pressure to afford a crude residue, which was used immediately without further purification.
A 0.5 M solution of crude aldehyde (1 equiv) in anhydrous acetonitrile was stirred with Na2SO4 (0.2 g/mmol) for 5 min before the addition of p-TSA (0.2 equiv) and subsequently the addition of CH(OBn)3 (2 equiv) in a dropwise manner. After complete addition, the reaction mixture was stirred at room temperature overnight; then, it was quenched with Et3N (2.0 mL), filtered, and concentrated under reduced pressure to afford a crude residue. Purification of this crude product by silica gel chromatography using 100% hexane to 5% ethyl acetate in hexane gave the dibenzylacetals.
Allyl 2,3,4,6,6-Penta-O-benzyl-β-d-galactopyranoside (3)
Prepared from compound 1 (3.67 g, 7.34 mmol) following general procedure GP1, eluting from silica gel with 5% ethyl acetate in hexanes. White amorphous solid (3.6 g, 71%) [α]D21 = +27.7 (c = 1.0, CHCl3). 1H NMR (500 MHz, CDCl3) δ 7.41–7.19 (m, 25H, ArH), 5.94 (ddt, J = 16.4, 10.9, 5.6 Hz, 1H, =CH), 5.29 (dq, J = 17.2, 1.8 Hz, 1H, =CH2a), 5.17 (dq, J = 10.5, 1.5 Hz, 1H, =CH2b), 5.02 (d, J = 7.4 Hz, 1H, H6), 5.00–4.92 (m, 2H, CH2), 4.82–4.73 (m, 3H, CH2), 4.74–4.63 (m, 3H, CH2), 4.48 (d, J = 11.5 Hz, 1H, CH2), 4.44–4.36 (m, 2H, H1, -OCH2a-), 4.26 (d, J = 11.3 Hz, 1H, CH2), 4.11 (dd, J = 13.0, 6.0 Hz, 1H, -OCH2b-), 4.03 (d, J = 2.9 Hz, 1H, H4), 3.88 (dd, J = 9.7, 7.7 Hz, 1H, H2), 3.52 (dd, J = 9.7, 2.9 Hz, 1H, H3), 3.49 (d, J = 7.3 Hz, 1H, H5); 13C{1H} NMR (126 MHz, CDCl3) δ 138.9 (C), 138.8 (C), 138.6 (C), 138.3 (C), 137.6 (C), 134.2 (=CH), 128.5 (Ar), 128.44 (Ar), 128.4 (Ar), 128.2 (Ar), 128.1 (Ar), 128.0 (Ar), 127.9 (Ar), 127.83 (Ar), 127.8 (Ar), 127.65 (Ar), 127.6 (Ar), 127.5 (Ar), 117.3 (=CH2), 103.2 (C1), 100.5 (C6), 82.4 (C3), 79.5 (C2), 75.7 (CH2), 75.3 (C5), 74.7 (CH2), 74.5 (C4), 73.3 (CH2), 70.3 (CH2), 69.7 (OCH2), 69.4 (CH2). HRMS (ESI) m/z: [M + Na]+ calcd for C44H46O7Na 709.3136; found, 709.3118.
Allyl 2,3,4,6,6-Penta-O-benzyl-β-d-glucopyranoside (4)
Prepared from compound 2 (3.52 g, 7.18 mmol) following general procedure GP1, eluting from silica gel with 5% ethyl acetate in hexanes. White amorphous solid (3.2 g, 65%) [α]D21 = +3.9 (c = 0.1, CHCl3). 1H NMR (500 MHz, CDCl3) δ 7.43–7.16 (m, 23H; Ar-H), 7.09 (dd, J = 7.0, 2.7 Hz, 2H; Ar-H), 5.96 (ddt, J = 17.3, 10.2, 5.5 Hz, 1H; =CH), 5.34 (dq, J = 17.3, 1.7 Hz, 1H; =CH2a), 5.20 (dq, J = 10.2, 1.7 Hz, 1H; =CH2b), 5.04 (d, J = 1.9 Hz, 1H; H6), 5.00 (d, J = 11.0 Hz, 1H; Ph-CH2-), 4.94 (d, J = 11.0 Hz, 1H; Ph-CH2-), 4.87–4.71 (m, 6H; Ph-CH2-), 4.59 (d, J = 12.0 Hz, 1H; Ph-CH2-), 4.47 (d, J = 7.7 Hz, 1H; H1), 4.43 (ddt, J = 13.1, 5.5, 1.7 Hz, 1H; -OCH2a-), 4.34 (d, J = 10.8 Hz, 1H; Ph-CH2-), 4.12 (ddt, J = 13.1, 5.5, 1.7 Hz, 1H; -OCH2b-), 3.73 (app t, J = 8.9 Hz, 1H; H4), 3.68 (t, J = 8.9 Hz, 1H; H3), 3.63 (dd, J = 8.9, 1.9 Hz, 1H; H5), 3.57 (dd, J = 8.9, 7.7 Hz, 1H; H2). 13C{1H} NMR (126 MHz, CDCl3) δ 138.46 (C), 138.4 (C), 138.22 (C), 138.2 (C), 137.5 (C), 134.0 (=CH), 128.34 (Ar), 128.30 (Ar), 128.24 (Ar), 128.2 (Ar), 128.10 (Ar), 128.07 (Ar), 127.77 (Ar), 127.7 (Ar), 127.59 (Ar), 127.56 (Ar), 127.54 (Ar), 127.5 (Ar), 127.4 (Ar), 117.2 (CH2), 102.7 (C1), 98.3 (C6), 84.6 (C3), 81.9 (C2), 78.1 (C4), 76.0 (C5), 75.6 (CH2), 74.6 (CH2), 74.4 (CH2), 70.1 (CH2), 68.8 (CH2), 68.4 (CH2). HRMS (ESI) m/z: [M + Na]+ calcd for C44H46O7Na 709.3136; found 709.3137.
General Procedure for the Syntheses of Monothioacetals 5a–d, (GP2)
Dibenzylacetal (3 or 4, 1 equiv) was coevaporated with toluene and dried overnight under high vacuum. The residue was dissolved in anhydrous toluene to give a 0.2 M solution and cooled down to −78 °C. Thiophenol (1.5 equiv) was added at the same temperature before. BF3·OEt2 (1.5 equiv) was added in a dropwise manner. After complete addition, the reaction mixture was stirred for 10 min at the same temperature and then gradually brought to −20 °C over a period of 3 h. The reaction mixture was stirred at −20 °C for 0.5 h and then quenched with saturated aqueous NaHCO3 (20 mL) and extracted with EtOAc (3 × 20 mL). The combined organic layers were collected, dried over anhydrous sodium sulfate, and concentrated under reduced pressure to afford a crude residue, which was purified by silica gel chromatography using 5–10% ethyl acetate and hexane as eluents to afford the corresponding monothioacetals.
Allyl (6R)-6-Phenylthio-2,3,4,6-tetra-O-benzyl-β-d-galactopyranoside 5a
Prepared from compound 3 (2.0 g, 2.91 mmol) following general procedure GP2, eluting from silica gel with 5% ethyl acetate in hexanes. White amorphous solid (0.68 g, 34%) [α]D23 = −32.1 (c = 1.0, CHCl3). 1H NMR (500 MHz, CD2Cl2) δ 7.57–7.48 (m, 2H, Ho-SPh), 7.39–7.19 (m, 24H, Ar-H), 6.02 (dddd, J = 17.1, 10.7, 6.0, 5.0 Hz, 1H, =CH), 5.37 (dq, J = 17.1, 1.7 Hz, 1H, =CH2a (trans)), 5.21 (dq, J = 10.7, 1.5 Hz, 1H, =CH2a (cis)), 5.14 (d, J = 11.0 Hz, 1H, CH2-O6), 5.00 (d, J = 8.9 Hz, 1H, H6), 4.95 (d, J = 11.0 Hz, 1H, CH2-O4), 4.88 (d, J = 11.0 Hz, 1H, CH2-O3), 4.71 (d, J = 11.0 Hz, 1H, CH2-O3), 4.68 (s, 2H, CH2-O2), 4.49 (ddt, J = 13.0, 5.0, 1.7 Hz, 1H, -O-CH2a-), 4.40 (d, J = 11.0 Hz, 1H, CH2-O4), 4.32 (d, J = 11.0 Hz, 1H, CH2-O6), 4.26 (d, J = 7.7 Hz, 1H, H1), 4.18 (ddt, J = 13.0, 6.0, 1.5 Hz, 1H, -O-CH2b-), 4.06 (dd, J = 3.0, 1.0 Hz, 1H, H4), 3.73 (dd, J = 9.7, 7.7 Hz, 1H, H2), 3.41 (dd, J = 9.7, 2.9 Hz, 1H, H3), 3.19 (dd, J = 8.9, 1.1 Hz, 1H, H5). 13C {1H} NMR (126 MHz, CD2Cl2) δ 139.1 (C), 139.0 (C), 138.7 (C), 137.2 (C), 134.7 (CHo-SPh), 134.5 (=CH), 131.8 (C), 128.9 (Ar), 128.5 (Ar), 128.4 (Ar), 128.28 (Ar), 128.26 (Ar), 128.19 (Ar), 128.18 (Ar), 128.08 (Ar), 128.01 (Ar), 127.6 (Ar), 127.54 (Ar), 127.50 (Ar), 127.4 (Ar), 116.8 (=CH2), 102.8 (C1), 87.0 (C6), 82.3 (C3), 80.0 (C2), 75.0 (CH2), 74.8 (CH2), 74.5 (C4), 73.4 (CH2), 73.0 (C5), 70.3 (CH2), 70.2 (OCH2). HRMS (ESI) m/z: [M + Na]+ calcd for C43H44O6SNa 711.2750; found 711.2734.
Allyl (6S)-6-Phenylthio-2,3,4,6-tetra-O-benzyl-β-d-galactopyranoside 5b
Prepared from compound 3 (2.0 g, 2.91 mmol) following general procedure GP2, eluting from silica gel with 5% ethyl acetate in hexanes. White amorphous solid (0.82 g, 41%). [α]D23 = +22.8 (c = 2.0, CHCl3) 1H NMR (500 MHz, C6D6) δ 7.47–7.41 (m, 2H, o-SPh), 7.37–7.31 (m, 2H, ArH), 7.32–7.21 (m, 6H, ArH), 7.15–6.97 (m, 12H, ArH), 6.96–6.85 (m, 3H, m,p-SPh), 5.87–5.70 (m, 1H, =CH), 5.37–5.27 (m, 2H, H6, CH2), 5.19 (dq, J = 17.4, 1.8 Hz, 1H, =CH2a), 4.98–4.89 (m, 2H, CH2, =CH2b), 4.77 (d, J = 11.7 Hz, 1H, CH2), 4.73 (d, J = 10.9 Hz, 1H, CH2), 4.65 (dd, J = 11.8, 5.3 Hz, 2H, CH2), 4.54 (d, J = 12.2 Hz, 1H, CH2), 4.43 (d, J = 11.7 Hz, 1H, CH2), 4.38 (d, J = 3.0 Hz, 1H, H4), 4.36–4.30 (m, 1H, -OCH2a-), 4.17 (d, J = 7.7 Hz, 1H, H1), 4.07 (dd, J = 9.7, 7.6 Hz, 1H, H2), 3.95 (ddt, J = 13.2, 5.9, 1.7 Hz, 1H, -OCH2b-), 3.41 (d, J = 8.8 Hz, 1H, H5), 3.27 (dd, J = 9.7, 2.9 Hz, 1H, H3). 13C{1H} NMR (126 MHz, C6D6) δ 139.5 (C), 139.3 (C), 139.0 (C), 138.0 (C), 134.6 (=CH), 133.5 (Ar), 132.7 (Ar), 128.9 (Ar), 128.4 (Ar), 128.29 (Ar), 128.2 (Ar), 128.1 (Ar), 128.0 (Ar), 127.97 (Ar), 127.91 (Ar), 127.86 (Ar), 127.78 (Ar), 127.73 (Ar), 127.68 (Ar), 127.6 (Ar), 127.5 (Ar), 127.4 (Ar), 127.2 (Ar), 116.3 (=CH2), 103.3 (C1), 87.2 (C6), 82.2 (C3), 79.7 (C2), 76.1 (C4), 76.0 (C5), 75.3 (CH2), 74.7 (CH2), 73.3 (CH2), 70.7 (CH2), 69.7 (OCH2). HRMS (ESI) m/z: [M + Na]+ calcd for C43H44O6SNa 711.2750; found 711.2750.
Allyl (6S)-6-Phenylthio-2,3,4,6-tetra-O-benzyl-β-d-glucopyranoside 5c
Prepared from compound 4 (1.6 g, 2.33 mmol) following general procedure GP2, eluting from silica gel with 5% ethyl acetate. Colorless syrup (0.923 g, 57%). [α]D20 = +6.3 (c = 2.0, CHCl3). 1H NMR (500 MHz, C6D6) δ 7.48–7.44 (m, 2H, Ho-SPh), 7.37–7.27 (m, 2H, Ar-H), 7.26–7.20 (m, 2H, Ar-H), 7.09–6.84 (m, 19H, Ar-H), 5.81 (ddt, J = 17.1, 10.7, 6.1, 5.0 Hz, 1H, =CH), 5.59 (d, J = 2.1 Hz, 1H, H6), 5.27 (dd, J = 17.1, 1.5 Hz, 1H, =CH2a), 5.03–4.98 (m, 2H, =CH2b, CH2), 4.93 (m, 2H, CH2), 4.83 (d, J = 11.3 Hz, 1H, CH2), 4.72 (d, J = 11.3 Hz, 1H, CH2), 4.66 (d, J = 11.3 Hz, 1H, CH2), 4.53 (d, J = 11.6 Hz, 1H, CH2), 4.47 (d, J = 11.6 Hz, 1H, CH2), 4.42 (d, J = 7.2 Hz, 1H, H1), 4.36 (ddt, J = 13.2, 5.0, 1.5 Hz, 1H, -OCH2a-), 4.04 (ddt, J = 13.2, 6.1, 1.5 Hz, 1H, -OCH2b-), 3.94 (dd, J = 9.7, 8.4 Hz, 1H, H4), 3.75 (dd, J = 9.7, 2.1 Hz, 1H, H5), 3.62 (m, 2H, H2, H3). 13C{1H} NMR (126 MHz, C6D6) δ 139.17 (C), 139.10 (C), 139.0 (C), 137.6 (C), 136.9 (C), 134.3 (=CH), 131.3 (SPh-CHo), 129.0 (Ar), 128.26 (Ar), 128.21 (Ar), 128.18 (Ar), 128.0 (Ar), 127.9 (Ar), 127.7 (Ar), 127.5 (Ar), 127.4 (Ar), 127.3 (Ar), 126.7 (Ar), 116.8 (=CH2), 103.3 (C1), 88.4 (C6), 84.8 (C3), 82.4 (C2), 79.6 (C5), 78.5 (C4), 75.3 (CH2), 74.6 (CH2), 74.5 (CH2), 69.9 (CH2), 69.8 (-OCH2-). HRMS (ESI) m/z: [M + Na]+ calcd for C43H44O6NaS 711.2751; found 711.2741.
Allyl (6R)-6-Phenylthio-2,3,4,6-tetra-O-benzyl-β-d-glucopyranoside 5d
Prepared from compound 4 (1.6 g, 2.33 mmol) following general procedure GP2, eluting from silica gel with 5% ethyl acetate in hexanes. White amorphous solid (0.192 g, 12%). [α]D20 = +6.6 (c = 0.2, CHCl3). 1H NMR (500 MHz, C6D6) δ 7.43–7.37 (m, 2H, Ho-SPh), 7.31–7.28 (m, 2H, Ar-H), 7.26–7.21 (m, 2H, Ar-H), 7.18–7.12 (m, 2H, Ar-H), 7.12–6.91 (m, 14H, Ar-H), 6.90–6.81 (m, 3H, Ar-H), 5.74 (ddt, J = 16.4, 10.7, 5.4 Hz, 1H, =C-H), 5.62 (d, J = 1.5 Hz, 1H, H6), 5.17 (dq, J = 16.4, 1.8 Hz, 1H, =CH2a), 5.00–4.93 (m, 3H, =CH2b, CH2), 4.89 (d, J = 11.4 Hz, 1H, CH2), 4.79 (d, J = 11.9 Hz, 1H, CH2), 4.71–4.68 (m, 2H, CH2), 4.64 (d, J = 11.4 Hz, 1H, CH2), 4.45 (d, J = 11.4 Hz, 1H, CH2), 4.38 (d, J = 7.5 Hz, 1H, H1), 4.25 (ddt, J = 13.0, 5.4, 1.8 Hz, 1H, -OCH2a-), 4.01 (dd, J = 9.6, 8.5 Hz, 1H, H4), 3.94–3.85 (m, 2H, H5, -OCH2b-), 3.67 (dd, J = 9.1, 8.5 Hz, 1H, H3), 3.62 (d, J = 9.1, 7.5 Hz, 1H, H2). 13C{1H} NMR (126 MHz, C6D6) δ 139.09 (C), 139.03 (C), 138.7 (C), 138.0 (C), 136.5 (Ci-SPh), 134.3 (=CH-), 131.4 (CoH-SPh), 128.9 (Ar-H), 128.3 (Ar), 128.24 (Ar), 128.21 (Ar), 128.0 (Ar), 127.8 (Ar), 127.6 (Ar), 127.5 (Ar), 127.45 (Ar), 127.42(Ar), 127.37 (Ar), 127.31 (Ar), 126.6 (Ar), 116.6 (=CH2), 102.8 (C1), 88.9 (C6), 84.6 (C3), 82.2 (C2), 79.0 (C4), 78.7 (C5), 75.1 (CH2), 74.6 (CH2), 74.5 (CH2), 69.8 (CH2), 69.5 (CH2). HRMS (ESI) m/z: [M + Na]+ calcd for C43H44O6NaS 711.2751; found 711.2741.
General Procedure for Deallylation, GP3
To a stirred solution of allyl glycoside (1.0 equiv) and RuCl2(PPh3)3 (0.1 equiv) in ethyl alcohol (0.1 M) was added DBU (1.0 equiv). The resulting mixture was heated to reflux for 5 h before the solvent was evaporated under reduced pressure to afford a crude residue that was taken up in CH2Cl2 (50 mL) and washed with water (50 mL). The organic layer was separated, and the aqueous layer was extracted with CH2Cl2 (2 × 50 mL). The organic layers were combined, dried over Na2SO4 addition, filtered, and concentrated under reduced pressure to give a crude mixture, which was purified with silica gel column chromatography using 10% ethyl acetate in hexane as an eluent to afford a colorless syrup.
A stirred 0.1 M solution of the compound prepared in the previous step in 1,4-dioxane/water (4:1) was treated with NMO (3.0 equiv) and OsO4 (2% in t-butanol, 2 drops) and stirred until completion (monitored by TLC) at room temperature. The reaction was quenched with saturated aqueous Na2S2O3, diluted with ethyl acetate, washed with brine, and dried over Na2SO4. The organic layer was filtered and concentrated under reduced pressure to afford a crude residue that was purified by silica gel column chromatography using 15–25% ethyl acetate in hexane as an eluent to afford a colorless syrup as a mixture of anomers.
(6R)-6-Phenylthio-2,3,4,6-tetra-O-benzyl-α,β-d-galactopyranose (6a)
Prepared using general protocol GP3 from 5a (0.7 g, 1.02 mmol) as a mixture of 2.4:1, ax:eq anomers after eluting from silica gel with 15–25% gradient of ethyl acetate in hexanes (0.53 g, 81%).
Mixture of anomers: colorless syrup. 1H NMR (500 MHz, CDCl3) δ 7.67–7.58 (m, 7H), 7.40–7.26 (m, 35H), 5.39 (d, J = 3.7 Hz, 2.4H), 5.19–5.12 (m, 3.5H), 5.09–4.99 (m, 7.1H), 4.93 (d, J = 11.0 Hz, 1H), 4.85–4.76 (m, 6.1H), 4.75–4.68 (m, 7.1H), 4.56 (dd, J = 7.4, 4.1 Hz, 1H), 4.48–4.40 (m, 3.5H), 4.32–4.26 (m, 3.5H), 4.17 (d, J = 2.7 Hz, 2.4H), 4.10–4.05 (m, 3.5H), 3.91 (d, J = 9.0 Hz, 2.4H), 3.88 (dd, J = 9.9, 2.7 Hz, 2.4H), 3.74 (d, J = 6.2 Hz, 1H), 3.45 (dd, J = 9.6, 2.8 Hz, 1H), 3.20 (d, J = 8.8 Hz, 1H), 2.77 (d, J = 2.5 Hz, 2.2H). 13C{1H} NMR (126 MHz, CDCl3) δ 139.10, 138.9, 138.8, 138.7, 138.5, 138.4, 137.3, 137.1, 135.4, 135.0, 131.9, 131.2, 129.0, 128.8, 128.68, 128.65, 128.5, 128.48, 128.47, 128.40, 128.38, 128.37, 128.35, 128.30, 128.16, 128.13, 128.10, 127.88, 127.8, 127.68, 127.66, 127.64, 127.61, 127.57, 127.55, 127.53, 127.51, 127.49, 97.9, 92.0, 87.1, 86.8, 82.6, 81.0, 79.2, 76.6, 75.4, 75.2, 74.8, 74.7, 74.3, 73.8, 73.5, 73.0, 70.4, 70.3, 70.1. HRMS (ESI) m/z: [M + Na]+ calcd for C40H40O6SNa 671.2438; found 671.2427.
(6S)-6-Phenylthio-2,3,4,6-tetra-O-benzyl-α,β-d-galactopyranose (6b)
Prepared using general protocol GP3 from 5b (0.9 g, 1.31 mmol) as a mixture of 2.4:1, ax:eq anomers after eluting from silica gel with 15–25% gradient of ethyl acetate in hexanes (0.74 g, 87%).
Mixture of anomers: colorless syrup. 1H NMR (500 MHz, CDCl3) δ 7.73–6.65 (m, 85H), 5.33 (t, J = 3.1 Hz, 2H), 5.18–5.09 (m, 6.5H), 4.99 (d, J = 11.6 Hz, 1H), 4.96–4.91 (m, 3.2H), 4.87 (d, J = 11.9 Hz, 2.3H), 4.85–4.59 (m, 16.9H), 4.48–4.42 (m, 3H), 4.35 (d, J = 2.8 Hz, 1H), 4.21 (d, J = 5.5 Hz, 1H), 4.14–4.03 (m, 4.2H), 3.88 (dd, J = 10.0, 2.7 Hz, 2H), 3.80 (dd, J = 9.7, 7.5 Hz, 1H), 3.65 (d, J = 2.8 Hz, 2H), 3.38 (d, J = 9.0 Hz, 1H), 3.33 (dd, J = 9.7, 2.8 Hz, 1H). 13C NMR (126 MHz, CDCl3) δ 138.99, 138.91, 138.90, 138.8, 138.6, 138.5, 137.2, 137.1, 133.6, 133.0, 132.7, 131.8, 129.17, 129.16, 128.58, 128.57, 128.56, 128.50, 128.49, 128.47, 128.46, 128.38, 128.33, 128.31, 128.2, 128.1, 128.04, 128.00, 127.9, 127.88, 127.86, 127.81, 127.79, 127.75, 127.74, 127.6, 127.53, 127.51, 127.0, 98.0, 91.9, 86.7, 86.1, 82.2, 80.42, 79.2, 76.3, 76.1, 75.6, 75.0, 74.97, 74.95, 74.8, 73.35, 73.33, 73.2, 71.5, 70.4, 69.9. HRMS (ESI) m/z: [M + Na]+ calcd for C40H40O6SNa 671.2438; found 671.2430.
(6S)-6-Phenylthio-2,3,4,6-tetra-O-benzyl-α,β-d-glucopyranose 6c
Prepared using general protocol GP3 from 5c (0.9 g, 1.30 mmol) as a mixture of 1.5:1, ax:eq anomers after eluting from silica gel with 15–25% gradient of ethyl acetate in hexanes (0.46 g, 54%).
Mixture of anomers: colorless syrup. 1H NMR (500 MHz, CDCl3) δ 7.55–7.49 (m, 5H), 7.39–7.33 (m, 7H), 7.32–7.08 (m, 40H), 7.06–7.01 (m, 5H), 5.35–5.31 (m, 3H), 5.30 (t, J = 2.9 Hz, 1.5H, H1 α anomer), 5.00–4.87 (m, 5H), 4.86–4.67 (m, 12.5H), 4.54 (d, J = 11.8 Hz, 1H, CH2), 4.43 (d, J = 11.8 Hz, 1.5H, CH2), 4.39 (d, J = 9.9 Hz, 1.5H, CH), 4.21 (d, J = 11.0 Hz, 1H, CH2), 4.17 (d, J = 11.3 Hz, 1.5H, CH2), 4.13 (t, J = 7.1 Hz, 1H, H1 β anomer), 4.02 (t, J = 9.3 Hz, 1.5H, CH), 3.83 (dd, J = 9.7, 1.7 Hz, 1H, CH), 3.75 (t, J = 9.2 Hz, 1H, CH), 3.70 (d, J = 9.5 Hz, 1H, CH), 3.66 (t, J = 9.2 Hz, 1.5H, CH), 3.61 (dd, J = 9.6, 3.5 Hz, 1.5H, CH), 3.45 (t, J = 8.6 Hz, 1.5H, CH), 3.37 (d, J = 5.2 Hz, 1.5H, CH), 3.12 (d, J = 2.9 Hz, 1.5H, CH). 13C{1H} NMR (126 MHz, CDCl3) δ 138.6 (C), 138.5 (C), 138.4 (C), 138.3 (C), 138.0 (C), 137.0 (C), 136.9 (C), 136.1 (C), 135.7 (C), 132.5 (Ar), 132.4 (Ar), 129.22 (Ar), 129.18 (Ar), 128.7 (Ar), 128.65 (Ar), 128.61 (Ar), 128.55 (Ar), 128.50 (Ar), 128.46 (Ar), 128.4 (Ar), 128.3 (Ar), 128.2 (Ar), 128.1 (Ar), 128.0 (Ar), 127.8 (Ar), 127.68 (Ar), 127.6 (Ar), 127.5 (Ar), 98.1 (C1 β anomer), 91.5 (C1 α anomer), 88.2 (CH), 87.2 (CH), 84.7 (CH), 83.2 (CH), 81.9 (CH), 80.0 (CH), 79.5 (CH), 78.4 (CH), 78.2 (CH), 76.0 (CH2), 75.8 (CH2), 75.3 (CH2), 74.8 (CH2), 74.7 (CH2), 73.4 (CH), 70.6 (CH2), 70.2 (CH2). HRMS (ESI) m/z: [M + Na]+ calcd for C40H40O6NaS 671.2438; found 671.2432.
(6R)-6-Phenylthio-2,3,4,6-tetra-O-benzyl-α,β-d-glucopyranose 6d
Prepared using general protocol GP3 from 5d (0.192 g, 0.28 mmol) as a mixture of 9.8:1, ax:eq anomers after eluting from silica gel with 15–25% gradient of ethyl acetate in hexanes (0.099 g, 55%).
Mixture of anomers: colorless syrup. 1H NMR (500 MHz, CDCl3) δ 7.41–7.13 (m, 23H, Ar-H), 7.01–6.98 (m, 2H, Ar-H), 5.38 (d, J = 1.4 Hz, 1H, H6), 5.24 (t, J = 3.2 Hz, 1H, H1), 4.97 (d, J = 11.0 Hz, 1H, CH2), 4.86 (d, J = 11.0 Hz, 1H, CH2), 4.83–4.74 (m, 3H, CH2), 4.71 (d, J = 12.0 Hz, 1H), 4.63 (d, J = 12.0 Hz, 1H, CH2), 4.43–4.36 (m, 2H, CH2, H5), 4.03 (app t, J = 9.2 Hz, 1H, H4), 3.79 (t, J = 9.2 Hz, 1H, H3), 3.60 (dd, J = 9.2, 3.2 Hz, 1H, H2), 2.91 (d, J = 3.2 Hz, 1H, OH). 13C{1H} NMR (126 MHz, CDCl3) δ 138.6 (C), 138.2 (C), 137.9 (C), 137.5 (C), 135.6 (C), 131.9 (Ar), 129.1 (Ar), 128.6 (Ar), 128.5 (Ar), 128.4 (Ar), 128.4 (Ar), 128.11 (Ar), 128.10 (Ar), 128.08 (Ar), 128.0 (Ar), 127.8 (Ar), 127.6 (Ar), 127.5 (Ar), 127.2 (Ar), 91.3 (C1), 89.0 (C6), 81.6 (C4), 80.2 (C2), 79.1 (C3), 75.7 (CH2), 74.9 (CH2), 74.1 (CH2), 73.3 (C5), 70.0 (CH2). HRMS (ESI) m/z: [M + Na]+ calcd for C40H40O6NaS 671.2438 found 671.2424.
General Procedure for Trichloroacetimidate Preparation, GP4
The hemiacetal (1.0 equiv) was dissolved in CH2Cl2 (0.2 M) and cooled down to 0 °C before trichloroacetonitrile (5.0 equiv) and then DBU (1 drop) were added, and the reaction mixture was stirred for 0.5 h at the same temperature. After completion, the reaction mixture was diluted with dichloromethane and washed with water. The aqueous layer was extracted with CH2Cl2 (2 × 20 mL). The organic layers combined were dried over Na2SO4 and concentrated under reduced pressure. The crude product was passed through a silica gel column, eluting with 10–20% ethyl acetate in hexane to give the trichloroacetimidate, which was used in the next step without further purification.
(6R)-6-Phenylthio-2,3,4,6-tetra-O-benzyl-α,β-d-galactopyranosyl Trichloroacetimidate (7a)
Prepared (0.55 g, 85%) by following the general protocol GP4 from hemiacetal 6a (0.53 g, 0.82 mmol) eluting with 10–20% ethyl acetate in hexane to give the trichloroacetimidate. Colorless syrup. HRMS (ESI) m/z: [M + Na]+ calcd for C42H40O6NCl3NaS 814.1534; found 814.1517.
(6S)-6-Phenylthio-2,3,4,6-tetra-O-benzyl-α,β-d-galactopyranosyl Trichloroacetimidate (7b)
Prepared (0.80 g, 89%) by following the general protocol GP4 from hemiacetal 6b (0.74 g, 1.14 mmol) eluting with 10–20% ethyl acetate in hexane to give the trichloroacetimidate. Colorless syrup. HRMS (ESI) m/z: [M + Na]+ calcd for C42H40O6NCl3NaS 814.1534; found 814.1506.
(6S)-6-Phenylthio-2,3,4,6-tetra-O-benzyl-α,β-d-glucopyranosyl Trichloroacetimidate (7c)
Prepared (0.48 g, 86%) by following the general protocol GP4 from hemiacetal 6c (0.46 g, 0.71 mmol) eluting with 10–20% ethyl acetate in hexane to give the trichloroacetimidate. Colorless syrup. HRMS (ESI) m/z: [M + Na]+ calcd for C42H40O6NCl3NaS 814.1534; found 814.1529.
(6R)-6-Phenylthio-2,3,4,6-tetra-O-benzyl-α,β-d-glucopyranosyl Trichloroacetimidate (7d)
Prepared (0.108 g, 90%) by following the general protocol GP4 from hemiacetal 6d (0.099 g, 0.15 mmol) eluting with 10–20% ethyl acetate in hexane to give the trichloroacetimidate. Colorless syrup. HRMS (ESI) m/z: [M + Na]+ calcd for C42H40O6NCl3NaS 814.1534; found 814.1511.
General Procedure for Glycosylation Reaction, GP5
A mixture of the donor (1.0 equiv) and acceptor (1.2 equiv) was coevaporated with toluene twice, then taken up in anhydrous CH2Cl2 (0.15 M) and stirred for 1 h with activated 4 Å AWMS (2 g/mmol of the donor) at room temperature under argon before cooling to −78 °C. After 0.5 h of continuous stirring, the reaction mixture was treated with TMSOTf (0.2 equiv) and stirred for 4–5 h at −78 °C before it was quenched with triethylamine (0.2 mL). The reaction mixture was diluted with dichloromethane (10 mL), filtered through a pad of celite, and washed with saturated aqueous NaHCO3. The organic layer was separated, dried over Na2SO4, filtered, and concentrated under reduced pressure. Purification by flash column chromatography on silica gel (hexane/ethyl acetate) afforded the corresponding α/β-glycopyranosides. The anomeric ratio of the products was determined by integration of the anomeric signals in the1H NMR spectra of the crude product mixtures unless otherwise stated.
Methyl (6R)-6-Phenylthio-2,3,4,6-tetra-O-benzyl-β-d-galactopyranosyl-(1→6)-2,3,4-tri-O-benzyl-α-d-glucopyranoside (8)
Compound 8β was obtained (31.1 mg, 75%) as a single isomer from the reaction of donor 7a (30.0 mg, 37.8 μmol) and acceptor (21.1 mg, 45.4 μmol) by following the general procedure GP5 for glycosylation (eluting with 10% ethyl acetate in hexanes).
Colorless syrup. [α]D20 = −14.0 (c = 1.9, CHCl3). 1H NMR (500 MHz, CDCl3) δ 7.50 (d, J = 7.5 Hz, 2H, ArH), 7.39–7.21 (m, 31H, ArH), 7.21–7.14 (m, 7H, ArH), 5.12 (d, J = 11.1 Hz, 1H, -CH2), 5.03–4.94 (m, 3H, H6′, -CH2), 4.91 (d, J = 10.9 Hz, 1H, CH2), 4.80 (m, 2H, CH2), 4.77–4.70 (m, 2H, CH2), 4.70–4.66 (m, 2H, CH2), 4.63 (d, J = 3.6 Hz, 1H, H1), 4.54 (d, J = 11.0 Hz, 1H, CH2), 4.40 (d, J = 11.4 Hz, 1H, CH2), 4.34 (dd, J = 10.9, 2.2 Hz, 1H, H6b), 4.25 (d, J = 11.0 Hz, 1H, CH2), 4.18 (d, J = 7.7 Hz, 1H, H1′), 4.05–3.99 (m, 2H, CH), 3.97–3.91 (m, 1H, CH), 3.87 (dd, J = 9.7, 7.7 Hz, 1H, H2′), 3.73 (dd, J = 10.9, 5.2 Hz, 1H, H6a), 3.58–3.50 (m, 2H, H2, CH), 3.38 (dd, J = 9.7, 2.8 Hz, 1H, H3′), 3.32 (s, 3H, OCH3), 3.13 (d, J = 8.8 Hz, 1H, H5′). 13C{1H} NMR (126 MHz, CDCl3) δ 139.04 (C), 139.02 (C), 138.8 (C), 138.7 (C), 138.5 (C), 138.4 (C), 138.3 (C), 137.1 (C), 134.8 (Ar), 132.5 (Ar), 131.9 (Ar), 128.8 (Ar), 128.6 (Ar), 128.5 (Ar), 128.45 (Ar), 128.43 (Ar), 128.40 (Ar), 128.3 (Ar), 128.28 (Ar), 128.24 (Ar), 128.13 (Ar), 128.11 (Ar), 128.08 (Ar), 128.04 (Ar), 128.00 (Ar), 127.9 (Ar), 127.65 (Ar), 127.60 (Ar), 127.58 (Ar), 127.53 (Ar), 127.51 (Ar), 127.46 (Ar), 127.40 (Ar), 104.3 (C1′), 98.1 (C1), 87.0 (C6′), 82.7 (CH), 82.2 (CH), 80.0 (CH), 79.4 (CH), 78.4 (CH), 75.8 (CH2), 75.3 (CH2), 75.1 (CH2), 74.7 (CH2), 74.3 (CH), 73.5 (CH), 73.4 (CH2), 73.0 (CH2), 70.3 (CH2), 70.0 (CH), 68.9 (C6), 55.3 (OCH3). HRMS (ESI) m/z: [M + Na]+ calcd for C68H70O11SNa 1117.4531; found 1117.4495.
(6R)-6-Phenylthio-2,3,4,6-tetra-O-benzyl-β-d-galactopyranosyl-(1→6)-1,2:3,4-O-diisopropylidene-α-d-galactopyranose (9)
Compound 9β was obtained (34.4 mg, 68%) as a single isomer from the reaction of donor 7a (45.0 mg, 56.7 μmol) and acceptor (17.7 mg, 68.0 μmol) by following the general procedure GP5 for glycosylation (eluting with 10% ethyl acetate in hexanes).
Colorless syrup. [α]D21 = −45.9 (c = 0.5, CHCl3). 1H NMR (500 MHz, CDCl3) δ 7.56–7.51 (m, 2H, ArH), 7.42–7.36 (m, 2H, ArH), 7.35–7.16 (m, 21H, ArH), 5.57 (d, J = 5.0 Hz, 1H, H1), 5.12 (d, J = 11.0 Hz, 1H, -CH2), 5.02–4.97 (m, 2H, -CH2, H6′), 4.94 (d, J = 11.4 Hz, 1H, -CH2), 4.74–4.66 (m, 2H, CH2), 4.65–4.57 (m, 2H, H3, -CH2), 4.40 (d, J = 11.4 Hz, 1H, CH2), 4.31 (dd, J = 5.0, 2.4 Hz, 1H, H2), 4.29–4.21 (m, 3H, CH2, CH), 4.19 (d, J = 7.4 Hz, 1H, H1′), 4.11 (dt, J = 7.4, 2.8 Hz, 1H, CH), 4.01 (d, J = 2.8 Hz, 1H, H4′), 3.79 (dd, J = 9.7, 7.4 Hz, 1H, H2′), 3.73 (dd, J = 10.9, 7.7 Hz, 1H, CH), 3.35 (dd, J = 9.7, 2.8 Hz, 1H, H3′), 3.08 (d, J = 8.8 Hz, 1H, H5), 1.50–1.46 (m, 6H, 2xCH3), 1.34 (s, 3H, CH3), 1.30 (s, 3H, CH3); 13C{1H} NMR (126 MHz, CDCl3) δ 139.06 (C), 139.0 (C), 138.7 (C), 137.1 (C), 135.2 (Ar), 131.4 (Ar), 128.8 (Ar), 128.66 (Ar), 128.60 (Ar), 128.4 (Ar), 128.3 (Ar), 128.26 (Ar), 128.20 (Ar), 128.1 (Ar), 128.0 (Ar), 127.8 (Ar), 127.46 (Ar), 127.40 (Ar), 127.38 (Ar), 109.4 (CMe2), 108.7 (CMe2), 104.7 (C1′), 96.5 (C1), 86.7 (C6′), 82.3 (C3′), 79.1 (C2′), 74.71 (CH2), 74.68 (CH2), 74.4 (C4′), 73.2 (C5), 71.6 (CH), 70.9 (CH), 70.6 (CH), 70.2 (C6), 70.1 (CH2), 67.8 (CH), 26.16 (CH3), 26.12 (CH3), 25.2 (CH3), 24.6 (CH3). HRMS (ESI) m/z: [M + Na]+ calcd for C52H58O11SNa 913.3592; found 913.3571.
Adamantyl (6R)-6-Phenylthio-2,3,4,6-tetra-O-benzyl-β-d-galactopyranoside (10)
Compound 10β was obtained (24.1 mg, 61%) as a single isomer from the reaction of donor 7a (40.0 mg, 50.4 μmol) and acceptor (9.2 mg, 60.5 μmol) by following the general procedure GP5 for glycosylation (eluting with 10% ethyl acetate in hexanes).
Colorless syrup. [α]D21 = −6.0 (c = 0.7, CHCl3). 1H NMR (500 MHz, CDCl3) δ 7.55–7.48 (m, 2H, ArH), 7.35–7.17 (m, 23H, ArH), 5.08–5.02 (m, 2H, H6, -CH2), 4.98 (d, J = 11.4 Hz, 1H, -CH2), 4.94 (d, J = 11.0 Hz, 1H, -CH2), 4.74–4.69 (m, 2H, CH2), 4.66 (d, J = 11.8 Hz, 1H, -CH2), 4.56 (d, J = 7.7 Hz, 1H, H1), 4.44 (d, J = 11.4 Hz, 1H, -CH2), 4.19 (d, J = 10.9 Hz, 1H, -CH2), 4.07 (dd, J = 2.8, 1.1 Hz, 1H, H4), 3.79 (dd, J = 9.7, 7.7 Hz, 1H, H2), 3.47–3.31 (m, 2H, H3, H5), 2.13 (dq, J = 6.4, 3.5, 3.0 Hz, 3H, Ada), 1.95–1.88 (m, 3H, Ada), 1.88–1.79 (m, 3H, Ada), 1.67–1.55 (m, 6H, Ada). 13C{1H} NMR (126 MHz, CDCl3) δ 139.1 (C), 138.9 (C), 138.7 (C), 137.1 (C), 134.0 (Ar), 132.7 (Ar), 128.7 (Ar), 128.5 (Ar), 128.35 (Ar), 128.30 (Ar), 128.27 (Ar), 128.21 (Ar), 128.1 (Ar), 127.9 (Ar), 127.6 (Ar), 127.5 (Ar), 127.48 (Ar), 127.45 (Ar), 127.29 (Ar), 127.23 (Ar), 96.67 (C1), 87.87 (C6), 82.93 (C3), 79.63 (C2), 75.27 (CH2), 75.23 (CH2), 74.73 (C4), 74.69 (CH2), 74.09 (C5), 73.20 (CH2), 70.37 (CH2), 42.79 (Ada), 42.36 (Ada), 36.43 (Ada), 30.82 (Ada), 30.74 (Ada). HRMS (ESI) m/z: [M + Na]+ calcd for C50H54O6SNa 805.3533; found 805.3537.
(6R)-6-Phenylthio-2,3,4,6-tetra-O-benzyl-α-d-galactopyranosyl-(1→3)-1,2:5,6-di-O-isopropylidene-α-d-glucofuranose (11)
Compounds 11α and 11β were obtained as a mixture of anomers (38.2 mg, 85%, α/β = 1:4.3) from the reaction of donor 7a (40.0 mg, 50.4 μmol) and acceptor (15.7 mg, 60.5 μmol) by following the general procedure GP5 for glycosylation (eluting with 10% ethyl acetate in hexanes). Repeated column chromatography gave pure samples of α- and β-isomers for full characterization.
α-Isomer
Colorless syrup. [α]D20 = +12.5 (c = 0.5, CHCl3). 1H NMR (500 MHz, CDCl3) δ 7.57–7.52 (m, 2H. ArH), 7.99–7.06 (m, 23H, ArH), 5.86 (d, J = 3.3 Hz, 1H, H1), 5.25 (d, J = 3.7 Hz, 1H, H1′), 5.03 (d, J = 9.11H, H6′), 4.99 (d, J = 11.0 Hz, 2H, -CH2), 4.94 (d, J = 3.6 Hz, 1H, H2), 4.83–4.74 (m, 2H, CH2), 4.73–4.66 (m, 2H, CH2), 4.50–4.42 (m, 1H, CH), 4.38 (d, J = 11.3 Hz, 1H, -CH2), 4.24 (d, J = 2.4 Hz, 1H, H4′), 4.20–4.14 (m, 4H, CH, CH2), 4.13–4.05 (m, 1H, H2′), 4.02–3.94 (m, 3H, CH, CH2), 3.89–3.83 (m, 1H, H3′), 1.47 (s, 3H, CH3), 1.40 (s, 3H, CH3), 1.24 (s, 3H, CH3), 1.22 (s, 3H, CH3). 13C{1H} NMR (126 MHz, CDCl3) δ 139.0 (C), 138.7 (C), 138.6 (C), 137.0 (C), 135.1 (C), 132.5 (Ar), 129.1 (Ar), 128.5 (Ar), 128.4 (Ar), 128.38 (Ar), 128.28 (Ar), 128.22 (Ar), 128.1 (Ar), 127.6 (Ar), 127.59 (Ar), 127.57 (Ar), 127.46 (Ar), 127.4 (Ar), 127.3 (Ar), 111.8 (CMe2), 108.9(CMe2), 105.3 (C1), 99.3 (C1′), 90.1 (C6′), 83.3 (C2), 82.1 (C4), 81.3 (CH), 78.7 (CH), 76.5 (CH), 75.6 (CH), 74.7 (CH2), 73.5 (CH2), 73.14 (CH2), 73.1 (CH), 72.6 (CH), 70.7 (CH2), 66.9 (CH2), 26.94 (CH3), 26.9 (CH3), 26.4 (CH3), 25.4 (CH3). HRMS (ESI) m/z: [M + Na]+ calcd for C52H58O11SNa 913.3592; found 913.3566.
β-Isomer
Colorless syrup. [α]D21 = −22.9 (c = 0.4, CHCl3). 1H NMR (500 MHz, CDCl3) δ 7.54–7.49 (m, 2H, ArH), 7.35–7.21 (m, 23H, ArH), 5.79 (d, J = 3.8 Hz, 1H, H1), 5.12 (d, J = 11.0 Hz, 1H, -CH2), 5.01 (d, J = 8.8 Hz, 1H, H6′), 4.96 (d, J = 11.5 Hz, 1H, -CH2), 4.73 (d, J = 11.0 Hz, 1H, -CH2), 4.67–4.62 (m, 3H, CH2), 4.51–4.45 (m, 3H, H2, -CH2), 4.42–4.38 (m, 2H, CH, CH2), 4.25 (d, J = 11.0 Hz, 1H, -CH2), 4.19 (d, J = 7.7 Hz, 1H, H1′), 4.11–4.05 (m, 2H, CH2), 4.03 (dd, J = 3.0, 1.1 Hz, 1H, H4′), 3.69 (dd, J = 9.7, 7.7 Hz, 1H, H2′), 3.37 (dd, J = 9.7, 2.9 Hz, 1H, H3′), 3.11 (dd, J = 8.9, 1.1 Hz, 1H, H5′), 1.51 (s, 3H, CH3), 1.45 (s, 3H, CH3), 1.37 (s, 3H, CH3), 1.25 (s, 3H, CH3). 13C{1H} NMR (126 MHz, CDCl3) δ 138.9 (C), 138.39 (C), 138.3 (C), 137.0 (C), 134.5 (Ar), 132.1 (Ar), 129.0 (Ar), 128.6 (Ar), 128.51 (Ar), 128.50 (Ar), 128.48 (Ar), 128.45 (Ar), 128.3 (Ar), 128.18 (Ar), 128.17 (Ar), 127.84 (Ar), 127.8 (Ar), 127.7 (Ar), 127.5 (Ar), 127.3 (Ar), 111.8 (CMe2), 108.3(CMe2), 105.2 (C1), 101.4 (C1′), 86.9 (C6′), 82.6 (C3′), 82.3 (CH), 80.4 (CH), 80.2 (CH), 79.4 (C2′), 75.4 (CH2), 74.7 (CH2), 74.3 (C4′), 74.0 (CH), 73.9 (C5′), 73.0 (CH2), 70.4 (CH2), 65.4 (CH2), 26.9 (CH3), 26.6 (CH3), 26.3 (CH3), 25.5 (CH3). HRMS (ESI) m/z: [M + Na]+ calcd for C52H58O11SNa 913.3592; found 913.3564.
Methyl (6R)-6-Phenylthio-2,3,4,6-tetra-O-benzyl-β-d-galactopyranosyl-(1→4)-2,3-O-isopropylidene-α-l-rhamnopyranoside (12)
Compound 12β was obtained (31.3 mg, 77%) as a single isomer from the reaction of donor 7a (38.0 mg, 47.9 μmol) and acceptor (12.5 mg, 57.5 μmol) by following the general procedure GP5 for glycosylation (eluting with 10% ethyl acetate in hexanes).
Colorless syrup. [α]D21 = −26.6 (c = 1.0, CHCl3). 1H NMR (500 MHz, CDCl3) δ 7.56–7.49 (m, 2H, ArH), 7.39 (dd, J = 7.9, 1.7 Hz, 2H, ArH), 7.34–7.17 (m, 21H, ArH), 5.09–5.03 (m, 2H, -CH2, H6′), 4.99 (d, J = 11.5 Hz, 1H, -CH2), 4.92 (d, J = 10.9 Hz, 1H, -CH2), 4.86 (s, 1H, H1), 4.78 (d, J = 7.7 Hz, 1H, H1′), 4.74–4.69 (m, 2H, -CH2), 4.66 (d, J = 11.8 Hz, 1H, -CH2), 4.42 (d, J = 11.5 Hz, 1H, -CH2), 4.26 (t, J = 6.0 Hz, 1H, CH), 4.20 (d, J = 11.0 Hz, 1H, CH2), 4.11 (dd, J = 5.9, 0.8 Hz, 1H, CH), 4.04 (dd, J = 3.1, 1.1 Hz, 1H, H4′), 3.77 (dd, J = 9.7, 7.7 Hz, 1H, H2′), 3.67 (m, 2H, H5), 3.49 (dd, J = 9.7, 2.9 Hz, 1H, H3′), 3.39 (s, 3H, OMe), 3.38–3.31 (m, 1H, CH), 1.54 (s, 3H, CH3), 1.40 (d, J = 5.7 Hz, 3H, CH3), 1.34 (s, 3H, CH3). 13C{1H} NMR (126 MHz, CDCl3) δ 139.2 (C), 138.9 (C), 138.7 (C), 137.1 (C), 133.6 (Ar), 133.5 (Ar), 133.2 (Ar), 128.9 (Ar), 128.5 (Ar), 128.3 (Ar), 128.29 (Ar), 128.25 (Ar), 128.0 (Ar), 127.6 (Ar), 127.57 (Ar), 127.53 (Ar), 127.52 (Ar), 127.47 (Ar), 127.42 (Ar), 127.37 (Ar), 127.34 (Ar), 109.3 (CMe2), 102.9 (C1′), 98.2 (C1), 88.0 (C6′), 82.6 (C3′), 82.4 (CH), 80.2 (CH), 79.8 (CH), 78.4 (CH), 75.9 (CH), 75.2 (CH2), 74.8 (C4′), 74.6 (CH2), 74.0 (CH), 73.2 (CH2), 70.3 (CH2), 64.5 (CH2), 54.9 (OCH3), 28.2 (CH3), 26.2 (CH3), 18.1 (CH3). HRMS (ESI) m/z: [M + Na]+ calcd for C50H56O10SNa 871.3486; found 871.3470.
Methyl (6R)-6-Phenylthio-2,3,4,6-tetra-O-benzyl-α/β-d-galactopyranosyl-(1→4)-2,3,4-tri-O-benzyl-α-d-glucopyranoside (13)
Compounds 13α and 13β were obtained as a mixture of anomers (47.9 mg, 76%, α/β = 1:1.1) from the reaction of donor 7a (50.0 mg, 63.0 μmol) and acceptor (35.1 mg, 76.6 μmol) by following the general procedure GP5 for glycosylation (eluting with 10% ethyl acetate in hexanes, 1:9). Repeated column chromatography gave pure samples of α- and β-isomers for full characterization.
α-Isomer
Colorless syrup. [α]D21 = +18.0 (c = 1.0, CHCl3). 1H NMR (500 MHz, CDCl3) δ 7.59–7.53 (m, 2H, ArH), 7.35–7.15 (m, 38H, ArH), 5.68 (d, J = 3.7 Hz, 1H, H1′), 5.07–4.99 (m, 2H, H6′, CH2), 4.96–4.89 (m, 2H, CH2), 4.82 (d, J = 11.4 Hz, 1H, CH2), 4.67 (d, J = 12.1 Hz, 1H, CH2), 4.65–4.61 (m, 2H, CH2), 4.61–4.55 (m, 3H, H1, CH2), 4.55–4.49 (m, 2H, CH2), 4.45 (d, J = 12.1 Hz, 1H, CH2), 4.37 (d, J = 11.2 Hz, 1H, CH2), 4.17 (d, J = 10.9 Hz, 1H, CH2), 4.07 (dd, J = 2.8, 1.4 Hz, 1H, H4′), 4.04–3.94 (m, 3H, H2′,CH), 3.90–3.81 (m, 2H, H5, CH), 3.76–3.68 (m, 2H, CH, H6b), 3.66 (dd, J = 10.4, 2.7 Hz, 1H, H6a), 3.55 (dd, J = 9.2, 3.6 Hz, 1H, CH), 3.38 (s, 3H, OCH3). 13C{1H} NMR (126 MHz, CDCl3) δ 139.2 (C), 139.0 (C), 138.8 (C), 138.4 (C), 138.2 (C), 138.2 (C), 137.4 (C), 135.6 (C), 132.7 (Ar), 129.0 (Ar), 128.5 (Ar), 128.4 (Ar), 128.37 (Ar), 128.35 (Ar), 128.32 (Ar), 128.31 (Ar), 128.29 (Ar), 128.27 (Ar), 127.99 (Ar), 127.96 (Ar), 127.94 (Ar), 127.7 (Ar), 127.59 (Ar), 127.53 (Ar), 127.4 (Ar), 127.3 (Ar), 127.2 (Ar), 127.1 (Ar), 127.0 (Ar), 97.8 (C1), 97.1 (C1′), 90.4 (C6′), 82.0 (CH), 79.9 (CH), 79.1 (CH), 76.0 (CH), 75.7 (C4′), 74.9 (CH2), 74.3 (CH2), 73.9 (CH), 73.5 (CH2), 73.4 (CH2), 73.1 (CH2), 72.6 (CH), 70.6 (CH2), 70.1 (C6), 69.6 (CH), 55.2 (OCH3). HRMS (ESI) m/z: [M + Na]+ calcd for C68H70O11SNa 1117.4531; found 1117.4503.
β-Isomer
Colorless syrup. [α]D21 = −22.0 (c = 1.0, CHCl3). 1H NMR (500 MHz, CDCl3) δ 7.59–7.55 (m, 2H, ArH), 7.47–7.41 (m, 2H, ArH), 7.34–7.16 (m, 30H, ArH), 7.16–7.06 (m, 4H, ArH), 7.05–6.98 (m, 2H, ArH), 5.17 (d, J = 11.0 Hz, 1H, -CH2), 5.03 (d, J = 11.0 Hz, 1H, -CH2), 4.99 (d, J = 11.3 Hz, 1H, -CH2), 4.84 (d, J = 12.2 Hz, 1H, -CH2), 4.80 (d, J = 8.8 Hz, 1H, H6′), 4.79–4.70 (m, 3H, -CH2), 4.69–4.64 (m, 3H, CH2), 4.57 (m, 2H, H1, -CH2), 4.38 (d, J = 11.3 Hz, 1H, -CH2), 4.33 (d, J = 12.2 Hz, 1H, -CH2), 4.21–4.14 (m, 2H, CH2), 4.07 (dd, J = 10.1, 9.0 Hz, 1H, CH), 4.04 (dd, J = 2.9, 1.1 Hz, 1H, H4′), 3.87 (t, J = 9.4 Hz, 1H, CH), 3.80–3.72 (m, 2H, H2′), 3.61 (dt, J = 10.0, 2.7 Hz, 1H, CH), 3.49 (dd, J = 9.7, 3.7 Hz, 1H, CH), 3.44 (dd, J = 10.8, 2.1 Hz, 1H, CH), 3.37 (s, 3H, OCH3), 3.26–3.14 (m, 2H, H3′, H5′). 13C{1H} NMR (126 MHz, CDCl3) δ 139.8 (C), 139.3 (C), 139.0 (C), 138.7 (C), 138.6 (C), 138.0 (C), 137.3 (C), 134.5 (Ar), 133.6 (Ar), 128.78 (Ar), 128.7 (Ar), 128.6 (Ar), 128.48, (Ar) 128.41 (Ar), 128.38 (Ar), 128.30 (Ar), 128.27 (Ar), 128.24 (Ar), 128.2 (Ar), 127.97 (Ar), 127.94 (Ar), 127.93 (Ar), 127.74 (Ar), 127.7 (Ar), 127.6 (Ar), 127.5 (Ar), 127.4 (Ar), 127.34 (Ar), 127.30 (Ar), 127.2 (Ar), 127.0 (Ar), 102.3 (C1′), 98.8 (C1), 88.8 (C6′), 82.6 (C3′), 80.6 (CH), 80.2 (CH), 78.5 (CH), 75.9 (CH), 75.7 (CH2), 75.2 (CH2), 74.7 (CH2), 74.5 (C4′), 74.4 (C5′), 73.8 (CH2), 73.5 (CH2), 72.8 (CH2), 70.26 (CH2), 70.2 (CH), 67.9 (CH2), 55.4 (OCH3). HRMS (ESI) m/z: [M + Na]+ calcd for C68H70O11SNa 1117.4531; found 1117.4529.
Methyl (6S)-6-Phenylthio-2,3,4,6-tetra-O-benzyl-α/β-d-galactopyranosyl-(1→6)-2,3,4-tri-O-benzyl-α-d-glucopyranoside (14)
Compounds 14α and 14β were obtained as a mixture of anomers (78.3 mg, 81%, α/β = 1:11.9) from the reaction of donor 7b (70.0 mg, 88.2 μmol) and acceptor (49.2 mg, 105.9 μmol) by following the general procedure GP5 for glycosylation (eluting with 10% ethyl acetate in hexanes). Repeated column chromatography gave pure samples of the α- and β-isomers for full characterization.
α-Isomer
Colorless syrup. [α]D20 = +23.2 (c = 2.0, CHCl3). 1H NMR (900 MHz, CDCl3) δ 7.42–7.12 (m, 40H, ArH), 5.14 (m, 2H, -CH2, H6′), 5.05 (d, J = 3.7 Hz, 1H, H1′), 4.95 (d, J = 11.0 Hz, 1H, -CH2), 4.85 (t, J = 11.2 Hz, 2H, -CH2), 4.82–4.67 (m, 6H, -CH2, -CH), 4.64–4.54 (m, 3H, -CH2), 4.49 (d, J = 11.1 Hz, 1H, -CH2), 4.45 (d, J = 3.5 Hz, 1H, H1′), 4.36 (s, 1H, H4′), 4.07 (d, J = 9.9 Hz, 1H, -CH), 3.94 (t, J = 9.5 Hz, 1H, -CH), 3.91 (d, J = 8.9 Hz, 1H, -CH), 3.84–3.81 (m, 1H, -CH), 3.71 (s, 3H), 3.50 (s, 1H, -CH), 3.38 (dd, J = 9.2, 4.0 Hz, 1H, -CH), 3.23 (s, 3H, -OCH3). 13C{1H} NMR (226 MHz, CDCl3) δ 138.9 (C), 138.8 (C), 138.4 (C), 138.2 (C), 137.9 (C), 132.9 (Ar), 128.9 (Ar), 128.4 (Ar), 128.36 (Ar), 128.34 (Ar), 128.3 (Ar), 128.19 (Ar), 128.13 (Ar), 128.0 (Ar), 127.97 (Ar), 127.94 (Ar), 127.9 (Ar), 127.8 (Ar), 127.6 (Ar), 127.58 (Ar), 127.53 (Ar), 127.50 (Ar), 127.37 (Ar), 127.34 (Ar), 97.7 (C1′), 97.6 (C1), 87.5 (C6′), 82.0 (CH), 80.1 (CH), 78.6 (CH), 78.1 (CH), 77.2 (CH), 76.36 (CH), 76.34 (CH), 75.7, 75.0, 74.9, 73.4 (CH2), 73.0 (CH2), 72.5 (CH2), 71.8 (CH2), 70.4 (CH2), 70.1 (CH2), 66.0 (CH2), 54.9 (OCH3). HRMS (ESI) m/z: [M + Na]+ calcd for C68H70O11SNa 1117.4531; found 1117.4532.
β-Isomer
Colorless syrup. [α]D21 = +27.4 (c = 1.0, CHCl3). 1H NMR (500 MHz, CDCl3) δ 7.39–7.15 (m, 38H, ArH), 7.11–7.06 (m, 2H, ArH), 5.14–5.06 (m, 2H, -CH2, H6′), 4.96–4.89 (m, 2H, -CH2), 4.84 (d, J = 11.6 Hz, 1H, -CH2), 4.80–4.69 (m, 5H, -CH2, -CH2), 4.68–4.55 (m, 5H, -CH2, H1), 4.45 (d, J = 11.1 Hz, 1H, -CH2), 4.29 (d, J = 2.8 Hz, 1H, H4′), 4.21–4.12 (m, 2H, H1′, H6b), 3.94 (t, J = 9.3 Hz, 1H, -CH), 3.86 (dd, J = 9.7, 7.7 Hz, 1H, H2′), 3.77 (ddd, J = 10.1, 4.5, 2.1 Hz, 1H, H5), 3.61 (dd, J = 11.0, 4.5 Hz, 1H, H6a), 3.54–3.44 (m, 2H, -CH, H2), 3.35 (dd, J = 9.7, 2.8 Hz, 1H, H3′), 3.33–3.27 (m, 4H, H5′,OCH3). 13C{1H} NMR (126 MHz, CDCl3) δ 139.0 (C), 138.9 (C), 138.8 (C), 138.52 (C), 138.5 (C), 138.3 (C), 137.5 (C), 133.7 (Ar), 131.9 (Ar), 129.0 (Ar), 128.53 (Ar), 128.50 (Ar), 128.41 (Ar), 128.39 (Ar), 128.34 (Ar), 128.31 (Ar), 128.23 (Ar), 128.19 (Ar), 128.03 (Ar), 128.01 (Ar), 127.99 (Ar), 127.97 (Ar), 127.96 (Ar), 127.8 (Ar), 127.78 (Ar), 127.74 (Ar), 127.71 (Ar), 127.57 (Ar), 127.52 (Ar), 127.44 (Ar), 127.41 (Ar), 104.2 (C1′), 98.1 (C1), 86.6 (C6′), 82.5 (C3′), 82.1 (-CH), 79.9 (CH), 79.2 (C2′), 77.9 (CH), 75.9 (C5′), 75.7 (CH2), 75.2 (C4′), 75.1 (CH2), 74.9 (CH2), 74.8 (-CH2), 73.4 (CH2), 73.3 (CH2), 71.0 (CH2), 69.9 (C5), 68.5 (C6), 55.2 (OCH3). HRMS (ESI) m/z: [M + Na]+ calcd for C68H70O11SNa 1117.4531; found 1117.4541.
(6S)-6-Phenylthio-2,3,4,6-tetra-O-benzyl-α/β-d-galactopyranosyl-(1→6)-1,2:3,4-O-diisopropylidene-α-d-galactopyranose (15)
Compounds 15α and 15β were obtained as a mixture of anomers (48.1 mg, 69%, α/β = 1:11.5) from the reaction of donor 7b (62.0 mg, 78.2 μmol) and acceptor (24.4 mg, 93.8 μmol) by following the general procedure GP5 for glycosylation (eluting with 10% ethyl acetate in hexanes, 1:9). The α-isomer was not obtained pure and was characterized in the mixture of anomers by the following diagnostic signals.
α-Isomer
1H NMR (500 MHz, CDCl3) δ 7.42–7.39 (m, 2H, ArH), 7.38–7.31 (m, 6H, ArH), 7.30–7.19 (m, 17H, ArH), 5.48 (d, J = 5.0 Hz, 1H, H1), 5.15–5.10 (m, 2H, H6′, -CH2), 5.07 (d, J = 10.8 Hz, 1H, -CH2), 4.88–4.82 (m, 2H, -CH2), 4.78–4.67 (m, 3H, -CH2), 4.63–4.57 (m, 2H, -CH2), 4.51 (dd, J = 7.9, 2.4 Hz, 1H, -CH), 4.39–4.35 (m, 1H, -CH), 4.27 (dd, J = 5.0, 2.4 Hz, 1H, H2), 4.14 (dd, J = 7.9, 1.9 Hz, 1H, -CH), 4.07 (dd, J = 10.0, 3.7 Hz, 1H, H6b), 3.99 (td, J = 6.9, 1.9 Hz, 1H, H5), 3.94–3.90 (m, 1H, -CH), 3.85 (dd, J = 10.1, 2.8 Hz, 1H, H2′), 3.77 (dd, J = 10.8, 7.1 Hz, 1H, H6a), 3.67 (dd, J = 10.8, 6.6 Hz, 1H, -CH), 1.51 (s, 3H, CH3), 1.37 (s, 3H, CH3), 1.31 (s, 3H, CH3), 1.25 (s, 3H, CH3).
Repeated column chromatography gave a pure sample of the β-isomer for full characterization.
β-Isomer
Colorless syrup. [α]D21 = +23.5 (c = 0.9, CHCl3). 1H NMR (500 MHz, CDCl3) δ 7.48–7.43 (m, 2H, ArH), 7.41–7.18 (m, 23H, ArH), 5.56 (d, J = 5.0 Hz, 1H, H1), 5.15–5.10 (m, 2H, -CH2, H6′), 5.05 (d, J = 11.2 Hz, 1H, -CH2), 4.91 (d, J = 11.4 Hz, 1H, -CH2), 4.86 (d, J = 12.1 Hz, 1H, -CH2), 4.75 (m, 2H, -CH2), 4.65 (m, 2H, -CH2), 4.51 (dd, J = 7.9, 2.4 Hz, 1H, H3), 4.34 (d, J = 7.7 Hz, 1H, H1′), 4.32 (d, J = 2.9 Hz, 1H, H4′), 4.29 (dd, J = 5.0, 2.4 Hz, 1H, H2), 4.18 (dd, J = 7.9, 1.9 Hz, 1H, -CH), 4.14 (dd, J = 10.8, 3.6 Hz, 1H, H6b), 4.09 (ddd, J = 7.4, 3.5, 1.8 Hz, 1H, H5), 3.86 (dd, J = 9.8, 7.6 Hz, 1H, H2′), 3.73 (dd, J = 10.8, 7.4 Hz, 1H, H6a), 3.40–3.34 (m, 2H, H3′, H5′), 1.50 (s, 3H, CH3), 1.41 (s, 3H, CH3), 1.32 (s, 3H, CH3), 1.29 (s, 3H, CH3). 13C{1H} NMR (126 MHz, CDCl3) δ 139.1 (C), 138.9 (C), 138.8 (C), 137.6 (C), 133.8 (Ar), 131.9 (Ar), 129.1 (Ar), 128.7 (Ar), 128.49 (Ar), 128.46 (Ar), 128.3 (Ar), 128.23 (Ar), 128.21 (Ar), 128.1 (Ar), 128.0 (Ar), 127.8 (Ar), 127.7 (Ar), 127.48 (Ar), 127.44 (Ar), 109.3 (CMe2), 108.7 (CMe2), 104.9 (C1′), 96.5 (C1), 86.9 (C6′), 82.0 (C3′), 78.9 (C2′), 75.8 (C5′), 75.3 (C4′), 75.0 (CH2), 74.6 (CH2), 73.5 (CH2), 71.5 (CH2), 71.1 (CH2), 70.8 (CH2), 70.6 (C2), 69.9 (C6), 67.7 (C5), 26.2 (CH3), 26.1 (CH3), 25.2 (CH3), 24.5 (CH3). HRMS (ESI) m/z: [M + Na]+ calcd for C52H58O11SNa 913.3592; found 913.3575.
Adamantyl (6S)-6-Phenylthio-2,3,4,6-tetra-O-benzyl-α-d-galactopyranoside (16)
Compounds 16α and 16β were obtained as a mixture of anomers (48.3 mg, 72%, α/β = 1:4.1) from the reaction of donor 7b (68.0 mg, 85.7 μmol) and adamantanol (15.6 mg, 102.9 μmol) by following the general procedure GP5 for glycosylation (eluting with 10% ethyl acetate in hexanes). Repeated column chromatography gave pure samples of α- and β-isomers for full characterization.
α-Isomer
Colorless syrup. [α]D20 = −6.6 (c = 0.6, CHCl3). 1H NMR (500 MHz, CDCl3) δ 7.46–7.42 (m, 2H, ArH), 7.41–7.38 (m, 2H, ArH), 7.38–7.33 (m, 4H, ArH), 7.32–7.19 (m, 17H, ArH), 5.33 (d, J = 3.8 Hz, 1H, H1), 5.13 (d, J = 10.9 Hz, 1H, CH2), 5.06 (d, J = 9.1 Hz, 1H, H6), 4.95 (d, J = 11.2 Hz, 1H, CH2), 4.86 (d, J = 11.7 Hz, 1H, CH2), 4.76 (d, J = 11.7 Hz, 1H, CH2), 4.72 (d, J = 12.0 Hz, 1H, CH2), 4.68 (d, J = 12.0 Hz, 1H, CH2), 4.61 (d, J = 10.9 Hz, 1H, CH2), 4.48 (d, J = 11.2 Hz, 1H, CH2), 4.42 (d, J = 2.8 Hz, 1H, H4), 4.08–4.00 (m, 2H, H2, H5), 3.85 (dd, J = 10.1, 2.8 Hz, 1H, H3), 2.00 (p, J = 3.3 Hz, 3H, Ada), 1.81–1.74 (m, 3H, Ada), 1.66 (dq, J = 11.7, 2.6 Hz, 3H, Ada), 1.59–1.52 (m, 3H, Ada), 1.51–1.45 (m, 3H, Ada). 13C{1H} NMR (126 MHz, CDCl3) δ 139.18 (C), 139.14 (C), 138.9 (C), 137.9 (C), 133.5 (Ar), 132.7 (Ar), 128.9 (Ar), 128.4 (Ar), 128.3 (Ar), 128.23 (Ar), 128.18 (Ar), 128.07 (Ar), 128.04 (Ar), 127.7 (Ar), 127.6 (Ar), 127.58 (Ar), 127.55 (Ar), 127.50 (Ar), 127.42 (Ar), 127.39 (Ar), 90.6 (C1), 87.9 (C6), 79.7 (C3), 77.3 (-CH2), 76.4 (C2), 76.3 (C4), 74.9 (CH2), 74.5 (CH2), 73.2 (CH2), 72.9 (CH2), 71.5 (CH2), 71.3 (C5), 42.4 (Ada), 36.3 (Ada), 30.7 (Ada). HRMS (ESI) m/z: [M + Na]+ calcd for C50H54O6SNa 805.3533; found 805.3520.
β-Isomer
Colorless syrup. [α]D21 = +21.6 (c = 1.0, CHCl3). 1H NMR (500 MHz, CDCl3) δ 7.40–7.18 (m, 25H, ArH), 5.15–5.08 (m, 2H, H6, CH2), 4.95 (d, J = 11.1 Hz, 1H, CH2), 4.87 (d, J = 11.0 Hz, 1H, CH2), 4.78 (d, J = 12.0 Hz, 1H, CH2), 4.74–4.68 (m, 2H, -CH2), 4.60 (d, J = 10.9 Hz, 1H, -CH2), 4.56 (d, J = 11.0 Hz, 1H, -CH2), 4.51 (d, J = 7.7 Hz, 1H, H1), 4.29 (dd, J = 2.9, 1.0 Hz, 1H, H4), 3.78 (dd, J = 9.8, 7.7 Hz, 1H, H2), 3.37 (dd, J = 9.8, 2.9 Hz, 1H, H3), 3.33 (dd, J = 8.8, 1.0 Hz, 1H, H5), 2.02 (t, J = 3.2 Hz, 3H, Ada), 1.85 (dq, J = 11.5, 2.4 Hz, 3H, Ada), 1.75 (dq, J = 11.6, 2.7 Hz, 3H, Ada), 1.61–1.47 (m, 6H, Ada). 13C{1H} NMR (126 MHz, CDCl3) δ 139.04 (C), 139.0 (C), 138.8 (C), 137.6 (C), 133.5 (Ar), 132.3 (Ar), 129.0 (Ar), 128.44 (Ar), 128.39 (Ar), 128.34 (Ar), 128.30 (Ar), 128.27 (Ar), 128.1 (Ar), 128.0 (Ar), 127.8 (Ar), 127.7 (Ar), 127.65 (Ar), 127.60 (Ar), 127.54 (Ar), 127.51 (Ar), 127.4 (Ar), 96.7 (C1), 87.3 (C6), 82.8 (C3), 79.4 (CH2), 75.9 (C5), 75.5 (C4), 75.16 (CH2), 75.1 (CH2), 74.9 (CH2), 73.5 (CH2), 71.5 (CH2), 42.8 (Ada), 36.3 (Ada), 30.7 (Ada). HRMS (ESI) m/z: [M + Na]+ calcd for C50H54O6SNa 805.3533; found 805.3525.
(6S)-6-Phenylthio-2,3,4,6-tetra-O-benzyl-α-d-galactopyranosyl-(1→3)-1,2:5,6-di-O-isopropylidene-α-d-glucofuranose (17)
Compounds 17α and 17β were obtained as a mixture of anomers (57.2 mg, 85%, α/β = 1:6.3) from the reaction of donor 7b (60.0 mg, 75.6 μmol) and acceptor (23.7 mg, 90.8 μmol) by following the general procedure GP5 for glycosylation (eluting with 10% ethyl acetate in hexanes). Repeated column chromatography gave pure samples of the α- and β-isomers for full characterization.
α-Isomer
Colorless syrup. [α]D23 = +36.3 (c = 1.0, CHCl3). 1H NMR (500 MHz, CDCl3) δ 7.43–7.20 (m, 26H, ArH), 5.20 (d, J = 3.5 Hz, 1H, H1), 5.13 (d, J = 10.9 Hz, 1H, -CH2), 5.08 (d, J = 3.8 Hz, 1H, H1), 5.01 (d, J = 11.6 Hz, 1H, -CH2), 4.96 (d, J = 9.3 Hz, 1H, H6′), 4.87 (d, J = 12.0 Hz, 1H, -CH2), 4.76–4.64 (m, 4H, -CH2), 4.62 (d, J = 11.6 Hz, 1H, -CH2), 4.53 (d, J = 3.5 Hz, 1H, H2), 4.44 (dd, J = 2.8, 1.1 Hz, 1H, H4′), 4.36 (td, J = 6.5, 5.2 Hz, 1H, -CH), 4.12 (dd, J = 6.5, 2.8 Hz, 1H), 4.05–3.97 (m, 3H, -CH2, CH), 3.89 (dd, J = 8.5, 5.3 Hz, 1H, -CH), 3.63 (dd, J = 10.2, 2.8 Hz, 1H, CH2), 3.57 (dd, J = 9.3, 1.1 Hz, 1H, CH), 1.39 (s, 3H, CH3), 1.38 (s, 3H, CH3), 1.19 (s, 3H, CH3), 0.86 (s, 3H, CH3). 13C{1H} NMR (126 MHz, CDCl3) δ 138.85 (C), 138.79 (C), 138.75 (C), 137.3 (C), 134.5 (C), 130.9 (Ar), 129.0 (Ar), 128.57 (Ar), 128.54 (Ar), 128.43 (Ar), 128.40 (Ar), 128.3 (Ar), 128.1 (Ar), 128.0 (Ar), 127.80 (Ar), 127.78 (Ar), 127.66 (Ar), 127.63 (Ar), 127.61 (Ar), 127.5 (Ar), 111.5 (CMe2), 108.8 (CMe2), 105.0 (C1), 99.7 (C1′), 86.2 (C6′), 82.8 (CH), 82.4 (CH), 80.9 (CH), 78.8 (CH), 76.6 (CH), 76.1 (CH), 75.1 (CH2), 73.5 (CH2), 73.2 (CH2), 72.8 (CH), 71.9 (CH2), 71.3 (CH2), 66.5 (C6), 26.9 (CH3), 26.8 (CH3), 25.8 (CH3), 25.3 (CH3). HRMS (ESI) m/z: [M + Na]+ calcd for C52H58O11SNa 913.3592; found 913.3596.
β-Isomer
Colorless syrup. [α]D21 = +26.1 (c = 1.0, CHCl3). 1H NMR (500 MHz, CDCl3) δ 7.38–7.23 (m, 23H, ArH), 7.23–7.18 (m, 2H, ArH), 5.72 (d, J = 3.8 Hz, 1H, H1), 5.10 (d, J = 11.1 Hz, 1H, CH2), 5.05 (d, J = 8.8 Hz, H6′), 4.85 (d, J = 11.5 Hz, 1H, -CH2), 4.79–4.72 (m, 3H, -CH2), 4.71–4.64 (m, 2H, -CH2), 4.61 (d, J = 11.2 Hz, 1H, -CH2), 4.45–4.37 (m, 2H, CH, H2), 4.33 (t, J = 3.7 Hz, 1H, CH), 4.31–4.25 (m, 3H, -CH, H1′), 4.04–3.97 (m, 2H, H6), 3.70 (dd, J = 9.7, 7.7 Hz, 1H, H2′), 3.35 (dd, J = 9.7, 2.8 Hz, 1H, H3′), 3.29 (dd, J = 8.8, 1.2 Hz, 1H, H5′), 1.44 (s, 3H, CH3), 1.36 (s, 3H, CH3), 1.26 (s, 3H, CH3), 1.18 (s, 3H, CH3). 13C{1H} NMR (126 MHz, CDCl3) δ 138.9 (C), 138.5 (C), 138.3 (C), 137.3 (C), 133.8 (Ar), 131.6 (Ar), 129.1 (Ar), 128.5 (Ar), 128.4 (Ar), 128.3 (Ar), 128.27 (Ar), 128.1 (Ar), 127.88 (Ar), 127.83 (Ar), 127.77 (Ar), 127.72 (Ar), 127.6 (Ar), 127.4 (Ar), 111.7 (CMe2), 108.4 (CMe2), 105.2 (C1), 101.7 (C1′), 86.2 (C6′), 82.8 (CH), 82.3 (C3′), 80.40 (CH), 80.3 (CH), 79.3 (C2′), 76.3 (C5′), 75.2(CH2), 75.1 (CH2), 74.8 (CH), 73.7 (CH), 73.3 (CH2), 71.3 (CH2), 65.7 (C6), 26.8 (CH3), 26.6 (CH3), 26.2 (CH3), 25.4 (CH3). HRMS (ESI) m/z: [M + Na]+ calcd for C52H58O11SNa 913.3592; found 913.3589.
Methyl (6S)-6-Phenylthio-2,3,4,6-tetra-O-benzyl-α/β-d-galactopyranosyl-(1→4)-2,3-O-isopropylidene-α-l-rhamnopyranoside (18)
Compounds 18α and 18β were obtained as a mixture of anomers (54.0 mg, 84%, α/β = 1:1.02) from the reaction of donor 7b (60.0 mg, 75.6 μmol) and acceptor (19.8 mg, 90.8 μmol) by following the general procedure GP5 for glycosylation (eluting with 10% ethyl acetate in hexanes). Repeated column chromatography gave pure samples of α- and β-isomers for full characterization.
α-Isomer
Colorless syrup. [α]D21 = +31.0 (c = 1.0, CHCl3). 1H NMR (500 MHz, CDCl3) δ 7.48–7.44 (m, 2H, ArH), 7.38–7.18 (m, 23H, ArH), 5.23 (d, J = 3.7 Hz, 1H, H1′), 5.15 (d, J = 7.4 Hz, 1H, H6′), 5.03 (d, J = 10.8 Hz, 1H, CH2), 4.84 (d, J = 11.5 Hz, 1H, CH2), 4.81–4.77 (m, 3H, CH2, H1), 4.74 (d, J = 11.8 Hz, 1H, CH2), 4.67 (m, 2H), 4.50 (d, J = 11.5 Hz, 1H, CH2), 4.37 (dd, J = 2.8, 1.3 Hz, 1H, H4′), 4.22–4.16 (m, 2H, H5′, H3), 4.13 (dd, J = 10.2, 3.6 Hz, 1H, H2′), 3.95 (d, J = 5.8 Hz, 1H, H2), 3.86 (dd, J = 10.2, 2.7 Hz, 1H, H3′), 3.62 (dq, J = 9.7, 6.3 Hz, 1H, H5), 3.37 (dd, J = 9.8, 7.1 Hz, 1H, H4), 3.33 (s, 3H, OCH3), 1.33 (s, 3H, CH3), 1.29 (d, J = 6.3 Hz, 3H, CH3), 1.13 (s, 3H, CH3). 13C{1H} NMR (126 MHz, CDCl3) δ 139.1 (C), 138.8 (C), 138.7 (C), 137.8 (C), 132.8 (Ar), 128.9 (Ar), 128.4 (Ar), 128.39 (Ar), 128.33 (Ar), 128.09 (Ar), 128.07 (Ar), 128.01 (Ar), 127.7 (Ar), 127.6 (Ar), 127.5 (Ar), 127.4 (Ar), 127.3 (Ar), 127.2 (Ar), 109.0 (CMe2), 98.3 (C1′), 98.1 (C1), 89.7 (C6′), 81.4 (C4), 79.3 (C3′), 77.0 (C3), 76.5 (C2′), 75.7 (C2), 75.4 (C4′), 74.6 (CH2), 73.8 (CH2), 72.9 (CH2), 71.5 (C5′), 71.2, 64.6 (C5), 54.8 (OCH3), 28.2 (CH3), 26.3 (CH3), 17.9 (CH3). HRMS (ESI) m/z: [M + Na]+ calcd for C50H56O10SNa 871.3486; found 871.3478.
β-Isomer
Colorless syrup. [α]D21 = +18.6 (c = 1.0, CHCl3). 1H NMR (500 MHz, CDCl3) δ 7.39–7.21 (m, 23H, ArH), 7.20–7.15 (m, 2H, ArH), 5.13–5.08 (m, 2H, H6′, CH2), 4.93–4.83 (m, 3H, H1, CH2), 4.80 (d, J = 12.1 Hz, 1H, CH2), 4.75 (dd, J = 7.8, 1.1 Hz, 1H, H1′), 4.73–4.67 (m, 3H, CH2), 4.61 (d, J = 11.1 Hz, 1H, CH2), 4.25 (d, J = 3.0 Hz, 1H, H4′), 4.17 (t, J = 6.4 Hz, 1H, H3), 4.04 (d, J = 5.7 Hz, 1H, H2), 3.78–3.70 (m, 2H, H2′, H4), 3.62 (m, 1H, H5), 3.42–3.36 (m, 4H, H3′, OCH3), 3.31–3.26 (m, 1H, H5′), 1.33 (d, J = 6.1 Hz, 3H, CH3), 1.29–1.22 (m, 6H, 2xCH3). 13C{1H} NMR (126 MHz, CDCl3) δ 139.2 (C), 138.8 (C), 137.4 (C), 133.6 (Ar), 132.1 (Ar), 128.9 (Ar), 128.5 (Ar), 128.26 (Ar), 128.24 (Ar), 128.21 (Ar), 128.1 (Ar), 127.9 (Ar), 127.86 (Ar), 127.80 (Ar), 127.73 (Ar), 127.69 (Ar), 127.46 (Ar), 127.41 (Ar), 109.3 (CMe2), 102.2 (C1′), 98.2 (C1), 86.3 (C6′), 82.5 (C3′), 79.7 (C4), 78.5 (C3), 78.1 (C2′), 76.0 (C2), 75.8 (C5′), 75.6 (C4′), 75.0 (CH2), 74.9 (CH2), 73.6 (CH2), 70.7 (CH2), 64.5 (C5), 54.9 (OCH3), 27.7 (CH3), 26.4 (CH3), 18.0 (CH3). HRMS (ESI) m/z: [M + Na]+ calcd for C50H56O10SNa 871.3486; found 871.3484.
Methyl (6S)-6-Phenylthio-2,3,4,6-tetra-O-benzyl-α/β-d-galactopyranosyl-(1→4)-2,3,4-tri-O-benzyl-α-d-glucopyranoside (19)
Compounds 19α and 19β were obtained as a mixture of anomers (47.9 mg, 56%, α/β = 4.2:1) from the reaction of donor 7b (62.0 mg, 78.2 μmol) and acceptor (43.6 mg, 93.8 μmol) by following the general procedure GP5 for glycosylation (eluting with 10% ethyl acetate in hexanes). Repeated chromatography gave a pure sample of the α-isomer for full characterization.
α-Isomer
Colorless syrup. [α]D21 = +35.8 (c = 0.8, CHCl3). 1H NMR (500 MHz, CDCl3) δ 7.44–7.40 (m, 2H, ArH), 7.25 (d, J = 2.1 Hz, 11H, ArH), 5.60 (d, J = 3.8 Hz, 1H, H1′), 5.10–5.03 (m, 2H, CH2, H6′), 4.90–4.84 (m, 2H, CH2), 4.82 (d, J = 11.4 Hz, 1H, CH2), 4.72 (d, J = 11.7 Hz, 1H, CH2), 4.68–4.61 (m, 3H, CH2), 4.59–4.54 (m, 2H, CH2), 4.53–4.49 (m, 2H, H1, CH), 4.47 (d, J = 11.3 Hz, 1H, CH2), 4.37–4.32 (m, 2H, CH, CH2), 4.22 (d, J = 12.0 Hz, 1H, CH2), 4.00 (dd, J = 10.3, 3.8 Hz, 1H, H2′), 3.94 (dd, J = 9.6, 8.1 Hz, 1H, CH), 3.82 (dd, J = 8.7, 1.3 Hz, 1H, H5′), 3.80–3.75 (m, 1H, CH), 3.76–3.70 (m, 2H, CH), 3.53 (dd, J = 10.9, 5.9 Hz, 1H, H6b), 3.43 (dd, J = 9.6, 3.5 Hz, 1H, CH), 3.37 (dd, J = 10.8, 2.1 Hz, 1H, H6a), 3.32 (s, 3H, OCH3). 13C{1H} NMR (126 MHz, CDCl3) δ 139.2 (C), 138.8 (C), 138.7 (C), 138.6 (C), 138.4 (C), 138.2 (C), 137.8 (C), 133.09 (Ar), 133.06 (C), 129.1 (Ar), 128.48 (Ar), 128.47 (Ar), 128.45 (Ar), 128.33 (Ar), 128.29 (Ar), 128.25 (Ar), 128.23 (Ar), 127.96 (Ar), 127.93 (Ar), 127.89 (Ar), 127.81 (Ar), 127.66 (Ar), 127.61 (Ar), 127.58 (Ar), 127.54 (Ar), 127.50 (Ar), 127.4 (Ar), 127.1 (Ar), 127.0 (Ar), 97.6 (C1′), 97.4 (C1), 88.3 (C6′), 81.9 (CH2), 80.0 (CH2), 79.2 (CH2), 76.2 (CH), 75.8 (CH2), 75.0 (CH2), 74.6 (CH2), 74.5 (CH2), 73.4 (CH2), 73.3 (CH2), 73.2 (CH2), 73.1 (CH2), 72.9 (C5′), 71.4 (CH), 70.2 (CH), 69.5 (C6), 55.0 (OCH3). HRMS (ESI) m/z: [M + Na]+ calcd for C68H70O11SNa 1117.4531; found 1117.4504.
The β-isomer was not obtained pure and was characterized in the mixture of anomers by the following diagnostic signals:
1H NMR (500 MHz, CDCl3) δ 4.94 (d, J = 8.7 Hz, 1H, H6′), 4.56 (d, J = 3.8 Hz, 1H, H1′), 3.25 (d, J = 8.7 Hz, 1H, H5′); 13C NMR (126 MHz, CDCl3) δ 139.4 (C), 139.1 (C), 139.0 (C), 138.69 (C), 138.67 (C), 138.2 (C), 137.9 (C), 133.1 (Ar), 132.8 (Ar), 129.0 (Ar), 128.97 (Ar), 128.91 (Ar), 128.49 (Ar), 128.48 (Ar), 128.45 (Ar), 128.43 (Ar), 128.33 (Ar), 128.30 (Ar), 128.26 (Ar), 128.23 (Ar), 128.1 (Ar), 128.09 (Ar), 128.06 (Ar), 127.97 (Ar), 127.94 (Ar), 127.91 (Ar), 127.90 (Ar), 127.7 (Ar), 127.69 (Ar), 127.67 (Ar), 127.64 (Ar), 127.63 (Ar), 127.59 (Ar), 127.56 (Ar), 127.55 (Ar), 127.51 (Ar), 127.4 (Ar), 127.2 (Ar), 127.1 (Ar), 127.07 (Ar), 127.01 (Ar), 102.4 (C1′), 98.7 (C1), 87.3 (C6′), 82.7 (CH), 80.2 (CH), 80.0 (CH), 79.1 (CH), 76.5 (CH2), 75.9 (CH), 75.7 (CH), 75.6 (CH2), 75.2 (CH2), 75.1 (CH2), 73.9 (CH2), 73.3 (CH2), 73.1 (CH2), 72.3 (CH2), 70.2 (CH2), 67.9 (C6), 55.3 (OCH3).
Methyl (6S)-6-Phenylthio-2,3,4,6-tetra-O-benzyl-α,β-d-glucopyranosyl-(1→6)-2,3,4-tri-O-benzyl-α-d-glucopyranoside (20)
Compounds 20α and 20β were obtained as a mixture of anomers (66.2 mg, 80%, α/β = 1:8.7) from the reaction of donor 7c (60.0 mg, 75.0 μmol) and acceptor (42.2 mg, 90.0 μmol) by following the general procedure GP5 for glycosylation (eluting with 10% ethyl acetate in hexanes). Repeated column chromatography gave a pure sample of β-isomer for full characterization.
β-Isomer
Colorless syrup. [α]D20 = +7.1 (c 0.3, CHCl3). 1H NMR (500 MHz, CDCl3) δ 7.48–7.45 (m, 2H, Ar-H), 7.34–7.14 (m, 36H, Ar-H), 7.06 (dd, J = 6.7, 2.9 Hz, 2H, CH2), 5.39 (d, J = 2.0 Hz, 1H, H6′), 4.97 (m, 2H, CH2), 4.89 (d, J = 11.0 Hz, 1H, CH2), 4.83 (d, J = 12.0 Hz, 1H, CH2), 4.78 (d, J = 11.0 Hz, 1H, CH2), 4.76–4.69 (m, 5H, CH2, CH), 4.64 (d, J = 12.0 Hz, 1H, CH2), 4.60 (d, J = 3.6 Hz, 1H, H1), 4.55–4.51 (m, 2H, CH2), 4.38–4.30 (m, 2H, H1′, CH2), 4.23 (dd, J = 10.9, 2.0 Hz, 1H, H5′), 4.00 (t, J = 9.2 Hz, 1H, CH), 3.86 (dd, J = 10.2, 5.0 Hz, 1H, H5), 3.75 (t, J = 9.4 Hz, 1H, CH), 3.71–3.64 (m, 2H, CH), 3.61 (t, J = 8.9 Hz, 1H, H3′), 3.54–3.45 (m, 3H, CH), 3.32 (s, 3H, OCH3). 13C{1H} NMR (126 MHz, CDCl3) δ 139.0 (C), 138.5 (C), 138.46 (C), 138.43 (C), 138.3 (C), 138.2 (C), 137.3 (C), 136.3 (C), 131.6 (Ar), 129.2 (Ar), 128.57 (Ar), 128.51 (Ar), 128.49 (Ar), 128.44 (Ar), 128.40 (Ar), 128.3 (Ar), 128.2 (Ar), 128.08 (Ar), 128.05 (Ar), 128.02 (Ar), 127.9 (Ar), 127.8 (Ar), 127.79 (Ar), 127.75 (Ar), 127.73 (Ar), 127.72 (Ar), 127.69 (Ar), 127.64 (Ar), 127.1 (Ar), 104.6 (C1′), 98.1 (C1), 88.0 (C6′), 84.8 (CH), 82.1 (CH), 82.0 (CH), 80.0 (CH), 79.4 (CH), 78.3 (CH), 78.2 (CH), 77.4 (CH2), 77.1 (CH2), 76.9 (CH2), 75.9 (CH2), 75.7 (CH), 75.0 (CH2), 74.9 (CH2), 74.9 (CH2), 73.4 (CH2), 70.0 (CH), 69.8 (CH2), 68.9, 55.3 (CH3). HRMS (ESI) m/z: [M + Na]+ calcd for C68H70O11NaS 1117.45310; found 1117.4521.
The α-isomer was identified in the mixture by the following diagnostic signals: 1H NMR (500 MHz, CDCl3) δ 5.31 (d, J = 1.7 Hz, 1H, H6′), 5.15 (d, J = 3.5 Hz, 1H, H1′), 3.48–3.42 (m, 1H), 3.38 (s, 3H, OCH3).
(6S)-6-Phenylthio-2,3,4,6-tetra-O-benzyl-α,β-d-glucopyranosyl-(1→6)-1,2:3,4-O-diisopropylidene-α-d-galactopyranose (21)
Compounds 21α and 21β were obtained as a mixture of anomers (57.0 mg, 89%, α/β = 1:4. Integration of H6′ from the donor moiety) from the reaction of donor 7c (57.0 mg, 71.8 μmol) and acceptor (22.4 mg, 86.0 μmol) by following the general procedure GP5 for glycosylation (eluting with 10% ethyl acetate in hexanes). Repeated column chromatography gave a pure sample of β-isomer for full characterization.
Synthesis of 21α and 21β on mmol Scale
A mixture of 1.056 g of donor 7c (1.33 mmol, 1.0 equiv) and 0.416 g of 1,2:3,4-di-O-isopropylidene-α-d-galactopyranose (1.60 mmol, 1.2 equiv) was coevaporated with toluene twice, then taken up in 8.8 mL of anhydrous CH2Cl2 (0.15 M), and stirred for 1 h with 2.6 g of activated 4 Å AWMS (2 g/mmol of the donor) at room temperature under argon before cooling to −78 °C. The reaction mixture was treated with 48 μL of TMSOTf (0.26 mmol, 0.2 equiv) and stirred for 2 h at −78 °C before it was quenched with triethylamine (0.4 mL). The reaction mixture was diluted with dichloromethane (50 mL), filtered through a pad of celite, and washed with 50 mL of saturated aqueous NaHCO3. The organic layer was separated, dried over Na2SO4, filtered, and concentrated under reduced pressure. Purification by flash column chromatography on silica gel eluting with 10% ethyl acetate in hexanes afforded 1.096 g of 21α and 21β in a 92% yield with a 1:3 α/β; 98 mg of 21α (8%) and 602 mg of 21β (51% yield) were isolated after repeated silica gel column chromatography eluting with 10% ethyl acetate in hexanes. The anomeric ratio of the products was determined by integration of the H6′ signals in the1H NMR spectra.
β-Isomer
Colorless syrup. [α]D20 = −16.8 (c 0.6, CHCl3). 1H NMR (500 MHz, CDCl3) δ 7.55–7.49 (m, 2H, Ar-H), 7.46–7.40 (m, 2H, Ar-H), 7.39–7.07 (m, 19H, Ar-H), 7.04–6.99 (m, 2H, Ar-H), 5.57 (d, J = 5.0 Hz, 1H, H1), 5.36 (d, J = 1.5 Hz, 1H, H6′), 5.07 (d, J = 11.2 Hz, 1H, CH2), 4.95 (d, J = 10.8 Hz, 1H, CH2), 4.86 (d, J = 11.9 Hz, 1H, CH2), 4.77–4.67 (m, 3H, CH2), 4.60 (dd, J = 7.9, 2.4 Hz, 1H, H3), 4.52 (d, J = 11.8 Hz, 1H, CH2), 4.47 (d, J = 7.8 Hz, 1H, H1′), 4.31 (dd, J = 5.0, 2.4 Hz, 1H, H2), 4.25 (dd, J = 7.9, 1.5 Hz, 1H, H4), 4.22 (d, J = 10.9 Hz, 1H, CH2), 4.15–4.04 (m, 2H, H5, H6a′), 3.74 (m, 3H, H4′, H5′, H6′b), 3.63 (t, J = 8.5 Hz, 1H, H3′), 3.49 (dd, J = 9.1, 7.8 Hz, 1H, H2′), 1.50 (s, 3H, CH3), 1.47 (s, 3H, CH3), 1.33 (s, 3H, CH3), 1.30 (s, 3H, CH3). 13C{1H} NMR (126 MHz, CDCl3) δ 138.8 (C), 138.6 (C), 138.3 (C), 137.1 (C), 136.3 (C), 131.9 (Ar), 129.2 (Ar), 128.78 (Ar), 128.74 (Ar), 128.5 (Ar), 128.4 (Ar), 128.39 (Ar), 128.36 (Ar), 128.30 (Ar), 128.0 (Ar), 127.9 (Ar), 127.8 (Ar), 127.68 (Ar), 127.66 (Ar), 127.5 (Ar), 127.2 (Ar), 109.4 (CMe2), 108.6 (CMe2), 105.0 (C1′), 96.5 (C1), 88.0 (C6), 84.7 (C3′), 81.5 (C2′), 79.3 (C4′), 78.2 (C5′), 75.8 (CH2), 74.7 (CH2), 74.3 (CH2), 71.5 (C4), 70.9 (CH2), 70.6 (C3), 70.1 (CH2), 69.9 (CH2), 67.6 (CH3), 26.17 (CH3), 26.14 (CH3), 24.5 (CH3). HRMS (ESI) m/z: [M + Na]+ calcd for C52H58O11NaS 913.3592; found 913.3573.
α-Isomer
Colorless syrup. [α]D20 = +2.1 (c 2.0, CHCl3). 1H NMR (500 MHz, CDCl3) δ 7.55–7.50 (m, 2H, Ar-H), 7.41–7.38 (m, 2H, Ar-H), 7.37–7.16 (m, 17H, Ar-H), 7.14–7.10 (m, 2H, Ar-H), 7.05 (dd, J = 6.4, 3.0 Hz, 2H, Ar-H), 5.56 (d, J = 5.1 Hz, 1H, H1), 5.37 (d, J = 1.7 Hz, 1H, H6′), 5.17 (d, J = 3.5 Hz, 1H, H1′), 5.01 (d, J = 10.7 Hz, 1H, CH2), 4.88–4.73 (m, 4H, CH2), 4.69 (d, J = 11.9 Hz, 1H, CH2), 4.62 (dd, J = 7.9, 2.4 Hz, 1H, H3), 4.44–4.37 (m, 2H, H4, CH2), 4.34 (dd, J = 5.1, 2.4 Hz, 1H, H2), 4.24–4.18 (m, 2H, H5′, CH2), 4.11 (ddd, J = 8.0, 6.2, 1.9 Hz, 1H, H5), 4.07–4.02 (m, 2H, H3′, H6), 3.84 (dd, J = 10.4, 7.9 Hz, 1H, H6), 3.72 (t, J = 9.4 Hz, 1H, H4′), 3.64 (dd, J = 9.6, 3.6 Hz, 1H, H2′), 1.54 (s, 3H, CH3), 1.44 (s, 3H, CH3), 1.32 (s, 3H, CH3), 1.30 (s, 3H, CH3). 13C{1H} NMR (126 MHz, CDCl3) δ 138.9 (C), 138.5 (C), 138.4 (C), 136.9 (C), 136.6 (C), 132.0 (Ar), 129.2 (Ar), 128.6 (Ar), 128.51 (Ar), 128.50 (Ar), 128.49 (Ar), 128.42 (Ar), 128.41 (Ar), 128.2 (Ar), 128.0 (Ar), 127.9 (Ar), 127.8 (Ar), 127.74 (Ar), 127.70 (Ar), 127.3 (Ar), 109.4 (C), 108.7 (C), 96.7 (C1′), 96.5 (C1), 88.5 (C6′), 82.1 (C3′), 79.8 (C2′), 78.4 (C4′), 75.9 (CH2), 75.5 (C5′) 74.8 (CH2), 72.2 (CH2), 70.9 (C4), 70.8 (C3), 70.78 (C2), 70.6 (CH2), 65.9 (C6), 65.4 (C5), 26.3 (CH3), 26.2 (CH3), 25.1 (CH3), 24.9 (CH3). HRMS (ESI) m/z: [M + Na]+ calcd for C52H58O11NaS 913.3592; found 913.3584.
Adamantyl (6S)-6-Phenylthio-2,3,4,6-tetra-O-benzyl-α,β-d-glucopyranoside (22)
Compounds 22α and 22β were obtained as a mixture of anomers (31.0 mg, 59%, α/β = 1:7.5) from the reaction of donor 7c (53.0 mg, 66.8 μmol) and acceptor (12.2 mg, 80.0 μmol) by following the general procedure GP5 for glycosylation (eluting with 10% ethyl acetate in hexanes). Repeated column chromatography gave pure samples of α- and β-isomers for full characterization.
α-Isomer
Colorless syrup. [α]D20 = +46.5 (c 0.1, CHCl3). 1H NMR (600 MHz, CDCl3) δ 7.55–7.50 (m, 2H, Ar-H), 7.41–7.15 (m, 18H, Ar-H), 7.11–7.07 (m, 2H, Ar-H), 7.05–7.01 (m, 2H, Ar-H), 5.42 (d, J = 3.5 Hz, 1H, H1′), 5.39 (d, J = 1.6 Hz, 1H, H6′), 5.00 (d, J = 10.7 Hz, 1H, CH2), 4.82–4.75 (m, 3H, CH2), 4.71 (m, 2H, CH2), 4.41 (d, J = 11.9 Hz, 1H, CH2), 4.37 (dd, J = 9.8, 1.6 Hz, 1H, H5′), 4.13 (d, J = 10.9 Hz, 1H, CH2), 4.06 (t, J = 9.8 Hz, 1H, H4′), 3.69 (t, J = 9.8 Hz, 1H, H3′), 3.58 (dd, J = 9.8, 3.5 Hz, 1H, H2′), 2.20–2.17 (m, 3H, CH), 2.02–1.98 (m, 3H, CH2), 1.88 (dd, J = 11.5, 3.0 Hz, 3H, CH2), 1.65 (d, J = 3.0 Hz, 6H, CH2). 13C{1H} NMR (156 MHz, CDCl3) δ 138.8 (C), 138.3 (C), 138.2 (C), 136.9 (C), 136.8 (C), 131.8 (Ar), 129.1 (Ar), 128.6 (Ar), 128.4 (Ar), 128.3 (Ar), 128.2 (Ar), 128.1 (Ar), 128.0 (Ar), 127.9 (Ar), 127.8 (Ar), 127.7 (Ar), 127.6 (Ar), 127.5 (Ar), 127.0 (Ar), 90.0 (C1′), 88.6 (C6′), 81.8 (C4′), 80.0 (C2′), 78.7 (C3′), 75.6 (CH2), 75.1 (C5′), 74.9 (CH2), 74.7 (CH2), 73.0 (CH2), 70.3 (CH2), 42.5 (CH2), 36.3 (CH2), 30.7 (CH). HRMS (ESI) m/z: [M + Na]+ calcd for C50H54O6NaS 805.3533; found 805.3537.
β-Isomer
Colorless syrup. [α]D20 = +5.7 (c 1.0, CHCl3). 1H NMR (500 MHz, CDCl3) δ 7.49 (dt, J = 8.1, 1.2 Hz, 2H, Ar-H), 7.37–7.33 (m, 2H, Ar-H), 7.32–7.11 (m, 19H, Ar-H), 7.05 (m, 2H, Ar-H), 5.43 (d, J = 1.3 Hz, 1H, H6′), 5.02 (d, J = 11.0 Hz, 1H, CH2), 4.90 (d, J = 11.0 Hz, 1H, CH2), 4.83 (d, J = 11.9 Hz, 1H, CH2), 4.77–4.66 (m, 4H, H1′, CH2), 4.56 (d, J = 11.9 Hz, 1H, CH2), 4.32 (d, J = 10.9 Hz, 1H, CH2), 3.78–3.69 (m, 2H, H4′, H5′), 3.63 (t, J = 8.6 Hz, 1H, H3′), 3.48 (dd, J = 8.6, 7.8, 1H, H2′), 2.22–2.16 (m, 3H, CH), 1.99 (dt, J = 11.6, 3.1 Hz, 3H, CH2), 1.91 (dt, J = 11.6, 3.1 Hz, 3H, CH2), 1.66 (d, J = 3.1 Hz, 6H, CH2). 13C{1H} NMR (126 MHz, CDCl3) δ 138.7 (C), 138.6 (C), 138.4 (C), 137.5 (C), 136.9 (C), 131.4 (Ar), 129.1 (Ar), 128.45 (Ar), 128.44 (Ar), 128.36 (Ar), 128.34 (Ar), 128.31 (Ar), 128.0 (Ar), 127.76 (Ar), 127.70 (Ar), 127.68 (Ar), 127.63 (Ar), 127.0 (Ar), 97.2 (C1′), 88.2 (C6′), 85.1 (C3′), 82.3 (C2′), 79.3 (C5′), 78.4 (C4′), 75.8 (CH2), 75.6 (C), 75.0 (CH2), 74.8 (CH2), 69.6 (CH2), 42.9 (CH2), 36.4 (CH2), 30.8 (CH). HRMS (ESI) m/z: [M + Na]+ calcd for C50H54O6NaS 805.3533; found 805.3537.
(6S)-6-Phenylthio-2,3,4,6-tetra-O-benzyl-α,β-d-glucopyranosyl-(1→3)-1,2:5,6-di-O-isopropylidene-α-d-glucofuranose (23)
Compounds 23α and 23β were obtained as a mixture of anomers (125.0 mg, 75%, α/β = 1:2.9) from the reaction of donor 7c (148.0 mg, 186.0 μmol) and acceptor (58.3 mg, 223.0 μmol) by following the general procedure GP5 for glycosylation (eluting with 10% ethyl acetate in hexanes). Repeated column chromatography gave pure samples of α- and β-isomers for full characterization.
α-Isomer
Colorless syrup. [α]D20 = +34.9 (c 0.9, CHCl3).1H NMR (500 MHz, CDCl3) δ 7.53–7.46 (m, 2H, Ar-H), 7.37–7.11 (m, 21H, Ar-H), 7.02–6.97 (m, 2H, Ar-H), 5.87 (d, J = 3.5 Hz, 1H, H1), 5.34–5.30 (m, 2H, H6′, H1′), 4.97–4.91 (m, 2H, CH2), 4.86 (d, J = 11.7 Hz, 1H, CH2), 4.76–4.72 (m, 3H, CH, CH2), 4.68 (d, J = 11.7 Hz, 1H, CH2), 4.47 (td, J = 8.4, 6.1, 5.1 Hz, 1H, H5), 4.42 (d, J = 11.7 Hz, 2H, CH2), 4.31 (d, J = 2.7 Hz, 1H, H3), 4.17 (dd, J = 8.4, 2.7 Hz, 1H, H4), 4.15–4.10 (m, 2H, CH), 4.06–4.04 (m, 2H, CH), 3.92 (t, J = 9.4 Hz, 1H, H4′), 3.68 (t, J = 9.4 Hz, 1H, H3′), 3.58 (dd, J = 9.4, 3.5 Hz, 1H, H2′), 1.49 (s, 3H, CH3), 1.41 (s, 3H, CH3), 1.25 (s, 6H, CH3). 13C{1H} NMR (126 MHz, CDCl3) δ 138.5 (C), 138.2 (C), 138.1 (C), 138.0 (C), 136.7 (C), 136.2 (C), 131.8 (Ar), 129.3 (Ar), 128.9 (Ar), 128.7 (Ar), 128.68 (Ar), 128.65 (Ar), 128.5 (Ar), 128.49 (Ar), 128.47 (Ar), 128.2 (Ar), 128.1 (Ar), 127.86 (Ar), 127.8 (Ar), 127.7 (Ar), 127.6 (Ar), 111.9 (CMe2), 109.1 (CMe2), 105.4 (C1), 98.4 (C1′), 88.5 (C6′), 83.7 (CH), 81.6 (CH), 81.4 (CH), 81.3 (CH), 80.0 (CH), 78.2 (CH), 76.3 (CH), 75.9 (CH2), 75.1 (CH2), 73.2 (CH2), 72.5 (CH), 70.7 (CH2), 67.2 (CH2), 27.1 (CH3), 26.9 (CH3), 26.3 (CH3), 25.6 (CH3). HRMS (ESI) m/z: [M + Na]+ calcd for C52H58O11NaS 913.3592; found 913.3596.
β-Isomer
Colorless syrup. [α]D20 = +6.2 (c 0.7, CHCl3).1H NMR (500 MHz, CDCl3) δ 7.50–7.44 (m, 2H, Ar-H), 7.35–7.18 (m, 21H, Ar-H), 7.13–7.06 (m, 2H, Ar-H), 5.78 (d, J = 3.7 Hz, 1H, H1), 5.40 (d, J = 1.7 Hz, 1H, H6′), 4.89 (d, 11.0 Hz, 1H, CH2), 4.84 (d, J = 11.7 Hz, 1H, CH2), 4.79–4.72 (m, 4H, CH2), 4.59 (d, J = 11.7 Hz, 1H, CH2), 4.48–4.45 (m, 3H, CH), 4.44–4.40 (m, 2H, CH), 4.27 (d, J = 3.2 Hz, 1H, H3), 4.06 (d, J = 6.3 Hz, 2H, CH), 3.80–3.70 (m, 2H, H4′, H5′), 3.63 (t, J = 8.6 Hz, 1H, H3′), 3.38 (dd, J = 8.6, 7.8 Hz, 1H, H2′), 1.50 (s, 3H, CH3), 1.45 (s, 3H, CH3), 1.28 (s, 3H, CH3), 1.25 (s, 3H, CH3). 13C{1H} NMR (126 MHz, CDCl3) δ 138.4 (C), 138.2 (C), 138.1 (C), 137.5 (C), 136.2 (C), 131.3 (Ar), 129.2 (Ar), 128.59 (Ar), 128.51 (Ar), 128.47 (Ar), 128.42 (Ar), 128.2 (Ar), 127.89 (Ar), 127.84 (Ar), 127.80 (Ar), 127.7 (Ar), 127.1 (Ar), 112.0 (CMe2), 108.4 (CMe2), 105.2 (C1), 102.0 (C1′), 87.5 (CH), 84.6 (CH), 82.8 (CH), 82.1 (CH), 80.8 (CH), 80.2 (CH), 79.6 (CH), 78.2 (CH), 77.3 (CH), 77.1 (CH), 76.8 (CH), 75.8 (CH2), 75.0 (CH2), 73.8 (CH), 69.4 (CH2), 65.6 (CH2), 26.6 (CH3), 26.2 (CH3), 25.2 (CH3). HRMS (ESI) m/z: [M + Na]+ calcd for C52H58O11NaS 913.3592; found 913.3596.
Methyl (6S)-6-Phenylthio-2,3,4,6-tetra-O-benzyl-α,β-d-glucopyranosyl-(1→4)-2,3-O-isopropylidene-α-l-rhamnopyranoside (24)
Compounds 24α and 24β were obtained as a mixture of anomers (53.7 mg, 79%, α/β = 1:1.1) from the reaction of donor 7c (63.0 mg, 79.4 μmol) and acceptor (20.8 mg, 95.3 μmol) by following the general procedure GP5 for glycosylation (eluting with 10% ethyl acetate in hexanes). Repeated column chromatography gave pure samples of α- and β-isomers for full characterization.
α-Isomer
Colorless syrup. [α]D20 = +20.6 (c 1.5, CHCl3). 1H NMR (500 MHz, CDCl3) δ 7.54–7.48 (m, 2H, Ar-H), 7.35–7.12 (m, 19H, Ar-H), 7.11–7.06 (m, 2H, Ar-H), 7.05–6.98 (m, 2H, Ar-H), 5.31 (d, J = 1.8 Hz, 1H, H6′), 5.28 (d, J = 3.4 Hz, 1H, H1′), 4.92 (d, J = 10.8 Hz, 1H, CH2), 4.83–4.70 (m, 5H, H1, CH2), 4.65 (d, J = 11.8 Hz, 1H, CH2), 4.48 (t, J = 6.4 Hz, 1H, H4), 4.38 (d, J = 11.8 Hz, 1H, CH2), 4.32 (dd, J = 9.8, 1.8 Hz, 1H, H5′), 4.19 (d, J = 10.8 Hz, 1H, CH2), 4.10 (dd, J = 6.3, 0.8 Hz, 1H, H2), 3.97 (t, J = 9.5 Hz, 1H, H3′), 3.72 (m, 2H, H4′, H5), 3.57 (dd, J = 9.5, 3.4 Hz, 1H, H2′), 3.42 (dd, J = 9.7, 6.3 Hz, 1H, H3), 3.34 (s, 3H, CH3), 1.48 (s, 3H, CH3), 1.35 (d, J = 6.3 Hz, 3H, CH3), 1.28 (s, 3H, CH3). 13C{1H} NMR (126 MHz, CDCl3) δ 138.7 (C), 138.4 (C), 138.3 (C), 137.0 (C), 136.9 (C), 132.1 (Ar), 129.1 (Ar), 128.6 (Ar), 128.54 (Ar), 128.50 (Ar), 128.3 (Ar), 128.1 (Ar), 127.94 (Ar), 127.90 (Ar), 127.7 (Ar), 127.6 (Ar), 127.1 (Ar), 109.1 (CMe2), 98.4 (C1), 97.5 (C1′), 89.5 (C6′), 82.8 (C3), 81.7 (C3′), 80.5 (C2′), 78.7 (CH), 77.0 (CH), 75.7 (CH2), 75.6 (CH), 75.5 (CH), 74.8 (CH2), 73.5 (CH2), 70.5 (CH2), 64.4 (CH), 54.8 (OCH3), 28.1 (CH3), 26.1 (CH3), 18.1 (CH3). HRMS (ESI) m/z: [M + Na]+ calcd for C50H56O10NaS 871.3486; found 871.3477.
β-Isomer
Colorless syrup. [α]D20 = −27.6 (c 1.3, CHCl3). 1H NMR (500 MHz, CDCl3) δ 7.47–7.43 (m, 2H, Ar-H), 7.40–7.36 (m, 2H), 7.35–7.15 (m, 19H, Ar-H), 7.13–7.09 (m, 2H), 5.48 (d, J = 2.0 Hz, 1H, H6′), 5.01–4.86 (m, 4H, CH, CH2), 4.85–4.65 (m, 6H, CH2), 4.48 (d, J = 10.9 Hz, 1H, CH2), 4.24 (dd, J = 7.3, 5.6 Hz, 1H, H3), 4.11 (d, J = 5.6 Hz, 1H, H2), 3.82–3.62 (m, 5H, CH), 3.44 (t, J = 8.5 Hz, 1H), 3.40 (s, 3H, OCH3), 1.59 (s, 3H, CH3), 1.42 (d, J = 6.2 Hz, 3H, CH3), 1.35 (s, 3H, CH3). 13C{1H} NMR (126 MHz, CDCl3) δ 138.7 (C), 138.6 (C), 138.4 (C), 137.8 (C), 136.7 (C), 130.9 (Ar), 129.1 (Ar), 128.44 (Ar), 128.41 (Ar), 128.3 (Ar), 128.1 (Ar), 127.9 (Ar), 127.8 (Ar), 127.7 (Ar), 127.6 (Ar), 126.8 (Ar), 109.6 (CMe2), 102.9 (C1′), 98.1 (C1), 87.8 (CH), 84.8 (CH), 82.4 (CH), 79.3 (CH), 79.2 (CH), 78.5 (CH), 78.4 (CH), 76.0 (CH), 75.8 (CH2), 74.9 (CH2), 74.8 (CH2), 68.9 (CH2), 64.4 (CH), 54.9 (CH3), 28.2 (CH3), 26.5 (CH3), 17.8 (CH3). HRMS (ESI) m/z: [M + Na]+ calcd for C50H56O10NaS 871.3486; found 871.3477.
Methyl (6S)-6-Phenylthio-2,3,4,6-tetra-O-benzyl-α,β-d-glucopyranosyl-(1→4)-2,3,6-tri-O-benzyl-α-d-glucopyranoside (25)
Compounds 25α and 25β were obtained as a mixture of anomers (34.0 mg, 80%, α/β = 1.3:1) from the reaction of donor 7c (30.5 mg, 38.4 μmol) and acceptor (21.4 mg, 46.1 μmol) by following the general procedure GP5 for glycosylation (eluting with 10% ethyl acetate in hexanes). Repeated column chromatography gave a pure sample of α-isomer for full characterization.
α-Isomer
Colorless syrup. [α]D20 = +29.5 (c 0.6, CHCl3). 1H NMR (500 MHz, CDCl3) δ 7.46–7.41 (m, 2H, Ar-H), 7.32–7.12 (m, 40H, Ar-H), 7.11–7.07 (m, 2H, Ar-H), 7.04–7.00 (m, 2H, Ar-H), 5.65 (d, J = 3.3 Hz, 1H, H1′), 5.34 (d, J = 1.7 Hz, 1H, H6′), 4.97 (d, J = 11.5 Hz, 1H, CH2), 4.86 (d, J = 11.5 Hz, 1H, CH2), 4.82 (d, J = 10.7 Hz, 1H, CH2), 4.78 (d, J = 11.8 Hz, 1H, CH2), 4.74 (d, J = 11.0 Hz, 1H, CH2), 4.70–4.65 (m, 2H, CH2), 4.60–4.57 (m, 2H, CH), 4.59–4.51 (m, 3H, CH), 4.45–4.41 (m, 2H, CH2), 4.17 (d, J = 11.05 Hz, 1H, CH2) 4.13 (dd, J = 9.50, 2.0 Hz, 1H, CH), 4.06 (t, J = 8.7 Hz, 1H, CH), 3.96–3.82 (m, 5H, CH, CH2), 3.63 (t, J = 9.4 Hz, 1H, H3′), 3.58 (dd, J = 9.4, 3.3 Hz, 1H, H2′), 3.45 (dd, J = 9.4, 3.2 Hz, 1H, H2), 3.37 (s, 3H, CH3). 13C{1H} NMR (126 MHz, CDCl3) δ 139.2 (C), 138.6 (C), 138.5 (C), 138.3 (C), 138.2 (C), 138.0 (C), 136.9 (C), 136.7 (C), 131.2 (Ar), 129.2 (Ar), 128.7 (Ar), 128.5 (Ar), 128.46 (Ar), 128.42 (Ar), 128.40 (Ar), 128.33 (Ar), 128.29 (Ar), 128.24 (Ar), 128.0 (Ar), 127.9 (Ar), 127.7 (Ar), 127.6 (Ar), 127.4 (Ar), 127.09 (Ar), 127.05 (Ar), 126.9 (Ar), 97.7 (C1), 96.9 (C1′), 88.0 (C6′), 81.9 (CH), 81.8 (CH), 79.9 (CH), 79.5 (CH), 78.4 (CH), 76.5 (CH), 75.7 (CH2), 74.9 (CH2), 74.8 (CH2), 74.5 (CH), 73.4 (CH2), 73.3 (CH2), 73.1 (CH2), 70.1 (CH2), 69.7 (CH), 55.1 (CH3). HRMS (ESI) m/z: [M + Na]+ calcd for C68H70O11NaS 1117.4531; found 1117.4506.
The β-isomer was not obtained pure and was characterized in the mixture of anomers by the following diagnostic signals.
β-Isomer
1H NMR (500 MHz, CDCl3) δ 5.35 (d, J = 1.7 Hz, 1H; H6′), 4.46 (d, J = 7.3 Hz, 1H; H1′), 3.38 (s, 3H; OCH3).
(6R)-6-Phenylthio-2,3,4,6-tetra-O-benzyl-α,β-d-glucopyranosyl-(1→6)-1,2:3,4-O-diisopropylidene-α-d-galactopyranose (26)
Compounds 26α and 26β were obtained as a mixture of anomers (32.0 mg, 73%, α/β = 1:1.3) from the reaction of donor 7d (38.5 mg, 48.0 μmol) and acceptor (15.2 mg, 58.0 μmol) by following the general procedure GP5 for glycosylation (eluting with 10% ethyl acetate in hexanes). Repeated column chromatography gave pure samples of α- and β-isomers for full characterization.
α-Isomer
Colorless syrup. [α]D20 = +3.2 (c 0.6, CHCl3). 1H NMR (500 MHz, CDCl3) δ 7.39–7.11 (m, 23H), 7.01 (m, 2H), 5.52 (d, J = 5.1 Hz, 1H, H1), 5.36 (d, J = 1.5 Hz, 1H, H6′), 5.08 (d, J = 3.5 Hz, 1H, H1′), 5.00 (d, J = 10.9 Hz, 1H, CH2), 4.86 (d, J = 11.0 Hz, 1H, CH2), 4.76 (m, 2H, CH2, CH), 4.68–4.62 (m, 2H, CH), 4.57 (dd, J = 8.0, 2.4 Hz, 1H, H5), 4.39 (d, J = 11.2 Hz, 1H, CH2), 4.34 (dd, J = 8.0, 2.0 Hz, 1H, H4), 4.31–4.27 (m, 2H), 4.07 (m, 2H, H3,), 3.90 (dd, J = 10.4, 6.4 Hz, 1H), 3.82 (d, J = 9.3 Hz, 1H, H3′), 3.78 (dd, J = 7.4, 3.0 Hz, 1H, H2), 3.61 (dd, J = 9.6, 3.5 Hz, 1H, H2′), 1.54 (s, 3H, CH3), 1.42 (s, 3H, CH3), 1.32 (s, 3H, CH3), 1.29 (s, 3H, CH3). 13C{1H} NMR (126 MHz, CDCl3) δ 138.8 (C), 138.4 (C), 138.3 (C), 137.4 (C), 135.9 (C), 131.8 (Ar), 129.1 (Ar), 128.5 (Ar), 128.47 (Ar), 128.41 (Ar), 128.39 (Ar), 128.03 (Ar), 128.00 (Ar), 127.83 (Ar), 127.79 (Ar), 127.6 (Ar), 127.1 (Ar), 109.3 (CMe2), 108.7 (CMe2), 96.8 (C1′), 96.4 (C1), 89.3 (CH), 81.8 (CH), 80.0 (CH), 79.1 (CH), 75.6 (CH2), 74.8 (CH), 74.0 (CH2), 72.4 (CH2), 70.9 (CH), 70.8 (CH), 70.7 (CH), 70.1 (CH), 66.3 (CH), 65.6 (CH2), 26.3 (CH3), 26.2 (CH3), 25.0 (CH3), 24.8 (CH3). HRMS (ESI) m/z: [M + Na]+ calcd for C52H58O11NaS 913.3592; found 913.3573.
β-Isomer
Colorless syrup. [α]D20 = −17.5 (c 0.3, CHCl3). 1H NMR (500 MHz, CDCl3) δ 7.44–7.39 (m, 4H, Ar-H), 7.32–7.15 (m, 19H, Ar-H), 7.02–6.98 (m, 2H, Ar-H), 5.56 (d, J = 5.0 Hz, 1H, H1), 5.40 (d, J = 1.3 Hz, 1H, H6′), 5.05 (d, J = 11.2 Hz, 1H, CH2), 4.96 (d, J = 11.0 Hz, 1H, CH2), 4.84–4.77 (m, 3H, CH2), 4.75–4.69 (m, 2H, CH2), 4.57 (dd, J = 7.9, 2.4 Hz, 1H, H4), 4.51 (d, J = 7.7 Hz, 1H, H1′), 4.41 (d, J = 10.9 Hz, 1H, CH2), 4.30 (dd, J = 5.0, 2.4 Hz, 1H, H2), 4.24 (dd, J = 7.9, 1.9 Hz, 1H, H5), 4.13 (dd, J = 10.7, 3.5 Hz, 1H, H6a), 4.08 (dt, J = 9.2, 2.5 Hz, 1H, H3), 3.87–3.79 (m, 2H, H4′, H5′), 3.73 (dd, J = 10.7, 7.5 Hz, 1H, H6b), 3.68 (t, J = 9.1 Hz, 1H, H3′), 3.49 (dd, J = 9.1, 7.7 Hz, 1H, H2′), 1.51 (s, 3H, CH3), 1.42 (s, 3H, CH3), 1.30 (m, 6H, CH3). 13C{1H} NMR (126 MHz, CDCl3) δ 138.76 (C), 138.7 (C), 138.1 (C), 137.7 (C), 136.0 (C), 131.5 (Ar), 129.1 (Ar), 128.7 (Ar), 128.44 (Ar), 128.41 (Ar), 128.38 (Ar), 128.3 (Ar), 128.0 (Ar), 127.89 (Ar), 127.8 (Ar), 127.65 (Ar), 127.6 (Ar), 127.0 (Ar), 109.4 (C), 108.7 (C), 104.2 (C1′), 96.5 (C1), 88.7 (C6′), 84.5 (CH), 81.7 (CH), 78.8 (CH), 78.4 (CH), 77.35 (CH), 77.30 (CH), 77.1 (CH), 76.8 (CH), 75.6 (CH2), 75.0 (CH2), 74.4 (CH), 71.5 (CH), 70.9 (CH), 70.6 (CH2), 69.8 (CH2), 69.6 (CH2), 67.6 (CH), 26.2 (CH3), 26.1 (CH3), 25.1 (CH3), 24.5 (CH3). HRMS (ESI) m/z: [M + Na]+ calcd for C52H58O11NaS 913.3592; found 913.3573.
Adamantyl (6R)-6-Phenylthio-2,3,4,6-tetra-O-benzyl-α,β-d-glucopyranoside (27)
Compounds 27α and 27β were obtained as a mixture of anomers (63.0 mg, 88%, α/β = 4.8:1) from the reaction of donor 7d (72.0 mg, 90.7 μmol) and acceptor (16.6 mg, 109.0 μmol) by following the general procedure GP5 for glycosylation (eluting with 10% ethyl acetate in hexanes). Repeated column chromatography gave a pure sample of α-isomer for full characterization.
α-Isomer
Colorless syrup. [α]D20 = +46.2 (c 1.1, CHCl3). 1H NMR (500 MHz, CDCl3) δ 7.46–7.14 (m, 24H, Ar-H), 7.08 (m, 2H, Ar-H), 5.33 (m, 2H, CH, H1′, H6′), 5.02 (d, J = 10.8 Hz, 1H, CH2), 4.90 (d, J = 11.0 Hz, 1H, CH2), 4.84–4.75 (m, 2H, CH2), 4.71–4.67 (m, 2H, CH2), 4.55–4.45 (m, 2H, H5′, CH2), 4.39 (d, J = 10.8 Hz, 1H, CH2), 4.10 (t, J = 9.5 Hz, 1H, H3′), 3.83 (dd, J = 9.5, 8.7 Hz, 1H, H4′), 3.58 (dd, J = 9.5, 3.6 Hz, 1H, H2′), 2.08 (t, J = 3.2 Hz, 3H, CH), 1.89 (m, 3H, CH2), 1.83 (m, 3H, CH2), 1.64–1.54 (m, 6H, CH2). 13C{1H} NMR (126 MHz, CDCl3) δ 138.9 (C), 138.4 (C), 138.3 (C), 137.6 (C), 136.1 (C), 131.8 (Ar), 129.1 (Ar), 128.5 (Ar), 128.4 (Ar), 128.3 (Ar), 128.2 (Ar), 128.0 (Ar), 127.9 (Ar), 127.8 (Ar), 127.7 (Ar), 127.68 (Ar), 127.62 (Ar), 127.5 (Ar), 90.4 (C1′), 89.9 (C6′), 82.0 (C3′), 80.4 (C2′), 79.5 (C4′), 75.5 (CH2), 75.0 (CH2), 74.9 (CH2), 73.3 (CH2), 73.0 (C5′), 71.0 (CH2), 42.5 (CH2), 36.3 (CH2), 30.7 (CH). HRMS (ESI) m/z: [M + Na]+ calcd for C50H54O6NaS 805.3533; found 805.3519.
The β-isomer was not obtained pure and was characterized in the mixture of anomers by the following diagnostic signals.
β-Isomer
1H NMR (500 MHz, CDCl3) δ 5.40 (d, J = 1.9 Hz, 1H; H6′), 3.71 (m, 1H, H3′), 3.50 (dd, J = 9.23, 7.61 Hz, 1H, H2′).
Methyl (6R)-6-Phenylthio-2,3,4,6-tetra-O-benzyl-α,β-d-glucopyranosyl-(1→4)-2,3-O-isopropylidene-α-l-rhamnopyranoside (28)
Compounds 28α and 28β were obtained as a mixture of anomers (27.3 mg, 68%, α/β = 2:1) from the reaction of donor 7d (37.0 mg, 46.7 μmol) and acceptor (12.2 mg, 56.0 μmol) by following the general procedure GP5 for glycosylation (eluting with 10% ethyl acetate in hexanes). Repeated column chromatography gave a pure sample of α-isomer for full characterization.
α-Isomer
Colorless syrup. [α]D20 = +7.2 (c 0.6, CHCl3) 1H NMR (500 MHz, CDCl3) δ 7.44–7.38 (m, 2H, Ar-H), 7.37–7.11 (m, 23H, Ar-H), 5.32 (d, J = 1.4 Hz, 1H, H6′), 5.12 (d, J = 3.4 Hz, 1H, H1′), 4.95 (d, J = 10.8 Hz, 1H, CH2), 4.85 (d, J = 10.8 Hz, 1H, CH2), 4.83–4.79 (m, 3H, H1, CH2), 4.77 (d, J = 11.6 Hz, 1H, CH2), 4.69 (d, J = 11.6 Hz, 1H, CH2), 4.64 (d, J = 12.1 Hz, 1H, CH2), 4.58 (d, J = 10.8 Hz, 1H, CH2), 4.51 (dd, J = 9.9, 1.4 Hz, 1H, H5′), 4.21 (dd, J = 7.0, 5.7 Hz, 1H, H3), 4.06 (t, J = 9.5 Hz, 1H, H3′), 3.99 (d, J = 5.7 Hz, 1H, H2), 3.89 (dd, J = 9.9, 9.5 Hz, 1H, H4′), 3.75–3.68 (m, 1H, H5), 3.57 (dd, J = 9.5, 3.4 Hz, 1H, H2′), 3.38 (dd, J = 9.9, 7.0 Hz, 1H, H4), 3.34 (s, 3H, OCH3), 1.41 (s, 3H, CH3), 1.33 (d, J = 6.3 Hz, 3H, CH3), 1.16 (s, 3H, CH3). 13C{1H} NMR (126 MHz, CDCl3) δ 138.7 (C), 138.3 (C), 138.1 (C), 137.8 (C), 136.8 (C), 131.5 (Ar), 129.0 (Ar), 128.53 (Ar), 128.50 (Ar), 128.3 (Ar), 128.0 (Ar), 127.95 (Ar), 127.93 (Ar), 127.8 (Ar), 127.7 (Ar), 127.6 (Ar), 127.56 (Ar), 126.9 (Ar), 109.2 (C), 98.1 (C1′), 98.0 (C1), 89.8 (C6′), 82.3 (C4), 81.8 (C3′), 80.6 (C2′), 78.9 (C4′), 77.2 (C3), 75.80 (CH2), 75.6 (CH2), 75.0 (CH2), 74.9 (CH), 73.9 (CH2), 70.6 (CH2), 64.6 (CH5), 54.7 (CH3), 28.0 (CH3), 26.2 (CH3), 17.8 (CH3). HRMS (ESI) m/z: [M + Na]+ calcd for C50H56O10NaS 871.3486; found 871.3455.
The β-isomer was not obtained pure and was characterized in the mixture of anomers by the following diagnostic signals.
β-Isomer
1H NMR (500 MHz, CDCl3) δ 5.35 (s, 1H, H6′), 4.99 (d, J = 8.3 Hz, 1H, H1′), 3.38 (s, 3H, CH3).
General Procedure for Desulfurization
A suspension of glycoside (1.0 equiv) in ethanol (0.1 M) was treated with Raney nickel (1 g/mmol) that had been previously washed with ethanol. The reaction mixture was stirred overnight under 1 atm of hydrogen at room temperature. The reaction mixture was diluted with ethanol and filtered through celite. The filter cake was washed with additional ethanol, and the combined filtrate was dried over Na2SO4, filtered through cotton, and concentrated under vacuum to afford a crude residue, which was purified by silica gel chromatography (0→10% CH3OH in CH2Cl2) to give the desired deprotected glycoside.
6-O-(β-d-Galactopyranosyl)-1,2:3,4-O-diisopropylidene-α-d-galactopyranose (29)
Obtained from the deprotection of compound 9β (15 mg) using the general procedure for desulfurization eluting from silica gel with 10% MeOH/ in CH2Cl2 to give 5.3 mg for a 75% yield, and spectral data consistent with the literature.50,51
Colorless syrup. [α]D23 = −36.5 (c = 0.3, MeOH) 1H NMR (500 MHz, CD3OD) δ 5.49 (d, J = 5.0 Hz, 1H), 4.60 (dd, J = 7.9, 2.4 Hz, 1H), 4.35 (dd, J = 5.0, 2.4 Hz, 1H), 4.28 (dd, J = 7.9, 1.6 Hz, 1H), 4.21 (d, J = 7.6 Hz, 1H), 4.07–3.98 (m, 2H), 3.80 (dd, J = 3.3, 1.1 Hz, 1H), 3.75–3.59 (m, 3H), 3.54–3.47 (m, 2H), 3.44 (dd, J = 9.7, 3.4 Hz, 1H), 1.49 (s, 3H), 1.37 (s, 3H), 1.32 (s, 3H), 1.30 (s, 3H). 13C {1H} NMR (126 MHz, CD3OD) δ 109.1 (C), 108.7 (C), 104.0 (CH), 96.4 (CH), 75.4 (CH), 73.4 (CH), 71.1 (CH), 70.65 (CH), 70.61 (CH), 68.9 (CH), 68.4 (CH), 67.6 (CH), 61.1 (CH2), 24.9 (CH2), 23.7 (CH3), 23.1 (CH3). HRMS (ESI) m/z: [M + Na]+ calcd for C18H30O11Na 445.1680; found 445.1674.
Adamantyl β-d-Galactopyranoside (30)
Obtained from the deprotection of compound 10β (9.0 mg) using general procedure for desulfurization eluting from silica gel with 10% MeOH in CH2Cl2 to give 2.7 mg for a 76% yield.
Colorless syrup. [α]D23 = +1.4 (c = 0.3, MeOH). 1H NMR (500 MHz, MeOD-d4) δ 4.52–4.43 (m, 1H, H1), 3.82–3.77 (m, 1H, H4), 3.72–3.61 (m, 2H, H6), 3.49–3.39 (m, 3H, H2, H3, H5), 2.10 (m, 3H, Ada), 1.93–1.87 (m, 3H, Ada), 1.83–1.74 (m, 3H, Ada), 1.70–1.59 (m, 6H, Ada). 13C {1H} NMR (126 MHz, MeOD-d4) δ 96.41 (C1), 74.91 (CH), 74.55 (CH), 73.76 (CH), 71.24 (CH), 68.84 (C4), 61.02 (C6), 42.31 (Ada), 36.07 (Ada), 30.87 (Ada). HRMS (ESI) m/z: [M + Na]+ calcd for C16H26O6Na 337.1621; found 337.1613.
Methyl 4-O-(β-d-Galactopyranosyl)-2,3-O-isopropylidene-α-l-rhamnopyranoside (31)
Obtained from the deprotection of compound 12β (12 mg) using the general procedure for desulfurization eluting from silica gel with 10% MeOH in CH2Cl2 to give 5.0 mg for a 93% yield.
Colorless syrup. [α]D23 = −27.6 (c = 0.7, MeOH). 1H NMR (500 MHz, MeOD-d4) δ 4.77 (s, 1H, H1), 4.73–4.65 (m, 1H, H1′), 4.23 (dd, J = 7.0, 5.7 Hz, 1H, CH), 4.07 (dd, J = 5.6, 0.8 Hz, 1H, CH), 3.84–3.80 (m, 1H, H4′), 3.75–3.57 (m, 4H, CH2, CH), 3.45–3.39 (m, 3H, H2′), 3.33 (s, 3H, OCH3), 1.47 (s, 3H, CH3), 1.30 (s, 3H, CH3), 1.24 (d, J = 6.0 Hz, 3H, CH3). 13C {1H} NMR (126 MHz, MeOD-d4) δ 109.0 (CMe2), 101.6 (C1′), 98.0 (C1), 78.1 (CH), 77.9 (CH), 75.9 (CH), 75.3 (CH), 73.7 (CH), 71.5 (CH), 68.8 (CH), 64.0 (CH), 60.8 (C6′), 53.8 (OCH3), 27.0 (CH3), 26.9 (CH3), 25.25 (CH3), 16.7 (CH3). HRMS (ESI) m/z: [M + Na]+ calcd for C16H28O10Na 403.1574; found 403.1567.
3-O-(β-d-Galactopyranosyl)-1,2:5,6-di-O-isopropylidene-α-d-glucofuranose (32)
Obtained from the deprotection of compound 11β (15 mg) using the general procedure for desulfurization eluting from silica gel with 10% MeOH in CH2Cl2 to give 6.5 mg for a 91% yield.
Colorless syrup. [α]D23 = −1.8 (c = 0.8, MeOH). 1H NMR (500 MHz, MeOD-d4) δ 5.91 (d, J = 3.7 Hz, 1H, H1), 4.68 (d, J = 3.7 Hz, 1H, H2), 4.46 (td, J = 6.3, 4.7 Hz, 1H, CH), 4.36 (d, J = 3.2 Hz, 1H, CH), 4.34–4.29 (m, 2H, H1′), 4.09 (dd, J = 8.5, 6.6 Hz, 1H, CH2), 4.02 (dd, J = 8.6, 6.1 Hz, 1H, CH2), 3.84–3.75 (m, 2H, H4′, H6b), 3.70 (dd, J = 11.4, 4.9 Hz, 1H, H6a), 3.56–3.49 (m, 1H, CH), 3.49–3.44 (m, 2H, CH), 1.46 (s, 3H, CH3), 1.40 (s, 3H, CH3), 1.33 (s, 3H, CH3), 1.31 (s, 3H, CH3); 13C {1H} NMR (126 MHz, CD3OD) δ 111.68 (CMe2), 108.31 (CMe2), 105.24 (C1), 102.32 (C1′), 83.13 (CH), 80.36 (CH), 80.12 (CH), 75.66 (CH), 73.71 (CH), 73.59 (CH), 70.67 (CH), 68.93 (CH), 65.45 (CH2), 61.25 (CH2), 25.77 (CH3), 25.41 (CH3), 25.11 (CH3), 24.08 (CH3). HRMS (ESI) m/z: [M + Na]+ calcd for C18H30O11Na 445.1680; found 445.1673.
Methyl α-d-Galactopyranosyl-(l-4)-α-d-glucopyranoside (33)
Obtained from the deprotection of compound 13α (15 mg) using the general procedure for desulfurization. The crude residue obtained was triturated with CHCl3 (3 × 2 mL), filtered, and concentrated to afford pure product; yield (4.6 mg, 94%), and spectral data consistent with the reported literature.52
Colorless syrup. [α]D23 = +30.7 (c = 0.4, MeOH). 1H NMR (600 MHz, D2O) δ 5.33 (s, 1H, H1), 4.74 (d, J = 3.9 Hz, 1H, H1′), 3.95–3.73 (m, 7H, CH, CH2), 3.70-3.66 (m, 3H, CH, CH2), 3.59–3.49 (m, 2H, CH), 3.35 (s, 3H, OCH3). 13C{1H} NMR (151 MHz, D2O) δ 99.9 (C1), 99.0 (C1′), 77.0 (CH), 73.5 (CH), 71.7 (CH), 71.0 (CH), 70.1 (CH), 69.3 (CH), 69.1 (CH), 68.6 (CH), 61.1 (CH2), 60.5 (CH2), 55.0 (OCH3). HRMS (ESI) m/z: [M + Na]+ calcd for C13H24O11Na 379.1210; found 379.1206.
Methyl β-d-Galactopyranosyl-(l-4)-α-d-glucopyranoside (34)
Obtained from the deprotection of compound 13β (16 mg) using the general procedure for desulfurization. The crude residue obtained was triturated with CHCl3 (3 × 2 mL), filtered, and concentrated to afford pure product; yield (4.6 mg, 89%).53,54
Colorless syrup. [α]D23 = +31.6 (c = 0.3, MeOH). 1H NMR (500 MHz, D2O) δ 4.84 (d, J = 3.8 Hz, 1H, H1), 4.47 (d, J = 7.8 Hz, 1H, H1′), 3.99–3.93 (m, 2H, CH), 3.90–3.73 (m, 6H, CH), 3.69 (dd, J = 9.8, 3.5 Hz, 2H, CH, CH2), 3.66–3.62 (m, 1H, CH), 3.57 (dd, J = 10.0, 7.8 Hz, 1H, CH2), 3.45 (s, 3H, OCH3). 13C{1H} NMR (126 MHz, D2O) δ 102.9 (C1′), 99.1 (C1), 78.46 (CH), 75.44 (CH), 72.63 (CH), 71.82 (CH), 71.05 (CH), 70.99 (CH), 70.34 (CH), 68.65 (CH), 61.12 (CH2), 59.99 (CH2), 55.18 (OMe). HRMS (ESI) m/z: [M + Na]+ calcd for C13H24O11Na 379.1210; found 379.1201.
Methyl 4-O-(α-d-Galactopyranosyl)-2,3-O-isopropylidene-α-l-rhamnopyranoside (35)
Obtained from the deprotection of compound 18α (14 mg) using the general procedure for desulfurization eluting from silica gel with 10% MeOH in CH2Cl2 to give 5.9 mg for a 94% yield.
Colorless syrup. [α]D23 = +46.6 (c = 0.6, MeOH). 1H NMR (500 MHz, CD3OD) δ 4.91 (d, J = 3.8 Hz, 1H, H1′), 4.77 (s, 1H, H1), 4.12–4.06 (m, 2H, CH), 4.06–4.01 (m, 1H, CH), 3.96 (dd, J = 3.3, 1.3 Hz, 1H, H4′), 3.77 (dd, J = 10.2, 3.8 Hz, 1H, H2′), 3.72 (s, 4H, H5′, CH2), 3.69–3.64 (m, 1H, CH), 3.34 (s, 3H, OCH3), 1.48 (s, 3H, CH3), 1.36–1.25 (m, 6H, 2xCH3). 13C{1H} NMR (126 MHz, CD3OD) δ 109.00 (CMe2), 100.46 (C1′), 97.95 (C1), 81.24 (CH), 77.11 (CH), 75.97 (CH), 70.21 (CH), 69.99 (CH), 69.76 (C4′), 69.06 (CH), 64.80 (CH), 61.03 (CH2), 53.81 (OMe), 27.02 (CH3), 25.15 (CH3), 16.67 (CH3). HRMS (ESI) m/z: [M + Na]+ calcd for C16H28O10Na 403.1574; found 403.1567.
6-O-β-d-Glucopyranosyl-1,2:3,4-O-diisopropylidene-α-d-galactopyranose (36)
Obtained from the deprotection of compound 21β (20 mg) using the general procedure for desulfurization eluting from silica gel with 10% MeOH in CH2Cl2 to give 6.4 mg for a 67% yield.
Obtained from the deprotection of compound 26β (12 mg) using the general procedure for desulfurization eluting from silica gel with 10% MeOH in CH2Cl2 to give 5.1 mg for an 83% yield.
602 mg Scale Desulfurization with Raney Nickel
To a suspension of 603 mg of 21β in 2 mL of ethanol while stirring was added 1 g of Raney nickel that had been previously washed with ethanol. The reaction mixture was stirred for 36 h under 1 atm of hydrogen at room temperature. The reaction mixture was diluted with 50 mL of ethanol and filtered through a pad of celite; the filter cake was washed with additional 50 mL of ethanol. The combined filtrate was dried over Na2SO4, filtered through cotton, and concentrated under reduced pressure to afford a crude residue. Two hundred and fifty-two milligrams were obtained after silica gel column chromatography eluting with 10% MeOH in CH2Cl2 for an 88% yield. The spectral data is consistent with the reported literature.55,56
Colorless syrup. [α]D20 = −41.8 (c 0.3, CH3CN) 1H NMR (500 MHz, CD3CN) δ 5.44 (d, J = 5.1 Hz, 1H, H1), 4.58 (dd, J = 8.0, 2.5 Hz, 1H, H3), 4.31 (dd, J = 5.1, 2.4 Hz, 1H, H2), 4.28–4.22 (m, 2H, H5, H1′), 3.97–3.88 (m, 2H, CH26a, CH26′a), 3.71 (d, J = 9.9 Hz 1H, CH26b), 3.63–3.51 (m, 3H, H4′, H5′, CH26′), 3.43 (br, 1H, CH26′b), 3.35 (s, 1H, OH), 3.30 (s, 1H, OH), 3.27–3.23 (m, 1H, H3′), 3.20 (s, 1H, OH), 3.07 (app t, J = 8.4 Hz, 1H, H2′), 2.80 (s, 1H, OH), 1.47 (s, 3H, CH3), 1.35 (s, 3H, CH3), 1.28 (s, 6H, CH3). 13C{1H} NMR (126 MHz, CD3CN) δ 108.9 (CMe2), 108.4 (CMe2), 103.4 (CH), 96.3 (CH), 76.7 (CH), 76.3 (CH), 73.7 (CH), 71.0 (CH), 70.6 (CH), 70.5 (CH), 68.4 (CH2), 67.2 (CH), 61.9 (CH2), 25.4 (CH3), 25.3 (CH3), 24.2 (CH3), 23.7 (CH3). HRMS (ESI) m/z: [M + Na]+ calcd for C18H30O11Na 445.1680; found 445.1680.
Adamantyl β-d-Glucopyranoside (37)
Obtained from the deprotection of compound 22β (25 mg) using the general procedure for desulfurization eluting from silica gel with 10% MeOH in CH2Cl2 to give 4.7 mg for a 46% yield. The spectral data is consistent with the literature.57
Colorless syrup. [α]D20 = −12.4 (c 0.3, CH3OH) 1H NMR (500 MHz, CD3OD) δ 4.53 (d, J = 7.8 Hz, 1H, H1), 3.79 (dd, J = 11.9, 2.3 Hz, 1H, H6a), 3.61 (dd, J = 11.9, 5.3 Hz, 1H, H6b), 3.33 (t, J = 8.7 Hz, 1H, H3), 3.24 (d, J = 8.2 Hz, 1H, H4), 3.26–3.19 (m, 1H), 3.09 (dd, J = 9.2, 7.8 Hz, 1H, H2), 2.11 (q, J = 3.3 Hz, 3H, Ada), 1.93–1.87 (m, 3H, Ada), 1.81–1.76 (m, 3H, Ada), 1.69–1.60 (m, 6H, Ada). 13C{1H} NMR (126 MHz, CD3OD) δ 95.8 (C1), 76.8 (CH), 76.2 (CH), 74.7 (CH), 73.7 (CH), 70.4 (CH), 61.5 (CH2), 42.2 (Ada), 36.0 (Ada), 30.8 (Ada). HRMS (ESI) m/z: [M + Na]+ calcd for C16H26O6Na 337.1622; found 337.1612.
Methyl 4-O-α-d-Glucopyranosyl-2,3-O-isopropylidene-α-l-rhamnopyranoside (38)
Obtained from the deprotection of compound 24α (23.5 mg) using the general procedure for desulfurization eluting from silica gel with 10% MeOH in CH2Cl2 to give 8.6 mg for a 76% yield.
Obtained from the deprotection of compound 28α (9.7 mg) using the general procedure for desulfurization eluting from silica gel with 10% MeOH in CH2Cl2 to give 3.8 mg for an 87% yield. The spectral data is consistent with the reported literature.58
White amorphous solid. [α]D20 = +41.7 (c 0.6, CH3OH) 1H NMR (500 MHz, CD3OD) δ 4.92 (d, J = 3.8 Hz, 1H, H1), 4.80 (s, 1H, H1′), 4.15–4.06 (m, 2H, H2, H3′), 3.84 (dt, J = 10.0, 3.1 Hz, 1H, H6′), 3.77 (m, 2H, CH), 3.70 (dq, J = 9.9, 6.3 Hz, 1H, H5), 3.65–3.60 (m, 1H, H4), 3.47–3.35 (m, 2H, H3, H4′), 3.36 (s, 3H, OCH3), 3.35–3.32 (m, 1H, CH) 1.51 (s, 3H, CH3), 1.33 (d, J = 6.4 Hz, 3H, CH3), 1.31 (s, 3H, CH3). 13C{1H} NMR (126 MHz, CD3OD) δ 110.3 (CMe2), 101.5 (C1′), 99.3 (C1), 82.54 (CH), 78.5 (CH), 77.3 (CH), 74.9 (CH), 73.7 (CH), 73.3 (CH), 71.3 (CH), 66.2 (CH), 62.1 (CH3), 55.1 (CH2), 28.4 (CH3), 26.5 (CH3), 17.9 (CH3). HRMS (ESI) m/z: [M + Na]+ calcd for C16H28O10Na 403.1575; found 403.1572.
Adamantyl α-d-Glucopyranoside (39)
Obtained from the deprotection of compound 27α (16.7 mg) using the general procedure for desulfurization eluting from silica gel with 10% MeOH in CH2Cl2 to give 4.3 mg for a 64% yield.
White amorphous solid. [α]D20 = +19.1 (c 0.5, CH3CN) 1H NMR (500 MHz, CD3OD) δ 5.17 (d, J = 3.9 Hz, 1H, H1), 3.75–3.70 (m, 2H, H4, CH26), 3.65 (dd, J = 12.3, 5.6 Hz, 1H, CH26), 3.62–3.58 (m, 1H, H5), 3.30 (d, J = 3.9 Hz, 1H, H2), 3.26 (br, 1H, H3), 2.15–2.08 (m, 3H, CHada), 1.94–1.84 (m, 3H, Ada), 1.85–1.78 (m, 3H, Ada), 1.65 (dd, J = 3.8 Hz, 6H, Ada). 13C{1H} NMR (126 MHz, CD3OD) δ 91.6 (C1), 74.0 (CH), 73.9 (CH), 72.1 (CH), 71.8 (CH), 70.7 (CH), 61.4 (CH2), 42.2 (Ada), 36.1 (Ada), 30.8 (Ada). HRMS (ESI) m/z: [M + Na]+ calcd for C16H26O6Na 337.1622; found 337.1608.
Acknowledgments
The authors thank the NIH (GM62160) for the support of this work. N.O.-M. thanks Fulbright Colombia with Colombia Cientifica for a fellowship.
Data Availability Statement
The data underlying this study are available in the published article and its online supplementary material.
Supporting Information Available
The Supporting Information is available free of charge at https://pubs.acs.org/doi/10.1021/acs.joc.2c02889.
Copies of 1H and 13C NMR spectra for all compounds (PDF)
Author Contributions
∥ K.U. and N.O.-M. contributed equally to this work.
The authors declare no competing financial interest.
Supplementary Material
References
- Pirrone M. G.; Gysin M.; Haldimann K.; Hobbie S. N.; Vasella A.; Crich D. Predictive Analysis of the Side Chain Conformation of the Higher Carbon Sugars: Application to the Preorganization of the Aminoglycoside Ring 1 Side Chain for Binding to the Bacterial Ribosomal Decoding A Site. J. Org. Chem. 2020, 85, 16043–16059. 10.1021/acs.joc.0c01836. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Upadhyaya K.; Bagul R.; Crich D. Influence of Configuration at the 4- and 6-positions on the Conformation and Anomeric Reactivity and Selectivity of 7-Deoxyheptopyranosyl Donors: Discovery of a Highly Equatorially Selective l-glycero-D-gluco-Heptopyranosyl Donor. J. Org. Chem. 2021, 86, 12199–12225. 10.1021/acs.joc.1c01535. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Siyabalapitiya Arachchige S.; Crich D. Side Chain Conformation and Its Influence on Glycosylation Selectivity in Hexo- and Higher Carbon Furanosides. J. Org. Chem. 2022, 87, 316–339. 10.1021/acs.joc.1c02374. [DOI] [PMC free article] [PubMed] [Google Scholar]
- The three staggered conformations of the exocyclic bond are defined as gauche, gauche (gg), gauche, trans (gt), and trans, gauche (tg), where the first descriptor refers to the relationship of O6 with O5 and the second to that between O6 and C4.
- Bock K.; Duus J. Ø. A Conformational Study of Hydroxymethyl Groups in Carbohydrates Investigated by 1H NMR spectroscopy. J. Carbohydr. Chem. 1994, 13, 513–543. 10.1080/07328309408011662. [DOI] [Google Scholar]
- Grindley T. B.Structure and Conformation of Carbohydrates. In Glycoscience: Chemistry and Chemical Biology I-III; Fraser-Reid B. O.; Tatsuta K.; Thiem J., Eds.; Springer, 2001; Vol. 1, pp 3–51. [Google Scholar]
- Rao V. S. R.; Qasba P. K.; Balaji P. V.; Chandrasekaran R.. Conformation of Carbohydrates; CRC Press, 1998. [Google Scholar]
- McMahon C. M.; Isabella C. R.; Windsor I. W.; Kosma P.; Raines R. T.; Kiessling L. L. Stereoelectronic Effects Impact Glycan Recognition. J. Am. Chem. Soc. 2020, 142, 2386–2395. 10.1021/jacs.9b11699. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Jeffrey G. A.; Kim H. S. Conformations of the Alditols. Carbohydr. Res. 1970, 14, 207–216. 10.1016/S0008-6215(00)80488-7. [DOI] [Google Scholar]
- Marchessault R. H.; Perez S. Conformations of the Hydroxymethyl Group in Crystalline Aldohexopyranoses. Biopolymers 1979, 18, 2369–2374. 10.1002/bip.1979.360180925. [DOI] [Google Scholar]
- Andreana P. R.; Crich D. Guidelines for O-Glycoside Formation from First Principles. ACS Cent. Sci. 2021, 7, 1454–1462. 10.1021/acscentsci.1c00594. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Demchenko A. V.Handbook of Chemical Glycosylation: Advances in Stereoselectivity and Therapeutic Relevance; Wiley-VCH, 2008. [Google Scholar]
- Adero P. O.; Amarasekara H.; Wen P.; Bohé L.; Crich D. The Experimental Evidence in Support of Glycosylation Mechanisms at the SN1–SN2 Interface. Chem. Rev. 2018, 118, 8242–8284. 10.1021/acs.chemrev.8b00083. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Jensen H. H.; Nordstrøm L. U.; Bols M. The Disarming Effect of the 4,6-Acetal Group on Glycoside Reactivity: Torsional or Electronic?. J. Am. Chem. Soc. 2004, 126, 9205–9213. 10.1021/ja047578j. [DOI] [PubMed] [Google Scholar]
- Moumé-Pymbock M.; Furukawa T.; Mondal S.; Crich D. Probing the Influence of a 4,6-O-Acetal on the Reactivity of Galactopyranosyl Donors: Verification of the Disarming Influence of the trans–gauche Conformation of C5–C6 Bonds. J. Am. Chem. Soc. 2013, 135, 14249–14255. 10.1021/ja405588x. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Dharuman S.; Crich D. Determination of the Influence of the Side-Chain Conformation on Glycosylation Selectivity Using Conformationally Restricted Donors. Chem. – Eur. J. 2016, 22, 4535–4542. 10.1002/chem.201505019. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Lin Y. A.; Chalker J. M.; Davis B. G. Olefin Cross-Metathesis on Proteins: Investigation of Allylic Chalcogen Effects and Guiding Principles in Metathesis Partner Selection. J. Am. Chem. Soc. 2010, 132, 16805–16811. 10.1021/ja104994d. [DOI] [PubMed] [Google Scholar]
- Ali A.; Vishwakarma R. A. Total Synthesis of The Fully Lipidated Glycosylphosphatidylinositol (GPI) Anchor of Malarial Parasite Plasmodium falciparum. Tetrahedron 2010, 66, 4357–4369. 10.1016/j.tet.2010.04.014. [DOI] [Google Scholar]
- Bellucci G.; Chiappe C.; D′Andrea F. Diastereoselective Bromination of Allyl Glycosides Using Tetrabutylammonium Tribromide. Tetrahedron: Asymmetry 1995, 6, 221–230. 10.1016/0957-4166(94)00378-O. [DOI] [Google Scholar]
- Dess D. B.; Martin J. C. A Useful 12-I-5 Triacetoxyperiodinane (the Dess-Martin Periodinane) for the Selective Oxidation of Primary or Secondary Alcohols and a Variety of Related 12-I-5 Species. J. Am. Chem. Soc. 1991, 113, 7277–7287. 10.1021/ja00019a027. [DOI] [Google Scholar]
- Peri F.; Deutman A.; Ferla B. L.; Nicotra F. Solution and Solid-Phase Chemoselective Synthesis of (1-6)-Amino(methoxy) Di- and Trisaccharide Analogues. Chem. Commun. 2002, 1504–1505. 10.1039/b203605c. [DOI] [PubMed] [Google Scholar]
- Kocienski P. J. A Formal Synthesis of 18-O-Methyl Mycalamide B. Synthesis 1999, 1999, 2087–2095. 10.1055/s-1999-3633. [DOI] [Google Scholar]
- Consistent with the design hypothesis in which the SPh moiety serves as a dummy sp3 carbon group, we assign configurations as d-glycero-d-galacto, l-glycero-d-galacto, l-glycero-d-gluco, and d-glycero-d-gluco as in the heptose series. Alternatively, these compounds can be named following Cahn-Ingold-Prelog rules to assign the side chain configuration.
- Nicolaou K. C.; Hummel C. W.; Bockovich N. J.; Wong C.-H. Stereocontrolled Synthesis of Sialyl LeX, The Oligosaccharide Binding Ligand to ELAM-1 (sialyl =N-acetylneuramin). J. Chem. Soc., Chem. Commun. 1991, 870–872. 10.1039/c39910000870. [DOI] [Google Scholar]
- Nicolaou K. C.; Mitchell H. J.; Fylaktakidou K. C.; Rodriguez R. M.; Suzuki H. Total Synthesis of Everninomicin 13,384-1 - Part 2: Synthesis of the FGHA2 Fragment. Eur. J. Chem. 2000, 6, 3116–3148. . [DOI] [PubMed] [Google Scholar]
- Schmidt R. R.; Michel J.; Roos M. Glycosylimidate, 12 Direkte Synthese von O-α- und O-β-Glycosyl-imidaten. Liebigs Ann. Chem. 1984, 1984, 1343–1357. 10.1002/jlac.198419840710. [DOI] [Google Scholar]
- Mori M.; Ito Y.; Ogawa T. Total Synthesis of the Mollu-Series Glycosyl Ceramides α-d-Manp-(1→3)-β-d-Manp-(1→4)-β-d-Glcp-(1→1)-Cer and α-d-Manp-(1→3)-[β-d-Xylp-(1→2)]-β-d-Manp-(1→4)-β-d-Glcp-(1→1)-Cer. Carbohydr. Res. 1990, 195, 199–224. 10.1016/0008-6215(90)84167-S. [DOI] [PubMed] [Google Scholar]
- Ban L.; Mrksich M. On-Chip Synthesis and Label-Free Assays of Oligosaccharide Arrays. Angew. Chem., Int. Ed. 2008, 47, 3396–3399. 10.1002/anie.200704998. [DOI] [PubMed] [Google Scholar]
- Sitz T.; Domey H.; Fischer J.; Rohn S. An Alternative Approach for the Synthesis of Sulfoquinovosyl Diacylglycerol. Molecules 2021, 26, 4275. 10.3390/molecules26144275. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Schmidt R. R.; W K. Anomeric-Oxygen Activation for Glycoside Synthesis: The Trichloroacetimidate Method. Adv. Carbohydr. Chem. Biochem. 1994, 50, 21–123. 10.1016/s0065-2318(08)60150-x. [DOI] [PubMed] [Google Scholar]
- Boutet J.; Mulard L. A. Synthesis of Two Tetra- and Four Pentasaccharide Fragments of Shigella flexneri Serotypes 3a and X O-Antigens from a Common Tetrasaccharide Intermediate. Eur. J. Org. Chem. 2008, 2008, 5526–5542. 10.1002/ejoc.200800693. [DOI] [Google Scholar]
- Meng X.; Pan Y.; Liu T.; Luo C.; Man S.; Zhang Y.; Zhang Y. Synthesis of Novel Diosgenyl Saponin Analogs and Evaluation Effects of Rhamnose Moeity on their Cytotoxic Activity. Carbohydr. Res. 2021, 506, 108359–108365. 10.1016/j.carres.2021.108359. [DOI] [PubMed] [Google Scholar]
- Schmidt R. R.; Behrendt M.; Toepfer A. Nitriles as Solvents in Glycosylation Reactions: Highly Selective β-Glycoside Synthesis. Synlett 1990, 1990, 694–696. 10.1055/s-1990-21214. [DOI] [Google Scholar]
- Imamura A.; Ando H.; Ishida H.; Kiso M. DTBS Effect: The Unique Sterically Driven Director for α-Galactosylation. Heterocycles 2008, 76, 883–908. 10.3987/REV-08-SR(N)4. [DOI] [Google Scholar]
- Van der Vorm S.; Hansen T.; Overkleeft H. S.; Van der Marel G. A.; Codée J. D. C. The Influence of Acceptor Nucleophilicity on the Glycosylation Reaction Mechanism. Chem. Sci. 2017, 8, 1867–1875. 10.1039/C6SC04638J. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Chang C.-W.; Lin M.-H.; Chan C.-K.; Su K.-Y.; Wu C.-H.; Lo W.-C.; Lam S.; Cheng Y.-T.; Liao P.-H.; Wong C.-H.; Wang C. Automated Quantification of Hydroxyl Reactivities: Prediction of Glycosylation Reactions. Angew. Chem., Int. Ed. 2021, 60, 12413–12423. 10.1002/anie.202013909. [DOI] [PubMed] [Google Scholar]
- Upadhyaya K.; Bagul R.; Crich D. Influence of Configuration at the 4- and 6-positions on the Conformation and Anomeric Reactivity and Selectivity of 7-Deoxyheptopyranosyl Donors: Discovery of a Highly equatorially selective L-glycero-D-gluco-Heptopyranosyl Donor. J. Org. Chem. 2021, 86, 12199–12225. 10.1021/acs.joc.1c01535. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Crich D.; Li L. Stereocontrolled Synthesis of D- and L-β-Rhamnopyranosides with 4-O-6-S-α-Cyanobenzylidene-Protected 6-Thiorhamnopyranosyl Thioglycosides. J. Org. Chem. 2009, 74, 773–781. 10.1021/jo8022439. [DOI] [PMC free article] [PubMed] [Google Scholar]
- McMillan T. F.; Crich D. Influence of 3-Thio Substituents on Benzylidene-Directed Mannosylation. Isolation of a Bridged Pyridinium Ion and Effects of 3-O-Picolyl and 3-S-Picolyl Esters. Eur. J. Org. Chem. 2022, 2022, 128–135. 10.1002/ejoc.202200320. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Somogyi L. Cyclic monothioacetals of aldoses: Synthesis and Desulphuration of 2-Poly-Acetoxyalkyl-1,3-oxathiolan-5-ones and -3,1-Benzoxathian-4-ones. Carbohydr. Res. 1987, 166, 166–169. 10.1016/0008-6215(87)80054-X. [DOI] [Google Scholar]
- Kim J.-H.; Yang H.; Park J.; Boons G.-J. A General Strategy for Stereoselective Glycosylations. J. Am. Chem. Soc. 2005, 127, 12090–12097. 10.1021/ja052548h. [DOI] [PubMed] [Google Scholar]
- Matwiejuk M.; Thiem J. New Method for Regioselective Glycosylation Employing Saccharide Oxyanions. Eur. J. Org. Chem. 2011, 2011, 5860–5878. 10.1002/ejoc.201100861. [DOI] [Google Scholar]
- Garegg P. J.; Hultberg H.; Wallin S. A Novel, Reductive Ring-Opening of Carbohydrates Benzylidene Acetals. Carbohydr. Res. 1982, 108, 97–101. 10.1016/S0008-6215(00)81894-7. [DOI] [Google Scholar]
- Garegg P. J.Regioselective Cleavage of O-Benzylidene Acetals to Benzyl Ethers. In Preparative Carbohydrate Chemistry; Hannessian S., Ed.; Marcel Dekker, 1996; pp 53–67. [Google Scholar]
- Chan J.; Lu A.; Bennet A. J. Turnover Is Rate-Limited by Deglycosylation for Micromonospora viridifaciens Sialidase-Catalyzed Hydrolyses: Conformational Implications for the Michaelis Complex. J. Am. Chem. Soc. 2011, 133, 2989–2997. 10.1021/ja109199p. [DOI] [PubMed] [Google Scholar]
- Frihed T. G.; Heuckendorff M.; Pedersen C. M.; Bols M. Easy Access to L-Mannosides and L-Galactosides by Using C-H Activation of the Corresponding 6-Deoxysugars. Angew. Chem., Int. Ed. 2012, 51, 12285–12288. 10.1002/anie.201206880. [DOI] [PubMed] [Google Scholar]
- Lipták A.; Imre J.; Nánási P. Preparation of Carbohydrate Isopropylidene Derivatives with 2,2-Dimethoxypropane in the Presence of Toluene-p-sulphonic Acid. Carbohydr. Res. 1981, 92, 154–156. 10.1016/S0008-6215(00)85991-1. [DOI] [Google Scholar]
- Nakahara Y.; Ogawa T. A Highly Stereocontrolled Synthesis of the Propyl Glycoside of a Decagalacturonic Acid, a Model Compound for the Endogenous Phytoalexin Elicitor-Active Oligogalacturonic Acids. Carbohydr. Res. 1989, 194, 95–114. 10.1016/0008-6215(89)85010-4. [DOI] [Google Scholar]
- Yamazaki T.; Sugawara F.; Ohta K.; Masaki K.; Nakayama K.; Sakaguchi K.; Sato N.; Sahara H.; Fujita T.. Sulforhamnosyl Acyglycerol Derivatives and Use Thereof as Medicaments. US Patent US006759522B2, 2004.
- Glaudemans C. P. J.; Zissis E.; Jolley M. E. Binding Studies on a Mouse-Myeloma Immunoglobulin a Having Specificity for β-d-(1→6)-Linked D-Galactopyranosyl Residues. Carbohydr. Res. 1975, 40, 129–135. 10.1016/S0008-6215(00)82675-0. [DOI] [PubMed] [Google Scholar]
- Borbás A.; Jánossy L.; Lipták A. Scope and Limitation of the Application of the (Methoxydimethyl)methyl Group in the Synthesis of 2′-O-, 6′-O- and 2′,6′-Di-O-(α-l-arabinofuranosyl)-β-d-galactopyranosyl-(1→6)-d-galactoses. Carbohydr. Res. 1999, 318, 98–109. 10.1016/S0008-6215(99)00094-4. [DOI] [PubMed] [Google Scholar]
- Dahmén J.; Frejd T.; Lave T.; Lindh F.; Magnusson G.; Noori G.; Pålsson K. 4-O-α-d-Galactopyranosyl-d-galactose: Efficient Synthetic Routes from “Polygalacturonic Acid”. Carbohydr. Res. 1983, 113, 219–224. 10.1016/0008-6215(83)88237-8. [DOI] [Google Scholar]
- Takeo K.; Okushio K.; Fukuyama K.; Kuge T. Synthesis of Cellobiose, Cellotriose, Cellotetraose, and Lactose. Carbohydr. Res. 1983, 121, 163–173. 10.1016/0008-6215(83)84014-2. [DOI] [Google Scholar]
- Farkas E.; Schmidt U.; Thiem J.; Kowalczyk J.; Kunz M.; Vogel M. Regioselective Synthesis of Galactosylated Tri- and Tetrasaccharides by Use of β-Galactosidase from Bacillus circulans. Synthesis 2003, 5, 699–706. 10.1055/s-2003-38075. [DOI] [Google Scholar]
- Parlato M. C.; Kamat M. N.; Wang H.; Stine K. J.; Demchenko A. Application of Glycosyl Thioimidates in Solid-Phase Oligosaccharide Synthesis. J. Org. Chem. 2008, 73, 1716–1725. 10.1021/jo701902f. [DOI] [PubMed] [Google Scholar]
- Hanessian S.; Bacquet C.; Lehong N. Chemistry of the Glycosidic Linkage. Exceptionally Fast and Efficient Formation of Glycosides by Remote Activation. Carbohydr. Res. 1980, 80, C17–C22. 10.1016/S0008-6215(00)84882-X. [DOI] [Google Scholar]
- Crich D.; Cai F. Stereocontrolled Glycoside and Glycosyl Ester Synthesis. Neighboring Group Participation and Hydrogenolysis of 3-(2′-Benzyloxyphenyl)-3,3-dimethylpropanoates. Org. Lett. 2007, 9, 1613–1615. 10.1021/ol070449y. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Salamone S.; Guerreiro C.; Cambon E.; Hargreaves J. M.; Tarrat N.; Remaud-Simeon M.; Andre I.; Mulard L. A. Investigation on the Synthesis of Shigella flexneri Specific Oligosaccharides Using Disaccharides as Potential Transglucosylase Acceptor Substrates. J. Org. Chem. 2015, 80, 11237–11257. 10.1021/acs.joc.5b01407. [DOI] [PubMed] [Google Scholar]
Associated Data
This section collects any data citations, data availability statements, or supplementary materials included in this article.
Supplementary Materials
Data Availability Statement
The data underlying this study are available in the published article and its online supplementary material.